

Abstract
Silmitasertib (CX-4945) as a potent and selective inhibitor of CK2 exhibited promising in vitro and in vivo anti-cancer activity. An assay employing cation-exchange solid phase extraction (SPE) followed by LC-MS/MS analysis was successfully developed and validated for the quantitation of silmitasertib in human plasma, brain tissue, and human cerebrospinal fluid (CSF). Reverse phase chromatographic separation was achieved using Synergi™ hydro-RP column (4 μm, 75 × 2.0 mm) and gradient elution with 5 mM ammonium formate aqueous solution (pH 6.5) as mobile phase A and 0.1% formic acid in acetonitrile as mobile phase B. Multiple reaction monitoring (MRM) transition of m/z 350.2→223.2 and m/z 316.2→223.2 were chosen for detection of silmitasertib and internal standard (CX-4786) respectively. Since silmitasertib concentration in patient plasma is expected to be in a wide range due to the study design, two calibration curves with range 0.2-125 ng/ml and 32-20,000 ng/ml were established. A different curve ranging from 2 to 40 ng/g was used for measurement of silmitasertib in brain tissue, while another calibration curve ranging from 0.2 to 20 ng/ml was established for CSF. All these calibration curves corresponding to different matrices showed good linearity (R2 > 0.99) over the concentration range. This assay demonstrated excellent precision below 15% and accuracies between 85-115% within-day and between-day for all the concentration levels in each matrix. This assay was also validated for each matrix for selectivity, sensitivity, matrix effects, recovery, and stability. We applied the validated method to the analysis of plasma silmitasertib for a clinical study.
Keywords: CX-4945, silmitasertib, CK2, human plasma, human CSF, brain tissue homogenate, LC-MS/MS, cation-exchange solid phase extraction
1. Introduction
Medulloblastoma, a heterogenous group of tumors, are among the most common malignant tumor in the pediatric population [1]. About 20-30% of these tumors are driven by activation of the Sonic Hedgehog (SHH) pathway [2]. Current treatment of pediatric SHH medulloblastoma consists of surgical resection, adjuvant chemotherapy, and craniospinal irradiation depending on age at diagnosis. However, tumor recurrence remains around 30% with no standard options and very poor outcomes, which pushes the need for developing new therapeutic approaches [3].
Silmitasertib (CX-4945), a potent and highly selective orally administered small-molecule inhibitor of casein protein kinase 2 (CK2), is involved in multiple cellular processes such as cell cycle progression, cell growth, and differentiation, and is overexpressed in multiple cancers [4, 5]. Preclinical studies showed that silmitasertib inhibited pro-survival and angiogenic signaling in different cancer cell lines including breast, leukemia, colon, lung, and prostate, and has exhibited antitumor activity in breast and pancreatic murine xenografts [5]. In adult clinical studies, silmitasertib is under development as a therapeutic agent for advanced solid tumors and hematological malignancies [6, 7]. Initial results suggest that silmitasertib exposure was related to inhibition of the CK2 pathway [6]. More recently, CK2 has also been identified as a specific driver of Hedgehog signaling [8]. Further studies showed that silmitasertib-induced CK2 inhibition led to a robust tumor stasis and long-term regression of mice with flank allografts of SHH medulloblastoma cells [8].
These findings paved the way to evaluate the safety and tolerability of silmitasertib in children with recurrent SHH medulloblastoma in a phase I/II and surgical study (PBTC053; NCT03904862). Pharmacokinetic studies will be performed in patients enrolled in the phase I, phase II and surgical component of this trial to determine silmitasertib concentrations in plasma, brain tumor tissue, and cerebrospinal fluid (CSF). Therefore, an accurate, precise, and robust bioanalytical method will be required for each matrix. The objective of this study was to develop and validate sensitive and robust liquid chromatography tandem mass spectrometry (LC-MS/MS) methods to measure silmitasertib in human plasma, brain tumor tissue, and CSF. The method for human plasma was then applied to measure the silmitasertib plasma samples collected for a patient enrolled in this clinical trial.
2. Materials and methods
2.1. Reagents and chemicals
Sodium 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylate (silmitasertib; Fig. 1a) and 5-(phenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid [CX-4786, internal standard (ISTD); Fig. 1b] were supplied by Senhwa Biosciences (San Diego, CA). Ammonium formate (for mass spectrometry, ≥99.0%) was purchased from Millipore Sigma (St. Louis, MO). Dimethyl sulfoxide (molecular biology grade), formic acid (LC-MS/MS grade, 99.5% purity), acetonitrile (HPLC grade), methanol (HPLC grade), ammonium hydroxide (Certified ACS Plus), O-phosphoric acid (85%, w/w, HPLC grade) were purchased from Fisher Scientific (Waltham, MA), and water was obtained from Millipore Q-advantage water purification system (Temecula, CA).
Fig.1.
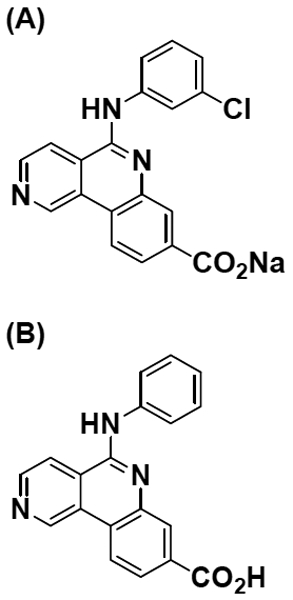
Structure of (A) silmitasertib and (B) ISTD, CX-4786.
Pooled human sodium EDTA plasma and whole blood and cynomolgus/rhesus brain tissue were purchased from BioIVT (Westbury, NY, USA). Mouse brains were collected from CD-1 nude mice that were anesthetized using isoflurane and perfused with 10 ml PBS. Then, the mice were decapitated and the brain was removed. The obtained brains were stored in plastic tubes at −80°C until analysis. The animal studies performed were approved by the St. Jude Institutional Animal Care and Usage Committee (IACUC).
2.2. Stock and working solutions
Stock solutions (1.00 mg/ml) of silmitasertib and CX-4786 (ISTD) were prepared in DMSO. Silmitasertib stock solution was serially diluted with ACN/H2O (4:1, v/v) to prepare the desired concentrations of working solutions. Two ISTD working solutions at 0.1 and 2 μg/ml were prepared by diluting the ISTD stock solution with ACN/H2O (4:1,v/v).
2.3. Calibration standards and quality controls
For the analysis of silmitasertib in human plasma, the lower calibration curve of 0.2-125 ng/ml was established by 8 calibration standards with silmitasertib concentration at 0.2, 1, 2, 5, 10, 25, 50, and 125 ng/ml, which were prepared by spiking 100 μl blank human plasma with 10 μl silmitasertib working solution of appropriate concentration. The higher calibration curve of 32-20,000 ng/ml in human plasma was constructed by 8 calibration standards at 32, 160, 320, 800, 1,600, 4,000, 8,000, and 20,000 ng/ml, which were prepared by adding 10 μl silmitasertib working solution of desired concentration into 25 μl human plasma. Human plasma QC samples at 0.6, 7.5, and 100 ng/ml for the calibration curve (0.2-125 ng/ml) and 96, 1,300, and 17,000 ng/ml for the curve (32-20,000 ng/ml) were prepared in a similar way, representing low QC (LQC), middle QC (MQC), and high QC (HQC), respectively.
For brain tissue, the calibrators (2, 6, 12, 20, and 40 ng/g) and QCs (4, 16, and 32 ng/g) were prepared in 100 μl of brain homogenate in human plasma. For human CSF, the calibrators (0.2, 0.4, 1.2, 2, 4, 10, 15, and 20 ng/ml) and QCs (0.6, 3, and 18 ng/ml) were prepared in 100 μl human CSF. The brain and CSF silmitasertib concentrations expected at the dosages studied in the clinical trial were expected to be low based on unpublished data. Thus, the concentration range for the brain and CSF calibrators were selected based on the lower calibration curve established in plasma.
The calibrators and QCs were prepared using the following sample preparation procedure.
2.4. Sample preparation
2.4.1. Human plasma and human CSF
For the quantitation using the lower calibration curve, 100 μl human plasma was mixed with 6 μl of 100 ng/ml ISTD working solution, while 25 μl human plasma with 6 μl of 2 μg/ml ISTD working solution if using the higher calibration curve. Ten microliter of ACN/H2O (4:1, v/v) was added to the study samples for volume correction. All samples including calibration standards and quality controls and study samples were diluted with 90 μl of 4% (w/w) O-phosphoric acid and then loaded into the MCX 96 well plate which was pretreated with 100 μl methanol and 200 μl H2O. Samples were washed with 200 μl of 2% formic acid and then 200 μl methanol. Finally, the analyte and ISTD were eluted twice using 100 μl of 5% (v/v) ammonium hydroxide in methanol. The eluate was collected in a 96 well collection plate and concentrated to dryness under a gentle stream of nitrogen at 45°C. The residue was reconstituted with methanol/H2O (1:1, v/v), followed by LC-MS/MS analysis. The reconstitution solution of 100 μl and injection volume of 5 μl was used for the lower calibration curve (0.2-125 ng/ml), while 300 μl reconstitution solution and 1 μl injection for the higher calibration curve (32-20,000 ng/ml).
Human CSF (100 μl) was processed and extracted in the same way as human plasma (100 μl).
2.4.2. Primate or mouse brain homogenate
In the surgical component of the clinical trial, it would be necessary to measure silmitasertib concentrations in human brain tumor tissue. However, access to this matrix would be exceedingly rare and none would be available for method development and validation. Thus, we chose to take an alternative approach reasoning that if silmitasertib could be measured in nonhuman primate brain tissue it could be measured in human brain tissue. Access to nonhuman primate brain tissue was limited, thus, we chose to begin by developing the method in mouse brain tissue, performing a full validation in that matrix, and then performing a partial validation in the nonhuman primate brain tissue matrix.
Primate or mouse brain homogenate was prepared by homogenizing 10 mg tissue in 90 μl human plasma for 3 min using a multi-sample homogenizer (Benchmark Scientific D2400) and 1.5 mm zirconium homogenization beads (Midwest Scientific D1032-15). To 100 μl of primate or mouse brain tissue homogenate was added 6 μl of 100 ng/ml ISTD working solution and then 90 μl of 4% (w/w) O-phosphoric acid. After mixing on vortex mixer for 30 s and centrifugation at 12,000 rpm for 5 min, 170 μl supernatant was transferred to a new tube. The remaining tissue pellet was further extracted by adding 100 μl methanol. After vortex mixing for 5 min and centrifugation again at 12,000 rpm for 5 min, 100 μl supernatant was transferred and combined with the supernatant obtained in the previous step. The combined supernatant was further purified in the same way as human plasma by using MCX 96 well plate. The dry residue from solid phase extraction was reconstituted with 100 μl methanol/H2O (1:1, v/v), followed by LC-MS/MS analysis with 5 μl injection.
2.5. Chromatographic conditions
The Shimadzu HPLC system (Kyoto, Japan) consisted of an online degasser (DGU-20A), two pumps (LC-30 AD), an autosampler (SIL-30AC), a controller (CBM-20A), and a column oven (CTO-20AC). The chromatographic separation was performed on a Synergi™ hydro-RP C18 column (75 × 2.0 mm, 4 μm) with column temperature maintained at 30°C. An optimized gradient of mobile phase A: 5 mM ammonium formate (pH 6.5) and mobile phase B: 0.1% formic acid in acetonitrile was used to elute silmitasertib and ISTD. The flow rate was set at 0.4 ml/min. Two types of gradient elution were employed in this assay. For analyzing human plasma and CSF samples, the gradient elution started with 50% mobile phase B, gradually increased to 80% B in 1.2 min, maintained constant for 3 mins, and then decreased to 50% in 0.3 min, followed by column equilibrium for 1.5 min. For the analysis of brain tissue homogenate, the gradient stated with 30% mobile phase B, reached 80% B in 1.2 min and held this composition for 3 mins, and then return to the initial condition in 0.3 min and held it for 1.5 min. The total analysis time for a single sample was 6 min for both gradients.
2.6. Mass spectrometric conditions
The mass spectrometric detection was performed on an AB SCIEX QTRAP 5500 system (Toronto, Canada) with an electrospray ionization source coupled with the above described HPLC system. The mass spectrometer was operated in positive ion mode with the MRM transitions set at m/z 350.2→223.2 for silmitasertib and m/z 316.2→223.2 for ISTD. Nitrogen was used as collision gas. The optimized ion source parameters for MS/MS analysis were set as follows: ionspray voltage (IS), 5,500 V; source temperature, 650°C; curtain gas (CUR), 40 psi; gas 1 (GS1), 65 psi; gas 2 (GS2), 55 psi; collision activated dissociation (CAD), medium. The compound parameters were as follows: declustering potential (DP), 95 V; entrance potential (EP), 10 V; collision energy (CE), 49 V for silmitasertib and 46 V for ISTD; collision exit potential (CXP), 13 V. Analyst software (Version, 1.6.2, AB SCIEX) was used to perform the data acquisition and quantification.
2.7. Method validation procedure
Using the Guidance for Industry, Bioanalytical Method Validation, May 2018, this LC-MS/MS method was validated in terms of linearity, precision, accuracy, sensitivity, lower limit of quantitation, limit of detection, matrix effects, recovery, and stability [9].
2.7.1. Linearity
Linearity was evaluated over two concentration ranges of 0.2-125 ng/ml and 32-20,000 ng/ml for the quantitation of silmitasertib in human plasma, one concentration range of 0.2-20 ng/ml for silmitasertib in human CSF, and another concentration range of 2-40 ng/g for silmitasertib in mouse brain. The calibration curves for each matrix were established using double blank (blank matrix with neither silmitasertib nor ISTD), zero blank (blank matrix with ISTD only), and nonblank calibration standards at a series of different concentrations in the corresponding matrix. The peak area ratio of silmitasertib to ISTD vs silmitasertib concentration (x) was plotted using linear regression with weighting factor of 1/x2. The linearity of each calibration curve was determined using the correlation coefficient (R2).
2.7.2. Precision, accuracy, and sensitivity
The accuracy and precision of assay across the calibration ranges were evaluated on a single day and also on three different days. For each matrix, six replicates of QC samples were prepared in the corresponding matrix at four concentrations levels (0.2, 0.6, 7.5, and 100 ng/ml for the calibration curve of 0.2-125 ng/ml in human plasma; 32, 96, 1,300, and 17,000 ng/ml for the curve of 32-20,000 ng/ml in human plasma; 0.2, 0.6, 3, and 18 ng/ml for the curve of 0.2-20 ng/ml in human CSF; 2, 4, 16, and 32 ng/g for the curve of 2-40 ng/g in brain tissue). They were analyzed and quantified by using a freshly prepared calibration curve each day. The precision and accuracy of the measurement were determined for each concentration level and were expressed as the percentage coefficient of variance (%CV) and the mean calculated concentration as percentage of the nominal concentration, respectively. The LLOQ was the lowest silmitasertib concentration in the calibration curve that can be quantified with sufficient accuracy and precision (%Nominal between 80 and 120%, precision less than 20%, and a chromatographic signal/noise (S/N) ratio greater than 5). The LOD was defined as the lowest silmitasertib concentration in the matrix with S/N at least 3 so that the chromatographic peak of silmitasertib can be reliably differentiated from the background noise.
2.7.3. Selectivity, matrix effects, and recovery
A selectivity study was performed by analysis of LLOQ and blank samples in six different lots of human plasma, pooled human CSF, and brain homogenate to confirm the absence of interfering peaks in blank matrices at or close to the retention time of silmitasertib or ISTD. Matrix effects, defined as the degree to which matrix components affect the signal of the analyte of interest by either suppression or enhancement, were determined by the ratio of the peak area of the post extraction spiked samples to that of the solvent spiked samples containing the same silmitasertib concentration. Recovery was determined by analysis of the pre-extraction and the post-extraction spiked samples, both of which were spiked with same amount of silmitasertib. Extraction efficiency of silmitasertib from each matrix were determined at two concentration levels.
2.7.4. Stability
The stability of silmitasertib in human plasma and human CSF and brain homogenate was investigated under different handling and storage conditions. Shortterm stability at room temperature and 4°C storage (up to 24h before extraction), long-term stability at −80°C (up to 7 days or longer), three cycles of freeze-thaw stability was assessed by analyzing three replicates of QC samples in the matrix at two concentration levels. The extract stability was evaluated by reinjection of the extracted samples which were stored at 4°C in the autosampler after analysis (up to 24 h or longer). Silmitasertib was considered to be stable in the matrices or extracts if the silmitasertib concentration measured after storage was within 85-115% of the initial value. Impact of the time interval from human blood sampling to plasma separation on silmitasertib concentration in plasma were also tested. Pools of human whole blood spiked with silmitasertib at 0.6 ng/ml and 100 ng/ml were prepared separately and stored at 4°C. At time 0 (immediately after mixing), 1h, and 24h, 0.5 ml whole blood from each pool was transferred to a centrifuge tube and centrifuged at 12,000 rpm for 1 min. The plasma was separated and analyzed for silmitasertib concentration.
The stability of master stock solution and working solution was evaluated by analyzing the old and fresh solutions diluted to a concentration appropriate for injection into the LC-MS/MS system.
2.8. Method application to patient pharmacokinetic plasma samples
Serial plasma samples for pharmacokinetic studies of silmitasertib were obtained from the first patient enrolled in a phase II pediatric clinical trial for patients with recurrent SHH medulloblastoma (PBTC053; NCT03904862). The patient received silmitasertib orally at a dose of 1000 mg twice a day for 28-day cycles. Pharmacokinetic samples were collected after a single-dose on day 1 and after repeated doses at steady-state on day 28 of the first cycle of therapy at the following times: pre-dose, 0.5, 1, 2, 4, 6, 8 (±1), and 24 (±4) hours post-dose. The second dose of silmitasertib on days 1 and 28 were held so that the 24-hour time point could be obtained prior to the administration of the next dose.
At each time-point, 1.0 ml of whole blood was collected in BD Vacutainer K2-EDTA tubes (BD, Franklin Lakes, NJ, USA), inverted several times to mix, immediately aliquoted to microcentrifuge tubes, and centrifuged at 10,000 rpm for two minutes. The plasma supernatants were stored at −80°C within one hour of collection. After bioanalysis, silmitasertib concentration-time data were analyzed using a compartmental pharmacokinetic approach with PKanalix (version 2019R1 Antony, France: Lixoft SAS, 2019. http://lixoft.com/products/PKanalix/). The pharmacokinetic parameters of interest (e.g., maximum concentration (Cmax), time of maximum concentration (Tmax), apparent oral clearance, and area under the concentration curve (AUC0-24h)) were derived, and the model predictions were visually compared with the observed data.
3. Results and discussion
3.1. Mass spectrometric and chromatography conditions
Appropriate MRM transitions for the analyte and ISTD are essential for the sensitivity and specificity of assay. The full MS scan shows the prominent molecular ion [M+H]+ of 350.2 for silmitasertib and 316.2 for ISTD. They were chosen for fragmentation and the corresponding product ion spectra are shown in Fig. 2. Due to the high similarity of their chemical structures, silmitasertib and ISTD show very similar product ion spectra. The predominant product ion with m/z of 223.2 was proposed as the loss of 3-chlorophenylamino group in silmitasertib and phenylamino group in ISTD. The MRM transitions of m/z 350.2→223.2 and m/z 316.2→223.2 were selected for quantification of silmitasertib and ISTD respectively. Compound parameters such as DP, CE and CXP were optimized manually for both MRM transitions to achieve the best sensitivity. The electrospray ionization efficiency is associated with ESI source parameters including IS, TEM, GS1, and GS2. These source parameters were fine-tuned to increase the signal of analyte and maintain low background noise, while the HPLC mobile phases and elution gradient were determined.
Fig.2.
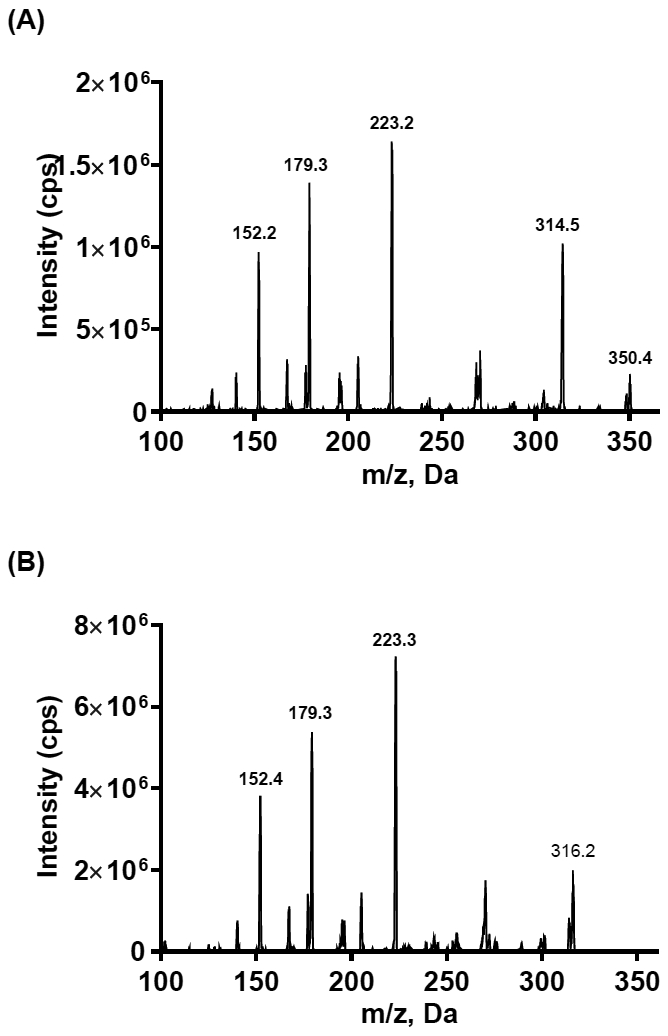
Product ion scans for (A) silmitasertib and (B) ISTD, CX-4786.
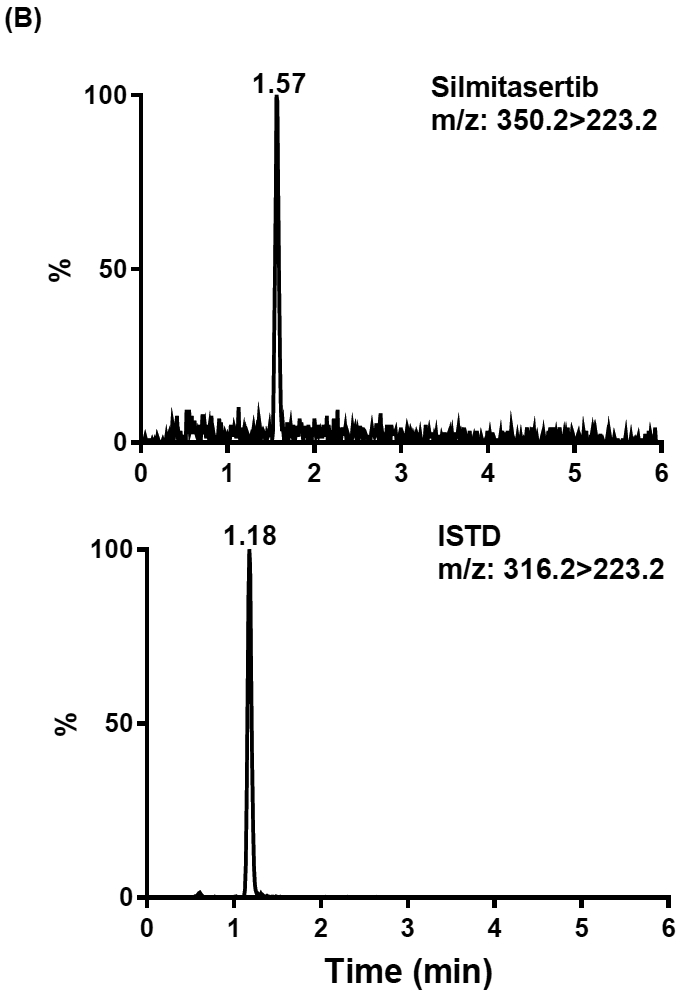
Reverse phase chromatographic separation was achieved on Synergi™ hydro-RP column (4 μm, 75 × 2.0 mm) using gradient elution with 5 mM ammonium formate aqueous solution as solvent A and 0.1% formic acid in acetonitrile as solvent B. In order to obtain the good peak shape and optimal retention time and good separation from matrix components for both analyte and ISTD, both gradient and isocratic elution were tested initially. A carryover was noticed after injection of a high silmitasertib concentration. The extent of carryover was related to the initial ratio of mobile phases used for elution. When gradient elution started with ≥50% of organic mobile phase, no carryover was observed. Therefore, the gradient starting with 50% of mobile phase B was used for the analysis of human plasma and human CSF. Under the optimized HPLC conditions, the retention times for silmitasertib and ISTD were 1.57 and 1.18 min respectively, while the total run time for one sample analysis was 6.0 min. In view of more complicated matrix components in brain tissue homogenate, the HPLC gradient starting with 30% of mobile phase B was employed to achieve the better separation from matrix components. Due to the low calibration range (2-40 ng/g) of brain tissue, no carryover was observed under this elution condition. The related retention times for silmitasertib and ISTD were 2.07 and 1.82 min respectively. Silmitasertib with a chlorine substituent is more polar than the ISTD and has a slightly stronger retention and longer retention time on this Synergi™ hydro-RP column.
3.2. Sample preparation
In order to achieve high sample throughput, a SPE 96-well plate was used for sample extraction. The first step was the selection of an appropriate sorbent for use in the development of an efficient extraction procedure. Based on the characteristics of the chemical structure and the functional group of silmitasertib, three type of SPE plates were tested including Oasis® HLB, MAX, and MCX.
Oasis® HLB packing has a reversed-phase polymeric sorbent, which can be applied for neutral, acidic, and basic analytes. MAX is a reversed-phase and strong anion-exchange plate. The functional group quaternary amine of its sorbent makes it selective for the retention of acidic analytes at the basic condition. MCX has a reversed-phase and strong-cation exchange sorbent, thus is selective for basic analytes due to its sulfonic group. Extraction with reverse phase HLB plate yielded about 10-27% recovery of silmitasertib from human plasma. Tuning the sample pH to increase the retention of silmitasertib on HLB sorbent in combination with various elution conditions failed to improve the recovery. It seemed that the MAX plate would afford better recovery of silmitasertib since the carboxylic group of silmitasertib can be deprotonated at the basic condition which would make this compound selectively retained by MAX sorbent. However, extraction efficiency of the MAX SPE plate for silmitasertib was still low with an average recovery of 28%. Dramatically increased recovery (≥ 77.4%) of silmitasertib from human plasma was achieved by using the MCX in combination with an optimized elution. To ensure the consistent and high recovery, 4% phosphoric acid was added to plasma samples before extraction to disrupt the protein-drug interaction and also to protonate the analyte. In some published methods, 0.1N hydrochloric acid has been used for sample pre-treatment for the cation-exchange SPE. In our case, using 0.1N hydrochloric acid instead of 4% phosphoric acid significantly decreased the extraction efficiency of MCX plate. The washing clean-up consisted of two steps with 200 μl of 2% FA in H2O and then 200 μl MeOH, which is demonstrated to be effective by the determined matrix factors for both silmitasertib and ISTD. Finally, analyte and ISTD were eluted from the MCX plate twice with 100 μl MeOH containing 5% NH4OH to disrupt the ionic interaction.
The same sample extraction procedure was also employed to extract silmitasertib from human CSF successfully. The mouse or primate brain tissue homogenate was extracted similarly except an additional extraction step before solid phase extraction. After the brain tissue was homogenized in human plasma and the supernatant was separated from the tissue residue, the tissue residue was further extracted by using methanol. The supernatant and methanol extract were combined together and subjected to solid phase extraction. Although the loaded samples contained a high percentage of methanol, no significant effects on binding of silmitasertib on the MCX sorbent was observed.
3.3. Method validation
3.3.1. Linearity
In order to analyze human plasma samples with silmitasertib concentration in a broad range, two calibration curves with range of 0.2-125 ng/ml and 32-20,000 ng/ml were established for human plasma and validated for linearity. Both curves were linear with correlation coefficients (R2) >0.99. The other two calibration curves (human CSF: 0.2-20 ng/ml and mouse brain homogenate: 2-40 ng/g) also showed linearity over the concentration range. The parameters for all calibration curves were summarized in Table 1. Initially, it was attempted to establish a calibration curve in human brain homogenate with an aim to measure silmitasertib in human brain tumor tissue obtained from patients undergoing neurosurgical procedures. However, the number of patients studied was anticipated to be few and the samples were rare, so we opted to utilize an alternative approach to develop and validate our brain tissue method.
Table 1.
Parameters for calibration curves in different matrices
| Linear range (ng/ml) |
Matrix | Mean slope ± SD | Mean intercept ± SD | R2 ± SD |
|---|---|---|---|---|
| 0.2-125 | Human plasma | 0.0936 ± 0.01 | 0.000903 ± 0.003 | 0.9987± 0.0016 |
| 32-20,000 | Human plasma | 0.00128 ± 0.00007 | 0.00218 ± 0.002 | 0.9976 ± 0.0003 |
| 0.2-20 | Human CSF | 0.111 ± 0.007 | 0.000334 ± 0.001 | 0.9988 ± 0.0007 |
| 2-40a | Mouse brain | 0.0837 ± 0.008 | 0.00128 ± 0.001 | 0.9956 ± 0.0045 |
The unit of concentration is ng/g.
3.3.2. Precision and accuracy and sensitivity
The intra-and inter-day precision and accuracy were evaluated for four concentration levels in human plasma, human CSF, and mouse brain homogenate. The results are depicted in Table 2. For both calibration curves of human plasma, the precision (%V) is ≤ 5.4 for within-run and ≤ 8.2 for between runs, whereas accuracy (%Nominal) ranged from 89.2 to 100.9 for within-run and from 91.1 to 102.4 for between runs. For the other two matrices (human CSF and mouse brain homogenate), this assay also meets the acceptance criteria for precision and accuracy. To explore if the mouse brain tissue is a suitable surrogate matrix for human brain tissue, QC samples at 4 and 32 ng/g were prepared in primate brain homogenate as the closest substitute for human brain homogenate and quantified by using the calibration curve (2-40 ng/g) prepared in mouse brain homogenate. The within-and between-run precisions for the analysis of primate brain homogenate using the calibration curve of mouse brain homogenate were ≤5.1, and the accuracies were ranged from 98.7 to 108.0%. Based on this result, it is promising that mouse brain tissue can be used as a surrogate matrix for the quantitation of silmitasertib in human brain tissue. The LLOQ for silmitasertib in human plasma and human CSF by this assay was determined to be 0.2 ng/ml and in mouse brain 2 ng/g with S/N all greater than 12. The LOD was 0.05 ng/ml in human plasma and human CSF and 0.5 ng/g in mouse brain with S/N all greater than 3.8.
Table 2.
Intra-and inter-day precision and accuracy for QC samples of silmitasertib in various matrices (Intra-day: n=6; Inter-day: n=18)
| Matrix | Nominal a(ng/ml) |
Mean conc. a(ng/ml) |
Within-day (n= 6) |
Mean conc. a(ng/ml) |
Between-day (n= 18) |
||
|---|---|---|---|---|---|---|---|
| %CV | %Nominal | %CV | %Nominal | ||||
| Human plasma (0.2-125 ng/ml) | 0.2 | 0.199 | 4.3 | 99.4 | 0.204 | 6.3 | 101.9 |
| 0.6 | 0.603 | 5.4 | 100.5 | 0.600 | 4.6 | 99.9 | |
| 7.5 | 7.57 | 2.9 | 100.9 | 7.32 | 3.3 | 97.6 | |
| 100 | 94.6 | 2.5 | 94.6 | 93.6 | 2.1 | 93.6 | |
| Human plasma (32-20,000 ng/ml) | 32 | 31.66 | 2.4 | 98.9 | 32.76 | 8.2 | 102.4 |
| 96 | 91.93 | 2.8 | 95.8 | 95.28 | 4.1 | 99.2 | |
| 1300 | 1260.4 | 2.5 | 97.0 | 1318.8 | 5.1 | 101.4 | |
| 17,000 | 15,168.6 | 1.9 | 89.2 | 15,482.8 | 3.0 | 91.1 | |
| Human CSF | 0.2 | 0.213 | 1.9 | 106.3 | 0.210 | 5.2 | 107.0 |
| 0.6 | 0.590 | 4.4 | 98.3 | 0.610 | 5.7 | 101.7 | |
| 3 | 2.890 | 3.5 | 96.5 | 2.93 | 4.8 | 97.5 | |
| 18 | 17.55 | 4.7 | 97.5 | 17.34 | 4.5 | 96.3 | |
| Mouse brain | 2 | 1.73 | 5.0 | 86.4 | 1.82 | 10.7 | 91.0 |
| 4 | 3.58 | 5.3 | 89.4 | 3.84 | 7.2 | 96.1 | |
| 16 | 15.01 | 3.1 | 93.8 | 15.92 | 6.3 | 99.5 | |
| 32 | 28.70 | 1.7 | 89.7 | 31.34 | 9.2 | 97.9 | |
| bPrimate brain | 4 | 4.32 | 3.2 | 108.0 | 4.29 | 4.4 | 107.2 |
| 32 | 31.59 | 2.3 | 98.7 | 32.42 | 5.1 | 101.3 | |
The unit of concentration is ng/g for QC samples in mouse brain and primate brain
QC samples for primate brain were analyzed by using the calibration curve of mouse brain homogenate.
3.3.3. Selectivity, matrix effects, and recovery
The selectivity of assay was evaluated by analyzing blank matrices and the LLOQ samples (i.e., 0.2 ng/ml). Depicted in Fig. 3 are a representative chromatogram of blank human plasma and LLOQ sample and 30 min plasma sample from a treated patient. Blank human plasma in both silmitasertib and ISTD scans showed low background noise. Some small peaks from blank human plasma eluted less than 1 min, however no interfering peaks co-eluted with silmitasertib or ISTD in blank human plasma. Silmitasertib and ISTD in the patient sample had the same retention time as in the spiked human plasma. Additionally, no interference was observed in the other matrices such as blank human CSF, mouse brain homogenate, and primate brain homogenate.
Fig.3.
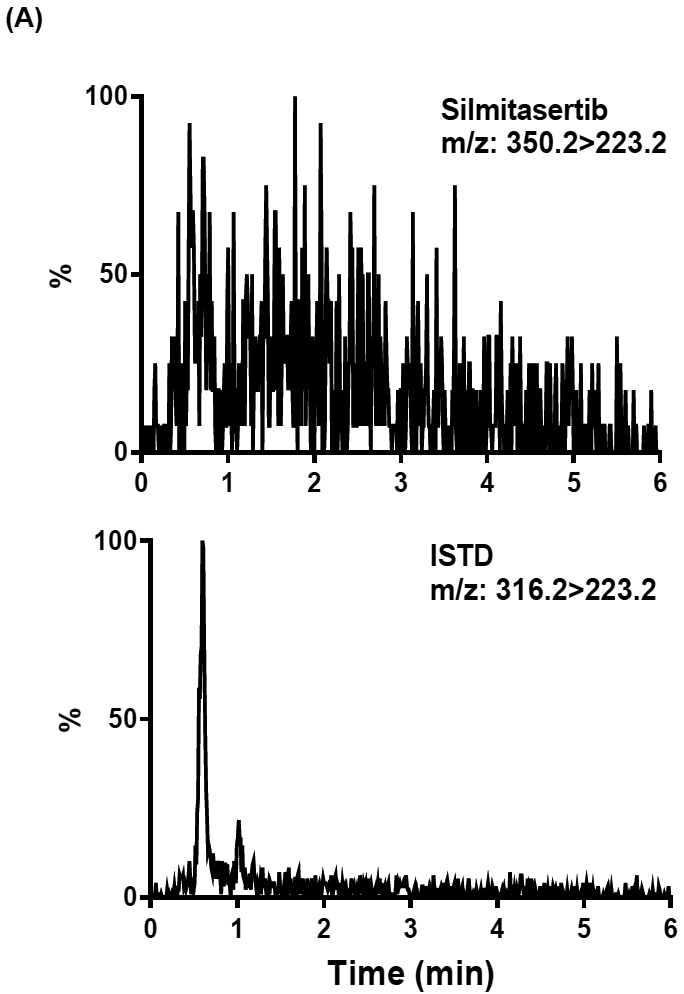
Representative extracted ion chromatograms of (A) human blank plasma, (B) silmitasertib spiked human plasma sample at LLOQ (0.2 ng/ml) with ISTD added, (C) 30 min plasma sample from a treated patient with ISTD added
The matrix effects and extraction efficiency from three matrices including human plasma, human CSF, and mouse brain homogenate were determined. No matrix effects from human plasma and CSF were observed for both silmitasertib and ISTD (see Table 3). The average recovery of silmitasertib is 84.2% at 0.6 ng/ml and 77.4% at 100 ng/ml from human plasma and nearly 100% at 0.6 and 18 ng/ml from human CSF, which indicates that within the calibration range the recovery of silmitasertib is irrespective of its concentration. The similar recovery rate was also observed for ISTD in human plasma and CSF. The absence of matrix effects and consistent extraction recovery for both silmitasertib and ISTD indicated that MCX SPE was an efficient way for the extraction of both matrices. The minor matrix effects for silmitasertib and ISTD were observed from mouse brain homogenate. The average matrix factor was 0.76 for silmitasertib and 1.12 for ISTD. The average recovery of silmitasertib from mouse brain homogenate was 70.7%, while the recovery of ISTD was 89.5%. It seems that in mouse brain homogenate ISTD as a structural analog behaved different from silmitasertib to some extent in terms of matrix effects and recovery efficiency. However, it did not affect the linearity of the calibration curve, or cause any quantitation biases as evidenced by the evaluation of precision and accuracy.
Table 3.
Matrix factors and recovery in various matrices
| Conc. e(ng/ml) |
aMatrix factor | %Recovery | |||||||
|---|---|---|---|---|---|---|---|---|---|
| silmitasertib | bISTD | silmitasertib | bISTD | ||||||
| cMean | CV% | cMean | CV% | dMean | CV% | dMean | CV% | ||
| Human | 0.6 | 1.06 | 5.7 | 0.97 | 2.1 | 84.2 | 4.2 | 87.6 | 5.3 |
| plasma | 100 | 1.06 | 1.9 | 1.00 | 3.0 | 77.4 | 2.0 | 79.4 | 3.0 |
| Human | 0.6 | 1.00 | 5.4 | 1.01 | 3.8 | 103.4 | 2.9 | 105.9 | 1.8 |
| CSF | 18 | 1.00 | 3.3 | 0.99 | 3.2 | 106.2 | 1.5 | 108.5 | 3.5 |
| Mouse brain | 4 | 0.70 | 8.3 | 1.12 | 9.8 | 74.1 | 6.1 | 83.8 | 3.5 |
| 32 | 0.83 | 3.4 | 1.12 | 1.1 | 67.4 | 2.4 | 95.3 | 4.7 | |
Matrix factor (MF) and recovery were evaluated for silmitasertib at two concentration levels for each matrix
ISTD is at the fixed concentration of 6 ng/ml
Mean MF of six lots of human plasma and CSF
Mean recovery from one lot of human plasma and CSF in triplicate
For mouse brain, the concentration unit is ng/g.
3.3.4. Stability
As depicted in Table 4, silmitasertib was stable up to 24 h in all the matrices at 4°C and room temperature. When stored at −80°C, silmitasertib was stable up to 185 days in human plasma, 7 days in human CSF, and 27 days in primate brain homogenate. Silmitasertib is also stable up to 3 cycles of freeze-thaw in human plasma and human CSF. The sample extracts of different matrices were all stable up to 24 h or even a longer time at 4°C in the autosampler, which makes sample reinjection feasible in case of instrument failure. The different time interval from human blood sampling to plasma separation did not show significant effects on the silmitasertib concentration in human plasma. At time 0, the measured plasma concentration was comparable to the spiked whole blood concentration for both levels. The plasma concentration remained almost unchanged after 1h at 4°C. After 24h at 4°C, it decreased by 12.2% at 0.6 ng/ml and 11.4% at 100 ng/ml, however it was still within 85-115% of the initial value. It was suggested that under the routine sampling procedure the impact of time interval (i.e., immediately till 24h) from blood sampling to plasma separation on the plasma concentration can be negligible.
Table 4.
Stability of silmitasertib in different handling and storage condition
| Matrix | Storage condition | LQC | HQC | ||
|---|---|---|---|---|---|
|
bMean (%CV) |
%change |
bMean (%CV) |
%change | ||
| Human plasma | 4°C, 24 h | 0.574 (5.0) | 2.3 | 94.2 (1.4) | −1.5 |
| R.T., 24 h | 0.582 (2.2) | −5.4 | 96.5 (4.1) | 0.2 | |
| −80°C, 185 days | 0.634 (2.3) | 5.9 | 96.2 (1.4) | 1.6 | |
| 3 aF/T cycles | 0.610 (4.4) | 0.0 | 92.2 (1.0) | −2.4 | |
| cExtracts, 4°C, 72 h | 0.569 (7.6) | −7.8 | 89.0 (5.4) | −6.1 | |
| Human whole blood | 4°C, 0h | 0.550 (3.4) | bN.D. | 90.7 (0.5) | cN.D. |
| 4°C, 1h | 0.547 (3.0) | −0.5 | 87.8 (1.4) | −3.2 | |
| 4°C, 24h | 0.483 (1.8) | −12.2 | 80.4 (5.5) | −11.4 | |
| Human CSF | 4°C, 24 h | 0.604 (0.9) | 2.5 | 18.5 (4.6) | 12.1 |
| R.T., 24 h | 0.641 (2.8) | 10.7 | 18.6 (2.2) | 11.4 | |
| −80°C, 7 days | 0.643 (5.7) | 11.2 | 18.8 (2.8) | 6.2 | |
| 3 F/T cycles | 0.593 (3.2) | −3.1 | 17.1 (2.3) | −2.8 | |
| cExtracts, 4°C, 48 h | 0.603 (4.8) | −7.5 | 18.0 (1.7) | −0.6 | |
| Mouse brain homogenate | 4°C, 24 h | 3.62 (10.6) | −9.0 | 32.2 (2.8) | 5.6 |
| R.T., 24 h | 3.84 (3.1) | −4.3 | 32.5 (5.9) | 1.4 | |
| cExtracts, 4°C, 24 h | 4.0 (6.1) | 3.4 | 32.1 (3.4) | 6.3 | |
| Primate brain homogenate | 4°C, 24 h | 4.16 (11.1) | −0.9 | 35.9 (3.3) | 6.1 |
| R.T., 24 h | 3.89 (8.0) | −5.4 | 35.3 (3.7) | 2.4 | |
| cExtracts, 4°C, 24 h | 3.84 (3.2) | −4.6 | 34.5 (2.7) | 2.3 | |
| −80°C, 27 days | 4.07 (5.6) | 5.5 | 33.2 (1.4) | 5.8 | |
Stability of silmitasertib in various matrices was evaluated at two concentration levels (LQC and HQC); the results are presented as mean concentration (% CV) of triplicate at the time of stability and average change of concentration with respective to the concentration at the time 0
F/T free-thaw cycles
Not determined
The unit of concentration is ng/g for samples in mouse brain homogenate and primate brain homogenate, while it is ng/ml in the other matrices
Sample extracts stored in an autosampler at 4°C for 24 hours.
The stability of silmitasertib master stock solution and working solution were evaluated (data not shown). Silmitasertib master stock solution and working solution was stable at −80°C up to 236 and 226 days respectively.
3.4. Application of method to clinical sample analysis
A total of 16 silmitasertib plasma samples were collected after single dose and at steady state from the first patient enrolled in the phase II component of the pediatric trial. Silmitasertib plasma concentrations were determined with our validated LC-MS/MS method, and analyzed using a compartmental approach. Silmitasertib plasma pharmacokinetics were best characterized by a two-compartment disposition model along with transit compartments to describe the absorption process. Fig. 4 depicts the observed and model-predicted silmitasertib plasma concentration-time profiles after single dose and at steady-state. The AUC0-24h were 9,532 h·ng/mL after single dose and 44,410 h·ng/mL at steady-state, showing an accumulation ratio of ~4.7 after multiple dosing. The quantification of the silmitasertib AUC0-24h in our population of children with medulloblastoma will allow for exploring the potential association between drug plasma exposure and drug-induced antitumor efficacy or toxicity. The characterization of a pharmacokinetic-pharmacodynamic association will help guiding the clinical development of silmitasertib in children with SHH medulloblastoma.
Fig.4.
Silmitasertib plasma concentration-time profile for a pediatric patient after administration of single and repeated oral doses of silmitasertib (1,000 mg twice a day). Observed and model-predicted data after single dose are shown by circles and dotted line, respectively. Observed and model-predicted data after repeated doses are shown by squares and dashed line, respectively.
4. Conclusion
The quantitation of silmitasertib in different matrices by cation-exchange solid phase extraction followed by LC-MS/MS detection was developed and validated for selectivity, linearity, accuracy, precision, matrix effects, recovery, and stability. This method is specific and sensitive with LLOQ as low as 0.2 ng/ml in human plasma and human CSF and 2 ng/g in brain tissue. This method also demonstrated excellent precision and accuracy for different matrices across the respective calibration range. The broad application of the method in various matrices are attributable to the use of selective cation-exchange solid phase extraction and column chromatography that can adequately separate silmitasertib from matrix components from different matrices. This LC-MS/MS method has utilized for measuring silmitasertib in human plasma in the ongoing clinical study. Base on the preliminary study using mouse brain homogenate as surrogate matrix for the quantitation of silmitasertib in primate brain homogenate, we may apply the same method to determine silmitasertib concentration in brain tumor tissue from the treated patients.
Highlights:
A sensitive, specific, and robust LC-MS/MS method for the quantitation of silmitasertib in human plasma, CSF, and brain tissue was developed and validated.
Silmitasertib was extracted by cation-exchange solid phase extraction.
This method demonstrated excellent precision, accuracy, reproducibility for routine analysis, and was utilized for an ongoing clinical study.
Acknowledgements
Research reported in this publication was supported by a Cancer Center Support (CORE) Grant CA 21765, 5UM1CA081457, and the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Conflict of interest
The authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, Rutkowski S, Ellison DW, Malkin D, Taylor MD, Gajjar A, Pfister SM, Medulloblastoma, Nat Rev Dis Primers, 5 (2019) 11. [DOI] [PubMed] [Google Scholar]
- [2].Northcott PA, Dubuc AM, Pfister S, Taylor MD, Molecular subgroups of medulloblastoma, Expert Rev Neurother, 12 (2012) 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Khatua S, Song A, Citla Sridhar D, Mack SC, Childhood Medulloblastoma: Current Therapies, Emerging Molecular Landscape and Newer Therapeutic Insights, Curr Neuropharmacol, 16 (2018) 1045–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Qiao Y, Chen T, Yang H, Chen Y, Lin H, Qu W, Feng F, Liu W, Guo Q, Liu Z, Sun H, Small molecule modulators targeting protein kinase CK1 and CK2, Eur J Med Chem, 181 (2019) 111581. [DOI] [PubMed] [Google Scholar]
- [5].Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O’Brien SE, Bliesath J, Omori M, Huser N, Ho C, Proffitt C, Schwaebe MK, Ryckman DM, Rice WG, Anderes K, CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy, Cancer Res, 70 (2010) 10288–10298. [DOI] [PubMed] [Google Scholar]
- [6].Marschke RF, Borad MJ, McFarland RW, Alvarez RH, Lim JK, Padgett CS, Von Hoff DD, O’Brien SE, Northfelt DW, Findings from the phase I clinical trials of CX-4945, an orally available inhibitor of CK2. , Journal of Clinical Oncology 29 (2011) 3087–3087. [Google Scholar]
- [7].Chon HJ, Bae KJ, Lee Y, Kim J, The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies, Front Pharmacol, 6 (2015) 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Purzner T, Purzner J, Buckstaff T, Cozza G, Gholamin S, Rusert JM, Hartl TA, Sanders J, Conley N, Ge X, Langan M, Ramaswamy V, Ellis L, Litzenburger U, Bolin S, Theruvath J, Nitta R, Qi L, Li XN, Li G, Taylor MD, Wechsler-Reya RJ, Pinna LA, Cho YJ, Fuller MT, Elias JE, Scott MP, Developmental phosphoproteomics identifies the kinase CK2 as a driver of Hedgehog signaling and a therapeutic target in medulloblastoma, Sci Signal, 11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Guidance for Industry, Bioanalytical Method Validation, in: F. US Department of Health and Human Services, CDER, CVM (Ed.), May 2018. [Google Scholar]