

Summary
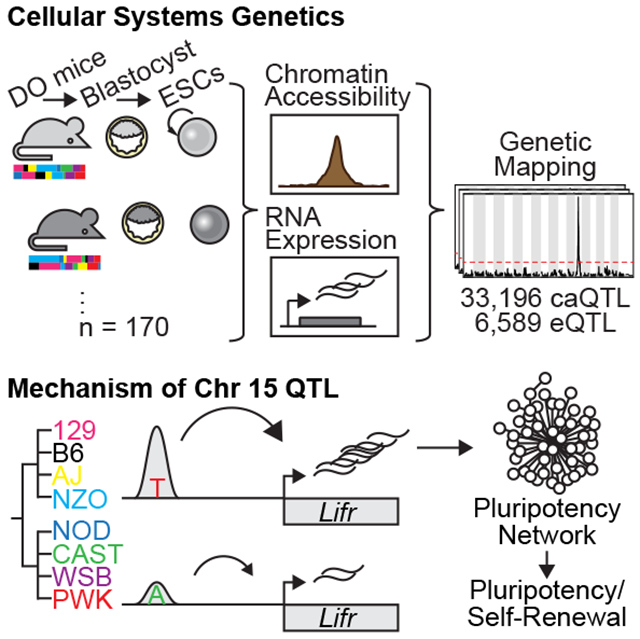
Mouse embryonic stem cells (mESCs) cultured in the presence of LIF occupy a ground state with highly active pluripotency-associated transcriptional and epigenetic circuitry. However, ground state pluripotency in some inbred strain backgrounds is unstable in the absence of ERK1/2 and GSK3 inhibition. Using an unbiased genetic approach, we dissected the basis of this divergent response to extracellular cues by profiling gene expression and chromatin accessibility in 170 genetically heterogeneous mESCs. We mapped thousands of loci affecting chromatin accessibility and/or transcript abundance, including ten QTL hotspots where genetic variation at a single locus coordinated regulation of genes throughout the genome. For one hotspot we identified a single enhancer variant ~10kb upstream of Lifr associated with chromatin accessibility and mediating a cascade of molecular events affecting pluripotency. We validated causation through reciprocal allele swaps, demonstrating the functional consequences of noncoding variation in gene regulatory networks that stabilize pluripotent states in vitro.
Keywords: ground state pluripotency, metastability, mESC, Diversity Outbred, gene expression, chromatin accessibility, genetic variation, Lifr
Graphical Abstract
eTOC:
Mouse ESCs occupy a highly stable pluripotent ground state, which can be unstable in some genetic backgrounds. Skelly et al. used an unbiased genetic approach to reveal how genetic variation influences chromatin state and gene expression, and validate a single enhancer variant upstream of Lifr that impacts ground state stability
Introduction
Derivation and in vitro propagation of pluripotent mouse embryonic stem cells (mESCs) are influenced by genetic background. Successful derivation of pluripotent mESC lines was first reported in 1981 using both inbred (substrains of 129, [C3HxC57BL/6] F1 hybrids) and outbred laboratory strains (Evans and Kaufman, 1981; Martin, 1981). These early advances demonstrated that in vitro mESCs sustain their capacity to contribute to all embryonic lineages, including the germ line (i.e. pluripotency), when propagated in media containing leukemia inhibitory factor [LIF]. However, these approaches were unsuccessful in other mouse strains like nonobese diabetic (NOD), which proved recalcitrant to ESC derivation (Kawase et al., 1994; Gardner and Brook, 1997). This recalcitrance was later surmounted through inhibition of ERK1/2 and GSK3 signaling (“2i”; Nichols et al., 2009; Ying et al., 2008).
mESCs grown in 2i media are relatively homogenous and exhibit a “ground” pluripotent state (Ying et al., 2008) characterized by epigenetic marks and transcriptional activity resembling the pre-implantation epiblast (Abranches et al., 2014; Marks et al., 2012; Nichols et al., 2009; Ying et al., 2008). However, the stability of this state varies across genetic backgrounds. Hanna et al. (2009) introduced the concept of metastability, defined as the interconversion of mESCs between ground state and a state more analogous to the post-implantation epiblast (EpiSC-like), depending on culture media. mESCs derived from NOD exhibit metastability, acquiring the EpiSC-like state in the absence of exogenous ERK1/2 and GSK3 inhibition. Ohtsuka and Niwa (2015) went on to show that mESCs from other recalcitrant (CBA, FVB) or intermediate (BALB) strains depend on ERK1/2 and GSK3 inhibition to maintain self-renewal due to attenuated LIF responsiveness. While differential response to LIF is genetic, the associated signaling pathways involve hundreds of genes collectively harboring multitudes of coding and regulatory variants. Therefore, the underlying genetic differences that drive interstrain variation in mESC responsiveness to the cell culture environment remain a mystery.
Here we leverage genetic diversity accumulated over ~500,000 years of evolution along the Mus musculus lineage to study ground state pluripotency in mESCs. A large subset of this diversity is captured by the eight parental strains of inbred mice used to develop the Collaborative Cross (CC) recombinant inbred mouse resource and complementary Diversity Outbred (DO) heterogeneous stock (Chesler et al., 2016; Churchill et al., 2004, 2012; Threadgill and Churchill, 2012). DO mice segregate >40 million genetic variants with reasonably balanced founder allele frequencies and a simple population structure that enables high-resolution genetic mapping with relatively small sample sizes. To determine the genetic and molecular mechanisms leading to pluripotent ground state metastability, we derived mESCs from hundreds of DO mice and measured molecular phenotypes in cells grown in the absence of ERK1/2 inhibition.
Results
Genetic background drives transcriptional variation in mESCs
We reasoned that the genetic variation driving attenuated responsiveness to LIF would exert effects through differences in transcription. To test this idea, we took advantage of germline-competent, euploid mESC lines derived from the eight inbred founder strains of the mouse CC (Czechanski et al., 2014). These include the classical laboratory strains C57BL/6J, A/J, 129S1/SvImJ, NZO/HILtJ, NOD/ShiLtJ (referred to here as B6, AJ, 129S1, NZO, and NOD, respectively); as well as inbred wild-derived strains representing three subspecies of Mus musculus including WSB/EiJ (WSB, M.m. domesticus), CAST/EiJ (CAST, M.m. castaneus), and PWD/PhJ (PWD, M.m. musculus; a consubspecific inbred wild-derived strain with similar geographic origin to the CC founder strain PWK/PhJ [PWK]). We profiled genome-wide gene expression by RNA-Seq in three male mESCs from each strain (N = 24 total). Unsupervised principal component analysis revealed that inbred strain background explained the majority of transcriptional variability (Figure 1A). Moreover, we found significant differences in expression of core pluripotency genes across genetic backgrounds (Figure 1B). Although core pluripotency transcription factors like Nanog and Sox2 were highly expressed in all cell lines (Figure 1B–C and Figure S1), their expression varied markedly (often >2-fold) across strains (Figure 1B and Figure S1). Among the top differentially expressed genes (false discovery rate [FDR] = 5%; Table S1) were members of cytokine receptor signaling pathways.
Figure 1: Molecular indicators of the pluripotent ground state vary in diverse mESCs.
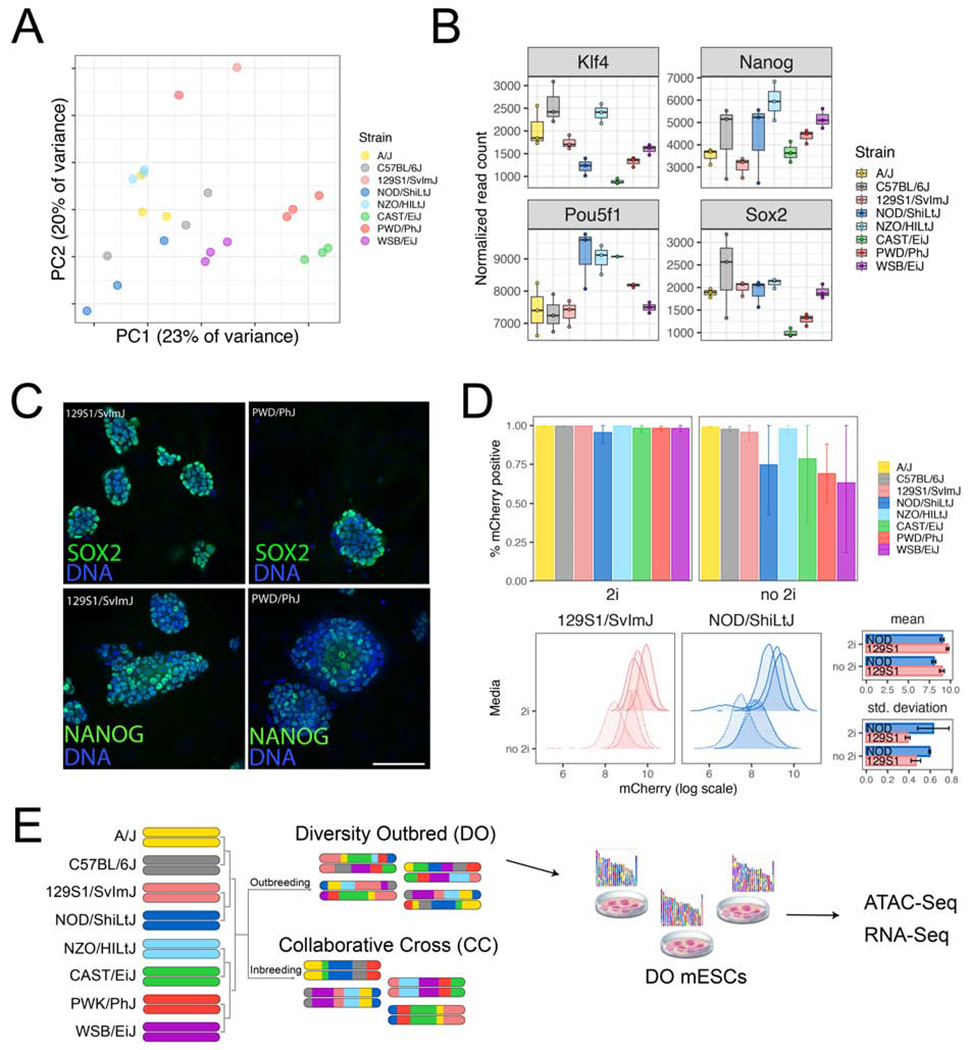
(A) Principal component analysis (PCA) of gene expression for mESCs from eight diverse backgrounds. Dots of the same color represent biological replicates derived from the same strain. (B) Expression of core pluripotency genes and early lineage markers shows both intrastrain (e.g. B6) and interstrain (e.g. NOD vs. WSB) variability. Points show normalized read counts for each biological replicate, with first quartile, median, and third quartile (boxes) and minimum/maximum (whiskers). (C, D) Quantification of NANOG expression using immunofluorescence and a Nanog-mCherry reporter knock-in. (C) Representative composite images of two mESC lines showing variable NANOG and SOX2 expression in 2i culture conditions. Scale bar = 50μm. (D) Nanog-mCherry expression in the presence and absence of 2i. Top bar graphs show percentage of mCherry positive cells in the two media conditions (mean ± standard error). Bottom histograms show the full distribution of mCherry fluorescence for strains 129S1 and NOD. Bar plots at right quantify distributions shown on left. (E) Overview of experimental design. See also Figure S1.
Based on this pattern of expression and previous work (Silva et al., 2009), we predicted that the removal of ERK1/2 and GSK3 inhibition would destabilize Nanog expression. These pathways act downstream of LIF and are proximal to genes that reinforce pluripotency. To test this, we introduced an in-frame self-cleaving mCherry reporter fused to the endogenous Nanog locus in all 24 inbred mESC lines. Upon removal of 2i, the fraction of cells expressing Nanog was reduced in CAST, NOD, PWD, and WSB mESCs (Figure 1D and Figure S1), demonstrating that mESCs from these backgrounds depend on ERK1/2 and GSK3 inhibition to maintain robust Nanog expression. We observed a similar pattern for SOX2 in CAST and PWD mESCs, while OCT-3/4 expression remained stable (Figure S1). These observations confirm nonpermissive strain dependence on ERK1/2 and GSK3 inhibition and extend this phenotype to additional strains (Hanna et al., 2009; Ohtsuka and Niwa, 2015).
A forward genetic approach to dissecting interstrain variation in mESC gene expression states
To understand the genetic control of these interstrain differences among mESCs, we leveraged the Diversity Outbred (DO) model (Figure 1E) by deriving ESCs from DO blastocysts. We selected 213 lines that met quality control criteria including availability of high-quality SNP genotypes (Methods). We expanded these mESCs under sensitized conditions in the absence of ERK1/2 inhibition to further expose transcriptional differences among the strains (Ying et al., 2008). To characterize our genetically diverse mESCs, we measured steady-state transcript abundance by RNA-Seq (N = 185 lines) and chromatin accessibility as an indicator of regulatory element activity by ATAC-Seq (N = 192 lines). Both assays were performed on a common set of 170 lines.
We quantified expression of 15,185 genes and chromatin accessibility at 102,173 genomic locations across the DO mESCs. Of these, 84% of transcripts (12,764) and 47% of open chromatin regions (47,786) showed heritable variation (FDR = 5%). While all cell lines had high expression of pluripotency-related genes, expression was highly variable among cell lines (Figure 2A). Variable activation of regulatory elements (i.e. promoters, enhancers, insulators), observed as changes in chromatin accessibility, may underlie variable gene expression. Indeed, we observed open chromatin near promoters (Figure S2A), and gene expression was correlated with variability in proximal chromatin accessibility (orange/red diagonal line in Figure 2B).
Figure 2: Genetic variation drives local and distant regulation of chromatin accessibility and gene expression.
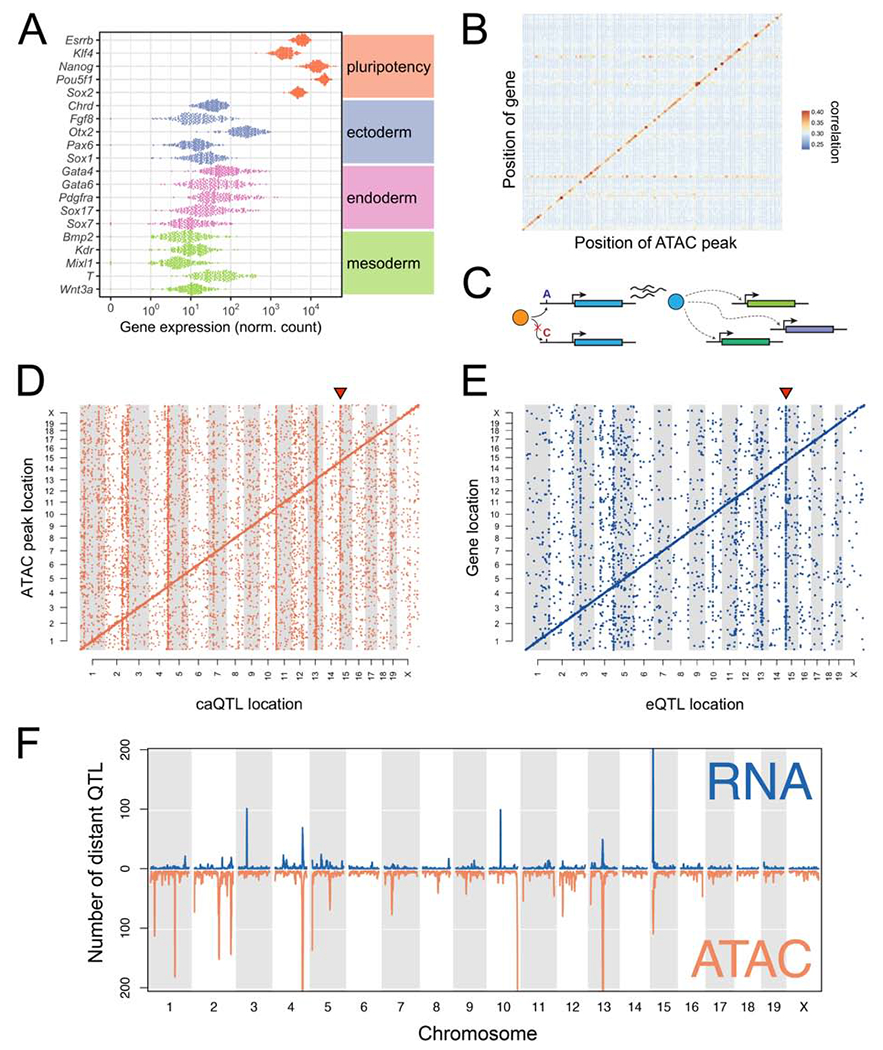
(A) Expression of core pluripotency genes and early lineage markers. Dots represent gene expression values for individual DO mESC lines. (B) Correlations between gene expression and chromatin accessibility across the genome. Only autosomal genes with a local eQTL (LOD >7.5) are shown. Genes/open chromatin regions were grouped in 20Mb bins. Correlations between all transcripts and accessibility of all chromatin regions in the bin were computed, and the maximum correlation retained for each transcript. Points are colored and sized according to the mean magnitude of correlation (i.e. average maximum transcript-to-peak correlation across the bin). (C) Schematic of an idealized eQTL hotspot. Transcript abundance of the blue gene is influenced by an upstream polymorphism (A/C) that alters binding of a transcription factor (orange). Protein (blue) directly or indirectly regulates the expression of other genes in trans (dotted lines with arrowhead). (D) Genomic locations of significant caQTL. Diagonal bands reflect the predominance of local caQTL. Triangle above caQTL shows an exemplary hotspot on Chr 15. (E) Genomic locations of significant eQTL. Diagonal bands and triangle above Chr 15 hotspot are as in (D). (F) Distinct and co-occuring caQTL and eQTL hotspots. See also Figure S2.
Molecular mapping identifies extensive distal regulation of chromatin and gene expression
To understand the genetic regulation of chromatin accessibility and gene expression in mESCs, we conducted quantitative trait locus (QTL) mapping. We mapped 33,196 chromatin accessibility QTL (caQTL) for 30,458 distinct open chromatin regions (Figure 2D; LOD > 7.5, corresponding to a permutation-based genome-wide P < 0.05) and 6,589 gene expression QTL (eQTL) for 5,853 distinct genes (Figure 2E; LOD > 7.6, permutation-based genome-wide P < 0.05). Most QTL mapped near their target open chromatin region or transcript (69% of caQTL and 63% of eQTL) as evident by the clear diagonal bands in Figure 2D–E; these local associations likely drive much of the observed high correlation between chromatin accessibility and transcript abundance of nearby genes (Figure 2B).
We also identified multiple instances of loci that control chromatin accessibility and/or gene expression at hundreds of distal sites. These QTL “hotspots” (Schadt et al., 2003), evident as vertical bands in Figure 2D–E, are of interest because they likely harbor genes that regulate many molecular features throughout the genome in trans. For example, a QTL hotspot would be evident if genetic variation that affects the expression or function of a transcription factor impacts many downstream target genes or open chromatin regions bound by that factor (schematic, Figure 2C). We identified thirteen such QTL hotspots, eight chromatin accessibility and five gene expression (Table S2), of which three impacted both molecular phenotypes (Figure 2F). Although we observed some evidence for aneuploidies in multiple DO mESC lines (Figure S2B), hotspot signals persisted after controlling for estimated aneuploidy (data not shown). Hotspot loci ranged in size from −1.5-10Mb and an average of 103 annotated genes were located inside or within 5Mb of hotspot boundaries (range 36-228; Table S2).
QTL hotspots regulated critical pathways driving pluripotent states. Two of the eQTL hotspots we identified were of particular interest as they independently regulate different gene networks related to maintenance of pluripotency. An eQTL hotspot on Chr 10 influenced the expression of 100 target genes with a striking overlap with genes that are upregulated in the rare 2-cell (2C)-like state (47/123; P < 1×10−16; Hendrickson et al., 2017). Cells in the 2C-like state are totipotent and present in mESC cultures derived from B6/129 substrain backgrounds at low frequency (~1%; Ishiuchi et al., 2015; Macfarlan et al., 2012). These cells are defined by their transcriptional similarity to totipotent early embryos, including characteristic activation of the endogenous retroviral element MERVL (Macfarlan et al., 2012). Expression of the MERVL long terminal repeat (MT2_mm) in our DO mESCs panel is strongly correlated with expression of Chr 10 QTL target genes (ρ = 0.96; Figure S2C). Additionally, we identified a QTL hotspot located on proximal Chr 15 (Figure 2D–E) that controls both chromatin state and gene expression of distant target genes. In this Chr 15 QTL hotspot, the 254 target transcripts showed strong enrichment for genes with known roles in pluripotency, including Klf4, Lin28a, and Cxxc1. We estimated the haplotype effects on each target gene at the Chr 15 locus and found that mESCs from permissive strains (129S1, AJ, B6, and NZO; denoted as “REF” haplotype) were similarly affected by the Chr 15 QTL genotype, while mESCs from nonpermissive strains (CAST, NOD, PWK, and WSB; denoted as “ALT” haplotype) exhibited opposite effects (Figure 3A).
Figure 3: QTL mapping to chromosome 15 implicates the cytokine receptor Lifr as a key gene regulating chromatin and gene expression at genomically distant loci.
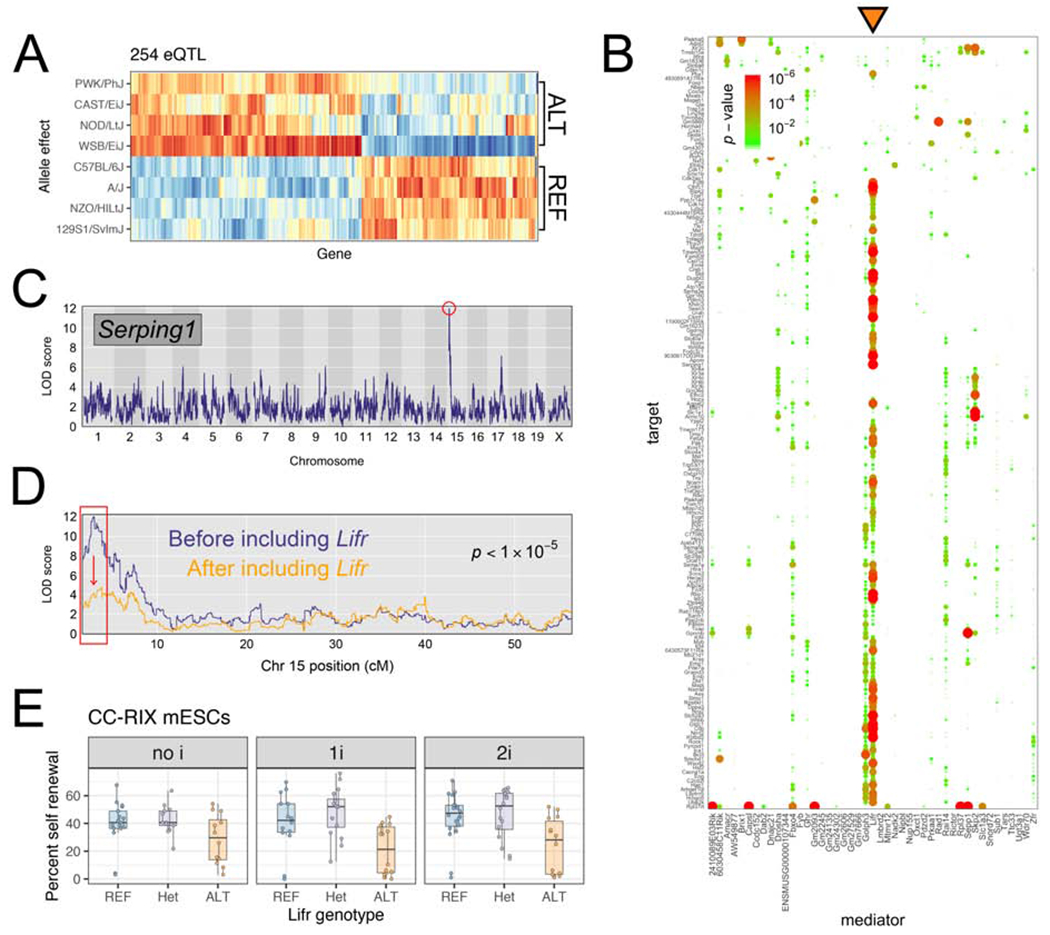
(A) Allelic effects for targets of the Chr 15 hotspot. Red/orange indicate higher expression of the target gene in mESCs carrying a haplotype derived from the founder listed at left. Blue/light blue) indicate the opposite. For each target gene, effects are scaled to have mean 0 and variance 1. (B) Mediation analysis reveals that Lifr is the best mediator (orange triangle) for hotspot targets. Dotplot shows candidate mediators located within Chr 15 hotspot along x-axis, and targets of the hotspot on y-axis. Change in LOD score is shown by points sized and colored proportional to the FDR-adjusted p-value as shown in the legend at top left. (C, D) Serping1 provides an example of a gene with expression mediated by Lifrexpression. (C) Serping1 is located on Chr 2 (~84Mb) and has a strong distant eQTL (LOD = 12) on Chr 15. (D) The LOD significance score for the Chr 15 QTL drops >6 LOD units (p-value is shown) when Lifr expression is included as a covariate in the genetic mapping model. (E) CC-RIX mESC self-renewal percentage measured in three media conditions shown in facets. Replicate measurements within each panel are connected by a line. Boxes show first quartile, median, and third quartile, while whiskers extend to a maximum of 1.5 times the inter-quartile range. See also Figure S3.
The identification of QTL hotspots presented an opportunity to test whether local genetic variation within the hotspot influences target gene expression indirectly through transcript abundance of a “mediator” gene (e.g. genetic variation → [mediator] → downstream targets; Figure 2C). We applied mediation analysis to each QTL hotspot (see Methods; Chick et al., 2016) to identify candidates most likely to confer the observed QTL effect. We identified candidate genes regulating eQTL and caQTL hotspots on Chr 3 (Exosc8 regulating genes involved in the chromatin remodeling 6 SWI/SNF complex) and Chr 5 (Steap2 regulating chromatin accessibility at 140 distant regions). We also found that abundance of the DUX transcript (Duxf3) was among the top two candidate mediators for the Chr 10 eQTL hotspot. This observation, in tandem with research identifying DUX as an activator of 2C-like genes (Hendrickson et al., 2017), suggest that variation in Duxf3 expression drives this hotspot. Rather than indicating differences in expression of 2C-like genes, this hotspot may reflect differences in cell state composition among DO mESC lines, with some lines having no/few cells in the 2C-like state and other lines having a higher frequency of cells in this state. Finally, we identified Lifr as the best mediator of the Chr 15 eQTL hotspot. Of the 175 Chr 15 targets genes with significant mediators (FDR = 5%), Lifr gene expression was the best mediator for 92 (53%) of them (Figure 3B–D).
Variation in Lifr expression drives a suite of gene expression changes with functional consequences for mESCs
Lifr encodes a protein that heterodimerizes with Glycoprotein 130 to form the LIF receptor. Given the known interstrain differences in LIF responsiveness, variation in Lifr expression is an attractive candidate regulating the ability to respond to extracellular LIF, putatively influencing metastability. QTL mapping of Lifr transcript abundance revealed a strong local eQTL. Therefore, we surmised that a cis-regulatory variant might directly influence Lifr expression and cause differential expression of downstream genes.
To validate Lifr expression as the direct effector of the Chr 15 QTL hotspot in an orthogonal genetic system, we derived mESCs from F1 intercrosses of recombinant inbred CC lines (CC-RIX) segregating the same genetic variation as DO mice. We quantified the expression of a subset of five Chr 15 eQTL target genes that were predicted to be mediated by Lifr expression (Rbp1, Cxcl12, Hap1, Klf4, and Socs3) by qRT-PCR in CC-RIX mESCs. The expression of each of these genes was correlated with Lifr expression in the CC-RIX lines, and both the magnitude and direction of this correlation agreed with our results in the DO mESCs (Figure S3), supporting our prediction that the locus containing Lifr is the primary causal mediator of genes in the Chr 15 QTL hotspot.
Self-renewal is an important property of pluripotent cells that is lost upon lineage commitment, and differences in self renewal underlies mESC permissiveness. To quantitatively examine the functional significance of Lifr genotype on self-renewal, we tested the ability of CC-RIX lines to proliferate from single cells in colony forming assays. Indeed, Lifr genotype was a good predictor of self-renewal, with lines homozygous for the low-expressing ALT allele showing abrogated self-renewal (Table S3, Figure 3E; p = 1.2×10−7; linear model testing contribution of Lifr genotype to self-renewal including a culture media covariate).
A non-coding single nucleotide variant explains differential responsiveness to LIF in mESCs.
To identify the causal genetic variant(s) within the Chr 15 QTL hotspot, we searched for genetic variants that matched the REF and ALT haplotype effects observed for the QTL targets (Fig. 3A). Although there are over 20,000 segregating genetic variants in DO mice within the 1.5Mb region surrounding Lifr, only 185 variants matched this unusual 4:4 strain pattern. Given that Lifr shows a local eQTL, and our observation that local chromatin accessibility correlates with gene expression (Fig. 2B), we used variation in regions of open chromatin within +/− 2Mb surrounding Lifr to identify putative regulatory elements. The best-mediating open chromatin region was located ~10kb upstream of the transcriptional start site of Lifr, and colocalizes with a DNaseI hypersensitive site that is unique to mESCs (Figure 4A). A single-nucleotide polymorphism (SNP) with the unusual 4:4 strain genotype split lies at the apex of this chromatin accessibility peak (SNP rs50454566, GRCm38/mm10 chr15:7116944, T/A). DO mESCs homozygous for the alternate allele (A/A, present on ALT haplotype of nonpermissive strains) had reduced chromatin accessibility and Lifr expression compared to lines homozygous for the mouse reference allele (T/T, present on REF haplotype of permissive strains; Figure 4B). RNA-Seq on founder inbred strain mESCs and qRT-PCR of CC-RIX lines confirmed the correlation between strain genotypes at this SNP and Lifr gene expression, with the reference genotype (T/T) associated with higher Lifr expression (Figure S4A–B). We also observed concordant patterns of LIFR protein expression by flow cytometry of 129S1 (REF) and NOD (ALT) mESCs (Figure S4C). Moreover, an ~500bp fragment of the open chromatin carrying the REF genotype drove higher reporter expression in vitro than the same fragment carrying the ALT genotype (Figure 4C).
Figure 4: A single variant upstream of Lifr influences mESC pluripotency.
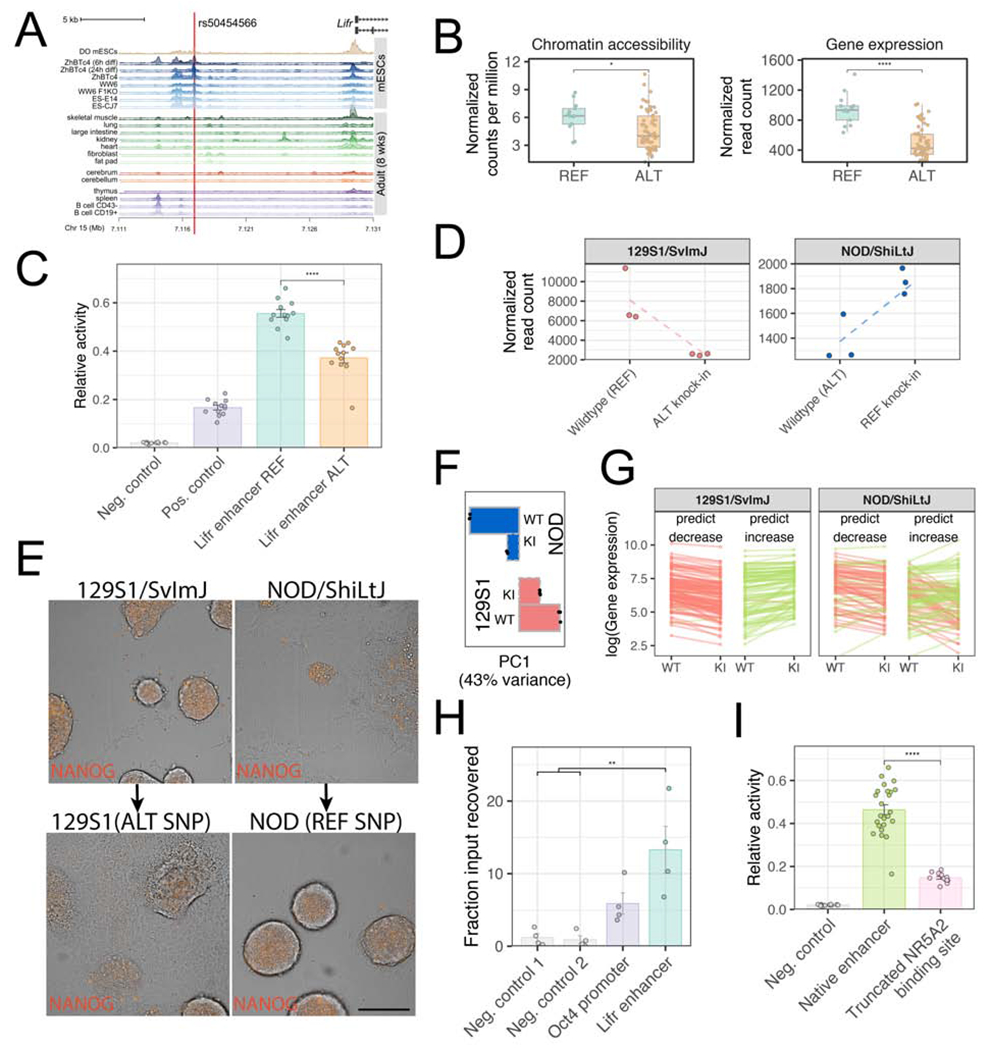
(A) 20kb surrounding the Lifr transcriptional start site, showing isoforms (Ensembl transcript support level 1). Top track shows the gene model and SNP rs50454566, chr15:7116944 (GRCm38/mm10). Second track shows a composite DO mESC ATAC-Seq track (median normalized sequencing depth across all DO mESC samples). Lower tracks show published DNaseI hypersensitive site data (Mouse ENCODE Consortium et al., 2012). (B) DO strains homozygous for the alternate (ALT) allele (A/A) have lower chromatin accessibility and Lifr expression compared to strains homozygous for the reference (REF) allele (T/T). *P < 0.05; ****p < 0.0001, Wilcoxon test. Boxes show first quartile, median, and third quartile, whiskers = 1.5 times inter-quartile range. (C) Enhancer activity quantified as relative Luciferase activity (Firefly/Renilla; bars show mean ± standard error). Both Lifr sequences consist of 509bp surrounding the SNP (REF = reference allele at SNP rs50454566, ALT = non-reference allele). Empty Firefly vector was used as negative control. Positive control consisted of 860bp containing the Oct4 promoter. ****p < 0.0001, Wilcoxon test. (D) RNA-Seq under 1i growth conditions shows that Lifr expression is higher in NOD mESC clones harboring the REF allele (T/T; REF knock-in) compared to the parental NOD mESC, and lower in 129S1 mESC clones harboring the ALT allele (A/A; ALT knock-in) compared to the parental 129S1 mESC line. (E) Colony morphology and Nanog-mCherry expression of 129S1 Nanog-mCherry mESC clones carrying the ALT allele worsens under 2i growth conditions with LIF depletion compared to the 129S1 clones carrying the native REF allele (left images). The converse is true for NOD mESC clones carrying the REF allele compared to NOD clones carrying the native ALT allele (right images). Phase contrast images are overlaid with fluorescent images of Nanog-mCherry expression (orange). Scale bar = 50 μm (F) Bar plot showing magnitude of the first PC calculated from gene expression in parental and allele swap lines. Points show three replicates of each line and bar indicates mean within each group (WT=parental genotype; KI=knock-in lines with the opposing allele at rs50454566 compared to the parental line). (G) Gene expression Chr 15 hotspot targets in parental and allele swap lines measured via RNA-Seq. Genes are divided into groups (“predict decrease” and “predict increase”) according to predictions of allele effects from eQTL mapping. Transcript abundance is plotted as the mean of three replicates (WT=parental genotype; KI=knock-in lines with the opposing allele at rs50454566 compared to the parental line). Lines connecting means are colored according to change from WT to KI (red=decrease; green=increase; genes not differentially expressed are omitted). (H) Chromatin immunoprecipitation (ChIP) – quantitative PCR assay to assess binding of NR5A2 (mean ± standard error). Negative controls were intergenic regions. Recovery of input using control IgG antibodies was negligible. **P < 0.01, Wilcoxon test. (I) Putative enhancer activity quantified as relative Luciferase activity (y-axis; mean ± standard error). Native Lifr enhancer sequences consist of 509bp surrounding the SNP (both REF and ALT haplotypes). Empty Firefly vector was used as negative control. “Truncated NR5A2 binding site” represents the REF Lifr enhancer with a 12bp deletion that removes 3bp of the putative NR5A2 binding site. ****P < 0.0001, Wilcoxon test. See also Figure S4.
To validate this SNP as a causal variant, founder strain Nanog-mCherry mESC lines with opposing rs50454566 genotypes (129S1 [T/T] vs. NOD and WSB [A/A]) were edited to create reciprocal allele swaps, as many hotspot targets are regulated by Nanog (Zheng et al., 2019). Replacing the reference allele in 129S1 with the alternate allele (129S1[A/A]) resulted in reduced Lifr transcript abundance (Figure 4D) and Nanog-mCherry expression (Figure S4D). Reciprocally, replacing the alternate allele with reference on the NOD genetic background (NOD[T/T]) increased Lifr expression (Figure 4D; comparable results for WSB allele swap lines, WSB[T/T], are shown in Figure S4D,F). In culture conditions depleted of LIF, the allele swap had a striking impact on colony morphology. Replacement of the single SNP corrected the poor colony morphology for both NOD and WSB backgrounds (Figure 4E and Figure S4E). Principal component analysis of genome-wide gene expression from these strains revealed that both allele swaps increased transcriptional similarity to parental lines carrying the swapped allele (Figure 4F). Focusing on the target genes of the Chr 15 hotspot, allele swapped 129S1[A/A] mESCs showed reduced expression of pluripotency-related genes compared to unaltered 129S1 mESCs, and allele swap gene expression changes were concordant with predictions made from eQTL mapping (Figure 4G; Fisher’s exact test; p < 2×10−16). In contrast, allele swapped NOD[T/T] mESCs exhibited higher expression of genes that promote pluripotency relative to the parent NOD mESCs, with overall gene expression patterns resembling 129S1 lines. Changes in Chr 15 hotspot targets in NOD allele swap mESCs were also concordant with predictions from eQTL mapping (Figure 4G; p = 0.0012), although weaker concordance compared to 129S1 lines.
A predicted transcription factor binding site (Lesurf et al., 2016) for the orphan nuclear receptor NR5A2 (also known as LRH-1) is located seven base pairs downstream of SNP rs50454566. During development NR5A2 maintains Oct4 expression during the epiblast stage (Gu et al., 2005), and can substitute for OCT4 during reprogramming from somatic cells to iPSCs (Heng et al., 2010). Using chromatin immunoprecipitation and quantitative PCR, we confirmed that NR5A2 binds to the identified Lifr enhancer (Figure 4H). In support of the role of NR5A2 in activating Lifr expression, sequences carrying either naturally occurring allele drove reporter gene expression higher than alleles lacking the NR5A2 binding site (Figure 4I), suggesting that NR5A2 directly activates expression of Lifr. Together, we leveraged natural genetic variation and unbiased genetic mapping to reconstruct a complete causal molecular chain linking hallmarks of pluripotency to a single variant that influences binding affinity of NR5A2, alters Lifr expression, and leads to downstream changes in a suite of genes that affect pluripotency.
Discussion
Genetic background strongly influences molecular phenotypes in ESCs, and can be harnessed to discover drivers of differences in ESC biology. These molecular phenotypes are reflections of a developmental continuum of cell states in vitro. Our genetic approach identified multiple loci that influence the dependence on ERK1/2 and GSK3 inhibition, and provides mechanistic details clarifying the recalcitrance of some inbred mouse strains to ESC derivation. We show that natural variation shapes the ability of mESCs to respond to exogenous signals, and why bypass of cytokine signaling through ERK1/2 and GSK3 inhibition stabilizes ground state pluripotency in mESCs from strain that carry the alternate Lifr allele. Natural variation in cellular response to exogenous cues also drives differences in in vitro differentiation capacity as has been demonstrated elsewhere in this issue (Ortmann et al., 2020).
Genetic background is a dominant source of inter-line variability in human (h)ESCs and hiPSCs (Burrows et al., 2016; Carcamo-Orive et al., 2017; Choi et al., 2015; DeBoever et al., 2017; Féraud et al., 2016; Kajiwara et al., 2012; Kyttälä et al., 2016; Osafune et al., 2008; Ramos-Mejia et al., 2010). QTL mapping in panels of differentiated hiPSCs revealed loci controlling transcript abundance and chromatin accessibility in lineage-committed cells (Alasoo et al., 2018; Schwartzentruber et al., 2018). In light of our results it is notable that LIF is dispensable for the maintenance of hESCs (Thomson, 1998). Nevertheless, compared to mESCs, hESCs are phenotypically more similar to mouse EpiSCs isolated from the post-implantation blastocyst (Brons et al., 2007; Tesar et al., 2007), which also do not require LIF. Moreover, hESCs can be coerced into a naive pluripotent state by ectopic expression of pluripotency factors including LIF (Buecker et al., 2010; Hanna et al., 2010), and LIFR is upregulated in naïve hESCs compared to primed, at lower passages (Sahakyan et al., 2017). These data demonstrate that in hESCs LIFR expression is correlated with pluripotent cell states, where higher LIFR expression may be a feature of naive hESCs. Since LIF signaling is involved in blastocyst implantation in humans (Aghajanova, 2004), shared regulatory circuits involving LIF and its receptor may contribute to transcriptional changes occurring in early development in both species. The unique ability of mouse ESCs to provide in vitro and in vivo access to early development, coupled with unbiased and high-resolution mapping enabled by genetic reference populations like the DO, underscores the utility of these resources for biological discovery.
Limitations of study
The Chr 15 QTL hotspot and causal Lifr upstream regulatory SNP only partially explain the transcriptional and epigenetic variation present in DO mESCs cultured in the absence of ERK1/2 inhibition. Our data reveal additional hotspot loci that likely impact ground state metastability and other cell states (e.g. Chrs 3, 5, and 10). For example, the Chr 10 QTL hotspot regulates many genes expressed in the early totipotent/2C-like cell state (Macfarlan et al., 2012). This finding suggests variation in the abundance of this rare and transient state among our DO mESCs, which has intriguing implications for the capture of cells that more persistently display their expanded developmental potential (Baker and Pera, 2018). Further studies of the causal variants underlying these QTL hotspots, as well as high resolution single cell profiling to disentangle heterogeneity of cell states, will be needed to reveal additional genes and pathways driving intraspecies variation in pluripotency and differentiation capacity.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to the lead contact, Laura Reinholdt (laura.reinholdt@jax.org).
Materials Availability
ESC lines derived from strains NOD/ShiLtJ (AC576/GrsJ, JAX #026874) and PWD/PhJ (AC401/GrsrJ, JAX #004660) are available from the Jackson Laboratory. The remaining founder and CC-RIX lines used in this study, as well as mESCs carrying a Nanog-mCherry knock-in and allele swap lines with variable genetic material at the Lifr locus are available from the Reinholdt laboratory with a completed Materials Transfer Agreement. There are restrictions on the availability of Diversity Outbred mESCs used in this study due to overlap with intellectual property claims for the Predictive Biology in vitro genetics platform. Predictive Biology, Inc. offers access to these and additional lines on a commercial basis through their genetic screening and stem cell biology services.
Data and Code Availability
RNA-Seq and ATAC-Seq data have been deposited in the ArrayExpress database at EMBL-EBI (https://www.ebi.ac.uk/arrayexpress/) with the following accession numbers: E-MTAB-7730 (founder inbred strain mESC RNA-Seq); E-MTAB-7728 (DO mESC RNA-Seq); E-MTAB-8759 (DO mESC ATAC-Seq); and E-MTAB-8695 (allele swap mESC RNA-Seq). Genotypes of DO mESCs are available in the Diversity Outbred Database at https://www.jax.org/research-and-faculty/genetic-diversity-initiative/tools-data/diversity-outbred-database (accession “Embryonic stem cell lines from Diversity Outbred mice”). Processed ATAC-Seq and RNA-Seq datasets and code are available at github.com/daskelly/CellStemCell_2020_diverse_mESCs. All data generated in this study, including processed functional genomics data, flow cytometry, qPCR, and cellular assays are available at https://doi.org/10.6084/m9.figshare.12233570.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Founder inbred strain mESCs
Euploid (>70%), germline competent, male mESCs were derived from C57BL/6J (The Jackson Laboratory strain ID JR#000664), 129S1/SvImJ (JR#002448), A/J (JR#000646), BALB/cByJ (JR#001026), NOD/ShiLtJ (JR#001976), NZO/HlLtJ (JR#002105), CAST/EiJ (JR#000928), PWD/PhJ (JR#004660), WSB/EiJ (JR#001145) as previously described. Note that PWD/PhJ is distinct from PWK/PhJ, which is a Collaborative Cross founder strain. Nevertheless, both are inbred strains of the M. m. musculus subspecies that are derived from wild mice trapped near Prague, Czech Republic. Derivation and characterization (mycoplasma testing, SNP genotyping, pluripotency marker expression, chromosome counting, germline testing) of the mESCs were as previously described (Czechanski et al., 2014). For each strain we initially derived 15-40 unique mESC lines from individual blastocysts and adapted lines to ESM, 2i+LIF (“2i”) media in the presence of mitotically inactivated mouse embryonic fibroblasts (MEFs, C57BL/6J) (ESM, 2i+LIF: Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 15% fetal bovine serum, 100 U/mL Penicillin-Streptomycin, 2mM GlutaMAX, 0.1mM non-essential amino acids, 1mM sodium pyruvate, 0.1mM 2-mercaptoethanol, 500pM LIF, 1uM PD0325901, and 3uM CHIR99021). Of these, three male lines from each genotype were selected for germline testing on the basis of robust expression of pluripotency markers and euploidy. We validated the ability of these ESCs to contribute to the germline in vivo, the gold standard for functionally defining pluripotency in mESCs. The resulting 24 germline-competent, euploid mESC lines (Supplementary Table 1) provide a panel of genetically distinct mESCs that differ at >40 million sites across the genome.
Diversity Outbred mESCs
Male and female Diversity Outbred mice (JR #009376) were obtained at approximately four weeks of age and maintained at Predictive Biology, Inc. for several rounds of breeding. At 24-26 days of age females were superovulated and subsequently mated to males aged 7-15 weeks. mESC lines were derived from random blastocysts using previously described protocols (Czechanski et al., 2014).
Blastocysts were transferred to 2i medium in 96 well round-bottom ultra-low attachment plates for 5-7 days. Those that had some inner cell mass outgrowth were dispersed and transferred onto MEF feeders in 96 well flat-bottom tissue culture plates in ES medium (ESM, 1i+LIF: Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 15% fetal bovine serum, 100 U/mL Penicillin-Streptomycin, 2mM GlutaMAX, 0.1mM non-essential amino acids, 1mM sodium pyruvate, 0.1mM 2-mercaptoethanol, approximately 2000U/ml LIF, and 3uM CHIR99021). Recombinant LIF protein was produced using a Chinese Hamster Ovary cell line. As the ES cells expanded, they were transferred into 24 well plates, followed by 6 well plates, and finally 10cM dishes, all without feeder cells but in the same media. Thus, as the cells expanded they were weaned off feeder cells by dilution.
CC-RIX mESCs
Collaborative Cross strains were selected on the basis of their genotype across the Lifr locus (those carrying the alternate allele [NOD, CAST, PWK, WSB] or those carrying the reference allele [AJ, B6, 129S1, NZO]) and mESCs were derived using our previously published method (Czechanski et al., 2014) from F1 (CC-RIX) embryos. 20 mESC lines were selected for further analysis as listed in Table S3.
METHOD DETAILS
Generation of Nanog-mCherry knock-in and allele swap lines
To construct Nanog-mCherry knock-in lines, a CRISPR/cas9 donor construct containing a Nanog-2A-mCherry reporter cassette designed and constructed by Yang et al. (2013) was obtained from Addgene (Plasmid #48680).-Targeting arms were shortened to 1kb so that the updated vector extended from Chr6:122,712,552-122,714,555 (GRCm38/mm10) with the insert at Chr6:122,713,552 (GRCm38/mm10). A guide targeting the last coding exon of Nanog (CCACTTTATACTCTGAATGC) was cloned into the pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene #62988). 5×105 cells seeded on a 12-well plate were co-transfected with equimolar amounts (0.5μg each) of donor and guide vector using Lipofectamine 3000 (Invitrogen #L3000) reagent. Following puromycin selection, positive knock-in cells were isolated by mCherry reporter expression using a FACSAria flow sorter and single mCherry expressing cells were plated individually in 96 well plates for clonal expansion. Standard and long range PCR were used to confirm targeted knock-in (KI) of the reporter cassette. Homozygous Nanog-mCherry KI cell lines or heterozygous KI cell lines with intact WT Nanog alleles were selected using these molecular data. Functional NANOG protein expression was confirmed by flow cytometry.
To construct allele swap lines, CRISPR/Cas9 donors and sgRNAs were designed to engineer reciprocal reference and alternate alleles into the caQTL Lifr locus in 129S1/SvImJ (129S1), WSB/EiJ (WSB), NOD/ShiLtJ (NOD), and C57BL6/J (B6) mESCs. A 128bp oligo donor was selected (Table S4). A 20bp guide (Table S4) was cloned into the pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene #62988). 2×106 cells seeded on a 60mm dish were co-transfected with equimolar amounts (2.5μg each) of donor and guide vector using Lipofectamine 3000 (Invitrogen #L3000) reagent. After 48 hours, cells underwent puromycin selection and after one week colonies were picked and expanded. Approximately 200bp upstream of Lifr was PCR amplified (see Table S4 for primer sequences) in individual clones and capillary sequencing of the resulting PCR products was used to identify correct, homozygous targeting.
Immunolabeling and imaging
For immunolabeling and imaging of founder ESC lines, cells were plated in triplicate at a density of 3,000/ well in 96 well plates and were grown with or without 2i supplementation for ~48hrs. Cells were then fixed using 4% paraformaldehyde (PFA) for 20 minutes at room temperature and washed 3x3 minutes with 1x phosphate buffered saline (PBS). Fixed cells were then blocked with 3% bovine serum albumin (BSA), 0.1% Triton-X, and .05% sodium azide for 45 minutes, incubated with primary antibody O/N @ 4C, washed 3x3 minutes in PBS, and then incubated with secondary antibody 1 hr prior to washing and staining with DAPI for 10 minutes at room temperature to stain nuclei. Primary antibodies included anti-NANOG (Invitrogen eBiosciences eBioMLC-51 660 conjugated catalog # 50-5761-82, 1:80 dilution), anti-Oct3/4 (Santa Cruz Biotechnology C-10 mouse monoclonal IgG2b Catalog # sc-5279, 1:100), anti-SOX2 (R&D Systems Anti-h/mo/rat Sox2 catalog # AF2018, 1:40 dilution). Secondary antibody from Life Technologies Alexafluor donkey-anti mouse 488 IgG (H + L) 1:2000 dilution). Immunolabeled cells and cells expressing the Nanog-mCherry reporter (see below) were imaged on the PerkinElmer Operetta High Content Imaging System at 20X magnification. Nine regions of interest were randomly selected from each well for quantification of fluorescence using Harmony high-content imaging and analysis software version 4.9 with PhenoLOGIC (PerkinElmer).
Flow cytometry for LIFR
Cells for allele swap mESC lines were thawed onto 60mm MEF dishes in ESM + LIF/1i (CHIR99021) and grown for 48 hours, with media replenished daily. Cells were trypsinized, washed with PBS, and fixed in 4% PFA at room temperature for 15 minutes. Cells were rinsed with PBS/1% FBS twice. Cells were blocked using PermWash (BD Biosciences 51-2091KZ), and then incubated with anti-Lifr antibody (R&D Systems FAB5990R), 5ul per sample, for 45 minutes at 30C. After rinsing 2x in Permwash, cells were analyzed on the LSRII cytometer (BD Biosciences).
Nanog-mCherry expression analysis
For each of the 8 Collaborative Cross founder strains, three independent Nanog-mCherry clones were passaged from ESM+2i (“2i”) conditions onto 24 well MEF plates in ESM+2i (“2i”) and in ESM (“no i”) for 48 hours with daily media changes until subconfluent. An additional well with a non-targeted mESC line was plated in both conditions to serve as a negative control for flow cytometry. Wells were then harvested, resuspended in 2 ml of PBS with DAPI and processed on the FACSymphony FlowJo version 10 was used for analysis and the background fluorescence of the negative control was used to adjust for background fluorescence. Immunolabeling and flow cytometry were used to correlate Nanog-mCherry expression with NANOG protein expression as previously described (Reinholdt et al., 2012).
Genotyping of Diversity Outbred mESCs
Cell lines were genotyped by Neogen Corp. (Lincoln, NE) using the Giga Mouse Universal Genotyping Array (GigaMUGA; Morgan et al., 2016). We used functions available in the argyle R package (Morgan, 2015) for quality control purposes. Specifically, we examined plots of B-allele frequency (BAF) and log2 intensity ratio (LRR) to scan for gross aneuploidies and did not move forward with any lines presenting such anomalies. We used the hidden Markov model implemented in DOQTL (Gatti et al., 2014) for haplotype reconstruction to obtain diplotype probabilities suitable for quantitative trait locus mapping.
Founder and allele swap mESC RNA-Seq
Prior to harvesting founder mESC cells (P6-P8) for RNA collection, MEFs were removed through sequential plating (2X, 1 hour) onto gelatin-coated dishes. For founder and allele swap mESCs, 1.5×104 cells were plated onto gelatinized 35mm tissue culture treated dishes. Cells were grown in 2i+LIF (founder lines) or 1i/LIF (CHIR99021; allele swap lines). Allele swap lines remained unfed until harvest, six days later. RNA was harvested using the RNeasy (Qiagen) RNA extraction kit. Poly(A) RNA-seq libraries were constructed using either the TruSeq Stranded mRNA Library Prep Kit (Illumina; founder lines) or KAPA mRNA HyperPrep Kit (KAPA Biosystems; allele swap lines). Founder mESC libraries were pooled and sequenced 125 bp paired-end on the HiSeq 2000 or 2500 (Illumina) using TruSeq SBS Kit v4 reagents (Illumina), while allele swap mESC libraries were sequenced 76bp single-end on the NextSeq 500 (Illumina) using NextSeq High Output Kit v2.5 reagents (Illumina).
For all data, to reduce spurious alignments, we constructed strain-specific genomes for the eight inbred founder strains using the software tool g2gtools and genetic variation data from Mouse Genomes Project SNP and indel release version 5 (ftp://ftp-mouse.sanger.ac.uk/REL-1505-SNPs_Indels/). We used Ensembl gene annotation version release-84 to extract strain-specific transcriptomes and annotations for these strains (ftp://ftp.ensembl.org/pub/release-84/gtf/mus_musculus). RNA-Seq reads from each strain were aligned to the strain-specific transcriptomes using bowtie (Langmead et al., 2009) aligner with parameters “--best --strata -a -m 100 -v 3”. These settings retain all read alignments with the best alignment score allowing up to 3 mismatches for further analysis. We used EMASE (Raghupathy et al., 2018) to quantify isoform-level and gene-level expression abundances. EMASE uses an EM algorithm to accounting for reads with multiple alignments and estimated read counts for isoforms and genes. Read counts were estimated in this manner on both raw and batch corrected data. Further analyses were conducted on both raw and batch corrected read count data to identify and mitigate batch effects. We used DESeq2 (Love et al., 2014) to test for variation in gene expression driven by genetic background (founder mESCs) or Lifr genotype (allele swap mESCs). When comparing gene expression to predictions made from DO eQTL mapping, we used upper quartile normalized read counts. To obtain predictions from eQTL mapping, we extracted QTL effects at the QTL peak for each target of the Chr 15 eQTL hotspot, using effects calculated using the scan1blup function in r/qtl2 (Broman et al., 2018).
Diversity Outbred mESC RNA-Seq
Total RNA was isolated from each of 183 DO mESC lines and quantitated by paired-end RNA sequencing. Briefly, for each mESC line, one 15cm dish of cells was grown to near confluence, washed 3x with PBS, and mechanically harvested to produce 80-150mg wet weight cell pellets (~50M cells). About 100k cells from each frozen cell pellet were used for RNA sequencing. Next, total RNA was extracted using the Quick-RNA 96 well format kit (Zymo Research) with in-column DNase treatment. Sequencing libraries were prepared by Akesogen using the TruSeq Stranded mRNA HT kit (Illumina, Cat no. 20020595) and included ribosomal RNA reduction and poly-A selection, enzymatic fragmentation, cDNA synthesis from random hexamer priming, adapter ligation and PCR amplification steps to generate indexed, stranded mRNA-seq libraries. Libraries were checked for quality and quantitated with the Agilent Bioanalyzer, and samples that failed QC were repeated starting from the cryovial stage. Finally, pooled libraries were sequenced on the NextSeq platform (Illumina) using the NextSeq 500/550 High Output v2 150-cycle kits (Illumina, Cat no. FC-404-2002). To minimize technical variation, samples were randomly assigned to lanes prior to sample processing steps, barcoded, and multiplexed at 16 samples per flow cell, yielding 6M-55M 2×75bp paired-end (PE) reads per sample. Two of the 183 lines were grown in replicate, resulting in a total of 185 samples with RNA-Seq data. For downstream genotyping, quantitation, and eQTL mapping analyses (see below), only the first read of the pair was used.
We aligned single-end 75bp reads with bwa v1.0.0 (Li and Durbin, 2009) to a pooled “8-way” transcriptome containing strain-specific isoform sequences from all eight DO founder strains. To construct the 8-way founder transcriptome, strain single nucleotide variants and short indels (release 1505) were downloaded from the Sanger Mouse Genomes Project (Keane et al., 2011) and incorporated into annotated transcripts (Ensembl release 82) using g2gtools v0.1.31 (https://github.com/churchill-lab/g2gtools). We inferred sample genotypes genome-wide from the RNA-Seq data using gbrs v0.1.6 (http://churchill-lab.github.io/gbrs/), and compared gbrs-derived genotypes to our DNA (GigaMUGA) genotypes to identify potential sample mix-ups. Typically, correlations between haplotype probabilities inferred from gbrs versus GigaMUGA on the same sample were on the order of 0.8-0.9, while correlations for these probabilities measured on different samples were <0.5. Using this procedure, we identified 10 DO mESC lines with incongruent genotypes, and were able to resolve nine of these errors. For the sample where a definitive genotype could not be ascertained, the gbrs genotype was used for QTL mapping.
We applied EMASE v0.10.16 (Raghupathy et al., 2018) to resolve multi-mapping reads and estimate transcript- and gene-level abundance for each sample. We filtered out genes for which the median TPM (transcripts per million) value was <0.5 or where more than half of the samples were zero (i.e. not expressed). This TPM filter was used for removal of genes expressed only at low levels. Next, we returned to raw counts, normalized gene-level counts to the upper quartile value in each sample to account for differences in library size, and then used ComBat (Leek et al., 2012) to ameliorate any potential batch effects stemming from library preparation. We chose upper quartile normalization rather than TPM normalization because the latter is influenced by variation in a small number of the most highly expressed genes (Bullard et al., 2010). Finally, we transformed upper quartile normalized, ComBat-adjusted values to rank normal scores using the ‘rankZ’ function in the DOQTL R package (Gatti et al., 2014).
ATAC-Seq
To measure chromatin accessibility, ~100,000 cryopreserved DO mESCs were used in the Fast-ATAC protocol (Corces et al., 2016). Cryopreserved cells were thawed in a 37°C water bath, an aliquot of ~100,000 cells removed, then spun and washed with cold 50 μl PBS to remove DMSO. The cell pellet was resuspended in the 50 μl transposase reaction mix (25μl of 2X TD buffer, 2.5 μl of TDE1,0.5 μl of 1% digitonin, and 22 μl of nuclease-free water). Transposition was carried out for 30 min at 37°C, and DNA purified using a Qiagen MinElute kit. Libraries were amplified for a total of 9 cycles and purified using 1.7X AMPure beads. Nucleosome banding was visualized using the Agilent Tapestation. Libraries were subject to 100 bp single-end sequencing on an Illumina HiSeq 2500.
We used Trimmomatic version 0.33 (Bolger et al., 2014) to trim Illumina adapters from 100bp ATAC-Seq reads with the following settings “ILLUMINACLIP:NexteraPE-PE.fa:2:30:7 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36”. We mapped trimmed reads to the mouse reference genome (GRCm38) using bwa version 0.7.9a (Li and Durbin, 2009). For each sample, we filtered out secondary alignments and reads with mapping quality equal to zero, and ran MACS version 1.4.2 (Zhang et al., 2008) on the remaining alignments with the following settings “-f BAM -g mm -p 1e-5”. We used the rtracklayer version 1.34.2 R package (Lawrence et al., 2009) to read in peaks called by MACS across each chromosome, saving only those genomic windows that were contained within peaks in at least 5 samples in order to define open chromatin regions that are unlikely to be spurious (yet allowing for the possibility of chromatin that is accessible in only some samples). To reduce the incidence of clusters of narrow open chromatin regions, we merged regions <20bp in width into the nearest region within 100bp. This set of open chromatin regions constituted our consensus set; we then used bedtools version 2.26.0 (Quinlan and Hall, 2010) to quantify ATAC-Seq read depth across regions in each sample. We removed regions that did not exceed one count per million in at least 20 samples (<1% of loci), and normalized at the sample level using the TMM method implemented in the edgeR version 3.16.5 R package (Robinson et al., 2010). Finally, we transformed normalized values to rank normal scores using the ‘rankZ’ function in the DOQTL R package (Gatti et al., 2014).
Quantitative RT-PCR
RNA was extracted from 1×106 CC-RIX mESCs according to the manufacturer’s instructions using RNeasy Plus Mini Kit (Qiagen, cat. No. 74134) after cellular homogenization using QIAshredder (Qiagen, cat. No. 79654). RNA was eluted in 50pl RNase free water and quantified with Nanodrop. cDNA was generated using High Capacity RNA to cDNA Kit (Thermofisher, cat. No. 4387406) with 500ng of RNA per sample. Resulting cDNA was diluted 1/10 for detection of target gene expression. Primer sequences and targets are provided in the Key Resources Table. Quantitative real-time PCR was performed with PowerUp SYBR Green Master Mix (Thermofisher, cat. No. A25742). Standard cycling conditions were performed on ViiA 7 Real-Time PCR System according to manufacturer’s instructions. Relative expression of target genes was determined using the ΔΔCt method with Gapdh as an internal control.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-Nanog (clone eBioMLC-51) | eBioscience | Cat#50-5761-82, RRID: AB_2574243 |
| Mouse monoclonal anti-Oct-3/4 (C-10) | Santa Cruz Biotechnology | Cat#sc-5279, RRID: AB_628051 |
| Goat polyclonal anti-SOX2 | R&D Systems | Cat#AF2018, RRID: AB_355110 |
| Goat polyclonal anti-LIF | R&D Systems | Cat# AB-449-NA, RRID:AB_354362 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse LIF protein | Isolated from Chinese Hamster Ovary (CHO) cell line | n/a |
| ESGRO Recombinant Mouse LIF protein | Millipore | Cat# ESG1107 |
| CHIR99021 GSK-3 inhibitor | Tocris | Cat# 4423, CAS: 252917-06-9 |
| PD0325901 MEK/ERK pathway inhibitor | STEMCELL Technologies | Cat# 72184, CAS: 391210-10-9 |
| Deposited data | ||
| RNA-Seq and ATAC-Seq | ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) | E-MTAB-7730 (founder inbred strain mESC RNA-Seq); E-MTAB-7728 (DO mESC RNA-Seq); E-MTAB-8759 (DO mESC ATAC-Seq); and E-MTAB-8695 (allele swap mESC RNA-Seq) |
| DO mESC genotypes | Diversity Outbred Database (https://www.jax.org/research-and-faculty/genetic-diversity-initiative/tools-data/diversity-outbred-database) | “Embryonic stem cell lines from Diversity Outbred mice” |
| Processed ATAC-Seq and RNA-Seq datasets and code | github.com/daskelly/CellStemCell_2020_diverse_mESCs | N/A |
| Experimental Models: Cell Lines | ||
| NOD/ShiLtJ mESC lines, JAX strain# 1976 | Laboratory of Laura Reinholdt | AC576, AC595, & AC601 |
| NZO/HiltJ mESC lines, JAX strain# 2105 | Laboratory of Laura Reinholdt | AC652, AC653, & AC671 |
| PWD/PhJ mESC lines, JAX strain# 4660 | Laboratory of Laura Reinholdt | AC398, AC401 & AC403 |
| WSB/EiJ mESC lines, JAX strain# 1145 | Laboratory of Laura Reinholdt | AC627, AC635 & AC660 |
| A/J mESC lines, JAX strain# 646 | Laboratory of Laura Reinholdt | AJ13, AJ27 & AJ28 |
| 129S1/SvImJ mESC lines, JAX strain# 2448. | Laboratory of Laura Reinholdt | AC677, AC680 & AC681 |
| Mouse embryonic fibroblasts (MEFs) derived from C57/Bl6J | Laboratory of Laura Reinholdt | N/A |
| 183 Diversity Outbred mESC lines | Predictive Biology | N/A |
| 20 CC:RIX mESC lines, see table_S3 | Laboratory of Laura Reinholdt / this paper | 0104CC01, 0104CC02, 0104CC09, 0218CC02, 0218CC03, 0351CC01, 0511CC01, 0625CC01, 1041CC01, 1041CC05, 1105CC02, 1105CC03, 2506CC07, 3240CC18, 3240CC19, 4360CC03, 4360CC04, 5103CC01, 6109CC03, 6109CC05 |
| C57BL/6J mESC lines, JAX strain# 664 | Cell Biology Department of the Jackson Laboratory | B6#49, B6#139, B6#146 |
| CAST/EiJ mESC lines, JAX strain# 928 | Laboratory of Laura Reinholdt | WM077, WM078, & WM080 |
| Experimental Models: Organisms/Strains | ||
| Mouse: A/J | The Jackson Laboratory | JAX: 000646 |
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: CAST/EiJ | The Jackson Laboratory | JAX: 000928 |
| Mouse: NOD/ShiLtJ | The Jackson Laboratory | JAX: 001976 |
| Mouse: NZO/HiLtJ | The Jackson Laboratory | JAX: 002105 |
| Mouse: PWD/PhJ | The Jackson Laboratory | JAX: 004660 |
| Mouse: WSB/EiJ | The Jackson Laboratory | JAX: 001145 |
| Mouse: 129S1/SvImJ | The Jackson Laboratory | JAX: 002448 |
| Mouse: J:DO | The Jackson Laboratory | JAX: 009376 |
| Mouse: CC001/UncJ | The Jackson Laboratory | JAX: 021238 |
| Mouse: CC002/UncJ | The Jackson Laboratory | JAX: 021236 |
| Mouse: CC003/UncJ | The Jackson Laboratory | JAX: 021237 |
| Mouse: CC004/TauUncJ | The Jackson Laboratory | JAX: 020944 |
| Mouse: CC005/TauUncJ | The Jackson Laboratory | JAX: 020945 |
| Mouse: CC006/TauUncJ | The Jackson Laboratory | JAX: 022869 |
| Mouse: CC010/GeniUncJ | The Jackson Laboratory | JAX: 021889 |
| Mouse: CC011/UncJ | The Jackson Laboratory | JAX: 018854 |
| Mouse: CC018/UncJ | The Jackson Laboratory | JAX: 021890 |
| Mouse: CC025/GeniUncJ | The Jackson Laboratory | JAX: 018857 |
| Mouse: CC032/GeniUncJ | The Jackson Laboratory | JAX: 020946 |
| Mouse: CC040/TauUncJ | The Jackson Laboratory | JAX: 023831 |
| Mouse: CC041/TauUncJ | The Jackson Laboratory | JAX: 021893 |
| Mouse: CC043/GeniUncJ | The Jackson Laboratory | JAX: 023828 |
| Mouse: CC051/TauUncJ | The Jackson Laboratory | JAX: 021897 |
| Mouse: CC060/UncJ | The Jackson Laboratory | JAX: 026467 |
| Mouse: CC061/GeniUncJ | The Jackson Laboratory | JAX: 023826 |
| Oligonucleotides | ||
| Primers for allele swap genome engineering, luciferase reporter, quantitative RTPCR, and Nr5a2 ChIP qPCR see table_S4 | this paper | N/A |
| Nanog guide sequence: CCACTTTATACTCTGAATGC | Yang et al., 2013 | N/A |
| Recombinant DNA | ||
| Nanog-2A-mCherry | Yang et al., 2013 | Addgene# 48680 |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Ran et al., 2013. | Addgene# 62988 |
| Software and Algorithms | ||
| argyle | Morgan 2015 | 10.1534/g3.115.023739 |
| bedtools v2.26.0 | Quinlan and Hall 2010 | 10.1093/bioinformatics/btq033 |
| bowtie | Langmead et al., 2009 | 10.1186/gb-2009-10-3-r25 |
| bwa v0.7.9a | Li and Durbin 2009 | 10.1093/bioinformatics/btp324 |
| ComBat | Leek et al., 2012 | 10.1093/bioinformatics/bts034 |
| DESeq2 | Love et al., 2014 | 10.1186/s13059-014-0550-8 |
| DOQTL | Gatti et al., 2014 | 10.1534/g3.114.013748 |
| edgeR | Robinson et al., 2010 | 10.1093/bioinformatics/btp616 |
| EMASE | Raghupathy et al., 2018 | 10.1093/bioinformatics/bty078 |
| g2gtools v0.1.31 | https://github.com/churchill-lab/g2gtools | N/A |
| GBRS v0.1.6 | https://github.com/churchill-lab/gbrs | N/A |
| intermediate | https://github.com/simecek/intermediate | N/A |
| MACS v1.4.2 | Zhang et al., 2008 | 10.1186/gb-2008-9-9-r137 |
| PANTHER | Mi et al., 2017 | 10.1093/nar/gkw1138 |
| qtl2 | Broman et al., 2018 | 10.1101/414748 |
| rtracklayer | Lawrence et al., 2009 | 10.1093/bioinformatics/btp328 |
| Trimmomatic v0.33 | Bolger et al., 2014 | 10.1093/bioinformatics/btu170 |
| Processed ATAC-Seq and RNA-Seq datasets and code | github.com/daskelly/CellStemCell_2020_diverse_mESCs | N/A |
Self-renewal assays
Suspensions of CC-RIX mESCs were stained with propidium iodide (PI) and flow cytometry was used to isolate and plate one cell per well x 2 96 well MEF feeder plates per cell line. After one week of growth, colonies visible under phase contrast microscopy were counted and percent cloning efficiency was estimated as the number of visible colonies divided by the number of cells plated.
LIF dose response of allele swap lines
mESCs were thawed onto gelatinized dishes into standard culture media containing Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 15% fetal bovine serum, 100 U/mL Penicillin-Streptomycin, 2mM GlutaMAX, 0.1mM non-essential amino acids, 1mM sodium pyruvate, 0.1mM 2-mercaptoethanol, 500pM LIF, 1μM PD0325901, and 3μM CHIR99021. After 48 hours cells were trypsinized, washed, and counted. 3×104 cells were plated per gelatinized 35mm dish in LIF concentrations ranging from 0.05–5000 pM. A control well containing 480 ng/ml of neutralizing anti-LIF antibody (R&D Systems AB-449-NA) was also plated. After 6 days cells were dissociated and the expression of mCherry was analyzed using the FACsymphony cytometer. Prior to flow cytometry analysis, the cells were stained with DAPI to determine viability.
Chromatin immunoprecipitation
For ChIP-qPCR, 129S1 mESCs were grown under LIF + 2i conditions on feeders. For each replicate ChIP, 3×106 cells were placed in 5ml of PBS pH 7.4 supplemented with 1mM MgCl2. A fresh 0.25M stock of Disuccinimidyl glutarate (DSG) was prepared in DMSO, added at a final concentration of 2mM, and cells were incubated at room temperature for 30 minutes with rotation. After incubation, fresh paraformaldehyde was added to a final concentration of 1% and cells were incubated for 5 minutes with rotation, and quenched with 125mM glycine. Cells were centrifuged and washed once with PBS, snap frozen, and stored at −80°C until use. Cell lysis, chromatin fragmentation, dialysis, and immunoprecipitation was performed as described (Baker et al., 2015). Immunoprecipitation was performed using 4μl anti-NR5A2 (Abcam ac189876) or normal mouse IgG (Millipore/Sigma #12-371, lot: 2880788) as negative control. qPCR was performed using PowerUp SYBR Green Master Mix (ThermoFisher #A25742) in a 20μl reaction using 2μl of purified ChIP or “input” DNA and run on the Viia7 Real-Time PCR System (ThermoFisher) for 40 cycles including a melting curve to determine primer specificity. Primers for qPCR are listed in Table S4. Each input DNA and ChIP reaction was run in triplicate and CT values were estimated using automatic threshold. Cycle threshold (Ct) values were calculated by averaging technical replicates and percent recovery was calculated by: 2(Ct input – Ct ChIP)*100.
Luciferase assay
Dual luciferase assay was performed using pGL4.23 (firefly, Promega #E8411) and pGL4.74 (Renilla, E6921) as transfection control. We cloned an 860bp region 5’ of the Oct4 locus (positive control, Chr17:35504584-35505443) and 509 bp Lifr enhancer element (Chr15:7116691-7117199) using genomic DNA from 129S1 mice. To generate the ALT allele of Lifr enhancer, we used the QuikChange Site-Directed Mutagenesis kit (Agilent #200523) to change T-to-A. The NR5A2 binding site deletion allele was generated by CRISPR/Cas9 and consisted of an 11 bp deletion (Chr15:7116963-7116974) overlapping 3bp of the putative NR5A2 binding site. The pGL4.23 empty vector was used for negative control. Primers for site-directed mutagenesis and cloning are listed in Table S4. 129S1 cells were transfected in solution with 100ng firefly and 10ng Renilla plasmid per well using Lipofectamine 3000 (Thermofisher #L3000008) following manufacturers recommendations, and seeded (1.3×104 cells/well) onto 96-well plates (Corning #3610) in 1i/LIF (CHIR99021) with feeders. After 24 hours growth, cells were lysed and luciferase activity was measured using the SpectraMax DuoLuc Reporter Assay (Molecular Devices #R8361) following manufacturers guidelines and detected using the SpectroMax i3x Microplate Reader (Molecular Devices).
QUANTIFICATION AND STATISTICAL ANALYSIS
Please refer to sections above (METHOD DETAILS) for details of quantitative and statistical analysis of ATAC-Seq and RNA-Seq data, which are integrated into sections describing the acquisition of these data. Statistical tests used for comparisons of data acquired via qRT-PCR, our self renewal assay, chromatin immunoprecipitation, or the luciferase assay are indicated in the main text and figure legends. All calculations were carried out using R version 3.5 (R Core Team, 2018).
caQTL and eQTL mapping
We performed caQTL and eQTL mapping on normalized, transformed gene-level expression values described above using the ‘scan1’ function in r/qtl2 (Broman et al., 2018). We included sex as an additive covariate in our mapping model for eQTL, and sex and sequencing plate for caQTL. To assess genome-wide significance, we applied a permutation strategy (1,000 permutations). Using this approach, we established a cutoff of LOD >7.5 for reporting significant eQTL and >7.6 for reporting significant caQTL.
We defined distant QTL as QTL where the location of the peak was greater than 10Mb from the genomic feature being mapped. To define hotspots, we started with lists of distant caQTL and eQTL with LOD scores exceeding a genome-wide permutation-based threshold (P < 0.05; LOD 7.5 for eQTL and 7.6 for caQTL). We tallied up distant links within overlapping 1cM bins (0.25cM shift) across the genome. We selected the top 0.5% of bins with most distant links (for each data type; thresholds corresponded to 33 for RNA and 137 for ATAC) and defined these loci as hotspots. We collapsed adjacent bins into a single region to obtain coordinates of hotspot loci. We used PANTHER (Mi et al., 2017) to perform gene ontology enrichment tests of hotspot targets.
Mediation analysis
We used the ‘intermediate’ package in R (https://github.com/simecek/intermediate) to perform mediation analysis to identify transcripts and regions of open chromatin in that region that were likely to be the causal mediator of distant eQTL and caQTL. Briefly, to perform mediation for a single distant QTL, we first identified expressed genes and open chromatin regions within +/−5Mb of the peak SNP. We then included the transcript abundance (or peak intensity) of these candidate mediators individually as additive covariates in the QTL mapping model, and compared LOD scores at the peak distant SNP with and without the addition of this covariate. In cases where the distant QTL effect is mediated by the chromatin state or transcript abundance of a gene near the locus, inclusion of that parameter as an additive covariate in the mapping model decreases the distant QTL effect, as evidenced by a decrease in LOD score. We calculated LOD scores using the ‘double-lod-diff’ method in r/intermediate to minimize the effects of missing data in our RNA-seq and ATAC-seq data sets. For mediation of caQTL and eQTL hotspots, we considered only transcript abundances as candidates to mediate the targets of each hotspot. We defined hotspot genomic coordinates as above and considered a gene to be a candidate mediator if it was located within or <5Mb from the hotspot boundaries. To calculate p-values to quantify the significance of observed decreases in LOD score after mediation, we built target-specific empirical distributions of LOD decreases using all transcripts outside the hotspot locus. We used these distributions to compute Z scores and calculate p-values for each candidate mediator-target pair. We adjusted for multiple testing using the FDR as implemented in the p.adjust function in R (method=”BH”).
Supplementary Material
Table S1, related to Figure 1. Inbred strain mESC lines and quality control measures, along with results of RNA-Seq differential expression tests using DESeq2.
Table S2, related to Figure 2. Genome coordinates of hotspot QTL and list of mediation results for QTL peaks that may be regulated by each hotspot.
Table S3, related to Figure 3. The Collaborative Cross strains, Chr 15 QTL region genotypes, and crosses used to generate CC-RIX mESCs.
Table S4, related to Figure 4. Oligonucleotides used in CRISPR/Cas9 genome engineering and primers used for luciferase reporter and ChIP qPCR assays.
Highlights:
Genetically diverse mouse ESCs have distinct molecular and functional properties
Genetic mapping reveals thousands of loci that affect chromatin accessibility
Ten QTL hotspots coordinate genome-wide chromatin and/or gene expression changes
A Chr15 QTL harbors an SNV that drives changes in Lifr transcript levels and function
Acknowledgements
We thank David Aylor and Thomas Konneker for early contributions in the analysis of the inbred strain transcriptome data. We thank Steve Murray, Kevin Peterson, and Susan Kales for sharing plasmids. We thank Martin Pera and Daniel Cortes-Perez for constructive feedback. We thank Richard Paules and Kristine Witt for contributing to the development of the DO mESCs and Alex Merrick of NIEHS for manuscript review. Funding sources included the NIH/NIEHS (AHH), grants HHSN273201500196P (NIEHS-NTP to TC), OD010921 (LGR), OD011102 (LGR), GM133724 (CLB), GM070683 (GAC), T32HD007065 (CB), DA039841-04 (LGR), GM133495 (SCM), and The Jackson Laboratory (CLB, SCM, LGR, GAC, DAS, AC, IG, NR). The University of Virginia Genome Analysis and Technology Core and The Jackson Laboratory Genome Technologies Service (JAX-GT), supported by P30CA034196, provided sequencing services. The views expressed in this manuscript do not represent the official views or endorsement of the NIH or the US government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
TC and DKP have an equity interest in Predictive Biology, Inc.
References
- Abranches E, Guedes AMV, Moravec M, Maamar H, Svoboda P, Raj A, and Henrique D (2014). Stochastic NANOG fluctuations allow mouse embryonic stem cells to explore pluripotency. Dev. Camb. Engl 141, 2770–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghajanova L (2004). Leukemia inhibitory factor and human embryo implantation. Ann. N. Y. Acad. Sci 1034, 176–183. [DOI] [PubMed] [Google Scholar]
- Alasoo K, Rodrigues J, Mukhopadhyay S, Knights AJ, Mann AL, Kundu K, HIPSCI Consortium, Hale C, Dougan G, and Gaffney DJ (2018). Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat. Genet 50, 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CL, and Pera MF (2018). Capturing Totipotent Stem Cells. Cell Stem Cell 22, 25–34. [DOI] [PubMed] [Google Scholar]
- Baker CL, Kajita S, Walker M, Saxl RL, Raghupathy N, Choi K, Petkov PM, and Paigen K (2015). PRDM9 drives evolutionary erosion of hotspots in Mus musculus through haplotype-specific initiation of meiotic recombination. PLoS Genet. 11, e1004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma. Oxf. Engl 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Broman KW, Gatti DM, Simecek P, Furlotte NA, Prins P, Sen S, Yandell BS, and Churchill GA (2018). R/qtl2: software for mapping quantitative trait loci with high dimensional data and multi-parent populations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brons IGM, Smithers LE, Trotter MWB, Rugg-Gunn P, Sun B, Chuva de Sousa Lopes SM, Howlett SK, Clarkson A, Ahrlund-Richter L, Pedersen RA, et al. (2007). Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448, 191–195. [DOI] [PubMed] [Google Scholar]
- Buecker C, Chen H-H, Polo JM, Daheron L, Bu L, Barakat TS, Okwieka P, Porter A, Gribnau J, Hochedlinger K, et al. (2010). A murine ESC-like state facilitates transgenesis and homologous recombination in human pluripotent stem cells. Cell Stem Cell 6, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, and Dudoit S (2010). Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows CK, Banovich NE, Pavlovic BJ, Patterson K, Gallego Romero I, Pritchard JK, and Gilad Y (2016). Genetic Variation, Not Cell Type of Origin, Underlies the Majority of Identifiable Regulatory Differences in iPSCs. PLoS Genet. 12, e1005793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carcamo-Orive I, Hoffman GE, Cundiff P, Beckmann ND, D’Souza SL, Knowles JW, Patel A, Papatsenko D, Abbasi F, Reaven GM, et al. (2017). Analysis of Transcriptional Variability in a Large Human iPSC Library Reveals Genetic and Non-genetic Determinants of Heterogeneity. Cell Stem Cell 20, 518–532.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ, Gatti DM, Morgan AP, Strobel M, Trepanier L, Oberbeck D, McWeeney S, Hitzemann R, Ferris M, McMullan R, et al. (2016). Diversity Outbred Mice at 21: Maintaining Allelic Variation in the Face of Selection. g3 Bethesda Md 6, 3893–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick JM, Munger SC, Simecek P, Huttlin EL, Choi K, Gatti DM, Raghupathy N, Svenson KL, Churchill GA, and Gygi SP (2016). Defining the consequences of genetic variation on a proteome-wide scale. Nature 534, 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Lee S, Mallard W, Clement K, Tagliazucchi GM, Lim H, Choi IY, Ferrari F, Tsankov AM, Pop R, et al. (2015). A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and eScs . Nat. Biotechnol 33, 1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, et al. (2004). The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet 36, 1133–1137. [DOI] [PubMed] [Google Scholar]
- Churchill GA, Gatti DM, Munger SC, and Svenson KL (2012). The Diversity Outbred mouse population. Mamm. Genome Off. J. Int. Mamm. Genome Soc 23, 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. (2016). Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet 48, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czechanski A, Byers C, Greenstein I, Schrode N, Donahue LR, Hadjantonakis A-K, and Reinholdt LG (2014). Derivation and characterization of mouse embryonic stem cells from permissive and nonpermissive strains. Nat. Protoc 9, 559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoever C, Li H, Jakubosky D, Benaglio P, Reyna J, Olson KM, Huang H, Biggs W, Sandoval E, D’Antonio M, et al. (2017). Large-Scale Profiling Reveals the Influence of Genetic Variation on Gene Expression in Human Induced Pluripotent Stem Cells. Cell Stem Cell 20, 533–546.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MJ, and Kaufman MH (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156. [DOI] [PubMed] [Google Scholar]
- Féraud O, Valogne Y, Melkus MW, Zhang Y, Oudrhiri N, Haddad R, Daury A, Rocher C, Larbi A, Duquesnoy P, et al. (2016). Donor Dependent Variations in Hematopoietic Differentiation among Embryonic and Induced Pluripotent Stem Cell Lines. PloS One 11, e0149291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RL, and Brook FA (1997). Reflections on the biology of embryonic stem (ES) cells. Int. J. Dev. Biol 41, 235–243. [PubMed] [Google Scholar]
- Gatti DM, Svenson KL, Shabalin A, Wu L-Y, Valdar W, Simecek P, Goodwin N, Cheng R, Pomp D, Palmer A, et al. (2014). Quantitative trait locus mapping methods for diversity outbred mice. G3 Bethesda Md 4, 1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu P, Goodwin B, Chung AC-K, Xu X, Wheeler DA, Price RR, Galardi C, Peng L, Latour AM, Koller BH, et al. (2005). Orphan nuclear receptor LRH-1 is required to maintain Oct4 expression at the epiblast stage of embryonic development. Mol. Cell. Biol 25, 3492–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Markoulaki S, Mitalipova M, Cheng AW, Cassady JP, Staerk J, Carey BW, Lengner CJ, Foreman R, Love J, et al. (2009). Metastable pluripotent states in NOD-mouse-derived ESCs. Cell Stem Cell 4, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Cheng AW, Saha K, Kim J, Lengner CJ, Soldner F, Cassady JP, Muffat J, Carey BW, and Jaenisch R (2010). Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc. Natl. Acad. Sci. U. S. A 107, 9222–9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson PG, Doráis JA, Grow EJ, Whiddon JL, Lim J-W, Wike CL, Weaver BD, Pflueger C, Emery BR, Wilcox AL, et al. (2017). Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet 49, 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng J-CD, Feng B, Han J, Jiang J, Kraus P, Ng J-H, Orlov YL, Huss M, Yang L, Lufkin T, et al. (2010). The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell 6, 167–174. [DOI] [PubMed] [Google Scholar]
- Hubley R, Finn RD, Clements J, Eddy SR, Jones TA, Bao W, Smit AFA, and Wheeler TJ (2016). The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiuchi T, Enriquez-Gasca R, Mizutani E, Bošković A, Ziegler-Birling C, Rodriguez-Terrones D, Wakayama T, Vaquerizas JM, and Torres-Padilla M-E (2015). Early embryonic-like cells are induced by downregulating replication-dependent chromatin assembly. Nat. Struct. Mol. Biol 22, 662–671. [DOI] [PubMed] [Google Scholar]
- Kajiwara M, Aoi T, Okita K, Takahashi R, Inoue H, Takayama N, Endo H, Eto K, Toguchida J, Uemoto S, et al. (2012). Donor-dependent variations in hepatic differentiation from human-induced pluripotent stem cells. Proc. Natl. Acad. Sci. U. S. A 109, 12538–12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase E, Suemori H, Takahashi N, Okazaki K, Hashimoto K, and Nakatsuji N (1994). Strain difference in establishment of mouse embryonic stem (ES) cell lines. Int. J. Dev. Biol 38, 385–390. [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyttälä A, Moraghebi R, Valensisi C, Kettunen J, Andrus C, Pasumarthy KK, Nakanishi M, Nishimura K, Ohtaka M, Weltner J, et al. (2016). Genetic Variability Overrides the Impact of Parental Cell Type and Determines iPSC Differentiation Potential. Stem Cell Rep. 6, 200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, and Carey V (2009). rtracklayer: an R package for interfacing with genome browsers. Bioinforma. Oxf. Engl 25, 1841–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, and Storey JD (2012). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinforma. Oxf. Engl 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesurf R, Cotto KC, Wang G, Griffith M, Kasaian K, Jones SJM, Montgomery SB, Griffith OL, and Open Regulatory Annotation Consortium (2016). ORegAnno 3.0: a community-driven resource for curated regulatory annotation. Nucleic Acids Res. 44, D126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, Firth A, Singer O, Trono D, and Pfaff SL (2012). Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks H, Kalkan T, Menafra R, Denissov S, Jones K, Hofemeister H, Nichols J, Kranz A, Stewart AF, Smith A, et al. (2012). The transcriptional and epigenomic foundations of ground state pluripotency. Cell 149, 590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GR (1981). Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. U. S. A 78, 7634–7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, and Thomas PD (2017). PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45, D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AP (2015). argyle: An R Package for Analysis of Illumina Genotyping Arrays. G3 Bethesda Md 6, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AP, Fu C-P, Kao C-Y, Welsh CE, Didion JP, Yadgary L, Hyacinth L, Ferris MT, Bell TA, Miller DR, et al. (2016). The Mouse Universal Genotyping Array: From Substrains to Subspecies. G3 Genes Genomes Genet. 6, 263–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse ENCODE Consortium, Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, Groudine M, Bender M, Kaul R, et al. (2012). An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol. 13, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Silva J, Roode M, and Smith A (2009). Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Dev. Camb. Engl 136, 3215–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka S, and Niwa H (2015). The differential activation of intracellular signaling pathways confers the permissiveness of embryonic stem cell derivation from different mouse strains. Dev. Camb. Engl 142, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osafune K, Caron L, Borowiak M, Martinez RJ, Fitz-Gerald CS, Sato Y, Cowan CA, Chien KR, and Melton DA (2008). Marked differences in differentiation propensity among human embryonic stem cell lines. Nat. Biotechnol 26, 313–315. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinforma. Oxf. Engl 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018). R: A Language and Environment for Statistical Computing (Vienna, Austria: R Foundation for Statistical Computing; ). [Google Scholar]
- Raghupathy N, Choi K, Vincent MJ, Beane GL, Sheppard KS, Munger SC, Korstanje R, Pardo-Manual de Villena F, and Churchill GA (2018). Hierarchical analysis of RNA-seq reads improves the accuracy of allele-specific expression. Bioinforma. Oxf. Engl 34, 2177–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Mejia V, Melen GJ, Sanchez L, Gutierrez-Aranda I, Ligero G, Cortes JL, Real PJ, Bueno C, and Menendez P (2010). Nodal/Activin signaling predicts human pluripotent stem cell lines prone to differentiate toward the hematopoietic lineage. Mol. Ther. J. Am. Soc. Gene Ther 18, 2173–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinholdt LG, Howell GR, Czechanski AM, Macalinao DG, Macnicoll KH, Lin C-S, Donahue LR, and John SWM (2012). Generating embryonic stem cells from the inbred mouse strain DBA/2J, a model of glaucoma and other complex diseases. PloS One 7, e50081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinforma. Oxf. Engl 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahakyan A, Kim R, Chronis C, Sabri S, Bonora G, Theunissen TW, Kuoy E, Langerman J, Clark AT, Jaenisch R, et al. (2017). Human Naive Pluripotent Stem Cells Model X Chromosome Dampening and X Inactivation. Cell Stem Cell 20, 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, Ruff TG, Milligan SB, Lamb JR, Cavet G, et al. (2003). Genetics of gene expression surveyed in maize, mouse and man. Nature 422, 297–302. [DOI] [PubMed] [Google Scholar]
- Schwartzentruber J, Foskolou S, Kilpinen H, Rodrigues J, Alasoo K, Knights AJ, Patel M, Goncalves A, Ferreira R, Benn CL, et al. (2018). Molecular and functional variation in iPSC-derived sensory neurons. Nat. Genet 50, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, Nichols J, Theunissen TW, Guo G, van Oosten AL, Barrandon O, Wray J, Yamanaka S, Chambers I, and Smith A (2009). Nanog is the gateway to the pluripotent ground state. Cell 138, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesar PJ, Chenoweth JG, Brook FA, Davies TJ, Evans EP, Mack DL, Gardner RL, and McKay RDG (2007). New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448, 196–199. [DOI] [PubMed] [Google Scholar]
- Thomson JA (1998). Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 282, 1145–1147. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, and Churchill GA (2012). Ten Years of the Collaborative Cross. Genetics 190, 291–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, and Jaenisch R (2013). One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q-L, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, and Smith A (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, Chen C-H, Brown M, Zhang X, Meyer CA, et al. (2019). Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 47, D729–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Figure 1. Inbred strain mESC lines and quality control measures, along with results of RNA-Seq differential expression tests using DESeq2.
Table S2, related to Figure 2. Genome coordinates of hotspot QTL and list of mediation results for QTL peaks that may be regulated by each hotspot.
Table S3, related to Figure 3. The Collaborative Cross strains, Chr 15 QTL region genotypes, and crosses used to generate CC-RIX mESCs.
Table S4, related to Figure 4. Oligonucleotides used in CRISPR/Cas9 genome engineering and primers used for luciferase reporter and ChIP qPCR assays.
Data Availability Statement
RNA-Seq and ATAC-Seq data have been deposited in the ArrayExpress database at EMBL-EBI (https://www.ebi.ac.uk/arrayexpress/) with the following accession numbers: E-MTAB-7730 (founder inbred strain mESC RNA-Seq); E-MTAB-7728 (DO mESC RNA-Seq); E-MTAB-8759 (DO mESC ATAC-Seq); and E-MTAB-8695 (allele swap mESC RNA-Seq). Genotypes of DO mESCs are available in the Diversity Outbred Database at https://www.jax.org/research-and-faculty/genetic-diversity-initiative/tools-data/diversity-outbred-database (accession “Embryonic stem cell lines from Diversity Outbred mice”). Processed ATAC-Seq and RNA-Seq datasets and code are available at github.com/daskelly/CellStemCell_2020_diverse_mESCs. All data generated in this study, including processed functional genomics data, flow cytometry, qPCR, and cellular assays are available at https://doi.org/10.6084/m9.figshare.12233570.