

Abstract
The SARS-CoV-2 virus is a major problem in the world right now. Currently, all the attention of research centers and governments globally are focused on the investigation of vaccination studies and the discovery of small molecules that inhibit the SARS-CoV-2 virus in the treatment of patients. The goal of this study was to locate small molecules to be used against COVID19 instead of favipiravir. Favipiravir analogues were selected as drug candidates from the PubChem web tool. The RNA dependent RNA polymerase (RdRp) protein was selected as the target protein as favipiravir inhibits this protein in the human body. Initially, the inhibition activity of the studied compounds against RdRp of different virus types was investigated. Then, the inhibition properties of selected drug candidates and favipiravir were examined in detail against SARS-CoV-2 RdRp proteins. It was found that 2-oxo-1H-pyrazine-3-carboxamide performed better than favipiravir in the results of molecular docking, molecular mechanics Poisson-Boltzmann surface area (MM-PSBA) calculations, and ADME analyses.
Communicated by Ramaswamy H. Sarma
Keywords: COVID19, RNA polymerase, favipiravir, MM-PBSA, ADME
Graphical Abstract

1. Introduction
The COVID19 pandemic, which has high mortality and infectivity, occurred in Wuhan Province of the People's Republic of China in December 2019 and subsequently become one of the world's major health problems. As of April 2020, approximately 1 million infected patients and 50,000 deaths have been observed. Its infectivity is very high, and there is an exponential increase in the number of infections (World Health Organization, 2020). Numerous treatment studies for COVID19, including medicines, anti-virals and herbal products originally used for different diseases are continuing all over the world. Many anti-virals, such as the lopinavir/ritonavir combination, favipiravir and arbidol have been experimented with in the treatment of this pandemic (Lim et al., 2020).
The coronavirus leading to COVID19 is the large and enveloped RNA virus with crown-shaped protrusions. Coronaviruses (CoVs) can cause respiratory, enteric, hepatic and neurological diseases. CoVs are divided into four subfamilies serologically and genotypically, which are α, β, γ and δ-CoVs. SARS coronavirus (SARS-CoVs) and MERS coronavirus (MERS-CoVs) are members of the β-CoV family. In genome analysis, SARS-CoV-2 exhibits 79.5% and 50% similarity with SARS-CoV and MERS-CoV, respectively (Jin et al., 2020).
The SARS-CoV-2 virus has a 30 kb genome size, similar to other β-CoVs, with a nucleocapsid consisting of a genomic RNA and a phosphorylated nucleocapsid (N) protein. The phospholipid double layers contain nucleocapsids that are surrounded by two different types of spike proteins. There are spike glycoprotein trimmers found in all CoVs. Also, some CoVs contain hemagglutinin-esterase. The membrane protein and the envelope protein are among the S proteins in the viral envelope (Elfiky, 2020). The SARS-CoV-2 cell representation is shown in Figure 1.
Figure 1.

SARS-CoV-2 genom and particles.
Like other RNA viruses, SARS-CoV-2 contains the RNA Polymerase 2 enzyme. It performs RNA replication, as well as virus replication (Furuta et al., 2017). Favipiravir is an anti-viral agent that selectively and strongly inhibits the RNA-dependent RNA polymerase (RdRp) of RNA viruses. Favipiravir is effective against a wide range of influenza viruses and subtypes, including strains resistant to existing anti-influenza drugs. Favipiravir also affects other RNA viruses, such as arenaviruses, bunyaviruses, filoviruses, and coronaviruses. Due to these effects, it is thought that favipiravir can be used in the treatment of diseases occurring not only with influenza infection, but also with other RNA viruses (Cai et al., 2020). The results of a study conducted with a total of 80 patients (the experimental group consisted of 35 patients given favipiravir, and the control group consisted of 45 patients given lopinavir/ritonavir) showed that favipiravir had a better anti-viral effect than lopinavir/ritonavir. The study showed that patients treated with favipiravir recover faster than the other treatments. The favipiravir group showed that the patient lungs healed better than in the control group. It was observed that patients receiving favipiravir gave positive results in terms of the virus within four days after treatment, while patients in the control group showed positive results only after 11 days. Finally, it was observed that 91.4% of those given favipiravir showed better results from imaging tests of the lungs, whereas only 62.2% of the control group showed an improvement. While no favourable side effects were observed in the treatment group, favipiravir was shown to have significantly fewer side effects than the lopinavir/ritonavir group (Nguyen & Haenni, 2003).
While viruses are replicating, an RNA with positive polarity is similar to mRNA and can be read directly by host cell ribosomes. RNA with negative polarity is the complement of mRNA; it must be converted to a positive-strand by the enzyme RNA polymerase for the host cell ribosomes to be able to read it (Fodor, 2013). In influenza viruses with negative polarity, during RNA polymerase transcription, viral RNA polymerase synthesizes mRNA using 5ρ-capped RNA primers. During replication, the viral RNA polymerase forms a complementary RNA (cRNA) replication intermediate, which is the complement of the RNA that acts as a pattern for the synthesis of new vRNA copies (Turkish Min. of Health, 2020). During replication, coronaviruses do not contain RNA-dependent RNA polymerase (RdRp) enzymes because they have a positive polarity, but they encode this enzyme into their genome. Coronaviruses have a large RNA genome encoded by viral replicase/polymerase (Bost et al., 2000). Similar to other positive-chain RNA viruses, the SARS-CoV RdRp is estimated to be the central enzyme that forms a replication complex responsible for replication of the viral RNA genome with other viral and cellular proteins (Brockway et al., 2003; Fudo et al., 2016).
In this article, the analogues of favipiravir, with a PubChem ID of 492405, are scanned from the PubChem website (https://pubchem.ncbi.nlm.nih.gov/). The selected whole molecules are minimized by the OPLS3e method using Maestro 12.2. At this stage, the acidity (pH) is determined in the range of 1 − 9. The possible state structures belonging to each compound is calculated at pH= 1–9. As for the target protein, RNA polymerase is investigated in the Protein Data Bank (PDB) web tool. PDB IDs of selected target proteins, which belong to different virus types, are 5FDD, 5I13, 5W44, 6CFP, 6E6V, 6FS8, 6QWL, 6QX3 and 6QX8 (Beylkin et al., 2017; Credille et al., 2019; Fan et al., 2019; Kirchdoerfer & Ward, 2019; Omoto et al., 2018). Some of these proteins are multi-chain while others are single chain. Molecular docking calculations are performed between the studied compounds and target proteins. Then, all results are compared with favipiravir due to favipiravir being selected as the reference material in this study.
As for the second stage of the study, the inhibition activities of the selected drug candidates are re-investigated against SARS-CoV RNA polymerase proteins which are 6NUR and 6NUS (Harder et al., 2016; Jin et al., 2020; Mesecar, 2020). All of these proteins were reported in 2020 and belong to the COVID virus.
The molecular mechanics Poisson-Boltzmann surface binding free energy values of area (MM-PBSA) calculations are completed to investigate the interaction stability between inhibitors and proteins, each at 100ps. Finally, absorption, distribution, metabolism and excretion/toxicity (ADME) analyses are performed for the related compounds and favipiravir.
2. Methods
2.1. Molecular docking and ADME analyses
The studied compounds were downloaded from the PubChem web tool. At this stage, the favipiravir was taken into account, and eleven compounds were determined as favipiravir analogues. The whole compounds are prepared as ligands under a pH condition of 5 ± 4.
As for the target proteins, all proteins were downloaded from the PDB web tool. Firstly, attention was paid to ensure that RNA depended RNA polymerase (RdRp) proteins are obtained from homosapiens. Then, 14 target proteins with the IDs of 5FDD, 5I13, 5W44, 6CFP, 6E6V, 6FS8, 6QWL, 6QX3, 6QX8, 6NUR, 6NUS, 6W63 and 6LU7 were selected. These proteins were minimized using the OPLS3e method to prepare them for docking calculations at pH = 5 ± 4. Active sites of these proteins were prepared using the Maestro 12.2 program (Beylkin et al., 2017; Credille et al., 2019; Fan et al., 2019; Harder et al., 2016; Jin et al., 2020; Kirchdoerfer & Ward, 2019; Mesecar, 2020; Omoto et al., 2018). Favipiravir and its analogues were then examined against different virus types and the better ones were selected for further analyses. Then, the inhibition activity of favipiravir and the selected samples were investigated against RdRp proteins of SARS-CoV. Finally, ADME analyses were performed for favipiravir and the selected samples. All these calculations were performed using the OPLS3e method with the Maestro 12.2 Schrödinger software program (Friesner et al., 2004, 2006; Schrödinger, 2019a, 2019b, 2020). Molecular docking calculations were completed at extra precision (XP). As for the ADME analyses, the Qik-prop module of the Maestro software is used to perform this analysis (Mermer et al., 2019).
2.2. Mm-PBSA binding free energy calculations
Molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) calculations were performed for favipiravir, CID 294642 and CID 76973015. Binding free energy, van der Waals energy, electrostatic energy, kinetic energy and potential energy changes were determined for these ligand–protein structures in 6NUR and 6NUS from 0 ns to 100 ns (Kollman et al., 2000). For these calculations, Nanoscale Molecular Dynamics (NAMD) (Nelson et al., 1996) and Visual Molecular Dynamics (VMD) (Humphrey et al., 1996) software programs were used.where; is the binding free energy, and demonstrates the total free energy of the protein-ligand complex and total free energies of the isolated protein and ligand, respectively. Each of the above terms is also used in the calculation of various energy components, including van der Waals energy, electrostatic energy, polar contribution to internal energy and solvent energy collected from molecular mechanics.
3. Results and discussion
3.1. Favipiravir and its analogues
The PubChem ID, name and structures of favipiravir and selected analogues are given in Table 1.
Table 1.
ID, name and structures of the studied compounds.
| PubChem ID | Name | Structure |
|---|---|---|
| CID 294642 | 2-oxo-1H-pyrazine-3-carboxamide | 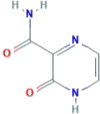 |
| CID 492405 (Favipiravir) | 5-fluoro-2-oxo-1H-pyrazine-3-carboxamide | 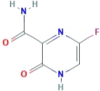 |
| CID 22674959 | 6-fluoro-2-oxo-1H-pyrazine-3-carboxamide | 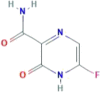 |
| CID 67534452 | N-methyl-2-oxo-1H-pyrazine-3-carboxamide | 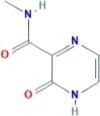 |
| CID 72188728 | 5,6-dideuterio-2-oxo-1H-pyrazine-3-carboxamide | 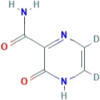 |
| CID 72201087 | 6-deuterio-5-fluoro-2-oxo-1H-pyrazine-3-carboxamide | 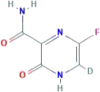 |
| CID 76973015 | 2-oxo-(1,4-15N2)1H-pyrazine-3-(15N)carboxamide | 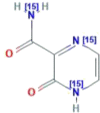 |
| CID 76973034 | 5,6-dideuterio-2-oxo-(5,6-13C2)1H-pyrazine-3-carboxamide | 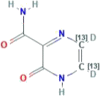 |
| CID 76973035 | 5,6-dideuterio-2-oxo-(1,4-15N2)1H-pyrazine-3-(15N)carboxamide | 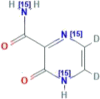 |
| CID 89869520 | 6-fluoro-4-methyl-3-oxopyrazine-2-carboxamide | 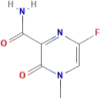 |
| CID 123273976 | 3-(aminomethyl)-5-fluoro-1H-pyrazin-2-one | 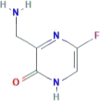 |
| CID 135001386 | 5-amino-2-oxo-1H-pyrazine-3-carboxamide | 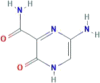 |
All compounds were minimized with the OPLS3e method, and a pH of 5 ± 4 was used in the calculations. The possible state structure at pH = 5 ± 4 was determined for all studied compounds. The number of total investigated structures with their possible states were determined as nearly 120 compounds.
3.2. Determination of active compounds
Molecular docking calculations were performed between the selected ligands and target proteins. It was found that CID 22674959 and CID 89869520 have no activity against RdRp proteins. As for the other samples, compounds and their possible states were docked with some of the selected proteins. Only the original structure of some of the studied compounds, such as favipiravir, were found to be active while both original and possible state structures, like CID 294642 were found to be active. The docking results are given in Table 2. The table lists each protein, its interaction with each drug candidate, and the interacting ligand structure types.
Table 2.
The ligandsa that interacted with the target proteins.
| Compound ID | 5FDD | 5I13 | 5W44 | 6CFP | 6E6V | 6FS8 | 6QWL | 6QX3 | 6QX8 |
|---|---|---|---|---|---|---|---|---|---|
| 294642 | – | – | OS, PS | – | – | OS, PS | – | – | OS |
| 492405b | – | – | OS | – | – | OS | – | – | OS |
| 22674959 | – | – | – | – | – | – | – | – | – |
| 67534452 | – | – | – | – | – | OS | OS, PS | – | – |
| 72188728 | OS | – | – | – | OS | – | – | – | – |
| 72201087 | – | – | OS | – | – | OS | – | OS | – |
| 76973015 | – | OS, PS | – | – | – | OS | – | – | OS |
| 76973034 | – | – | – | – | – | OS, PS | – | – | – |
| 76973035 | – | OS | – | – | – | – | – | – | – |
| 89869520 | – | – | – | – | – | – | – | – | – |
| 123273976 | – | OS | – | – | – | – | – | – | – |
| 135001386 | OS, PS | OS | OS | – | – | – | – | – | – |
aOS: Original Structure; PS: Possible State.
bCID 492405 is favipiravir.
According to Table 2, the 6CFP protein is not inhibited by any ligands. Additionally, the CID 22674959 and CID 89869520 compounds are found to be inactive as they failed to interact with the proteins. As for favipiravir, it interacted with 5W44, 6FS8 and 6QX8. CID 294642 and favipiravir inhibit the same proteins. Therefore, it can be said that these structure have similar properties. However, the original structure and possible state structures of CID 294642 interacted with 5W44, while only the original structure of favipiravir inhibits the protein. Considering the total number of interacting structures, CID 294642 has an advantage in respect to favipiravir. In terms of the total number of interacting molecules, CID 294642 exhibits better properties than the others. The results of CID 76973015 and CID 1350011386 are better than that of favipiravir because the possible state structures of them both inhibit the target proteins too. The docking score (DS), van der Waals energy (Evdw), coulomb energy (ECoul) and total interaction energy (ETotal) of CID 294642, CID 76973015, CID 1350011386 and favipiravir are given in Table 3.
Table 3.
The calculated docking score, van der Waals energy and coulomb energy for selected drug candidates.
| Compound ID | DSa | Evdwa | ECoula | ETotala |
|---|---|---|---|---|
| 492405 | −2.804 | −12.669 | −10.216 | −22.885 |
| 294642 | −3.279 | −14.495 | −12.784 | −27.280 |
| 76973015 | −3.320 | −15.385 | −6.524 | −21.910 |
| 1350011386 | −2.223 | −14.031 | −11.372 | −25.404 |
a in kcal/mol.
To determine the better drug candidate, the first criteria is the docking score, and the total energy is the second criteria. This is because, key-lock harmony is vitally important in drug protein interactions. According to Table 3, the docking score of CID 1350011386 is worse than that of favipiravir, and therefore, is excluded from further analyses. Because the docking score implies the key-lock compatibility in the ligand-receptor complex, it is desired that the docking score of any ligand should be better than that of the reference substance. As for the other compounds, the docking and van der Waals energy scores are better than that of favipiravir. Although total interaction energy in CID 76973015 is smaller than favipiravir, they are closely similar to each other. As a result, CID 294642 and 76973015 are selected for further analyses. The docking structures for favipiravir and CID 294642 with the 6QX8 protein are represented in Figure 2.
Figure 2.

Docking structures of favipiravir and CID 294642 with 6QX8.
3.3. The interaction with COVID19 RNA depends on the RNA polymerase protein
RNA polymerase proteins with PDB IDs of 6NUR and 6NUS were reported in late 2019 and early 2020 as belonging to SARS-CoV (Jin et al., 2020; Kirchdoerfer & Ward, 2019; Mesecar, 2020). The active region of related proteins was calculated by using the Maestro 12.2 software program, and this region of 6NUR is represented in Figure 3.
Figure 3.

The active region of the 6NUR protein.
According to Figure 3, the active sites of 6NUR are represented with a dummy ball and different colored (blue, red and yellow) surfaces. These regions are the active sites of the RdRp proteins for inhibitors. The three colors red, blue and yellow imply the hydrogen bond acceptor, hydrogen bond donor and hydrophobic interaction regions, respectively. Thus, the selected compounds were to dock with this region in each target protein. The inhibition properties of the selected compounds and favipiravir are determined from the docking calculations, and the active and passive structures against 6NUR and 6NUS are expressed in Table 4.
Table 4.
The active and passive compounds against 6NUR and 6NUS proteins.
| Compound ID | 6NUR | 6NUS |
|---|---|---|
| Favipiravir | NO | YES |
| 294642 | YES | YES |
| 76973015 | YES | YES |
According to Table 4, favipiravir inhibited only the 6NUS protein while CID 294642 and CID 76973015 inhibited both proteins. The complex (ligand–receptor) structures and the ligand binding domain electrostatic potential map of 6NUS are represented in Figure 4. Additionally, DS, Evdw, ECoul and ETotal are reported in Table 5 for each target protein.
Figure 4.

Electrostatic potential map of the active site in 6NUS and ligand-receptor complexes.
Table 5.
Molecular docking results.
| Compound ID | DS a | Evdwa | ECoula | ETotala |
|---|---|---|---|---|
| 6NUR | ||||
| 294642 | −4.063 | −13.498 | −11.839 | −25.336 |
| 76973015 | −4.063 | −13.498 | −11.839 | −25.336 |
| 6NUS | ||||
| Favipriavir | −5.001 | −16.755 | −10.427 | −27.182 |
| 294642 | −5.878 | −16.612 | −11.739 | −28.350 |
| 76973015 | −5.345 | −16.218 | −11.205 | −27.423 |
ain kcal/mol.
According to Table 5, docking results of CID 294642 and CID 76973015 are better than that of favipiravir. If the results are examined in detail, especially the docking score and total interaction energy, it can be easily said that CID 294642 shows better results than the other molecules. For 6NUR, CID 294642 and CID 76973015 have similar properties. However, the calculated results of CID 294642 are superior compared to the calculated results of the others. As a result, CID 294642 and CID 76973015 have the potential to be good alternatives of favipiravir in the treatment of patients with COVID19.
The interaction scheme of related ligands against 6NUS is represented in Figure 5.
Figure 5.

The interaction schema at the ligand-receptor complexes in the 6NUS protein.
According to Figure 5, the hydrogen bond is especially significant in the interaction between the receptors of the ligands. Furthermore, hydrophobic, polar, charged (negative and positive), glycine and solvent exposure plays a significant role in the interaction.
3.4. Sar studies
3.4.1. Mm-PSBA calculations
Molecular docking has some disadvantages in the analysis of binding stability at the nano-second level of the ligand-protein complex. The protein accepts a rigid structure in the docking calculations while the ligand is flexible. Molecular mechanics-Poisson-Boltzmann surface area (MM-PSBA) calculations should be made for a more detailed examination of the stability between the ligand and protein. In this calculation, the ligand and protein are flexible and solvent molecules surround the entire structure. In this study, the binding stabilities of protein-ligand structures are investigated in each 100ps. Gibbs free energy of protein and ligand-protein structures are represented in Figures 6 and 7 for 6NUR and 6NUS proteins, respectively.
Figure 6.

Change of Gibbs free energy values of protein and inhibitors in every five ns intervals.
Figure 7.

Change of Gibbs free energy values of protein and inhibitors in every five ns intervals.
The binding free energy changes and their deviations in each five ns intervals are given in Table 6. The van der Waals energy, electrostatic energy, kinetic energy and potential energy changes are given in supp. Table S1-S4, respectively.
Table 6.
Representation of ΔG of the binding free energy (kcal/mol) and standard deviation values of proteins and inhibitors.
| Second | 6NUR-CID 294642 | 6NUR-CID 76973015 | 6NUS-CID 294642 | 6NUS-CID 76973015 | 6NUS- Favipiravir | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 369.9 | ±510.8 | −89.5 | ±1586.5 | −249.8 | ±214.7 | 392.1 | ±399.8 | −174.6 | ±174.9 |
| 10 | −92.0 | ±377.5 | −776.8 | ±409.7 | −65.0 | ±181.3 | −349.4 | ±252.6 | 166.8 | ±466.1 |
| 15 | 291.7 | ±321.2 | −457.2 | ±332.1 | 166.7 | ±314.5 | −208.4 | ±149.2 | 88.4 | ±215.5 |
| 20 | 101.6 | ±183.7 | −45.8 | ±314.5 | 402.2 | ±301.7 | 373.7 | ±234.3 | −417.0 | ±266.9 |
| 25 | −288.4 | ±255.4 | −379.5 | ±418.3 | 150.4 | ±138.0 | −179.4 | ±349.9 | −583.2 | ±270.1 |
| 30 | 105.3 | ±274.7 | −886.7 | ±204.1 | 296.1 | ±191.6 | −34.4 | ±204.4 | −253.4 | ±263.7 |
| 35 | −153.7 | ±289.1 | 232.9 | ±580.1 | −356.1 | ±428.6 | −839.2 | ±189.6 | −463.4 | ±209.8 |
| 40 | −644.5 | ±334.6 | −307.9 | ±303.5 | −190.7 | ±224.5 | 53.6 | ±355.1 | 301.7 | ±356.5 |
| 45 | 221.2 | ±262.8 | 326.1 | ±363.7 | −736.9 | ±319.2 | 215.9 | ±279.9 | −735.2 | ±220.4 |
| 50 | −1015.9 | ±404.0 | −502.1 | ±369.3 | −696.1 | ±178.9 | −609.2 | ±314.5 | −594.7 | ±200.4 |
| 55 | 1226.8 | ±585.2 | 949.4 | ±468.8 | −974.3 | ±244.3 | −300.5 | ±225.9 | −481.9 | ±218.8 |
| 60 | 174.1 | ±335.3 | 238.7 | ±275.0 | −834.9 | ±480.5 | 362.1 | ±241.0 | 674.2 | ±343.9 |
| 65 | 377.0 | ±186.5 | −366.5 | ±357.5 | −112.0 | ±531.3 | −587.2 | ±425.0 | −399.1 | ±443.1 |
| 70 | 585.1 | ±275.5 | 485.8 | ±269.8 | −578.3 | ±330.2 | −77.2 | ±234.2 | 82.6 | ±317.6 |
| 75 | 17.5 | ±501.3 | 365.9 | ±369.3 | −230.1 | ±152.3 | −16.9 | ±264.7 | 48.5 | ±209.9 |
| 80 | −862.8 | ±349.6 | −1107.2 | ±392.5 | 249.1 | ±327.5 | 115.2 | ±278.9 | 457.3 | ±234.6 |
| 85 | −113.7 | ±487.8 | 241.0 | ±441.6 | 880.7 | ±481.2 | 793.9 | ±272.6 | 1018.6 | ±307.1 |
| 90 | −532.9 | ±331.9 | −540.7 | ±332.5 | 273.7 | ±265.9 | 124.7 | ±208.2 | 231.4 | ±188.3 |
| 95 | −707.4 | ±231.0 | 211.3 | ±268.7 | 941.9 | ±202.8 | 551.7 | ±316.8 | 284.0 | ±130.5 |
| 100 | −266.2 | ±265.3 | −452.3 | ±277.5 | −74.4 | ±318.7 | −176.1 | ±307.3 | −46.3 | ±260.7 |
Inhibitor activities of favipiravir and its analogues against RNA dependent RNA polymerase (RdRp) proteins were compared. With this comparison, calculations were made to support the estimation of the free energies of binding using the molecular mechanics-Poisson-Boltzmann surface area (MM-PBSA) method. The negative value of the related parameters indicates better binding (Gupta et al., 2020). As a result of the calculations, the average values of Gibbs free energies were −36.6 kcal/mol for 6NUR-CID294642; and 43.7 kcal/mol for 6NUR-CID76973015. According to these results, CID294642 exhibits better inhibiting properties for the 6NUR protein. Moreover, CID294642 becomes more stable with 6NUR more quickly. As for 6NUS, CID294642 and favipiravir exhibit better properties than CID76973015 due to the Gibbs free energy changes. When these results are examined in more detail, the negative Gibbs free energy in the CID294642-6NUS complex is more than that of the favipiravir-6NUS complex. As a result, CID294642 is selected as the best drug candidate for the SARS-CoV-2 virus via MM-PSBA calculations.
3.4.2. Adme analysis
Absorption, distribution, metabolism, and excretion (ADME) is related to the pharmacokinetics and pharmacology properties of drug candidates. Absorption is related to the uptake into the bloodstream of the compound to reach the target tissue. Distribution is related to the carrying of the compounds in the circulatory system. Metabolism is related to the breaking down of compounds as soon as entering the body and excretion is related to the removing of the compound from the body. These properties are vitally important for drug candidates because ADME analyses give us significant clues about the likely success of the drug. There are some parameters to investigate the ADME properties of drug candidates such as the solute total SASA, QPlogHERG, RuleOfFive, etc.
ADME analyses of CID 294642, CID 76973015 and favipiravir were completed and all results were compared with reference ranges. In this study, thirty-one parameters, which are the most commonly used, are calculated by the Maestro program. The ADME results of selected compounds are given in Table 7.
Table 7.
ADME properties of molecules.
| CID 294642 | Favipiravir | CID 76973015 | Reference Range | |
|---|---|---|---|---|
| Solute Molecular Weight | 139.1 | 157.1 | 139.1 | 130 − 725 |
| Solute Dipole Moment (D) | 8.1 | 6.1 | 7.0 | 1.0 − 12.5 |
| Solute Total SASA | 308.9 | 315.6 | 309.1 | 300 − 1000 |
| Solute Hydrophobic SASA | 0 | 0 | 0 | 0 − 750 |
| Solute Hydrophilic SASA | 207.3 | 204.0 | 206.2 | 7.0 − 330 |
| Solute Carbon Pi SASA | 101.5 | 62.7 | 103.0 | 0 − 450 |
| Solute Weakly Polar SASA | 0 | 48.889 | 0 | 0 − 175 |
| Solute Molecular Volume (A3) | 461 | 475 | 463 | 500 − 2000 |
| Solute as Donor-Hyrogen Bonds | 1 | 1 | 1 | 0.0 − 6.0 |
| Solute as Acceptor-Hydrogen | 4 | 4 | 4 | 2.0 − 20.0 |
| Solute Globularity (Sphere =1) | 0.9 | 0.9 | 0.9 | 0.75 − 0.95 |
| QP Polarizability (A3) | 12.7 | 12.8 | 12.7 | 13.0 − 70.0 |
| QP log p for hexadecane/gas | 5.0 | 4.5 | 5.1 | 4.0 − 18.0 |
| QP log p for octanol/gas | 9.6 | 9.0 | 9.1 | 8.0 − 35.0 |
| QP log p for water/gas | 7.7 | 7.5 | 7.8 | 4.0 − 45.0 |
| QP log p for octanol/water | −0.6 | −0.4 | −0.6 | −2 − 6.5 |
| QP log S aqueous solubility | −0.6 | −1.2 | −0.6 | −6.5 − 0.5 |
| QP log S-conformation independent | −1.0 | −1.3 | −1.0 | −6.5 − 0.5 |
| QPlogHERG | -3.1 | -2.9 | -3.0 | (concern below −5) |
| QPPCaco (nm/sec) | 107.1 | 115.2 | 109.9 | 25 < X < 500 |
| QPlogBB | −1.1 | −0.9 | −1.1 | −3 − 1.2 |
| QPPMDCK (nm/sec) | 44.2 | 88.7 | 45.5 | 25 < X < 500 |
| QPlogKp | −4.9 | −5.0 | −4.9 | (-8.0) – (-1.0) |
| IP (ev) | 9.4 | 9.6 | 9.6 | 7.9 − 10.5 |
| EA (eV) | 1.1 | 1.5 | 1.0 | 0.9 − 1.7 |
| #metab | 2 | 1 | 2 | 1.0 − 8.0 |
| QPlogKhsa | −0.8 | −0.7 | −0.8 | −1.5 − 1.5 |
| % Human Oral Absorption | 59.7 | 61.4 | 60.0 | <25% is poor >80% is high |
| PSA | 105.1 | 105.0 | 105.4 | 7 − 200 |
| RuleOfFive | 0 | 0 | 0 | Maximum is 3 |
| RuleOfThree | 0 | 0 | 0 | Maximum is 3 |
According to Table 7, the calculated parameters of CID 264642, CID 76973015 and favipiravir are mainly in the desired range. There are three deviations, which are, solute molar volume, QP polarizability and QPlogHERG. In these parameters, the results of selected compounds are out of the desired range. The meanings of the investigated parameters are given below.
Many parameters obtained from the calculations have been examined for the future probability of ligand molecules to be drugs. The first of these parameters is the Solute Molecular Weight of the molecules, which is desired to be between 130-725 g/mol. Molecules are required to be between the Solute Dipole Moment of 1.0-12.5 Debye. Many other parameters are indicated as follows - Solute Total SASA: total solvent-accessible surface area; Solute Carbom Pi SASA: Van der Waals surface area of polar nitrogen and oxygen atoms and carbonyl carbon atoms; QPlogPo/w: Predicted octanol/water partition coefficient; QPlogS: Predicted aqueous solubility; log S: S in mol dm-3 is the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid; QPlogBB: brain/blood partition coefficient; QPlogKhsa: binding to human serum albumin; Apparent Caco-2 Permeability: Predicted apparent Caco-2 cell permeability (nm/s) where Caco-2 cells are a model for the gut–blood barrier; QP log BB for brain/blood: Predicted brain/blood partition coefficient for orally delivered drugs; Apparent MDCK Permeability: Predicted apparent MDCK cell permeability (nm/s) where MDCK cells are considered to be a good mimic for the blood-brain barrier; % Human Oral Absorption in Gl: Human oral absorption on a 0-100% scale (>80% is high, <25% is poor); Solute as Donor-Hydrogen Bonds: Predicted number of donor hydrogen bonds; Solute as Acceptor-Hydrogen: Predicted number of acceptor hydrogen bond; QP log p for X/Y: Predicted X/Y partition coefficient; IC50 value for the blockage of HERG K + channels (acceptable range: above 25.0); Solute CdW Polar SA (PSA): total polar surface area (Acar et al., 2019; Bicak et al., 2019; Budama-Kilinc et al., 2018; Ertas et al., 2019; Kecel-Gunduz et al., 2020; Menteşe et al., 2019; Mermer et al., 2019; Sari et al., 2019; Singh & Bast, 2014); Lipinski Rule of 5 Violations: Number of violations of Lipinski's rule of five where the rules are: mol_MW (molecular weight of the molecule) < 500; QPlogPo/w (predicted octanol/water partition coefficient) < 5; donorHB (hydrogen-bond donor atoms) ≤ 5; accptHB (hydrogen-bond acceptor atoms) ≤ 10 where compounds that provide these rules are considered as drug-like molecules (Saglık et al., 2019a; Saglık et al., 2019 b; Lipinski et al., 1997; Jorgensen & Duffy, 2002); and Jorgensen Rule of 3 Violations: Number of violations of Jorgensen's rule of three where the three rules are: QPlogS (predicted aqueous solubility) > −5.7, QPPCaco (predicted apparent Caco-2 cell permeability in nm/s) > 22 nm/s, # Primary Metabolites < 7 where compounds with fewer (and preferably no) violations of these rules are more likely to be orally available agents. When the numerical values of these parameters are very close to each other, as can be seen from Table 7, it can be concluded that the results obtained show the potential future use of these three molecules as drugs, and will not cause a problem according to ADME analyses.
4. Conclusion
Currently countries worldwide are taking precautions against the COVID19 epidemic, and active molecule research and vaccination studies against the SARS-CoV-2 virus are of great importance. Most research has focused on COVID19. Favipiravir is one of the drugs used in the treatment of the virus. In this study, a target molecule investigation is completed against RNA polymerase of different virus types and RNA polymerase proteins of the SARS-CoV-2 virus. The drug candidates are identified as analogues of favipiravir from PubChem. All analyzes are performed in silico, and the target proteins selected from the protein data bank are 5FDD, 5I13, 5W44, 6CFP, 6E6V, 6FS8, 6QWL, 6QX3, 6QX8, 6NUR and 6NUS. The last four of the selected proteins belong to the SARS-CoV virus. As a result of docking analysis, drug candidates which may be more effective than favipiravir are identified. The PubChem IDs of these drug candidates are CID 294642 and CID 76973015. The ADME of related compounds of the two drug candidates and favipiravir are performed. Additionally, MM-PSBA calculations are performed and it is found that CID 294642 exhibits superior activity. As a result, CID 294642 can be a good candidate as an antiviral drug against SARS-CoV-2. Accordingly, in vitro and in vivo research is certainly recommended.
Supplementary Material
Acknowledgements
This work was supported by the Scientific Research Project Fund of Sivas Cumhuriyet University (CUBAP) under the project number RGD-020. We thank all reviewers who helped improve this article.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Acar, M. F., Sari, S., & Dalkara, S. (2019). Synthesis, in vivo anticonvulsant testing, and molecular modeling studies of new nafimidone derivatives. Drug Development Research, 80(5), 606–616. 10.1002/ddr.21538 [DOI] [PubMed] [Google Scholar]
- Beylkin, D., Kumar, G., Zhou, W., Park, J., Jeevan, T., Lagisetti, C., Harfoot, R., Webby, R. J., White, S. W., & Webb, T. R. (2017). Protein-structure assisted optimization of 4, 5-dihydroxypyrimidine-6-carboxamide inhibitors of influenza virus endonuclease. Scientific Reports, 7(1), 1–12. 10.1038/s41598-017-17419-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bıçak, B., Gündüz, S. K., Kökcü, Y., Özel, A. E., & Akyüz, S. (2019). Molecular docking and molecular dynamics studies of L-glycyl-L-glutamic acid dipeptide. Bilge International Journal of Science and Technology Research, 3(1), 1–9. 10.30516/bilgesci.476841 [DOI] [Google Scholar]
- Bost, A. G., Carnahan, R. H., Lu, X. T., & Denison, M. R. (2000). Four proteins processed from the replicase gene polyprotein of mouse hepatitis virus colocalize in the cell periphery and adjacent to sites of virion assembly. Journal of Virology, 74(7), 3379–3387. 10.1021/acs.biochem.5b01087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockway, S. M., Clay, C. T., Lu, X. T., & Denison, M. R. (2003). Characterization of the expression, intracellular localization, and replication complex association of the putative mouse hepatitis virus RNA-dependent RNA polymerase. Journal of Virology, 77(19), 10515–10527. 10.1128/jvi.77.19.10515-10527.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budama-Kilinc, Y., Cakir-Koc, R., Kecel-Gunduz, S., Zorlu, T., Kokcu, Y., Bicak, B., Karavelioglu, Z., & Ozel, A. E. (2018). Papain loaded poly(ε-caprolactone) nanoparticles: In-silico and in-vitro studies. Journal of Fluorescence, 28(5), 1127–1142. 10.1007/s10895-018-2276-6 [DOI] [PubMed] [Google Scholar]
- Cai, Q., Yang, M., Liu, D., Chen, J., Shu, D., Xia, J., Liao, X., Gu, Y., Cai, Q., Yang, Y., Shen, C., Li, X., Peng, L., Huang, D., Zhang, J., Zhang, S., Wang, F., Liu, J., Chen, L., … Liu, L. (2020). Experimental treatment with favipiravir for COVID-19: An open-label control study. Engineering. 10.1016/j.eng.2020.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Credille, C. V., Morrison, C. N., Stokes, R. W., Dick, B. L., Feng, Y., Sun, J., Chen, Y., & Cohen, S. M. (2019). SAR exploration of tight-binding inhibitors of influenza virus PA endonuclease. Journal of Medicinal Chemistry, 62(21), 9438–9449. 10.1021/acs.jmedchem.9b00747 [DOI] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020). Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sciences, 253, 117592. 10.1016/j.lfs.2020.117592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertas, M., Sahin, Z., Bulbul, E. F., Bender, C., Biltekin, S. N., Berk, B., Yurttas, L., Nalbur, A. M., Celik, H., & Demirayak, Ş. (2019). Potent ribonucleotide reductase inhibitors: Thiazole‐containing thiosemicarbazone derivatives. Archiv Der Pharmazie, 352(11), 1900033. 10.1002/ardp.201900033 [DOI] [PubMed] [Google Scholar]
- Fan, H., Walker, A. P., Carrique, L., Keown, J. R., Martin, I. S., Karia, D., Sharps, J., Hengrung, N., Pardon, E., Steyaert, J., Grimes, J. M., & Fodor, E. (2019). Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature, 573(7773), 287–290. 10.1038/s41586-019-1530-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor, E. (2013). The RNA polymerase of influenza a virus: Mechanisms of viral transcription and replication. Acta Virologica, 57(2), 113–122. 10.4149/av_2013_02_113 [DOI] [PubMed] [Google Scholar]
- Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., Repasky, M. P., Knoll, E. H., Shelley, M., Perry, J. K., Shaw, D. E., Francis, P., & Shenkin, P. S. (2004). Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of Medicinal Chemistry, 47(7), 1739–1749. 10.1021/jm0306430 [DOI] [PubMed] [Google Scholar]
- Friesner, R. A., Murphy, R. B., Repasky, M. P., Frye, L. L., Greenwood, J. R., Halgren, T. A., Sanschagrin, P. C., & Mainz, D. T. (2006). Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. Journal of Medicinal Chemistry, 49(21), 6177–6196. 10.1021/jm051256o [DOI] [PubMed] [Google Scholar]
- Fudo, S., Yamamoto, N., Nukaga, M., Odagiri, T., Tashiro, M., & Hoshino, T. (2016). Two distinctive binding modes of endonuclease inhibitors to the N-terminal region of influenza virus polymerase acidic subunit. Biochemistry, 55(18), 2646–2660. 10.1021/acs.biochem.5b01087 [DOI] [PubMed] [Google Scholar]
- Furuta, Y., Komeno, T., & Nakamura, T. (2017). Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences, 93(7), 449–463. 10.2183/pjab.93.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, M. K., Vemula, S., Donde, R., Gouda, G., Behera, L., & Vadde, R. (2020). In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.2020.1751300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder, E., Damm, W., Maple, J., Wu, C., Reboul, M., Xiang, J. Y., Wang, L., Lupyan, D., Dahlgren, M. K., Knight, J. L., Kaus, J. W., Cerutti, D. S., Krilov, G., Jorgensen, W. L., Abel, R., & Friesner, R. A. (2016). OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. Journal of Chemical Theory and Computation, 12(1), 281–296. 10.1021/acs.jctc.5b00864 [DOI] [PubMed] [Google Scholar]
- Humphrey, W., Dalke, A., & Schulten, K. (1996). VMD: Visual molecular dynamics. Journal of Molecular Graphics, 14(1), 33–38. 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- Hunt, R. (2020). Coronaviruses, colds and sars. Microbiology and immunology. https://www.microbiologybook.org/virol/coronaviruses.htm
- Jin, Y., Yang, H., Ji, W., Wu, W., Chen, S., Zhang, W., & Duan, G. (2020). Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses, 12(4), 372. 10.3390/v12040372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., … Yang, H. (2020). Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature, 582, 289–293. 10.1101/2020.02.26.964882 [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., & Duffy, E. M. (2002). Prediction of drug solubility from structure. Advanced Drug Delivery Reviews, 54(3), 355–366. 10.1016/S0169-409X(02)00008-X [DOI] [PubMed] [Google Scholar]
- Kecel-Gunduz, S., Budama-Kilinc, Y., Cakir-Koc, R., Zorlu, T., Bicak, B., Kokcu, Y., Ozel, A. E., & Akyuz, S. (2020). In Silico design of AVP (4–5) peptide and synthesis, characterization and in vitro activity of chitosan nanoparticles. DARU: Journal of Faculty of Pharmacy, Tehran University of Medical Sciences, 28(1), 139–157. 10.1007/s40199-019-00325-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchdoerfer, R. N., & Ward, A. B. (2019). Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nature Communications, 10(1), 1–9. 10.1038/s41467-019-10280-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman, P. A., Massova, I., Reyes, C., Kuhn, B., Huo, S., Chong, L., Lee, M., Lee, T., Duan, Y., Wang, W., Donini, O., Cieplak, P., Srinivasan, J., Case, D. A., Cheatham, T. E. 3rd., &, (2000). Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Accounts of Chemical Research, 33(12), 889–897. 10.1021/ar000033j [DOI] [PubMed] [Google Scholar]
- Lim, J., S., Jeon, S., Shin, H. Y., Kim, M. J., Seong, Y. M., Lee, W. J., Choe, K. W., Kang, Y. M., Lee, B., & Park, S. J. (2020). Case of the index patient who caused tertiary transmission of COVID-19 infection in Korea: The application of lopinavir/ritonavir for the treatment of COVID-19 infected pneumonia monitored by quantitative RT-PCR. Journal of Korean Medical Science, 35(6), e79. 10.3346/jkms.2020.35.e79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 23(1–3), 3–25. 10.1016/S0169-409X(96)00423-1 [DOI] [PubMed] [Google Scholar]
- Menteşe, E., Emirik, M., & Sökmen, B. B. (2019). Design, molecular docking and synthesis of novel 5,6-dichloro-2-methyl-1H-benzimidazole derivatives as potential urease enzyme inhibitors. Bioorganic Chemistry, 86, 151–158. 10.1016/j.bioorg.2019.01.061 [DOI] [PubMed] [Google Scholar]
- Mermer, A., Demirbas, N., Uslu, H., Demirbas, A., Ceylan, S., & Sirin, Y. (2019). Synthesis of novel Schiff bases using green chemistry techniques; antimicrobial, antioxidant, antiurease activity screening and molecular docking studies. Journal of Molecular Structure, 1181, 412–422. 10.1016/j.molstruc.2018.12.114 [DOI] [Google Scholar]
- Mesecar, A. D. (2020). Structure of COVID-19 main protease bound to potent broad-spectrum non-covalent inhibitor X77. 10.2210/pdb6w63/pdb [DOI]
- Nelson, M. T., Humphrey, W., Gursoy, A., Dalke, A., Kalé, L. V., Skeel, R. D., & Schulten, K. (1996). NAMD: A parallel, object-oriented molecular dynamics program. The International Journal of Supercomputer Applications and High Performance Computing, 10(4), 251–268. 10.1177/109434209601000401 [DOI] [Google Scholar]
- Nguyen, M., & Haenni, A. L. (2003). Expression strategies of ambisense viruses. Virus Research, 93(2), 141–150. 10.1016/S0168-1702(03)00094-7 [DOI] [PubMed] [Google Scholar]
- Omoto, S., Speranzini, V., Hashimoto, T., Noshi, T., Yamaguchi, H., Kawai, M., Kawaguchi, K., Uehara, T., Shishido, T., Naito, A., & Cusack, S. (2018). Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Scientific Reports, 8(1), 1–15. 10.1038/s41598-018-27890-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sağlık, B. N., Çavuşoğlu, B. K., Osmaniye, D., Levent, S., Çevik, U. A., Ilgın, S., Özkay, Y., Kaplancıklı, Z. A., & Öztürk, Y. (2019. b). In vitro and in silico evaluation of new thiazole compounds as monoamine oxidase inhibitors. Bioorganic Chemistry, 85, 97–108. 10.1016/j.bioorg.2018.12.019 [DOI] [PubMed] [Google Scholar]
- Sağlık, B. N., Çevik, U. A., Osmaniye, D., Levent, S., Çavuşoğlu, B. K., Demir, Y., Ilgın, S., Özkay, Y., Koparal, A. S., Beydemir, Ş., & Kaplancıklı, Z. A. (2019. a). Synthesis, molecular docking analysis and carbonic anhydrase I-II inhibitory evaluation of new sulfonamide derivatives. Bioorganic Chemistry, 91, 103153. 10.1016/j.bioorg.2019.103153 [DOI] [PubMed] [Google Scholar]
- Sari, S., Barut, B., Özel, A., Kuruüzüm-Uz, A., & Şöhretoğlu, D. (2019). Tyrosinase and α-glucosidase inhibitory potential of compounds isolated from Quercus coccifera bark: In vitro and in silico perspectives. Bioorganic Chemistry, 86, 296–304. 10.1016/j.bioorg.2019.02.015 [DOI] [PubMed] [Google Scholar]
- Schrödinger . (2019. a). Schrödinger Release 2019-4: LigPrep. Schrödinger, LLC. [Google Scholar]
- Schrödinger . (2019. b). Schrödinger Release 2019-4: Maestro. Schrödinger, LLC. [Google Scholar]
- Schrödinger . (2020). Schrödinger Release 2020-1: QikProp. Schrödinger, LLC. [Google Scholar]
- Singh, P., & Bast, F. (2014). In silico molecular docking study of natural compounds on wild and mutated epidermal growth factor receptor. Medicinal Chemistry Research, 23(12), 5074–5085. 10.1007/s00044-014-1090-1 [DOI] [Google Scholar]
- Turkish Min. of Health. (2020). COVID-19 (SARS-CoV-2) infection guide science committee work. Republic of Turkey Ministry of Health Directorate General of Public Health. https://covid19bilgi.saglik.gov.tr/depo/rehberler/COVID-19_Rehberi.pdf [Google Scholar]
- World Health Organization . (2020). Coronavirus disease 2019 (COVID-19): Situation report, 67.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.