

Abstract
Microglia play important roles in the pathogenesis of Alzheimer’s disease (AD), in part, by affecting the clearance of amyloid-β (Aβ) peptides. Most studies, however, used synthetic soluble Aβ (sAβ) at higher concentrations. The exact mechanisms underlying microglia-mediated clearance of physiological sAβ at very low concentrations remain unclear. Here we reported that there were much more Iba-1- and CD68-positive microglia and significantly less sAβ left in the brain of adult mice 5 days after the surgery of sAβ microinjection compared to 2 h after the surgery (p < 0.05). However, very few Iba-1- and CD68-positive microglia co-localized with microinjected fluorescently labeled sAβ (FLsAβ42) 5 days after the surgery. Also, there was no co-localization of FLsAβ42 with a lysosomal marker (LAMP-1) 5 days after the surgery. There was no significant difference in the percentage of Aβ+/PE-CD11b+/APC-CD45low microglia between the control group and the group microinjected with TBS-soluble Aβ extracted from the brains of AD patients (p > 0.05). The degradation of physiological sAβ was prevented by a highly selective insulin-degrading enzyme inhibitor (Ii1) but not by a phagocytosis inhibitor (polyinosinic acid) or pinocytosis inhibitor (cytochalasin B) in vitro. Furthermore, the reduction of synthetic and physiological sAβ in the brain was partially prevented by the co-injection of Ii1 in vivo (p < 0.05). Our results demonstrate that microglia do not take up synthetic or physiological sAβ, but partially degrade it via the secretion of insulin-degrading enzyme, which will be beneficial for understanding how sAβ is removed from the brain by microglia.
Keywords: Alzheimer’s disease, microglia, soluble Aβ, insulin-degrading enzyme
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder of the brain and the most common form of dementia among the elderly. Currently, there are an estimated 5.8 million Americans age 65 or older suffering from AD (2020 Alzheimer’s Disease Facts and Figures). The characteristic pathological features of AD are extracellular amyloid plaques and intracellular neurofibrillary tangles, both of which are associated with activated microglia and reactive astrocytes (Selkoe, 1991). The pathogenesis of AD is considered to be due, in part, to alterations in Aβ homeostasis i.e. overproduction or inefficient clearance of Aβ, resulting in an accumulation of Aβ peptides in the brain (Lane et al., 2018; Tanzi and Bertram, 2005; Wang et al., 2006). Many studies have focused on investigating how Aβ is generated based on the genetic findings in familial AD, which accounts for less than 1% AD patients (Bateman et al., 2011; Bekris et al., 2010). However, understanding how Aβ is taken up and cleared might be more beneficial for “sporadic” AD cases in which Aβ generation is apparently normal but it accumulates excessively in the brain (Henstridge et al., 2019; Qiu et al., 1998).
Reactive microglia co-localize with fibrillar Aβ (fAβ)-containing neuritic plaques in AD and may be involved in either the removal or the production of amyloid plaques (Giulian et al., 1996; Henstridge et al., 2019; Keren-Shaul et al., 2017; Wang et al., 2015). Microglia are resident immune cells of the brain and express many pattern recognition receptors to detect exogenous pathogen-associated molecular patterns (PAMPs) or endogenous danger-associated molecular patterns (DAMPs) (Lucin and Wyss-Coray, 2009) in order to patrol the brain for injury. Among them, scavenger receptors, Toll-like receptors, purinergic G protein-coupled receptors (P2Y2 and P2Y6), complement components and their associated receptors as well as the recent discovery of triggering receptor expressed on myeloid cells 2 (TREM2) have been implied in microglia-mediated phagocytosis of fAβ (Brazil et al., 2000; Choucair-Jaafar et al., 2011; El Khoury et al., 1996; Fu et al., 2012; Keren-Shaul et al., 2017; Kim et al., 2012; Liu et al., 2005; Maier et al., 2008; Paresce et al., 1996; Richard et al., 2008; Wang et al., 2015; Webster et al., 2000; Wyss-Coray et al., 2002). Soluble form of Aβ (sAβ), especially oligomeric forms, have been shown to be more toxic than fAβ and to play a crucial role in the learning and memory impairments during early stages of AD (Dahlgren et al., 2002; Fu et al., 2006; Loo et al., 1993; Lue et al., 1999; McLean et al., 1999; Shankar et al., 2008). In contrast to the receptor-mediated phagocytosis of fAβ, there is some evidence that both receptor-mediated endocytosis and receptor-independent fluid-phase pinocytosis might be involved in the uptake of synthetic sAβ by microglia (Chung et al., 1999; Frenkel et al., 2013; Kim et al., 2012; Li et al., 2013; Mandrekar et al., 2009; Yang et al., 2011; Yeh et al., 2016; Zhao et al., 2018). Internalized Aβ might be transported to the late endosomes/lysosomes for degradation. In addition to their ability to endocytose Aβ, microglia might also clear Aβ by degradation via secreting Aβ-degrading enzymes such as insulin-degrading enzyme (IDE), neprilysin (NEP), matrix metalloproteinase 2 (MMP2) and MMP9, endothelin-converting enzyme (ECE-1 and ECE-2) and tissue plasminogen activator (Chu et al., 1998; Chung et al., 1999; Czirr et al., 2017; Eckman et al., 2001; Farris et al., 2007; Hernandez-Guillamon et al., 2010; Leissring et al., 2003; Mandrekar et al., 2009; Melchor et al., 2003; Pacheco-Quinto and Eckman, 2013; Qiu et al., 1998; Qiu et al., 1997; Sikanyika et al., 2019; Yan et al., 2006). However, the exact mechanisms by which microglia take up and clear sAβ, remain unclear and controversial, especially in vivo.
To date, most published studies used a very high concentration (~ 1 μM) of synthetic Aβ, whereas concentrations of Aβ in physiological fluids are only 0.1 – 10 nM (Podlisny et al., 1995; Seubert et al., 1992; Suzuki et al., 1994). Therefore, the exact mechanisms underlying microglia-mediated uptake and clearance of sAβ need to be re-examined or confirmed with natural and biological sources of Aβ at very low, physiologically relevant concentrations. In the present study, we sought to investigate the molecular mechanisms underlying microglia-mediated uptake and clearance of both synthetic and physiological sAβ. Here we demonstrate that very few microglia take up microinjected synthetic and physiological sAβ, but most surrounded and degraded it in vivo. The degradation of sAβ was reversed by Ii1, a novel and highly-selective inhibitor of IDE (Leissring et al., 2010). Elucidating the exact mechanisms underlying microglia-mediated uptake and clearance of different sources of sAβ will be useful for understanding the role of microglia in the pathogenesis of AD and developing novel therapeutics against AD.
EXPERIMENTAL PROCEDURES
Mice
C57BL/6J mice were originally obtained from The Jackson Laboratory (Stock No: 000664) and maintained in a virus antibody-free animal facility at the Harvard Medical School. All animals were housed under standard conditions, including 12 hour light/dark cycle, 20-22°C, 50-60% humidity, access to food and water ad libitum, no physical enrichment, and 4-5 mice/cage with regular cleaning twice a week. Adult female mice (8-9 months old, 25-30 g, n = 5 per group) were used for this study. The reasons of focusing on female mice and its limitation were detailed in the second last paragraph of Discussion. The number of mice used for each experiment was indicated in the Figure legend. The mice were randomized to the experimental conditions. Sample sizes were chosen primarily on the basis of experience with similar types of experiments. All animal protocols were approved by the Harvard Medical Area Standing Committee on Animals, and studies were performed in accordance with all state and federal regulations. The Harvard Medical School animal management program is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International and meets all National Institutes of Health standards as demonstrated by an approved Assurance of Compliance (A3431-01) filed at the Office of Laboratory Animal Welfare.
Microglia cell culture
N9 microglial cells were cultured as described (Hickman et al., 2008). Briefly, cells were grown in Rosewell Park Memorial Institute (RPMI-1640) medium containing 10% fetal bovine serum (FBS). The medium was changed every three days and replaced with serum-free medium the day before experiments. All cell culture-related materials were purchased from Invitrogen.
Soluble Aβ preparation
HiLyte Fluor 488-labeled human Aβ1-42 (Anaspec, CA) peptide was dissolved in 0.1% NH4OH at 2.5 μg/μl and diluted in 1x PBS at 0.5 μg/μl immediately before each experiment. This preparation is herein referred to as FLsAβ42, which was characterized by Western blot assay (Supplemental Figure 1). In order to keep the soluble and oligomeric status of FLsAβ42, we immediately injected the prepared peptide into the mouse brains after 1 h of incubation at 37°C. Secreted soluble Aβ was obtained from the conditioned medium (CM) of a CHO cell line (7PA2) stably transfected with human APP751 containing the V717F AD mutation (Li et al., 2009; Podlisny et al., 1995). The secreted soluble Aβ is referred to as 7PA2 CM. Human AD soluble brain extract (AD-TBS) was prepared as described previously (Shankar et al., 2008). Brain specimens from deceased human AD subjects (n = 2 males and 2 females, age = 78-82 years old, PMI < 12 h) at autopsy were collected after obtaining informed consent from the next of kin under protocols approved by the Partners Human Research Committee at Brigham and Women’s Hospital. Frozen human frontal cortices containing white and gray matter were weighted, and freshly prepared, ice-cold TBS consisting of 20 mM Tris-HCl, 150 mM NaCl, pH 7.4 was added to the frozen cortex at 4:1 (TBS volume / brain wet weight). Brains were homogenized and centrifuged at 175,000 g in a TLA100.2 rotor on a Beckman TL 100 centrifuge. The supernatants (called AD-TBS extract) from 4 AD subjects were pooled, aliquoted, and stored at −80°C. Both 7PA2 CM and AD-TBS were provided by our colleague, Dr. Dennis Selkoe. The concentration of soluble Aβ in 7PA2 CM and AD-TBS is very low (~ 1 nM).
Stereotaxic brain microinjection
Eight-to-nine mo-old adult female mice were anesthetized by inhalation of isoflurane. 2 μl FLsAβ42 or the same fluorescent dye-labeled scrambled peptide at 0.5 mg/ml or 2 μl 10x AD-TBS extraction in the presence or absence of 100 μM Ii1 was microinjected into the frontal cortex bilaterally using a stereotaxic apparatus (Stoelting, IL) via a 5 μl Hamilton syringe (Hamilton Company, NV). The coordinates, with respect to bregma, were +2.8 mm anterior, 1.2 mm lateral, and −1.3 mm ventral to the skull. The rate of injection was 0.5 μl/min. After injection, the cannula was left in place for an additional 5 min to allow for diffusion. Animals were kept on a warming pad until they had fully recovered from anesthesia and were kept in individual cages until they were sacrificed for tissue processing to prevent damage to the scalp sutures. All appropriate measures were taken to minimize pain and discomfort in experimental animals according to the animal surgery procedures approved by the Harvard Medical Area Standing Committee on Animals. Buprenorphine (subcutaneous, 0.05 - 0.1 mg/kg) was administered as an analgesic to reduce the pain before the surgery and every 6-12 h for a total of 24 h after the surgery and then as needed. Any mice died after the surgery were excluded for this study, but no surviving animals were excluded for analysis.
Immunohistochemistry
Two hours or five days after the surgery, the mice were perfused with PBS and the brains were removed and fixed in 4% PFA for 24 h before being placed into 30% sucrose for cryoprotection. Brains were sectioned coronally at 20 μm on a cryostat and mounted on glass slides. Frozen mouse sections (n=8 sections/animal, 80 μm interval, covering most of sections with obvious FLsAβ42 signal) were blocked with 8% goat serum and 3% BSA in PBS containing 0.3% Triton X-100, and then incubated with anti-Iba-1 (1:200; Wako), anti-CD68 (1:200; Bio-rad), and anti-LAMP-1 (1D4B, 1:100; DSHB) antibodies overnight at 4 °C. After washing with PBS, sections were incubated with suitable Alexa Fluor second antibodies for 1 h at room temperature and nuclei were visualized by TO Pro-3 staining. Images of the injected frontal cortex with FLsAβ42 signal were obtained as a z-series stack (1 μm) using a Zeiss LSM 510 confocal microscope. The imaging and quantitation were performed by the investigator who were blinded to the experimental groups. The mean intensities of microinjected FLsAβ42 on each section at 2 h and 5 days after surgery were analyzed.
Isolation of adult microglia
Five days after the surgery, adult microglia were isolated as described previously (Cardona et al., 2006). Briefly, the mice were perfused with ice-cold 1x HBSS and brain regions including the cortex and hippocampus were homogenized in the same above buffer. After centrifuging, the cell suspension was gently rocked at room temperature for 20 min. Then, a discontinuous gradient density centrifugation step using Percoll (GE healthcare) was performed. The cells were resuspended in 70% stock isotonic Percoll (SIP) in HBSS and carefully overlaid with 37% SIP and centrifuged at 200 g for 40 min at room temperature with slow acceleration and no brake. The 70%-37% interphase containing adult microglia was transferred to a new tube and washed twice with ice-cold 1x HBSS. Finally, the isolated adult microglia were resuspended in FACS buffer (HBSS containing 0.2% BSA and 0.09% sodium azide) for flow cytometry immediately.
Flow cytometry
Isolated adult microglia were incubated with rat anti-mouse CD16/CD32 (Mouse FcR blocker, 1:100, Bio-rad) at 4 °C for 15 min before incubating with PE-Cy7-CD11b (1:100, BD Biosciences) or anti-Aβ antibody (6E10, 1:400, Signet) and APC-CD45 (1:100, BD Biosciences) at 4 °C for 20 min in the dark. The appropriate goat anti-mouse FITC-IgG (1:500, Biolegend) was added later for samples with the injection of AD-TBS. Cells were washed three times with cold FACS buffer and subjected to a BD LSR II flow cytometer (BD Biosciences, CA). Ramified parenchymal microglia have been demonstrated to possess the phenotype CD11b+/CD45low, whilst other CNS macrophages and peripheral macrophages exhibit the phenotype CD11b+/CD45hlgh (Becher and Antel, 1996; Ford et al., 1995). The percentage of CD11b+/CD45low gated microglia was recorded and compared between different groups.
Immunoprecipitation and Western blot assay
N9 microglial cells were incubated with sAβ from 7PA2 CM or AD-TBS in the presence or absence of different inhibitors. Conditioned media were collected. Cells were washed with cold PBS and lysed using RIPA buffer (Thermo Scientific, IL) containing protease inhibitor cocktail (Roche, IN). The collected media and cell lysates were pre-cleared with protein A-Sepharose (Sigma, MO), and immunoprecipitated overnight with anti-Aβ antiserum (R1282, 1:100) as described previously (Podlisny et al., 1995). Aβ species in the immunoprecipitate were detected by Western blot assay as described previously (Maier et al., 2008). Briefly, media and cell lysates from cells exposed to different treatments were separated on 12% Bis-Tris gels (Invitrogen). Proteins on the gels were transferred to membranes and detected with anti-Aβ antibody (6E10, 1:1000). Detailed information of all primary antibodies used in this study is described in Table 1.
Table 1.
Primary antibodies used in this study
| Antibody name |
Structure of immunogen | Manufacturer, Catalog number |
Research Resource Identifiers |
Species raised in |
Polyclonal/monoclonal; concentration used |
|---|---|---|---|---|---|
| IBA-1 | Synthetic peptide (C-terminal of Iba1) | WAKO, 019-19741 | RRID:AB_839504 | Rabbit | Polyclonal, 1:200 |
| CD68 | Purified Concanavalin A acceptor glycoprotein from P815 cell line | Bio-Rad, MCA1957GA | RRID:AB_324217 | Rat | Monoclonal, 1:200 |
| LAMP-1 | NIH/3T3 mouse embryo fibroblast tissue culture cell membranes | DSHB1D4B | RRID:AB_2134500 | Rat | Monoclonal, 1:100 |
| CD16/CD32 | PU5 1.8 IOE7 Balb/c mouse cell line | Bio-Rad, MCA2305GA | RRID:AB_2262717 | Rat | Monoclonal, 1:100 |
| PE-Cy7- | CD1lb | Human monocytes | BD Biosciences, 557,743 | RRID: | AB_396849 |
| Mouse | Monocional, 1:100 | ||||
| 6E10 | The epitope lies within amino acids 3–8 of beta amyloid (EFRHDS) | Covance, SIG-39300-200 | RRID:AB_662803 | Mouse | Monoclonal, 1:400 |
| APC-CD45 | Mouse Thymus/Spleen | BD Biosciences, 559864 | RRID:AB_398672 | Rat | Monoclonal, 1:100 |
| R1282 | Synthetic peptide NH2-CYS-NH-CH2- (CH2)5-CO- GLMVGGVVIA-COOH | Dennis Selkoe | N/A | Rabbit | Polyclonal, 1:100 |
ELISA
Two hours or five days after the surgery, the mice were perfused with ice-cold 1x PBS and removed brains including the cortex and hippocampus were homogenized in 1 x RIPA buffer containing protease inhibitor cocktail. The protein concentration of the homogenates was measured by BCA assay and the Aβ42 in the homogenates was measured according to the instructions in the human Aβ42 ultrasensitive ELISA kit (Invitrogen, Catalog # KHB3544).
Statistical analysis
Data were expressed as mean ± SEM. Significance was assessed with Student’s t-test if the data can pass the D'Agostino & Pearson omnibus normality test or nonparametric Mann Whitney test if the sample size is too small for normality test using GraphPad Prism software. We did not perform multiple comparisons simultaneously due to the potential problem of false discovery rate. A value of p < 0.05 was considered significant. Detailed statistical results are shown in Table 2.
Table 2.
Statistical results for Student’s t-test and nonparametric Mann Whitney test
| Figure | Test | t | P value | DF | Mann-Whitney U |
|---|---|---|---|---|---|
| 1B | t-test | 12.57 | <0.0001 | 78 | |
| 3C | Mann Whitney test | 0.0117 | 0.0000 | ||
| 4C | Mann Whitney test | 0.0952 | 4.000 | ||
| 5B | Mann Whitney test | 0.0294, 0.4857, 0.8857, 0.0286, 0.3094, 0.3429, 0.0571 | 0.0000, 5.000, 7.000, 0.0000, 4.000, 4.000, 1.000 | ||
| 5D | Mann Whitney test | 0.0294, 0.0286, 0.3429, 0.3429, 0.6857, 0.8857, 0.6857, 0.4857 | 0.0000, 0.0000, 4.000, 4.000, 6.000, 7.000, 6.000, 5.000 | ||
| 6A | Mann Whitney test | 0.0079 | 0.0000 | ||
| 6B | Mann Whitney test | 0.0079 | 0.0000 |
RESULTS
Microinjected synthetic sAβ induced microglial activation and was degraded in vivo
To investigate whether and how microglia clear sAβ in vivo, we stereotaxically microinjected 1 μg FLsAβ42 into the frontal cortex of the C57BL/6J mouse brain. Immunofluorescent labeling in brain sections showed abundant Iba-1 (resident microglia/macrophage marker) and CD68 staining (activated microglia/macrophage marker) closely surrounding the microinjected FLsAβ42 5 d after surgery compared to 2 h after surgery (Figure 1A), suggesting that microglia are significantly activated 5 days after surgery. We further quantified the FLsAβ42 mean intensity in the microinjected mice 2 h and 5 d after surgery and found that the FLsAβ42 mean intensity significantly decreased 5 d after surgery (p < 0.05, Figure 1B). These observations indicate that microglia clear sAβ in vivo.
Figure 1. C57BL/6J mice exhibit a significant reduction of microinjected FLsAβ42 and abundant microglial activation 5 days after surgery.

(A and B) Two hours or five days after surgery, the brains were removed and fixed. Brains were sectioned coronally at 20 μm on a cryostat. Cryosections were blocked and incubated with anti-Iba-1 or anti-CD68 antibodies. Nuclei were visualized by TO Pro-3 staining. Images with microinjected FLsAβ42 in the frontal cortex were obtained using a Zeiss LSM 510 confocal microscope. (B) The mean intensities of microinjected FLsAβ42 2 h and 5 days after surgery were analyzed (n = 5 females per group, 8 sections per mouse). The data, expressed as percentages of control (i.e. 2 h), represent the means ± SEM of 5 microinjected mice (40 sections) from A; ** p < 0.01 versus control (Student’s t-test). Detailed statistical results are shown in Table 2. Scale bar = 50 μm.
Microglia did not take up microinjected synthetic sAβ or transport it to the lysosomes in vivo
Next, we asked if microglia could degrade the microinjected synthetic sAβ by taking it up and transporting it to lysosomes in vivo. Using confocal microscopy, we found, unexpectedly, the microinjected FLsAβ42 was not taken up by Iba-1/CD68 positive microglial cells nor did it colocalize with the lysosomal marker (LAMP-1) inside microglia. Instead, microglia were clustered in the center of microinjected FLsAβ42 (Figures 2A-C). To further confirm the inability of microglia to uptake FLsAβ42, we isolated adult microglia from the injected mice and incubated them with antibodies specific for activated microglia/macrophage (CD11b and CD45) for flow cytometry analysis. Although the results showed a higher percentage of Aβ+/PE-CD11b+/APC-CD45low microglia in the FLsAβ42-treated group compared with scrambled peptide-treated controls (p < 0.05, Figures 3A-C), only a very small fraction (less than 1.5%) of microglia in the FLsAβ42-treated animals were Aβ-positive. Taken together, these data suggest that most microglia cannot take up synthetic sAβ or transport it to lysosomes for degradation, although they are activated in vivo.
Figure 2. Microglia do not take up microinjected synthetic sAβ or transport it to the lysosomes 5 days after the surgery.

(A-C) Five days after surgery, the brains were removed and fixed (n = 5 females per group). Brain cryosections were blocked and incubated with anti-Iba-1 (A), anti-CD68 (B) or LAMP-1 (C) antibodies. Nuclei were visualized by TO Pro-3 staining. Images were obtained as a z-series stack using a Zeiss LSM 510 confocal microscope. White scale bar = 100 μm; red scale bar = 20 μm.
Figure 3. The uptake of microinjected synthetic sAβ by microglia was measured by flow cytometry.

(A-C) Five days after surgery, adult primary microglia were isolated and stained with PE-Cy7-CD11b and APC-CD45. The CD11b+/CD45low microglia were gated and the percentage of Aβ+ cells among those gated microglia was analyzed. Representative Aβ+/CD11b+/CD45low microglia were recorded in the control group microinjected with fluorescence-labeled scrambled peptide (A) and in the treatment group microinjected with FLsAβ42 (B). (C) The percentage of Aβ+/CD11b+/CD45low microglia in both groups was analyzed (n= 5 females per group); p < 0.05 (nonparametric Mann Whitney test). Detailed statistical results are shown in Table 2. Only a very small fraction (less than 1.5%) of microglia in the FLsAβ42-treated group were Aβ-positive.
Microglia did not take up physiological sAβ in vivo and in vitro
Most previous studies used a very high concentration (~ 1 μM) of synthetic sAβ, whereas concentrations of sAβ in physiological fluids are 0.1 – 10 nM (Podlisny et al., 1995; Seubert et al., 1992; Suzuki et al., 1994). Therefore, in this study, we also investigated whether and how microglia clear physiological sAβ (7PA2 CM and AD-TBS) at very low concentration (~ 1 nM). Five days after the surgery, it was found that there was no significant difference of the percentage of Aβ+/PE-CD11b+/APC-CD45low microglia between the TBS-control group and the group microinjected with AD-TBS (p > 0.05, Figures 4A-C), indicating that microglia do not take up sAβ at a physiologically relevant concentration. Next, using N9 microglia cultures in combination with immunoprecipitation and Western blotting, we found that monomeric, but not oligomeric forms of Aβ species were reduced in the culture medium when N9 microglia were incubated with 7PA2 CM or AD-TBS for 3 h compared to the same medium without exposure to N9 microglia (p < 0.05, Figures 5A-D). However, we did not detect any Aβ species in the cell lysates of the N9 microglia (Supplemental Figure 2). Overall, these data suggest that microglia might not take up physiological sAβ in vivo and in vitro, although they can degrade the monomers in vitro.
Figure 4. Microglia do not take up microinjected physiological sAβ 5 days after the surgery.

(A-C) Five days after surgery, adult primary microglia were isolated and stained with PE-Cy7-CD11b, APC-CD45 or anti-Aβ (6E10) antibodies. Following incubation with FITC-anti-mouse secondary antibody, CD11b+/CD45low microglia were gated and the percentage of FITC-Aβ+ cells among those gated microglia was analyzed. Representative Aβ+/CD11b+/CD45low microglia were recorded in the control group microinjected with TBS (A) and in the treatment group microinjected with AD-TBS brain extracts (B). (C) The percentage of Aβ+/CD11b+/CD45low microglia in both groups was analyzed (n= 5 females per group); p > 0.05 (nonparametric Mann Whitney test). Detailed statistical results are shown in Table 2.
Figure 5. Natural sources of sAβ cannot be taken up by N9 microglial cells but monomer degradation can be reversed by an insulin degrading enzyme inhibitor.
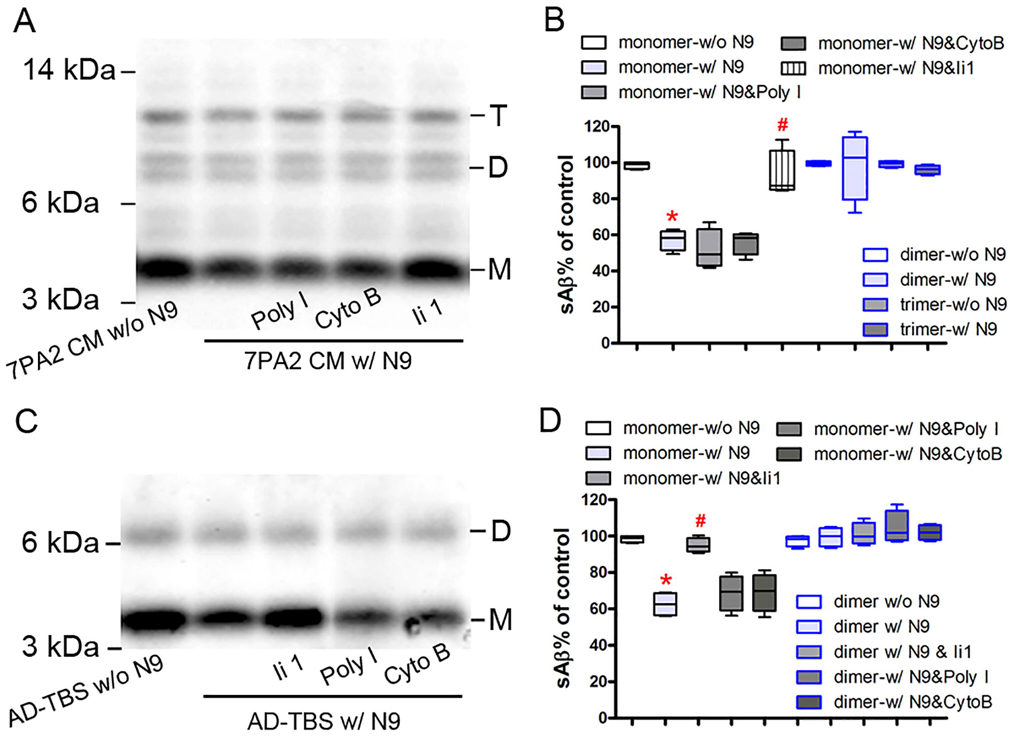
(A) and (C) N9 microglial cells were pretreated with or without 50 μg/ml polyinosinic acid (Poly I), 20 μg/ml cytochalasin B (Cyto B), or 10 μM Ii 1 and then incubated with or without soluble Aβ (sAβ from 7PA2 CM (A) or AD-TBS brain homogenate (C) for 3 h. Conditioned medium were collected, pre-cleared with protein A-Sepharose, and immunoprecipitated overnight with anti-Aβ antiserum (R1282). Aβ was detected in the immunoprecipitate by an anti-Aβ antibody (6E10) using Western blot assay. (B) and (D) The percentage of different forms of sAβ in panel A and C was quantified, respectively. The data, expressed as percentages of control (i.e. w/o N9), represent the means ± SEM of four separate experiments from each A and C; * p < 0.05 versus control, # p < 0.05 versus the group w/ N9 alone (nonparametric Mann Whitney test). Detailed statistical results are shown in Table 2.
The degradation of physiological sAβ by N9 microglia was prevented by an IDE inhibitor, but not by other phagocytosis or pinocytosis inhibitors in vitro
The reduction of Aβ monomer was not affected by pretreatment of a phagocytosis inhibitor (polyinosinic acid, Poly I) or a pinocytosis inhibitor (cytochalasin B, Cyto B) (p > 0.05), which had been proven to significantly inhibit the uptake of synthetic sAβ in vitro (El Khoury et al., 1996; Mandrekar et al., 2009). However, pretreatment of N9 microglia with Ii1, a highly selective inhibitor of IDE, almost completely prevented the reduction of Aβ monomer in both 7PA2 CM and AD-TBS brain extract (p < 0.05, Figures 5A-D). Together, these data suggest that microglia can degrade Aβ monomer extracellularly by secreting IDE without actually taking up any of the physiological sAβ.
An IDE inhibitor significantly prevented the degradation of microinjected synthetic and physiological sAβ in vivo
To further confirm the role of IDE in vivo, we measured the Aβ42 levels in the brain homogenates of C57BL/6J mice microinjected with FLsAβ42 or AD-TBS brain extract in the presence or absence of Ii1. We found that there was significantly less Aβ42 5 days after surgery compared to 2 h after surgery. Furthermore, the reduction of Aβ42 was significantly prevented by the co-injection of Ii1 (p < 0.05, Figures 6A and B). These results indicate that the degradation of both microinjected synthetic and physiological sAβ can be prevented by co-treatment with an IDE inhibitor in vivo.
Figure 6. The IDE inhibitor prevents the degradation of the microinjected synthetic and physiological sAβ in vivo.
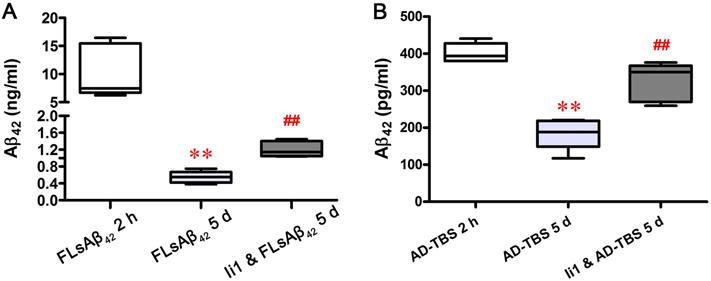
(A and B) Two hours or five days after surgery, mouse brains were removed and homogenized. The Aβ42 level in the homogenates was measured by ELISA as described in the Methods. The data represent the means ± SEM of all mice in each group (n = 5 females per group); ** p < 0.01 versus the group of FLsAβ42 2 h (A) or the group of AD-TBS 2 h (B); ## p < 0.01 versus the group of FLsAβ42 5 d (A) or the group of AD-TBS 5 d (B) (nonparametric Mann Whitney test). Detailed statistical results are shown in Table 2.
DISCUSSION
Based on the fact that activated microglia and reactive astrocytes are frequently associated with the amyloid core and/or within the senile plaques in AD (Itagaki et al., 1989), numerous studies have been conducted to investigate the relationship between microglia and fAβ. Many receptors have been found to mediate the uptake and clearance of fAβ by microglia, including scavenger receptors, Toll-like receptors, purinergic G protein-coupled receptors (P2Y2 and P2Y6), TREM2, complement components and their associated receptors (Brazil et al., 2000; Choucair-Jaafar et al., 2011; El Khoury et al., 1996; Fu et al., 2012; Kim et al., 2012; Liu et al., 2005; Maier et al., 2008; Paresce et al., 1996; Richard et al., 2008; Webster et al., 2000; Wyss-Coray et al., 2002; Yeh et al., 2016; Zhao et al., 2018). However, little is known about the underlying mechanisms by which microglia take up and clear sAβ, especially physiological sAβ, which are found to be more toxic and critical in the cognitive dysfunction during the early stage of AD. The concentration of physiological sAβ is similar to that in physiological fluids and much lower than that of synthetic Aβ used in most previous studies (Podlisny et al., 1995; Seubert et al., 1992; Suzuki et al., 1994). In addition, previous studies have suggested that Aβ aggregation states may impact microglial uptake of Aβ. For example, resting microglia have been shown to react to fAβ but do not detect sAβ oligomers or oligomer-induced neuronal damage (Ferrera et al., 2014). Another example is that microglia are found to internalize Aβ protofibrils more extensively than Aβ monomers (Gouwens et al., 2016).
First, we wanted to answer one very important question: can microglia be activated and clear sAβ in vivo? Using stereotaxic microinjection technique, we microinjected fluorescently labeled synthetic sAβ into the frontal cortex of mouse brains and found that there were many more Iba-1 and CD68 positive cells and lower fluorescent intensity of injected sAβ 5 d but not 2 h after surgery. Using ELISA, we confirmed that there were much less synthetic or physiological sAβ remaining in the brain homogenates 5 days after the surgery compared to 2 h after the surgery. These data suggest that microglia become activated and clear sAβ in vivo. Individual examination of the microglial cells clustered at the injection site using the Z-stack function of confocal microscopy led to the unexpected finding that the synthetic sAβ was not taken up by Iba-1/CD68 positive microglia or transported to the lysosomes evidenced by no obvious co-localization of sAβ and lysosomal marker LAMP-1. Instead, microglia were clustered in the center of microinjected FLsAβ42. The pattern of Iba-1, CD68 and LAMP-1 staining induced by FLsAβ42 microinjection was different from that induced by fAβ (Krauthausen et al., 2015). Furthermore, the results of flow cytometry showed that most microglia (CD11b+/CD45low) did not take up the injected synthetic or physiologically sAβ. Together, these data suggest that microglia can degrade sAβ in vivo, but mainly not by taking up and transporting sAβ to lysosomes for degradation. However, it should be noticed that we cannot exclude the possibility that such low concentration of sAβ in the current study might be out of the limitation of detection for Flow cytometry, although sAβ species in human AD synaptosomes can be detected using this technique (Sokolow et al., 2012).
Next, we aimed to investigate how microglia degrade sAβ in vivo. IDE, one of most important Aβ-degrading enzymes, is present in human brain and cerebrospinal fluid and can effectively degrade Aβ in vitro and in vivo (Farris et al., 2003; Kurochkin and Goto, 1994; McDermott and Gibson, 1997; Miller et al., 2003; Qiu et al., 1998; Qiu et al., 1997). Microglia have been found to degrade extracellular Aβ by secreting IDE (Qiu et al., 1997). In this study, we found that although there is a significant reduction in Aβ monomers when physiological sAβ was incubated with N9 microglia, no significant changes of Aβ dimers or trimers were found in the presence of microglia. None of the Aβ species were detected in the cell lysate (Supplemental Figure 2). The reduction of Aβ monomer was independent of phagocytosis and pinocytosis, both of which have been found to mediate the uptake and clearance of synthetic sAβ by microglia in vitro (Chung et al., 1999; Mandrekar et al., 2009; Yang et al., 2011). This discrepancy may be due in part to the differences in the sAβ concentrations (~1 μM VS. ~ 1 nM) and/or the culture conditions of different microglia cell lines used among these studies. In contrast, the reduction of physiological Aβ monomer was completely reversed by pretreatment of microglia with a highly selective IDE inhibitor (Ii1). Together, these in vitro data suggest that microglia may only degrade extracellular Aβ monomer by secreting IDE without taking up any conformational species of physiological sAβ.
Finally, we confirmed the role of IDE in degrading sAβ in vivo. Previous research showed that microglia reacted to fAβ much stronger than sAβ (Ferrera et al., 2014), and a robust activation of microglia was observed 2 days after fAβ injection (Krauthausen et al., 2015). Since we were using sAβ, we chose a longer time duration (i.e. 5 days) as our examination time point in order to allow microglia enough time to react to the injected sAβ. Five days after the surgery, both of the microinjected synthetic and physiological sAβ were significantly reduced, compared to the Aβ levels at 2 h after the surgery. The co-injection of Ii1 and synthetic or physiological sAβ partially reversed the Aβ levels 5 days after the surgery compared to the group injected with sAβ alone. This finding demonstrates that IDE plays a significant role in the degradations of both synthetic and physiological sAβ in vivo. We noticed that the reduction of FLsAβ42 could only be partially reversed by Ii1, although it almost completely reversed the reduction of sAβ in the AD-TBS extraction. This indicates that our current study cannot totally exclude the possibility of microglial uptake of recombinant sAβ before 5 days. More detailed concentration-dependent and time-course studies in vivo are needed for elucidating the role of microglia in the clearance of sAβ in the future.
Since astrocytes are also reported to be involved in the degradation of sAβ in vitro and in vivo via matrix metalloproteinases (Yin et al., 2006), IDE and NEP (Dorfman et al., 2010; Son et al., 2016), we cannot exclude the possibility that astrocytes might also participate the degradation of the microinjected sAβ via secreting IDE in our model. In addition, we only chose 8-9 mo-old adult female mice for this study. This choice is based on four reasons: 1) AD has a higher prevalence in women above 65 years old (1.6–3:1 ratio compared to men) and progresses with a greater cognitive deterioration (Plassman et al., 2011; Seshadri et al., 1997); 2) Aβ accumulation and decreased expression of IDE are more profound in female AD-like mice compared with male counterparts (Gallagher et al., 2013; Hirata-Fukae et al., 2008; Wang et al., 2003); 3) The ε4 variant of APOE gene is the strongest genetic risk factor for developing late-onset AD and is more abundant in females compared to males (Altmann et al., 2014; Cambronero et al., 2018; Seshadri et al., 1997). Microglia are the main origin of plaque-associated APOE (Parhizkar et al., 2019), and female aged mice-derived microglia demonstrate a significant upregulation of genes that are involved in the APOE network (Kang et al., 2018), suggesting that APOE is a gene that could partially explain increased AD susceptibility in females; 4) Sex differences in microglial gene signatures have been revealed. Male microglia express genes that are more associated with pro-inflammatory responses, while female microglia have higher phagocytic capacity and higher gene expression of cell repair and inflammatory control genes (Villa et al., 2019; Villa et al., 2018). Given that there are potential gender and age differences in the uptake and degradation of Aβ by microglia as well as the decrease of IDE in aging and AD (Floden and Combs, 2011; Hefendehl et al., 2014; Hickman et al., 2008; Kochkina et al., 2015; Stephen et al., 2019), it will be extremely interesting and important to investigate if the identified mechanism in this study can be applied to adult male mice and aged mice as well as AD-like mice in the future.
In conclusion, we provide evidence that microglia can partially clear different sources of sAβ mainly by secreting IDE, but not by taking up and transporting them to lysosomes for degradation at least in our experimental model. This study provides useful insights regarding how microglia clear sAβ, especially physiologically sAβ, and may identify new therapeutic targets for the prevention and/or treatment of AD, the mostly common form of dementia.
Supplementary Material
HIGHLIGHTS:
Although being activated by sAβ, microglia do not take up synthetic and physiological sAβ in vivo.
Microglia degrade physiological sAβ through secreting IDE in vitro, independently of phagocytosis and pinocytosis.
Pretreatment of IDE inhibitor prevents microglia-mediated degradation of both synthetic and physiological sAβ in vivo.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health under Award Number R01 AG020159 and RF1 AG058657 awarded to CAL, AG056673 awarded to HF, the Alzheimer’s Association (AARF-17-505009) awarded to HF, and the Neuroscience Research Institute Pilot Award and the Chronic Brain Injury Pilot Award from The Ohio State University awarded to HF. We sincerely thank Dr. Min Jin for helping with the confocal microscopy; Dr. Soyon Hong for helping with the microglia culture and participating in the discussion of this study; Dr. Malcolm A. Leissring for providing the IDE inhibitor; Dr. Dennis J. Selkoe for providing the physiologically soluble Aβ and participating in the discussion of this study; Drs. Joseph El Khoury and Paola Ricciardi-Castagnoli for providing N9 microglial cells. The 1D4B monoclonal antibody developed by Dr. J Thomas August was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
REFERENCES
- Alzheimer’s Association (2020) 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s & Dementia 2020:16(3):391+. [Google Scholar]
- Altmann A, Tian L, Henderson VW, Greicius MD, Alzheimer's Disease Neuroimaging Initiative I (2014) Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol 75, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Aisen PS, De Strooper B, Fox NC, Lemere CA, Ringman JM, Salloway S, Sperling RA, et al. (2011) Autosomal-dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimers Res Ther 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Antel JP (1996) Comparison of phenotypic and functional properties of immediately ex vivo and cultured human adult microglia. Glia 18, 1–10. [DOI] [PubMed] [Google Scholar]
- Bekris LM, Yu CE, Bird TD, Tsuang DW (2010) Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 23, 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazil MI, Chung H, Maxfield FR (2000) Effects of incorporation of immunoglobulin G and complement component C1q on uptake and degradation of Alzheimer's disease amyloid fibrils by microglia. J Biol Chem 275, 16941–16947. [DOI] [PubMed] [Google Scholar]
- Cambronero FE, Liu D, Neal JE, Moore EE, Gifford KA, Terry JG, Nair S, Pechman KR, et al. (2018) APOE genotype modifies the association between central arterial stiffening and cognition in older adults. Neurobiol Aging 67, 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Huang D, Sasse ME, Ransohoff RM (2006) Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat Protoc 1, 1947–1951. [DOI] [PubMed] [Google Scholar]
- Choucair-Jaafar N, Laporte V, Levy R, Poindron P, Lombard Y, Gies JP (2011) Complement receptor 3 (CD11b/CD18) is implicated in the elimination of beta-amyloid peptides. Fundam Clin Pharmacol 25, 115–122. [DOI] [PubMed] [Google Scholar]
- Chu T, Tran T, Yang F, Beech W, Cole GM, Frautschy SA (1998) Effect of chloroquine and leupeptin on intracellular accumulation of amyloid-beta (Aβ) 1-42 peptide in a murine N9 microglial cell line. FEBS Letters 436, 439–444. [DOI] [PubMed] [Google Scholar]
- Chung H, Brazil MI, Soe TT, Maxfield FR (1999) Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta-peptide by microglial cells. J Biol Chem 274, 32301–32308. [DOI] [PubMed] [Google Scholar]
- Czirr E, Castello NA, Mosher KI, Castellano JM, Hinkson IV, Lucin KM, Baeza-Raja B, Ryu JK, et al. (2017) Microglial complement receptor 3 regulates brain Abeta levels through secreted proteolytic activity. J Exp Med 214, 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB Jr., Baker LK, Krafft GA, LaDu MJ (2002) Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem 277, 32046–32053. [DOI] [PubMed] [Google Scholar]
- Dorfman VB, Pasquini L, Riudavets M, Lopez-Costa JJ, Villegas A, Troncoso JC, Lopera F, Castano EM, et al. (2010) Differential cerebral deposition of IDE and NEP in sporadic and familial Alzheimer's disease. Neurobiol Aging 31, 1743–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckman EA, Reed DK, Eckman CB (2001) Degradation of the Alzheimer's amyloid beta peptide by endothelin-converting enzyme. J Biol Chem 276, 24540–24548. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD (1996) Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 382, 716–719. [DOI] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, et al. (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 100, 4162–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris W, Schutz SG, Cirrito JR, Shankar GM, Sun X, George A, Leissring MA, Walsh DM, et al. (2007) Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am J Pathol 171, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrera D, Mazzaro N, Canale C, Gasparini L (2014) Resting microglia react to A β 42 fibrils but do not detect oligomers or oligomer-induced neuronal damage. Neurobiology of aging 35, 2444–2457. [DOI] [PubMed] [Google Scholar]
- Floden AM, Combs CK (2011) Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J Alzheimers Dis 25, 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, Sedgwick JD (1995) Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol 154, 4309–4321. [PubMed] [Google Scholar]
- Frenkel D, Wilkinson K, Zhao L, Hickman SE, Means TK, Puckett L, Farfara D, Kingery ND, et al. (2013) Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nat Commun 4, 2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Li W, Lao Y, Luo J, Lee NT, Kan KK, Tsang HW, Tsim KW, et al. (2006) Bis(7)-tacrine attenuates beta amyloid-induced neuronal apoptosis by regulating L-type calcium channels. J Neurochem 98, 1400–1410. [DOI] [PubMed] [Google Scholar]
- Fu H, Liu B, Frost JL, Hong S, Jin M, Ostaszewski B, Shankar GM, Costantino IM, et al. (2012) Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Abeta by microglia. Glia 60, 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JJ, Minogue AM, Lynch MA (2013) Impaired performance of female APP/PS1 mice in the Morris water maze is coupled with increased Abeta accumulation and microglial activation. Neurodegener Dis 11, 33–41. [DOI] [PubMed] [Google Scholar]
- Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick J, Kuo LM, et al. (1996) Specific domains of beta-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci 16, 6021–6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouwens LK, Makoni NJ, Rogers VA, Nichols MR (2016) Amyloid-β 42 protofibrils are internalized by microglia more extensively than monomers. Brain research 1648, 485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefendehl JK, Neher JJ, Suhs RB, Kohsaka S, Skodras A, Jucker M (2014) Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 13, 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henstridge CM, Hyman BT, Spires-Jones TL (2019) Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat Rev Neurosci 20, 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Guillamon M, Mawhirt S, Fossati S, Blais S, Pares M, Penalba A, Boada M, Couraud PO, et al. (2010) Matrix metalloproteinase 2 (MMP-2) degrades soluble vasculotropic amyloid-beta E22Q and L34V mutants, delaying their toxicity for human brain microvascular endothelial cells. J Biol Chem 285, 27144–27158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J (2008) Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci 28, 8354–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata-Fukae C, Li HF, Hoe HS, Gray AJ, Minami SS, Hamada K, Niikura T, Hua F, et al. (2008) Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res 1216, 92–103. [DOI] [PubMed] [Google Scholar]
- Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D (1989) Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 24, 173–182. [DOI] [PubMed] [Google Scholar]
- Kang SS, Ebbert MTW, Baker KE, Cook C, Wang X, Sens JP, Kocher JP, Petrucelli L, et al. (2018) Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J Exp Med 215, 2235–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, et al. (2017) A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell 169, 1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Ajit D, Peterson TS, Wang Y, Camden JM, Gibson Wood W, Sun GY, Erb L, et al. (2012) Nucleotides released from Aβ1-42-treated microglial cells increase cell migration and Aβ1-42 uptake through P2Y2 receptor activation. J Neurochem 121, 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochkina EG, Plesneva SA, Vasilev DS, Zhuravin IA, Turner AJ, Nalivaeva NN (2015) Effects of ageing and experimental diabetes on insulin-degrading enzyme expression in male rat tissues. Biogerontology 16, 473–484. [DOI] [PubMed] [Google Scholar]
- Krauthausen M, Kummer MP, Zimmermann J, Reyes-Irisarri E, Terwel D, Bulic B, Heneka MT, Muller M (2015) CXCR3 promotes plaque formation and behavioral deficits in an Alzheimer’s disease model. The Journal of clinical investigation 125, 365–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurochkin IV, Goto S (1994) Alzheimer's β - amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS letters 345, 33–37. [DOI] [PubMed] [Google Scholar]
- Lane CA, Hardy J, Schott JM (2018) Alzheimer's disease. Eur J Neurol 25, 59–70. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ (2003) Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40, 1087–1093. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Malito E, Hedouin S, Reinstatler L, Sahara T, Abdul-Hay SO, Choudhry S, Maharvi GM, et al. (2010) Designed inhibitors of insulin-degrading enzyme regulate the catabolism and activity of insulin. PLoS One 5, e10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HQ, Chen C, Dou Y, Wu HJ, Liu YJ, Lou HF, Zhang JM, Li XM, et al. (2013) P2Y4 receptor-mediated pinocytosis contributes to amyloid beta-induced self-uptake by microglia. Mol Cell Biol 33, 4282–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D (2009) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, Heine H, Penke B, et al. (2005) LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain 128, 1778–1789. [DOI] [PubMed] [Google Scholar]
- Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW (1993) Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci U S A 90, 7951–7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T (2009) Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 64, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue L-F, Kuo Y-M, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, et al. (1999) Soluble Amyloid β Peptide Concentration as a Predictor of Synaptic Change in Alzheimer's Disease. The American Journal of Pathology 155, 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA (2008) Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci 28, 6333–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE (2009) Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J Neurosci 29, 4252–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott JR, Gibson AM (1997) Degradation of Alzheimer's beta-amyloid protein by human and rat brain peptidases: involvement of insulin-degrading enzyme. Neurochem Res 22, 49–56. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Konrad V, Bush AI, Masters CL (1999) Soluble pool of A? amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Annals of Neurology 46, 860–866. [DOI] [PubMed] [Google Scholar]
- Melchor JP, Pawlak R, Strickland S (2003) The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-beta (Abeta) degradation and inhibits Abeta-induced neurodegeneration. J Neurosci 23, 8867–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, Hersh LB, Thiele DL (2003) Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci U S A 100, 6221–6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco-Quinto J, Eckman EA (2013) Endothelin-converting enzymes degrade intracellular beta-amyloid produced within the endosomal/lysosomal pathway and autophagosomes. J Biol Chem 288, 5606–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paresce DM, Ghosh RN, Maxfield FR (1996) Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron 17, 553–565. [DOI] [PubMed] [Google Scholar]
- Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, Nuscher B, Xiong M, et al. (2019) Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci 22, 191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassman BL, Langa KM, McCammon RJ, Fisher GG, Potter GG, Burke JR, Steffens DC, Foster NL, et al. (2011) Incidence of dementia and cognitive impairment, not dementia in the United States. Ann Neurol 70, 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ (1995) Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem 270, 9564–9570. [DOI] [PubMed] [Google Scholar]
- Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, et al. (1998) Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem 273, 32730–32738. [DOI] [PubMed] [Google Scholar]
- Qiu WQ, Ye Z, Kholodenko D, Seubert P, Selkoe DJ (1997) Degradation of amyloid beta-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J Biol Chem 272, 6641–6646. [DOI] [PubMed] [Google Scholar]
- Richard KL, Filali M, Prefontaine P, Rivest S (2008) Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer's disease. J Neurosci 28, 5784–5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (1991) The molecular pathology of Alzheimer's disease. Neuron 6, 487–498. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Wolf PA, Beiser A, Au R, McNulty K, White R, D'Agostino RB (1997) Lifetime risk of dementia and Alzheimer's disease. The impact of mortality on risk estimates in the Framingham Study. Neurology 49, 1498–1504. [DOI] [PubMed] [Google Scholar]
- Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, et al. (1992) Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature 359, 325–327. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, et al. (2008) Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikanyika NL, Parkington HC, Smith AI, Kuruppu S (2019) Powering Amyloid Beta Degrading Enzymes: A Possible Therapy for Alzheimer's Disease. Neurochem Res 44, 1289–1296. [DOI] [PubMed] [Google Scholar]
- Sokolow S, Henkins KM, Bilousova T, Miller CA, Vinters HV, Poon W, Cole GM, Gylys KH (2012) AD synapses contain abundant Abeta monomer and multiple soluble oligomers, including a 56-kDa assembly. Neurobiol Aging 33, 1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son SM, Cha MY, Choi H, Kang S, Choi H, Lee MS, Park SA, Mook-Jung I (2016) Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy 12, 784–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen TL, Cacciottolo M, Balu D, Morgan TE, LaDu MJ, Finch CE, Pike CJ (2019) APOE genotype and sex affect microglial interactions with plaques in Alzheimer's disease mice. Acta Neuropathol Commun 7, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L Jr., Eckman C, Golde TE, Younkin SG (1994) An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 264, 1336–1340. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. [DOI] [PubMed] [Google Scholar]
- Villa A, Della Torre S, Maggi A (2019) Sexual differentiation of microglia. Front Neuroendocrinol 52, 156–164. [DOI] [PubMed] [Google Scholar]
- Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, Lolli F, Marcello E, et al. (2018) Sex-Specific Features of Microglia from Adult Mice. Cell Rep 23, 3501–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tanila H, Puolivali J, Kadish I, van Groen T (2003) Gender differences in the amount and deposition of amyloidbeta in APPswe and PS1 double transgenic mice. Neurobiol Dis 14, 318–327. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, et al. (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Zhou HD, Zhou XF (2006) Clearance of amyloid-beta in Alzheimer's disease: progress, problems and perspectives. Drug Discov Today 11, 931–938. [DOI] [PubMed] [Google Scholar]
- Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ (2000) Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp Neurol 161, 127–138. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E (2002) Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci U S A 99, 10837–10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan P, Hu X, Song H, Yin K, Bateman RJ, Cirrito JR, Xiao Q, Hsu FF, et al. (2006) Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J Biol Chem 281, 24566–24574. [DOI] [PubMed] [Google Scholar]
- Yang CN, Shiao YJ, Shie FS, Guo BS, Chen PH, Cho CY, Chen YJ, Huang FL, et al. (2011) Mechanism mediating oligomeric Abeta clearance by naive primary microglia. Neurobiol Dis 42, 221–230. [DOI] [PubMed] [Google Scholar]
- Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M (2016) TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 91, 328–340. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, et al. (2006) Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J Neurosci 26, 10939–10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, Sun Y, Zhu B, et al. (2018) TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron 97, 1023–1031 e1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.