

Abstract
Drug re-purposing might be a fast and efficient way of drug development against the novel coronavirus disease 2019 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). We applied a bioinformatics approach using molecular dynamics and docking to identify FDA-approved drugs that can be re-purposed to potentially inhibit the non-structural protein 9 (Nsp9) replicase and spike proteins in SARS-CoV-2. We performed virtual screening of FDA-approved compounds, including antiviral, anti-malarial, anti-parasitic, anti-fungal, anti-tuberculosis, and active phytochemicals against the Nsp9 replicase and spike proteins. Selected hit compounds were identified based on their highest binding energy and favorable absorption, distribution, metabolism and excretion (ADME) profile. Conivaptan, an arginine vasopressin antagonist drug exhibited the highest binding energy (-8.4 Kcal/mol) and maximum stability with the amino acid residues present at the active site of the Nsp9 replicase. Tegobuvir, a non-nucleoside inhibitor of the hepatitis C virus, also exhibited maximum stability along with the highest binding energy (-8.1 Kcal/mol) at the active site of the spike proteins. Molecular docking scores were further validated by molecular dynamics using Schrodinger, which supported the strong stability of ligands with the proteins at their active sites through water bridges, hydrophobic interactions, and H-bonding. Our findings suggest Conivaptan and Tegobuvir as potential therapeutic agents against SARS-CoV-2. Further in vitro and in vivo validation and evaluation are warranted to establish how these drug compounds target the Nsp9 replicase and spike proteins.
Keywords: SARS-CoV-2, Nsp9 replicase, spike proteins, molecular docking, drug designing, drug repurposing
Graphical Abstract

Introduction
The current coronavirus pandemic has caused 3,87,911 deaths across the globe and has infected around 6 million people as of 5th June 2020. Initial studies on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) stated that it was closely related to SARS-CoV. Coronaviruses (CoVs) are enveloped viruses from the family of Coronaviridae with a positive-sense single-stranded RNA genome (Fehr & Perlman, 2015). The genome size of CoVs is relatively large, ranging from approximately 27 to 37 kilobases. The envelope of the virus contains a lipid bilayer with three structural proteins: membrane (M), envelope (E), and spike (S) (Boopathi et al., 2020). The multiple copies of nucleocapsid protein present are associated with the positive sense single stranded RNA genome and are responsible for the formation of the nucleocapsid present inside the envelope (Bride et al., 2014). Viral protection outside the host is provided by the lipid bilayer, nucleocapsid, and membrane proteins (Lenard, 2008). The CoV infection is initiated by the attachment of the S glycoprotein to the complementary host receptor (Young & Alexandra, 2019). The entry of viral particles and its attachment to host membrane is mediated through either direct fusion of the viral envelope or endocytosis using the host membrane (Huang et al., 2020). The single positive-stranded RNA genome of COVs has the capacity to replicate their own genomic information as well as translate proteins in the cytoplasm of host (Nakagawa et al., 2016). Polymerase is synthesized by the virus and used to subsequently synthesize the minus strand using the positive strand as a template (Elfiky, 2020). This positive sense genomic RNAs generated through replication develops into the viral progeny. The genomic RNA is attached to the N glycoprotein. The M glycoprotein integrates into the endoplasmic reticulum (ER) membrane exactly as the S and hemagglutinin esterase (HE) proteins. The ER is the location for the translation of RNA and viral structural proteins. The M protein assists the protein-protein interactions that help in the assembly of viral particles, which is followed by its binding to the nucleocapsid. These viral particles are then released from the host cell via exocytosis (Mousavizadeh & Ghasemi, 2020. The main protease domain (Mpro) is a conserved target in SARS-CoV-2, and thus provide the opportunity to design new inhibitors throughout the entire Coronaviridae subfamily (Mahanta et al., 2020). Two-third region of the 5′ end of the CoV genome consists of open reading frame I (ORFI), which encodes two large polypeptides involved in the replicase machinery: pp1a and pp1ab1. Two proteases encoded in the 5′ region of ORF 1: 3 C-like protease (3CL or Nsp5) and papain-like protease (PLP), co-translationally cleave the two polypeptides into mature non-structural proteins (NSPs) (Lim et al., 2000; Kumar, Sharma, et al., 2020). The 3CL protease, also referred to as Mpro due to its dominant role in the post-translational machinery of the replicase protein (Kanchan et al., 2003). Both these proteins have a substrate-binding pocket where at P1, glutamine is the substrate and at the P2, either leucine or methionine are the source of substrates. This strong structural basis provides a loophole for the design of a wide spectrum of anti-CoV inhibitors. In general, there are no treatment options for corona based viral diseases that occur suddenly and spread at a higher frequency.
The spike proteins (PDB ID-6LZG) form a crown shape on the surface of the novel virus and are of major research interest as little is known about how they attach, fuse and gain entry into the host cell (Walls et al., 2020). There are mainly two subunits in the spike protein, namely S1 and S2. The S1-portion has diverged sequences even with the same coronavirus species, whereas the S2 subunit is highly conserved. The S1 subunit has 2 domains N and C terminal domains. These domains mainly function as receptor-binding domains and bind to various proteins and sugar molecules. These spike proteins contain heptad repeats of hydrophobic domains that help in fusing into the host. The cell entry program is mediated by the spike proteins, mainly through binding to the ACE-2 receptor on the host surface and subsequently mediating the viral infection. The major role played by the spike proteins in the host entry and attachment illustrates the large possibilities of targeted and to find effective vaccines and anti-bodies to neutralize the viral infection (Li, 2016; Robson, 2020).
The Nsp9 (PDB ID-6W4B) replicase is a non-structural protein encoded by ORF1a, which has no eminent function, but is related to viral RNA synthesis (Dene et al., 2020). This protein contains a single folded beta-barrel, which is unique, unlike the single domain proteins. This fold is related to the OB-fold having an extended C-terminal in the subdomains of both SARS-CoV-2 and 3 C-like protease that belongs to the serine protease superfamily. The crystal structure of Nsp9 replicase emphasizes it as a dimeric protein. Nsp9 replicase specifically binds to the RNA, further interacting with the nsp8 protein and activating it, which is essential for its function (Sutton et al., 2004). As Nsp9 replicase plays a major role in viral replication, it can be a unique target for the discovery of novel drugs against this protein, enabling the inhibition of the viral progression.
As many vaccines and drugs are undergoing clinical trials globally, drug repurposing has been one of the effective approaches taken by the scientists across the globe to bring out an effective medicine for the eradication of the novel coronavirus. Anti-viral drugs such as chloroquine and hydroxychloroquine, used to treat malaria and arthritis respectively, were approved in the USA to treat SARS-CoV-2 patients (Touret & Lamballerie, 2020). Some of the other drugs such as Remdesivir, Actemra, and Galidesivir are currently undergoing clinical trials, but there has been no vaccine or therapeutic drug currently approved by the FDA for the prevention or treatment of SARS-CoV-2 (Hendaus, 2019; Chaudhuri et al., 2018; Das et al., 2020). Therefore, structure-based drug repurposing through targeting the Nsp9 replicase and spike proteins of SARS-CoV-2 can provide successful therapeutic strategies against this virus. Our study used a computational approach towards structure-based drug repurposing of different anti-viral, anti-malarial, anti-parasitic, anti-fungal, anti-tuberculosis and active phytochemicals against major target proteins such as Nsp-9 replicase and the spike protein of SARS-CoV-2.
Material and methods
Data sources
In our study, a dataset of 2000 FDA-approved compounds, including antiviral, anti-malarial, anti-parasitic, anti-fungal, anti-tuberculosis, and active phytochemicals from FDA and Indian Medicinal Plants, Phytochemistry and Therapeutic database were obtained (Mohanraj et al., 2018; Morris et al., 2009).
Preparation of receptor
The atomic coordinates of the protein crystal structures of Nsp9 replicase (PDB ID-6W4B) and the spike protein (PDB ID-6LZG) were downloaded from the RCSB-PDB (protein data bank) database. Prior to docking or analysis, the solvation parameters, charge assignment, fragmental volumes, and protein optimization were checked using Autodock Tool 4 (ADT) (Chandel et al., 2020; Kumar, Chandel, et al., 2020; Raj et al., 2020; O’Boyle et al., 2011).
Preparation of ligands
The 3 D SDF structures of all the compounds were downloaded from the PubChem database (Chandel et al., 2020). The 2 D ligand structures of the compounds were designed using Chemdraw. The optimization of the ligands was done using Avogadro and the data converted into the PDB file format using Open Babel software.
Compound screening
Molecular screening of the compounds was performed using PyRx virtual screening tool-python prescription 0.8 software and Autodock wizard as the engine for molecular docking (Dallakyan & Olson, 2015; Khan et al., 2020; Pagadala et al., 2017; Seeliger & Groot, 2010). The ligands were minimized to their stable form. During the period of docking, the protein was considered to be rigid and the ligands were considered to be flexible. Auto Grid engine in PyRx was used to generate the configuration file for the grid parameters. The application was also used to identify/predict the amino acids in the active site of the protein that interact with the ligands. A result of positional root-mean-square deviation (RMSD) less than 1.0Å was considered ideal for finding the favorable binding. The ligand with the highest binding energy (most negative) was considered as the ligand with maximum binding affinity.
Analysis and visualization
Pymol version 2.3.4 and ADT were used for visual analysis of the docking site and the results were validated using Autodock-Vina (Seeliger & Groot, 2010).
ADME analysis
ADME analysis of the selected ligands obtained from PubChem was done on the basis of canonical SMILES using Swiss-ADME programme (Daina et al., 2017). The ADME properties of the chosen compounds were calculated. The major ADME associated parameters such as Lipinski’s rule of five, drug likeliness, pharmacokinetic properties, the solubility of the drug, were considered. The values of the observed properties are presented in Tables 1 and 2.
Table 1.
Molecular docking analysis of antiviral compounds against Nsp9 replicase (6W4B) of SARS-CoV-2.
| Protein | Compound | Binding Energy (Kcal/mol) | Amino acid residues |
|---|---|---|---|
| Nsp9 replicase | Conivaptan | −8.4 | CYS74, LEU107, LEU113, ALA108, LEU5, ASN34, ASN96, LEU98, PHE41, THR36, ALA9, LEU104, VAL8, VAL42, ASN99, SER6 |
| Telmisatan | −8.1 | ARG100, LEU98, PHE9, MET102, PHE41, ASN34, THR36, LEU113, LEU107, ALA108, VAL8, PRO7, LEU104, PHE76, LEU5, GLU4, SER6, CYS74, PHE91 | |
| Phaitanthrin D | −7.9 | PHE76, CYS74, LEU89, LEU104, LEU107, GLY105, MET102, SER6, VAL8, PRO7, ALA108, LEU113 | |
| Phytosterols | −7.8 | PHE41, THR36, ASN34, ASN99, SER6, VAL8, LEU5, CYS74, PHE76, LEU89, LEU104, MET102, LEU113, ALA108, LEU107, | |
| Withanolide R | −7.8 | PHE76, CYS74, LEU89, PHE91, VAL103, MET102, ASN99, GLN105, LEU104, LEU107, PHE41, ASN34, THR36, LEU113, LEU5, VAL8, ALA108 | |
| Withanolide G | −7.7 | ASN96, ASN99, MET102, LEU98, LEU104, ALA108, PHE41, ASN34, VAL8, LEU113, LEU5, CYS74, THR36, SER6 | |
| 17-alpha-hydroxywithanolide D | −7.7 | GLU4, PHE76, LEU5, SER6, PRO7, LEU113, LEU107, ALA108, LEU104, VAL8, ALA9, MET102, ASN34, LEU98, ALA99, PHE41 | |
| Stigmasta-5, 22-dien-3-ol | −7.7 | PHE41, ASN96, ASN99, ASN34, VAL8, LEU5, LEU113, PHE76, LEU107, LEU89, CYS74, ALA108, MET102, SER6 | |
| Gedunin | −7.6 | THR36, ASN34, LEU113, VAL8, ALA108, SER6, LEU5, GLU4, CYS74, LEU107, MET102, ASN99, LEU98, LEU104 | |
| Ciclesonide | −7.5 | LEU113, SER6, LEU5, GLU4, ASN3, CYS7, VAL8, THR36, THR35, ASN34, ALA108, PHE41, MET102, ASN99, LEU98, PHE91, ASN100, | |
| Ezetimibe | −7.5 | ASN34, MET102, LEU104, PHE91, GLY105, ASN3, CYS74, LEU89, PHE76, SER6, LEU113, PRO7, VAL8, ALA108, ALA9, | |
| Meldenin | −7.5 | ASN99, MET102, PHE41, THR36, SER6, VAL8, LEU113, LEU107, ALA108, THR35, LEU5 | |
| Magnolol | −7.4 | MET102, GLY105, ARG100, LEU104, LEU107, ALA108, PHE91, LEU89, CYS74, PHE76, SER6, LEU113, VAL8, PRO7 | |
| Pioglitazone | −7.4 | MET102, ASN99, ASN34, ALA108, LEU104, LEU107, LEU113, LEU85, VAL8, THR36, PHE41, THR35, SER6, | |
| Gloriosine | −7.4 | LYS102, PHE103, VAL104, ARG105, ILE106, GLN107, GLN110, PHE294, PHE8, ASN151, TYR154, ASP153 |
Table 2.
Molecular docking analysis of antiviral compounds against spike protein (6LZG) of SARS-CoV-2.
| Protein | Compound | Binding Energy (Kcal/mol) | Amino acid residues |
|---|---|---|---|
| Spike Protein | Tegobuvir | −8.1 | PRO337, ALA344, ARG355, ARG466, ASN343, PHE342, PHE347, PHE338, GLY339, GLU340, VAL341 |
| Bromocriptine | −7.7 | ASN450, LEU452, ILE468, TYR351, ALA352, SER349, LYS356, GLU340, ASN354, VAL341, THR345, ARG346, PHE347, ALA348, SER349 | |
| Baicalin | −7.6 | ASN450, ARG346, ALA344, PHE342, GLU340, VAL341, SER399, ASN354, TRP353, ARG466, ILE468, ALA352, PHE400, PHE347 | |
| Deleobuvir | −7.6 | ARG466, TRP353, ASN354, PRO463, PHE464, PRO426, TYR396, GLU516, ARG355 | |
| Dantrolene | −7.6 | TYR351, ALA352, ASN354, SER399, LYS356, ALA348, SER349, LEU452, ASN450 ARG346, PHE347, ALA344, VAL341, PHE342, GLU340 | |
| Cassameridine | −7.4 | ARG355, PHE464, PRO463, TYR396, GLU516, SER514, PRO426, PHE515, PHE429, ASP428, PRO426 | |
| Chrysin-7-O-glucuronide | −7.4 | LYS356, ASN354, ALA352, TYR351, SER349, ASN450, ARG346, ALA344, GLU340, PHE347, ALA348, PHE400, SER399, VAL341 | |
| Conivaptan | −7.4 | PHE464, GLU465, ARG466, AG355, ASN354, TRP353, ALA352 | |
| Phaitanthrin D | −7.2 | PRO463, PHE464, ARG355, TYR396, SER514, THR430, PHE515, GLU516 | |
| Telmisartan | −7.2 | ILE468, ARG466, ASN354, PHE347, ARG346, ALA352, ARG355, LYS356, TYR396, ARG357 | |
| Troglitazone | −7.2 | ARG466, GLU465, PRO463, PHE464, TRP353, ARG355, PRO426, ASP428, PHE429, THR430, SER514, TYR396, PHE515, GLU516 | |
| Raltegravir | −7.1 | ALA352, SER349, ALA348, PHE347, ASN354, SER399, ARG346, ALA344, GLU340, VAL341, ARG357, LYS356, ARG355 | |
| Ceferoperazone | −7.1 | LEU452, TYR451, ASN450, SER349, ARG346, PHE347, ALA344, ALA352, TRP353, SER399, ASN354, VAL341, GLU340, ARG355, LYS356, ARG477, PHE347, ALA344 | |
| Dasatinib | −7.0 | ARG357, LYS356, TYR396, ARG355, ARG346, PHE347, ALA348, ASN450, TYR451, SER349, ALA332, TYR351, ILE468. ARG466, TRP353, ASN354, SER349 | |
| Dolutegravir | −7.0 | ARG355, TYR396, PHE515, SER514, GLU516, THR430, PHE429, PRO426, PRO463, GLU465, ARG466, TRP353, PHE464 |
Molecular dynamics & simulation
The complete study was performed on different modules of Schrodinger suite 2020-1 trial version. Both complexes were prepared prior to MD simulation in the protein preparation wizard and Prime module of Schrodinger suite to remove defects such as missing hydrogen atoms, incorrect bond order assignments, charge states, orientations of various groups and missing side chains suite (Schrödinger, 2020, 2016). Removal of steric clashes and strained bonds/angles were done by performing a restrained energy minimization, allowing movement in heavy atoms up to 0.3 Å. Extensive 100 ns MD simulation was carried out for both complexes through Desmond, D. E. Shaw Research, New York, NY, 2015 (Schrödinger, 2020) to access the binding stability of query molecule with respect to nelfinavir in the complex. Both complex systems were solvated in TIP3P water model and 0.15 M NaCl to mimic a physiological ionic concentration. The full system energy minimization step was done for 100 ps. The MD simulation was run for 100 ns at 300 K temperature, standard pressure (1.01325 bar), within an orthorhombic box with buffer dimensions 10 × 10 × 10 Å3 and NPT ensemble. The energy (kcal/mol) was recorded at intervals of 1.2 ps. The protein-ligand complex system was neutralized by balancing the net charge of the system by adding Na + or Cl- counter ions. The Nose-Hoover chain and Martyna-Tobias-Klein dynamic algorithm was used maintain the temperature of all the systems at 300 K and pressure 1.01325 bar, respectively.
Results and discussion
Our study focused on drug repurposing against the structural proteins Nsp9 replicase (PDB ID-6W4B) and the spike protein (PDB ID-6LZG) (Figure 1) of SARS-CoV-2 in combination as potential therapeutic targets for the treatment of coronavirus. In this study, we applied a computational approach of structure-based drug repurposing to identify specific therapeutic agents against SARS-CoV-2. We created a database of 2000 FDA approved compounds, including antiviral, anti-malarial, anti-parasitic, anti-fungal, anti-tuberculosis and active phytochemicals from FDA and Indian Medicinal Plants, Phytochemistry and Therapeutic database (Figure 2). These compounds were screened using a virtual screening tool PyRx, based on which 15 hits were selected depending on their best binding energy. Further, molecular docking was performed for hits against Nsp9 replicase and the spike protein (Tables 1 and 2).
Figure 1.

Crystal structure of protein/receptor (A) Nsp9 replicase (PDB ID-6W4B) of SARS-CoV-2. The structure is shown in ribbon representation, coloured from the N-terminus to the C-terminus with colours changing from blue through green and yellow to red. (B) Spike protein (PDB ID-6LZG) of SARS-CoV-2 shows ribbon structure representation, coloured from the N-terminus to the C-terminus with colours changing from red through yellow and green to blue Ribbon structure.
Figure 2.

Docking analysis and visualisation of protein-ligand complex (A) Nsp9 replicase (pink) and Conivaptan (yellow), (B) Nsp9 replicase (pink) and Telmisartan (green), (C) Nsp9 replicase (pink) and Phaitanthrin D (salmon), (D) Spike protein (green) and Tegobuvir (pink), (E) Spike protein (green) and Bromocriptine (blue), (F) Spike protein (green) and Baicalin (orange). The binding site of the Nsp9 replicase and spike protein is depicted using surface representation. The ligands are depicted using stick model representation. The ligand is interacting at the active site pocket of the protein/receptor.
Molecular docking is a computational approach that aims to identify non-covalent binding between (ligand/inhibitor) and protein (receptor). Docking predicts the mode of interaction between a receptor and the ligand for an established binding site. Binding energy suggests the affinity and strength of a specific ligand to which a compound binds and interacts at the active site pocket of a target protein. To understand the effect of active antiviral, anti-malarial, anti-parasitic, anti-fungal, anti-tuberculosis, anti-bacterial and active phytochemical compounds on SARS-CoV-2 molecular docking of 15 active compounds against each target selected after screening from PyRx, was performed. Further, based on their binding energy and best ADME properties, the top three compounds were selected (Tables 3 and 4).
Table 3.
ADME Properties of selected inhibitors of Nsp9 replicase.
| S. No | Compound | Molecular formula | ADME Properties (Lipinki’s Rule of Five) |
Structure | Drug likeliness | |
|---|---|---|---|---|---|---|
| Properties | Value | |||||
| 1. | Conivaptan | C32H26N4O2 | Molecular weight (<500Da) | 498.57 | 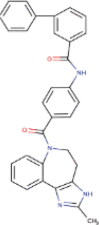 |
Yes |
| LogP (<5) | 5 | |||||
| H-Bond donor (5) | 2 | |||||
| H-bond acceptor (<10) | 3 | |||||
| Violations | 0 | |||||
| 2. | Telmisartan | C33H30N402 | Molecular weight (<500Da) | 514.62 | 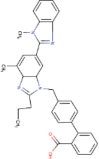 |
Yes |
| LogP (<5) | 5.9 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 4 | |||||
| Violations | 2 | |||||
| 3. | Phaitanthrin D | C17H12N203 | Molecular weight (<500Da) | 292.29 | 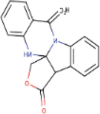 |
Yes |
| LogP (<5) | 1.7 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 3 | |||||
| Violations | 0 | |||||
| 4. | Phytosterols | C29H50O | Molecular weight (<500Da) | 414.71 | 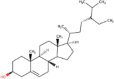 |
Yes |
| LogP (<5) | 7.1 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 1 | |||||
| Violations | 1 | |||||
| 5. | Withanolide R | C28H38O6 | Molecular weight (<500Da) | 454.60 |  |
Yes |
| LogP (<5) | 3.9 | |||||
| H-Bond donor (5) | 2 | |||||
| H-bond acceptor (<10) | 5 | |||||
| Violations | 0 | |||||
| 6. | Withanolide G | C28H38O5 | Molecular weight (<500Da) | 480.64 |  |
Yes |
| LogP (<5) | 4.1 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 6 | |||||
| Violations | 0 | |||||
| 7. | 17-alpha-hydroxywithanolide D | C28H40O7 | Molecular weight (<500Da) | 488.61 |  |
Yes |
| LogP (<5) | 2.1 | |||||
| H-Bond donor (5) | 4 | |||||
| H-bond acceptor (<10) | 7 | |||||
| Violations | 0 | |||||
| 8 | Stigmasta-5, 22-dien-3-ol | C29H48O | Molecular weight (<500Da) | 412.69 |  |
Yes |
| LogP (<5) | 6.9 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 1 | |||||
| Violations | 1 | |||||
| 9. | Gedunin | C28H34O7 | Molecular weight (<500Da) | 482.57 | 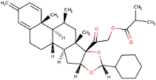 |
Yes |
| LogP (<5) | 3.7 | |||||
| H-Bond donor (5) | 0 | |||||
| H-bond acceptor (<10) | 7 | |||||
| Violations | 0 | |||||
| 10. | Ciclesonide | C32H44O7 | Molecular weight (<500Da) | 540.69 | 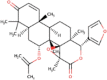 |
Yes |
| LogP (<5) | 4.4 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 7 | |||||
| Violations | 1 | |||||
| 11. | Ezetimibe | C24H21F2NO3 | Molecular weight (<500Da) | 409.43 | 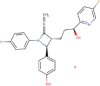 |
Yes |
| LogP (<5) | 4.3 | |||||
| H-Bond donor (5) | 2 | |||||
| H-bond acceptor (<10) | 5 | |||||
| Violations | 1 | |||||
| 12 | Meldenin | C28H38O5 | Molecular weight (<500Da) | 454.60 | 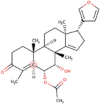 |
Yes |
| LogP (<5) | 4.4 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 5 | |||||
| Violations | 0 | |||||
| 13 | Magnolol | C18H18O2 | Molecular weight (<500Da) | 266.33 | 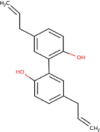 |
Yes |
| LogP (<5) | 4.2 | |||||
| H-Bond donor (5) | 2 | |||||
| H-bond acceptor (<10) | 5 | |||||
| Violations | 0 | |||||
| 14 | Pioglitazone | C19H20N2O3S | Molecular weight (<500Da) | 356.44 | 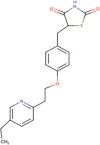 |
Yes |
| LogP (<5) | 3.1 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 4 | |||||
| Violations | 0 | |||||
| 15. | Gloriosine | C21H23NO6 | Molecular weight (<500Da) | 385.41 | 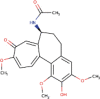 |
Yes |
| LogP (<5) | 2.1 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 6 | |||||
| Violations | 0 | |||||
Table 4.
ADME Properties of selected inhibitors of spike protein.
| S. No | Compound | Molecular formula | ADME Properties (Lipinki’s Rule of Five) |
Structure | Drug likeliness | |
|---|---|---|---|---|---|---|
| Properties | Value | |||||
| 1. | Tegobuvir | C25H14F7N5 | Molecular weight (<500Da) | 517.40 | 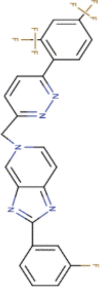 |
Yes |
| LogP (<5) | 5.8 | |||||
| H-Bond donor (5) | 0 | |||||
| H-bond acceptor (<10) | 11 | |||||
| Violations | 2 | |||||
| 2. | Baicalin | C21H18011 | Molecular weight (<500Da) | 446.36 | 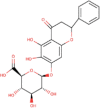 |
Yes |
| LogP (<5) | 0.2 | |||||
| H-Bond donor (5) | 6 | |||||
| H-bond acceptor (<10) | 11 | |||||
| Violations | 2 | |||||
| 3. | Cassameridine | C18H9NO5 | Molecular weight (<500Da) | 319.27 | 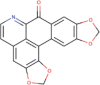 |
Yes |
| LogP (<5) | 2.7 | |||||
| H-Bond donor (5) | 0 | |||||
| H-bond acceptor (<10) | 6 | |||||
| Violations | 0 | |||||
| 4. | Deleobuvir | C34H33BrN6O3 | Molecular weight (<500Da) | 653.57 | 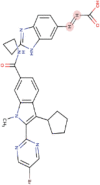 |
Yes |
| LogP (<5) | 5.1 | |||||
| H-Bond donor (5) | 2 | |||||
| H-bond acceptor (<10) | 6 | |||||
| Violations | 1 | |||||
| 5. | Bromocriptine | C32H40BrN5O5 | Molecular weight (<500Da) | 654.59 | 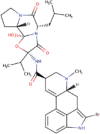 |
Yes |
| LogP (<5) | 3.1 | |||||
| H-Bond donor (5) | 3 | |||||
| H-bond acceptor (<10) | 6 | |||||
| Violations | 1 | |||||
| 6. | Chrysin-7-O-glucuronide | C21H18O10 | Molecular weight (<500Da) | 430.36 | 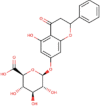 |
Yes |
| LogP (<5) | 0.64 | |||||
| H-Bond donor (5) | 5 | |||||
| H-bond acceptor (<10) | 10 | |||||
| Violations | 0 | |||||
| 7. | Phaitanthrin D | C29H48O | Molecular weight (<500Da) | 412.69 | 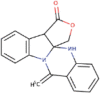 |
Yes |
| LogP (<5) | 6.9 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 1 | |||||
| Violations | 1 | |||||
| 8 | Dantrolene | C14H10N4O5 | Molecular weight (<500Da) | 314.25 | 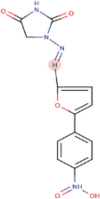 |
Yes |
| LogP (<5) | 0.72 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 4 | |||||
| Violations | 0 | |||||
| 9. | Telmisartan | C24H21F2NO3 | Molecular weight (<500Da) | 409.43 | 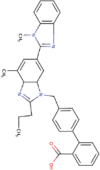 |
Yes |
| LogP (<5) | 4.3 | |||||
| H-Bond donor (5) | 2 | |||||
| H-bond acceptor (<10) | 5 | |||||
| Violations | 1 | |||||
| 10. | Conivaptan | C28H38O5 | Molecular weight (<500Da) | 454.60 | 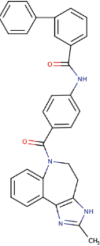 |
Yes |
| LogP (<5) | 4.4 | |||||
| H-Bond donor (5) | 1 | |||||
| H-bond acceptor (<10) | 5 | |||||
| Violations | 0 | |||||
| 11. | Ceferoperazone | C18H18O2 | Molecular weight (<500Da) | 266.33 | 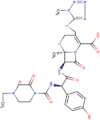 |
Yes |
| LogP (<5) | 4.2 | |||||
| H-Bond donor (5) | 2 | |||||
| H-bond acceptor (<10) | 5 | |||||
| Violations | 0 | |||||
| 12 | Troglitazone | C24H27NOS5 | Molecular weight (<500Da) | 441.54 | 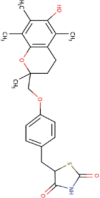 |
Yes |
| LogP (<5) | 4.1 | |||||
| H-Bond donor (5) | 2 | |||||
| H-bond acceptor (<10) | 5 | |||||
| Violations | 0 | |||||
| 13 | Dolutegravir | C20H19F2N3O5 | Molecular weight (<500Da) | 419.38 | 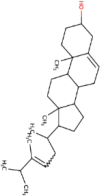 |
Yes |
| LogP (<5) | 1.8 | |||||
| H-Bond donor (5) | 2 | |||||
| H-bond acceptor (<10) | 7 | |||||
| Violations | 0 | |||||
| 14 | Raltegravir | C20H21FN6O5 | Molecular weight (<500Da) | 444.42 | 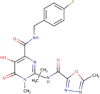 |
Yes |
| LogP (<5) | 1.4 | |||||
| H-Bond donor (5) | 3 | |||||
| H-bond acceptor (<10) | 9 | |||||
| Violations | 1 | |||||
| 15. | Dasatinib | C22H26CIN7O2S | Molecular weight (<500Da) | 488.01 | 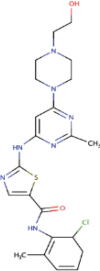 |
Yes |
| LogP (<5) | 2.8 | |||||
| H-Bond donor (5) | 3 | |||||
| H-bond acceptor (<10) | 6 | |||||
| Violations | 0 | |||||
The three selected compounds (Conivaptan, Telmisartan, and Phaitanthrin D) showed the best docking scores and were found to be best molecules against the target site of the Nsp9 replicase. Out of these, Conivaptan exhibited the best binding energy (-8.4 Kcal/mol) with Nsp9 replicase, interacting with the CYS74, LEU107, LEU113, ALA108, LEU5, ASN34, LEU98, ASN96, LEU98, PHE41, THR36, ALA9, LEU104, VAL8, ALA108, ASN99 and SER6 amino acid residues at the active site (Figure 3A). Moreover, Conivaptan showed strong interaction with Nsp9 replicase at the active site through H-bond with VAL42 amino acid (Figure 5A). Conivaptan was the first of this class of FDA-approved arginine vasopressin antagonists for the management of hypervolemic and euvolemic hyponatremia (Ghali et al., 2009). However, the most common side effects of Conivaptan include allergic reactions, fluid or electrolyte problems, signs of high or low blood pressure, headache and throat pain (Ghali et al., 2009). Telmisartan exhibited (-8.1 Kcal/mol) binding affinity with Nsp9 replicase interacting with the ARG100, LEU98, PHE9, MET102, PHE41, ASN34, THR36, LEU113, LEU107, ALA108, VAL8, PRO7, LEU104, PHE76, LEU5, GLU4, SER6, CYS74 and PHE91 amino acid residues (Figure 3B). Telmisartan, an antagonist of angiotensin II receptor is highly selective for angiotensin II receptors type 1. It is a useful therapeutic choice in the management of patients suffering from hypertension. (Miura et al., 2011). Phaitanthrin D showed (-7.9 Kcal/mol) binding energy with Nsp9 replicase and interacted with the 6W4B. PHE76, CYS74, LEU89, LEU104, LEU107, GLY105, MET102, SER6, VAL8, PRO7, ALA108 and LEU113 amino acid residues (Figure 3C). Phaitanthrin D is natural alkaloid found that exhibits potent anti-tubercular activity against MDR-TB (Kamal et al., 2015). The molecular docking analysis in our study showed the inhibition potential of the top three compounds against Nsp9 replicase ranked by binding energy and best ADME properties as being: Conivaptan > Telmisartan > Phaitanthrin D.
Figure 3.

Figure shows interaction between the active site residues of the Nsp9 replicase protein and ligands with their respective binding energies. (A) Nsp9 replicase (green) with conivaptan (carbon in gray) (Binding energy -8.4 Kcal/mol), (B) Nsp9 replicase (hot-pink) with Telmisartan (carbon in green) (Binding energy -8.1 Kcal/mol), (C) Nsp9 replicase (yellow) with Phaitanthrin D (carbon in dark pink) (Binding energy -7.9 Kcal/mol). The protein backbone is depicted using ribbon structure representation and ligands are depicted using stick model representation. Bond length is depicted in Angstrom. Figure represents strong binding affinity between the hydrophobic pocket of the protein and ligand.
Figure 5.

Molecular interaction between protein and ligand. (A) Represents the Nsp9 replicase interaction with Conivaptan through H-bonding with VAL42 aminoacid (B) Represents the Spike protein interaction with Tegobuvir through H-bonding with ARG355 and ARG466 aminoacid.
Docking results of spike protein of SARS-CoV-2 with another three compounds (Tegobuvir, Bromocriptine and Baicalin) showed best binding energy and were found to be best molecules at the target site of the protein. Out of the 15 compounds, Tegobuvir exhibited the binding energy (-8.1 Kcal/mol) interacting with the PRO337, ALA344, ASN343, PHE342, PHE347, PHE338, GLY339, GLU340 and VAL341 amino acid residues of spike protein (Figure 4A). Tegobuvir showed strong interaction with spike protein at the active site through H-bond with the ARG355 and ARG466 amino acids (Figure 5B). Tegobuvir is a non-nucleoside inhibitor of hepatitis C virus (HCV) RNA replication with proven antiviral activity in the patients suffering from chronic genotype 1 HCV infection. Tegobuvir is an analog of imidazopyridine class inhibitors that selectively targets HCV (Vliegen et al., 2015). However, the most common side effects of Tegobuvir includes cough, dizziness, fatigue and dry mouth (Vliegen et al., 2015). Bromocriptine functions as a serotonin modulator and postsynaptic dopamine receptor clinically used to treat Parkinson’s disease (Kato et al., 2016). Bromocriptine has also shown antiviral activity against dengue virus replication (Kato et al., 2016). Bromocriptine exhibited (-7.7 Kcal/mol) binding affinity with the 6LZG, ASN450, LEU452, ILE468, TYR351, ALA352, SER349, LYS356, GLU340, ASN354, VAL341, THR345, ARG346, PHE347, ALA348 and SER349 amino acid residues of spike protein (Figure 4B). Baicalin exhibited (-7.6 Kcal/mol) binding affinity interacting with the 6LZG, ASN450, ARG346, ALA344, PHE342, GLU340, VAL341, SER399, ASN354, TRP353, ARG466, ILE468, ALA352, PHE400 and PHE347 amino acid residues of spike protein (Figure 4C). Baicalin is a flavonoid derived from Scutellaria baicalensis. Baicalin also exhibits a potent inhibitory effect against viruses such as anti-influenza virus and against chikungunya virus (Chu et al., 2015).
Figure 4.

Figure shows interaction between the active site residues of the spike protein and ligands with their respective binding energies. (A) Spike protein (purple) with Tegobuvir (carbon in yellow) (Binding energy -8.1 Kcal/mol), (B) Spike protein (wheat) with Bromocriptine (carbon in light-blue) (Binding energy -7.7 Kcal/mol), (C) Spike protein (grey) with Baicalin (carbon in light-green) (Binding energy -7.6 Kcal/mol). The protein backbone is depicted using ribbon structure representation and ligands are depicted using stick model representation. Bond length is depicted in Angstrom. Figure represents strong binding affinity between the hydrophobic pocket of the protein and ligand.
The molecular docking analysis ranked the three compounds based on their binding energy and ADME properties as follows: Tegobuvir > Bromocriptine > Baicalin.
In addition to the three best compounds, Conivaptan (-7.4 Kcal/mol), Phaitanthrin D (-7.2 Kcal/mol) and Telmisartan (-7.2 Kcal/mol) also exhibited good docking scores against the spike protein suggesting that these compounds could potentially target both Nsp9 replicase as well as the spike protein.
Molecular dynamics study of Conivaptan with Nsp9 replicase (Figure 6) and Tegobuvir with spike protein (Figure 7) for 100 ns showed strong interactions and stability between proteins and their ligands at the active domain of proteins interacting through water bridges, hydrophobic interactions, and H-bonds. The molecular dynamic study strongly validated the molecular docking data of protein ligand interaction.
Figure 6.

Molecular dynamics and simulation of Conivaptan with Nsp9 replicase complex. (A) Ramachandran plot of Conivaptan-Nsp9 replicase complex represents 102 (95.2%) residues lie in favoured region 4 (3.7%) residues lie in allowed regionand 1 (0.9%) outlier residues. (B) RMSF plot of residue number and C-alpha of Nsp9 replicase at 100 ns simulation. It predicts the fluctuations of the C-alpha atoms; Residues are shown in three letter code with their respective number in green color belong to binding site residues interacting to compound shown in green line. (C) RMSD plot for Cα of Nsp9 replicase in complex with Conivaptan. (D) A timeline representation of the interactions and contacts (H-bonds, hydrophobic, ionic, water bridges) with compound. The top panel shows the total number of specific contacts the protein makes with the ligand over the course of the trajectory. The bottom panel shows which residues interact with the ligand in each trajectory frame.
Figure 7.

Molecular dynamics and simulation of Tegobuvir with spike protein complex. (A) Ramachandran plot of Tegobuvir-spike protein complex represents 179 (92.7%) residues lie in favoured region 13 (6.7%) residues lie in allowed region and 1 (0.5%) outlier residues. (B) RMSF plot of residue number and C-alpha of spike protein at 100 ns simulation. It predicts the fluctuations of the C-alpha atoms; Residues are shown in three letter code with their respective number in green color belong to binding site residues interacting to compound shown in green line. (C) RMSD plot for Cα of spike protein in complex with Tegobuvir. (D) A timeline representation of the interactions and contacts (H-bonds, hydrophobic, ionic, water bridges) with compound. The top panel shows the total number of specific contacts the protein makes with the ligand over the course of the trajectory. The bottom panel shows which residues interact with the ligand in each trajectory frame.
The major criteria for the evaluation of the likeliness of the drug is “Lipinski’s rule of five” suggesting that if a specific ligand with a certain pharmacological and biological activity has chemical and physical properties that would make it as a chosen option for orally active drug for humans (Brito, 2011). Lipinski’s rule describes the molecular properties that are crucial for pharmacokinetics of a drug in the human body for example; absorption, distribution, metabolism, and excretion (ADME) (Brito, 2011). If three or more of Lipinski’s rule of five are violated, the rule of drug likeliness is discarded and the drug is not considered further for use in treatment. ADME studies of the selected 15 compounds showed that out of 15, all virtual hits were successful at passing the test filters.
Conclusion
In view of the current outbreak of the novel coronavirus and rising death toll scenario, novel drug discovery is a challenge constrained by time. Drug repurposing could greatly aid the development of therapeutic drugs for the effective management of COVID-19. Structure-based drug design approaches have developed into valuable drug discovery tools, owing to their synergy and versatility. Here, we described structure-based drug repurposing of a collection of FDA-approved antiviral, anti-malarial, anti-parasitic, anti-fungal, anti-tuberculosis and active phytochemical compounds against Nsp9 replicase and spike protein of SARS-CoV-2. Several molecules were identified as potent inhibitors of Nsp9 replicase (Conivaptan, Telmisartan and Phaitanthrin D) and spike protein (Tegobuvir, Bromocriptine, and Baicalin) of SARS-CoV-2. Interestingly, Conivaptan, Phaitanthrin D and Telmisartan showed good binding affinity with both Nsp9 replicase and the spike protein, suggesting the potential of utilizing these compounds for the inhibition of multiple molecular targets in SARS-CoV-2. We propose that these compounds might be applicable in the management of COVID-19 and should be investigated as potential leads in the drug development against SARS-CoV-2. Further in vitro and in vivo validation of these compounds is warranted.
Acknowledgements
Authors acknowledge Department of Science and Technology-Science and Engineering Research Board (DST- SERB), Government of India for partial financial support.
Funding Statement
Department of Science & Technology- Science and Engineering Research Board (DST-SERB) funded research grant (ECR/2016/001489), Govt. of India.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Boopathi, S., Poma, A. B., & Kolandaivel, P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1758788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bride, M. C., Marjorie, V. Z., & Burtram, C. F. (2014). The coronavirus Nucleocapsid is a multifunctional protein. Viruses, 6, 2991–3018. 10.3390/v6082991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito, M. A. (2011). Pharmacokinetic study with computational tools in the medicinal chemistry course. Brazilian Journal of Pharmaceutical SciencesSciences, 47(4), 797–805. 10.1590/S1984-82502011000400017 [DOI] [Google Scholar]
- Chandel, V., Srivastava, M., Srivastava, A., Asthana, S., & Kumar, D. (2020). In-silico interactions of active phytochemicals with c-Myc EGFR and ERBB2 oncoproteins. Chemical Biology Letters, 7(1), 47–54. [Google Scholar]
- Chaudhuri, S., Symons, J. A., & Deval, J. (2018). Innovation and trends in the development and approval of antiviral medicines: 1987–2017 and beyond. Antiviral Researchres, 155, 76–88. 10.1016/j.antiviral.2018.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, M., Xu, L., Zhang, M., Chu, Z., & Wang, Y. D. (2015). Role of Baicalin in anti- influenza virus A as a potent inducer of IFN-gamma. BioMed rResearch Internationalinternational, 2015, 263630–263611. 2014). 10.55/2015/263630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: a A free web tool to evaluate pharmacokinetics, drug- likeness and medicinal chemistry friendliness of small molecules. Scientific ReportsReports, 7(1), 42717. 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan, S., & Olson, A. J. (2015). Small-molecule library screening by docking with PyRx. Methods in mMolecular bBiology (Clifton, N.J.)N.J.), 1263, 243–250. 10.1007/978-1-4939-2269-7_19 [DOI] [PubMed] [Google Scholar]
- Das, S., Sarmah, S., Lyndem, S., & Roy, A. S. (2020). An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1763201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dene, R. L., Benjamin, S. G., Rhys, N. C., & Jamie, R. (2020). Crystal structure of the SARS-CoV-2 non-structural protein 9. Nsp9. iscience, 28. 10.1016/j.isci.2020.101258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: aAn in silico perspective. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1761882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr, A. R., & Perlman, S. (2015). Coronaviruses: An overview of their replication and pathogenesis. Coronaviruses: Methods and Protocols, 1282, 1–23. 10.1007/978-1-4939-2438-7_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghali, J. K., Farah, J. O., Daifallah, S., Zabalawi, H. A., & Zimmy, H. D. (2009). Conivaptan and its role in the treatment of hyponatremia. Drug Design, Development and Therapy, 3, 253–268. 10.2147/dddt.s4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendaus, M. A. (2019). Remdesivir in the treatment of coronavirus disease 2019 (COVID- 19): a A simplified summary. Journal of Biomolecular Structure and Dynamics, 1–6. 10.1080/07391102.2020.1767691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., Zhang, L., Fan, G., Xu, J., Gu, X., Cheng, Z., Yu, T., Xia, J., Wei, Y., Wu, W., Xie, X., Yin, W., Li, H., Liu, M., … Cao, B. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan. Lancet (London, England)China. Lancet, 395(10223), 497–506. 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal, A., Reddy, B. V. S., Sridevi, B., Ravikumar, A., Venkateswarlu, A., Sravanthi, G., Sridevi, J. P., Yogeeswari, P., & Sriram, D. (2015). Synthesis and biological evaluation of phaitanthrin congeners as anti-mycobacterial agents. Bioorganic & mMedicinal cChemistry Lettersletters, 25(18), 3867–3872. 10.1016/j.bmcl.2015.07.057 [DOI] [PubMed] [Google Scholar]
- Kanchan, A., John, Z., Parvesh, W., Jeroen, R. M., & Rolf, H. (2003). Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science, 300, 1763–1767. 10.1126/science.1085658 [DOI] [PubMed] [Google Scholar]
- Kato, F., Ishida, Y., Oishi, S., Fujii, N., Watanabe, S., Vasudevan, S. G., Tajima, S., Takasaki, T., Suzuki, Y., Ichiyama, K., Yamamoto, N., Yoshii, K., Takashima, I., Kobayashi, T., Miura, T., Igarashi, T., & Hishiki, T. (2016). Novel antiviral activity of bromocriptine against dengue virus replication. Antiviral ResearchRes, 131, 141–147. 10.1016/j.antiviral.2016.04.014 [DOI] [PubMed] [Google Scholar]
- Khan, R. J., Jha, R. K., Amera, G. M., Jain, M., & Singh, E. (2020). Trageting SARS-CoV-2: a A systemic drug repurposing approach to identify promising inhibitors against 3C-protease and 2'-O-ribose methyltransferase. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1753577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S., Sharma, P. P., Shankar, U., Kumar, D., Joshi, S. K., Pena, L., Durvasula, R., Kumar, A., Kempaiah, P., Poonam, & Rathi, B. (2020). Discovery of new hydroxyethylamine analogs against 3CLpro protein target of SARS-CoV-2: Molecular docking, molecular dynamics simulation and structure-activity relationship studies. Journal of Chemical Information and Modeling, 1–17. 10.1021/acs.jcim.0c00326 [DOI] [PubMed] [Google Scholar]
- Kumar, D., Chandel, V., Raj, S., & Rathi, B. (2020). In silico identification of potent FDA approved drugs against coronavirus COVID-19 main protease: A drug repurposing approach. Chemical Biology Letters, 7(3), 166–175. [Google Scholar]
- Lenard, J. (2008). Viral membranes. Encyclopedia of vVirology, 308–314. 10.1016/B978-012374410-4.00530-6 [DOI] [Google Scholar]
- Li, F. (2016). Structure, function, and evolution of coronavirus spike proteins. Annual Review of VirologyVirology, 3(1), 237–261. 10.1146/annurev-virology-110615-042301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, K. P., Ng, L. F. P., & Liu, D. X. (2000). Identification of a novel cleavage activity of the first papain-like proteinase domain encoded by open reading frame 1a of the coronavirus avian infectious bronchitis virus and characterization of the cleavage products. Journal of Virologyvirology, 74(4), 1674–1685. 10.1128/JVI.74.4.1674-1685.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta, S., Chowdhury, P., Gogoi, N., Goswami, N., Borah, D., Kumar, R., Chetia, D., Borah, P., Buragohain, A. K., & Gogoi, B. (2020). Potential anti-viral aFctivity of approved repurposed drug against main protease of SARS-CoV-2: aAn in silico based approach. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.2020.1768902 [DOI] [PubMed] [Google Scholar]
- Miura, S. I., Karnik, S. S., & Saku, K. (2011). Angiotensin II type 1 receptor blockers: cClass effects vs. Molecular effects. Journal of the Renin-Angiotensin-Aldosterone SystemSystem, 12(1), 1–7. 10.1177/1470320310370852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanraj, K., Karthikeyan, B. S., Vivek-Ananth, R. P., Chand, R. P. B., Aparna, S. R., Mangalapandi, P., & Samal, A. (2018). IMPAAT: A curated database of Indian Medicinal Plants. Scientific Reportsreports, 8(1), 4329. 10.1038/s41598-018-22631-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational ChemistryChemistry, 30(16), 2785–2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavizadeh, L., & Ghasemi, S. (2020). Genotype and phenotype of COVID-19: Their roles in pathogenesis. Journal of Microbiology, Immunology and Infection, 1–5. 10.1016/j.jmii.2020.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, K., Lokugamage, K. G., & Makino, S. (2016). Viral and cellular mRNA translation in Coronavirus-infected cells. Advances in vVirus Researchresearch, 96, 165–192. 10.1016/bs.aivir.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle, N. M., Banck, M., & James, C. A. (2011). Open Babel: An open chemical toolbox. J. Cheminform, 33, 1–14. 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagadala, N. S., Syed, K., & Jack, T. (2017). Software for molecular docking: A review. Biophysical Reviewsreviews, 9(2), 91–102. 10.1007/s12551-016-0247-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj, S., Chandel, V., Rathi, B., & Kumar, D. (2020). Understanding the Molecular Mechanism (s) of SARS-CoV2 Infection and Propagation in Human to Discover Potential Preventive and Therapeutic Approach. Coronaviruses, 1(1), 1–13. [Google Scholar]
- Robson, B. (2020). COVID-19 Coronavirus spike protein analysis for synthetic vaccines, a peptidomimetic antagonist, and therapeutic drugs, and analysis of a proposed Achilles’ heel conserved region to minimize probability of escape mutations and drug resistance. Computers in bBiology and Medicinemedicine, 121, 103749. 10.1016/j.compbiomed.2020.103749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2020-1: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY . (2020). Maestro-desmond interoperability tools. Schrödinger. [Google Scholar]
- Schrödinger Release 2020-1: Maestro, Schrödinger, LLC, New York, NY, 2020. Schrödinger Release 2020-1: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY. (2016). Impact, Schrödinger, LLC. Prime, Schrödinger, LLC, New York, NY, 2020. [Google Scholar]
- Seeliger, D., & Groot, B. L. D. (2010). Ligand docking and binding site analysis with PyMOL and Autodock/vina. Journal of cComputer-aAided mMolecular Designdesign, 24(5), 417–422. 10.1007/s10822-010-9352-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton, G., Fry, E., Carter, L., Sainsbury, S., Walter, T., Nettleship, J., Berrow, N., Owens, R., Gilbert, R., Davidson, A., Siddell, S., Poon, L. L. M., Diprose, J., Alderton, D., Walsh, M., Grimes, J. M., & Stuart, D. I. (2004). The nsp9 Replicase Protein of SARS-coronavirus. Structure, 12(2), 341–353. 10.1016/j.str.2004.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touret, F., & Lamballerie, X. d. (2020). Of chloroquine and COVID-19. Antiviral Researchresearch, 177. 104762. 10.1016/j.antiviral.2020., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vliegen, I., Paeshuyse, J., Zhong, W., & Neyts, J. (2015). In vitro combinations containing Tegobuvir are highly efficient in curing cells from HCV replicon and in delaying/preventing the development of drug resistance. Antiviral ResearchRes, 120, 112–121. 10.1016/j.antiviral.2015.05.011 [DOI] [PubMed] [Google Scholar]
- Walls, A. C., Park, Y. J., Tortorici, M. A., Wall, A., McGuire, A. T., & Veesler, D.Alexandra, C. W. (2020). Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell, 181, 281–292. 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, J. P., & Alexandra, C. W. (2019). Structure of MERS-CoV spike glycoprotein in complex with sialoside attachment receptors. nNature sStructural & mMolecular bBiology, 26, 1151–1157. 10.1038/s41594-019-0334-7 [DOI] [PMC free article] [PubMed] [Google Scholar]