

Abstract
Background & Aims:
Studies are needed to determine the mechanism by which Barrett’s esophagus (BE) progresses to esophageal adenocarcinoma (EAC). Notch signaling maintains stem cells in the gastrointestinal tract and is dysregulated during carcinogenesis. We explored the relationship between Notch signaling and goblet cell maturation, a feature of BE, during EAC pathogenesis.
Methods:
We measured goblet cell density and levels of Notch mRNAs in BE tissues from 164 patients, with and without dysplasia or EAC, enrolled in a multicenter study. We analyzed the effects of conditional expression of an activated form of NOTCH2 (pL2.Lgr5.N2IC), conditional deletion of NOTCH2 (pL2.Lgr5.N2fl/fl), or loss of NF-κB (pL2.Lgr5.p65fl/fl), in Lgr5+ (progenitor) cells in L2-IL1B mice (overexpresses interleukin 1 beta in esophagus and squamous forestomach and are used as a model of BE). We collected esophageal and stomach tissues and performed histology, immunohistochemistry, flow cytometry, transcriptome, and real-time PCR analyses. Cardia and forestomach tissues from mice were cultured as organoids and incubated with inhibitors of Notch or NF-kB.
Results:
Progression of BE to EAC was associated with a significant reduction in goblet cell density, compared to non-dysplastic regions of tissues from patients; there was an inverse correlation between goblet cell density and levels of NOTCH3 and JAG2 mRNA. In mice, expression of the activated intracellular form of NOTCH2 in Lgr5+ cells reduced goblet-like cell maturation, increased crypt fission, and accelerated development of tumors in the squamocolumnar junction. Mice with deletion of NOTCH2 from Lgr5+ cells had increased maturation of goblet-like cells, reduced crypt fission, and developed fewer tumors. Esophageal tissues from in pL2.Lgr5.N2IC mice had increased levels of RelA (encodes the p65 unit of NF-κB) compared to tissues from L2-IL1B mice, and we found evidence of increased NF-κB activity in Lgr5+ cells. Esophageal tissues from pL2.Lgr5.p65fl/fl mice had lower inflammation and metaplasia scores than pL2.Lgr5.N2IC mice. In organoids derived from pL2-IL1B mice, the NF-κB inhibitor JSH-23 reduced cell survival and proliferation.
Conclusions:
Notch signaling contributes to activation of NF-κB and regulates differentiation of gastric cardia progenitor cells in a mouse model of BE. In human esophageal tissues, progression of BE to EAC associated with reduced goblet cell density and increased levels of Notch expression. Strategies to block this pathway might be developed to prevent EAC in patients with BE.
Keywords: Barrett’s esophagus, esophageal adenocarcinoma, goblet cells, carcinogenesis
Lay Summary:
We identified a pathway in esophageal tissues of patients with BE that mediates cell differentiation and promotes progression to EAC.
INTRODUCTION
Barrett’s esophagus (BE) is a precursor lesion to esophageal adenocarcinoma (EAC), and is characterized by replacement of squamous epithelium in the distal esophagus by metaplastic columnar epithelium.1 BE can progress to low-grade (LGD) and high-grade dysplasia (HGD) and ultimately to EAC. Prevalence estimates of BE range from 1% to 5%, and BE progresses to EAC at a rate of 0.1%-0.3% per year.2, 3, 4 In light of the low rates of progression to EAC, a detailed understanding of the biological processes underlying neoplastic progression is critical to develop personalized strategies for patient management.
Based on various gastroenterology society guidelines,5, 6 the presence of columnar-lined epithelium with goblet cells (GC) is the hallmark of BE. However, there is evidence to suggest that decreased GC density in BE could potentially be associated with an increased risk of EAC.7, 8 In fact, a recent study reported worse outcomes in EAC when not associated with GC intestinal metaplasia at the time of surgical resection.9 The underlying molecular mechanisms of GC differentiation and maturation during BE to EAC progression remain enigmatic.4 Notch signaling plays a key role in regulating stem cell differentiation in the gastro-intestinal tract and may be particularly relevant to BE and progression to EAC, as previously demonstrated by pharmacological pan-Notch inhibition.10 Stem cells of the intestinal epithelium control crypt homeostasis and GC maturation through morphogen-like gradient of Wnt and Notch signaling. Binding of Notch ligands results in an activated form of the Notch receptor, intracellular domain of Notch (Notch-IC), which is translocated into the nucleus to form a transcriptionally active complex with RBP/CSL. This in turn may physically interact with other transcription factors including NF-κB, which appears to be an essential mediator of signaling from the tumor microenvironment. Aberrant activation of Notch signaling and its impact on differentiation events has been suggested in histologic observations of BE patients and cell lines as well as in the L2-IL1B mouse model of BE.10-12
We hypothesized that increased Notch signaling contributes to EAC pathogenesis and is manifested phenotypically by decreased GC density within the BE segment. In order to understand the functional role of Notch signaling and assess its relationship to GC differentiation, we conducted parallel studies in humans and in mice. In humans, we carried out a multi-center prospective cross-sectional study of patients with BE, with and without dysplasia or EAC. We utilized the L2-IL1B mouse model of BE that expresses human interleukin-1β in the esophagus and forestomach squamous epithelium, resulting in inflammation with development of metaplasia and progression to dysplasia at the squamocolumnar junction.12, 13 Using these mice, we genetically engineered abolished or activated Notch signaling in progenitor cells that reside in the gastric cardia to gain insight as to how aberrant Notch signaling and the inflammatory microenvironment alter stem cell function and contribute to disease progression to EAC.
MATERIALS AND METHODS
Human Subjects
We performed a prospective, multi-center cross-sectional study of 164 patients presenting for upper endoscopy with non-dysplastic BE (NDBE), low grade dysplasia (LGD), high grade dysplasia (HGD), or EAC, as well as non-BE controls. Inclusion criteria included age ≥18 years and (for BE subjects) history of histologically confirmed BE, defined as endoscopic BE length ≥2 cm and presence of intestinal metaplasia on esophageal biopsies. We collected data with regard to demographics, anthropometrics (height, weight, and waist and hip circumference), medical history, family history of BE or EAC, medications (including specific notation of proton pump inhibitors, aspirin and other non-steroidal anti-inflammatory medications, and statins), history of GERD symptoms, and exposures (including smoking history). Characteristics of these patients are shown in Supplementary Table 1. During the endoscopy, biopsies were taken for clinical purposes, and additional biopsies were taken from the mid-BE segment, avoiding any visible nodules or other abnormalities, placed in Qiagen AllProtect® and frozen at −80°C. Approval for this study was obtained from the Institutional Review Boards at Columbia University Medical Center, the University of Pennsylvania, and Mayo Clinic (Rochester).
Statistics
Categorical variables were compared between groups using chi-squared tests, and continuous variables were compared using t-tests or rank sum tests as appropriate. Kruskal-Wallis tests were performed to compare continuous variables across three or more groups. Multivariable linear regression was performed to assess for the association between histology and GC density, adjusting for potential confounders. The final model included variables associated with GC density on univariate analyses with a p-value <.05. Statistical significance was defined as p<.05.
Mouse Models
Our genetic mouse model of BE (Tg(ED-L2-IL1RN/IL1B)#Tcw mice (a model of BE, called L2-IL1B) that overexpresses interleukin-1β in the mouse esophagus and squamous forestomach12 as well as the Lgr5-EGFP-ires-CreERT214 Notch2-IC,15 Notch2 fl/fl,16 p65/RelA fl/fl17 and Rosa26-LacZ18 mouse models have been described previously. All mice were intercrossed to obtain the genotypes L2-IL1B.Lgr5-CreERT2-EGFP (pL2-IL1B), L2-IL1B.Lgr5-CreERT2-EGFP.Notch2fl/fl (pL2. Lgr5. N2fl/fl), L2-IL1B.Lgr5-CreERT2-EGFP.Rosa26-Notch2-IC (pL2.Lgr5.N2IC) and L2-IL1B.Lgr5-CreERT2-EGFP.p65fl/fl (pL2.Lgr5.p65fl/fl) mice. Tamoxifen (6 mg, T5648-5G, Sigma) was administered to Lgr5CreERT2 mice (and all comparison groups) starting at 4 months, 3 times within one week and then every 2 months, resulting in respective expression levels of Notch2 in Lgr5+ cells. As this dose of tamoxifen would be expected to induce gastric injury due to parietal cell loss19, all comparison groups received the same tamoxifen treatment. After induction, mice were carried on for another 3, 6, 9, or 12 months, termed time points are 7, 10, 13 and 16 months. Genotyping was routinely performed. All animal experiments were approved by the District Government of Upper Bavaria and performed in accordance with the German Animal Welfare and Ethical Guidelines of the Klinikum rechts der Isar, TUM, Munich, Germany.
Additional details on macroscopic scoring as well as experiments using immunohistochemistry, flow cytometry, organoid culture, transcriptional profiling, and real-time PCR analyses (Supplementary Table 3) are provided in the Supplementary Methods.
RESULTS
Upregulation of Notch signaling occurs with progression to EAC and inversely correlates with goblet cell differentiation in humans
In the non-dysplastic regions of the BE segment in humans, there was a significant decline in GC density with progression from an overall diagnosis of no dysplasia to EAC (Figure 1A and B). In multivariable linear regression analyses, the presence of HGD or EAC remained significantly associated with decreased GC density in non-dysplastic regions of the BE segment (p=.001), while male sex (p=.04) and longer BE segment length (p=.005) were independently associated with increased GC density. There was a significant correlation within individuals between GC density and both TFF3 (rho=0.54, p<.001) and MUC2 expression (rho=0.59, p<.001) (Figure 1C and D), supporting the use of these genes as markers of GC density within the BE segment.
Figure 1.
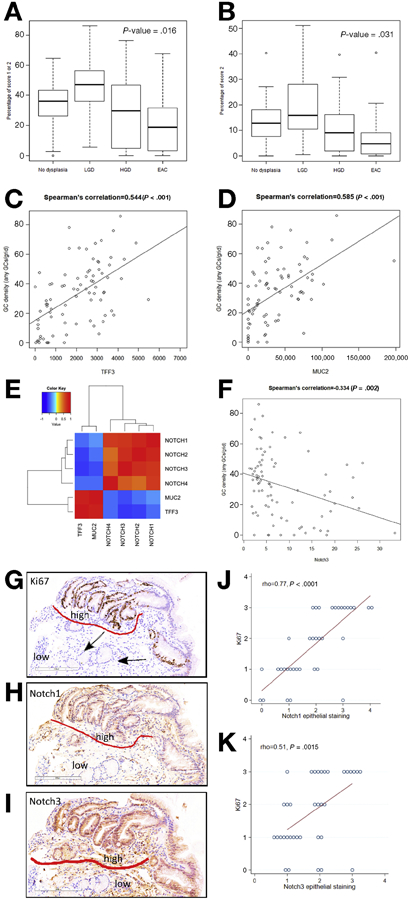
There was a significant decrease in GC density from non-dysplastic regions of BE epithelium with neoplastic progression in patients without dysplasia, low grade dysplasia (LGD), high grade dysplasia (HGD), and adenocarcinoma (EAC), measured for each subject as proportion of evaluable grids with any GCs (A) and proportion of evaluable grids with >3 GCs (B). Expression of TFF3 (C) and MUC2 (D) were also correlated with GC density Correlogram of the expression of TFF3 and MUC2 and NOTCH1-4 (E). Significant inverse correlation between NOTCH3 and GC density (F). Representative IHC images (G-I) demonstrating that glands with high Notch1 and Notch3 expression on IHC contained few GCs and also showed high levels of proliferation (e.g. for Ki67, “high” and “low” refer to regions of high and low expression, respectively, GCs are labeled with an arrow). IHC analyses demonstrated significant correlation between Notch1 and Notch3 with Ki67 (J, K).
As Notch signaling is involved in the regulation of GC differentiation in intestinal epithelium, we also assessed for a relationship between Notch signalling and GC gene markers. All of the Notch receptors (NOTCH 1-4) were significantly, inversely associated with expression of both TFF3 and MUC2 (Figure 1E and Supplementary Table 4). Furthermore, there was significant positive correlation between expression of all the Notch receptors with each other. NOTCH2 and NOTCH3 were the receptors most strongly, inversely associated with TFF3 expression (NOTCH2) rho=−0.56, p<2.5x10−9; NOTCH3 rho=−0.60, p<2.2x10−16). These two receptors were highly correlated with each other (rho=0.71, p<2.2x10−16) and also inversely correlated with GC density (Figure 1F). On immunohistochemistry analyses, Notch1 and Notch3 expression were both observed in the epithelium and the stroma, although overall Notch1 expression was greater in the epithelium and Notch3 was greater in the stroma. There was no significant correlation between global goblet density score and Notch expression by IHC; however, upon visual inspection, there was both increased epithelial Notch expression and proliferation seen together with decreased GC differentiation within corresponding glands (Figure 1G-I).
As Notch receptors and ligands that may be involved in the development of EAC have not been well described, we then assessed gene expression of all Notch receptors and ligands during progression from BE to EAC. (Supplementary Table 5) Comparing HGD/EAC to NDBE, NOTCH3 (log-fold change 0.61, FDR .08) and JAG2 (log-fold change .91, FDR .04) were significantly upregulated. There was significant correlation between expression of JAG2 and Notch target genes HES1B (rho=.56, p=1.5x10−9) and HEY1 (rho=0.47, p=1.0x10−6). JAG2 also correlated inversely with TFF3 and MUC2 expression (rho=−.60, p<2.2x10−16, and rho=−.55, p=5.4x10−9, respectively), and had a non-significant inverse correlation with GC density (rho=−.19, p=.08).
As NOTCH3 and JAG2 were most upregulated in HGD and EAC, we then assessed for relationships between these two markers of Notch signalling and expression of other markers potentially associated with esophageal carcinogenesis (Supplementary Table 2). There was a strong correlation between NOTCH3 and TGFB1, and moderate correlation with VEGFA, WNT7B, and MYC (Supplementary Figure 1D). JAG2 expression was strongly correlated with EGFR, WNT7A, and WNT7B, and moderately correlated with MYC and TGFB1 (Supplementary Figure 1E). Both NOTCH3 and JAG2 were significantly inversely correlated with CASP3 and CTTNB1. On immunohistochemistry analyses, epithelial Notch1 and Notch3 expression both significantly correlated with Ki67 expression (rho=0.77, p<0.0001, and rho=0.51, p=0.0015, respectively) (Figure 1J, K). In sum, these data suggest that Notch signalling in BE involves multiple Notch genes, is inversely correlated with GC differentiation within BE, increases with progression to EAC, and correlates with other markers that may also promote esophageal neoplasia.
Notch signalling in progenitor cells regulates cell proliferation and reduces survival in the L2-IL1B mouse model
We previously demonstrated that pharmacological Notch inhibition in the L2-IL1B mouse leads to increased GC differentiation.12 We re-analyzed gene expression data from the SCJ in this mouse BE model and assessed for Notch signatures by comparing the transcriptome of SCJ tissue from L2-IL1B mice and WT mice via GSEA, and found an enrichment of Notch signature genes in L2-IL1B mice (NES-value: 2.41; p-value<.01; Figure 2A). Single gene expression analyses pointed to Notch2 as the main mediator of Notch signalling in the BE tissue at the SCJ of the mouse (Figure 2B). The comparison of murine cancerous and BE tissue that was treated with bile acid from previous experiments12 resulted in the enrichment of another Notch signature (NES-value: 1.99; p-value<.01) in the cancerous tissue, indicating involvement of Notch signaling in later stages of esophageal carcinogenesis as well (Figure 2C).
Figure 2.
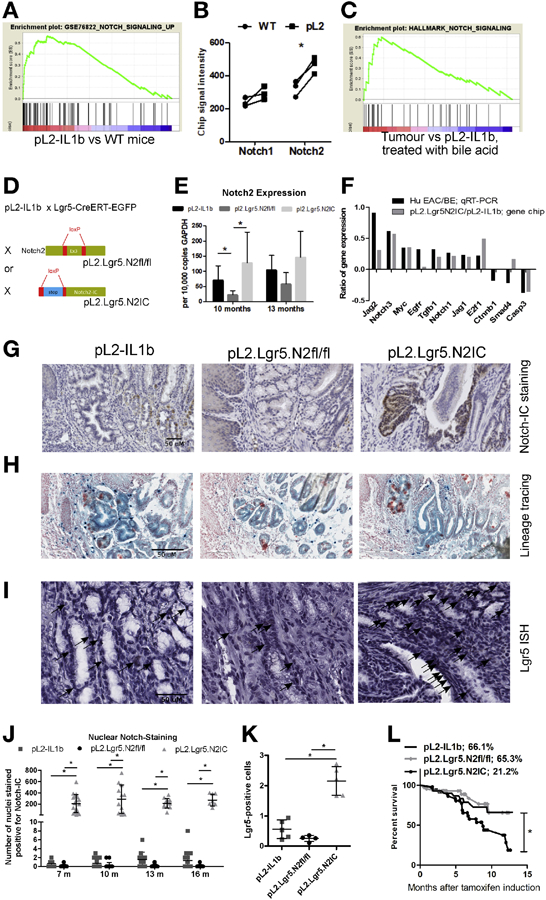
A BE mouse model with engineered Notch2IC expression in gastric Lgr5+ stem cells lead to dysplasia, increased Notch-IC staining and decreased survival rates. (A) GSEA comparing L2-IL1B versus WT mice with a set of Notch dependent genes. (B) single gene expression analysis of Notch1 and Notch2. (C) GSEA of a Notch signature comparing tumor bearing mice and L2-IL1B mice that were previously treated with bile acid. (D) Generating mouse models with the conditional knockout of Notch2 and overexpression of activated Notch2-IC in Lgr5+ cells on top of the inflammatory background of L2-IL1B mice. (E) qRT-PCR validates Notch2 expression levels in genetically modified L2-IL1B mice. (F) differential gene expression pattern of human donors (HGD/EAC vs. non-dysplastic BE) and our mouse models (mice overexpressing IL1B and NOTCH2 in Lgr5 cells vs. mice only overexpressing IL1B). (G) IHC staining of Notch-IC. Shown are representative examples at 13 months after induction. (H) Lineage tracing experiments of our mouse strains that were crossed to Rosa26-LacZ mice indicating the presence of Lgr5 expressing progenitor cells in dysplastic tissue. (I) In situ hybridization (ISH) for Lgr5 depicting Lgr5+ progenitor cells at the SCJ in areas of BE. (J) Intranuclear Notch-IC reactivity in the three mouse strains at indicated time points as positive cells in the BE region in 10 high-power fields. (K) Statistical quantification of Lgr5+ cells per 10 high -power fields as shown as representative images in (I). (L) Kaplan-Meier curves showing decreased survival of pL2.Lgr5.N2IC mice compared to the other two strains. Data is presented as means ± standard deviation. Statistical analysis were performed using one-way ANOVA and Tukey’s multiple comparison test; *p<.05.
As noted above, in the human BE data we found a strong correlation between NOTCH2 and NOTCH3 gene expression. Notch2 appeared to be the more relevant receptor in our mouse model of EAC (Figure 2B), and we thus combined our transgenic mouse model, L2-IL1B, with both a conditional knockout of Notch2 (pL2.Lgr5.N2fl/fl) as well as with overexpression of the intracellular domain (the activated form) of Notch2 specifically in Lgr5+ stem cells (pL2.Lgr5.N2IC), which we previously defined as one of the potential cells of origin for BE and EAC (Figure 2D). qRT-PCR analyses of SCJ tissue from the three mouse strains validated the engineered gene expression of Notch2 in whole tissue samples from the SCJ, with pL2.Lgr5.N2fl/fl mice displaying the lowest and pL2.Lgr5.N2IC displaying the highest mRNA levels, especially at early time points (Figure 2E). GSEA showed that the classic Notch signalling downstream targets correlated with induced Notch2 receptor expression. Moreover, the transcriptome of SCJ tissue from pL2.Lgr5.N2IC mice was enriched with gene signatures that are associated with cell proliferation, Wnt signalling, stem cell biology, migration, and cancer (Supplementary Figure 3). Next, we compared mouse and human BE gene expression, comparing the top 11 human differentially expressed genes (HGD/EAC vs. NDBE) with the corresponding mouse data (pL2.Lgr5.N2IC vs. pL2.Lgr5). The gene expression patterns in the SCJ tissue from the mouse models were similar to that from the biopsies from human BE patients (Figure 2F), consistent with the concept that increased Notch signalling drives esophageal carcinogenesis. Immunohistochemistry of Notch intracellular domain (Notch-IC) showed a significant increase in Notch-IC in the SCJ of pL2.Lgr5.N2IC mice in comparison to pL2-IL1B and pL2.Lgr5.N2fl/fl mice over time after tamoxifen induction (Figure 2G and Supplementary Figure 2A). Importantly, Notch-IC staining was associated with Lgr5+ lineage tracing at the SCJ in pL2.Lgr5.N2IC mice, highlighting Lgr5+ lineage specific Notch expression and expansion (Figure 2G). Indeed, intercrossing all three mouse strains with Rosa26-LacZ expressing mice (pL2.Lgr5.LacZ, pL2.Lgr5.N2fl/fl.LacZ, pL2.Lgr5.N2IC.LacZ) confirmed the lineage tracing ability of Lgr5+ progenitor cells in metaplasia and dysplasia (Figure 2H), although the purpose of this experiment was not specifically to provide a detailed Notch dependent lineage tracing analysis and characterization of cell types. Further, in situ hybridization (ISH) for Lgr5 displayed an increase in Lgr5+ progenitor cells in pL2.Lgr5.N2IC mice (Figure 2I, K), of which all identifiable Lgr5+ cells were also Ki67+ in serial sections (data not shown). Additionally, there was significant activation of Notch intranuclear domain reactivity measured by Notch-IC staining in pL2.Lgr5.N2IC mice (Figure 2J). These results indicated that the engineered expression of Notch2 in Lgr5 progenitor cells was reliably translated into Notch signal activation at the BE region at the SCJ. Interestingly, survival of Notch overexpressing mice was significantly decreased compared to the pL2-IL1B and pL2.Lgr5.N2fl/fl mice (Figure 2L). We did not observe any Notch-induced tumors or dysplasia in the colon or small intestine in pL2.Lgr5.N2IC mice, with the exception of late-onset increased proliferation and changes in degree of GC differentiation in pL2.Lgr5.N2IC mice (Supplementary Figure 5).
Notch signalling activation accelerates dysplasia independently of the inflammatory microenvironment
Notch activation in pL2.Lgr5.N2IC mice resulted in higher macroscopic tumor scores at the SCJ compared to L2-IL1B and pL2.Lgr5.N2fl/fl mice (Figure 3A and B). While there were no significant differences between the three strains in microscopic amount of metaplasia development prior to 16 months, there was significantly increased dysplasia as early as 10 months in pL2.Lgr5.N2IC mice compared to L2-IL1B and pL2.Lgr5.N2fl/fl mice (Figure 3C and D). On low coverage genome sequencing on macroscopically dissected tissue from the SCJ of pL2-IL-1B and pL2.Lgr5.N2IC mice, there was increased genomic alteration and copy number changes in pL2.Lgr5.N2IC mice, consistent with an accelerated dysplastic phenotype (Supplementary Figure 4). Thus, in these mouse models of BE with variable Notch activity, accelerated dysplasia formation was attributable to increased cell intrinsic Notch signalling.
Figure 3.
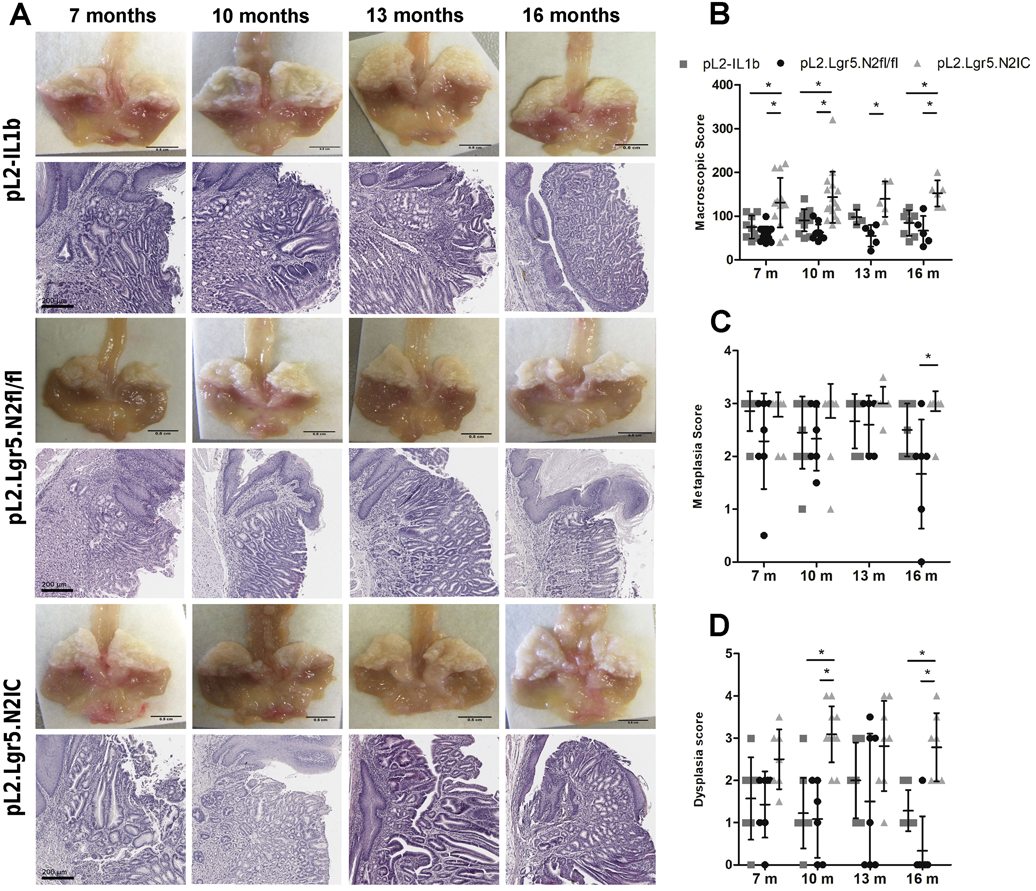
Overexpression of NOTCH2 in Lgr5+ progenitor cells accelerates the development of metaplasia, dysplasia, and esophageal tumors in L2-IL1B mice. (A) (upper panel) Macroscopic images of the distal esophagus and gastric cardia, sliced along the sagittal plane, and (lower panel) representative H&E staining of pL2.Lgr5 control mice, pL2.Lgr5N2fl/fl, pL2.Lgr5.N2IC mice at indicated time points. Statistical analysis of the (B) macroscopic tumor score, and histopathological scoring for (C) metaplasia, and (D) dysplasia. Data is presented as means ± standard deviation. Statistical analysis were performed using one-way ANOVA and Tukeys multiple comparison test.*p<.05.
As induction of chronic inflammation is a crucial factor for development of the BE-like phenotype in mice, we investigated whether changes in Notch signalling activation resulted in increased or decreased inflammation. Interestingly, the morphological differences between pL2.Lgr5.N2IC and pL2.Lgr5.N2fl/fl mice did not become apparent until 16 months of age (Supplementary Figure 6A). This was in line with FACS analyses of SCJ immune cells in the three groups of mice at 7 and 10 months of age, where none of the analysed immune cell populations displayed significant changes in cell frequency. This suggested that accelerated BE dysplasia with Notch activation was mainly due to cell intrinsic mechanisms, rather than to an enhanced immune cell response (Supplementary Figure 6B and C).
Notch signalling activation induces crypt fission events and inversely correlates with goblet cell differentiation
As with our analysis of human BE tissue, we also assessed in murine BE tissue the relationship between Notch signalling activation and goblet-like cell density. In a series of independent experiments we observed that the percentage of PAS-positive cells, mucus cell maturation, and GC-like ratios were significantly reduced in pL2.Lgr5.N2IC mice and somewhat increased in pL2.Lgr5.N2fl/fl mice (Figure 4A-C). Of note, elimination of NOTCH2 signalling in the pL2.Lgr5.N2fl/fl mice resulted in a non-significant decrease in the number of Lgr5+ progenitor cells at the SCJ. A diminished population of mucin producing cells was also reflected by GSEA of the SCJ from pL2.Lgr5.N2IC mice compared to L2-IL1B and pL2.Lgr5.N2fl/fl strains (Supplementary Figure 3). Additional single gene expression analyses demonstrated an inverse correlation between Notch2 and Muc5b as well as Notch3 and Muc5b/ac (Supplementary Figure 7). Furthermore, the GC maturation marker MUC5AC was also significantly inhibited and correlated with increased macroscopic tumor score in pL2.Lgr5.N2IC mice (Supplementary Figure 8). These data indicate that Notch driven inhibition of goblet-like cell differentiation is associated with an increase in macroscopic tumor growth (Figure 4D).
Figure 4.
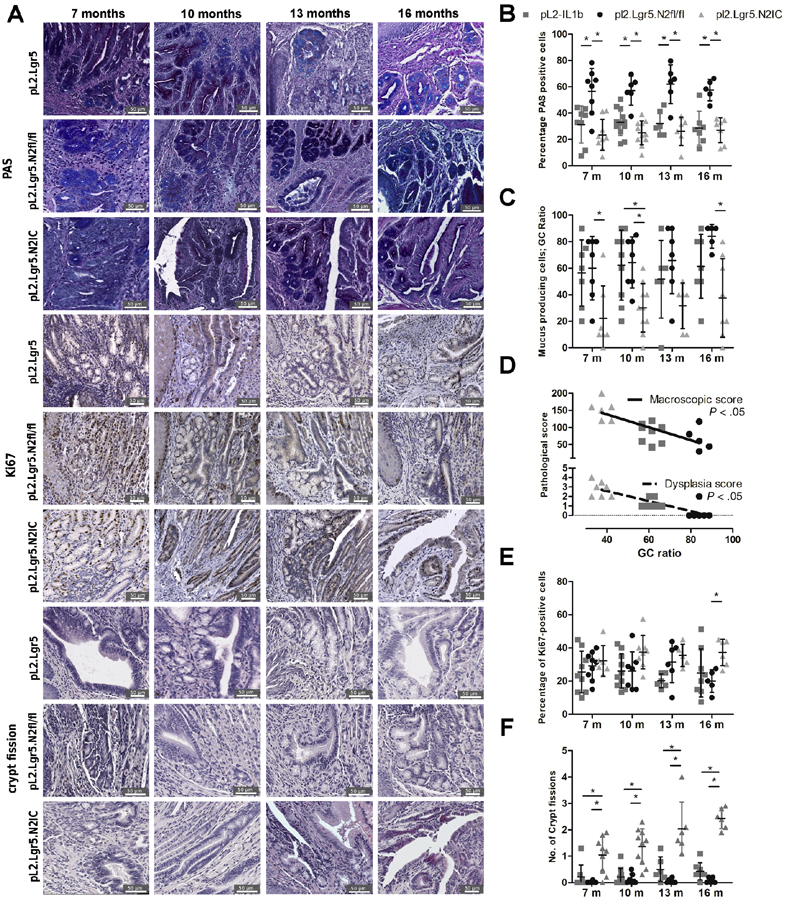
Notch2 overexpression in Lgr5+ cells leads to increased crypt fission and impedes the differentiation of mucus producing cells. (A) Representative images for goblet-like cell maturation and crypt fission. (B) Statistical summary of PAS staining indicating goblet-like cell maturation and (C) goblet-like cells frequencies indicated as GC ratio. (D) Linear regression of pathological progression correlating the macroscopic and dysplasia score with GC ratio. (E) Cell proliferation was evaluated by Ki67 reactivity and (F) crypt fission events. Analysis was performed in the BE region in 10 high-power fields. Data is presented as means ± standard deviation. Statistical analysis were performed using one-way ANOVA and Tukey’s multiple comparison test. *p<.05.
We also assessed whether Notch signalling regulates cell proliferation. An increase in Ki67+ cells was not observed in pL2.Lgr5.N2IC mice until 16 months. Crypt fission events, however, were characteristic in the BE region of pL2.Lgr5.N2IC mice throughout their lifetime (Figure 4E and F). Crypt fission is a mechanism producing multiple crypts by branching and is of major importance in early intestinal development and in regenerative states following intestinal damage.20, 21 The finding of increased crypt fission in pL2.Lgr5.N2IC mice was further corroborated by gene expression profiling. Expression of genes upregulated during cell cycle was enriched in pL2.Lgr5.N2IC mice compared to pL2-IL1B mice. Similar analyses with stem cell-associated gene sets, such as Wnt downstream targets, were also enriched in pL2.Lgr5.N2IC mice (Supplementary Figure 3). Thus, structural changes in the BE region, including crypt fission events, were functionally linked to enhanced Notch signalling and increased stem cell expansion.
In vivo, crypt growth can be initiated by Lgr5+ progenitor cells. We performed 3D organotypic culture of murine BE crypts to assess in a surrogate model whether progenitor cells isolated from the three mouse strains differed in their capacity to proliferate or differentiate based on the degree of Notch signalling activation. As seen by IHC, organoids derived from pL2.Lgr5.N2IC mice displayed significantly greater Notch-IC and Ki67 and significantly reduced PAS compared to pL2.Lgr5.N2fl/fl derived organoids. (Figure 5A). qPCR results showed similar findings for Notch2, Ki67, and Muc2 (Figure 5B). Comparing pL2.Lgr5.N2fl/fl and pL2.Lgr5.N2IC derived organoids; we observed the highest organoid survival rate over time, a measure of stemness, in the latter (Figure 5C). In contrast, wall thickness per overall organoid size, a surrogate measure for cell differentiation, was significantly greater in the organoids derived from Notch2 knockout BE samples compared to pL2.Lgr5.N2IC derived organoids (Figure 5D). In order to evaluate further whether the observed effects were Notch signalling-dependent, we exposed pL2.Lgr5.N2IC derived organoids to the gamma-secretase inhibitor DAPT, which resulted in reduced Notch expression, and increased mucin expressing cells. The latter was true for cells of mouse and human origin (Figure 5E and F). Representative examples for Notch-IC, Ki67 and PAS staining are displayed in Supplementary Figure 2C. Furthermore, DAPT exposure led to reduced Notch1, Notch2, Jag2 and Egfr mRNA levels, while Muc2 levels increased (Figure 5G). In sum, 3D culture of BE derived mouse organoids reflected the dependence of cell proliferation and mucus producing cell lineage fate on Notch signalling.
Figure 5.
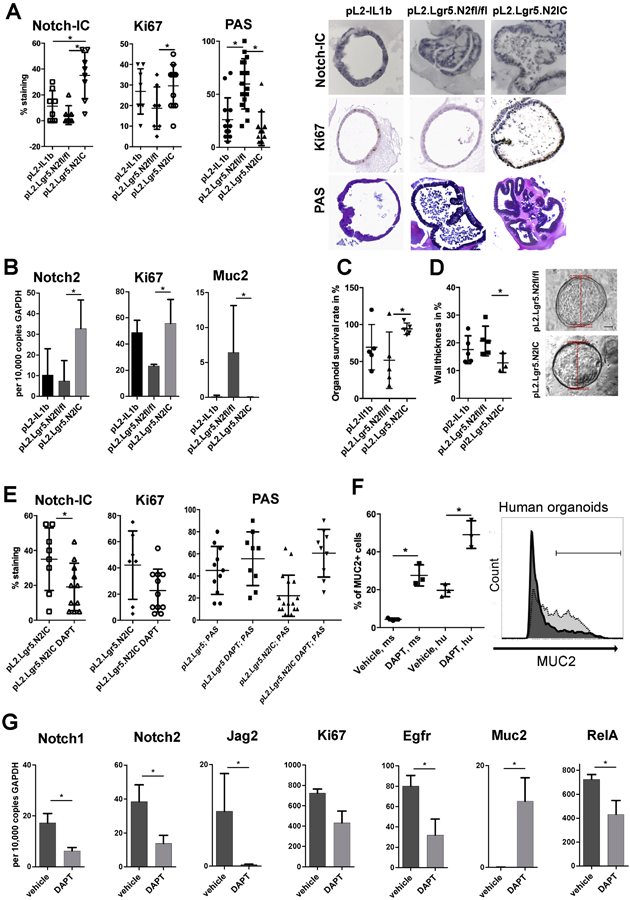
Notch activity in progenitor cells determines cell fate and growth rates of ex vivo cultured organoids. (A) IHC results of 3D cultured organoids. Organoids were isolated from indicated mouse strains and analysed for Notch-IC, Ki67 and PAS reactivity with representative images at passages 3-5 after isolation. (B) Differential gene expression of organoid cells was performed via qRT-PCR. (C) Organoid survival rates and (D) organoid wall thickness (relative to the organoid diameter) that serves an indicator of goblet-like maturation. Representative examples are displayed (right). (E-G) pL2.Lgr5.N2IC or pL2.Lgr5 mice derived organoids were treated with the gamma-secretase inhibitor of Notch signaling, DAPT (50μM). (E) Summarized IHC results of 3D cultured organoids. (F) Flow cytometry of singularized organoid cells derived from mouse (ms; L2-IL1B) and human (hu, BE) samples. The representative histogram indicates the range of Muc2+ cells for vehicle (dark, 19.4%) and inhibitor (50μM; light grey, 56.8%) treated conditions quantified via flow cytometry after enzymatic cell separation of organoids 3D culture. Data is presented as means ± standard deviation of at least three independent experiments. Statistical analysis was performed using Student's T-Test, *p<.05 (G) qRT-PCR of pL2.Lgr5.N2IC derived organoids. At least three independent experiments were performed; Data are presented as mean standard deviation. (A-E, G) Data is presented as means ± standard deviation. Statistical analysis was performed using one-way ANOVA and Tukey’s multiple comparison test or student t-test. *p<.05.
Notch signalling correlates with activation of NF-κB in Lgr5+ progenitor cells
While IL-1B is known to be an upstream activator of NF-κB, GSEA in pL2.Lgr5.N2IC mice revealed a number of gene signatures that implicate NF-κB activation (Figure 6A). RT-qPCR analyses showed a pronounced upregulation of RelA in pL2.Lgr5.N2IC mice compared to L2-IL1B mice (Figure 6B). In the 3D organoid cultures we also observed Notch dependent regulation of RelA, the transcriptionally active subunit of NF-κB (Figure 5G). Single gene chip-based analyses of murine transcriptomes showed an upregulation of Nf-kb1 and RelA that correlated with genes associated with Notch signalling (Supplementary Figure 7). At the protein level, an increased frequency of cells positive for the NF-κB activator pIKKα/β (Figure 6C-E) was found. Evidence of increased NF-κB activity was predominantly observed in Lgr5+ progenitor cells, as evidenced by co-expression of intrinsic Lgr5-driven GFP expression and pIKKα/β IHC (Figure 6D and E). Interestingly, NF-κB activation was not observed in GCs, as combined staining of pIKKα/β and Alcian blue in pL2.Lgr5.N2IC BE tissue sections was absent. These findings suggest that NF-κB activity correlates with Notch signaling activation in progenitor cells and does not lead to GC differentiation.
Figure 6.
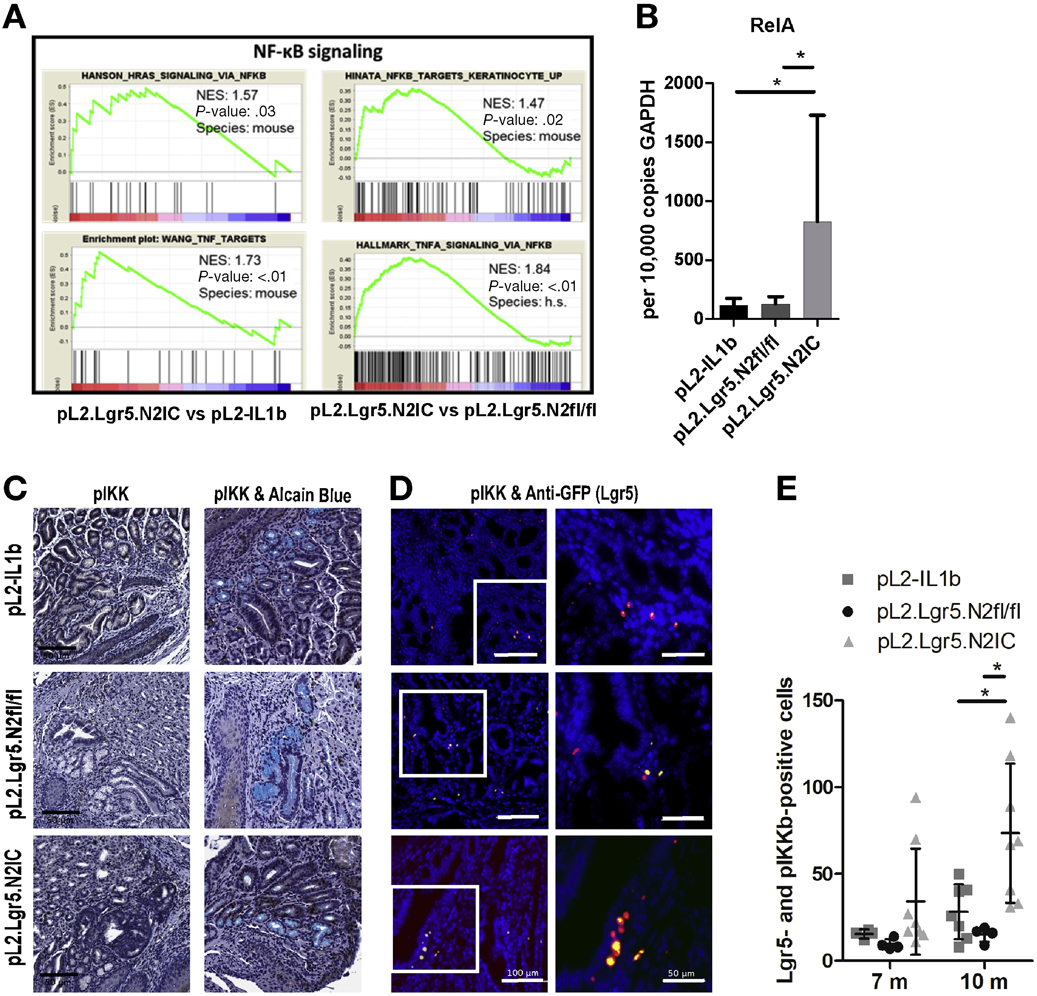
Aberrant Notch expression results in increased NF-κB in progenitor and progenitor derived cells. (A) GSEA display an enrichment of NF-κB associated gene sets in mice with overexpressed Notch2 versus mice with less Notch signalling. (B) qRT-PCR of RelA expression levels of indicated mouse strains. (C) Representative IHC images of pIKK (left) and a combinatory staining of pIKK and Alcain Blue (right) derived from mice at 7 months. (D) Exemplary immunofluorescence images of pIKK (red) and anti-GFP (Lgr5; green) staining derived from mice at 10 months. (E) Corresponding statistically summarized results in the BE region as number of positive cells in 10 high-power fields. Data is presented as means ± standard deviation. Statistical analysis was performed using one-way ANOVA and Tukey’s multiple comparison test. *p<.05.
Elimination of RelA in Lgr5+ cells attenuates Notch driven tumorigenesis
To address NF-κB activation, we generated an additional mouse model with a conditional knockout of the NF-κB subunit p65, RelA, in Lgr5+ stem cells on the chronic inflammatory background of pL2.Lgr5 mice (pL2.Lgr5.p65fl/fl) (Figure 7A). Elimination of RelA specifically in epithelial Lgr5+ progenitor cells resulted in a diminished reactivity of pIKK and reduced inflammation scores compared to pL2.Lgr5.N2IC and L2-IL1B mice within the BE region at the SCJ (Figure 7A-C). Metaplasia and in particular dysplasia scores were strongly abrogated in pL2.Lgr5.p65fl/fl mice compared to pL2.Lgr5.N2IC mice (Figure 7D and E). In line with these findings, cell proliferation was decreased and mucus cell differentiation was increased in pL2.Lgr5.p65fl/fl mice (Figure 7F-H). To further evaluate the significance of NF-κB activation in BE independent of its microenvironment, we disrupted NF-κB activation pharmacologically in pL2-IL1B mouse derived organoids using the NF-κB inhibitor JSH-23, demonstrated by effective downregulation of RelB as one of the NF-κB target genes (Figure 7I). Similar to our in vivo findings, reduced NF-κB activation in vitro resulted in decreased cell survival and proliferation (Figure 7J and K). Moreover, treatment of human and mouse 3D BE organoid cultures with an NF-κB inhibitor led to an increase in GC formation (Figure 7L). Overall, these results suggest a key role for NF-κB activation in Lgr5+ cardia progenitor cells.
Figure 7.
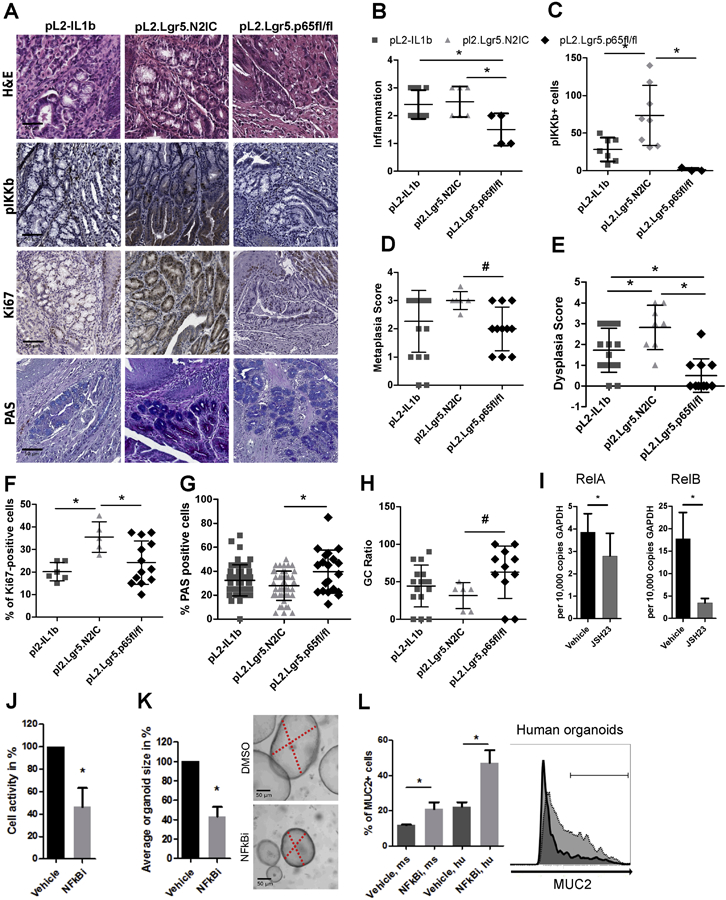
A RelA knockout reduces the pathology observed in mice with induced inflammation and Notch signaling. A) Representative IHC images of RelA KO mice that are depicted next to L2-IL1B and pL2.Lgr5.N2IC mice. (B-H) Histopathological evaluations for metaplasia, dysplasia, inflammation, GC ratio, and numbers of pIKK+, Ki67+ and PAS+ cells in the BE region in 10 high-power fields. Statistical analysis was performed using one-way ANOVA and Tukeys multiple comparison test, *p<.05, or the Kruskal-Wallis test #p<.05. All indicated IHC based results were derived from mice ranging from 10 to 12 months of age. Only mice with the same age were compared. (I-L) L2-IL1B mice derived organoid cultures were treated with the NF-κB inhibitor, JSH-23 (10μM), or vehicle control. (I) qRT-PCR of L2-IL1B mice derived organoids. (J) Cell activity was measured via MTT assay and (K) organoid growth was determined microscopically. (L) Flow cytometry of organoid cells after enzymatic disaggregation to determine the proportion of Muc2+ cells, derived from (ms; L2-IL1B) and human (hu, BE) samples. The representative histogram indicates the range of MUC/Muc2+ cells for vehicle (dark, 22.5%) and inhibitor (light grey, 41.1%) treated conditions. Data is presented as means ± standard deviation of at least three independent experiments. Statistical analysis was performed using Student's t-test, *p<.05
DISCUSSION
Despite an increasing incidence of EAC in western countries over the past half century, the important signaling pathways driving development of this highly lethal cancer remain poorly understood. Notch plays a key role in regulating stem cell fate in the gastrointestinal tract, and aberrant Notch has been shown to promote various types of cancer.22, 23 In parallel studies in humans and mice, we now demonstrate that Notch signaling within proliferating gastric cardia progenitor cells inhibits GC differentiation in BE and serves as a marker of progression. Furthermore, increased Notch signaling directly promotes the development of dysplasia and is closely linked to NF-κB activation during this process. Nevertheless, it might be possible that progression of intestinal type of BE to EAC is a dynamic process, including a phase of likely Notch driven dedifferentiation towards a less differentiated phenotype.
These findings have potential clinical implications, as Notch activation in BE may lead to a GC-poor phenotype, more rapid progression to dysplasia, and development of more aggressive tumors. BE defined as intestinal metaplasia with goblet cells has a clearly increased risk of progression to EAC. However, an emerging body of literature suggests that a major subset of EACs are characterized by genomic “catastrophes”, such as genome doubling or chromothripsis24-26, there is an inverse association between aneuploidy and GC density in BE27, and patients with BE and increased chromosomal alterations progress very rapidly to EAC.28 A recent published analysis of two separate large cohorts demonstrated that, even after adjusting for tumor stage, EAC patients without associated intestinal metaplasia have a significantly worse prognosis compared to EAC patients with associated intestinal metaplasia.9 We previously reported that BE patients with HGD or EAC have significantly decreased GC density and TFF2 expression,8 and markers of reduced GC differentiation and increased Notch signaling in patients with BE may serve as biomarkers of increased EAC risk.
Gastric cardia stem cells, such as Lgr5+ progenitor cells, can become activated in response to chronic inflammation such as that which occurs in the setting of GERD29. These cardia stem cells either proliferate and expand into the esophagus, or alternatively differentiate and form specialized tissue such as GC containing intestinal metaplasia. We have shown that Notch gene expression correlates with disease progression in humans and in mice. Here we used the BE mouse model to specifically address the role of aberrant Notch signaling in Lgr5+ progenitor cells. We found that the Notch pathway is a critical determinant of either progenitor cell proliferation and expansion with increased progression to dysplasia at the SCJ, or development of GC-rich epithelium associated with decreased progression. These findings were further supported by the human BE biopsies, which showed upregulation of Notch signaling with increased proliferation and progression to EAC and an inverse correlation with GC differentiation. In intestinal epithelial stem cells, Notch signaling supports an undifferentiated and proliferative stem cell phenotype.30-32 Elimination of Notch2 in mice resulted in a mild but not florid increase in GCs, likely due to a corresponding reduction in Lgr5+ cells at the SCJ in these mice. Activating Notch in gastric cardia Lgr5+ cells resulted in lineage-traced accelerated proliferation, loss of differentiation and expansion of dysplastic tissue, likely through increased crypt fission. In contrast, eliminating Notch in the identical cell population led to decreased proliferation, increased cellular differentiation with formation of goblet-like cells, in line with prior work in BE cell lines.33 Thus, BE is likely a cardia stem cell-driven disease, with metaplasia and tumor formation controlled by regulators of stem cell maintenance such as the Notch pathway.
Consistent with the established function of Notch signaling in gastrointestinal progenitor cells,30, 31, 33 forced overexpression of Notch in pL2.Lgr5.N2IC mice led to reduced GC differentiation and a shift instead towards cell proliferation, as reflected by a pronounced increase in crypt fission events and reduced survival; we did not observe a severe intestinal Notch-driven phenotype without additional inflammation in the colon or small intestine. These histopathological observations in mice resemble the type of human BE with more proliferation and rapid progression, and suggest a Notch dependent mechanism by which gastric cardia epithelium clonally expands through enhanced crypt fission, resulting in expanded proliferating columnar epithelium.34, 35 In isolated human and mouse 3D BE organoids, we recapitulated this Notch driven phenotype, and were able to rescue the loss of differentiation by Notch inhibition, suggesting a potential strategy for cancer prevention. The fact that treatment with a gamma secretase inhibitor in the Notch overexpressing organoids resulted in increased differentiation suggests that Notch2 may suppress GC differentiation by regulating other Notch receptor paralogs and ligands. This can be accounted for by positive feedback loops between Notch receptor paralogs.36, 37 A Notch driven mechanism was also supported by observations from patient biopsies, with increased expression of Notch signaling genes and decreased GC maturation with progression from BE to EAC.
Interestingly, the observed Notch driven acceleration of esophageal neoplasia is closely paralleled by previous observations regarding Notch signaling in gastric cancer, in which activation of Notch signaling results in decreased GC maturation and an unfavorable prognosis.8, 38-40 Intestinal-type gastric and esophageal adenocarcinoma share fundamental features in that they emerge in the setting of chronic inflammation. Accumulating data suggests that Notch signaling is a major driver of gastro-esophageal neoplasia, and a role for Notch signaling in the regulation of inflammatory mediators has been documented in a number of malignancies.41 While there may be differences between specific receptors and ligands responsible for Notch activation in BE in humans and in mice, we have noted a consistent finding of increased Notch activation in all analyzed settings. Of note, in the pL2.Lgr5.N2IC mouse model we did not observe tumors in the stomach as previously described with a different Notch1 transgene42, which might be due to a lack of a stabilized and prolonged Notch signal, IL-1B overexpression being restricted to the squamous foregut, and distinct housing and corresponding microbiome.13
Perhaps the major pathway that sustains the chronic inflammatory microenvironment is the NF-κB signalling cascade, which can be triggered through a variety of soluble factors and cytokines, such as TNF-α and IL-1B.43 In general, a strong correlation has been observed between IL-1B and NF-κB activation in both EAC patients and in the L2-IL1B mouse model.12 In patients, NF-κB increases steadily in the sequence from BE to EAC, with high levels seen in late stage disease.44 Our gene expression analyses indicated that while NF-κB is upregulated in L2-IL1B mice, activation of the NF-κB is further enhanced in Notch activated pL2.Lgr5.N2IC mice. Histopathologic analysis also supported Notch-triggered NF-κB activation, as pIKK-positive cells were more abundant in pL2.Lgr5.N2IC SCJ tissue and, importantly, co-localized with Notch2 overexpressing, proliferating Lgr5+ cells. In the various mouse BE strains, we did not observe a significant alteration in the immune microenvironment, including IMCs, neutrophils or NK cells, which suggests that increased inflammation was not primarily responsible for NF-κB activation and accelerated disease in pL2.Lgr5.N2IC mice. This cell intrinsic interaction with Notch was underlined by genetic elimination of RelA, which attenuated the dysplastic phenotype in our mouse model. This also resulted in increased goblet-like cell differentiation, highlighting that inflammatory stimuli through NF-κB can also affect cellular plasticity. Cell intrinsic activation of NF-κB was also supported by in vitro 3D BE organoid experiments lacking any environmental stimuli (IL-1b is expressed in the squamous compartment of the mouse esophagus), where NF-κB inhibition reduced cell proliferation and increased mucus cell differentiation. It has been reported that Notch signalling activates NF-κB in several hematopoietic malignancies as well as in other cell types such as progenitor and pancreatic cells.45-47 In immune cells, NOTCH1 directly augments NF-κB activity by facilitating its nuclear retention.48 Although in this study we were not able to address in this study the possibility of direct interaction between the two pathways, we have shown that both Notch and NF-κB closely regulate cellular differentiation, proliferation, and expansion at the stem cell level.
In conclusion, Notch signaling plays a key role in both cellular differentiation and in the development of EAC. These processes appear to be regulated by a close relationship between Notch and NF-κB activation. The current study also further supports the notion that metaplasia and dysplasia are distinct processes, yet both originate from stem cells within the gastric cardia. A population of Notch-high and NF-κB-high cells rapidly progresses to dysplasia and EAC; in contrast a Notch-low and NF-κB-low cell population forms classical intestinal metaplasia with increased GC differentiation in BE patients, and is associated with slower progression to EAC. Consideration should be given to incorporating markers of increased Notch and NF-κB activation in both BE and the gastric cardia into future surveillance strategies, as these patients may be at highest risk for development of esophageal and gastro-esophageal junction adenocarcinoma.
Supplementary Material
What you need to know:
BACKGROUND AND CONTEXT: Barrett’s esophagus (BE) progresses to esophageal adenocarcinoma (EAC) by unknown mechanisms. Notch signaling maintains stem cells in the gastrointestinal tract and is dysregulated during carcinogenesis, so it might be involved.
NEW FINDINGS: Notch signaling results in activation of NF-κB and reduces differentiation of gastric cardia progenitor cells in a mouse model of BE. In human esophageal tissues, progression of BE to EAC associated with reduced goblet cell density and increased levels of Notch expression.
LIMITATIONS: This study was performed in mice and on human tissue samples.
IMPACT: Strategies to block this pathway might be developed to prevent EAC in patients with BE.
Acknowledgments
Funding: Supported by the Deutsche Forschungsgemeinschaft (Grant Agreement No. SFB 824; DFG), the Deutsche Krebshilfe within the Max Eder Program (MQ), a U54 award from the NCI (Barrett’s Esophagus Translational Research Network (BETRNet), U54 CA163004; PIs TCW, AKR, KKW), and a R01 from the NCI (R01 CA208711; ARS).
Abbreviations:
- BE
Barrett’s esophagus
- EAC
esophageal adenocarcinoma
- FDR
false discovery rate
- GC
goblet cell
- GEJ
gastroesophageal junction
- GERD
gastro-esophageal reflux disease
- GF
germfree
- HFD
high fat diet
- HGD
high grade dysplasia
- LGD
low grade dysplasia
- NDBE
non-dysplastic Barrett’s esophagus
- NK cells
natural killer cells
- SCJ
squamocolumnar junction
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: none to report
Gene Expression Analysis:
Raw sequencing data have been deposited in National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) (GSE103616).
References
- 1.Quante M, Graham TA, Jansen M. Insights Into the Pathophysiology of Esophageal Adenocarcinoma. Gastroenterology 2018;154:406–420. [DOI] [PubMed] [Google Scholar]
- 2.Bhat S, Coleman HG, Yousef F, et al. Risk of Malignant Progression in Barrett's Esophagus Patients: Results from a Large Population-Based Study. J Natl Cancer Inst 2011;103:1049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandrasekar VT, Hamade N, Desai M, et al. Significantly lower annual rates of neoplastic progression in short- compared to long-segment non-dysplastic Barrett's esophagus: a systematic review and meta-analysis. Endoscopy 2019;51:665–672. [DOI] [PubMed] [Google Scholar]
- 4.Hvid-Jensen F, Pedersen L, Drewes AM, et al. Incidence of adenocarcinoma among patients with Barrett's esophagus. N Engl J Med 2011;365:1375–83. [DOI] [PubMed] [Google Scholar]
- 5.Shaheen NJ, Falk GW, Iyer PG, et al. ACG Clinical Guideline: Diagnosis and Management of Barrett's Esophagus. Am J Gastroenterol 2016;111:30–50; quiz 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.AGA. American Gastroenterological Association Medical Position Statement on the Management of Barrett's Esophagus. Gastroenterology 2011; 140:1084–91. [DOI] [PubMed] [Google Scholar]
- 7.Tamagawa Y, Ishimura N, Uno G, et al. Bile acids induce Delta-like 1 expression via Cdx2-dependent pathway in the development of Barrett's esophagus. Lab Invest 2016;96:325–37. [DOI] [PubMed] [Google Scholar]
- 8.Schellnegger R, Quante A, Rospleszcz S, et al. Goblet Cell Ratio in Combination with Differentiation and Stem Cell Markers in Barrett Esophagus Allow Distinction of Patients with and without Esophageal Adenocarcinoma. Cancer Prev Res (Phila) 2017;10:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawas T, Killcoyne S, Iyer PG, et al. Identification of prognostic phenotypes of esophageal adenocarcinoma in two independent cohorts. Gastroenterology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menke V, van Es JH, de Lau W, et al. Conversion of metaplastic Barrett's epithelium into post-mitotic goblet cells by gamma-secretase inhibition. Dis Model Mech 2010;3:104–10. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Jiang K, Fan Z, et al. Aberrant expression of Wnt and Notch signal pathways in Barrett's esophagus. Clin Res Hepatol Gastroenterol 2012;36:473–83. [DOI] [PubMed] [Google Scholar]
- 12.Quante M, Bhagat G, Abrams JA, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012;21:36–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munch N, Fang H-Y, Ingermann J, et al. High-fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett’s Esophagus via IL8 and Alterations to the Gut Microbiome. Gastroenterology 2019;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449:1003–7. [DOI] [PubMed] [Google Scholar]
- 15.Hampel F, Ehrenberg S, Hojer C, et al. CD19-independent instruction of murine marginal zone B-cell development by constitutive Notch2 signaling. Blood 2011;118:6321–31. [DOI] [PubMed] [Google Scholar]
- 16.Besseyrias V, Fiorini E, Strobl LJ, et al. Hierarchy of Notch-Delta interactions promoting T cell lineage commitment and maturation. J Exp Med 2007;204:331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Algul H, Treiber M, Lesina M, et al. Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J Clin Invest 2007;117:1490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soriano P Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 1999;21:70–1. [DOI] [PubMed] [Google Scholar]
- 19.Saenz JB, Burclaff J, Mills JC. Modeling Murine Gastric Metaplasia Through Tamoxifen-Induced Acute Parietal Cell Loss. Methods Mol Biol 2016;1422:329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin G, Ramanathan V, Quante M, et al. Inactivating cholecystokinin-2 receptor inhibits progastrin-dependent colonic crypt fission, proliferation, and colorectal cancer in mice. J Clin Invest 2009;119:2691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He XC, Yin T, Grindley JC, et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet 2007;39:189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meurette O, Mehlen P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018;34:536–548. [DOI] [PubMed] [Google Scholar]
- 23.Nowell CS, Radtke F. Notch as a tumour suppressor. Nat Rev Cancer 2017;17:145–159. [DOI] [PubMed] [Google Scholar]
- 24.Nones K, Waddell N, Wayte N, et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat Commun 2014;5:5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stachler MD, Taylor-Weiner A, Peng S, et al. Paired exome analysis of Barrett's esophagus and adenocarcinoma. Nat Genet 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross-Innes CS, Becq J, Warren A, et al. Whole-genome sequencing provides new insights into the clonal architecture of Barrett's esophagus and esophageal adenocarcinoma. Nat Genet 2015;47:1038–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srivastava A, Golden KL, Sanchez CA, et al. High Goblet Cell Count Is Inversely Associated with Ploidy Abnormalities and Risk of Adenocarcinoma in Barrett's Esophagus. PLoS One 2015;10:e0133403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X, Paulson TG, Galipeau PC, et al. Assessment of Esophageal Adenocarcinoma Risk Using Somatic Chromosome Alterations in Longitudinal Samples in Barrett's Esophagus. Cancer Prev Res (Phila) 2015;8:845–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee Y, Urbanska AM, Hayakawa Y, et al. Gastrin stimulates a cholecystokinin-2-receptor-expressing cardia progenitor cell and promotes progression of Barrett's-like esophagus. Oncotarget 2017;8:203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vooijs M, Liu Z, Kopan R. Notch: architect, landscaper, and guardian of the intestine. Gastroenterology 2011;141:448–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim TH, Shivdasani RA. Notch signaling in stomach epithelial stem cell homeostasis. J Exp Med 2011;208:677–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hibdon ES, Razumilava N, Keeley TM, et al. Notch and mTOR Signaling Pathways Promote Human Gastric Cancer Cell Proliferation. Neoplasia 2019;21:702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamagawa Y, Ishimura N, Uno G, et al. Notch signaling pathway and Cdx2 expression in the development of Barrett's esophagus. Lab Invest 2012;92:896–909. [DOI] [PubMed] [Google Scholar]
- 34.McDonald SA, Lavery D, Wright NA, et al. Barrett oesophagus: lessons on its origins from the lesion itself. Nat Rev Gastroenterol Hepatol 2015;12:50–60. [DOI] [PubMed] [Google Scholar]
- 35.Hayakawa Y, Fox JG, Wang TC. The Origins of Gastric Cancer From Gastric Stem Cells: Lessons From Mouse Models. Cell Mol Gastroenterol Hepatol 2017;3:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen KY, Srinivasan T, Tung KL, et al. A Notch positive feedback in the intestinal stem cell niche is essential for stem cell self-renewal. Mol Syst Biol 2017;13:927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohashi S, Natsuizaka M, Yashiro-Ohtani Y, et al. NOTCH1 and NOTCH3 Coordinate Esophageal Squamous Differentiation Through a CSL-Dependent Transcriptional Network. Gastroenterology 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Demitrack ES, Samuelson LC. Notch as a Driver of Gastric Epithelial Cell Proliferation. Cell Mol Gastroenterol Hepatol 2017;3:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim SJ, Lee HW, Baek JH, et al. Activation of nuclear PTEN by inhibition of Notch signaling induces G2/M cell cycle arrest in gastric cancer. Oncogene 2016;35:251–60. [DOI] [PubMed] [Google Scholar]
- 40.Kang H, An HJ, Song JY, et al. Notch3 and Jagged2 contribute to gastric cancer development and to glandular differentiation associated with MUC2 and MUC5AC expression. Histopathology 2012;61:576–86. [DOI] [PubMed] [Google Scholar]
- 41.Fazio C, Ricciardiello L. Inflammation and Notch signaling: a crosstalk with opposite effects on tumorigenesis. Cell Death Dis 2016;7:e2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Demitrack ES, Gifford GB, Keeley TM, et al. Notch signaling regulates gastric antral LGR5 stem cell function. EMBO J 2015;34:2522–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdel-Latif MM, O'Riordan J, Windle HJ, et al. NF-kappaB activation in esophageal adenocarcinoma: relationship to Barrett's metaplasia, survival, and response to neoadjuvant chemoradiotherapy. Ann Surg 2004;239:491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Riordan JM, Abdel-latif MM, Ravi N, et al. Proinflammatory cytokine and nuclear factor kappa-B expression along the inflammation-metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Gastroenterol 2005;100:1257–64. [DOI] [PubMed] [Google Scholar]
- 45.D'Altri T, Gonzalez J, Aifantis I, et al. Hes1 expression and CYLD repression are essential events downstream of Notch1 in T-cell leukemia. Cell Cycle 2011;10:1031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu ZS, Zhang JS, Zhang JY, et al. Constitutive activation of NF-kappaB signaling by NOTCH1 mutations in chronic lymphocytic leukemia. Oncol Rep 2015;33:1609–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng P, Zlobin A, Volgina V, et al. Notch-1 regulates NF-kappaB activity in hemopoietic progenitor cells. J Immunol 2001;167:4458–67. [DOI] [PubMed] [Google Scholar]
- 48.Shin HM, Minter LM, Cho OH, et al. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. EMBO J 2006;25:129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.