

Abstract
Dopamine (DA) transporter (DAT) is a major target for psychostimulant drugs of abuse such as cocaine that competitively binds to DAT, inhibits DA reuptake, and consequently increases synaptic DA levels. In addition to the central binding site inside DAT, the available experimental evidence suggests the existence of alternative binding sites on DAT, but detection and characterization of these sites are challenging by experiments alone. Here we integrate multiple computational approaches to probe the potential binding sites on the wild-type Drosophila melanogaster DAT and identify a new allosteric site that displays high-affinity for cocaine. This site is located on the surface of DAT, and binding of cocaine is primarily dominated by interactions with hydrophobic residues surrounding the site. We show that cocaine binding to this new site allosterically reduces the binding of DA/cocaine to the central binding pocket, and simultaneous binding of two cocaine molecules to a single DAT seems infeasible. Furthermore, we find that binding of cocaine to this site stabilizes the conformation of DAT but alters the conformational population and thereby reduces the accessibility by DA, providing molecular insights into the inhibitory mechanism of cocaine. In addition, our results indicate that the conformations induced by cocaine binding to this site may be relevant to the oligomerization of DAT, highlighting a potential role of this new site in modulating the function of DAT.
Introduction
Dopamine (DA) transporter (DAT) is a member of the neurotransmitter:sodium symporter (NSS) transporters, which plays an important role in regulating neurotransmission by sodium-driven reuptake of neurotransmitters from the synapse into the presynaptic neuron.1 The dysfunction of DAT and other related NSS members has been implicated in the neuropsychiatric disorders including depression, attention-deficit hyperactivity disorder, epilepsy, and Parkinson’s disease.2, 3 The NSS transporters are also the primary targets for many psychostimulants such as cocaine and amphetamines.4 The architecture of the NSS involves 12 transmembrane (TM) helices, which displays significant sequence identities (~50%‒70%) among the eukaryotic NSS members but only about 20% sequence identity in the prokaryotic amino acid transporters such as the bacterial leucine transporter (LeuT).5 Despite the varying sequence identity, DAT shares a conserved fold as the other NSS transporters, containing two helical bundles, TMs 1‒5 and TMs 6‒10.5 DAT works through the proposed alternating access mechanism, in which the transporter switches between the outward-open and inward-open conformational states, to alternate the substrate access between the extracellular and intracellular sides of the membrane.6 In addition, the alternating accessibility may be related to the switching between alternative conformations of the two helical bundles (TMs 1‒5 and TMs 6‒10).6 The transition from the outward-open to the inward-open state may also involve the outward-occluded state.7
DAT efficiently removes neurotransmitter DA from the synaptic space.3 The widely abused psychostimulant cocaine is a competitive inhibitor of DAT and enhances extracellular DA concentrations by locking the transporter in an inactive conformation.8 One of the fundamental questions regarding the function of DAT is the identification of the binding sites for the substrate and the inhibitor as well. The X-ray crystal structures of the Drosophila melanogaster DAT (dDAT) in complex with DA and cocaine in an outward-open conformation show that DA and cocaine bind to the central binding site, surrounded by TMs 1, 3, 6, and 8.8 This observation confirms that the binding site for DA and cocaine in DAT overlaps, a finding obtained based on the structural model of bacterial homologue LeuT.9 In addition to the primary binding site, a secondary binding site for leucine located in the extracellular vestibule (~11 Å above the primary binding site) has been suggested for LeuT.10 This secondary binding site overlaps with the binding site of tricyclic antidepressant clomipramine in the cocrystal structure of LeuT where the substrate leucine occupies the primary binding site.11 In addition to clomipramine, binding of a tryptophan,12 a sertraline,13 and an octylglucoside detergent molecule14 in the secondary site of LeuT was also observed in the crystal structures. Like DAT and LeuT, the serotonin transporter (hSERT), and multi-hydrophobic amino acid (MhsT) transporters are also NSS homologues. The binding of S-citalopram molecules in both the primary and secondary sites of hSERT has been observed in the crystal structure,15 and the simultaneous binding of two tryptophan substrates in MhsT were also indicated.16 Although these evidence support the existence of a secondary substrate binding site in NSSs, further experiments using a variety of binding measurements,17 as well as solid-state nuclear magnetic resonance (NMR) technique18 suggested that only one leucine binds to the central/primary site of LeuT. This apparent controversy could be ascribed to the different experimental conditions—the binding of leucine in the secondary site can be impaired by the protein preparation procedures used for crystallography and for some functional studies.19, 20
The X-ray crystal structures of DA- and cocaine-bound dDAT suggest the presence of a single, high-affinity substrate/inhibitor binding site in dDAT.8 However, the existence of a secondary binding site in DAT has also been proposed. Using the X-ray crystal structure of the bacteria LeuT21 as a template, a human DAT structure (23% sequence identity) was homolog-modeled, and a second DA binding site surrounded by residues from TMs 1, 3, 10, and extracellular loops (ELs) 2 and 4 was identified by performing steered molecular dynamics (MD) simulations.22 In another computational study of human DAT, a model based on the crystal structure of dDAT (52% sequence identity)8 indicated that the DA transiently binds to the secondary site from the extracellular medium before entering into the primary binding site, but cocaine and other inhibitors were precluded from this secondary binding site.23 Interestingly, experimental study of human DAT mutants identified a phenylalanine-to-alanine mutant in TM3 F154A (F154 corresponds to F122 in dDAT) that significantly lowers cocaine affinity, implying that cocaine may not bind to the primary site as shown in the crystal structure of dDAT.24, 25 Another experimental study using mouse DAT indicated that F155Y (F155 corresponds to F123 of the dDAT) showd different effects on the binding of cocaine and cocaine analogs.26 Together, these mutation studies seem to support the presence of a binding site for cocaine in TM3. However, the exact location remains elusive. Moreover, the study of binding of bivalent compounds to DAT suggested the existence of multiple substrate binding sites in a single DAT.27 Identification of these potential binding sites is challenging by experiments alone considering different methods and conditions applied to characterize proteins.20 In addition, the NSS transporters have been shown to oligomerize at the plasma membrane,28, 29, 30 which may also affect the recognition of substrates/inhibitors by DAT.
In this study, we investigated the potential binding sites for cocaine on dDAT by performing molecular docking, MD simulations, and binding energy calculations. In addition to the primary binding site, our random docking results show different binding patterns between cocaine and DA. MD simulations of cocaine-bound dDAT complexes obtained from docking provide evidence for the binding of cocaine in a site formed by TMs 3, 9, 10, and 12 of dDAT. The predicted binding affinity of cocaine in this site is even higher than that in the central binding site, and the binding of cocaine in this site allosterically reduces the binding affinity of cocaine in the central site. Our results show that the dDAT mutant F122A lowers the binding affinity of cocaine in both binding sites, in agreement with previous experimental study.24 Contrary to the increased stability of dDAT when accommodating two DA molecules in the central and extracellular site simultaneously, the stability of dDAT decreases when two cocaine molecules bind to the central and the new site respectively, suggesting that the binding of cocaine to this new site significantly alters the conformation of dDAT. As a consequence, dDAT becomes less favorable for the binding of DA.
Methods
Modeling of the Wild-Type dDAT
The X-ray structure of DA-bound dDAT (PDB ID: 4XP1)8 was used to construct the wild-type dDAT structure. Mutations in the crystal structure were reverted to the wild-type amino acids. The missing residues Ser162‒Val202 were not determined because of high flexibility, and modeled using MODELLER 9.23.31 After model building, the region Ser162‒Val202 in the best model was subjected to loop refinement by using the newer DOPE-based loop modeling protocol.32 The N-terminal (residues 1‒24) and C-terminal (residues 601‒645) were not added in the above modeling procedure, but were capped by an acetyl and amide group, respectively. The two sodium ions (Na+) and one chloride (Cl−) ion found in the crystal structure were kept in the final modeled structure. The best model after loop refinement was selected for subsequent molecular docking and simulations. The position of the DA in the crystal structure was used as the binding site of DA in the central binding pocket. The position of the cocaine in the model structure was determined by the alignment of cocaine-bound crystal structure (PDB ID: 4XP4) with the DA-bound crystal structure (PDB ID: 4XP1). These two structures are almost identical, with the root-mean-square deviation (RMSD) of heavy atoms being about 0.25 Å. The structures of DA and cocaine are positively charged in the quaternary ammonium motif.33
Molecular Docking
Molecular docking was carried out using Autodock 4.2 program.34 Instead of using the default partial charges for protein and ligand, the partial charges from the CHARMM36m force field parameters35 were applied to the structure of dDAT. The partial charges for the cocaine and DA molecules were derived using the RESP method36 (Fig. S1). In each docking, the structure of dDAT was treated as rigid but the ligand was considered as flexible. Random docking was performed, that is, the grid space used to define the searching space of a protein includes the whole surface of the protein, and the ligand (cocaine or DA) randomly searches the potential binding sites available to the protein. A longer search with 25 million evaluations was used to provide adequate searching. Lamarckian genetic algorithm37 was applied to search 100 ligand conformations in each docking experiment. Our docking results show that the above docking protocol is capable of predicting the binding conformation of DA and cocaine in the central site as found in the crystal structures (Fig. 1). The stability of the docked conformations is further evaluated by performing MD simulations.
Fig. 1.
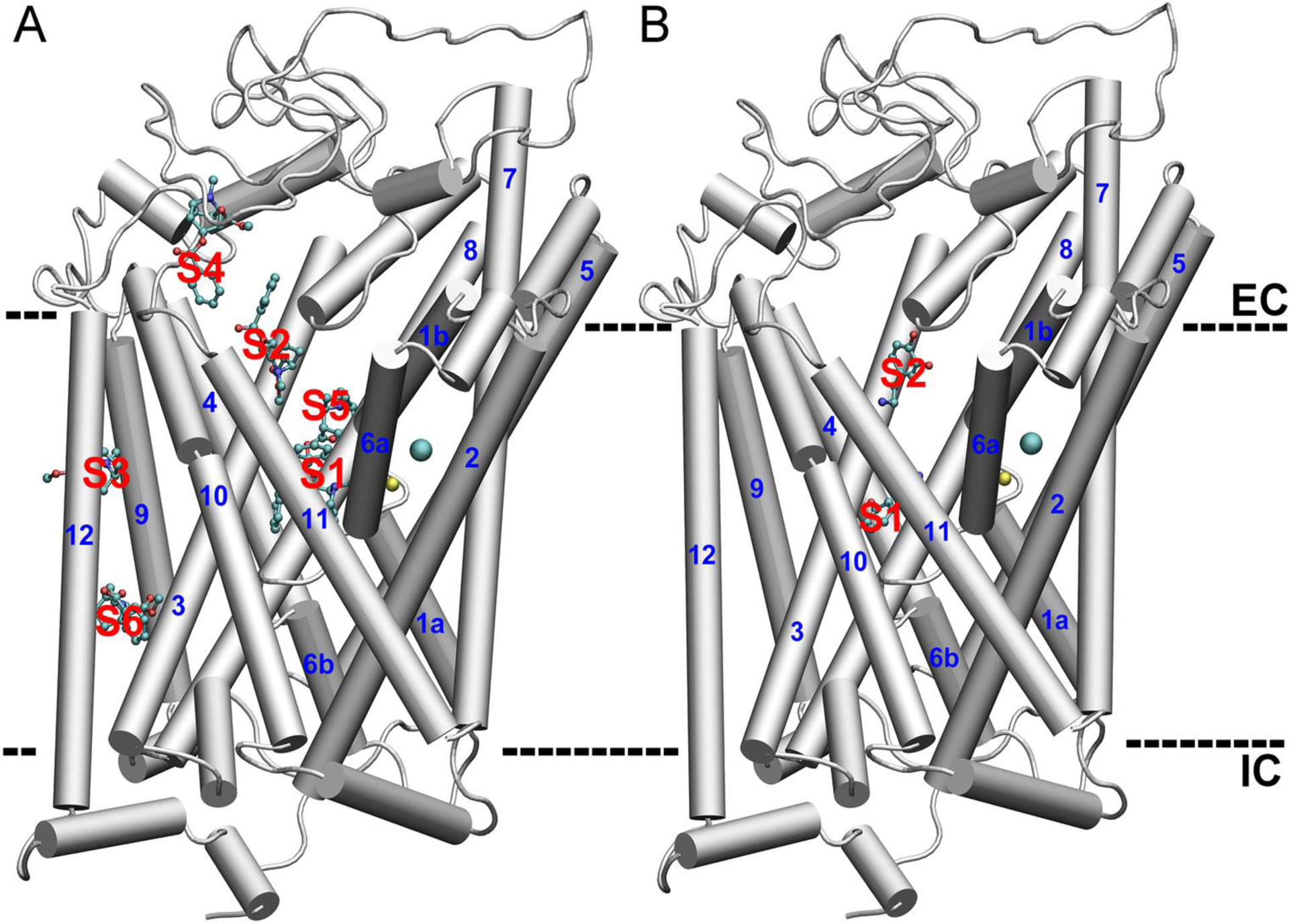
The representative docked conformations of cocaine (A) and DA (B) on the structure of dDAT. The TMs are in cartoon representation and the cocaine and DA molecules were in CPK representations. The bound Na+ and Cl− are shown in yellow and cyan spheres, respectively. S1 to S6 refers to different binding pockets predicted by random docking. EC and IC indicate the extracellular and intracellular space, respectively.
MD Simulations
To model the ligand-bound dDAT in membrane environment, the cholesteryl hemisuccinate molecule found at the interface among TMs 2, 7, and 11 of the crystal structure was modeled by a cholesterol (CHOL) molecule, together with another CHOL molecule (among TMs 1a, 5, and 7) and water molecules found in the crystal structure were included in the model dDAT structure. The binding conformation of cocaine and DA in the central site of dDAT was taken from the crystal structure, and other binding conformations were obtained from the docked conformations with the best predicted binding affinity using Autodock 4.2.34 The structure of dDAT was represented with the CHARMM36m force field parameters,35 and the CHARMM General Force Field (CGenFF)38 was used to generate force field parameters for the ligands, with the partial charges replaced by the RESP charges as used in docking studies. The above model of dDAT was embedded into a mixed lipid bilayer consisting of total 420 lipids (POPC:POPE:POPG:CHOL = 3:1:1:1).39 This lipid bilayer has been shown to be a suitable membrane mimic for the study of the conformational dynamics of dDAT.39 A different type of lipid bilayer consisting of 420 lipids (POPE:CHOL = 5: 1) was also used to reproduce the results of some systems. The orientation of the dDAT model relative to the membrane was defined by aligning to the dDAT structure (PDB ID: 4M48) in the Orientation of Protein in Membrane database.40, 41The CHARMM36 lipid force field parameters42 were used to represent all lipids. The system was solvated by adding a 20 Å thick water layer (TIP3P water molecules) below and above the lipid bilayer, and each system contains 0.15 M NaCl. The CHARMM-GUI web server43 was used to generate the starting structures and configuration files for MD simulations.44
MD simulations were performed using NAMD2.13 program.45 Each system was first subjected to energy minimization for 10, 000 steps, followed by six stages of equilibration with the harmonic constraints exerted on lipid head groups, protein and ligand heavy atoms. The force constant was gradually decreased from 50 to 0.5 kcal/(mol·Å2) during the equilibration procedures. The temperature was kept constant at 300 K using the Langevin dynamics method with a damping coefficient of 5 ps, the pressure was controlled at 1.0 bar by applying the Nosé-Hoover Langevin piston method.46 The use of flexible cell was enabled to allow the system to fluctuate independently in three dimensions, along with the ratio of the system cell in the x-y plane kept constant. A cutoff of 12 Å was applied for the van der Waals interactions and the long-range electrostatic interactions were treated using the PME method. The integration time step is 2 fs and the trajectory was saved every 10 ps. The production dynamics were performed at the constant temperature (300 K) and pressure (1.0 bar) without any positional constraints. The simulation time varies from 250 to 300 ns for each system after equilibration, and the last 50-ns trajectory of each system was used for analysis.
Conformational Energy and Binding Energy
The relative stability of dDAT conformations (including two bound Na+ and one Cl− in the central binding pocket) was evaluated by the conformational energy, which was calculated using the Generalized Born using Molecular Volume (GBMV) implicit solvent model implemented in the CHARMM (v44b1) program.47 The single point energy was calculated after a 200-step minimization of each conformation using the GBMV II algorithm.48, 49 Other energy terms including bonded energy, van der Waals energy, electrostatic energy, and solvation energy were also obtained with the GB implicit solvent model. The block-average method was used to estimate the mean value (500 frames) and standard deviations.
The binding free energy of cocaine/DA to dDAT was calculated using the g_mmpbsa program,50 which implements the Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) method51, 52 to predict the binding affinity for protein-ligand complex. The entropy contribution was not included in the current binding energy calculations. To be consistent, the block average method was used to estimate the mean value (1,000 frames) and standard deviations rather than applying the default bootstrap analysis method.
Results
Cocaine and DA display different docking patterns.
In the docking experiment, 100 conformers of each ligand were obtained and clustered according to their position on the structure of dDAT. The distribution of 100 docked conformations of cocaine and DA on dDAT is shown in Fig. S2. For each cluster, the docked conformer with the highest binding affinity was selected and shown in Fig. 1. Six distinct binding sites of cocaine on dDAT were predicted by docking analysis. The site 1 (S1) surrounded by TMs 1, 3, 6, and 8 corresponds to the central binding site of cocaine as observed in the cocaine-bound dDAT crystal structure.8 The S2 is exposed to the extracellular side and is about 12 Å (the distance of center of mass between two cocaine molecules in the S1 and S2, respectively, Fig. 1B) away from the S1. In S2, the docked cocaine interacts with Trp51, Gly385, Pro386, Phe471, His472, Asp475, Phe468, and Tyr547 (Fig. S3), similar to the proposed secondary substrate binding site in LeuT.10 Among the remaining binding sites, the hydrophobic binding pocket S3 is particularly interesting as the docked cocaine interacts with Phe122 in TM3, a residue relevant for the binding affinity of cocaine in a previous mutation study of DAT.24 In addition to Phe122, the cocaine in S3 also interacts with Phe118 and Tyr119 in TM3, Leu454 and Val458 in TM9, Phe470 and Leu474 in TM10, and Ile563 in TM12 (Fig. S4). Different from cocaine, results from docking of DA to dDAT show that DA mainly occupies the central and secondary binding sites, and the space between these two sites (Figs. 1B and S2B). The binding sites for DA in the extracellular loops were excluded because the protein was treated as rigid in docking, but in fact the loop regions are rather flexible during MD simulations. As a result, the extracellular loops are not expected to be able to accommodate DA stably.
Cocaine binds to a new site S3 on dDAT in an outward-open conformation.
The multiple cocaine binding sites predicted by docking need further validation by MD simulations because of the limitations in docking such as rigid receptor and fixed bond angles in the ligand, as well as the simplified scoring function based on empirical free energy of binding.53 MD simulations were performed on dDAT with the cocaine-bound conformations taken from docked complexes. Results from MD simulations show that if one cocaine initially binds to S2, S4, S5, and S6, the docked cocaine dissociates from these sites either quickly or slowly, depending on the specific site to which cocaine binds. Specifically, cocaine moves away quickly from S2 (within 2 ns) to extracellular loops, but is transiently trapped there for 350 ns before entering into solution (Fig. S5A). When a cocaine initially binds to S4, S5 or S6, it is observed to diffuse into solution after 4 ns, 30 ns, and 200 ns, respectively (Figs. S5B, S5C, and S5D). In contrast, the binding of cocaine to S3 is stable over the 300-ns MD simulations. Taken together, these results indicate that S3 could be an alternative binding site for cocaine on dDAT.
The effect of binding of cocaine to S3 on the conformation of dDAT was compared with the binding of cocaine to S1. It is found that the two Na+ ions, the Cl− ion, cocaine, and the cholesterol molecules taken from the crystal structure are stable in their binding sites over the 300-ns MD simulations. The RMSDs of the dDAT relative to the starting dDAT crystal structure are below 2.5 Å, suggesting that the cocaine-bound dDAT is stable in the lipid bilayer (Fig. 2A). The simultaneous binding of two cocaine molecules to S1 and S3 slightly enhances the conformational dynamics of the dDAT. The calculated root-mean-square fluctuations (RMSFs) reveal no significant structural dynamics in the TM domains of cocaine-bound dDAT (Fig. 2B), which display the lowest RMSF values. The large fluctuations in the EL2 provide a rationale for the truncation of segment from Ser162 to Val 202 in the crystal structure.
Fig. 2.

MD simulations of cocaine-bound dDAT complexes inserted in a lipid bilayer. COC (1) and COC(3) suggest the binding of a cocaine molecule to S1 and S3, respectively. (A) Change in the root-mean-square deviation (RMSD) of the dDAT structure relative to the crystal structure (PDB ID: 4XP1). (B) Residue-based root-mean-square fluctuation (RMSF) of the dDAT structure. (C) Distance between residues Arg52 (atom Cζ) and Asp475 (atom Cγ) of the dDAT structure. (D) Distance between gating residues Tyr124 (atom Cα) and Phe319 (atom Cα).
To further examine the influence of binding of cocaine to S3 on the conformational change of dDAT, we calculated the distance between two pairs of residues: Arg52-Glu475 and Tyr124-Phe319. The former pair is able to form a salt-bridge, and the latter aromatic pair serves as the extracellular gate. The binding of cocaine decreases the distances between residues Arg52 and Glu475 from the initial 11.5 Å to an average distance of 6.5 Å, 7.0 Å, and 4.6 Å for cocaine in S1, S3, and in both S1 and S3, respectively, suggesting that the formation of the salt-bridge is feasible only when cocaine occupies S1 and S3 on dDAT simultaneously. However, this salt-bridge seems unstable as the distance between Arg52 and Glu475 fluctuates significantly (Fig. 2C). On the other hand, the distance between the gating residues Tyr124 and Phe319 decreases slightly from 14.4 Å to an average value of 13.7 Å, 12.3 Å, and 13.8 Å for cocaine in S1, S3, and in both S1 and S3, respectively (Fig. 2D), implying that the structure of dDAT remains in the outward-open state and the binding of cocaine may not trigger the conformational transition during MD simulations.
For comparison, we also simulated ligand-free and DA-bound dDAT under the same conditions. The structure of dDAT remains stable in the simulations of one DA in S1 and two DAs in both S1 and S2 initially. In fact, it appears that the structural dynamics of dDAT decrease slightly with the increasing number of bound DA molecules (Fig. S6), suggesting an overall increase in the stability of dDAT upon DA binding. Similar to cocaine-bound systems, the DA-bound dDAT also remains in the outward-open conformation. Different from the dynamics of cocaine in S2 where cocaine escapes from S2 and dissociates from dDAT eventually (Fig. S5A), the DA in S2 moves closer to the central pocket and is trapped in a location above S1 (Fig. S7) over the 250-ns MD simulations. The time scale required for the DA to reorient and occupy the same binding site as observed in the crystal structure seems far beyond the time scale of simulations presented here. Interestingly, when one DA binds to S1, a second DA bound to S2 initially was also observed to move toward S1 and interact with the DA bound to S1 (Fig. S8, and discussion below). To test if this phenomenon is independent of simulation conditions, we inserted the DA-bound dDAT into a different lipid bilayer consisting of POPE and CHOL (POPE:CHOL = 5: 1). DA was also found to diffuse toward S1 in the absence or presence of another DA in S1 (data not shown). Collectively, these results imply that S2 may not provide a high-affinity binding site for DA, but most likely act as a pathway connecting S1 and the extracellular space.
S3 is a high-affinity binding site for cocaine and F122A mutation lowers cocaine binding affinity.
The interacting residues with the bound cocaine in S1 is shown in Fig. 3B, including Phe43 (100%, interaction frequency over the last 50-ns trajectory), Ala44 (97%), Asp46 (100%), Ala48 (80%), Ala117 (83%), Val120 (100%), Asp121 (99%), Tyr123 (69%), Tyr124 (100%), Phe319 (100%), Ser320 (100%), Gly322 (100%), Phe325 (90%), Ser421 (100%), Ser422 (94%), Gly425 (76%), Ser426 (50%), and Ile429 (78%). Except Gly322, Ser422, Gly425, and Ile429, the other residues interfacing with cocaine are the same as those identified in the crystal structure,8 confirming the stable binding of cocaine to S1. Compared with the initial orientation when binding to S3 (Fig. S4), the cocaine molecule changes its orientation and displays enhanced interactions with its surrounding residues over the 300-ns MD simulations (Fig. 3A). In addition to Tyr119 (99%), Phe122 (76%), Phe470 (100%), and Ile563 (100%), more hydrophobic residues are involved, including Ile229 (81%), Leu473 (100%), Ala478 (81%), Trp555 (82%), Ala556 (99%), and Leu559 (76%). In addition, strong interactions with Tyr477 (93%) and Gly560 (94%) were also identified.
Fig. 3.

Residues interacting with cocaine in the specific binding site identified from different MD simulations. (A) Binding of cocaine to S3. (B) Binding of cocaine to S1. (C) Binding of cocaine to S3 in the simulations of the dDAT complexed with two cocaine molecules in S1 and S3, respectively. (D) Binding of cocaine to S1 in the simulations of the dDAT complexed with two cocaine molecules in S1 and S3, respectively. Residues are colored based on their type: blue for basic, red for acidic, green for polar, and white for nonpolar residues. The bound Na+ and Cl− are shown in yellow and cyan spheres, respectively. A contact occurs if any atom of the ligand is within 3 Å of any atom of protein residues. For clarity, hydrogen atoms are not shown.
To investigate the mutual effects of simultaneous binding of two cocaine molecules to S1 and S3, residues interacting with cocaine in S1 or S3 in the presence of a second cocaine are also shown in Fig. 3. The binding of cocaine to S3 appears to show little effect on the binding of cocaine to S1. Most interacting residues in S1 are preserved, and only interactions with Tyr123 and Ser426 are weakened to a negligible extent (Fig. 3D). By contrast, the binding of cocaine to S1 has notable impacts on the interactions of cocaine in S3 with dDAT (Fig. 3C), which diminishes interactions with Ala478, Ala556, and Gly560, weakens interactions with Tyr119 (64%), Leu473 (81%), Trp555 (68%), and Ile563(77%), but enhances interactions with Phe122(97%) and Leu559(96%), and establishes new interactions with Val458 (88%). Therefore, the binding of cocaine to S1 seems to have more significant influence on the binding of cocaine to S3.
The binding affinity of cocaine in different binding sites of dDAT was predicted in terms of the MM-PBSA approach implemented in the tool g_mmpbsa,50 and results were summarized in Table 1. The most striking finding is that the binding affinity of S3 for cocaine is higher than that of the central binding site S1 when only one cocaine binds to dDAT. In the case of two cocaine molecules separately binding to S1 and S3, the binding energy increases for both sites, indicating that binding of two cocaine molecules becomes less favorable. Also, the binding energy of cocaine in S1 increases 19%, and the binding energy of cocaine in S3 increases 54%, suggesting that binding of cocaine to S3 is more prone to be allosterically modulated by binding of cocaine to S1, in line with the above analyses of interacting residues.
Table 1.
Summary of calculated binding energies (in kcal/mol) for different systems.
| System | Binding Site | Binding Energy I | Binding Energy II | Binding Energy III |
|---|---|---|---|---|
| dDAT + 1 COC | S1 | −24.88 (1.34) | ||
| dDAT + 1 COC | S3 | −30.38 (0.04) | −46.43 (0.25) | |
| dDAT + 2 COC | S1 | −27.74 (0.93) | −20.25 (0.49) | |
| S3 | −21.42 (0.22) | −30.20 (0.75) | −13.93 (0.16) | |
| dDATF122A + 1 COC | S1 | −28.77 (0.32) | ||
| dDATF122A + 1 COC | S3 | −19.75 (5.83) | −40.04 (3.18) | |
| dDATF122A + 2 COC | S1 | −24.42 (0.33) | −17.26 (0.48) | |
| S3 | −17.24 (3.69) | −29.37 (4.12) | −10.00 (3.47) | |
| dDAT + 1 COC + 1 DA | S1 (DA) | −11.16 (0.51) | −4.40 (0.46) | |
| S3 (COC) | −22.10 (0.37) | −35.81 (0.93) | −15.33 (0.32) | |
| dDAT + 1 DA | S1 | −14.68 (1.00) | ||
| dDAT + 2 DA | S1 | −23.77 (5.78) | −14.13 (3.36) | |
| S2* | −21.77 (1.08) | −11.86 (1.39) |
Notes:-
dDAT, COC, and DA denote dopamine transporter, cocaine, and dopamine, respectively;
Because DA in S2 is unstable, S2* is used to indicate the final binding site for that DA;
S1 (DA) and S3 (COC) indicate the binding of DA to S1 and binding of COC to S3, respectively;
For two ligand-bound systems, Binding Energy I was calculated with a dielectric constant ε(solvent) = 80 after removing another bound ligand from the trajectory; Binding Energy II was calculated with a dielectric constant ε(membrane) = 3 after removing another bound ligand from the trajectory; and Binding Energy III was calculated with a dielectric constant ε(solvent) = 80 in the presence of another bound ligand.
The binding affinity of DA with respect to dDAT was also calculated (Table 1). As expected, the binding affinity of cocaine to S1 is higher than that of DA, supporting a competitive mechanism for cocaine inhibition of DA uptake. As shown in Figs. S7 and S8, DA moves from the more extracellularly positioned site S2 toward S1 regardless of the absence (Fig. S7) or presence (Fig. S8) of a second DA in S1. When one DA occupies S1, the secondary incoming DA does not alter the stability of dDAT (Fig. S6) by interacting with residues in the centrally located S1. (Fig. S9) Direct interactions between the two bound DA molecules were also found, but such interactions are not intense as the frequency of occurrence is about 39%. Because the binding energy for individual DA was calculated in the presence of another DA, direct comparison with the result of only one DA binding to dDAT is not straightforward. Therefore, the binding energy was also calculated without considering the presence of a second DA (Table 1), and a lower binding energy was obtained. However, the binding energy calculated in this way may not reflect the real binding environment as the effect of binding of a second DA was not taken into account. Nevertheless, the finding that the binding affinity of the second DA is on the same magnitude as that of the first one may suggest the possibility of simultaneous binding of two DA molecules on a single dDAT.
To further explore the effect of cocaine binding on DA binding, MD simulations of dDAT inserted in the same lipid bilayer were performed (Fig. S11), along with one DA bound to S1 and one cocaine bound to S3 (Fig. 4). Compared with DA-bound dDAT system (Fig. S7), in addition to strong interactions with Phe43 (99%), Asp46 (100%), Tyr124 (97%), Ser421 (100%), and Gly425 (92%), the frequency of interactions with Ala117 and Ser422 by DA is increased by 37% and 97%, respectively, whereas interaction of DA with Phe325 is decreased by 26%. The greatly enhanced interactions with Ser421 and Ser422 might imply a shrink of the binding pocket S1. On the other hand, in the presence of DA in S1, cocaine largely maintains the initial docking orientation, different from those systems where one cocaine binds to S1 (Fig. 3A) or two cocaine molecules bind to S1 and S3, respectively (Fig. 3C). As a result, interactions of cocaine with dDAT become weaker in general. In particular, interactions with Phe122, Ile229, Leu473, Tyr477, Ala478, Trp555, Ala556, and Gly560 were not observed. Instead, the cocaine establishes stable interactions with Val458 (96%), Ala461 (85%), and Ser462 (96%). Taken one DA- or cocaine-bound dDAT as a reference, the calculated binding energy of DA to S1 and cocaine to S3 increases by 3.52 kcal/mol and 8.28 kcal/mol, respectively (Table 1), suggesting a relatively lower binding affinity of DA with respect to S1 and cocaine with respect to S3. In addition, the greater change in the binding energy of cocaine could indicate that the binding of DA to S1 affects the binding of cocaine to S3 more significantly.
Fig. 4.

Residues interacting with cocaine (A) and DA (B) in the specific binding site identified from MD simulations of the dDAT complexed with one cocaine and DA molecule in S3 and S1, respectively. Residues are colored based on their type: blue for basic, red for acidic, green for polar, and white for nonpolar residues. The bound Na+ and Cl− are shown in yellow and cyan spheres, respectively. For clarity, hydrogen atoms are not shown. Residues R52, D475, and F319, which do not interact with DA, are shown as thin Licorice representations.
The human DAT F154A mutant was reported to lower cocaine affinity.24 The corresponding F122 in dDAT is located in S3 and strongly interacts with cocaine (Fig. 3). To examine the impact of F122A mutant on cocaine binding, we performed MD simulations on F122A mutant bound with one cocaine in S1 or S3, and in both S1 and S3. No significant structural changes were observed and dDAT remains in the outward-open conformation (Fig. S12). The notable difference in the ligand-protein interactions is the decreased number of residues that interfere with cocaine bound to S3 and S1 simultaneously, compared to the interacting residues when only cocaine binds to S3 (Fig. S13). Because the contribution of residue F122/A122 to the binding energy is less than 4% in all simulated systems (Table S2), it seems applicable to direct compare the change in the binding energy between the wild-type and the F122A mutant. Because of the mutation, the binding energy of cocaine to S1 decreases by −3.89 kcal/mol, suggesting that the F122A mutation enhances the binding affinity of cocaine to S1. On the other hand, the binding energy of cocaine to S3 increases by 10.63 kcal/mol, resulting in a net increase in the binding energy and thereby a net decrease in the binding affinity of cocaine to dDAT. Similar results were also obtained for the binding energies calculated using ε(membrane) = 3. Moreover, the binding of two cocaine molecules reduces the binding affinity of both sites in the F122A mutant.
Binding of cocaine to S3 reduces binding of DA to dDAT.
The influence of ligand binding on the conformational stability of dDAT was estimated based on the calculated conformational energy of protein structure (Fig. 5). The ligand was removed from the binding site but the bound Na+ and Cl− ions were kept in all calculations. For comparison, all-atom MD simulations were also performed on the ligand-free dDAT embedded in the same lipid bilayer, and the calculated conformational energy of the dDAT was used as a reference. Relative to the ligand-free dDAT, binding of one or two DA molecules decreases the conformational energy of dDAT by about 45 kcal/mol and 126 kcal/mol, respectively, suggesting that binding of DA increases the stability of dDAT. However, the conformational energy of dDAT with one cocaine bound to S1 is comparable to that of dDAT in free state, indicating no obvious energy barrier to the conformational transition from cocaine-free to cocaine-bound state. Interestingly, binding of one cocaine to S3 results in a decrease of about 27 kcal/mol in the conformational energy of dDAT, but binding of two cocaine molecules to S1 and S3 leads to an increase of about 47 kcal/mol in the conformational energy of dDAT. A similar trend was also found in the dDAT with F122A mutation. Such a destabilizing effect reveals that simultaneous binding of cocaine to both S1 and S3 seems unstable and infeasible. This finding is in line with the results of binding energy, showing that the binding affinity of both sites decreases when cocaine molecules bind to S1 and S3 simultaneously. Another interesting finding is that the dDAT displays stabilization upon binding of DA to S1 and cocaine to S3. The conformational energy decreases by about 31 kcal/mol, comparable to the system where one cocaine binds to S3 of dDAT (~27 kcal/mol). This result may indicate the coexistence of DA bound to S1 and cocaine bound to S3, but such a state appears unstable and tends to shift toward the more stable state in which only one DA binds to S1. This conclusion is also consistent with the calculated binding energy, which shows a decrease in the binding affinity of DA to S1 and cocaine to S3. However, the calculated binding energy implies that the binding affinity of cocaine to S3 is higher than that of DA to S1, in that the calculations are based on the ligand-bound states.
Fig. 5.

Conformational energies calculated for different systems. dDAT refers to the dDAT in free state; dDAT + DA refers to DA-bound dDAT; dDAT + COC refers to cocaine-bound dDAT; dDATF122A + COC refers to the cocaine-bound dDAT with F122A mutation; and dDAT + DA +COC refers to the dDAT complexed with both DA and cocaine. DA(1), COC(1), and COC(3) indicate the binding of DA to S1, cocaine to S1, and cocaine to S3, respectively.
To study the impact of binding of cocaine to S3 on the binding of DA, the change in the volume of S1 during simulations was calculated using MDpocket program,54 and results are shown in Fig. 6. No significant fluctuations were observed for those systems where S1 is occupied by DA and cocaine. The volume of S1 varies between 418 to 438 Å3 for cocaine-bound systems, and a smaller value of ~291 Å3 was obtained for DA-bound system. By contrast, in the wild-type dDAT and F122A mutant, the binding of cocaine to S3 results in striking fluctuations in the volume of S1, and the averaged volume of S1 decreases to ~100 Å3 and ~190 Å3 for the wild-type and the F122 mutant, respectively, which may reduce the binding DA to dDAT as the volume of S1 is smaller than that needed for accommodating DA (~291 Å3), or a large conformational change of the protein induced by DA binding is required. Of interest, no significant alterations in the helical structures were observed along with the large fluctuations in the pocket volume (Fig. S14), suggesting that the changes in the pocket volume may involve the movement of domain that consists of several transmembrane helices.
Fig. 6.

Changes in the volume of binding site S1 calculated for different systems. dDAT + COC(1) refers to the dDAT complexed with cocaine in S1; dDATF122A + COC(1) refers to the dDAT F122A mutant complexed with cocaine in S1; dDAT + COC(1) + COC(3) refers to the dDAT complexed with two cocaine molecules in S1 and S3, respectively; dDATF122A + COC(1) + COC(3) refers to the dDAT F122A mutant complexed with two cocaine molecules in S1 and S3, respectively; dDAT + DA(1) + COC(3) refers to the dDAT complexed with DA and cocaine in S1 and S3, respectively; dDAT + COC (3) refers to the dDAT complexed with cocaine in S3; and dDATF122A + COC(3) refers to the dDAT F122A mutant complexed with cocaine in S3.
Discussion
The bacterial homologues LeuT is a model transporter used to study the molecular mechanism of substrate binding and transporting in NSS family. The crystal structures of LeuT in complex with one substrate and one other ligand provide evidence to support the two binding site model of NSS transporters.55 However, in addition to the primary binding site at the center of the protein, the existence of an alternative substrate binding site in LeuT remains controversial.10, 17, 19 The DAT is also a member of NSS family, but the X-ray crystal structures of dDAT in complex with DA and cocaine determine that both ligands bind to the central binding site S1,8 excluding the existence of a secondary high-affinity binding site in the protein. In addition to the binding sites in the interior of a protein, binding pockets on the surface of a protein may also be suitable for binding of a ligand.56 However, characterization of such binding pockets on the surface of membrane proteins such as DAT is challenging because most of cavities on the protein surface could be buried by surrounding lipid or cholesterol molecules. Identification of potential binding sites on the surface of DAT allows us to investigate the difference in the binding propensity between substrate DA and inhibitor cocaine and, accordingly, provide molecular insights into the conformational dynamics induced by ligand binding.
In the present study, the combination of molecular docking and MD simulations enables us to identify different binding modes of DA and cocaine on dDAT (Fig. 1). In addition to S1, an alternative DA binding site S2, which has been proposed by early steered MD simulations,22 was also found by the present docking study. Further MD simulations show that DA diffuses from S2 toward S1 (Figs. S7 and S8), suggesting that S2 is not a high-affinity binding site for DA. In a previous study of human DAT modeled based on the structure of dDAT, DA was also found to transiently bind to S2.23 The finding that cocaine escapes from S2 and translocates into solution (Fig. S5A) is also consistent with the above MD simulations of human DAT which showed that S2 precluded cocaine to penetrate into inner extracellular vestibule.23 The residues Arg52 and Asp475 were found to form a salt-bridge intermittently, but this salt-bridge is not sufficient to close the extracellular gate because the distance between the pair of gating residues Tyr124 and Phe319 is reduced by only ~1–2Å. Therefore, the structure of dDAT remains in the outward-open conformation (Fig. 2). In addition, the present simulations reproduced the interactions between DA/cocaine and dDAT as observed in the crystal structures (Fig. 3 and Fig. S9A), confirming that S1 is a high-affinity binding site for both DA and cocaine. Note that the calculated binding energy seems very sensitive to the alterations of conformations of dDAT. For example, the same residues that interact with DA in S1 were observed for the system where only one DA binds to S1, as well as for the system where two DA molecules bind to S1 and S2, respectively (Figs. S9 and S10, and Table S3). However, the residue contributions to the corresponding binding energy vary significantly, which could be attributed to the different conformational spaces sampled by dDAT. A large difference in the conformational energy between these two dDAT systems in complex with DA was observed (Fig. 5).
Our docking studies identified multiple cocaine binding pockets on the surface of dDAT (Fig. S2A), but only one site S3 located between TMs 3, 9, 10, and 12 (Fig.S4) is capable of binding cocaine stably. Interactions with more hydrophobic residues from different TMs contribute to the association of cocaine with S3 on dDAT (Fig. 3). Extending our current 300-ns MD simulations of cocaine-bound dDAT to 1 μs, we did not observe any dissociation of cocaine from S3 (Fig. S15), and the dDAT still remains in the outward-open state, suggesting that S3 might be an alternative high-affinity binding site for cocaine on the surface of dDAT. Moreover, the calculated binding energies imply that the binding affinity of S3 to cocaine is even higher than that of S1. However, different from S1 that is in the interior of the protein, S3 is on the surface of dDAT, and cocaine is positively charged, the actual binding affinity could be even higher than the calculations in which a dielectric constant of ε=80 was used to model solvent effect in the solvation energy calculations.50 To test this, we calculated the binding energy for several systems with cocaine bound to S3 using a dielectric constant of ε=3 to mimic the lipid bilayers in aqueous solutions57 (Table 1). Indeed, the calculated binding energies decreased by ~9–20 kcal/mol, depending on the systems, suggesting an increase in the binding affinity of cocaine to S3 in the membrane environment.
Our results show that upon simultaneous binding of two cocaine molecules to S1 and S3, the binding affinity of S3 reduced, independent of environment. This finding indicates a coupling effect between these two sites. Because S1 is buried inside the protein and S3 is located on the surface of the protein, their distinct property prevents us from directly comparing the binding affinity between the two sites. The calculated binding energy in an aqueous solution predicted that S1 is a more favorable binding site, but on the other hand, S3 was expected to have a lower binding energy and accordingly a higher binding affinity than in solution. Nevertheless, our results showed a greater change in the binding energy of S3, implying that the binding of cocaine to S1 has a more significant impact on the binding of cocaine to S3. The experimental finding that the F122A mutant lowers cocaine affinity24 could be due to a net reduction in the binding affinity of cocaine to different sites. Compared to the wild-type dDAT, the increase in the binding affinity of S1 was overwhelmed by the decrease in the binding affinity of S3. Similar to the wild-type dDAT, a larger change in the calculated binding energy of cocaine to S3 was observed when two cocaine molecules bound to the F122A mutant when compared to the corresponding systems where only one cocaine molecule binds to the protein. Overall, these results indicate that the binding of cocaine to S1 destabilizes the binding of cocaine to S3 significantly.
Cocaine acts as a competitive inhibitor of DA uptake by locking the DA transporter in the outward-open conformation. In this way, the binding of cocaine reduces the availability of the inward-open conformation that facilitates the transport of DA from an extracellular space to an intracellular space.8, 58 Our results show that the binding energy of cocaine to S1 is much lower than that of DA to S1, confirming the higher binding affinity of cocaine than that of DA to the same central binding pocket. Of interest, our results show that the binding of two DA molecules in a single dDAT seems possible, and the interactions between the two DA molecules may accelerate the conformational transition and therefore facilitate the efficient transport of DA. For cocaine-bound dDAT, the stabilization effect is more prominent when cocaine binds to S3 only, whereas the finding that binding of a second cocaine to S1 destabilizes the conformation of dDAT, along with the decreased binding affinity, collectively suggests that cocaine either binds to S1 or S3, but simultaneous binding to S1 and S3 appears infeasible. Furthermore, the binding of cocaine to S3 could also affect the binding affinity of DA to S1, providing an alternative mechanism on the competitive inhibitor of DA by cocaine. In addition, the binding of cocaine to S3 induces significant fluctuations on the volume of S1, but no significant changes in the helical structures were found. Although the plasticity of S1 allows it to accommodate ligands of varying sizes,8 but on the other hand, the large fluctuations in the binding pocket would make ligand recognition by dDAT less favorable as ligand-protein association is modulated by the relation between the time scale of accessibility of the binding site and the time scale for ligand binding.56 Large pocket dynamics could inevitably influence the binding of ligands with high specificity. Therefore, in addition to directly compete for binding to S1 and inhibit DA binding, our results imply that binding to S3 could also allosterically reduce the binding of DA to S1, providing an alternative mechanism on the multiple inhibitory patterns of cocaine.
In the DA- or cocaine-bound crystal structures, a CHOL molecule is found at the interface of TMs 1a , 5, and 7 and a CHOL analogue, cholesteryl hemisuccinate, is found at the interface between TMs 2 and 7.8 Because no cholesterol molecules occupy this allosteric binding site, S3 seems free for cocaine binding. However, the binding of cocaine to S3 could not be identified by crystallization because the bound cocaine could dissociate from dDAT during the preparation of the crystals such as washing out the impurities. Moreover, the dDAT has been suggested to form oligomers,30 and cocaine has been reported to induce the formation of DAT oligomers in mouse neuroblastoma cells,59 resulting in cocaine addiction. Our results show that the binding of cocaine to S3 results in a more stable conformation of the dDAT, suggesting that this conformational state is most likely to associate and form dimer and other oligomers. Because no structures of DAT oligomers are solved to date, we used the LeuT dimer as a template (PDB ID: 3TT1)7 to model a dDAT dimer with the dimer interface involving TMs 9 and 12 (Fig. 7). Note that this interface overlaps with the cocaine binding site S3. Although a clear relation between the formation of DAT dimer and the cocaine binding to S3 remains to be determined, our work highlights the important role of S3 in cocaine binding and oligomerization of DAT. Overall, the present work provides novel insights into the molecular mechanism on the inhibitory effect of cocaine on DAT.
Fig. 7.

(A) The LeuT dimer (PDB ID: 3TT1). (B) The dDAT dimer modeled based on the LeuT dimer.
The TMs 9, and 12 in the interface are also shown in surface representation. Residue Ile106 in LeuT, which corresponds to Phe122 in dDAT, as well as Phe122 in dDAT, are shown as van der Waals spheres. Na+ and Cl− ions are shown as yellow and cyan spheres, respectively.
Supplementary Material
Acknowledgements
The authors acknowledge support the Texas Advanced Computing Center Frontera computing project (Frontera is made possible by National Science Foundation award OAC-1818253).
Funding
The authors acknowledge support from the NIH (Grant #GM121275).
Footnotes
The authors declare no competing financial interest.
Supporting Information
Figures S1 to S14, and Table S1-S3. RESP partial charges for cocaine and DA molecules (Fig. S1); Distribution of the docked conformations of cocaine and DA on the structure of dDAT (Fig. S2); Residues interacting with the docked cocaine in binding pocket S2 (Fig. S3); Residues interacting with the docked cocaine in binding pocket S3 (Fig. S4); Movement of cocaine molecules from their initial docked positions (Fig. S5); MD simulations of DA-bound dDAT complexes (Fig. S6); Movement of DA from its initial docked position in dDAT complexed with one DA in S1 (Fig. S7); Movement of DA from its initial docked position in dDAT complexed with two DA molecules (Fig. S8); Residues interacting with DA in the DA-bound dDAT complexes (Fig. S9); Residue contributions to the binding energy of DA in the central binding site S1 (Fig. S10); MD simulations of dDAT complexed with DA in S1 and cocaine in S3 (Fig. S11); MD simulations of cocaine bound dDAT F122A mutant (Fig. S12); Residues interacting with cocaine in the dDAT complexed with cocaine (Fig. S13); the fluctuations of secondary structures (Fig. S14); One-microsecond MD simulations of dDAT complexed with cocaine in S3 (Fig. S15); summary of calculated binding energies (in kcal/mol) for different systems (Table S1); the contribution of F122/A122 and the second ligand to the binding energy (Table S2), and the contribution of contacting residues to the binding energy of DA to S1 in different systems (Table S3). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cheng MH; Bahar I Monoamine Transporters: Structure, Intrinsic Dynamics and Allosteric Regulation. Nat. Struct. Mol. Biol 2019, 26, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leviel V Dopamine Release Mediated by the Dopamine Transporter, Facts and Consequences. J. Neurochem 2011, 118, 475–489. [DOI] [PubMed] [Google Scholar]
- 3.Vaughan RA; Foster JD Mechanisms of Dopamine Transporter Regulation in Normal and Disease States. Trends Pharmacol. Sci 2013, 34, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verma V Classic Studies on the Interaction of Cocaine and the Dopamine Transporter. Clin. Psychopharmacol. Neurosci 2015, 13, 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joseph D; Pidathala S; Mallela AK; Penmatsa A Structure and Gating Dynamics of Na+/Cl– Coupled Neurotransmitter Transporters. Front. Mol. Biosci 2019, 6 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forrest LR; Zhang YW; Jacobs MT; Gesmonde J; Xie L; Honig BH; Rudnick G Mechanism for Alternating Access in Neurotransmitter Transporters. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 10338–10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krishnamurthy H; Gouaux E X-ray Structures of LeuT in Substrate-free Outward-Open and Apo Inward-Open States. Nature 2012, 481, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang KH; Penmatsa A; Gouaux E Neurotransmitter and Psychostimulant Recognition by the Dopamine Transporter. Nature 2015, 521, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beuming T; Kniazeff J; Bergmann ML; Shi L; Gracia L; Raniszewska K; Newman AH; Javitch JA; Weinstein H; Gether U; Loland CJ The Binding Sites for Cocaine and Dopamine in the Dopamine Transporter Overlap. Nat. Neurosci 2008, 11, 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi L; Quick M; Zhao Y; Weinstein H; Javitch JA The Mechanism of a Neurotransmitter:Sodium Symporter—Inward Release of Na+ and Substrate Is Triggered by Substrate in a Second Binding Site. Mol. Cell 2008, 30, 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh SK; Yamashita A; Gouaux E Antidepressant Binding Site in a Bacterial Homologue of Neurotransmitter Transporters. Nature 2007, 448, 952–956. [DOI] [PubMed] [Google Scholar]
- 12.Singh SK; Piscitelli CL; Yamashita A; Gouaux E A Competitive Inhibitor Traps LeuT in an Open-to-Out Conformation. Science 2008, 322, 1655–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Z; Zhen J; Karpowich NK; Law CJ; Reith MEA; Wang D-N Antidepressant Specificity of Serotonin Transporter Suggested by Three LeuT–SSRI Structures. Nat. Struct. Mol. Biol 2009, 16, 652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quick M; Winther AML; Shi L; Nissen P; Weinstein H; Javitch JA Binding of an Octylglucoside Detergent Molecule in the Second Substrate (S2) Site of LeuT Establishes an Inhibitor-Bound Conformation Proc. Natl. Acad. Sci U. S. A: 2009, 106, 5563–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coleman JA; Green EM; Gouaux E X-ray Structures and Mechanism of the Human Serotonin Transporter. Nature 2016, 532, 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quick M; Abramyan AM; Wiriyasermkul P; Weinstein H; Shi L; Javitch JA The LeuT-Fold Neurotransmitter:Sodium Symporter MhsT Has Two Substrate Sites Proc. Natl. Acad. Sci U. S. A. 2018, 115, E7924–E7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piscitelli CL; Krishnamurthy H; Gouaux E Neurotransmitter/Sodium Symporter Orthologue LeuT Has a Single High-Affinity Substrate Site. Nature 2010, 468, 1129–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erlendsson S; Gotfryd K; Larsen FH; Mortensen JS; Geiger M-A; van Rossum B-J; Oschkinat H; Gether U; Teilum K; Loland CJ Direct Assessment of Substrate Binding to the Neurotransmitter:Sodium Symporter LeuT by Solid State NMR. eLife 2017, 6, e19314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quick M; Shi L; Zehnpfennig B; Weinstein H; Javitch JA Experimental Conditions Can Obscure the Second High-Affinity Site in LeuT. Nat. Struct. Mol. Biol 2012, 19, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reyes N; Tavoulari S To be, or Not to Be Two Sites: That Is the Question About LeuT Substrate Binding. J. Gen. Physiol 2011, 138, 467–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita A; Singh SK; Kawate T; Jin Y; Gouaux E Crystal Structure of a Bacterial Homologue of Na+/Cl--Dependent Neurotransmitter Transporters. Nature 2005, 437, 215–223. [DOI] [PubMed] [Google Scholar]
- 22.Shan J; Javitch JA; Shi L; Weinstein H The Substrate-Driven Transition to an Inward-Facing Conformation in the Functional Mechanism of the Dopamine Transporter. PLoS ONE 2011, 6, e16350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng MH; Block E; Hu F; Cobanoglu MC; Sorkin A; Bahar I Insights into the Modulation of Dopamine Transporter Function by Amphetamine, Orphenadrine, and Cocaine Binding. Front. Neurol. 2015, 6, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin Z; Uhl GR Dopamine Transporter Mutants with Cocaine Resistance and Normal Dopamine Uptake Provide Targets for Cocaine Antagonism. Mol. Pharmacol 2002, 61, 885–891. [DOI] [PubMed] [Google Scholar]
- 25.Uhl GR; Lin Z, The Top 20 Dopamine Transporter Mutants: Structure–Function Relationships and Cocaine Actions. Eur. J. Pharmacol 2003, 479, 71–82. [DOI] [PubMed] [Google Scholar]
- 26.Hill ER; Huang X; Zhan C-G; Ivy Carroll F; Gu HH Interaction of Tyrosine 151 in Norepinephrine Transporter with the 2β Group of Cocaine Analog RTI-113. Neuropharmacology 2011, 61, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmitt KC; Mamidyala S; Biswas S; Dutta AK; Reith MEA Bivalent Phenethylamines as Novel Dopamine Transporter Inhibitors: Evidence for Multiple Substrate-Binding Sites in a Single Transporter. J. Neurochem 2010, 112, 1605–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sitte HH Sodium-Dependent Neurotransmitter Transporters: Oligomerization as a Determinant of Transporter Function and Trafficking. Mol. Interv 2004, 4, 38–47. [DOI] [PubMed] [Google Scholar]
- 29.Zhen J; Antonio T; Cheng S-Y; Ali S; Jones KT; Reith MEA Dopamine Transporter Oligomerization: Impact of Combining Protomers with Differential Cocaine Analog Binding Affinities. J. Neurochem 2015, 133, 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cecchetti C; Pyle E; Byrne B Transporter Oligomerisation: Roles in Structure and Function. Biochem. Soc. Trans 2019, 47, 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webb B; Sali A Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinformatics 2016, 15, 5.6.1–5.6.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fiser A; Do RKG; Šali A Modeling of Loops in Protein Structures. Protein Sci. 2009, 9, 1753–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berfield JL; Wang LC; Reith MEA Which Form of Dopamine Is the Substrate for the Human Dopamine Transporter: the Cationic or the Uncharged Species? J. Biol. Chem 1999, 274, 4876–4882. [DOI] [PubMed] [Google Scholar]
- 34.Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem 2009, 30 (16), 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD CHARMM36m: an Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J; Cieplak P; Kollman PA How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J. Comput. Chem 2000, 21, 1049–1074. [Google Scholar]
- 37.Morris GM; Goodsell DS; Halliday RS; Huey R; Hart WE; Belew RK; Olson AJ Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem 1998, 19, 1639–1662. [Google Scholar]
- 38.Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nielsen AK; Möller IR; Wang Y; Rasmussen SGF; Lindorff-Larsen K; Rand KD; Loland CJ Substrate-Induced Conformational Dynamics of the Dopamine Transporter. Nat. Commun 2019, 10, 2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lomize MA; Lomize AL; Pogozheva ID; Mosberg HI OPM: Orientations of Proteins in Membranes Database. Bioinformatics 2006, 22, 623–625. [DOI] [PubMed] [Google Scholar]
- 41.Lomize MA; Pogozheva ID; Joo H; Mosberg HI; Lomize AL OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40, D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD; Pastor RW Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jo S; Kim T; Iyer VG; Im W CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- 44.Lee J; Cheng X; Swails JM; Yeom MS; Eastman PK; Lemkul JA; Wei S; Buckner J; Jeong JC; Qi Y; Jo S; Pande VS; Case DA; Brooks CL; MacKerell AD; Klauda JB; Im W CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput 2015, 12, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable Molecular Dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feller SE; Zhang Y; Pastor RW; Brooks BR Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys 1995, 103, 4613–4621. [Google Scholar]
- 47.Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem 1983, 4, 187–217. [Google Scholar]
- 48.Lee MS; Salsbury FR; Brooks CL Novel Generalized Born Methods. J. Chem. Phys 2002, 116, 10606–10614. [Google Scholar]
- 49.Feig M; Onufriev A; Lee MS; Im W; Case DA; Brooks CL Performance Comparison of Generalized Born and Poisson Methods in the Calculation of Electrostatic Solvation Energies for Protein Structures. J. Comput. Chem 2004, 25, 265–284. [DOI] [PubMed] [Google Scholar]
- 50.Kumari R; Kumar R; Lynn A g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model 2014, 54, 1951–1962. [DOI] [PubMed] [Google Scholar]
- 51.Kollman P Free Energy Calculations: Applications to Chemical and Biochemical Phenomena. Chem. Rev 1993, 93, 2395–2417. [Google Scholar]
- 52.Kollman PA; Massova I; Reyes C; Kuhn B; Huo S; Chong L; Lee M; Lee T; Duan Y; Wang W; Donini O; Cieplak P; Srinivasan J; Case DA; Cheatham TE Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res 2000, 33, 889–897. [DOI] [PubMed] [Google Scholar]
- 53.Forli S; Huey R; Pique ME; Sanner MF; Goodsell DS; Olson AJ Computational Protein–Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc 2016, 11, 905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidtke P; Bidon-Chanal A; Luque FJ; Barril X MDpocket: Open-Source Cavity Detection and Characterization on Molecular Dynamics Trajectories. Bioinformatics 2011, 27, 3276–3285. [DOI] [PubMed] [Google Scholar]
- 55.Nyola A; Karpowich NK; Zhen J; Marden J; Reith ME; Wang D-N Substrate and Drug Binding Sites in LeuT. Curr. Opin. Struc. Biol 2010, 20, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stank A; Kokh DB; Fuller JC; Wade RC Protein Binding Pocket Dynamics. Acc. Chem. Res 2016, 49, 809–815. [DOI] [PubMed] [Google Scholar]
- 57.Gramse G; Dols-Perez A; Edwards MA; Fumagalli L; Gomila G Nanoscale Measurement of the Dielectric Constant of Supported Lipid Bilayers in Aqueous Solutions with Electrostatic Force Microscopy. Biophys. J 2013, 104, 1257–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen N; Justice JB Cocaine Acts as an Apparent Competitive Inhibitor at the Outward-Facing Conformation of the Human Norepinephrine Transporter: Kinetic Analysis of Inward and Outward Transport. J. Neurosci 1998, 18, 10257–10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siciliano CA; Saha K; Calipari ES; Fordahl SC; Chen R; Khoshbouei H; Jones SR Amphetamine Reverses Escalated Cocaine Intake via Restoration of Dopamine Transporter Conformation. J. Neurosci 2018, 38, 484–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.