

Abstract
Rationale:
Cardiotoxic β1 adrenergic receptor (β1AR)-CaMKII signaling is a major and critical feature associated with development of heart failure. Synapse-associated protein 97 (SAP97) is a multi-functional scaffold protein that binds directly to the C-terminus of β1AR and organizes a receptor signalosome.
Objective:
We aim to elucidate the dynamics of β1AR-SAP97 signalosome and its potential role in chronic cardiotoxic β1AR-CaMKII signaling that contributes to development of heart failure.
Methods and Results:
The integrity of cardiac β1AR-SAP97 complex was examined in heart failure. Cardiac specific deletion of SAP97 was developed to examine β1AR signaling in ageing mice, after chronic adrenergic stimulation, and in pressure overload hypertrophic heart failure. We show that the β1AR-SAP97 signaling complex is reduced in heart failure. Cardiac specific deletion of SAP97 yields an ageing-dependent cardiomyopathy and exacerbates cardiac dysfunction induced by chronic adrenergic stimulation and pressure overload, which are associated with elevated CaMKII activity. Loss of SAP97 promotes PKA-dependent association of β1AR with arrestin2 and CaMKII and turns on an Epac-dependent activation of CaMKII, which drives detrimental functional and structural remodeling in myocardium. Moreover, we have identified that GRK5 is necessary to promote agonist-induced dissociation of SAP97 from β1AR. Cardiac deletion of GRK5 prevents adrenergic-induced dissociation of β1AR-SAP97 complex and increases in CaMKII activity in hearts.
Conclusions:
These data reveal a critical role of SAP97 in maintaining the integrity of cardiac β1AR signaling and a detrimental cardiac GRK5-CaMKII axis that can be potentially targeted in heart failure therapy.
Keywords: Adrenergic signaling, SAP97, GRK5, CaMKII, heart failure, cardiac myocyte, calcium/calmodulin-dependent protein kinase II, heart failure, signaling pathways, Basic Science Research, Cardiomyopathy, Cell Signaling/Signal Transduction, Heart Failure, Myocardial Biology
Graphical Abstract
INTRODUCTION
Adrenergic stimulation is the key regulatory mechanism to modulate cardiac contractile function in stress response. Activation of β1AR, the major adrenergic receptor subtype in the heart, leads to Gs-dependent activation of adenylyl cyclases, which produce cAMP to activate protein kinase A (PKA). 1, 2 PKA plays a key role in enhancing cardiac contractility by promoting protein phosphorylation involved in excitation-contraction (E-C) coupling. 3, 4 However, under elevated sympathetic drive, chronic stimulation of β1AR also induces detrimental CaMKII activity in cardiac diseases, 5–7 promoting cardiac maladaptation and development of HF. 8, 9 Until today, the mechanism governing the switching of β1AR signaling from physiological cAMP-PKA activity to pathological CaMKII activity remains poorly understood. Literature shows divergent pathways can take part in adrenergic-induced CaMKII activity in heart. For example, overexpression of a PKA specific peptide inhibitor PKI in cardiac myocytes abolishes adrenergic-induced CaMKII activity in mouse hearts.10 Conversely, inhibition of another cAMP effector, exchange protein directly activated by cAMP (Epac) also blocks adrenergic-induced CaMKII in mouse hearts. 7 These data indicate additional factors can critically modulate different adrenergic pathways to stimulate CaMKII in (patho)physiological conditions.
Recently, precise and localized modulation of agonist-induced β1AR signaling is emerging as a potential therapeutic strategy through regulating receptor complexes with signaling partners and subcellular distributions. 11 For example, A-kinase anchoring proteins (AKAPs) tether PKA to the β1AR and facilitate receptor signaling to local downstream effectors such as ion channels on the plasma membrane, therefore regulating cell physiology. 12, 13 β1AR also binds to a series of scaffold proteins via the C-terminal PDZ motif including membrane associated guanylate kinases inverted (MAGIs), GIPC PDZ domain containing family members (GIPCs), and synaptic associated proteins (SAPs). 14 Among them, SAP97 is emerging as a critical regulator of cardiac β1AR distribution and signaling in myocytes. 15–17 Earlier studies show that SAP97 regulates agonist-induced β1AR trafficking in myocytes; disruption of the binding of SAP97 to β1AR facilitates agonist-induced receptor internalization. 15,16 More recent studies show that SAP97 controls agonist-induced β1AR trafficking including recycling of receptors from endosomes, which could anchor receptors on the plasma membrane. 16 In addition, SAP97 also connects β1AR to phosphodiesterase 4D, a negative regulator that controls cAMP magnitude and distribution in myocytes. 15, 17 Silencing SAP97 or disrupting SAP97 binding to β1AR in neonatal myocytes enhances cAMP signal and myocyte contraction rates after adrenergic stimulation. 15, 17 Nevertheless, the roles of SAP97 in adult hearts remain unclear.
In this study, we applied a cardiac specific deletion of SAP97 to examine its roles in adrenergic signaling in heart. Our data show that deletion of SAP97 switches on a β1AR-CaMKII signaling, which promotes spontaneous HF in ageing mice and exacerbates cardiac hypertrophy in the transverse aortic constriction (TAC) model of HF. Both models are associated with increased fibrosis and myocyte apoptosis. Moreover, we show that in human HF, the SAP97-β1AR complex is reduced in the presence of increased expression of GRKs, mimicking a state of loss of SAP97 in cardiac adrenergic regulation. Deletion of cardiac GRK5 abolishes agonist-induced dissociation of SAP97 from β1AR and inhibits chronic β1AR stimulation of CaMKII in mouse hearts. The mechanisms underlying the GRK5-mediated and SAP97-dependent modulation of β1AR-CaMKII signaling are explored. Our data indicate that GRK5-dependent loss of SAP97 in β1AR complex switches on receptor signaling to detrimental CaMKII in myocardium, contributing to HF development.
METHODS
The authors declare that all supporting data are available within the article and its online supplementary files.
SAP97-cKO mice were generated through crossing SAP97-f/f 18 with MHC-cre mice (Stock # 009074) from Jackson laboratory. Cardiac specific deletion of GRK2 and GRK5 and flox controls were gifts from Walter Koch (Temple University, Philadelphia, PA) and Gerald Dorn (Washington University, St Louis, MO), respectively. All animal experiments followed the NIH guide for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committees at the University of California, Davis, CA. Osmotic minipumps were implanted and transverse aortic constriction (TAC) were performed on 10–12 weeks old SAP97-f/f and SAP97-cKO male littermates for 3 and 4 weeks, respectively. Cardiac function was monitored with echocardiograph with Vevo 2100. Adult ventricular cardiomyocytes (AVMs) from male mice (8 to 12 weeks old) were used for cellular and biochemical analysis, electrophysiology, E-C coupling, or myocyte death measurement. 19 FRET based biosensors for cAMP (ICUE3), PKA (AKAR3) and CaMKII (camui) were expressed in AVMs to measure the changes in response to adrenergic stimulation. 20, 21
The data, analytic methods, study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedures. Expanded detailed materials and methods can be found in the Online Expanded Materials & Methods.
RESULTS
Genetic loss of SAP97 causes an ageing-dependent mouse heart failure.
GRKs are elevated in HF and implicated in agonist-induced phosphorylation of the β1AR C-terminal PDZ motif to promote receptor dissociation from scaffold proteins such as SAP97. 22 To explore the role of SAP97 in adrenergic regulation of cardiac function, we generated a cardiac specific deletion of SAP97 gene (SAP97-cKO) mouse to mimic loss of SAP97 in β1AR complex. SAP97-cKO mice were grossly normal with equivalent cardiac fractional shortening at 2-month of age compared to SAP97-f/f controls (Figure 1A and Online Table I). The cardiac fractional shortening of SAP97-cKO mice displayed a gradual decline and became severely depressed at 10-month of age compared to those of SAP97-f/f controls (Figure 1A). SAP97-cKO mice had similar heart/body weight ratios at 2- and 6-month of ages relative to SAP97-f/f but developed cardiac hypertrophy at 10-month of age with significant increases in cardiac fibrosis and apoptosis (Figure 1B–1E and Online Figure IA-C). In comparison, SAP97-cKO mice had normal brain and kidney weights at 10-month of age (Online Figure ID-E). In ageing SAP97-cKO hearts, there were increases in phosphorylation of CaMKII at threonine 286 and cleaved caspase 3 compared to SAP97-f/f controls (Figure 1F–1G). These data indicate that cardiac specific deletion of SAP97 leads to an ageing-dependent development of HF associated with elevated CaMKII and pro-apoptotic caspase activity in myocardium.
Figure 1. Cardiac deletion of SAP97 promotes spontaneous heart failure in ageing mice.

A) Cardiac fraction shortening from SAP97-f/f and SAP97-cKO mice were measured with echocardiogram at different ages. Data were from 11 SAP97-f/f and 9 SAP97-cKO mice. ## p < 0.01 by two-way ANOVA followed by Tukey’s test. B) Cardiac LVID and LV volume on 10-month old mice are plotted. ***p < 0.001 between SAP97-cKO and SAP97-f/f mice at 10-month age. C-E) Representative images show H & E staining (Scale bar 500 μm), TUNEL staining (Scale bar 10 μm), Masson’s trichrome staining (Scale bar 100 μm) of heart tissues from SAP97-cKO and SAP97-f/f at 10-month old. Nuclei are shown with DAPI staining. The quantification of the images are plotted below. For H &E staining, * p < 0.05 by one-way ANOVA followed by Tukey’s test. For TUNEL staining, data were 2–3 repeated measurements from 5 2-month and 6 10-month old mice; and Masson’s trichrome staining, data were 2–3 repeated measurements of 5 2-month and 7 10-month old mice. * p < 0.05 and *** p < 0.001 by one-way nested ANOVA followed by Tukey’s test. F-G) Western blots show total and phosphorylated CaMKII and total and cleaved caspase 3 in 10-month old SAP97-cKO and SAP97-f/f heart tissues. ***p < 0.001 by unpaired student t-test.
Loss of SAP97 promotes cardiac β1AR-CaMKII signaling to enhance E-C coupling and ejection fraction in young mice.
We sought to understand the mechanisms underlying elevated CaMKII activity in SAP97-cKO hearts. Previous studies indicate that stimulation of β1AR leads to two independent pathways for activation of CaMKII: PKA- and Epac-dependent pathways. 710 Interestingly, SAP97-cKO hearts displayed increased β1AR binding to CaMKII and arrestin2 despite minimal changes in membrane expression of the receptor compared to SAP97-f/f controls (Figure 2A–2B). The binding of β1AR to Epac2 displayed a small but not significant increase in SAP97-cKO hearts (Figure 2B). The phosphorylation of CaMKII at threonine 286 was significantly elevated in SAP97-cKO hearts compared to SAP97-f/f controls (Figure 2C). These data indicate that the binding of CaMKII to β1AR may enhance activation of CaMKII in SAP97-cKO hearts leading to development of HF. Accordingly, SAP97-cKO mice displayed increases in phosphorylation of ryanodine receptor 2 (RyR2) at serine 2808 and 2814, but little change in expression in sarcoplasmic reticulum Ca2+ ATPase 2 (serca2) and in phosphorylation of phospholamban at serine 16 and threonine 17 and troponin I at serine 23/24 in hearts (Online Figure IIA-D). The 2-month old SAP97-cKO mice also displayed elevated cardiac ejection fraction after injection of isoproterenol relative to SAP97-f/f controls (Figure 2D).
Figure 2. Cardiac deletion of SAP97 enhances adrenergic stimulation of CaMKII and E-C coupling.

A) The expression of membrane β1AR in 2-month old SAP97-f/f and SAP97-cKO hearts was quantified with radioligand binding assay. B) Cardiac β1AR was immunoprecipitated in 10-month old SAP97-f/f and SAP97-cKO heart lysates. The pulled down proteins were subjected to detection of CaMKII, Epac2, arrestin2, and β1AR. ** p < 0.01 by t-test. C) Western blots show total and phosphorylated CaMKII at Threonine 286 in 2-month old SAP97-f/f and SAP97-cKO heart lysates. * p < 0.05 by student t-test. D) Cardiac ejection fraction was measured at baseline and after stimulation with 2 μg/kg and 200 μg/kg of isoproterenol in 2-month old SAP97-f/f (n = 4) and SAP97-cKO (n = 5) mice. * p < 0.05 by t-test compared to SAP97-f/f controls. E) SAP97-f/f and SAP97-cKO AVMs from 2-month old mice were stimulated with 100 nmol/L of isoproterenol, total and phosphorylated CaMKII were detected in western blots. * p < 0.05 by one-way ANOVA followed by Tukey’s test. F-G) SAP97-f/f and SAP97-cKO AVMs from 2-month old mice were pretreated with 1 μmol/L of H89 and KN93 as indicated before stimulation with 100 nmol/L of isoproterenol. Myocyte contractile shortening was recorded before and after isoproterenol stimulation. Data were 2–5 repeated measurements of cells from 5–6 mice. * p < 0.05, **p < 0.01, and ***p < 0.001 by one-way nested ANOVA followed by Tukey’s test.
We then isolated adult ventricular myocytes (AVMs) from 2-month old mice for analysis of β1AR signaling. Consistent with in vivo observations, loss of SAP97 enhanced CaMKII activity, calcium transient, and myocyte contractile shortening but without significant increase in SR calcium load after stimulation with isoproterenol in SAP97-cKO AVMs compared to SAP97-f/f controls (Figure 2E–2G and Online Figure IIIA-E). Inhibition of PKA with H89 completely abolished isoproterenol-induced responses in SAP97-f/f AVMs (Figure 2F and Online Figure IIIA and C). Interestingly, inhibition of either PKA or CaMKII partially reduced isoproterenol-induced increases in fractional shortening (Figure 2G) but neither of them affected increases in calcium transient in SAP97-cKO AVMs (Online Figure IIIB and D). Yet, in SAP97-cKO AVMs with PKA inhibition, isoproterenol was able to enhance contractility over baseline. Only simultaneous inhibition of both kinases abolished isoproterenol-induced responses in fractional shortening (Figure 2G). They appear additive in the figure further highlighting the point that, while PKA dependent signaling is present, there is a shift toward CaMKII signaling.
Deletion of SAP97 switches from PKA to CaMKII-dependent SR leakage and arrhythmia in myocytes.
Consistent with our observations in E-C coupling, deletion of SAP97 enhanced isoproterenol-induced increases in phosphorylation of PLB at both PKA site of serine 16 and CaMKII site of threonine 17 in 2-month old AVMs (Figure 3A). Deletion of SAP97 also enhanced phosphorylation of RyR2 at PKA site of serine 2808 and CaMKII site of serine 2814 after stimulation with isoproterenol (Figure 3B). The elevated phosphorylation of RyR2 can enhance spontaneous calcium release from the SR. Accordingly, SAP97-cKO AVMs displayed increases in calcium sparks after stimulation with isoproterenol compared to SAP97-f/f controls (Figure 3C). Consequently, calcium loads in the SR after spontaneous leakages were reduced in SAP97-cKO myocytes relative to SAP97-f/f controls (Figure 3D). Inhibition of CaMKII significantly reduced calcium sparks in both SAP97-f/f and SAP97-cKO AVMs whereas inhibition of PKA was not effective (Figure 3C). We also observed significant increases in irregular calcium cycling in paced SAP97-cKO AVMs compared to SAP97-f/f controls (Figure 3E–3F). Additionally, we examined the impact of deletion of SAP97 on adrenergic regulation of L-type calcium channels (LTCCs) in AVMs. Deletion of SAP97 reduced LTCC current density at the baseline and after stimulation with isoproterenol (Online Figure IVA-F). To assess how changes observed in SAP97-cKO myocytes impact calcium handling and myofilament contractility and to reconcile mechanistically our experimental findings, we used our established mathematical model of E-C coupling in the mouse ventricular myocytes.23,24 Our simulations allowed reconciling the mechanisms leading to enhanced calcium transients and contractility in SAP97-cKO AVMs, despite reduced LTCC current and SR calcium load (Online Figure VA-C). Indeed, we showed that decreased peak LTCC current and increased SR calcium leak, which have negative inotropic effects, were opposed by action potential prolongation 25 and decreased PDE activity in adrenergic signaling.17 Thus it is conceivable that LTCC downregulation and increased SR calcium leak in SAP97-cKO are counteracted by potentially compensatory changes to preserve contractility.
Figure 3. Cardiac deletion of SAP97 promotes CaMKII-dependent SR calcium leakage and arrhythmia in myocytes.

A-B) SAP97-f/f and SAP97-cKO AVMs were stimulated with 100 nmol/L of isoproterenol for 10 minutes, total and phosphorylated PLB at serine 16 and threonine 17 and total and phosphorylated RyR2 at serine 2808 and serine 2814 were examined in western blots. * p < 0.05 and ***p < 0.001 by one-way ANOVA followed by Tukey’s test. C-D) Calcium sparks were measured with fluo-4 calcium dye in SAP97-f/f and SAP97-cKO AVMs before and after adrenergic stimulation with isoproterenol (100 nmol/L) in the presence of H89 or KN93 (1 μmol/L for 10 minutes). Data were 1–3 repeated measurements of cells from 6 mice. * p < 0.05 and ** p < 0.01 by one-way nested ANOVA followed by Tukey’s test. Calcium load was measured with caffeine (20 mmol/L) after the period of calcium sparks recording. * p < 0.05 by unpaired t-test. E-F) SAP97-f/f and SAP97-cKO AVMs were paced at 1Hz. Calcium transients were measured with fluo-4 dye before and after stimulation with isoproterenol (100 nmol/L). The percentage of cells showing arrhythmia were counted. ** p < 0.01 and ***p < 0.001 by one-way ANOVA followed by Tukey’s test.
Loss of SAP97 promotes PKA-dependent recruitment of CaMKII to β1AR for CaMKII activation.
We further dissected the mechanisms underlying elevated CaMKII activity in SAP97-cKO hearts. Our previous studies show that deletion of SAP97 promotes βAR-induced cAMP-PKA activity by reducing phosphodiesterase 4D8 association with the receptor complex, which controls the magnitude and distribution of cAMP induced by receptor activation.17 Deletion of SAP97 enhanced phosphorylation of the receptor at the PKA site of serine 312 at baseline in 2-month old hearts although the levels of cAMP in SAP97-cKO hearts were not different from those in SAP97-f/f controls (Figure 4A–4B and Online Figure VIIA). Moreover, deletion of SAP97 enhanced β1AR- but not β2AR-induced cAMP signal and PKA activity detected by FRET biosensors ICUE3 and AKAR3 in SAP97-cKO AVMs (Online Figure VIIB-C). Deletion of SAP97 enhanced phosphorylation of the receptor at the PKA site of serine 312 in hearts after stimulation with isoproterenol (Figure 4B). These data indicate that a local elevation of cAMP-PKA activity promotes phosphorylation of β1AR in SAP97-cKO hearts. Both GRK and PKA phosphorylation of β1AR drive the recruitment of arrestin to the receptor. 26 We suspected that a phosphorylated β1AR might promote recruitment of arrestin2 and CaMKII to facilitate Epac-dependent activation of CaMKII. In comparison to SAP97-f/f controls, SAP97-cKO AVMs displayed significantly more puncta PLA staining with antibodies against β1AR and CaMKII and with antibodies against β1AR and arrestin2 (Figure 4C–4E). Inhibition of PKA with PKI significantly reduced PLA staining of these proteins in SAP97-cKO AVMs whereas inhibition of Epac with ESI09 did not (Figure 4C–4E). In comparison, PLA staining with antibodies against β1AR and Epac2 was marginally increased in SAP97-cKO AVMs compared to SAP97-f/f controls (Figure 4F). Inhibition of Epac2 but not PKA significantly reduced PLA \ staining of β1AR and Epac2 in both SAP97-f/f and SAP97-cKO AVMs (Figure 4F). These data indicate that in SAP97-cKO hearts, PKA enhances phosphorylation of β1AR and promotes recruitment of arrestin2 and CaMKII but not Epac2 to the receptor.
Figure 4. Cardiac deletion of SAP97 promotes PKA-dependent recruitment of CaMKII and arrestin2 and Epac2-dependent activation of CaMKII in myocytes.

A) The antibody against β1AR was validated with tissues from WT and β1AR-KO hearts. A phospho-specific antibody against PKA phosphorylation site at serine 312 of mouse β1AR was validated with wild type and mutant β1AR lacking serine 312 expressed in HEK293 cells. B) SAP97-f/f and SAP97-cKO 2-month old mice were subjected to I.P. injection of saline or isoproterenol (2 mg/kg) for 10 minutes. Heart tissues were lysed for western blot to examine total and phosphorylated β1AR. *p < 0.05 and ***p < 0.001 by one-way ANOVA followed by Tukey’s test. C-F) PLA assays were performed with antibodies against β1AR and CaMKII, β1AR and arrestin2, and β1AR and Epac2 in SAP97-f/f and SAP97-cKO AVMs after pretreating cells with PKI or ESI09 for 30 minutes. The panel C shows images of puncta PLA staining from antibodies against β1AR and CaMKII. Dot plots show the puncta PLA staining quantified per cell. Scale bar 10 μm. Data were 1–3 repeated measurements of cells from 6 mice in panel D and F and from 5 mice in panel E. *p < 0.05 and **p < 0.01 by one-way nested ANOVA followed by Tukey’s test. G-H) CaMKII biosensor camui was expressed in SAP97-f/f and SAP97-cKO AVMs before stimulation with 100 nmol/L of isoproterenol. Data show the time courses of FRET ratios before and after isoproterenol stimulation. Dot plot shows the maximal changes in FRET ratio. Data were 2–5 repeated measurements from 5 SAP97-f/f and 6 SAP97-cKO mice. *p < 0.05 and **p < 0.01 by one-way nested ANOVA followed by Tukey’s test.
We further examined the dynamic CaMKII activity after adrenergic stimulation in living AVMs with FRET-based biosensor camui. 21 Stimulation of 1AR but not β2AR induced significant increases in CaMKII activity in both SAP97-f/f and SAP97-cKO AVMs (Online Figure VID and Figure 4G). Deletion of SAP97 further enhanced the β1AR-induced CaMKII activity in AVMs. Inhibition of PKA abolished isoproterenol stimulation of CaMKII activity in SAP97-f/f but only partially reduced adrenergic stimulation of CaMKII activity in SAP97-cKO AVMs (Figure 4H). In comparison, inhibition of Epac completely abolished isoproterenol stimulation of CaMKII activity in SAP97-cKO AVMs (Figure 4H). Together with biochemical data, these data indicate that deletion of SAP97 leads to recruitment of CaMKII to β1AR and switches on an Epac-dependent activation of CaMKII after receptor stimulation.
Deletion of SAP97 promotes β1AR-CaMKII signaling and myocyte death under chronic adrenergic stimulation and TAC.
Chronic stimulation of cardiac β1AR can drive a CaMKII-mediated increase in myocyte apoptosis, 5 contributing to HF development. Stimulation of β1AR but not β2AR promoted cell death in both SAP97-f/f and SAP97-cKO AVMs, in which SAP97-cKO myocytes displayed higher cell death rates than SAP97-f/f controls (Figure 5A–5D, Online Figure VIIA-D). Inhibition of Epac with ESI-09 and CaMKII with KN93 significantly attenuated cell death in SAP97-cKO AVMs whereas inhibition of PKA was not effective, indicating that a critical role of the Epac-CaMKII cascade in β1AR-induced cell death in SAP97-cKO myocytes (Figure 5D). We further assessed the role of elevated β1AR-CaMKII signaling in myocyte apoptosis in SAP97-cKO hearts. Chronic infusion of isoproterenol via osmotic minipump significantly attenuated cardiac fraction shortening in SAP97-cKO relative to SAP97-f/f mice, which was associated with deleterious cardiac remodeling including higher levels of cell apoptosis and fibrosis (Figure 5E–5G and Online Figure VIIIA-B). Together, these data indicate that deletion of SAP97 enhances β1AR-Epac-CaMKII signaling and promotes detrimental cardiac remodeling in animal hearts.
Figure 5. Cardiac deletion of SAP97 promotes β1AR-induced and Epac-CaMKII-dependent cell death in hearts.

A-B) Representative images show SAP97-f/f and SAP97-cKO AVMs before and after stimulation with isoproterenol (100 nmol/L) for 48 hrs. Scale bar 100 μm. C) Dot plot shows quantification of dead cells from panel A and B. Data are from 4–5 mice as indicated. * p < 0.05 by one-way nested ANOVA followed by Tukey’s test. D) SAP97-cKO AVMs were pretreated with H89 (1 μmol/L), KN93 (1 μmol/L), and ESI-09 (20 nmol/L) as indicated before stimulation with isoproterenol (100 nmol/L) for 24 and 48 hrs. Dot plot shows quantification of dead cells. Data were 2–3 repeated measurements from 5 SAP97-f/f and 4 SAP97-cKO mice. * p < 0.05 by one-way nested ANOVA followed by Tukey’s test. E) SAP97-f/f and SAP97-cKO mice were subjected to infusion of isoproterenol (60mg/kg/day) for 3 weeks. Cardiac fraction shortening was measured with echocardiogram. F) Images show representative TUNEL staining of heart tissues after minipump infusion of saline and isoproterenol. The quantification is plotted below. Scale bar 10 μm. Nuclei are shown with DAPI staining. Data were measurements from 6 Saline and 7 ISO treated mice. * p < 0.05 and ***p < 0.001 by one-way ANOVA followed by Tukey’s test. G) Images show representative Masson’s trichrome staining of heart tissues after minipump infusion of saline and isoproterenol; the quantification is plotted below. Scale bar 100 μm. Data were 1–3 repeated measurements from 6 Saline and 5 ISO treated mice. * p < 0.05 and ** p < 0.01 by one-way nested ANOVA followed by Tukey’s test.
Next, we explored the role of SAP97-dependent β1AR signaling in cardiac hypertrophy induce by pressure overload with 4-week of TAC. Loss of cardiac SAP97 enhanced TAC-induced cell apoptosis and fibrosis associated with increased cardiac hypertrophy (Figure 6A–6C). After TAC, the expression of SAP97 in SAP97-f/f hearts did not change in TAC mice relative to SHAM controls while there was a slight decrease in β1AR expression in TAC hearts (Online Figure VIIIC). However, SAP97-cKO hearts displayed significantly higher levels of CaMKII and caspase 3 activity than SAP97-f/f controls (Figure 6D–6F). Cardiac fractional shortening was further depressed in SAP97-cKO mice relative to SAP97-f/f controls (Figure 6G and Online Table II); this observation was absent in female SAP97-cKO mice relative to SAP97-f/f controls (Online Figure VIII D). These data indicate that deletion of SAP97 promotes CaMKII and caspase activity after pressure overload, which exacerbates cardiac remodeling and HF development.
Figure 6. Cardiac deletion of SAP97 exacerbates cardiac remodeling and dysfunction induced by TAC.

A-C) Representative H & E staining (Scale bar 500 μm), TUNEL staining (Scale bar 20 μm), and Masson’s trichrome staining (Scale bar 100 μm) images of SAP97-f/f and SAP97-cKO hearts after 4 weeks of SHAM and TAC. Nuclei are shown with DAPI staining. Dot plots represent image quantification. Data were 1–3 repeated measurements from 5 SHAM and 6 TAC mice. * p < 0.05, ** p < 0.01, and *** p < 0.001 by one-way ANOVA or one-way nested ANOVA followed by Tukey’s test. D-F) Total and cleaved caspase 3 and total and phosphorylated CaMKII in SAP97-f/f and SAP97-cKO hearts after 4 weeks of SHAM and TAC were detected in western blots. * p < 0.05, ** p < 0.01, and *** p < 0.001 by one-way ANOVA followed by Tukey’s test. G) Cardiac fractional shortening was measured with echocardiogram in SAP97-f/f and SAP97-cKO mice after 4 weeks of SHAM and TAC. * p < 0.05 and *** p < 0.001 by one-way ANOVA followed by Tukey’s test.
GRK5 promotes dissociation of β1AR-SAP97 complex and CaMKII signaling associated with heart failure.
We examined the integrity of β1AR-SAP97 complex in human HF. The expression levels of β1AR, SAP97, and PDE4B were not different between healthy subjects and HF with dilated cardiomyopathy and ischemic cardiomyopathy (Figure 7A-7B). The expression levels of major cardiac GRK2, GRK5, and PDE4D were increased in human HF compared to healthy subjects, consistent with data in the literature. 27–29 GRKs are implicated in agonist-induced phosphorylation of the β1AR C-terminal PDZ motif to modulate receptor binding to scaffold proteins. 22 Accordingly, the binding between SAP97 and β1AR was reduced in human HF compared to healthy subjects (Figure 7C and Online Figure IX). We examined the potential role of cardiac GRK2 and GRK5 on the integrity of β1AR-SAP97 complex. Stimulation of β1AR with isoproterenol promoted dissociation of SAP97 from the receptor in WT mouse hearts. Cardiac specific deletion of GRK5 but not GRK2 blocked dissociation of SAP97 from the receptor in hearts (Figure 7D). Previous studies shown that cardiac deletion of GRK5 protects against HF induced by chronic adrenergic stress with infusion of isoproterenol 30. We suspected that deletion of GRK5 blocks cardiotoxic β1AR-CaMKII activity under chronic stress. Deletion of cardiac GRK5 significantly attenuated phosphorylation of CaMKII at threonine 286 in hearts after chronic infusion of isoproterenol, which was associated with ameliorated cardiac contractile dysfunction (Figure 7E-7F and Online Table III). Together, these data indicate that GRK5 is necessary for agonist-induced dissociation of SAP97 from cardiac β1AR, which promotes activation of CaMKII for detrimental cardiac remodeling and HF development.
Figure 7. GRK5 promotes dissociation of cardiac SAP97-β1AR complex and β1AR-CaMKII signaling in heart failure.

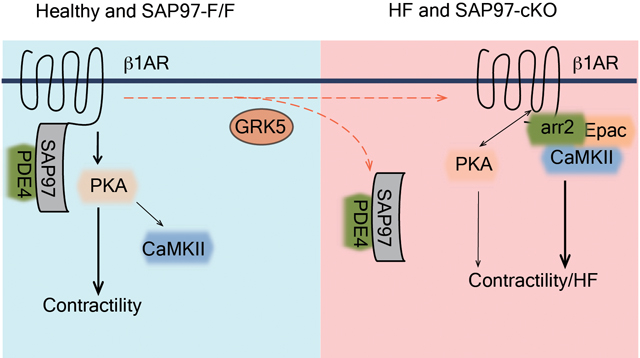
A-B) Cardiac expression of β1AR, SAP97, PDE4D, PDE4B, GRK2 and GRK5 were detected in human heart tissues from healthy subjects and HF with either ischemic cardiomyopathy or dilated cardiomyopathy. * p < 0.05 by one-way ANOVA followed by Tukey’s test. C) The β1AR was pulled down from human heart lysates from healthy and HF subjects. The pulled down β1AR and SAP97 were detected by western blot. D) GRK2-cKO, GRK5-cKO and flox control mice were subjected to I.P. injection with isoproterenol (2 mg/kg) for 10 mins. The cardiac β1AR was pulled down from heart lysates; and both β1AR and SAP97 were detected in western blots. E) GRK5-f/f and GRK5-cKO mice were subjected to chronic infusion of isoproterenol (60 mg/kg/day) for 1 week. Cardiac fractional shortening was monitored with echocardiogram after isoproterenol infusion. ** p < 0.01 by one-way ANOVA followed by Tukey’s test. F) Total and phosphorylated CaMKII in GRK5-f/f and GRK5-cKO hearts after one-week infusion of isoproterenol were detected in western blot and quantified. ** p < 0.01 by unpaired t-test. G) Cartoon depicts the β1AR signaling in healthy (SAP97-f/f) and failing (SAP97-cKO) hearts.
DISCUSSION
In this study, we have revealed a critical role of SAP97 in safeguarding β1AR signaling integrity in heart. Our data show that the binding of SAP97 to β1AR is reduced in HF, which is accompanied with increased expression of GRK5. Genetic deletion of cardiac SAP97 in male mice leads to spontaneous development of cardiomyopathy and exacerbates cardiac dysfunction and hypertrophy induced by TAC, both of which are associated with elevated cardiac fibrosis and myocyte apoptosis. Deletion of SAP97 in female mice did not exacerbate TAC-induced cardiac dysfunction and hypertrophy, indicating a gender dependent-compensation. Loss of SAP97 enhances β1AR stimulation-induced cAMP signal and PKA activity, promotes PKA phosphorylation of β1AR, and recruits arrestin2 and CaMKII to the receptor to facilitate Epac-dependent CaMKII activity. Consequently, loss of SAP97 switches on β1AR signaling to CaMKII to regulate E-C coupling and promote SR calcium leakage, irregular calcium cycling, and myocyte apoptosis. Moreover, cardiac specific deletion of GRK5 blocks agonist-induced dissociation of SAP97 from β1AR, inhibits cardiotoxic β1AR-CaMKII signaling, and prevents cardiac remodeling in myocardium. Together, these data reveal the master role of SAP97 in dictating cardiac β1AR signaling to different downstream PKA and CaMKII kinases. GRK5 acts as a switch to turn on β1AR-induced CaMKII activation in pathological HF development.
SAP97 is a multi-functional scaffold protein with three PDZ domains, one of which directly binds to the C-terminal PDZ motif of β1AR. 14, 16 SAP97 also binds to phosphodiesterase 4D8. 17 The connection of β1AR to phosphodiesterase 4D8 together with the receptor-associated adenylyl cyclases 31 form a signaling circuitry for a tight regulation of cAMP signal in space and time. 1, 17 This is essential to coordinate downstream activation of PKA for phosphorylation of specific substrates necessary for rhythmic beat-to-beat contractile response. Using PLA and FRET-based living cell imaging assays, we showed that the β1AR-bound SAP97 played an essential role in modulating activation of CaMKII, another critical downstream signaling molecule in heart. SAP97 has been shown to bind to CaMKII in heart, 32 which may form a complex with β1AR to facilitate receptor-induced activation of CaMKII. In SAP97-f/f AVMs, the β1AR-induced CaMKII activation is inhibited by the PKA inhibitor, supporting a necessary role of PKA in promoting CaMKII activation under physiological stimulation. This is likely due to an increase in LTCC-dependent calcium influx and RyR2-dependent calcium release to promote local activation of CaMKII. 6, 33 In agreement, overexpression of PKI, a specific PKA peptide inhibitor, abolishes β1AR-induced CaMKII activity in feline AVMs in vitro and transgenic mouse hearts in vivo. 10 In comparison, deletion of SAP97 leads to elevated cAMP signal and promotes PKA phosphorylation of β1AR in SAP97-cKO AVMs. The activated β1AR recruits arrestin2 and CaMKII in a PKA activity-dependent fashion and promotes Epac-dependent activation of CaMKII via activation of Epac with higher levels of cAMP 34. Inhibition of PKA abolishes the increased binding of β1AR to arrestin2 and CaMKII and attenuates isoproterenol-induced CaMKII activation in SAP97-cKO myocytes. In comparison, inhibition of Epac completely blocks the β1AR-induced CaMKII activation in SAP97-cKO myocytes. There is still significant difference in CaMKII activity between Epac inhibitor- and PKA inhibitor-treated AVMs, indicating that there is a PKA-independent but Epac-dependent component in isoproterenol-induced CaMKII activity. Together, these data for the first time reveal that both PKA and Epac can lead to CaMKII activation depending on the orchestra of the receptor signaling complexes, the concentrations of cAMP, and the accessibility to downstream effectors.
A recent report shows that agonist stimulation of β1AR promotes GRK-dependent recruitment of Epac with arrestin and CaMKII, which facilitates Epac-dependent activation of CaMKII in mouse hearts.7 Notably, the difference between this report and current study lies in the ability of β1AR to recruit Epac. The GRK-phosphorylated β1AR is able to recruit Epac whereas our current data show that the PKA-phosphorylated β1AR marginally does so. Biochemical and structural studies show that after binding to GPCRs, arrestin can present different conformational changes, which may facilitate binding to distinct signaling partners. 35, 36 While both PKA and GRK phosphorylation can promote agonist-induced recruitment of arrestin2 to the β1AR, 26 the GRK-phosphorylated β1AR may transduce a specific arrestin2 conformation to recruit Epac. In both scenarios, the β1AR-arrestin2-CaMKII complexes facilitate Epac-dependent activation of CaMKII. Our data show that inhibition of PKA blocks β1AR-induced activation of CaMKII detected with FRET-based biosensor, indicating that PKA is a major mechanism for CaMKII activation under acute adrenergic stimulation. However, the CaMKII activity induced by prolonged adrenergic stimulation is likely involved in both PKA and GRK-dependent mechanisms. 7 Studies show that inhibition of PKA does not block prolonged adrenergic stimulation of myocyte apoptosis. 5 Our data suggest that once the β1AR signaling integrity is disrupted, PKA is dispensable in β1AR-induced myocyte apoptosis. Nevertheless, loss of SAP97 in the β1AR complex drastically changed cellular properties in myocytes. With elevation of CaMKII activity, the β1AR-induced stimulation of E-C coupling becomes less dependent on PKA; only simultaneous inhibition of both PKA and CaMKII is able to block adrenergic-induced increases in myocyte contractile shortening.
Interestingly, loss of SAP97 leads to reduction in LTCC current together with elevation of RyR2-mediated SR calcium release, which would be expected to diminish rather than enhance E-C coupling. We sought to reconcile these counterintuitive observations using computational modeling. Our simulated data show that in SAP97-cKO AVMs, decreased calcium influx through LTCC, along with increased SR calcium leakage, would lead to diminished SR calcium load, calcium transient amplitude, and myofilament shortening. But these effects are counteracted by reduced PDE concentration associated with β1AR 17 leading to increased serca2 function for SR reuptake and loading, and by action potential prolongation 25 leading to prolonged calcium influx. As result of these potentially adaptive changes, we observed an increased calcium transient and contractility without an increase in SR calcium load in SAP97-cKO myocytes. Similarly, a compensatory increase of RyR2-dependent SR calcium release has been observed in cardiac heterozygous deletion of LTCC, to maintain positive inotropy despite a reduction in calcium influx via LTCCs.37 How SAP97 affects LTCC activity remains to be addressed. Additionally, inhibition of CaMKII effectively blocked ISO-induced contractile shortening in SAP97-cKO; it did not affect the ISO-induced increases in calcium transient. These data indicate overlapping or redundant roles of PKA and CaMKII in calcium handling in adrenergic stimulation in SAP97-cKO. The precise role(s) of PKA and CaMKII involved in E-C coupling in SAP97-cKO myocytes remains to be addressed. Meanwhile, deletion of SAP97 also leads to abnormal sodium and potassium channel activity. 25 All these channel abnormalities can contribute to irregular calcium cycling and cardiac arrhythmia in SAP97-cKO mice 25. Together, SAP97 acts as an essential coordinator to maintain the integrity of ion channel currents to ensure rhythmic contractile responses. However, in the long term, the CaMKII-dependent calcium release can lead to chronic elevation of cytoplasmic calcium in SAP97-cKO AVMs. The abnormal high levels of calcium signal and CaMKII activity can further exacerbate adrenergic-induced SR leakage associated with reduced SR calcium load. 38, 39 The elevated CaMKII activity and cytoplasmic calcium are also critically involved in β1AR-induced and mitochondria-dependent cell apoptosis in SAP97-cKO hearts. 5 Moreover, the elevated intracellular calcium signal and CaMKII activity can promote calcineurin and NFAT-dependent cardiac hypertrophy and remodeling, as reported in the LTCC heterozygous knockout hearts 37. While overexpression of CaMKII by more than 10 folds in mouse hearts leads to development of HF in 3 months, 40 loss of SAP97 in the β1AR complex leads to a mild activation of CaMKII (less than 2 folds) to promote detrimental cardiac adaptation. This mild adrenergic stress not only promotes spontaneous development of HF in 10-month-old mice but also accelerates HF induced by chronic perfusion of isoproterenol and TAC.
Importantly, we show in HF, the binding between β1AR and SAP97 is reduced, which could facilitate Epac-dependent activation of CaMKII similar to those observed in SAP97-cKO myocytes. The β1AR PDZ motif has a serine residue that can undergo phosphorylation by GRKs after agonist stimulation, which promotes dissociation of the phosphorylated receptor from scaffold proteins such as SAP97. 14 Despite a prominent role of GRK2 in desensitization of cardiac βARs under chronic stress,28, 29 our data show that cardiac GRK5 but not GRK2 is necessary to promote agonist-induced dissociation of SAP97 from β1AR. Genetic deletion of cardiac GRK5 effectively reduces activation of CaMKII in myocardium after chronic infusion of isoproterenol. These data reveal a novel role of GRK5 in preventing cardiotoxic β1AR-CaMKII signaling, consistent with the necessary roles of these two kinases in TAC-induced HF development. 8, 9, 30 The expression levels of GRK5 are elevated in human HF; therefore, inhibition of GRK5 can prevent dissociation of SAP97-β1AR complex and CaMKII activation in HF development.
Taken together, we have revealed a cardiac signaling paradigm, in which GRK5 promotes dissociation of SAP97 from β1AR to facilitate receptor signaling to cardiotoxic CaMKII and detrimental remodeling in HF development. The binding of SAP97 to β1AR, which is accompanied with elevated GKR5 expression, is reduced in HF. Therefore, targeting of the GRK5-CaMKII signaling axis could be a promising strategy to treat HF.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Chronic stimulation of β1AR induces detrimental CaMKII activity, promoting cardiac maladaptation and development of heart failure.
SAP97 is a multi-functional scaffold protein that binds to β1AR and organizes a β1AR signalosome.
The β1AR-SAP97 signalosome can be modulated by adrenergic stimulation and controls cAMP signaling.
What New Information Does This Article Contribute?
Loss of SAP97 promotes CaMKII association with β1AR and increases CaMKII activity in an Epac-dependent manner.
Loss of SAP97 drives detrimental functional and structural remodeling in the myocardium.
Deletion of GRK5 prevents agonist-induced dissociation of SAP97 from β1AR and increase in CaMKII activity in hearts. The β1AR-SAP97 signalosome is reduced in human heart failure.
Chronic activation of cardiac β1AR drives toxic intracellular CaMKII and the development of heart failure. Both PKA and Epac are implicated in β1AR-induced activation of CaMKII, however, the mechanisms that control each pathway in the development of heart failure remain unclear. Here, we identified a molecular machinery by which a multi-functional scaffold protein SAP97 binds to cardiac β1AR and facilitates the receptor induced PKA-dependent activation of CaMKII. Moreover, GRK5 switches β1AR signaling to Epac-dependent activation of CaMKII via promoting dissociation of SAP97 from the receptor. These data reveal a novel cardiac GRK5-CaMKII axis that can be potentially targeted in heart failure therapy.
ACKNOWLEDGEMENT
We thank Joseph M. Martinez for manuscript editing.
SOURCE of FUNDING
This work was supported by National Institutes of Health grants HL127764 and HL147263 (YKX), R01HL131517 (EG), and K99HL138160 (SM), a VA Merit grant 01BX002900 (YKX), and an American Heart Association grant 15SDG24910015 (EG). QS and AS are recipients of American Heart Association postdoctoral fellowship. YKX is an established American Heart Association investigator.
Nonstandard Abbreviations and Acronyms:
- CaMKII
Calmodulin-dependent kinase II
- β1AR
β1-adrenergic receptor
- SAP97
Synapse-associated protein 97
- RT-qPCR
Quantitative reverse transcription PCR
- TUNEL
Terminal deoxynucleotidyl transferase (TdT) dUTP nick-end
- AVM
Adult ventricular cardiomyocyte
- GRK
G-protein receptor kinase
- PKA
protein kinase A
- PLA
proximity ligation assay
- AKAP
A-kinase anchoring proteins
- Epac
exchange protein directly activated by cAMP
- TAC
transverse aortic constriction
- FRET
Förster resonance energy transfer
- HF
heart failure
- E-C coupling
excitation-contraction coupling
Footnotes
DISCLOSURE
None
REFERENCES
- 1.Xiang YK. Compartmentalization of beta-adrenergic signals in cardiomyocytes. Circ Res. 2011;109:231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perrino C and Rockman HA. Reversal of cardiac remodeling by modulation of adrenergic receptors: a new frontier in heart failure. Curr Opin Cardiol. 2007;22:443–9. [DOI] [PubMed] [Google Scholar]
- 3.Xiao RP, Cheng H, Zhou YY, Kuschel M and Lakatta EG. Recent advances in cardiac beta(2)-adrenergic signal transduction. Circ Res. 1999;85:1092–100. [DOI] [PubMed] [Google Scholar]
- 4.Hell JW. Beta-adrenergic regulation of the L-type Ca2+ channel Ca(V)1.2 by PKA rekindles excitement. Sci Signal. 2010;3:pe33. [DOI] [PubMed] [Google Scholar]
- 5.Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, Devic E, Kobilka BK, Cheng H and Xiao RP. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. 2003;111:617–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP and Cheng H. Sustained beta1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 2004;95:798–806. [DOI] [PubMed] [Google Scholar]
- 7.Mangmool S, Shukla AK and Rockman HA. beta-Arrestin-dependent activation of Ca(2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J Cell Biol. 2010;189:573–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D and Brown JH. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson ME, Brown JH and Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY and Chen X. Cardiotoxic and cardioprotective features of chronic beta-adrenergic signaling. Circ Res. 2013;112:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shcherbakova OG, Hurt CM, Xiang Y, Dell’Acqua ML, Zhang Q, Tsien RW and Kobilka BK. Organization of beta-adrenoceptor signaling compartments by sympathetic innervation of cardiac myocytes. J Cell Biol. 2007;176:521–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McConnachie G, Langeberg LK and Scott JD. AKAP signaling complexes: getting to the heart of the matter. Trends Mol Med. 2006;12:317–23. [DOI] [PubMed] [Google Scholar]
- 13.Nichols CB, Rossow CF, Navedo MF, Westenbroek RE, Catterall WA, Santana LF and McKnight GS. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ Res. 2010;107:747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He J, Bellini M, Inuzuka H, Xu J, Xiong Y, Yang X, Castleberry AM and Hall RA. Proteomic analysis of beta1-adrenergic receptor interactions with PDZ scaffold proteins. J Biol Chem. 2006;281:2820–7. [DOI] [PubMed] [Google Scholar]
- 15.Xiang Y, Devic E and Kobilka B. The PDZ binding motif of the beta 1 adrenergic receptor modulates receptor trafficking and signaling in cardiac myocytes. J Biol Chem. 2002;277:33783–90. [DOI] [PubMed] [Google Scholar]
- 16.Nooh MM, Chumpia MM, Hamilton TB and Bahouth SW. Sorting of beta1-Adrenergic Receptors Is Mediated by Pathways That Are Either Dependent on or Independent of Type I PDZ, Protein Kinase A (PKA), and SAP97. J Biol Chem. 2014;289:2277–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu Q, Kim S, Soto D, De Arcangelis V, DiPilato L, Liu S, Xu B, Shi Q, Zhang J and Xiang YK. A long lasting beta1 adrenergic receptor stimulation of cAMP/protein kinase A (PKA) signal in cardiac myocytes. J Biol Chem. 2014;289:14771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou W, Zhang L, Guoxiang X, Mojsilovic-Petrovic J, Takamaya K, Sattler R, Huganir R and Kalb R. GluR1 controls dendrite growth through its binding partner, SAP97. J Neurosci. 2008;28:10220–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbagallo F, Xu B, Reddy GR, West T, Wang Q, Fu Q, Li M, Shi Q, Ginsburg KS, Ferrier W, Isidori AM, Naro F, Patel HH, Bossuyt J, Bers D and Xiang YK. Genetically Encoded Biosensors Reveal PKA Hyperphosphorylation on the Myofilaments in Rabbit Heart Failure. Circ Res. 2016;119:931–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soto D, De Arcangelis V, Zhang J and Xiang Y. Dynamic protein kinase a activities induced by beta-adrenoceptors dictate signaling propagation for substrate phosphorylation and myocyte contraction. Circ Res. 2009;104:770–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reddy GR, West TM, Jian Z, Jaradeh M, Shi Q, Wang Y, Chen-Izu Y and Xiang YK. Illuminating cell signaling with genetically encoded FRET biosensors in adult mouse cardiomyocytes. J Gen Physiol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao TT, Deacon HW, Reczek D, Bretscher A and von Zastrow M. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the beta2-adrenergic receptor. Nature. 1999;401:286–90. [DOI] [PubMed] [Google Scholar]
- 23.Surdo NC, Berrera M, Koschinski A, Brescia M, Machado MR, Carr C, Wright P, Gorelik J, Morotti S, Grandi E, Bers DM, Pantano S and Zaccolo M. FRET biosensor uncovers cAMP nano-domains at beta-adrenergic targets that dictate precise tuning of cardiac contractility. Nat Commun. 2017;8:15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morotti S, Edwards AG, McCulloch AD, Bers DM and Grandi E. A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J Physiol. 2014;592:1181–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gillet L, Rougier JS, Shy D, Sonntag S, Mougenot N, Essers M, Shmerling D, Balse E, Hatem SN and Abriel H. Cardiac-specific ablation of synapse-associated protein SAP97 in mice decreases potassium currents but not sodium current. Heart rhythm : the official journal of the Heart Rhythm Society. 2015;12:181–92. [DOI] [PubMed] [Google Scholar]
- 26.Rapacciuolo A, Suvarna S, Barki-Harrington L, Luttrell LM, Cong M, Lefkowitz RJ and Rockman HA. Protein kinase A and G protein-coupled receptor kinase phosphorylation mediates beta-1 adrenergic receptor endocytosis through different pathways. J Biol Chem. 2003;278:35403–11. [DOI] [PubMed] [Google Scholar]
- 27.Wang Q, Liu Y, Fu Q, Xu B, Zhang Y, Kim S, Tan R, Barbagallo F, West T, Anderson E, Wei W, Abel ED and Xiang YK. Inhibiting Insulin-Mediated beta2-Adrenergic Receptor Activation Prevents Diabetes-Associated Cardiac Dysfunction. Circulation. 2017;135:73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iaccarino G, Tomhave ED, Lefkowitz RJ and Koch WJ. Reciprocal in vivo regulation of myocardial G protein-coupled receptor kinase expression by beta-adrenergic receptor stimulation and blockade. Circulation. 1998;98:1783–9. [DOI] [PubMed] [Google Scholar]
- 29.Brinks H and Koch WJ. Targeting G protein-coupled receptor kinases (GRKs) in Heart Failure. Drug Discov Today Dis Mech. 2010;7:e129–e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gold JI, Gao E, Shang X, Premont RT and Koch WJ. Determining the absolute requirement of G protein-coupled receptor kinase 5 for pathological cardiac hypertrophy: short communication. Circ Res. 2012;111:1048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW and Scott JD. Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell. 2006;23:925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El-Haou S, Balse E, Neyroud N, Dilanian G, Gavillet B, Abriel H, Coulombe A, Jeromin A and Hatem SN. Kv4 potassium channels form a tripartite complex with the anchoring protein SAP97 and CaMKII in cardiac myocytes. Circ Res. 2009;104:758–69. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Zhang H, Gao H, Kubo H, Berretta RM, Chen X and Houser SR. {beta}1-Adrenergic receptor activation induces mouse cardiac myocyte death through both L-type calcium channel-dependent and -independent pathways. Am J Physiol Heart Circ Physiol. 2010;299:H322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A and Bos JL. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem. 2000;275:20829–36. [DOI] [PubMed] [Google Scholar]
- 35.Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, Lohse MJ and Hoffmann C. beta-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature. 2016;531:661–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, Dosey AM, Su M, Liang CR, Gu LL, Shan JM, Chen X, Hanna R, Choi M, Yao XJ, Klink BU, Kahsai AW, Sidhu SS, Koide S, Penczek PA, Kossiakoff AA, Woods VL, Jr., Kobilka BK, Skiniotis G and Lefkowitz RJ. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goonasekera SA, Hammer K, Auger-Messier M, Bodi I, Chen X, Zhang H, Reiken S, Elrod JW, Correll RN, York AJ, Sargent MA, Hofmann F, Moosmang S, Marks AR, Houser SR, Bers DM and Molkentin JD. Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J Clin Invest. 2012;122:280–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu L, Lai D, Cheng J, Lim HJ, Keskanokwong T, Backs J, Olson EN and Wang Y. Alterations of L-type calcium current and cardiac function in CaMKII{delta} knockout mice. Circ Res. 2010;107:398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, Wehrens XH, Chen J and Bers DM. Epac2 mediates cardiac beta1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation. 2013;127:913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH and Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.