

Abstract
Background and Purpose
No therapy is approved for vascular calcification or calcific uraemic arteriolopathy (calciphylaxis), which increases mortality and morbidity in patients undergoing dialysis. Deposition of hydroxyapatite (HAP) crystals in arterial walls is the common pathophysiologic mechanism. The mechanism of action of SNF472 to reduce HAP deposition in arterial walls was investigated.
Experimental Approach
We examined SNF472 binding features (affinity, release kinetics and antagonism type) for HAP crystals in vitro, inhibition of calcification in excised vascular smooth muscle cells from rats and bone parameters in osteoblasts from dogs and rats.
Key Results
SNF472 bound to HAP with affinity (K D) of 1–10 μM and saturated HAP at 7.6 μM. SNF472 binding was fast (80% within 5 min) and insurmountable. SNF472 inhibited HAP crystal formation from 3.8 μM, with complete inhibition at 30.4 μM. SNF472 chelated free calcium with an EC50 of 539 μM. Chelation of free calcium was imperceptible for SNF472 1–10 μM in physiological calcium concentrations. The lowest concentration tested in vascular smooth muscle cells, 1 μM inhibited calcification by 67%. SNF472 showed no deleterious effects on bone mineralization in dogs or in rat osteoblasts.
Conclusion and Implications
These experiments show that SNF472 binds to HAP and inhibits further HAP crystallization. The EC50 for chelation of free calcium is 50‐fold greater than a maximally effective SNF472 dose, supporting the selectivity of SNF472 for HAP. These findings indicate that SNF472 may have a future role in the treatment of vascular calcification and calcific uraemic arteriolopathy in patients undergoing dialysis.
Keywords: calcific uraemic arteriolopathy, calciphylaxis, inositol hexaphosphate, IP6, peripheral arterial calcification, SNF472, vascular calcification

Abbreviations
- ALP
alkaline phosphatase
- cbfa1
core‐binding factor alpha 1
- CI
confidence interval
- CR
concentration ratio
- DMEM/F12
DMEM/Nutrient Mixture F‐12
- EC50
effective concentration 50% maximum response
- Emax
maximum response
- FAAS
flame atomic absorption spectrometry
- FCS
fetal calf serum
- HAP
hydroxyapatite
- ICP‐OES
inductively coupled plasma optical emission spectrometry
- pA2
antagonist potency
- STS
sodium thiosulfate
- TRAP
tartrate‐resistant acid phosphatase
What is already known
Central and peripheral arterial calcification contribute to increased mortality and morbidity in patients undergoing dialysis.
No therapy is approved for the treatment of vascular calcification or calcific uraemic arteriolopathy (calciphylaxis).
What does this study add
This series of experiments examined the mechanism of action of the calcification inhibitor, SNF472.
SNF472, 1‐10 μM, selectively inhibited calcification by binding to hydroxyapatite crystals without chelating free calcium.
What is the clinical significance
These findings support the clinical investigation of SNF472 for the treatment of vascular calcification.
1. INTRODUCTION
Mortality in patients undergoing dialysis is approximately 8 times higher than in the general population (de Jager et al., 2009) and 2–4 times higher than in patients with other chronic conditions such as diabetes, cancer or cardiovascular diseases (United States Renal Data System, 2018). Patients under 80 years of age who are undergoing dialysis live less than one‐third as long as age‐matched patients not undergoing dialysis (United States Renal Data System, 2018). For example, life expectancy among adults 30–34 years of age is 45.6 years for men and 49.8 years for women in the general population, compared with 14.1 years for men and 12.7 years for women who are undergoing dialysis. Mortality rates are highest in the first year of dialysis (17%) and half of the deaths in patients undergoing dialysis are due to cardiovascular disease (United States Renal Data System, 2018). Conventional therapies for cardiovascular disease such as lipid‐lowering drugs do not reduce mortality or morbidity in patients undergoing dialysis (Wanner et al., 2005).
Cardiovascular calcification of the coronary arteries, heart valves or aorta has been identified as a major contributor to increased mortality and morbidity in patients undergoing dialysis (Mizobuchi, Towler, & Slatopolsky, 2009). Progression of cardiovascular calcification , which predicts increased risk of future cardiovascular events and mortality (Budoff et al., 2010; Noordzij et al., 2011), is more aggressive in patients undergoing dialysis than in non‐dialysis patients (Sarnak et al., 2003). In patients undergoing dialysis, vascular calcification occurs when calcium and phosphate aggregate into hydroxyapatite crystals and deposit into the media layer of blood vessels, leading to medial calcification (Mönckeberg's medial sclerosis) (Lanzer et al., 2014; Yiu, Callaghan, Sultana, & Bandyopadhyay, 2015). Osteogenic mechanisms also play a role. Some cells and especially vascular smooth muscle cells in the vessels undergo changes into bone and cartilage‐like phenotypes and calcify when exposed to elevated calcium and phosphorus (Jono et al., 2000; Yang, Curinga, & Giachelli, 2004).
Calcification of small peripheral arteries can also result in painful, necrotic skin ulcers, known as calciphylaxis or calcific uraemic arteriolopathy, a rare disease predominantly observed in the dialysis population with a prevalence of 1–4% (Angelis, Wong, Myers, & Wong, 1997; Budisavljevic, Cheek, & Ploth, 1996; Nigwekar, Thadhani, & Brandenburg, 2018). One‐year mortality for calcific uraemic arteriolopathy has been reported to be between 45% and 80% and ulcerated lesions are associated with higher mortality than non‐ulcerated lesions (Nigwekar et al., 2018).
There are several exogenously administered compounds that may affect vascular calcification, including bisphosphonates, sodium thiosulfate, vitamin K, phosphate binders and calcimimetics (O'Neill & Lomashvili, 2010). None of these selectively target hydroxyapatite crystal formation and none have been approved for the treatment of cardiovascular calcification or calcific uraemic arteriolopathy. Myo‐inositol hexaphosphate (IP6, phytate), a naturally occurring substance in foods with high fibre content, binds to hydroxyapatite crystals and can thus prevent calcification (Cao, Dong, & Chen, 2011; Chen, Chen, Ma, Cao, & Chen, 2007; Grases et al., 2000; Grases et al., 2001; Grases, Simonet, Prieto, & March, 2001; López‐González et al., 2013; Muñoz, López‐Mesas, & Valiente, 2010; Muñoz & Valiente, 2003). While phytate provides natural protection against cardiovascular calcification related to aging, phytate is not absorbed well from dietary sources and parenteral administration is required to achieve supraphysiologic levels with a potential to treat cardiovascular calcification (Joubert, Ketteler, Salcedo, & Perello, 2016). SNF472, the hexasodium salt of phytate, is being developed as an intravenously administered form of phytate for clinical use. Results from phase 1 clinical studies (Perelló et al., 2018; Salcedo et al., 2019) and phase 2 clinical studies (Brandenburg et al., 2019; Raggi et al., 2020) support the continued investigation of SNF472 in patients receiving dialysis with either cardiovascular calcification or calcific uraemic arteriolopathy. In a previous report, we used in vivo animal models to show that SNF472 inhibited development of cardiovascular calcification by 60–70% in non‐uraemic rats and by 80% in uraemic rats and SNF472 completely inhibited further progression of cardiovascular calcification in rats with established cardiovascular calcification (Ferrer et al., 2018). This report describes a series of in vitro and in vivo experiments conducted to examine the mechanism of action of SNF472.
2. METHODS
For each experiment, we prepared SNF472 using lot DS1 (Azopharma, Miramar, FL, USA) for non‐clinical use. To quantify bound SNF472 to hydroxyapatite, we recovered hydroxyapatite via filtration and dissolved it in 0.08% HNO3 (Sigma Aldrich, St. Louis, MO, USA). We transferred the solution to a chromatographic column containing an AG 1‐X8 (Bio‐Rad, Hercules, CA, USA) anion exchange resin to separate SNF472 from inorganic phosphate (Grases, Perelló, Isern, & Prieto, 2004). We quantified SNF472 using inductively coupled plasma optical emission spectrometry (ICP‐OES; Optima 5300DV spectrometer, Perkin‐Elmer SL, Waltham, MA, USA). Concentrations of SNF472 for each experiment are reported in μM (660 ng·ml−1 = 1 μM). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010) and with the recommendations made by the British Journal of Pharmacology.
2.1. Affinity of SNF472 to hydroxyapatite
To examine affinity of SNF472 for hydroxyapatite, we incubated different hydroxyapatite concentrations (25, 75, 130 and 300 mg) in a final volume of 400 ml each of 0.05‐M Tris buffer (Sigma‐Aldrich, St. Louis, Missouri, USA), pH 7.4, for 8 h at 37°C with constant agitation using a magnetic stirrer in the presence of SNF472 at 0.076, 0.15, 0.76, 1.5, 7.6 and 15 μM. We quantified the amount of SNF472 attached to hydroxyapatite for each combination. We prepared curves for SNF472 bound to hydroxyapatite with a non‐linear fitting program (GraphPad Prism; GraphPad Software, La Jolla, CA, USA). The values were adjusted with a one‐site binding hyperbola analysis to calculate K D (SNF472 concentration that binds to half the hydroxyapatite at equilibrium) and Bmax (maximum adsorption to hydroxyapatite) for each hydroxyapatite concentration, with the following equation:
We performed each experiment for binding of SNF472 to hydroxyapatite in duplicate.
2.2. Release kinetics of SNF472 from hydroxyapatite
To examine release kinetics of SNF472, we incubated hydroxyapatite 130 mg in 400 ml of 0.05‐M Tris buffer, pH 7.4, at 37°C with constant agitation using a magnetic stirrer, with SNF472 at 7.6 μM for 2 h. After this period, we recovered hydroxyapatite by filtration and started a new incubation with the recovered material in fresh 0.05‐M Tris buffer at pH 7.4 for 15 or 30 min, 1, 2, 4 or 8 h and 1, 2, 3 or 7 days. At each time point, we measured the amount of remaining SNF472 bound to hydroxyapatite. We performed each experiment for release kinetics in triplicate.
2.3. SNF472 inhibition of hydroxyapatite crystal formation
To examine inhibition of hydroxyapatite crystal formation, we induced hydroxyapatite crystal formation using physiological calcium (2.5‐mM CaCl2, Sigma‐Aldrich, St. Louis, Missouri, USA) and supraphysiologic phosphate (5.5‐mM Na2HPO4·12H2O, Panreac Quimica, Barcelona, Spain) concentrations in 0.15‐M NaCl (Sigma‐Aldrich, St. Louis, Missouri, USA) at pH 7.4 (Grases et al., 2008). We measured hydroxyapatite crystal formation by turbidimetry in 12‐well plates by reading absorbance at 550 nm every 75 s, with constant agitation in a plate spectrophotometer (PowerWave XS; Biotek Instruments, Winooski, VT, USA) for 4 h. We incubated each solution for 4 h at room temperature with SNF472 concentrations of 0 (control), 0.19, 0.38, 0.76, 1.14, 1.52, 3.79, 7.58, 11.36, 15.15, 30.3, 45.5 and 60.6 μM, adjusted at pH 7.4. For each crystallization curve, two linear equations were obtained: one line representing the basal absorbance with a slope near to 0 and a second line representing the maximum slope of increase in absorbance. Induction time for each sample was calculated as the intersection between these two lines (basal absorbance and maximum slope of increase in absorbance), thus representing the time needed for hydroxyapatite crystals to form in these conditions. The induction time for each SNF472 concentration was measured and compared with the control induction time. We prepared curves for hydroxyapatite with a non‐linear fitting program (GraphPad Prism; GraphPad Software, La Jolla, CA, USA). We performed each experiment for inhibition of hydroxyapatite crystal formation in duplicate.
2.4. SNF472 inhibition of hydroxyapatite crystallization
To examine inhibition of hydroxyapatite crystallization, we induced hydroxyapatite crystal formation using a physiological concentration of phosphate (1‐mM Na2HPO4·12H2O, Panreac Quimica, Barcelona, Spain) and variable concentrations of calcium (1.0‐ to 6.4‐mM CaCl2, Sigma‐Aldrich, St. Louis, Missouri, USA) in adapted synthetic fluid containing 2‐mM MgCl2 (Panreac Quimica, Barcelona, Spain), 8‐mM KCl (Panreac Quimica, Barcelona, Spain), 32‐mM NaHCO3 (Panreac Quimica, Barcelona, Spain), 28‐mM Na2CO3 (Probus, Barcelona, Spain) and 91‐mM NaCl (Sigma‐Aldrich, St. Louis, Missouri, USA) at pH 7.4. Each solution of phosphate and calcium was incubated for 10 min at room temperature with SNF472 concentrations of 0 (control), 0.76, 1.5, 3.8, 7.6, 22.7 or 30.3 μM. Crystallization was assessed by turbidimetry at 550 nm in 24‐well plates in an orbital plate shaker at 300 g. We prepared calcium concentration curves with a non‐linear fitting program (GraphPad Prism; GraphPad Software, La Jolla, CA, USA) and semi‐logarithmic concentration response curves to determine half‐maximal effective concentration (EC50). We determined the concentration ratio (CR) from pEC50 in the presence or absence of SNF472. Antagonist potency (pA2) was defined as the negative logarithm base 10 of the molar concentration of antagonist necessary to double the concentration of agonist needed to elicit the original submaximal response, determined from the x‐axis intercept from a Schild plot with the following equations:
We expressed pA2 as mean with 95% confidence interval (CI) and performed each experiment for inhibition of hydroxyapatite crystallization in duplicate.
2.5. Effect of SNF472 on calcification, apoptosis and gene expression in vascular smooth muscle cells
To examine calcium deposition in vascular smooth muscle cells, we killed Wistar rats (body weight 250–300 g, both sexes) by an overdose of isoflurane and exsanguination. We perfused the thoracic aortas with PBS, excised the aortas, removed the adventitia under a microscope and cut the aortas into rings to produce cell cultures. The control medium was DMEM/Nutrient Mixture F‐12 (DMEM/F12) with 10% fetal calf serum (FCS). We prepared fresh aqueous stock solutions of CaCl2 and Na2HPO4 filtered on a 0.22‐μM sterile filter, added calcium (CaCl2 at 3 mM) and phosphate (Na2HPO4 at 3 mM) to generate Ca3(PO4)2 to initiate calcification and then added nothing, SNF472 (1, 10, 30, or 100 μM) or sodium thiosulfate (sodium thiosulfate; 25, 50, or 100 mM; Sigma‐Aldrich, St. Louis, Missouri, USA). After 3, 5, 7 and 10 days, we assayed cell cultures for calcium concentration. After 3 and 5 days, we examined cell cultures for apoptosis with TUNEL staining using a cell death detection kit (Roche #11684795910) according to the manufacturer's instructions. Apoptosis was analysed after 3 and 5 days of treatment by TUNEL staining (POD Cell Death Detection Kit; Roche, Basel, Switzerland). Cells were counterstained with DAPI (10 μg·ml−1; Roche, Basel, Switzerland). Images were taken on a KEYENCE BZ Analyzer microscope. Aortas of rats were harvested in accordance with the German law on animal welfare (Tierschutzgesetz §4 (1a)) organ harvest for scientific purpose (Institute of Laboratory Animal Science, RWTH Aachen, Germany).
We used real‐time PCR to measure up‐regulation of gene expression indicating a switch from contractile cells to osteoblast‐like cells. We isolated RNA by the Qiagen RNeasy Kit, following the manufacturers' instructions. We synthesized cDNA by the RT core kit (RT‐RTCK‐05; Eurogentec, Liège, Belgium). For qPCR, we used the qPCR Core Kit for SYBR Green I (RT SN10‐05; Eurogentec, Liège, Belgium) and measured in duplicates on the 7500 Real‐Time PCR System (Applied Biosystems®, Foster City, CA, USA). The primers we used were alpha smooth muscle actin (SMA): sense 5′‐CCCGCTCTGTCTCTAGCAC‐3′ antisense 5′‐CACACGAGTAACAAATCAAAGC‐3′; GAPDH: sense 5′‐ACAGATGGTGAAGGTCGGTA‐3′ antisense 5′‐AGAAGGCAGCCCTGGTAACC‐3′; alkaline phosphatase (ALP): sense 5′‐CCTTGAAAAATGCCCTGAAA‐3′ antisense 5′‐CTTGGAGAGAGCCACAAAGG‐3′; core‐binding factor alpha 1 (cbfa1): sense 5′‐AGACACAGAGCCTGTGGG‐3′ antisense 5′‐CTTGGAGAGAGCCACAAAGG‐3′.
We performed each vascular smooth muscle cell experiment in six replicates. We performed statistical analyses using a one‐way ANOVA with Bartlett's method. We analysed differences between groups with Tukey's multiple comparison method and performed D'Agostino and Pearson normality tests to check for Gaussian distribution. We set the level for significance at P < 0.05.
2.6. Binding of SNF472 to free calcium
To examine chelation of free calcium, we exposed a physiological calcium (2.5‐mM CaCl2, Sigma‐Aldrich, St. Louis, Missouri, USA) concentration in 0.15‐M NaCl (Sigma‐Aldrich, St. Louis, Missouri, USA) solution at pH 7.4 to increasing concentrations of SNF472 (0.15 to 75.8 μM) or sodium thiosulfate (0.63 to 575 μM; Sigma‐Aldrich, St. Louis, Missouri, USA). We measured ionized calcium with a selective calcium electrode (DX240‐Ca; Mettler Toledo, Columbus, OH, USA) and a potentiometer (micropH 2002; Crisson, Barcelona, Spain), quantified using a calibration curve. We calculated chelated calcium by subtracting the concentration of ionized calcium in the presence of chelator (SNF472 or sodium thiosulfate) from the experimental concentration of ionized calcium without chelator. Ionized calcium curves were analysed by using a non‐linear fitting program (GraphPad Prism; GraphPad Software, La Jolla, CA, USA). We used a semi‐logarithmic concentration–response curve to determine EC50, hillslope and maximum response (Emax) with the following equation:
where “Bottom” was the baseline, “Top” was the Emax and “hillslope” was a measure of the steepness of the curve. We performed each experiment for binding of SNF472 to free calcium in duplicate.
2.7. Effects of SNF472 on bone
In a repeated‐dose toxicology study, we administered SNF472 (0 or 25 mg·kg−1) using 15 min intravenous infusion three times weekly for 9 months in two groups of eight (4 male and 4 female) healthy Beagle dogs (Beijing Marshall Biotechnology, Co. Ltd., Beijing, China). Previous toxicology studies (14‐day, 28‐day and 3‐month toxicology studies in dogs) examined the effects of 3, 10 or 25 mg·kg−1, which cover the expected therapeutic concentrations in humans. In this 9 month study, results from the control group and the highest dose tested were sent for histomorphometry evaluation. If an effect on bone had been detected at the highest dose of 25 mg·kg−1, then we would have tested the lower doses in sequence (10 mg·kg−1 then 3 mg·kg−1). We analysed femur samples by histomorphometry for trabecular parameters, including thickness, separation and number and cortical parameters including cortical area fraction and thickness. We performed tartrate‐resistant acid phosphatase staining to highlight osteoclasts and evaluated mineralized tissue on slides with Von Kossa staining. Pharmaron's Institutional Animal Care and Use Committee reviewed the protocol and approved the animal care and use application.
Bone samples were fixed and preserved in 10% neutral buffered formalin for at least 1 week prior to microtomographic analysis. We performed microtomographic analysis using a SkyScan 1076 Desktop X‐ray Microtomograph (Bruker, Kontich, Belgium) with SkyScan, NRecon, DataViewer and CTAn software. For histological analyses, we sectioned samples using a RM 2255 microtome (Leica, Wetzlar, Germany) and analysed them using an Eclipse 80i fluorescence microscope (Nikon, Minato, Tokyo, Japan) and NIS‐Elements D (Nikon, Minato, Tokyo, Japan) and ImageJ software.
We washed bone samples in PBS solution and performed micro‐CT analysis of architectural variables at room temperature using the SkyScan‐1076. We set the X‐ray source to a tube voltage of 100 kV and a tube current of 100 μA, using an isotropic voxel size of 18 μm. We obtained acquisition files with a rotation of 0.50°, two frames averaging and using an aluminium filter (1 mm). After scanning, we transferred image data to a workstation and reconstructed the full data set for each sample as a series of 2D binarized cross‐sections (reconstruction step), which we then used for the calculations using the SkyScan analysis system.
For the analysis of the trabecular properties of the femurs, we calculated the distance between the epiphysis and the end of femoral trochlea and determined the reference slice to be located at three‐quarters of this distance, proximally. We used 50 transverse axial‐cut slices in both sides of the reference slice for calculations. On the 50 slices, we determined a region of interest including only the trabeculae. For the evaluation of cortical properties, we chose 100 transverse axial‐cut slices on both sides of the midpoint of the bone.
We dehydrated the samples in an ascending alcohol series at room temperature, then embedded samples in a resin specifically developed for mineralized tissues (Technovit 9100, Kulzer, Hanau, Germany). We pre‐permeated specimens before the permeation step at 4°C, embedded in resin at −11°C. We cut 5‐μm sagittal sections (six sections per sample) using a Leica RM2255 sawing microtome (Leica, Wetzlar, Germany), stretched the sections on slides by immersion in an alcohol solution and covered them with polyethylene films. We deplasticized sections and performed the following staining protocols:
tartrate‐resistant acid phosphatase staining procedure: We post‐fixated sections in 4% formalin solution. After washing, we immersed sections in tartrate‐resistant acid phosphatase solution and washed them again. We counterstained with haematoxylin solution and mounted sections in aqueous mounting solution.
Von Kossa staining procedure: We incubated sections in 1% silver nitrate solution under UV light, then washed out unspecific staining by 5% sodium thiosulfate solution. We counterstained sections with Van Gieson picrofuschin stain. After the washing step, we dehydrated sections with ethanol and xylene solutions and mounted them with resin mounting solution.
For each group, we calculated the median values and 95% confidence intervals per dose (total: females + males, n = 8) and per dose and sex (n = 4). We used a statistical test (Kruskall–Wallis followed by Dunn's post hoc test) to compare median values between groups, using PRISM software (GraphPad Prism; GraphPad Software, La Jolla, CA, USA).
2.8. Effects of SNF472 on in vitro osteoblast calcification
Primary rat osteoblasts were cultured according to a previously optimized procedure (Verberckmoes, De Broe, & D'Haese, 2003). In short, we collected the femur, tibia and humerus of healthy 10‐week old Wistar rats and put them into cell culture medium (α‐MEM with 10% fetal bovine serum, 10−8 M dexamethasone, 1% penicillin/streptomycin and 50 mg·L−1 ascorbic acid) at 4°C. We removed the epiphyses and collected bone marrow cells by spurting sterile cell culture medium through the diaphysis with a sterile syringe. The collected cells were cultured in the same culture medium in 75 cm2 flasks at 37°C and 5% CO2. Cell culture medium was replaced three times weekly until confluence was reached. After confluence, cells were trypsinized and plated onto 12‐well plates at a density of 5 to 10 × 104 cells·cm−2. Again, cell culture medium was replaced three times weekly until confluence.
When confluence was reached in the 12‐well plates, we replaced the medium with calcifying medium (same as above +0.59 g·L−1 β‐glycerolphosphate), supplemented with SNF472 at concentrations of 0, 1, 10 and 30 μM. Three times per week, we replaced cell culture medium with the calcifying medium containing SNF472. We stored samples of replaced cell culture medium at −20°C pending further analysis. We measured calcium content of the cell medium with flame atomic absorption spectrometry (FAAS) using methods developed in‐house. When osteoblasts calficied, calcium is removed from the medium and assumed to be incorporated into hydroxyapatite crystals. Hence, the proportion of calcium removed is used as a measure of hydroxyapatite deposition (Verberckmoes et al., 2003). After 2 weeks, we processed cell cultures for Von Kossa and alkaline phosphatase staining. We extracted mRNA using the PureLink RNA Mini Kit (Thermo Fisher) and stored extracts at −80°C until further processing. Each parameter was quantified in at least three wells per condition and obtained from three separate experiments.
Real‐time qPCR was performed using the TaqMan assay with commercially available primers on a QuantStudio 3 system (Applied Biosystems). We analysed the following genes: GAPDH, alkaline phosphatase, bone sialoprotein (BSP), Osterix, RANK ligand (RANKL), sclerostin (SOST), osteoprotegerin (OPG), osteocalcin, osteopontin, Bax and Bcl2. Using the software provided by the manufacturer and the ΔΔCt method, we calculated relative expression levels and corrected for GAPDH.
The study was approved by the Ethical Committee for Animal Testing from the University of Antwerp (approval number 2017‐72). The animals were obtained from Charles River (France).
2.9. Data and analysis
Studies were designed to generate groups of equal size, using randomization and blinded analysis. Statistical analysis was undertaken for studies where each group size was at least n = 5, except for bone parameters in dogs, in which n = 4 after splitting the treatment groups by sex. The declared group size for each experiment or study is the number of independent values and statistical analysis was done using these independent values (i.e. not treating technical replicates as independent values). In multigroup studies with parametric variables, post hoc tests were conducted only if F in ANOVA achieved statistical significance (P < 0.05) and there was no significant variance in homogeneity. The threshold for statistical significance for determining whether groups differed was P < 0.05. We selected all group sizes based on previously published data for similar experiments; we did not conduct sample size calculations for these experiments. No outliers were removed in these experiments. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018).
2.10. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018) and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Affinity of SNF472 for hydroxyapatite
SNF472 binding to hydroxyapatite was concentration‐dependent and was found to be saturated when hydroxyapatite was incubated with SNF472 at concentrations above 7.6 μM for 8 h (Figure 1a). The amount of SNF472 required to bind 50% of hydroxyapatite binding sites was linear with hydroxyapatite concentration, with a K D of 0.45, 1.18, 2.20 and 5.20 μM for 25, 75, 130 and 300 mg hydroxyapatite, respectively (Figure 1b). Calculated values for Bmax were between 6 and 7 μM in all cases. These data show that SNF472 binds hydroxyapatite concentration‐dependently and that binding is saturated at approximately 7 μM.
FIGURE 1.
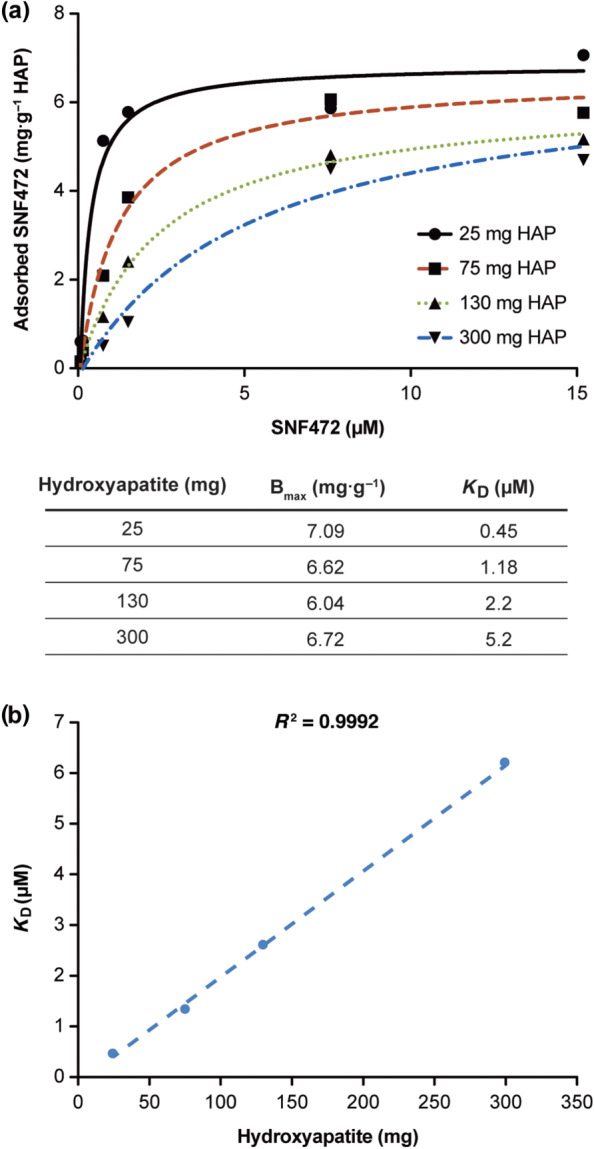
Affinity of SNF472 to hydroxyapatite (HAP) crystals after incubation for 8 h. (a) Adsorption of HAP 25, 75, 130 or 300 mg was saturated at a SNF472 concentration of 7.6 μM. Calculated values for maximum adsorption to HAP (Bmax) were between 6 and 7 μM in all cases. (b) The calculated K D for adsorption was linear for HAP
3.2. Release kinetics of SNF472 from hydroxyapatite
After incubating SNF472 and hydroxyapatite (130 mg) for 2 h, followed by filtration to fresh Tris buffer, the amount of SNF472 still bound to hydroxyapatite was evaluated up to 7 days. No SNF472 release from hydroxyapatite through day 7 (Figure 2) was observed. These data show that SNF472 binds to hydroxyapatite crystals for at least 7 days.
FIGURE 2.
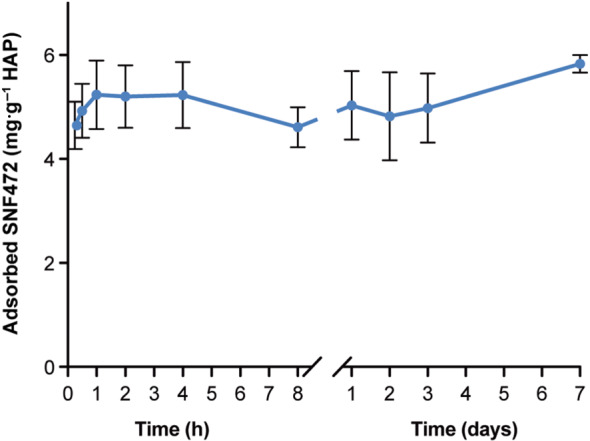
Kinetics of SNF472 adsorption to hydroxyapatite (HAP) crystals after incubation for 2 h in the initial solution, followed by up to 7 days in fresh Tris buffer. Results are mean ± SE
3.3. SNF472 inhibition of hydroxyapatite crystal formation
In control conditions, without SNF472, hydroxyapatite crystals formed after 7.96 min, which was considered the control induction time (Figure 3). SNF472 concentration‐dependently increased the induction time to form hydroxyapatite crystals. SNF472 at 7.6 μM increased hydroxyapatite crystal induction time 2.6‐fold mean of the control, while SNF472 concentrations greater than 15.2 μM totally inhibited hydroxyapatite crystal formation throughout the 4‐h duration of the experiment. These data show that SNF472 inhibits the formation of hydroxyapatite crystals concentration‐dependently.
FIGURE 3.

Effect of SNF472 on in vitro hydroxyapatite (HAP) crystal formation. Induction time for HAP crystals in a solution of 2.5‐mM calcium and 5.5‐mM phosphate at pH 7.4 in the presence of SNF472 at various concentrations. *The final measurement was at 240 min; standard error (SE) was not determined for these values
3.4. SNF472 inhibition of hydroxyapatite crystallization
In a control solution, hydroxyapatite crystals were formed by increasing the calcium concentration from 1.0 to 6.4 mM in a fixed phosphate concentration of 1 mM (Figure 4). The EC50 of calcium required to produce hydroxyapatite crystal formation was 3.57 mM. As the concentration of SNF472 increased, a higher calcium concentration was required to obtain the same effect. The calcium curves in the presence of SNF472 were concentration‐dependently displaced to the right. SNF472 at 7.6 μM almost doubled the calcium EC50 required to obtain hydroxyapatite crystal formation. SNF472 also affected the Emax concentration‐dependently (Figure 4). These data show that SNF472 inhibits hydroxyapatite crystallization concentration‐dependently. Substantial decreases in Emax suggest that binding of SNF472 to hydroxyapatite is insurmountable and when the calcium concentration is increased, SNF472 is not displaced from hydroxyapatite. Although the binding profile was insurmountable, we calculated the pA2 of SNF472 by using a variation of adjustment for competitive and reversible antagonism. The pA2 of SNF472 for in vitro hydroxyapatite crystal formation in synthetic fluid, pH 7.4, with 1‐mM phosphate is 9.29 μM. The Schild plots were linear with slopes around 4–5.
FIGURE 4.
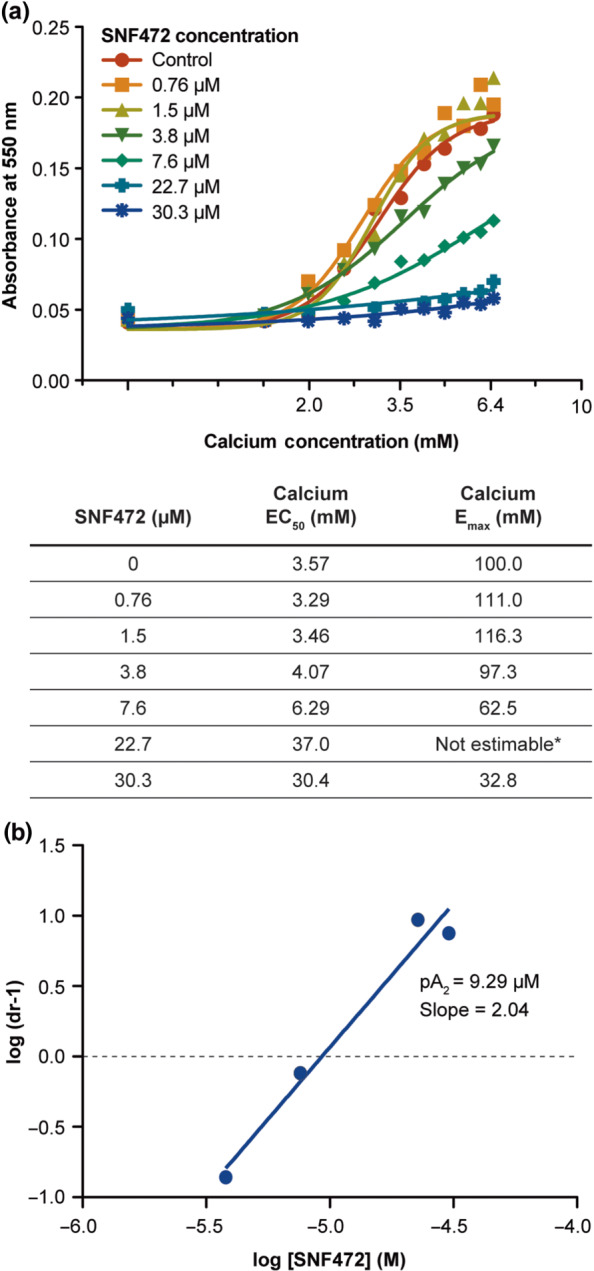
Effect of SNF472 on hydroxyapatite (HAP) crystal formation. (a) Absorbance of 550 nm after 10 min of incubation is shown for a range of calcium concentrations and a range of SNF472 concentrations (or control), with a fixed, physiological phosphate concentration. (b) After plotting the logarithm of (dose ratios [dr] – 1) versus the log of SNF472, pA2 was calculated as the x‐intercept of the fitted regression line. *Due to the asymptotic shape of the curve for SNF472 at 22.7 μM, maximum response (Emax) could not be estimated for this concentration. EC50, half‐maximal effective concentration for HAP crystal formation; Emax, concentration required for maximal HAP crystal formation
3.5. Effect of SNF472 on calcification, apoptosis and gene expression in vascular smooth muscle cells
Using TUNEL staining, no significant increase in apoptosis was seen with exposure to SNF472 for 5 days (Figure 5a). Exposure to sodium thiosulfate at 50 or 100 mM was associated with high levels of apoptosis (77%).
FIGURE 5.
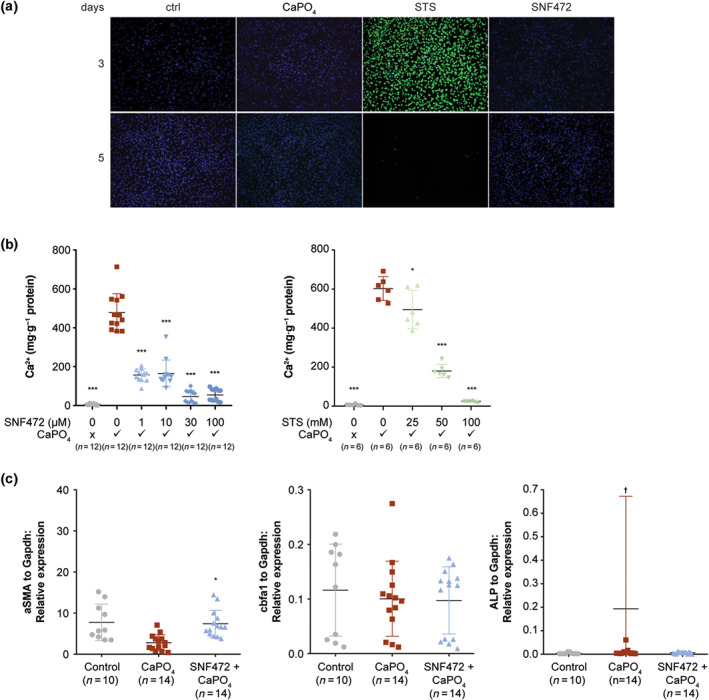
Effect of SNF472 on calcification, apoptosis and gene expression in vascular smooth muscle cells. Cell cultures containing rat aorta sections in DMEM/Nutrient Mixture F‐12 with 10% fetal calf serum were exposed to calcium (CaCl2 at 3 mM) and phosphate (Na2HPO4 at 3 mM) to generate Ca3(PO4)2 to initiate calcification, then exposed to nothing (control [ctrl]), SNF472 (1, 10, 30 or 100 μM), or sodium thiosulfate (STS; 25, 50 or 100 mM). (a) Apoptosis, shown by green staining, of TUNEL‐stained cells after 3 and 5 days. (b) Calcification, shown as the ratio of calcium (Ca2+) to protein, after 5 days. (c) Markers for gene expression, relative to GAPDH, including smooth muscle actin (SMA), core‐binding factor alpha 1 (Cbfa1) and alkaline phosphatase (ALP). Gene expression was not summarized in the STS group, which provided only two pooled samples after apoptosis of STS‐treated cells. *P < 0.05, compared with Ca3(PO4)2 alone. ***P < 0.001, compared with Ca3(PO4)2 alone. †One value at 1.6 is not shown in the plot
After incubation of rat vascular smooth muscle cells with Ca3(PO4)2 to initiate calcification, incubation with SNF472 at 1, 10, 30 or 100 μM for 5 days significantly inhibited calcification (Figure 5b). At the first concentrations tested of 1 μM, SNF472 already inhibited calcification by 67%. Calcification levels with Ca3(PO4)2 and SNF472 at 30 or 100 μM were similar to those for the control, which was not exposed to Ca3(PO4)2. Exposure to sodium thiosulfate at 50 or 100 mM significantly inhibited calcification; exposure to sodium thiosulfate at 25 mM had only a small effect on calcification.
Expression of alpha smooth muscle actin, a marker of muscle phenotype, decreased in vascular smooth muscle cells that were exposed to Ca3(PO4)2 (Figure 5c). SNF472 prevented this down‐regulation. Markers for the osteoblast phenotype, such as core‐binding factor alpha 1 and alkaline phosphatase, were not up‐regulated in calcified vascular smooth muscle cells. Genetic changes were not summarized in the sodium thiosulfate group, which had only two pooled samples for real‐time PCR after apoptosis of a majority of sodium thiosulfate‐treated cells.
These data show that SNF472 significantly inhibits calcium deposition in vascular smooth muscle cells and normalizes genetic expression of alpha smooth muscle actin at concentrations that do not induce cell death. sodium thiosulfate also inhibits calcification in vascular smooth muscle cells, but at concentrations that are associated with substantial cell death.
3.6. Binding of SNF472 to free calcium
A semi‐logarithmic dose–response curve showed a sigmoid profile for chelation of ionized calcium by SNF472 (Figure 6). Statistically significant chelation occurred with SNF472 at 379 μM or greater and was saturated at 3,788 μM, with an EC50 of 539 μM. For sodium thiosulfate, 15.8 mM was required for significant chelation and saturation was attained from 253 mM, with an EC50 of 54.3 mM. These data show that the SNF472 concentrations required for chelation of free calcium in saline solution are approximately 50‐fold greater than the concentrations of SNF472 that maximally inhibit formation and growth of hydroxyapatite crystals in adapted synthetic fluid.
FIGURE 6.
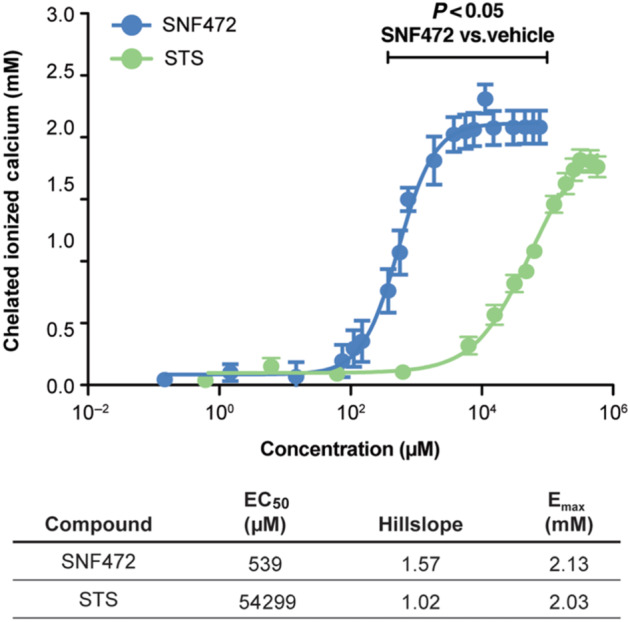
Chelation of ionized calcium by SNF472 and by sodium thiosulfate (STS). We added increasing concentrations of SNF472 or STS to a physiological (2.5 mM) calcium solution in 0.15‐M NaCl at pH 7.4 and recorded ionized calcium levels. Chelation is shown as the concentration of ionized calcium in the absence of chelator minus the observed concentration in the presence of chelator. A semi‐logarithmic concentration–response curve was used to determine EC50, hillslope and maximum response (Emax)
3.7. Effects of SNF472 on bone
To evaluate the potential effects of SNF472 on bone, we exposed healthy dogs to SNF472 by 15‐min intravenous infusion three times weekly for 9 months. Bone histomorphometry and histological analyses after exposure to SNF472 at 25 mg·kg−1 (maximum concentration, 384 μM) showed comparable trabecular and cortical parameters to the control group (Figure 7, Table 1). We did not observe tartrate‐resistant acid phosphatase‐positive cells on the edge of trabeculae in any specimen (Figure 8), suggesting that bone resorption by osteoclasts was not enhanced by SNF472. The mineralized tissue fraction was similar between tissue specimens in the SNF472 and control groups (Figure 9), confirming that SNF472 did not have an adverse effect on bone mineralization in dogs.
FIGURE 7.
Representative images of bone samples from dogs treated with 25 mg·kg−1 intravenous SNF472 for up to 9 months. (a) Trabecular bone samples. (b) Cortical bone samples
TABLE 1.
Trabecular and cortical parameters in bones from dogs treated with 25 mg·kg−1 intravenous SNF472 for up to 9 months
| Control (N = 4 male/4 female) | SNF472 (N = 4 male/4 female) | ||
|---|---|---|---|
| Trabecular parameters | |||
| Bone volume fraction (%) | Female | 18.7 (15.39–23.21) | 19.8 (16.7–22.9) |
| Male | 19.3 (15.9–23.8) | 19.3 (16.5–24.4) | |
| Trabecular thickness (mm) | Female | 0.123 (0.111–0.140) | 0.122 (0.107–0.134) |
| Male | 0.119 (0.107–0.136) | 0.128 (0.118–0.137) | |
| Trabecular separation (mm) | Female | 0.425 (0.397–0.455) | 0.403 (0.362–0.450) |
| Male | 0.390 (0.350–0.453) | 0.416 (0.349–0.486) | |
| Trabecular number (mm−1) | Female | 1.49 (1.34–1.73) | 1.67 (1.48–1.81) |
| Male | 1.68 (1.39–1.87) | 1.57 (1.34–1.88) | |
| Cortical parameters | |||
| Cortical area fraction (%) | Female | 61.9 (55.1–69.9) | 67.0 (57.5–75.5) |
| Male | 60.2 (52.1–64.8) | 62.8 (58.2–67.4) | |
| Cortical thickness (mm) | Female | 1.19 (1.06–1.31) | 1.29 (0.97–1.50) |
| Male | 1.27 (1.11–1.41) | 1.24 (1.04–1.37) |
Note: Results are presented as median (95% confidence interval) per dose and sex (n = 4). A Kruskall–Wallis followed by Dunn's post hoc test was used to compare median values between groups. No significant differences were evident between groups (P > 0.05).
FIGURE 8.
Representative images of tartrate‐resistant acid phosphatase (TRAP) staining in trabecular bone samples from dogs treated with 25 mg·kg−1 intravenous SNF472 for up to 9 months. (a) Control female. (b) Control male. (c) SNF472 female. (d) SNF472 male
FIGURE 9.
Representative images of Von Kossa staining in bone samples from dogs treated with 25 mg·kg−1 intravenous SNF472 for up to 9 months. (a) Control female. (b) Control male. (c) SNF472 female. (d) SNF472 male
3.8. Effects of SNF472 on in vitro osteoblast calcification
Fresh, unused cell culture medium (with all supplements) contained 94.5 mg·L−1 Ca. After 1 day of incubation with the calcifying medium, 74.0 ± 0.97% of the Ca was removed from the medium in control cultures, indicating a rapid onset of calcification. Cultures containing SNF472 showed no statistically significant difference in Ca removal (74.7 ± 3.67%, 74.4 ± 1.57% and 73.1 ± 2.06% from the cultures containing 1, 10 and 30 μM, respectively). From day 5 onward, only 5% to 10% of the Ca was removed from the medium, indicating that no further calcification occurred. No statistically significant differences were found between the different SNF472 concentrations nor compared with vehicle during the 14‐day follow‐up period (Figure 10).
FIGURE 10.

Percentage of calcium removed from the cell culture medium. After confluency, β‐glycerolphosphate and SNF472 were added to osteoblast culture medium and Ca levels were analysed in spent medium
After 2 weeks of incubation with calcifying medium containing SNF472, cultures were used to stain for calcification (Von Kossa) and osteoblast activity (ALP). No differences were noted between the controls and the various SNF472 concentrations used (Figure 11).
FIGURE 11.
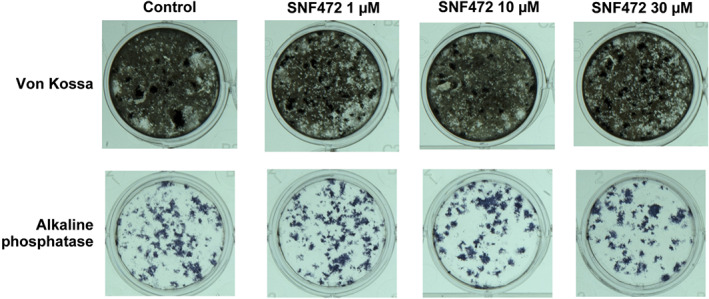
Representative images of Von Kossa staining and alkaline phosphatase staining of rat primary osteoblast cultures after 2 weeks of treatment with β‐glycerolphosphate and SNF472
Using a one‐way ANOVA with Bonferroni correction, SOST, TNAP, Osteocalcin and Bax genes did not show any statistically significant differences between groups (Figure 12). OPG and Bcl2 both showed a similar pattern, an increase versus controls at the lowest dose of SNF472, followed by a steady decline towards control levels for SNF472 10 and 30 μM. Osterix showed an increase for SNF472 10 and 30 μM versus controls. RANKL showed an increase up to the SNF472 10 μM dose versus controls. Osteopontin showed a decrease for SNF472 1 and 10 μM versus control.
FIGURE 12.
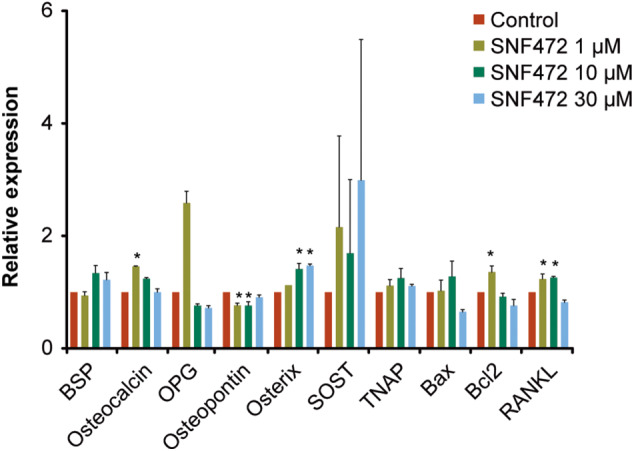
Relative mRNA expression of various osteoblast marker/function genes using real time qPCR, after 2 weeks of treatment with β‐glycerolphosphate and SNF472. BSP, bone sialoprotein; OPG, osteoprotegerin; RANKL, receptor activator for NF‐κB ligand; SOST, sclerostin; TNAP, tissue non‐specific alkaline phosphatase
4. DISCUSSION
Calcification is an active and complex process that involves numerous mechanisms responsible for calcium and phosphate deposition in arterial walls (Karwowski, Naumnik, Szczepanski, & Mysliwiec, 2012). The initiation of this calcification requires the presence of triggers that can vary but share a common pathway:‐ hydroxyapatite crystals build from calcium and phosphate aggregation and the crystals are deposited into the media layer of blood vessels, leading to medial calcification (Mönckeberg's medial sclerosis) (Lanzer et al., 2014; Yiu et al., 2015). In patients undergoing dialysis, vascular smooth muscle cells also have been described as responsible for hydroxyapatite formation in the vessels. These vascular smooth muscle cells can undergo changes into bone and cartilage‐like phenotypes and calcify when exposed to elevated calcium and phosphate by releasing hydroxyapatite into the extracellular space or in vesicles or exosomes containing hydroxyapatite‐like material (Jono et al., 2000; Kapustin et al., 2015; Schurgers et al., 2007; Yang et al., 2004).
In this series of in vitro experiments, we examined the mechanism of action of SNF472 to inhibit hydroxyapatite formation and growth as a new therapeutic target to treat calcification disorders. Calcification is especially relevant for cardiovascular calcification and calcific uraemic arteriolopathy in patients undergoing dialysis and hydroxyapatite formation is the common pathophysiologic mechanism for both.
The experiments included in this report show that SNF472 is able to block the formation and growth of hydroxyapatite crystals and SNF472 inhibits calcification in cells that are induced to calcify. These experiments also show that SNF472 binds to hydroxyapatite in a physicochemical interaction not a membrane‐receptor interaction. Thus, although we applied pharmacological concepts to evaluate this interaction, these should be interpreted with caution.
SNF472 showed high affinity for hydroxyapatite crystals, with a K D ranging from 1 to 10 μM. The amount of SNF472 bound to hydroxyapatite was linear versus hydroxyapatite concentrations in the milligram range, which is the range that was observed in uraemic animal models with administration of adenine and vitamin D to induce vascular calcification (Ferrer et al., 2018). This is the expected behaviour for a physicochemical interaction, without the effect of membrane‐receptor features such as spare receptor effect or cross‐binding to other receptors. Binding of hydroxyapatite was saturated by SNF472 at approximately 7 μM, regardless of the hydroxyapatite concentration, which could mean that hydroxyapatite was always in excess in these preparations, even at the lower concentration of 25 mg and SNF472 was then able to block hydroxyapatite binding sites maximally. In another experiment, we showed that SNF472 was bound to the surface of hydroxyapatite very rapidly (within 5 min) and with a long‐lasting effect (for up to 7 days, the last measurement in the assay). In a previous publication with similar findings, binding of SNF472 to hydroxyapatite occurred quickly, reaching nearly 80% by 1 min and more than 90% by 15 min and adsorption was insurmountable for 3 days (the last measurement reported) (Ferrer et al., 2018). Based on both reports, the inhibitory profile of SNF472 for hydroxyapatite is compatible with fast association to the crystal and slow or even insurmountable dissociation. However, the observed lack of release of SNF472 from the hydroxyapatite crystals might also be related to a situation of equilibrium reached in the assayed conditions. A total of 130 mg of hydroxyapatite contained 0.78 mg of SNF472 in a volume of 400 ml. If SNF472 was totally free, a concentration of approximately 3 μM would be found, which is a concentration above the K D of SNF472 for these conditions and so the equilibrium is displaced towards binding of SNF472 to hydroxyapatite.
In other experiments in this report, SNF472 inhibited both the formation and growth of hydroxyapatite crystals. SNF472 at 7.6 μM delayed time to hydroxyapatite crystal formation by 2.6‐fold and doubled the calcium concentration required to induce crystal formation. On the other hand, SNF472 at 7.6 to 22.7 μM inhibited hydroxyapatite crystallization concentration‐dependently and SNF472 increased up to 10‐fold the concentration of calcium needed to induce hydroxyapatite crystal formation. Moreover, the Emax was significantly decreased, showing a profile of SNF472 compatible with an insurmountable antagonist as calcium was not able to displace SNF472 from its binding site. These findings, together with the results showing binding of SNF472 to hydroxyapatite until the maximum time point evaluated at 7 days, could confirm the insurmountable interaction. These results suggest that SNF472 concentrations greater than 7 μM ensure effective “sealing” of the hydroxyapatite crystal to stop its growth.
We also examined the effects of SNF472 in a cell model for medial calcification induced by high concentrations of calcium and phosphate. SNF472 concentrations ranging from 1 to 100 μM decreased calcium deposition in rodent vascular smooth muscle cells without inducing apoptosis and SNF472 restored expression of genes that maintain the contractile phenotype of these cells. SNF472 at 1 μM was able to block >50% of the calcification. The lack of apoptosis with exposure to SNF472 in this experiment is important because previous research has shown that apoptosis is linked to calcification of vascular smooth muscle cells (Leopold, 2015), but in this case, SNF472 blocked the calcification process without affecting cell survival. sodium thiosulfate was included in this study as a comparator due to its well‐known anti‐calcification properties in vitro and in vivo (O'Neill & Hardcastle, 2012; Pasch et al., 2008). Our results demonstrated that sodium thiosulfate is also effective in reducing mineralization of vascular smooth muscle cells (at higher concentrations than SNF472), but at the same time, it induces apoptosis in vascular smooth muscle cells, which was not observed when cells were treated with SNF472. When we exposed vascular smooth muscle cells to Ca3(PO4)2, expression of alpha smooth muscle actin, a marker of muscle phenotype, decreased; adding SNF472 prevented this down‐regulation to normalize genetic expression of alpha smooth muscle actin. We cannot interpret from the current data if SNF472 has other effects that may influence expression of alpha smooth muscle actin mRNA. Our experiment was not designed to investigate whether alpha smooth muscle actin levels were reduced by exposure to increased concentrations of ionic calcium or phosphate or if it was due to formation of hydroxyapatite crystals in the culture medium. This question deserves investigation in future research. Previous evidence from animal models of vascular calcification also showed that SNF472 inhibited calcification in the aorta and the heart. SNF472 between 8.5 and 20 μM (free concentration) completely inhibited cardiovascular calcification in rats (Ferrer et al., 2018), which is consistent with SNF472 concentrations associated with significant inhibition of hydroxyapatite crystal formation in this report (between 3.8 to 30.4 μM).
Concentrations of SNF472 required to inhibit hydroxyapatite crystal formation in these experiments were evaluated in early clinical studies. In a phase 1 dose‐finding study, healthy volunteers received a single dose of SNF472 at 5, 9 or 12.5 mg·kg−1 resulting in a mean maximum SNF472 concentration of 17.6, 37.1 and 64.1 μM, respectively (Perelló et al., 2018). In the same study, the mean maximum SNF472 concentration in patients undergoing dialysis receiving a single dose of SNF472 at 9 mg·kg−1 was 44.1 μM (11.6 μM free concentration) (Perelló et al., 2018). Using a pharmacodynamic assay to measure the crystal formation potential of blood (Ferrer et al., 2017), SNF472 at 9 mg·kg−1 inhibited hydroxyapatite formation by 80.2% in the plasma of patients undergoing dialysis. In a phase 1b trial, eight patients undergoing dialysis received ascending SNF472 doses from 1 to 20 mg·kg−1 and then eight patients received SNF472 at 10 mg·kg−1, infused over 4 h, during dialysis, three times per week for 1 month (Salcedo et al., 2019). Maximum SNF472 concentration was 101 μM for a single dose of 20 mg·kg−1 and 62.9 μM after 12 doses of 10 mg·kg−1 (Salcedo et al., 2019). Thus, concentrations of SNF472 that were associated with rapid, selective and insurmountable binding of hydroxyapatite to prevent formation and growth of hydroxyapatite crystals in our experiments were achieved by SNF472 doses that were well tolerated in phase 1 trials.
Given the high affinity of SNF472 for hydroxyapatite crystals, we examined the affinity of SNF472 for circulating ionized calcium. Chelation of ionized calcium by 50% in saline occurred at SNF472 concentrations of 539 μM, nearly 50‐fold greater than concentrations that inhibited hydroxyapatite crystallization in adapted synthetic fluid and >40‐fold greater than free concentrations of SNF472 associated with a single dose of 9 mg·kg−1 in the phase 1 clinical trial described above (Perelló et al., 2018). Doses of SNF472 in clinical development to treat cardiovascular calcification or calcific uraemic arteriolopathy (Brandenburg et al., 2019; Raggi et al., 2020) are not expected to chelate circulating calcium and hypocalcaemia was not reported in the phase 1b trial (Salcedo et al., 2019).
Previously published studies for phytate suggest that SNF472 is unlikely to have a negative effect on bone health and may even show a positive effect on bone restoration (del Mar Arriero, Ramis, Perello, & Monjo, 2012a; del Mar Arriero, Ramis, Perello, & Monjo, 2012b; López‐González et al., 2013). In cell cultures, phytate decreased gene expression of osteoblast markers and mineralization in osteoblast cells without negatively affecting cell viability (del Mar Arriero et al., 2012a). Other cell cultures showed that phytate inhibited primary osteoclasts and non‐committed osteoclast precursors but did not inhibit committed, mature osteoclasts (del Mar Arriero et al., 2012b). In 157 postmenopausal women, those with low (≤0.76 μM) phytate concentrations had significantly greater bone mass loss in the lumbar spine than those with high (≥1.42 μM) phytate concentrations (López‐González et al., 2013). In our experiments, the 9‐month repeated dosing toxicology study in dogs showed no differences in full bone histomorphology or tartrate‐resistant acid phosphatase, an osteoclast biomarker, between control animals and animals treated with SNF472.
In the experiment using primary rat osteoblasts, SNF472 was added at the same time that β‐glycerolphosphate was started. This latter compound induces mineralization by acting as a phosphate source. Thus, effects of SNF472 on mineralization would most likely be seen at this time (i.e. during development of calcification). However, in our cultures, we did not observe any significant effect of SNF472 on the mineralization process (as shown by the Von Kossa stain) and osteoblast activity (as shown by alkaline phosphatase staining). Furthermore, SNF472 was present in the cultures for 2 weeks following the start of mineralization and no toxic effects or overt effects on cell viability were seen in any of the doses tested. Changes in gene expression were minor and point to a neutral to slightly positive effect as reflected by markers of bone formation. Additional studies of SNF472 in animal models for a dynamic bone disease would be needed to examine the effects of SNF472 on diseased bone.
Medications such as bisphosphonates, sodium thiosulfate, vitamin K, phosphate binders and calcimimetics have been used experimentally in patients with calcifications (O'Neill & Lomashvili, 2010), but they do not selectively target hydroxyapatite crystallization. SNF472, was specifically developed for this purpose. Another experimental approach that has been examined is nanoparticle‐based EDTA chelation therapy (Karamched, Nosoudi, Moreland, Chowdhury, & Vyavahare, 2019). Conjugating EDTA‐loaded albumin nanoparticles with elastin antibody is designed to deliver the nanoparticles to the site of injury, where EDTA can resorb mineral deposits. Injection of EDTA‐loaded albumin nanoparticles in rats reversed calcification; injection of blank nanoparticles or systemic EDTA without nanoparticles was not effective.
We acknowledge the following potential limitations of our experiments. The analysis used was based on the accepted pharmacology theory for drug‐receptor interaction, but SNF472 does not target a membrane or intracellular receptor. The mechanism of action of SNF472 is to block the formation and growth of the inorganic crystal hydroxyapatite in the extracellular compartment. To examine binding of SNF472 to hydroxyapatite, we agitated the solutions for 8 h; to examine release kinetics, we agitated the solutions for 2 h. This was determined based on another study of binding kinetics for SNF472 to hydroxyapatite in which we showed that maximum binding is attained by 2 h (Ferrer et al., 2018). We examined SNF472 chelation of free calcium in saline, which provided a controlled environment for the experiment and allowed us to examine the relative binding of hydroxyapatite crystals or free calcium in solution. The experiment did not provide direct evidence about chelation of free calcium in human blood, but phase 1 clinical studies have shown no evidence that SNF472 chelates free calcium.
In summary, these experiments show that SNF472 has high affinity for hydroxyapatite but not for free calcium. SNF472 quickly binds the face of a growing hydroxyapatite crystal on the surface of a forming hydroxyapatite nucleus to inhibit calcification specifically. This inhibitory effect is observed when the crystal is being formed or after it is already formed, preventing further growth. At doses selected for clinical investigation, SNF472 inhibits the final common pathway in cardiovascular calcification and calcific uraemic arteriolopathy by binding to hydroxyapatite, without binding appreciably to ionized calcium. SNF472 acts independently of calcium or phosphate plasma levels and is not expected to affect calcium and phosphate homeostasis. These experimental results describe the mechanism of action of SNF472 based on selective physicochemical inhibition of its extracellular target, hydroxyapatite. The data provide a scientific rationale for the ongoing clinical investigation of SNF472 as a potential treatment for cardiovascular calcification and calcific uraemic arteriolopathy in patients undergoing dialysis, for which SNF472 has shown potential efficacy in clinical studies (Brandenburg et al., 2019; Raggi et al., 2020).
AUTHOR CONTRIBUTIONS
J.P., V.B., P.D.H., B.I., and C.S. conceived and designed the study. M.D.F., M.dM.P., N.K. and G.J.B. did the experiments. M.D.F. and C.S. wrote the manuscript. All authors interpreted the data. All authors reviewed and revised the manuscript and approved the final manuscript.
CONFLICT OF INTEREST
J.P. and B.I. are founders, employees and stockholders of Sanifit Therapeutics and co‐inventors of patents owned or controlled by Sanifit Therapeutics. M.D.F. has received research grants and travel support from Sanifit Therapeutics, is a stockholder of Sanifit Therapeutics and is a co‐inventor of a patent owned or controlled by Sanifit Therapeutics. M.dM.P. and A.G. are employees and stockholders of Sanifit Therapeutics. N.K. has no conflicts of interest to declare for this work. V.B. has received consulting fees from Pharmacosmos, Vifor and FMC; lecture fees from Amgen, Pfizer, Bayer, Pharmacosmos, Vifor, FMC, Servier, Novartis, Daiichi‐Sankyo and Cardiobridge; and research grants from Amgen, Pfizer, Bayer, Sanifit Therapeutics, Pharmacosmos, Vifor, FMC, Servier, Novartis, Daiichi‐Sankyo and Cardiobridge. G.J.B. has no conflict of interest to declare for this work. P.D.H. received research grants from Vifor Pharma, Cycle Pharma, Sanifit, Shire (Takeda), Inositec, Merck KGaA, Fresenius Kabi and Fresenius Medical Care. R.G. was an employee and stockholder of Sanifit Therapeutics at the time this work was done. M.W. has received consulting fees from Akebia, Amag, Amgen, Ardelyx, Diasorin, Luitpold,and Pharmacosmos; and research grants from SHIRE. C.S. is an employee and stockholder of Sanifit Therapeutics and a co‐inventor of patents owned or controlled by Sanifit Therapeutics.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design and Analysis and Animal Experimentation and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
ACKNOWLEDGEMENTS
This work was supported by Sanifit Therapeutics. The authors thank Ayshe Hyusein for her contributions to the examination of calcium deposition in vascular smooth muscle cells. Medical writing support from Jonathan Latham of PharmaScribe, LLC was funded by Sanifit.
Perelló J, Ferrer MD, del Mar Pérez M, et al. Mechanism of action of SNF472, a novel calcification inhibitor to treat vascular calcification and calciphylaxis. Br J Pharmacol. 2020;177:4400–4415. 10.1111/bph.15163
REFERENCES
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , … CGTP Collaborators . (2019). The concise guide to pharmacology 2019/20: Introduction and other protein targets. British Journal of Pharmacology, 176(Suppl 1), S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelis, M. , Wong, L. L. , Myers, S. A. , & Wong, L. M. (1997). Calciphylaxis in patients on hemodialysis: A prevalence study. Surgery, 122, 1083–1089. discussion 1089‐1090. 10.1016/S0039-6060(97)90212-9 [DOI] [PubMed] [Google Scholar]
- Brandenburg, V. M. , Sinha, S. , Torregrosa, J. V. , Garg, R. , Miller, S. , Canals, A. Z. , … Perelló, J. (2019). Improvement in wound healing, pain, and quality of life after 12 weeks of SNF472 treatment: A phase 2 open‐label study of patients with calciphylaxis. Journal of Nephrology, 32, 811–821. 10.1007/s40620-019-00631-0 [DOI] [PubMed] [Google Scholar]
- Budisavljevic, M. N. , Cheek, D. , & Ploth, D. W. (1996). Calciphylaxis in chronic renal failure. Journal of American Society of Nephrology, 7, 978–982. [DOI] [PubMed] [Google Scholar]
- Budoff, M. J. , Hokanson, J. E. , Nasir, K. , Shaw, L. J. , Kinney, G. L. , Chow, D. , … Raggi, P. (2010). Progression of coronary artery calcium predicts all‐cause mortality. JACC: Cardiovascular Imaging, 3, 1229–1236. 10.1016/j.jcmg.2010.08.018 [DOI] [PubMed] [Google Scholar]
- Cao, S. , Dong, N. , & Chen, J. (2011). Synchronous fluorescence determination of phytic acid in foodstuffs and urine based on replacement reaction. Phytochemical Analysis, 22, 119–123. 10.1002/pca.1254 [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Chen, J. , Ma, K. , Cao, S. , & Chen, X. (2007). Fluorimetric determination of phytic acid in urine based on replacement reaction. Analytica Chimica Acta, 605, 185–191. 10.1016/j.aca.2007.10.041 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jager, D. J. , Grootendorst, D. C. , Jager, K. J. , van Dijk, P. C. , Tomas, L. M. , Ansell, D. , … Dekker, F. W. (2009). Cardiovascular and noncardiovascular mortality among patients starting dialysis. JAMA, 302, 1782–1789. 10.1001/jama.2009.1488 [DOI] [PubMed] [Google Scholar]
- del Mar Arriero, M. , Ramis, J. M. , Perello, J. , & Monjo, M. (2012a). Differential response of MC3T3‐E1 and human mesenchymal stem cells to inositol hexakisphosphate. Cellular Physiology and Biochemistry, 30, 974–986. 10.1159/000341474 [DOI] [PubMed] [Google Scholar]
- del Mar Arriero, M. , Ramis, J. M. , Perello, J. , & Monjo, M. (2012b). Inositol hexakisphosphate inhibits osteoclastogenesis on RAW 264.7 cells and human primary osteoclasts. PLoS ONE, 7, e43187 10.1371/journal.pone.0043187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer, M. D. , Ketteler, M. , Tur, F. , Tur, E. , Isern, B. , Salcedo, C. , … Perelló, J. (2018). Characterization of SNF472 pharmacokinetics and efficacy in uremic and non‐uremic rats models of cardiovascular calcification. PLoS ONE, 13, e0197061 10.1371/journal.pone.0197061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer, M. D. , Perez, M. M. , Canaves, M. M. , Buades, J. M. , Salcedo, C. , & Perello, J. (2017). A novel pharmacodynamic assay to evaluate the effects of crystallization inhibitors on calcium phosphate crystallization in human plasma. Scientific Reports, 7, 6858 10.1038/s41598-017-07203-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grases, F. , March, J. G. , Prieto, R. M. , Simonet, B. M. , Costa‐Bauza, A. , Garcia‐Raja, A. , & Conti, A. (2000). Urinary phytate in calcium oxalate stone formers and healthy people—Dietary effects on phytate excretion. Scandinavian Journal of Urology and Nephrology, 34, 162–164. 10.1080/003655900750016526 [DOI] [PubMed] [Google Scholar]
- Grases, F. , Perelló, J. , Isern, B. , & Prieto, R. M. (2004). Determination of myo‐inositol hexakisphosphate (phytate) in urine by inductively coupled plasma atomic emission spectrometry. Analytica Chimica Acta, 510, 41–43. [Google Scholar]
- Grases, F. , Sanchis, P. , Costa‐Bauza, A. , Bonnin, O. , Isern, B. , Perello, J. , & Prieto, R. M. (2008). Phytate inhibits bovine pericardium calcification in vitro. Cardiovascular Pathology, 17, 139–145. 10.1016/j.carpath.2007.08.005 [DOI] [PubMed] [Google Scholar]
- Grases, F. , Simonet, B. M. , Prieto, R. M. , & March, J. G. (2001). Phytate levels in diverse rat tissues: Influence of dietary phytate. The British Journal of Nutrition, 86, 225–231. 10.1079/bjn2001389 [DOI] [PubMed] [Google Scholar]
- Grases, F. , Simonet, B. M. , Vucenik, I. , Prieto, R. M. , Costa‐Bauza, A. , March, J. G. , & Shamsuddin, A. M. (2001). Absorption and excretion of orally administered inositol hexaphosphate (IP(6) or phytate) in humans. BioFactors, 15, 53–61. 10.1002/biof.5520150105 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jono, S. , McKee, M. D. , Murry, C. E. , Shioi, A. , Nishizawa, Y. , Mori, K. , … Giachelli, C. M. (2000). Phosphate regulation of vascular smooth muscle cell calcification. Circulation Research, 87, E10–E17. 10.1161/01.res.87.7.e10 [DOI] [PubMed] [Google Scholar]
- Joubert, P. , Ketteler, M. , Salcedo, C. , & Perello, J. (2016). Hypothesis: Phytate is an important unrecognised nutrient and potential intravenous drug for preventing vascular calcification. Medical Hypotheses, 94, 89–92. 10.1016/j.mehy.2016.07.005 [DOI] [PubMed] [Google Scholar]
- Kapustin, A. N. , Chatrou, M. L. , Drozdov, I. , Zheng, Y. , Davidson, S. M. , Soong, D. , … Shanahan, C. M. (2015). Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circulation Research, 116, 1312–1323. 10.1161/CIRCRESAHA.116.305012 [DOI] [PubMed] [Google Scholar]
- Karamched, S. R. , Nosoudi, N. , Moreland, H. E. , Chowdhury, A. , & Vyavahare, N. R. (2019). Site‐specific chelation therapy with EDTA‐loaded albumin nanoparticles reverses arterial calcification in a rat model of chronic kidney disease. Scientific Reports, 9, 2629 10.1038/s41598-019-39639-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwowski, W. , Naumnik, B. , Szczepanski, M. , & Mysliwiec, M. (2012). The mechanism of vascular calcification—A systematic review. Medical Science Monitor, 18, RA1–RA11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzer, P. , Boehm, M. , Sorribas, V. , Thiriet, M. , Janzen, J. , Zeller, T. , … Shanahan, C. (2014). Medial vascular calcification revisited: Review and perspectives. European Heart Journal, 35, 1515–1525. 10.1093/eurheartj/ehu163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold, J. A. (2015). Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends in Cardiovascular Medicine, 25, 267–274. 10.1016/j.tcm.2014.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐González, A. A. , Grases, F. , Monroy, N. , Marí, B. , Vicente‐Herrero, M. T. , Tur, F. , & Perelló, J. (2013). Protective effect of myo‐inositol hexaphosphate (phytate) on bone mass loss in postmenopausal women. European Journal of Nutrition, 52, 717–726. 10.1007/s00394-012-0377-6 [DOI] [PubMed] [Google Scholar]
- Mizobuchi, M. , Towler, D. , & Slatopolsky, E. (2009). Vascular calcification: The killer of patients with chronic kidney disease. Journal of American Society of Nephrology, 20, 1453–1464. 10.1681/ASN.2008070692 [DOI] [PubMed] [Google Scholar]
- Muñoz, J. A. , López‐Mesas, M. , & Valiente, M. (2010). Minimum handling method for the analysis of phosphorous inhibitors of urolithiasis (pyrophosphate and phytic acid) in urine by SPE‐ICP techniques. Analytica Chimica Acta, 658, 204–208. 10.1016/j.aca.2009.11.003 [DOI] [PubMed] [Google Scholar]
- Muñoz, J. A. , & Valiente, M. (2003). Determination of phytic acid in urine by inductively coupled plasma mass spectrometry. Analytical Chemistry, 75, 6374–6378. 10.1021/ac0345805 [DOI] [PubMed] [Google Scholar]
- Nigwekar, S. U. , Thadhani, R. , & Brandenburg, V. M. (2018). Calciphylaxis. The New England Journal of Medicine, 378, 1704–1714. 10.1056/NEJMra1505292 [DOI] [PubMed] [Google Scholar]
- Noordzij, M. , Cranenburg, E. M. , Engelsman, L. F. , Hermans, M. M. , Boeschoten, E. W. , Brandenburg, V. M. , … for the NECOSAD Study Group . (2011). Progression of aortic calcification is associated with disorders of mineral metabolism and mortality in chronic dialysis patients. Nephrology, Dialysis, Transplantation, 26, 1662–1669. 10.1093/ndt/gfq582 [DOI] [PubMed] [Google Scholar]
- O'Neill, W. C. , & Hardcastle, K. I. (2012). The chemistry of thiosulfate and vascular calcification. Nephrology, Dialysis, Transplantation, 27, 521–526. 10.1093/ndt/gfr375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill, W. C. , & Lomashvili, K. A. (2010). Recent progress in the treatment of vascular calcification. Kidney International, 78, 1232–1239. 10.1038/ki.2010.334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasch, A. , Schaffner, T. , Huynh‐Do, U. , Frey, B. M. , Frey, F. J. , & Farese, S. (2008). Sodium thiosulfate prevents vascular calcifications in uremic rats. Kidney International, 74, 1444–1453. 10.1038/ki.2008.455 [DOI] [PubMed] [Google Scholar]
- Perelló, J. , Joubert, P. H. , Ferrer, M. D. , Canals, A. Z. , Sinha, S. , & Salcedo, C. (2018). First‐time‐in‐human randomized clinical trial in healthy volunteers and haemodialysis patients with SNF472, a novel inhibitor of vascular calcification. British Journal of Clinical Pharmacology, 84, 2867–2876. 10.1111/bcp.13752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raggi, P. , Bellasi, A. , Bushinsky, D. , Bover, J. , Rodriguez, M. , Ketteler, M. , … Chertow, G. M. (2020). Slowing progression of cardiovascular calcification with SNF472 in patients on hemodialysis: Results of a randomized Phase 2b study. Circulation, 141, 728–739. 10.1161/CIRCULATIONAHA.119.044195 [DOI] [PubMed] [Google Scholar]
- Salcedo, C. , Joubert, P. H. , Ferrer, M. D. , Canals, A. Z. , Maduell, F. , Torregrosa, V. , … Perelló, J. (2019). A phase 1b randomized, placebo‐controlled clinical trial with SNF472 in haemodialysis patients. British Journal of Clinical Pharmacology, 85, 796–806. 10.1111/bcp.13863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnak, M. J. , Levey, A. S. , Schoolwerth, A. C. , Coresh, J. , Culleton, B. , Hamm, L. L. , … American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention . (2003). Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension, 42, 1050–1065. 10.1161/01.HYP.0000102971.85504.7c [DOI] [PubMed] [Google Scholar]
- Schurgers, L. J. , Spronk, H. M. , Skepper, J. N. , Hackeng, T. M. , Shanahan, C. M. , Vermeer, C. , … Proudfoot, D. (2007). Post‐translational modifications regulate matrix Gla protein function: Importance for inhibition of vascular smooth muscle cell calcification. Journal of Thrombosis and Haemostasis, 5, 2503–2511. 10.1111/j.1538-7836.2007.02758.x [DOI] [PubMed] [Google Scholar]
- United States Renal Data System . (2018). 2018 USRDS annual data report: Epidemiology of kidney disease in the United States. https://www.usrds.org/2018/view/Default.aspx. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD.
- Verberckmoes, S. C. , De Broe, M. E. , & D'Haese, P. C. (2003). Dose‐dependent effects of strontium on osteoblast function and mineralization. Kidney International, 64, 534–543. 10.1046/j.1523-1755.2003.00123.x [DOI] [PubMed] [Google Scholar]
- Wanner, C. , Krane, V. , Marz, W. , Olschewski, M. , Mann, J. F. , Ruf, G. , & Ritz, E. (2005). Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. The New England Journal of Medicine, 353, 238–248. 10.1056/NEJMoa043545 [DOI] [PubMed] [Google Scholar]
- Yang, H. , Curinga, G. , & Giachelli, C. M. (2004). Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney International, 66, 2293–2299. 10.1111/j.1523-1755.2004.66015.x [DOI] [PubMed] [Google Scholar]
- Yiu, A. J. , Callaghan, D. , Sultana, R. , & Bandyopadhyay, B. C. (2015). Vascular calcification and stone disease: A new look towards the mechanism. Journal of Cardiovascular Development and Disease, 2, 141–164. 10.3390/jcdd2030141 [DOI] [PMC free article] [PubMed] [Google Scholar]