

Abstract
The transcription factor forkhead box Q1 (FoxQ1), which is overexpressed in different solid tumors including breast cancer, but the mechanism underlying its oncogenic function is still not fully understood. In this study, we compared RNA-seq data from FoxQ1 overexpressing SUM159 cells with that of empty vector-transfected control cells to identify novel mechanistic targets of this transcription factor. Analysis of The Cancer Genome Atlas (TCGA) dataset revealed significantly higher expression of FoxQ1 in black breast cancer patients compared to white women with this disease. On the other hand, expression of FoxQ1 was comparable in ductal and lobular carcinomas in the breast cancer TCGA dataset. Complementing our published findings in basal-like subtype, immunohistochemistry revealed upregulation of FoxQ1 protein in luminal-type human breast cancer tissue microarrays when compared to normal mammary tissues. Many previously reported transcriptional targets of FoxQ1 (e.g., E-cadherin, N-cadherin, fibronectin 1, etc.) were verified from the RNA-seq analysis. FoxQ1 overexpression resulted in downregulation of genes associated with cell cycle checkpoints, M phase, and cellular response to stress/external stimuli as evidenced from the Reactome pathway analysis. Consequently, FoxQ1 overexpression resulted in mitotic arrest in basal-like SUM159 and HMLE cells, but not in luminal-type MCF-7 cells. Finally, we show for the first time that FoxQ1 is a direct transcriptional regulator of interleukin (IL)-1α, IL-8, and vascular endothelial growth factor in breast cancer cells as evidenced by chromatin immunoprecipitation assay. In conclusion, the present study reports novel mechanistic targets of FoxQ1 in human breast cancer cells.
Keywords: Breast Cancer, FoxQ1, Cell Cycle, Interleukin-1α, Interleukin-8
1. INTRODUCTION
Breast cancer remains an alarming health concern for women worldwide reflected by more than 40,000 deaths each year in the United States alone.1 A challenging aspect in clinical management of breast cancer relates to molecular heterogeneity of the disease that is characterized by distinct gene expression signatures and overexpression of driver oncogenic proteins.2-5 Majority of the invasive mammary ductal carcinomas are broadly grouped into four subtypes, including luminal A type [estrogen receptor positive (ER+), progesterone receptor positive (PR+), and human epidermal growth factor receptor 2 negative (HER2−)], luminal B type (ER+/PR+/HER2− or HER2+), HER2-enriched, and basal-like.2,3 Nearly 75% of basal-like breast tumors are triple-negative due to lack of ER, PR, and HER2 expression.6 Further characterization of the disease subtype-independent oncogenic dependencies in breast cancer is necessary to identify novel druggable targets to broaden therapeutic options for different subtypes of breast cancer.
The transcription factor forkhead box Q1 (FoxQ1) has recently emerged as a key player in the pathogenesis of breast cancer.7-9 Zhang et al.7 were the first to demonstrate a role for FoxQ1 in epithelial-mesenchymal transition (EMT) in breast cancer metastasis using a cross-species gene expression profiling strategy. Forced expression of FoxQ1 in a human mammary epithelial cell line (HMLE) and EpRas cells increased their ability to migrate and invade in vitro, and these effects were reversible by knockdown of this protein in 4T1 mouse mammary carcinoma cells.7 Moreover, FoxQ1 overexpressing EpRas cells exhibited increased propensity for pulmonary metastasis in vivo.7 Promotion of the EMT phenotype by FoxQ1 overexpression was due to direct repression of E-cadherin (CDH1) protein expression.7 In another study, FoxQ1 expression was shown to correlate with high-grade basal-like breast cancers and associated with poor clinical outcomes.8 RNA interference of FoxQ1 in a highly invasive basal-like human breast cancer cell line (MDA-MB-231) attenuated EMT phenotype and decreased its invasion ability.8 Ectopic expression of FoxQ1 in a human mammary epithelial cell line immortalized by hTERT and SV40 large T antigen (HMLER) also promoted stem-like phenotype characterized by increased mammosphere multiplicity.8 Importantly, suppression of FoxQ1 expression in MDA-MB-231 cells resulted in increased active caspase-3 level in response to treatment with several chemotherapy drugs, including 5-fluorouracil, taxol, and camptothecin.8 Transcriptional inactivation of CDH1 by FoxQ1 overexpression was also demonstrated in this study.8 Subsequently, platelet-derived growth factor receptor (PDGFR) α/β were identified as additional downstream targets of FoxQ1 in promotion of breast cancer stem cell-like phenotype as well as chemoresistance.9 We have also shown previously that FoxQ1 promotes breast cancer stem-like phenotype in a luminal-type (MCF-7) and a basal-like (SUM159) cell line by causing direct transcriptional repression of the tumor suppressor dachshund homolog 1 (DACH1).10
Published studies thus far clearly indicate that FoxQ1 regulates expression of multiple cancer-relevant genes to promote cell invasion/migration, stem-like phenotype, and chemotherapy resistance in vitro and metastasis in vivo in breast cancer cell lines, but the possibility of additional functionally important targets of this transcription factor can’t be fully ignored. To address this question, we compared RNA-seq data from FoxQ1 overexpressing SUM159 (hereafter abbreviated as FoxQ1 cells) cells with that of empty vector transfected control cells (hereafter abbreviated as EV cells) to identify its additional mechanistic targets. The present study reveals novel downstream targets of FoxQ1 in breast cancer including cell cycle control, interleukin (IL)-1α, IL-8, and vascular endothelial growth factor (VEGF).
2. Materials and Methods
2.1. Reagents and cell lines
Cell culture reagents including fetal bovine serum, cell culture media, phosphate-buffered saline (PBS), and antibiotic mixture were purchased from Life Technologies-Thermo Fisher Scientific (Waltham, MA). An antibody against FoxQ1 was from Santa Cruz Biotechnology (Dallas, TX). An antibody specific for detection of phospho-(Ser10) histone H3 was from Cell Signaling Technology (Danvers, MA). Anti-β-Actin antibody was from Sigma-Aldrich (St. Louis, MO). The MCF-7 cell line was purchased from the American Type Culture Collection and authenticated by us in 2015 and 2017. The SUM159 cell line was purchased from Asterand (Detroit, MI) and authenticated by us in 2015 and 2017. Details of stable transfection of MCF-7 and SUM159 cells with pCMV6 empty vector and the same vector encoding FoxQ1 and their culture conditions have been described by us previously.10 The HMLE cells stably transfected with FoxQ1 and EV cells were generously provided by Dr. Guojun Wu (Karmanos Cancer Institute, Departments of Oncology and Pathology, Wayne State University, Detroit, MI) and maintained as recommended by the provider.7
2.2. The Cancer Genome Atlas (TCGA) data analysis for FoxQ1 expression in breast cancer
RNA-seq data from TCGA database for breast cancer (n= 1,097) was analyzed using the University of California Santa Cruz Xena Browser (https://xena.ucsc.edu/public/). The correlation coefficient and statistical significance was determined by Pearson test.
2.3. Immunohistochemistry
Expression of FoxQ1 protein in tissue microarrays of human luminal-type breast cancers (US Biomax, Rockville, MD; catalog # BR1508) and normal human mammary tissues (US Biomax, Rockville, MD; catalog # BRN801a) was determined by immunohistochemistry essentially as described by us previously.10 At least three randomly-selected and non-overlapping fields on each core of the tissue microarray were examined using nuclear algorithm of the Aperio ImageScope software (Leica Biosystems, Buffalo Grove, IL). Data is expressed as H-score that is based on intensity (0, 1+, 2+, and 3+) and % positivity (0-100%), and then calculated using the following formula: (% of negative cells × 0) + (% of 1+ cells × 1) + (% of 2+ cells × 2) + (% of 3+ cells × 3). Some specimens for normal breast tissue (n=10) and luminal-type breast cancer (n=2) were not available for analysis due to poor staining and/or less than optimal sectioning on the tissue microarray (cracked or folded).
2.4. RNA-seq analysis
FoxQ1 overexpressing SUM159 cells and EV cells (n=3 for each) were used for RNA-seq analysis. Prior to RNA-seq analysis, overexpression of FoxQ1 was confirmed as done in our previous study.10 Cells (5×105 cells/6-cm dish) were harvested by trypsinization and used for RNA isolation and RNA-seq analysis. The RNA-seq analysis was performed by Novogene (Sacramento, CA). Other details of RNA-seq analysis were essentially same as described by us previously.11 Analysis was performed with a combination of software programs including STAR, HTseq, Cufflink, and wrapped scripts. Tophat program was used for alignments and DESeq2 was used for analysis of differential gene expressions. RNA-seq data presented in this study have been submitted to the Gene Expression Omnibus of NCBI (GSE151059).
2.5. Western blotting
Details of western blotting have been described by us previously.12 The blots were stripped and re-probed with β-Actin antibody for normalization. Immunoreactive bands were detected by the enhanced chemiluminescence method. Densitometric quantitation was done using UN-SCAN-IT software (Silk Scientific, Orem, UT).
2.6. Flow cytometry for determination of mitotic fraction
The cells were plated (SUM159 and HMLE cells - 3 × 105 cells/6-cm dish and MCF-7 cells - 6 × 105 cells/6-cm dish) in triplicate, incubated overnight, and then replaced with fresh medium and incubated for 24 hours. The cells were collected by trypsinization and fixed in 70% ethanol at 4 °C for 2 hours. Subsequently, the cells were permeabilized with 0.25% Triton X-100 for 15 minutes, and incubated with Alexa Fluor 488-conjugated phospho-(Ser10) histone H3 antibody for 1 hour followed by staining with PI (50 μg/mL) and RNase A (80 μg/mL) for 30 minutes at room temperature. Stained cells were analyzed using BD Accuri™ C6 flow cytometer (BD Biosciences).
2.7. Quantitative real-time polymerase chain reaction (qRT-PCR)
The qRT-PCR was performed as described by us previously11 and relative gene expression was calculated using the method of Livak and Schmittgen13. PCR was performed using 2× SYBR green qPCR kit (Thermo Fisher Scientific) with 95°C (15 sec), 60°C (30 sec), and 72°C (20 sec) for 40 cycles. Primers for IL-8 and IL-1α were purchased from GeneCopoeia (Rockville, MD). VEGFA was amplified (60°C, 1 min for 40 cycles) with the following primers and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a normalization control. The primers used were as follows: for VEGFA Forward: 5’-ATCTTCAAGCCATCCTGTGTG-3’; Reverse; 5’-CAAGGCCCACAGGGATTTTC-3’ and for GAPDH Forward: 5’- GGACCTGACCTGCCGTCTAGAA-3’; Reverse; 5’- GGTGTCGCTGTTGAAGTCAGAG-3’.
2.8. Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed according to the manufacturers’ protocol (Magnetic Chip kit, Pierce, Rockford, IL) using normal mouse IgG and FoxQ1 antibodies. Other details of ChIP assay have been described by us previously.10 Putative FoxQ1 binding sites at the IL-8, IL-1α, and VEGFA promoters were amplified (60°C, 1 min, 40 cycles) with the following region-specific primers: for IL-8 site #1, 5′-ATGCACTGTGTTCCGTATGC -3′ (forward) and 5′-GCTTTGCTAGTACAGGACAGG-3𠌒 (reverse); site #3, 5′-TGCTTTCTTCTTCTGATAGACCA-3′ (forward) and 5′-TGTTAACAGAGTGAAGGGGCA-3′ (reverse); for IL-1α, 5’-TTCTTTGGTGAACTGAGGCA-3’ (forward) and 5’-GTGAGCTGCTATGGAGATGC-3’ (reverse); for VEGFA site #1, 5’-AAGGTGAGGCCCTCCAAG-3’ (forward) and 5’-ACCTAGCAGATTGGGGGAAG-3’(reverse); site #2, 5’-GGAGGACAGTTGGCTTATGG-3’(forward) and 5’-GCCAACAGACCTGAAAGAGC-3’(reverse); site #3, 5’-TCCAGATGGCACATTGTCAG-3’(forward) and 5’-TCTGGCTAAAGAGGGAATGG-3’(reverse). Fold enrichment was normalized to the input.
2.9. Measurement of IL-8, IL-1α, and VEGFA levels
Cells were plated in 10-cm dishes at a density of 6 × 105 (SUM159) or 1 × 106 (MCF-7) cells per dish. The secretion of IL-1α, IL-8, and VEGFA in medium was determined using kits from R & D System (Minneapolis, MN) according to the instructions provided by the supplier. The value of IL-1α in EV cells was below detection limit, and therefore a value of 1 was assigned for comparison with FoxQ1 overexpressing SUM159 cells.
2.10. Statistical analysis
Statistical tests were performed using GraphPad Prism (version 7.02). Student’s t test was performed for statistical comparisons between two groups.
3. RESULTS
3.1. FoxQ1 protein is overexpressed in luminal-type human breast cancers compared to normal mammary tissues
We have shown previously that the level of FoxQ1 protein and mRNA is relatively higher in basal-like human breast cancer cell lines (e.g., MDA-MB-231) in comparison with a normal mammary epithelial cell line (MCF-10A) or luminal-type cells (e.g., MCF-7 and MDA-MB-361).10 Moreover, basal-like human breast cancers exhibited higher level of FoxQ1 protein when compared to normal mammary tissues.10 Initially, we compared the expression of FoxQ1 gene expression in white versus black breast cancer patients and between ductal and lobular carcinomas in breast cancer TCGA dataset. The expression of FoxQ1 was modestly but statistically significantly higher in the mammary tumors of black women when compared to white women (Figure 1A). On the other hand, the expression of FoxQ1 was comparable between ductal and lobular breast cancers (Figure 1A). In this study, we also examined the expression of FoxQ1 protein in tissue microarrays consisting of luminal-type breast cancers and normal mammary tissues. Figure 1B shows immunohistochemical staining for FoxQ1 in representative normal mammary tissues and luminal-type breast cancers. The H-score for FoxQ1 expression was higher by 1.6-fold in luminal-type breast cancers when compared to normal mammary tissues (Figure 1C). These results indicated overexpression of the FoxQ1 protein in luminal-type human breast cancers.
Figure 1.

FoxQ1 expression was significantly higher in luminal-type human breast cancer specimens compared to normal human mammary tissues. A, Expression of FoxQ1 in mammary tumors of black and white women, and in ductal and lobular carcinomas. B, Representative immunohistochemical images (200× magnification; scale bar = 100 μm) for FoxQ1 expression in normal human mammary tissues and luminal-type human breast cancer tissues. C, Quantification of FoxQ1 expression in normal human mammary tissues and luminal-type human breast cancer tissues. Results shown are mean ± SD. *P < 0.05 by two-sided Student's t-test. D, Volcano plot for differential gene expression. E, Heatmap of the differentially expressed genes in empty vector transfected cells (EV) and FoxQ1 overexpressing SUM159 cells. F, Venn diagram showing unique and overlapping genes between EV and FoxQ1 overexpressing SUM159 cells.
3.2. FoxQ1-regulated transcriptome in SUM159 cells
Next, we performed RNA-seq analysis using SUM159 cells to identify additional targets of FoxQ1. The mapping results are summarized in Table S1. The Volcano plot in Figure 1D depicts distribution of differentially expressed genes between FoxQ1 overexpressing SUM159 cells and EV cells at an adjusted p value of < 0.05 (Figure 1D). The heatmaps of three replicates of each group exhibiting highly consistent transcriptional changes are shown in Figure 1E. The Venn diagram shown in Figure 1F shows unique and overlapping gene expression between FoxQ1 overexpressing SUM159 cells and EV cells.
3.3. Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis
The top 5 pathways with upregulated genes from the KEGG analysis included pathways in cancer (84 genes), mitogen-activated protein kinase signaling (56 genes), regulation of actin cytoskeleton (49 genes), focal adhesion (50 genes), and axon guidance (37 genes) (Figure 2A). The top 5 pathways with downregulated genes from the KEGG pathway analysis were cell cycle (41 genes), oocyte meiosis (38 genes), progesterone-mediated oocyte maturation (29 genes), lysine degradation (18 genes), and protein processing in endoplasmic reticulum (46 genes) (Figure 2B).
Figure 2.

Functional analysis of the differential expressed genes by FoxQ1 overexpression in SUM159 cells. Scatter plots for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses showing upregulation (A) or downregulation (B) by FoxQ1 overexpression. Scatter plots for gene ontology (GO) enrichment analyses showing upregulation (C) or downregulation (D) by FoxQ1 overexpression. Scatter plots for Reactome pathway analyses showing upregulation (E) or downregulation (F) by FoxQ1 overexpression. padj, adjusted p-value.
3.4. The gene ontology (GO) and Reactome pathway analyses
All three ontologies including cellular component, molecular function, and biological processes are included in the GO enrichment analysis. The GO enrichment analysis revealed that FoxQ1 overexpression caused upregulation of genes associated with positive regulation of locomotion, axon development, positive regulation of cellular component movement, cell substrate/adherens junction, focal adhesion, positive regulation of cell motility/migrations and a few other pathways (Figure 2C). The downregulated genes following FoxQ1 overexpression in SUM159 cells were mostly associated with the GO pathway terms related to cell cycle regulation (GO terms: regulation of cell cycle phase transition, regulation of mitotic cell cycle phase transition, nuclear division, nuclear chromosome segregation, microtubule, cell cycle G2/M phase transition, centromeric region, G2/M transition of mitotic cell cycle, etc.) (Figure 2D).
The Reactome database contains annotations for diverse set of molecular and cell biological topics such as cell cycle, metabolism, signaling, transport, cell motility, immune function, host-virus interaction, and neural function. Genes associated with phospholipid metabolism, VEGF/VEGF receptor pathway, transforming growth factor β pathway were significantly upregulated in FoxQ1 overexpressing SUM159 cells (Figure 2E). Like KEGG and GO pathway analyses, most downregulated genes from the Reactome analyses were associated with cell cycle regulation (Figure 2F). Genes associated with cellular response to stress/external stimuli were also significantly downregulated by FoxQ1 overexpression (Figure 2F).
3.5. Comparison of RNA-seq data with published literature
Published studies have revealed multiple downstream targets of FoxQ1 in breast cancer.7-10,14,15 For example, Zhang et al.7 showed downregulation of CDH1 in highly metastatic breast cancer cell lines (e.g., MDA-MB-435, MDA-MB-231, SUM149, etc.) in comparison with breast cancer cell lines with low metastatic capacity (e.g., MCF-7, MDA-MB-361, BT20, etc.) as well as upregulation of mesenchymal markers such as N-cadherin (CDH2) and fibronectin (FN1) and downregulation of CDH1 in FoxQ1 overexpressing HMLE cells when compared to EV cells. As shown in Figure S1A, the RNA-seq results from the present study were consistent with the findings of Zhang et al..7 Meng et al.9 showed downregulation of CST6, SEMA3A, ADAM9, THBS1, FOXA1, and EDN1 but upregulation of PDGFRA, JAM3, ZEB2, and CD44 in FoxQ1 overexpressing HMLE when compared to EV cells, and these changes were also observed in the RNA-seq data in the present study (Figure S1B). We have shown previously that promotion of stem-like phenotype in FoxQ1 overexpressing SUM159 cell line is associated with suppression of DACH1 and ZEB1 expression and upregulation of mRNA levels of MYC and TWIST210 and RNA-seq results from the present study were consistent with these published observations (Figure S1C). At the same, some published changes in genes downstream of FoxQ17,9,14 were not validated in the RNA-seq data from the present study, including PLD1, SNAI1, CTNNB1, JUP, S100A4, DCN, PDGFRB, CADM3, COL1A1, COL6A1, DSG2, and TWIST1 (Figure S2A-C). Two possibilities exist to explain the inconsistencies between published data and RNA-seq results. The RNA-seq results were obtained using SUM159 cells and this cell line was not used in other published studies7,9,14 except for our own published study.10 Secondly, the possibility of regulatory functions of other transcription factors for these genes can’t be excluded.
3.6. Effect of FoxQ1 overexpression of cell cycle progression
Because the Reactome pathway analysis revealed downregulation of genes associated with terms regulation of mitotic cell cycle progression, M phase, mitotic spindle checkpoints, etc. upon FoxQ1 overexpression, we explored the possibility whether it affected mitotic fraction. We performed western blotting and flow cytometry to quantitate mitotic fraction in FoxQ1 overexpressing cells and corresponding EV cells. Opposite effects were observed in basal-like SUM159 cells and luminal-type MCF-7 cells. FoxQ1 overexpression in SUM159 cells resulted in increased Ser10 phosphorylation of histone H3, which is a marker of mitotic cells (Figure 3A). In the MCF-7 cell line, Ser10 phosphorylation of histone H3 was significantly lower in FoxQ1 overexpressing cells than in EV cells (Figure 3B). These results were confirmed by flow cytometry (Figure 3B,C). Because of the cell line-specific differences, we also performed similar experiments in another basal-like cell line (HMLE). The mitotic arrest by FoxQ1 overexpression was also observed in the HMLE cells similar to SUM159 (Figure 3B,C). These results indicated different role for FoxQ1 in basal-like versus luminal-type breast cancer cells.
Figure 3.
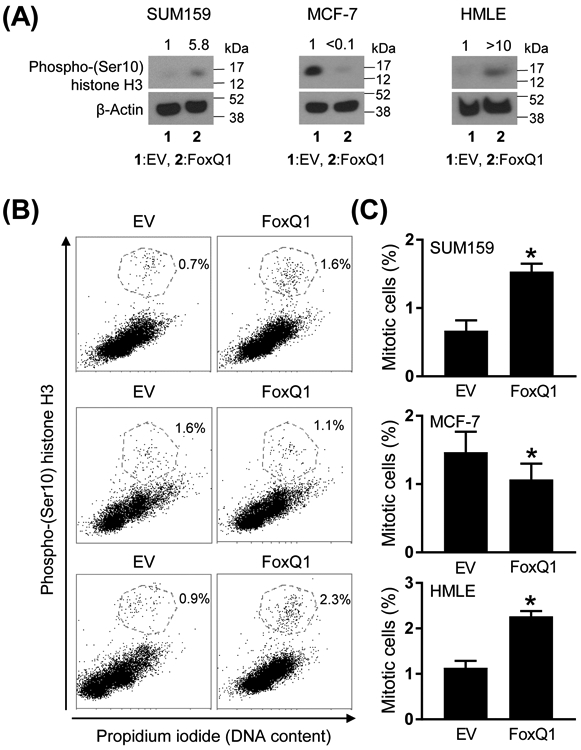
FoxQ1 overexpression resulted in mitotic arrest. A, Western blot analysis for phospho-(Ser10) histone H3 protein using lysates from EV cells or FoxQ1 overexpressing SUM159, MCF-7, and HMLE cells. The numbers on top of the bands represent changes in protein level compared to corresponding EV cells. B, Representative flow histograms showing mitotic fraction in EV cells or FoxQ1 overexpressing SUM159, MCF-7, and HMLE cells. C, Quantitation of mitotic fraction. Data shown are mean ± SD (n = 3-6). *P < 0.05 by two-sided Student's t-test. Experiments were done twice with comparable results.
3.7. IL-1α, IL-8, and VEGF are novel downstream transcriptional targets of FoxQ1
The Reactome pathways analysis also showed alterations in expression of genes associated with cellular response to stress upon FoxQ1 overexpression in the SUM159 cell line (Figure 2F). Table S2 lists genes associated with cellular response to stress whose expression was altered upon FoxQ1 overexpression. We compared the correlations observed from the RNA-seq data (present study) with TCGA dataset. Positive correlation was observed for several upregulated or downregulated genes from the RNA-seq data and the breast cancer TCGA analysis (Table S2). Because FoxQ1 has oncogenic function, we focused on IL-1α for further investigation. RNA-seq data indicated a 23-fold increase in expression of IL-1α in FoxQ1 overexpressing SUM159 cells compared with EV cells (Figure 4A). The breast cancer TCGA analysis revealed a significant positive association between expression of IL-1α and that of FoxQ1 (Figure 4B). qRT-PCR confirmed overexpression of IL-1α mRNA upon forced expression of FoxQ1 in SUM159 cells (Figure 4C). Secretion of IL-1α in the media was also significantly higher in the FoxQ1 overexpressing SUM159 cells when compared to EV cells (Figure 4D). The promoter of IL-1α contains a single FoxQ1 binding consensus sequence of TGTTTA.9 ChIP confirmed recruitment of FoxQ1 at the promoter of IL-1α in SUM159 cells (Figure 4E). The expression of IL-1α was very low in MCF-7 cells when compared to SUM159 and thus these experiments were not performed in the former cell line. These results indicated that IL-1α is a direct transcriptional target of FoxQ1 at least in the SUM159 cell line.
Figure 4.
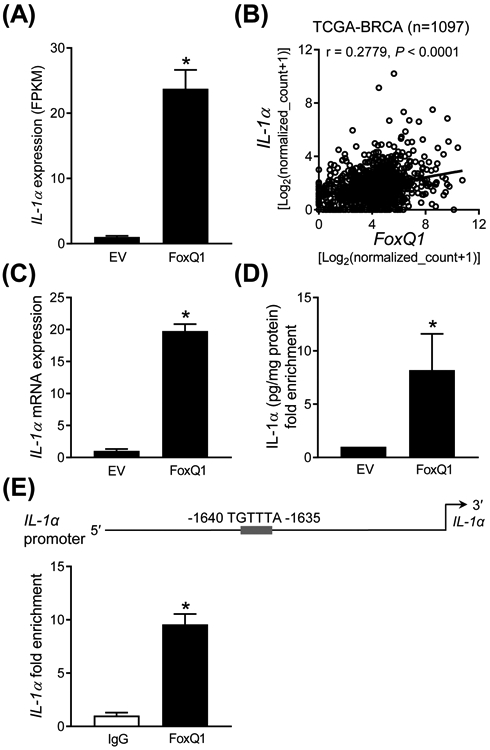
FoxQ1 is a novel regulator of IL-1α. A, RNA-seq analysis of IL-1α gene expression in EV cells FoxQ1 overexpressing SUM159 cells. Data shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t-test. B, Positive correlation between FoxQ1 and IL-1α expression in breast tumor TCGA dataset (n = 1097). Pearson's test was used to determine statistical significance of the correlation. C, Quantification of IL-1α mRNA expression in EV and FoxQ1 overexpressing SUM159 cells. Data shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t-test. D, Quantification of IL-1α secretion in the media of EV or FoxQ1 overexpressing SUM159 cells. Data shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t-test. E, Identification of a putative FoxQ1 binding site in the promoter region of IL-1α in SUM159 cells.. The bar graph shows recruitment of FoxQ1 at the IL-1α promoter by ChIP analysis. The results shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t-test. Experiments were done twice with similar results.
The RNA-seq data indicated a significantly higher level of IL-8 mRNA in FoxQ1 overexpressing SUM159 cells compared to EV cells (Figure 5A). A significant positive association between expression of IL-8 and that of FoxQ1 was also observed in the breast cancer TCGA dataset (Figure 5B). Overexpression of IL-8 in FoxQ1 overexpressing SUM159 cell was confirmed by qRT-PCR (Figure 5C). The level of IL-8 in the media was also significantly higher in the FoxQ1 overexpressing SUM159 cells when compared to EV cells (Figure 5D). Three FoxQ1 binding consensus sequences were observed at the promoter of IL-8. ChIP assay confirmed recruitment of FoxQ1 at two of those sites of IL-8 promoter (Figure 5E). Similarly, FoxQ1 overexpressing MCF-7 cells exhibited a significantly higher level of IL-8 secretion in comparison with EV cells (Figure S3A). Furthermore, FoxQ1 was recruited at two sites of the IL-8 promoter in MCF-7 cells (Figure S3B).
Figure 5.
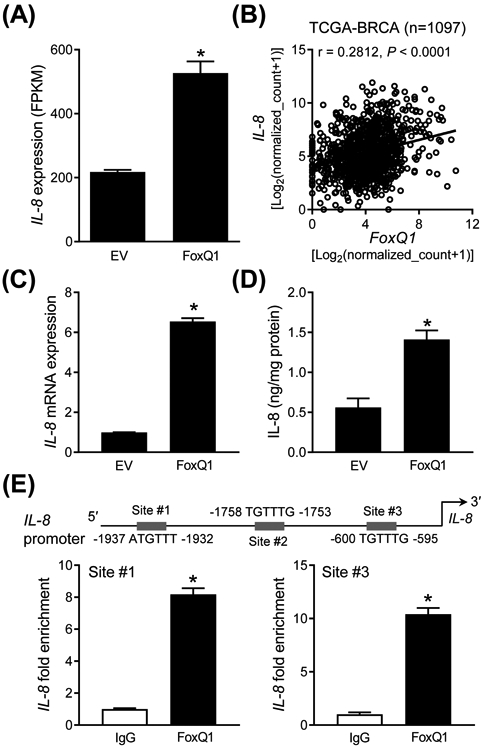
IL-8 is a novel downstream target of FoxQ1 in SUM159 cells. A, RNA-seq analysis for IL-8 gene expression in EV cells and FoxQ1 overexpressing SUM159 cells. Data shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t-test. B, Positive correlation between FoxQ1 and IL-8 expression in breast tumor TCGA dataset (n = 1097). Pearson's test was used to determine statistical significance of the correlation. C, Quantification of IL-8 mRNA expression by qRT-PCR in EV and FoxQ1 overexpressing SUM159 cells. Data shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t-test. D, Quantification of IL-8 secretion in the media of EV and FoxQ1 overexpressing SUM159 cells. Data shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t-test. E, Identification of putative FoxQ1 binding sites at the promoter region of IL-8 in SUM159 cells. The bar graph shows recruitment of FoxQ1 at the IL-8 promoter by ChIP analysis. The results shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t-test. Experiments were repeated with similar results.
The Reactome pathway analysis also revealed upregulation of genes associated with VEGF/VEGFR2 pathway in FoxQ1 overexpressing SUM159 cells. The RNA-seq data revealed upregulation of VEGFA mRNA in FoxQ1 overexpressing SUM159 cells when compared to EV cells (Figure 6A). Analysis of the breast cancer TCGA dataset revealed a positive correlation between expression of FoxQ1 and that of VEGF (Figure 6B). The VEGF mRNA level (Figure 6C) and/or secretion of the protein (Figure 6D) were increased upon FoxQ1 overexpression. ChIP assay indicated recruitment of FoxQ1 at all 3 sites of VEGFA promoter (Figure 6E). Because of weak association between expression of FoxQ1 and VEGFA in MCF-7 cells, ChIP assay was not performed in this cell line.
Figure 6.
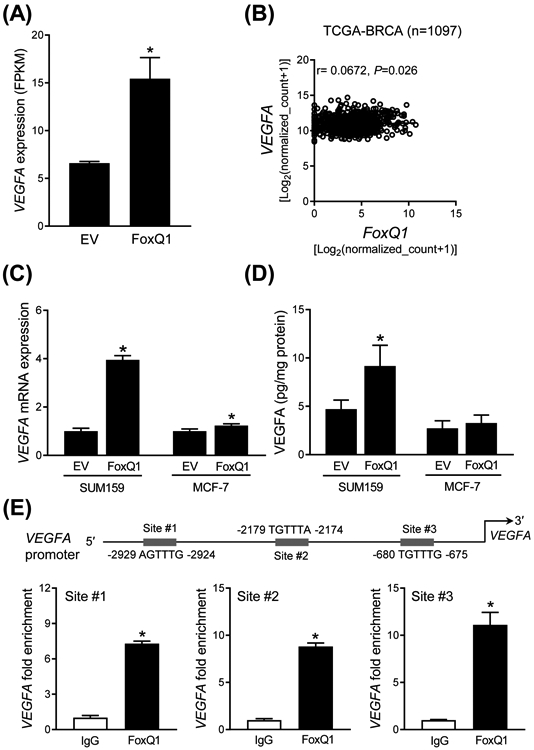
FoxQ1 regulates VEGFA expression in SUM159 cells. A, RNA-seq analysis of VEGFA gene expression difference between EV and FoxQ1 overexpressing SUM159 cells. Data shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t test. B, TCGA dataset showed the correlation between FoxQ1 and VEGFA in breast tumor (n = 1097). Pearson's correlation coefficient was used for the analysis. C, Quantification of VEGFA mRNA expression by quantitative real-time PCR in EV or FoxQ1 overexpressing SUM159 and MCF-7 cells. Data shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t test. Experiments were done twice with comparable results. D, Quantification of VEGFA secretion in EV or FoxQ1 overexpressing SUM159 and MCF-7 cells after 16 hours of serum-starvation. Data shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t test. Experiments were done twice with comparable results. E, Identification of putative FoxQ1 binding sites in the promoter region of VEGFA in SUM159 cells. The bar graph shows recruitment of FoxQ1 in the VEGFA promoter by ChIP analysis. The results shown are mean ± SD (n = 3). *P < 0.05 by two-sided Student's t test. Experiments were done twice with similar results.
4. Discussion
FoxQ1, also known as HNF-3/fkh homolog-1 or HFH-1, belongs to the forkhead family of transcription factors and its normal physiological functions include regulation of hair differentiation and control of mucin expression and granule content in stomach surface mucous cells. 15-18 The FoxQ1 gene is located at chromosome 6p25.3 that encodes a 403-amino acid protein.19,20 Structurally, the FoxQ1 is characterized by alanine- and glycine-rich regions, the forkhead box domain (Winged-helix or DNA-binding domain), and proline-rich region.20 The FoxQ1 is an evolutionary conserved protein with 100% amino acid sequence similarity in the DNA binding domain between human, mouse, and rat.20,21 Overexpression of protein or mRNA levels of FoxQ1 has been reported in different cancers, including gastric cancer, hepatocellular carcinoma, laryngeal carcinoma, pancreatic cancer, colorectal cancer, and breast cancer.8,10,22-27 We showed previously that the level of FoxQ1 protein is higher by 18.4-fold in basal-like human breast cancers compared to normal mammary tissue.10 Because luminal-type disease accounts for majority of human breast cancers, in this study we analyzed the level of FoxQ1 protein in this subtype. Like basal-like breast cancers, the expression of FoxQ1 protein was significantly higher in the luminal-type human breast cancers in comparison with normal breast tissues. These results suggest that oncogenic function of FoxQ1 may span across disease subtypes in human breast cancers.
In breast cancer, FoxQ1 is well-known for its role in promotion of EMT, cell migration and invasion, self-renewal of breast cancer stem like cells, and metastasis.7-10 Consistent with these published results,7-10 the GO pathways analysis of the RNA-seq data also indicated upregulation of genes associated with positive regulation of locomotion, positive regulator of cellular component movement, positive regulation of cell motility, and positive regulation of cell migration. At the same time, the RNA-seq data also suggests additional functions of FoxQ1 based on GO and Reactome pathway analyses, including regulation of cell morphogenesis, regulation of actin cytoskeleton organization, peptidyl-tyrosine dephosphorylation, and so forth. Further work is necessary to systematically test the role of FoxQ1 in these processes.
Studies have revealed direct regulation of multiple cancer-relevant genes by FoxQ1.7-10 FoxQ1 functions to repress CDH1 expression in breast cancer cell lines.8 For example, the promoter activity of CDH1 was repressed by overexpression of FoxQ1 in HMLER cells but restored by knockdown of FoxQ1 in MDA-MB-231 cells.8 Another study confirmed these findings in 293FT and MCF-7 cells.7 The three regulatory E-boxes of the CDH1 promoter were shown to cooperate in its repression mediated by FoxQ1 overexpression.7 Additional direct targets of FoxQ1 in breast cancer include transcription factors (TWIST1 and ZEB2) and PDGFRα/β growth factor receptors to confer resistance to chemotherapy drugs like doxorubicin, paclitaxel, and imatinib in vitro and in vivo.9 The DACH1 homolog gene was originally identified as a critical regulator of eye and limb development in Drosophila.28 Studies have pointed to a tumor suppressor function of DACH1 in breast cancer.29-33 For example, overexpression of DACH1 inhibited breast cancer stem-like fraction in vitro and in vivo.30 Colony formation in vitro and xenograft growth in vivo was also significantly suppressed by DACH1 overexpression in MDA-MB-231 cells.29 DACH1 was also shown to inhibit EMT in breast cancer cells in vitro and repress metastatic potential of 4T1/Luc murine breast carcinoma cells in vivo in syngeneic mice.31 Our own group was the first to implicate FoxQ1 in direct transcriptional repression of DACH1.10 In one study, DACH1 was shown to repress IL-8 level.33 Collectively, these studies indicate that FoxQ1 is a druggable target in breast and other cancers but a selective inhibitor of this transcription factor is yet to be identified.
We found that downregulation of genes associated with cell cycle regulation was the most prominent pathway enrichment by FoxQ1 overexpression from KEGG, GO, and Reactome pathway analyses (Figure 2). Consequently, FoxQ1 overexpression in SUM159 cells, but not in MCF-7, results in mitotic arrest. The mechanistic basis for differential effect of FoxQ1 overexpression on mitotic fraction in basal-like versus luminal-type breast cancer cell lines is yet to be elucidate but it could be attributable to p53 expression. From the cell cycle distribution changes at least in SUM159 cells, one would expect a pronounced effect of FoxQ1 on cell proliferation and cell survival but the published in vitro data do not support this possibility at least in breast cancer7,8 To the contrary, overexpression of FoxQ1 in non-small cell lung cancer cell lines (A549 and HCC827) results in increased proliferation when compared to corresponding EV cells.34 Cell proliferation is decreased upon FoxQ1 knockdown in a laryngeal carcinoma cell line.24 Because the full spectrum of the FoxQ1-regulated transcriptome is still not fully appreciated, further work may shed light as to why FoxQ1 overexpression is unable to increase breast cancer cell proliferation.
The present study, for the first time demonstrates that IL-1α, IL-8, and VEGFA are direct transcriptional targets of FoxQ1. FoxQ1 is recruited at the promoters of IL-1α, IL-8, and VEGFA genes. Moreover, protein levels of IL-1α, IL-8, and/or VEGFA are increased by FoxQ1 overexpression in SUM159 and/or MCF-7 cells. In breast cancer patients, IL-1α protein secretion is correlated with malignant phenotype.35 IL-1α-derived from cancer cells was shown to promote autocrine and paracrine induction of pro-metastatic genes in breast cancer cells.36 Interestingly, IL-1α protein expression in breast cancer correlated with that of IL-837, and both these cytokines are direct targets of FoxQ1 (present study). Similar to the IL-1α, studies have established a pro-tumorigenic function of IL-8 in breast cancer.38-43 For example, the IL-8 expression in breast cancer cells is proportional to their invasion potential.40,41 The invasiveness of breast cancer cells is increased by overexpression of IL-8 as well as treatment with recombinant IL-8.40,41 RNA interference of IL-8 in MDA-MB-231 cells diminishes its ability to invade.42 Studies have also indicated that IL-8 can promote stemness and EMT in breast cancer cells.44 These results indicate that promotion of EMT and stem-like phenotypes in breast and, possibly other cancers by FoxQ1 overexpression is partly mediated by direct regulation of the IL-1α and/or IL-8 expression.
More than 40,000 American women succumb to metastatic breast cancer every year.1 Increased tumor angiogenesis is considered critical for tumor growth.45,46 The VEGFA, routinely known as VEGF, is one of the major mediators of tumor neo-angiogenesis.47 The VEGF binds to VEGF receptor to promote vascular endothelial growth, migration, survival, and lymphangiogenesis.47,48 The present study shows that VEGFA is a direct transcriptional target of FoxQ1. However, this regulation seems more pronounced in the SUM159 than in MCF-7 cells. Thus, it is reasonable to conclude that VEGFA regulation by FoxQ1 is partly responsible for its pro-migratory and metastatic effects.
In conclusion, the present study reveals novel targets and functions of FoxQ1 in breast cancer. The RNA-seq data also reveals additional direct regulatory targets of FoxQ1 at least in SUM159 cells that require further verification.
Supplementary Material
Acknowledgments
Funding: This study was supported by the USPHS grant RO1 CA219180 and RO1 CA142604 awarded by the National Cancer Institute. This research used the Flow Cytometry Facility and the Tissue and Research Pathology Facility supported in part by Cancer Center Support Grant from the National Cancer Institute (P30 CA047904).
Abbreviations:
- ADAM9
ADAM metallopeptidase domain 9
- CADM3
cell adhesion molecule 3
- CDH1
E-cadherin
- CDH2
N-cadherin
- ChIP
chromatin immunoprecipitation
- COL1A1
collagen type I alpha 1
- COL6A1
collagen Type VI alpha 1
- CST6
cystatin 6
- CTNNB1
catenin beta 1
- DACH1
dachshund homolog 1
- DCN
decorin
- DSG2
desmoglein 2
- EDN1
endothelin 1
- EMT
epithelial-mesenchymal transition
- ER
estrogen receptor
- EV
empty vector transfected
- FN1
fibronectin 1
- FOXA1
forkhead box A1
- FoxQ1
forkhead box Q1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GO
gene ontology
- HER2
human epidermal growth factor receptor 2
- HMLE
human mammary epithelial cell line
- HMLER
human mammary epithelial cell line immortalized by hTERT and SV40 large T antigen
- IL
interleukin
- JAM3
junctional adhesion molecule 3
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- PBS
phosphate-buffered saline
- PDGFR
platelet-derived growth factor receptor
- PI
propidium iodide
- PLD1
phospholipase D1
- PR
progesterone receptor
- qRT-PCR
quantitative real-time polymerase chain reaction
- S100A4
S100 calcium binding protein A4
- SEMA3A
semaphorin 3A
- SNAI1
snail family transcriptional repressor 1
- TCGA
The Cancer Genome Atlas
- THBS1
thrombospondin 1
- TWIST
twist basic helix-loop-helix transcription factor
- VEGF
vascular endothelial growth factor
- ZEB
zinc finger E-box binding homeobox
Footnotes
Data Availability: The data and the material generated for this study are available upon request to the corresponding author.
Conflict of interest: None of the authors has any conflict of interest.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–752. [DOI] [PubMed] [Google Scholar]
- 3.Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001;98:10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer 2007;7:659–672. [DOI] [PubMed] [Google Scholar]
- 5.Sørlie T Molecular classification of breast tumors: toward improved diagnostics and treatments. Methods Mol Biol 2007;360:91–114. [DOI] [PubMed] [Google Scholar]
- 6.Saha P, Nanda R. Concepts and targets in triple-negative breast cancer: recent results and clinical implications. Ther Adv Med Oncol 2016;8:351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang H, Meng F, Liu G, Zhang B, Zhu J, Wu F et al. Forkhead transcription factor Foxq1 promotes epithelial-mesenchymal transition and breast cancer metastasis. Cancer Res 2011;71:1292–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qiao Y, Jiang X, Lee ST, Karuturi RK, Hooi SC, Yu Q. FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res 2011;71:3076–3086. [DOI] [PubMed] [Google Scholar]
- 9.Meng F, Speyer CL, Zhang B, Zhao Y, Chen W, Gorski DH, et al. PDGFRα and β play critical roles in mediating Foxq1-driven breast cancer stemness and chemoresistance. Cancer Res 2015;75:584–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SH, Kaschula CH, Priedigkeit N, Lee AV, Singh SV. Forkhead Box Q1 is a novel target of breast cancer stem cell inhibition by diallyl trisulfide. J Biol Chem 2016;291:13495–13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SH, Hahm ER, Singh KB, Shiva S, Stewart-Ornstein J, Singh SV. RNA-seq reveals novel mechanistic targets of withaferin A in prostate cancer cells. Carcinogenesis 2020;in press. doi: 10.1093/carcin/bgaa009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao D, Srivastava SK, Lew KL, Zeng Y, Hershberger P, Johnson CS, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis 2003;24:891–897. [DOI] [PubMed] [Google Scholar]
- 13.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 14.Hardy K, Wu F, Tu W, Zafar A, Boulding T, McCuaig R, et al. Identification of chromatin accessibility domains in human breast cancer stem cells. Nucleus 2016;7:50–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol 2002;250:1–23. [DOI] [PubMed] [Google Scholar]
- 16.Hoggatt AM, Kriegel AM, Smith AF, Herring BP. Hepatocyte nuclear factor-3 homologue 1 (HFH-1) represses transcription of smooth muscle-specific genes. J Biol Chem 2000;275:31162–311670. [DOI] [PubMed] [Google Scholar]
- 17.Martinez-Ceballos E, Chambon P, Gudas LJ. Differences in gene expression between wild type and Hoxa1 knockout embryonic stem cells after retinoic acid treatment or leukemia inhibitory factor (LIF) removal. J Biol Chem 2005;280:16484–16498. [DOI] [PubMed] [Google Scholar]
- 18.Potter CS, Peterson RL, Barth JL, Pruett ND, Jacobs DF, Kern MJ, et al. Evidence that the satin hair mutant gene Foxq1 is among multiple and functionally diverse regulatory targets for Hoxc13 during hair follicle differentiation. J Biol Chem 2006:281:29245–29255. [DOI] [PubMed] [Google Scholar]
- 19.Katoh M, Katoh M. Human FOX gene family (Review). Int J Oncol 2004;25:1495–1500. [PubMed] [Google Scholar]
- 20.Li Y, Zhang Y, Yao Z, Li S, Yin Z, Xu M. Forkhead box Q1: A key player in the pathogenesis of tumors (Review). Int J Oncol 2016;49:51–58. [DOI] [PubMed] [Google Scholar]
- 21.Abba M, Patil N, Rasheed K, Nelson LD, Mudduluru G, Leupold JH, et al. Unraveling the role of FOXQ1 in colorectal cancer metastasis. Mol Cancer Res 2013;11:1017–1028. [DOI] [PubMed] [Google Scholar]
- 22.Bieller A, Pasche B, Frank S, Gläser B, Kunz J, Witt K, et al. Isolation and characterization of the human forkhead gene FOXQ1. DNA Cell Biol 2001;20:555–561. [DOI] [PubMed] [Google Scholar]
- 23.Liang SH, Yan XZ, Wang BL, Jin HF, Yao LP, Li YN, et al. Increased expression of FOXQ1 is a prognostic marker for patients with gastric cancer. Tumour Biol 2013;34:2605–2609. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Li W, Dai S, Tai X, Jia J, Guo X. FOXQ1 is overexpressed in laryngeal carcinoma and affects cell growth, cell cycle progression and cell invasion. Oncol Lett 2015;10:2499–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W, He S, Ji J, Huang J, Zhang S, Zhang Y. The prognostic significance of FOXQ1 oncogene overexpression in human hepatocellular carcinoma. Pathol Res Pract 2013;209:353–358. [DOI] [PubMed] [Google Scholar]
- 26.Cao D, Hustinx SR, Sui G, Bala P, Sato N, Martin S, et al. Identification of novel highly expressed genes in pancreatic ductal adenocarcinomas through a bioinformatics analysis of expressed sequence tags. Cancer Biol Ther 2004;3:1081–1089. [DOI] [PubMed] [Google Scholar]
- 27.Kaneda H, Arao T, Tanaka K, Tamura D, Aomatsu K, Kudo K, et al. FOXQ1 is overexpressed in colorectal cancer and enhances tumorigenicity and tumor growth. Cancer Res 2010;70:2053–2063. [DOI] [PubMed] [Google Scholar]
- 28.Mardon G, Solomon NM, Rubin GM Dachshund encodes a nuclear protein required for normal eye and leg development in Drosophila. Development 1994;120:3473–3486. [DOI] [PubMed] [Google Scholar]
- 29.Wu K, Li A, Rao M, Liu M, Dailey V, Yang Y, et al. DACH1 is a cell fate determination factor that inhibits cyclin D1 and breast tumor growth. Mol Cell Biol 2006;26:7116–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu K, Jiao X, Li Z, Katiyar S, Casimiro MC, Yang W, et al. Cell fate determination factor Dachshund reprograms breast cancer stem cell function. J Biol Chem 2011;286:2132–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao F, Wang M, Li S, Bai X, Bi H, Liu Y, et al. DACH1 inhibits SNAI1-mediated epithelial-mesenchymal transition and represses breast carcinoma metastasis. Oncogenesis 2015;4:e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu K, Chen K, Wang C, Jiao X, Wang L, Zhou J, et al. Cell fate factor DACH1 represses YB-1-mediated oncogenic transcription and translation. Cancer Res 2014;74:829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu K, Katiyar S, Li A, Liu M, Ju X, Popov VM, et al. Dachshund inhibits oncogene-induced breast cancer cellular migration and invasion through suppression of interleukin-8. Proc Natl Acad Sci USA 2008;105:6924–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng J, Xu L, Ni S, Gu J, Zhu H, Wang H, et al. Involvement of FoxQ1 in NSCLC through regulating EMT and increasing chemosensitivity. Oncotarget 2014;5:9689–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singer CF, Kronsteiner N, Hudelist G, Marton E, Walter I, Kubista M, et al. Interleukin 1 system and sex steroid receptor expression in human breast cancer: interleukin 1α protein secretion is correlated with malignant phenotype. Clin Cancer Res 2003;9:4877–4883. [PubMed] [Google Scholar]
- 36.Nozaki S, Sledge GW Jr, Nakshatri H. Cancer cell-derived interleukin 1alpha contributes to autocrine and paracrine induction of pro-metastatic genes in breast cancer. Biochem Biophys Res Commun 2000;75:60–62. [DOI] [PubMed] [Google Scholar]
- 37.Kurtzman SH, Anderson KH, Wang Y, Miller LJ, Renna M, Stankus M, et al. Cytokines in human breast cancer: IL-1α and IL-1β expression. Oncol Rep 1999;6:65–70. [DOI] [PubMed] [Google Scholar]
- 38.Singh JK, Simões BM, Howell SJ, Farnie G, Clarke RB. Recent advances reveal IL-8 signaling as a potential key to targeting breast cancer stem cells. Breast Cancer Res 2013; 15:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Todorović-Raković N, Milovanović J. Interleukin-8 in breast cancer progression. J Interferon Cytokine Res 2013;33:563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freund A, Chauveau C, Brouillet JP, Lucas A, Lacroix M, Licznar A, et al. IL-8 expression and its possible relationship with estrogen-receptor-negative status of breast cancer cells. Oncogene 2003;22:256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin Y, Huang R, Chen L, Li S, Shi Q, Jordan C, et al. Identification of interleukin-8 as estrogen receptor-regulated factor involved in breast cancer invasion and angiogenesis by protein arrays. Int J Cancer 2004;109:507–515. [DOI] [PubMed] [Google Scholar]
- 42.Yao C, Lin Y, Chua MS, Ye CS, Bi J, Li W, et al. Interleukin-8 modulates growth and invasiveness of estrogen receptor-negative breast cancer cells. Int J Cancer 2007; 121:1949–1957. [DOI] [PubMed] [Google Scholar]
- 43.Clark-Lewis I, Schumacher C, Baggiolini M, Moser B. Structure-activity relationships of interleukin-8 determined using chemically synthesized analogs. Critical role of NH2-terminal residues and evidence for uncoupling of neutrophil chemotaxis, exocytosis, and receptor binding activities. J Biol Chem 1991;266:23128–23134. [PubMed] [Google Scholar]
- 44.Fernando RI, Castillo MD, Litzinger M, Hamilton DH, Palena C. IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res 2011;71:5296–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bielenberg DR, Zetter BR. The contribution of angiogenesis to the process of metastasis. Cancer J 2015;21:267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Folkman J Role of angiogenesis in tumor growth and metastasis. Semin Oncol 2002;29(suppl. 16):15–18. [DOI] [PubMed] [Google Scholar]
- 47.Ferrara N VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer 2002;2:795–803. [DOI] [PubMed] [Google Scholar]
- 48.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 2005;23:1011–1027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.