

Abstract
Background and aims:
Cardiovascular disease (CVD) is the leading cause of death in chronic kidney disease (CKD) patients, however, the underlying mechanisms that link CKD and CVD are not fully understood and limited treatment options exist in this high-risk population. microRNAs (miRNA) are critical regulators of gene expression for many biological processes in atherosclerosis, including endothelial dysfunction and inflammation. We hypothesized that renal injury-induced endothelial miRNAs promote atherosclerosis. Here, we demonstrate that dual inhibition of endothelial miRNAs inhibits atherosclerosis in the setting of renal injury.
Methods:
Aortic endothelial miRNAs were analyzed in apolipoprotein E-deficient (Apoe−/−) mice with renal damage (5/6 nephrectomy, 5/6Nx) by real-time PCR. Endothelial miR-92a-3p and miR-489–3p were inhibited by locked-nucleic acid (LNA) miRNA inhibitors complexed to HDL.
Results:
Renal injury significantly increased endothelial miR-92a-3p levels in Apoe−/−;5/6Nx mice. Dual inhibition of miR-92a-3p and miR-489–3p in Apoe−/−;5/6Nx with a single injection of HDL + LNA inhibitors significantly reduced atherosclerotic lesion area by 28.6% compared to HDL + LNA scramble (LNA-Scr) controls. To examine the impact of dual LNA treatment on aortic endothelial gene expression, total RNA sequencing was completed, and multiple putative target genes and pathways were identified to be significantly altered, including the STAT3 immune response pathway. Among the differentially expressed genes, Tgfb2 and Fam220a were identified as putative targets of miR-489–3p and miR-92a-3p, respectively. Both Tgfb2 and Fam220a were significantly increased in aortic endothelium after miRNA inhibition in vivo compared to HDL + LNA-Scr controls. Furthermore, Tgfb2 and Fam220a were validated with gene reporter assays as direct targets of miR-489–3p and miR-92a-3p, respectively. In human coronary artery endothelial cells, over-expression and inhibition of miR-92a-3p decreased and increased FAM220A expression, respectively. Moreover, miR-92a-3p overexpression increased STAT3 phosphorylation, likely through direct regulation of FAM220A, a negative regulator of STAT3 phosphorylation.
Conclusions:
These results support endothelial miRNAs as therapeutic targets and dual miRNA inhibition as viable strategy to reduce CKD-associated atherosclerosis.
Keywords: Atherosclerosis, Chronic kidney disease, Endothelium, microRNAs, HDL
1. Introduction
Clinical and experimental studies have firmly established that CKD dramatically increases the risk for CVD, including atherosclerosis [1–3]. Although traditional risk factors, e.g. hypertension and diabetes, are highly prevalent in CKD, statistical adjustments do not fully explain this predisposition and CKD itself is now considered a major risk factor for CVD mortality [1–3]. Despite these significant associations, the mechanistic link(s) between CKD and CVD remain to be fully defined, and thus, limits our ability to develop targeted risk-reduction interventions. Atherosclerosis is characterized by endothelial dysfunction, which promotes inflammatory responses to cholesterol and lipid accumulation within the arterial wall. Evidence suggests that CKD is associated with endothelial dysfunction, which provides a potential systemic link between CKD and CVD [4]; however, much of the underlying biology remains to be determined. Endothelial dysfunction encompasses a spectrum of biological processes and miRNAs have emerged as critical regulators of endothelial gene regulatory networks [5–7]. miRNAs are small non-coding RNAs that post-transcriptionally regulate gene expression through inhibition of protein translation and destabilization/degradation of targeted mRNAs [8,9]. For example, miR-92a-3p, has been identified as an important regulator of both angiogenesis and endothelial dysfunction through targeted suppression of protective endothelial genes, including kruppel-like factor 2 (KLF2) and 4 (KLF4) [7,10–14]. miR-92a-3p was also identified as a pro-atherogenic miRNA in endothelial cells. Inhibition of miR-92a-3p in low-density lipoprotein receptor null (Ldlr−/−) mice was demonstrated to reduce atherosclerotic burden [14]. Nevertheless, the impact of miR-92a-3p inhibition on atherosclerosis in other models of hypercholesterolemia, e.g. Apoe−/− mice, has not been tested. The potential of miR-92a-3p as a therapeutic target for CKD-associated atherosclerosis has also not been investigated. This is especially intriguing since miR-92a-3p was recently linked to CKD. miR-92a-3p levels were found to be increased in serum from CKD subjects and aortic miR-92a-3p levels were reported to be increased in rats with adenine-induced CKD [15]. Thus, inhibition of endothelial miR-92a-3p has great potential as a therapeutic target to treat atherosclerosis in the context of CKD.
In addition to miR-92a-3p, miR-489–3p has also been linked to kidney damage. Urinary miR-489–3p levels were elevated in rats with gentamicin kidney injury even before detectable increases in creatinine and blood urea nitrogen (BUN) suggesting that urinary miR-489–3p may serve as a novel biomarker for renal injury [16,17]. Another model of kidney damage, renal ischemia-reperfusion injury, was shown to have increased intra-renal miR-489–3p levels together with increased urinary levels of miR-489–3p [17]. These studies link extracellular and cellular levels of miR-92a-3p and miR-489–3p to cardiovascular and kidney diseases; however, the mechanistic roles of miR-92a-3p and miR-489–3p in vascular endothelium and their therapeutic potential in CKD-associated atherosclerosis have not been directly investigated.
In this study, we utilized HDL delivery of LNA miRNA inhibitors to aortic endothelium in vivo and demonstrated that dual inhibition of miR-92a-3p and miR-489–3p reduced atherosclerosis in a model of renal damage (Apoe−/−;5/6Nx). We found that endothelial levels of the pro-atherogenic miRNA, miR-92a-3p, were significantly increased in Apoe−/−;5/6Nx mice compared to Apoe−/− control mice with intact kidneys. Results from high-throughput sequencing, pathway analysis, and in silico prediction suggest that miR-92a-3p and miR-489–3p regulate multiple genes in aortic endothelial cells that may contribute to atherosclerosis, as LNA treatments significantly altered many genes (mRNA) within critical endothelial pathways. Gene reporter assays support that Tgfb2 and Fam220a are directly regulated by miR-489–3p and miR-92a-3p, respectively. Furthermore, a novel miR-92a-3p/FAM220A/STAT3 axis was identified in human coronary artery endothelial cells (HCAEC) that may contribute to the reduced atherosclerosis observed in mouse models of renal injury. Overall, these results demonstrate that simultaneous inhibition of multiple endothelial miRNAs is a potential strategy to reduce atherosclerosis in the setting of CKD.
2. Materials and methods
2.1. Animal studies
Apolipoprotein E−/− (Apoe−/−) B6.129P2-Apoetm1Unc/J mice from The Jackson Laboratory were studied under active Vanderbilt University Institutional Animal Care and Usage Committee protocols. Subtotal 5/6 nephrectomy (5/6Nx) was performed under isoflurane inhalation anesthesia on 10–12 wk old female Apoe−/− mice, as previously described [18]. Female Apoe−/− mice were utilized due to the increased atherosclerosis compared to males and mice were maintained on regular chow diet throughout the study. Seven wks after 5/6Nx surgery, mice received a single intravenous (retro-orbital) injection of saline, 4 mg of HDL alone, or 20 mg/kg (body weight) LNA miRNA inhibitors against miR-92a-3p (LNA-92a), miR-489–3p (LNA-489), or LNA-scramble (LNA-Scr) individually, or combination of 20 mg/kg of each LNA-92a and LNA-489 complexed to 4 mg HDL. Apoe−/− mice received intravenous (retro-orbital) injection of 4 mg of HDL alone, 20 mg/kg LNA-92a, or 20 mg/kg LNA-92a complexed with 4 mg of HDL. All mice were sacrificed 7 days post-injection by isoflurane inhalation followed by cervical dislocation. The surgical procedure is detailed in the Online Supplement Methods.
2.2. Transcriptomics
Total RNA was isolated from aortic endothelium lysates that were sheared from intact (excised) aortae by passing 200 μL of Qiazol (Qiagen) through the dissected aorta, as previously described [19]. Total RNA was isolated from aortic endothelium and HCAEC by miRNEasy mini kits (Qiagen) following manufacturer’s instructions and RNA levels were quantified by spectrophotometry using Take3 micro-volume plates with a Synergy Mx plate-reader. Total RNA was reverse transcribed using microRNA RT kits with individual miRNA RT primers and high-capacity cDNA RT kit with random hexamers for miRNAs and mRNAs, respectively (Life Technologies). Individual transcript levels were quantified by real-time PCR using TaqMan probes for candidate miRNAs and mRNAs on the Quantstudio real-time PCR System (Life Technologies). Relative quantitative values (RQV) were determined by the delta Ct method (RQV = 2−dCt) with normalization to U6 for miRNAs and peptidylprolyl isomerase A (PPIA) or ribosomal protein S9 (RPS9) for mRNAs (Supplementary Table 1).
2.3. Cell culture
Primary HCAEC (Lonza) were cultured in EGM-2MV media containing 5% fetal bovine serum (FBS) and HEK293 cells were cultured in DMEM media containing 10% FBS and 1% penicillin/streptomycin were maintained at 37 °C with 5% CO2. For transient transfection, cells were plated at 1–1.5 × 105 cells/ml 24 h prior to transfection with miRIDIAN miRNA mimetics (50 nM, Dharmacon), FAM220A ON-TARGETplus siRNA pool (50 nM, Dharmacon), or miRCURY LNA inhibitors (10 nM, Exiqon) using DharmaFECT4 transfection reagent (Dharmacon). For transfection experiments that required low basal levels of STAT3 phosphorylation, HCAEC were cultured in EBM-2 media that lacked growth factors. To over-express FAM220A, HCAEC were transfected with pAdtrack-CMV-FAM220A vector (or an empty control pAdtrack vector) using Dharmafect Duo transfection reagent (Dharmacon).
2.4. Statistical analysis
Data are presented as mean ± S.E.M. Mann-Whitney non-parametric tests were used to compare between two groups and p-values < 0.05 were considered significant. One-way ANOVA tests were used for multiple comparisons and significance was determined using Bonferroni (correction) alpha based on the number of comparisons performed. Statistical outliers were identified using Grubbs tests and were excluded. Although we presented data from many control groups, the HDL-LNA-Scr group was selected as the negative control group since it accounts for the effects of the vehicle and LNA chemistry. For miRNA real-time PCR results, 3 comparisons (α = 0.017) were made between treatments including HDL to HDL + LNA-Scr, HDL to HDL + single LNA, and HDL to HDL + dual LNA. For all physiological assays, 6 comparisons (α = 0.008) were made including Sham to 5/6Nx; Saline, 5/6Nx; Saline to HDL, HDL to HDL + LNA-Scr, HDL + LNA-Scr to HDL + LNA-92a, HDL + LNA-Scr to HDL + LNA-489, and HDL + LNA-Scr to HDL + LNA-92a + LNA-489. For mRNA real-time PCR results, 5 comparisons (α = 0.01) were made including Sham to 5/6Nx; Saline, 5/6Nx; Saline to HDL, HDL to HDL + LNA-Scr, HDL + LNA-Scr to HDL + single LNA, and HDL + LNA-Scr to HDL + dual LNA. Data analysis and graphing were performed with GraphPad Prism. MAplot was generated with R using the data.frame package. Raw and processed sequencing data have been deposited in NCBI’s Gene Expression Omnibus: GSE120038.
3. Results
3.1. Aortic endothelial miR-92a-3p levels increase with renal injury
To identify potential molecular mechanisms that link CKD and CVD, specifically endothelium dysfunction and atherosclerosis, a well-established mouse model of renal damage was used in hypercholesterolemic Apoe−/− mice [18,20]. Seven wks after renal parenchymal reduction, total RNA was isolated from aortic endothelium, as previously reported (Supplementary Fig. 1A) [19]. To confirm that the isolated endothelial RNA was enriched for endothelial cells over smooth muscle cells, mRNA levels of platelet endothelial cell adhesion molecule 1 (Pecam1) and smooth muscle α-actin 2 (Acta2) were quantified by real-time PCR. The endothelial RNA was enriched for Pecam1, an endothelial cell marker, while the remaining aortic tissue was enriched for Acta2, as reported by Pecam1/Acta2 ratio (Supplementary Fig. 1B). Using real-time PCR, endothelial miR-92a-3p levels were found to be significantly increased in Apoe−/−;5/6Nx mice compared to Apoe−/− sham control mice with intact kidneys (p=0.02, N = 23–30, Fig. 1A). Likewise, miR-489–3p levels were also elevated in these mice (p=0.08, N = 23–30, Fig. 1B). To inhibit miR-92a-3p and miR-489–3p activity in vivo, LNA miRNA inhibitors against miR-92a-3p and miR-489–3p were complexed with human HDL (4 mg) individually (20 mg/kg) or in combination (20 mg of each LNA/kg) and intravenously injected into Apoe−/−;5/6Nx mice (Fig. 1C). To confirm that endothelial miRNA levels were decreased following LNA treatments in vivo, miR-92a-3p and miR-489–3p levels were quantified in aortic endothelium by real-time PCR. Seven days after a single-dose of LNA-92a, endothelial miR-92a-3p levels were significantly reduced in mice that received either the individual LNA-92a treatment (HDL + LNA-92a, p=0.0001) or the combined LNA treatment (HDL + LNA-92a + LNA-489, p=0.002) compared to control vehicle (HDL only) treated mice (Fig. 1D). Likewise, endothelial miR-489–3p levels were significantly reduced in mice that received either individual LNA-489 treatment (HDL + LNA-489, p=0.0001) or the combined LNA treatment (p=0.0001) compared to control vehicle (HDL) only treated mice (Fig. 1E). Of note, we failed to detect significant changes in endothelial miR-92a-3p or miR-489–3p levels in mice treated with negative control LNA-Scr (HDL + LNA-Scr) compared to HDL only treatments (Fig. 1D and E). To evaluate the delivery efficiency of LNA-92a with and without complexing to HDL as a carrier vehicle, Apoe−/− mice were injected with LNA-92a alone, HDL alone, and HDL + LNA-92a complex and endothelial miR-92a-3p levels were quantified by real-time PCR after 7 days. Endothelial miR-92a-3p levels were significantly reduced with both the LNA-92a only treatment and the HDL + LNA-92a complex treatment compared to the HDL only control treatment (Supplementary Fig. 2). The miR-92a-3p inhibition was not significantly different between treatments of LNA-92a alone or complexed with HDL. Furthermore, our LNA treatment strategy was designed to target miR-92a-3p and miR-489–3p in the vascular endothelium, and we demonstrated that aortic endothelial levels of miR-92a-3p and miR-489–3p were significantly reduced, but this strategy may also suppress miR-92a-3p and miR-489–3p levels in other tissues and cell types. Therefore, miR-92a-3p and miR-489–3p levels were also quantified by real-time PCR in the remaining aortic tissue (after endothelium lysis) and liver in treated mice. We found that LNA treatments significantly reduced miR-92a-3p levels in the remaining aortic tissue and liver in LNA-92a and dual LNA treated mice, as compared to LNA-Scr treated mice (Supplementary Figs. 3A and B). LNA-489 treatments failed to significantly decrease miR-489–3p levels in the remaining aortic tissue and were not detectable in the liver (Supplementary Fig. 3C). Collectively, these results support that a single (intravenous) injection of LNA is sufficient to suppress aortic endothelial miRNA expression after 7 days.
Fig. 1.
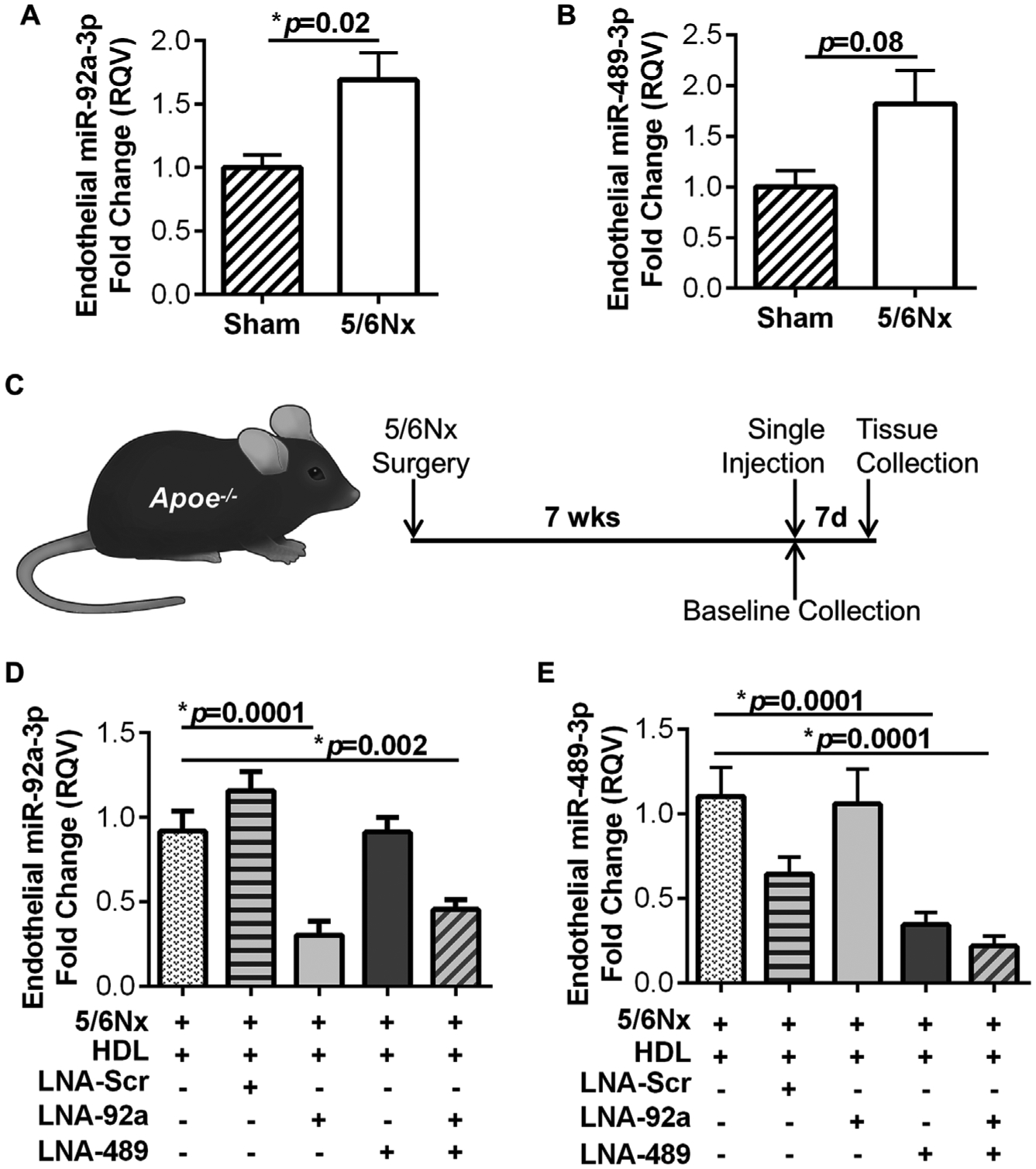
Aortic endothelial miR-92a-3p levels are increased in CKD-associated atherosclerosis. Real-time PCR quantification of (A) miR-92a-3p and (B) miR-489–3p in aortic endothelium of Apoe−/− mice 7 wks post sham or 5/6Nx surgery. Mann-Whitney non-parametric test. N = 23–30 (C) Schematic of experimental timeline for in vivo studies. Real-time PCR quantification of (D) miR-92a-3p and (E) miR-489–3p in the aortic endothelium of Apoe−/−;5/6nx mice after single treatment with HDL, HDL + LNA-scramble (LNA-Scr), HDL + LNA-92a, HDL + LNA-489, and HDL + LNA-92a + LNA-489. One-way ANOVA with Bonferroni α = 0.017. N = 11–17.
3.2. Dual miRNA inhibition reduces atherosclerosis in Apoe−/− mice with renal injury
To assess the impact of endothelial miRNA inhibition on atherosclerosis in Apoe−/−;5/6Nx mice, aortae were harvested from both individual LNA treated and dual LNA treated mice, as well as HDL + LNA-Scr, HDL alone, and saline control mice for histological analyses. To quantify atherosclerotic lesion area, cross-sections of the ascending aorta were stained for neutral lipids (Oil-Red-O, ORO). Renal injury (5/6Nx) increased, albeit not significantly, atherosclerotic lesion area in Apoe−/− mice compared to Apoe−/−;Sham mice (p=0.04, Fig. 2A and B). Apoe−/−;5/6Nx mice treated with HDL alone were found to have reduced lesion area compared to saline-treated control mice (p=0.002, Fig. 2A and B). Nevertheless, dual LNA treatment (HDL + LNA-92a + LNA-489) further reduced atherosclerotic lesion area in Apoe−/−;5/6Nx mice. We observed a significant 28.6% reduction of atherosclerotic lesion area in Apoe−/−;5/6Nx mice treated with dual LNAs (HDL + LNA-92a + LNA-489) compared to HDL + LNA-Scr treated mice (p=0.003, Fig. 2A and B). We found no significant difference in lesion area between HDL + LNA-Scr and HDL (only) treatments in Apoe−/−;5/6Nx mice (Fig. 2A and B). These results suggest that the LNA-92a and LNA-489 are biologically active in reducing atherosclerotic lesion area and that the observed changes are not simply related to vehicle (HDL only) or non-specific LNA chemistry (LNA-Scr). To determine if the reduction of neutral lipid in the lesions coincided with other changes in the lesion characteristics, we also assessed collagen content, macrophage content and necrosis. Aortic cross-sections stained with Aniline blue (Fig. 3A and B) and Masson’s trichrome (Supplementary Fig. 4) showed that LNA treatments failed to significantly alter lesion collagen content compared to LNA-Scr treated Apoe−/−;5/6Nx mice (Fig. 3A and B). Monocyte/macrophage content assessed by anti-CD68 (Fig. 3C and D) and anti-MOMA-2 (Supplementary Figs. 5A and B) immunohistochemistry showed that renal injury significantly increased CD68 + cells (macrophages) in aortic lesions, i.e. Apoe−/−;5/6Nx mice compared to Apoe−/−;Sham mice (p=0.0007); however, HDL or LNA treatments did not affect these results (Fig. 3C and D). To determine if LNA treatments altered lesion necrotic area, aortic cross-sections were stained with Hematoxylin and Eosin (H&E), and necrotic areas were identified by acellular regions within the lesion. Neither individual LNA treatments nor the dual LNA treatment were found to significantly reduce lesion necrotic area compared to LNA-Scr treated Apoe−/−;5/6Nx mice (Fig. 3E and F). Of note, renal injury in the setting of hypercholesterolemia significantly increased necrotic area in aortic lesions compared to Sham control mice (Apoe−/−;5/6Nx vs. Apoe−/−;Sham mice, p=0.001) (Fig. 3E and F). Moreover, HDL (only) treatments were found to significantly reduce lesion necrotic area compared to Apoe−/−;5/6Nx mice (p=0.0008, Fig. 3E and F). Overall, the histological analyses of the atherosclerotic lesions suggest that dual LNA treatments significantly reduced neutral lipid content compared to LNA-Scr treatments in absence of significantly altering plaque characteristics including collagen, macrophage content, or necrotic area.
Fig. 2.
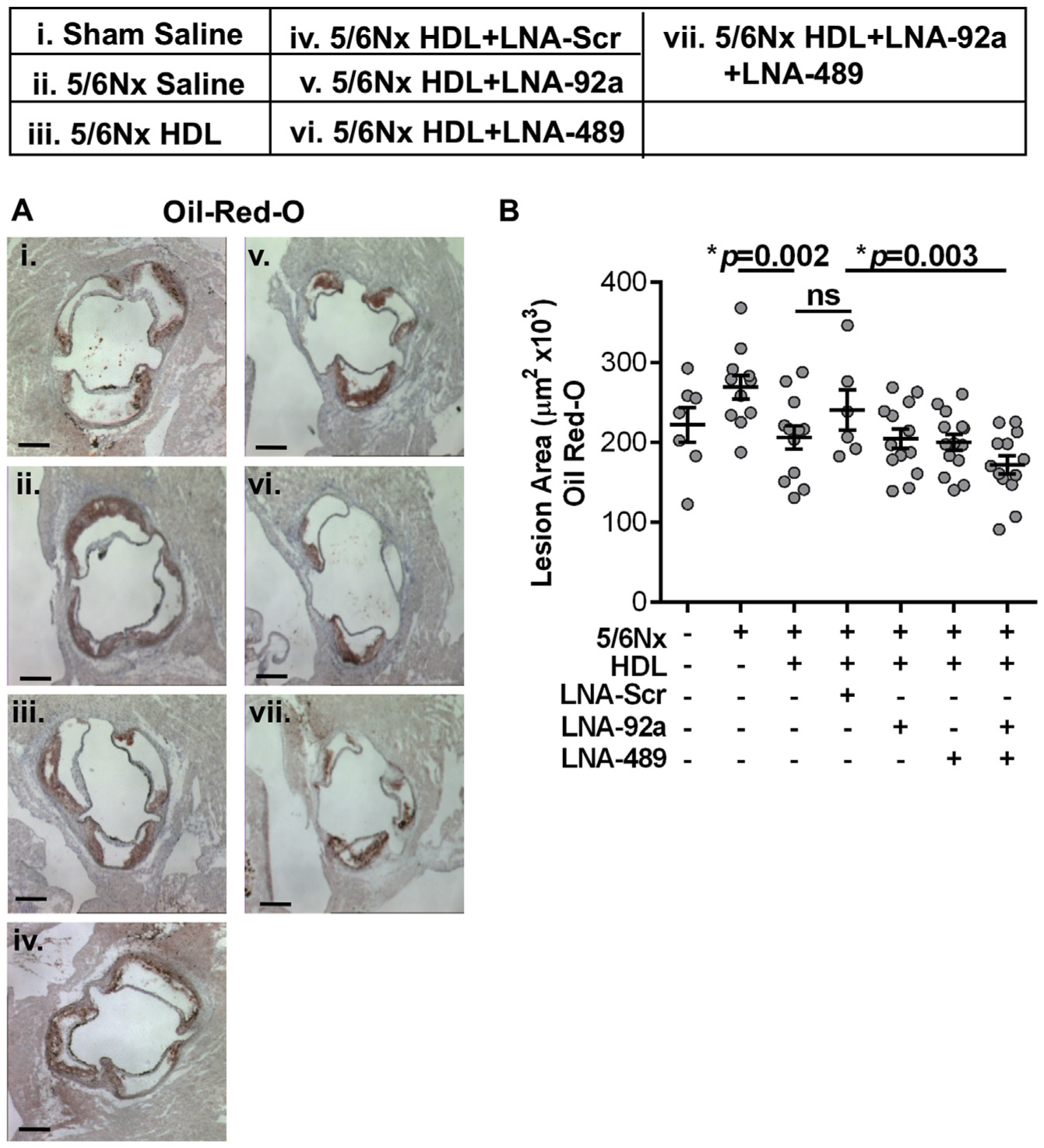
Dual inhibition of miR-92a-3p and miR-489–3p significantly reduces CKD-associated atherosclerosis. (A) Representative images and (B) quantification of aortic cross-sections stained with ORO from Apoe−/−;5/6Nx mice 7 days post-treatment. One-way ANOVA with Bonferroni α = 0.008. N = 6–15. Scale bar represents 500 μm.
Fig. 3.
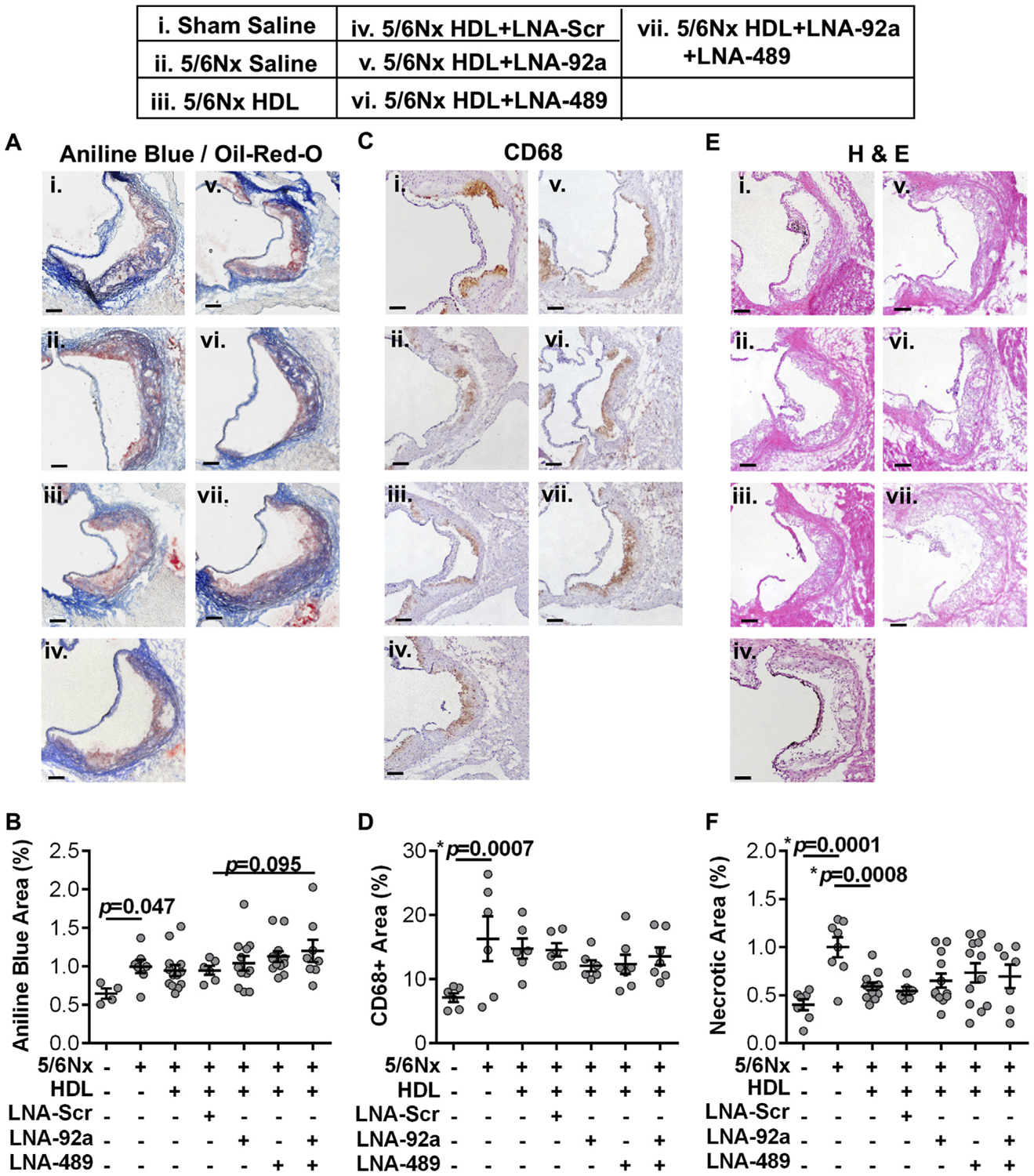
Characterization of atherosclerotic lesions. (A and B) Histological images of aortic cross-sections from treated Apoe−/−;5/6Nx mice and quantification of collagen with Aniline Blue normalized to lesion area (Oil-Red-O, ORO). N = 4–13. (C and D) Immunohistochemical images of aortic cross-sections from treated Apoe−/−;5/6Nx mice and quantification of CD68+ inflammatory cells (e.g. macrophages). N = 6–7. (E and F) Histological images of aortic cross-sections from treated Apoe−/−;5/6Nx mice and quantification of necrotic area by H&E staining. N = 6–12. One-way ANOVA with Bonferroni correction α = 0.008. Scale bars represent 100 μm.
To assess the potential effects of LNA on systemic parameters we measured systolic blood pressure (SBP), kidney function by blood urea nitrogen (BUN) and plasma lipid profile. SBP was not significantly altered by renal injury, LNA, or HDL treatments (Supplementary Fig. 6A). BUN increased with renal injury in Apoe−/− mice compared to sham controls (p=0.0001, Supplementary Fig. 6B). While HDL-only treatment did not affect BUN, a significant reduction in BUN was observed with HDL + LNA-92a (p=0.002) and HDL + LNA-92a + LNA-489 (p=0.003) treatments compared to HDL + LNA-Scr (Supplementary Fig. 6B). While these results suggest the possibility that LNA-92a treatment may improve kidney function, the possibility requires further investigation with more sensitive analysis such as creatinine clearance or sinistrin glomerular filtration rate (GFR). Total plasma cholesterol levels were significantly increased with renal injury; however, HDL and LNA treatments did not reduce the plasma cholesterol levels (Supplementary Fig. 7). Together, these results suggest that dual LNA treatments significantly reduced atherosclerotic (lesion) burden in Apoe−/−;5/6Nx mice without affecting SBP or circulating cholesterol levels.
3.3. Dual LNA treatments alter the aortic endothelium transcriptome
To identify gene regulatory networks and potential biological mechanisms underlying the impact of miRNA inhibition, high-throughput total RNA (ribosomal RNA-depleted) sequencing was completed on RNA isolated from the aortic endothelium of Apoe−/−;5/6Nx mice treated with individual LNAs, dual LNAs, vehicle (HDL) only, and saline (control). The HDL + LNA-92a treatment in mice resulted in 996 significantly altered endothelial genes (595 down, 401 up) (Supplementary Fig. 8A, Supplementary Table 2). Likewise, we observed that the HDL + LNA-489 treatment resulted in 404 significantly altered endothelial genes (120 down; 284 up) (Supplementary Fig. 8B, Supplementary Table 2). Most interestingly, dual inhibition of endothelial miR-92a-3p and miR-489–3p resulted in 241 significant differentially expressed genes compared to saline-injected control mice (132 down; 109 up) (Fig. 4A, Supplementary Table 2). Vehicle (HDL) alone resulted in 348 differentially expressed genes compared to saline-treated control mice (223 down; 125 up) (Supplementary Fig. 8C, Supplementary Table 2). Of the 125 genes that were significantly up-regulated with HDL alone, 72 (57.6%) were unique to HDL only treatments with no overlap with any of the other treatments despite the use of HDL as a delivery vehicle for the LNAs (Supplementary Fig. 8D). Furthermore, 117 of the 223 (52.5%) down-regulated genes with vehicle (HDL) alone were also unique to the HDL only treatment (Supplementary Fig. 8E). These results suggest that while treatments with HDL alone altered many genes, dual LNA treatment, with HDL as a delivery vehicle, significantly altered a completely new set of genes. Furthermore, there was overlap in significantly altered genes with individual LNA treatment and dual LNA treatment, e.g. we found 38 up-regulated genes and 30 down-regulated genes that were in common with the dual LNA treatment and the individual LNA treatments (Supplementary Fig. 8D and E). Therefore, the results support that LNA-mediated gene regulation likely contributed to the observed reduction in atherosclerosis in LNA-treated mice. In silico prediction studies were used to parse putative miR-92a-3p and miR-489–3p target genes from the list of significantly increased genes (mRNA) in response to individual and/or dual LNA treatments. Using this strategy, we determined that 16.5%–31.2% of the significantly increased genes after individual or dual LNA treatments were predicted targets of the specific miRNAs (Table 1). These results suggest that the endothelial gene expression response to miRNA inhibition in vivo is likely a function of some direct effects. Nevertheless, most of gene expression changes in response to individual and dual miRNA inhibition were indirect changes (Table 1).
Fig. 4.
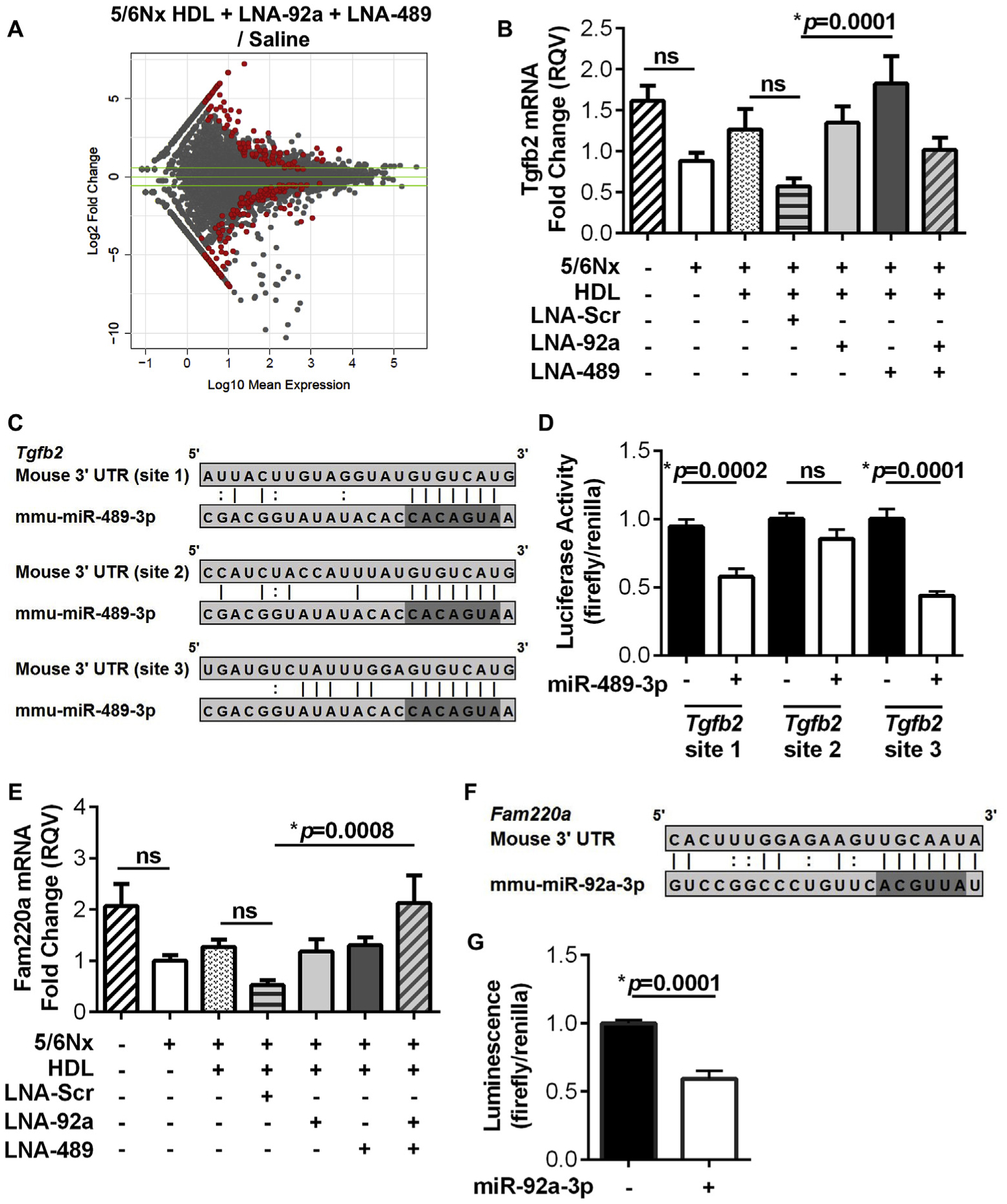
Dual inhibition of miR-92a-3p and miR-489–3p alter endothelial gene expression in CKD-associated atherosclerosis. (A) MA plot of the log10 DeSeq2 normalized mean expression and the log2 fold change of aortic endothelial gene expression for HDL + LNA-92a + LNA-489 compared to saline control with significantly altered genes highlighted in red. Absolute fold change ≥1.5 indicated by the green lines. N = 4. (B) Real-time PCR quantification of aortic endothelial Tgfb2 mRNA levels in Apoe−/−;5/6Nx cohort. N = 11–16. (C) Putative mmu-miR-489–3p target sites within Tgfb2 3′ UTR. (D) Gene reporter (luciferase) assays with dual transfection of miR-489–3p mimics and luciferase reporter plasmids harboring the predicted miR-489–3p target sites from the mouse Tgfb2 3′ UTR in HEK293 cells. N = 11–12. (E) Real-time PCR quantification of aortic endothelial Fam220a mRNA levels in Apoe−/−;5/6Nx cohort. N = 11–16. (F) Putative miR-92a-3p target sites within Fam220a 3′ UTR. (G) Gene reporter (luciferase) assay with dual transfection of miR-92a-3p mimics and luciferase reporter plasmids harboring the predicted miR-92a-3p target sites from the mouse Fam220a 3′ UTR in HEK293 cells. N = 11–12. For real-time PCR analysis, one-way ANOVA with Bonferroni α = 0.01 were used. For luciferase assays, Mann-Whitney non-parametric tests were used.
Table 1.
Aortic endothelium gene (mRNA) changes in response to treatments.
| Treatment | Total gene changes | Down-regulated | Up-regulated | miR-92a-3p | miR-489-3p | ||||
|---|---|---|---|---|---|---|---|---|---|
| MMU predicted | Percent up-regulated | Human conserved | MMU predicted | Percent up-regulated | Human conserved | ||||
| HDL | 348 | 223 | 125 | n/a | n/a | n/a | n/a | n/a | n/a |
| HDL + LNA-92a | 996 | 595 | 401 | 66 | 16.46 | 36 | n/a | n/a | n/a |
| HDL + LNA-489 | 404 | 120 | 284 | n/a | n/a | n/a | 73 | 25.70 | 28 |
| HDL + LNA-92a + LNA-489 | 242 | 132 | 109 | 14 | 12.84 | 7 | 20 | 18.35 | 4 |
3.4. Dual LNA treatment targets key endothelial signaling pathways
To further characterize the impact of dual LNA treatments on aortic endothelial gene regulation within CKD-associated atherosclerosis, significantly altered genes were analyzed at the pathway level using MetaCore analysis (GeneGo). In dual LNA treated mice, 45 pathways were significantly altered based on both increased and decreased gene (mRNA) changes (Supplementary Table 3). These significantly altered pathways include multiple immune processes and cytokine signaling cascades (Supplementary Table 3). Furthermore, 12 biological process networks were significantly altered, including calcium transport, cell-cell interactions, signal transduction, and vessel morphogenesis (Supplementary Table 3). Within the MetaCore database we identified 51 transcription factors with significant enrichment of transcriptional targets within the differentially expressed genes after dual LNA treatment (Supplementary Table 4). These data suggest that the observed indirect effects may be the result of altered endothelial transcription factor activity. To identify novel miRNA regulatory networks in the aortic endothelium that may contribute to the reduction in atherosclerotic lesion area in LNA-treated mice, we investigated potential direct targets of miR-92a-3p and miR-489–3p. Putative mRNA targets of miR-92a-3p and miR-489–3p that were significantly increased with individual and/or dual LNA treatments were filtered (Supplementary Table 2). From this list of candidate genes, a single gene predicted to be targeted by both miRNAs was not confirmed by real-time PCR; however, we identified and confirmed regulation of putative targets for each miRNA individually. From this list of candidate genes, transforming growth factor beta 2 (Tgfb2) was selected as a candidate putative target of miR-489–3p, and real-time PCR was used to confirm that Tgfb2 mRNA levels were significantly increased in aortic endothelium of LNA-treated Apoe−/−;5/6Nx mice compared to LNA-Scr-treated mice (p=0.0001, Fig. 4B). Furthermore, we failed to find a significant difference in endothelial Tgfb2 mRNA levels between LNA-Scr and vehicle (HDL) only treated Apoe−/−;5/6Nx mice, or between HDL only and saline treated control mice (Fig. 4B). In mice, Tgfb2 3′ untranslated region (3′ UTR) harbors 3 predicted miR-489–3p target sites (Fig. 4C). To experimentally validate that miR-489–3p directly targets and regulates Tgfb2 mRNA within the 3′ UTR, gene reporter (luciferase) constructs were generated for each putative miR-489–3p target site (Fig. 4C). Upon dual transfection of the Tgfb2 3’ UTR luciferase reporter and miR-489–3p mimetics in HEK293 cells, normalized luciferase activity (Firefly/Renilla) was significantly decreased for site 1 (p=0.0002) and site 3 (p=0.0001), but not site 2, compared to control cells without miR-489–3p mimetics (Fig. 4D). These results suggest that miR-489–3p directly regulates Tgfb2, and the observed increase in endothelium Tgfb2 expression with LNA-489 treatment in mice is likely due to decreased miR-489–3p activity. TGF-β signaling is linked to many anti-atherogenic biological processes; therefore, decreased miR-489–3p activity and increased Tgfb2 expression may contribute to the observed LNA-mediated reduction in lesion area (Fig. 2A,B) [21–23].
From the list of candidate putative targets, Fam220a was identified as a novel candidate gene that may antagonize CKD-associated atherosclerosis. By real-time PCR, aortic endothelial Fam220a mRNA levels were confirmed to be significantly increased upon dual LNA treatment compared to LNA-Scr treatment in mice (p=0.0008, Fig. 4E). We failed to find a significant difference in Apoe−/−;5/6Nx treated with LNA-Scr compared to vehicle (HDL) only treated Apoe−/−;5/6Nx mice, or between HDL only and saline-treated (control) Apoe−/−;5/6Nx mice (Fig. 4E). The Fam220a 3′ UTR is predicted to harbor one miR-92a-3p target site (Fig. 4F). To experimentally validate that miR-92a-3p directly regulates Fam220a in the 3’ UTR, the predicted miR-92a-3p target site was cloned downstream of Firefly luciferase (Fig. 4F). Upon dual transfection of the Fam220a luciferase reporter and miR-92a-3p mimetics in HEK293 cells, normalized luciferase activity (Firefly/Renilla) was significantly decreased compared to control cells without miR-92a-3p mimetics (p=0.0001, Fig. 4G). Although very little is currently known about Fam220a, the protein product of this gene likely serves as an adapter for nuclear phosphatases that regulate the activity of STAT3, which is a well-known pro-inflammatory transcription factor that accelerates atherosclerosis development [24–28]. These data suggests that miR-92a-3p inhibition of Fam220a expression may contribute to CKD-associated atherosclerosis and the observed decrease in lesion area with dual LNA inhibition may be mediated through altered STAT3 activity.
3.5. miR-92a-3p regulates STAT3 phosphorylation in human endothelial cells
Previously, FAM220A was demonstrated to interact with a nuclear phosphatase and STAT3, which resulted in changes to STAT3 phosphorylation in human mast cells [25–27]. Nevertheless, FAM220A regulation of STAT3 in endothelial cells has not been investigated. Therefore, we examined the connection between miR-92a-3p, FAM220A, and STAT3 in human endothelial cells since FAM220A is a conserved (putative) miR-92a-3p target gene in mice and humans. To experimentally validate miR-92a-3p regulation of FAM220A in human endothelial cells, LNAs against miR-92a-3p were transiently transfected into HCAEC, and FAM220A mRNA levels were significantly increased (at 48 h) in HCAEC that received either LNA-92a (p=0.002) or a combination of LNA-92a and LNA-489 (p=0.0001, Fig. 5A, Supplementary Figs. 9A and B). In the animal study, dual LNA treatment (20 mg/kg for each LNA) was compared to the LNA-Scr treatment (20 mg/kg), thus the dual LNA treatment had twice as much LNA than the LNA-Scr condition. To determine whether the observed differential effects of dual LNA compared to LNA-Scr were due to differences in LNA concentration, we tested the effects of different LNA concentrations in vitro. We found that transfection of HCAEC with LNA-92a at 10, 20, and 50 nM all significantly increased FAM220A expression (Supplementary Fig. 9C). Transfection of LNA-92a at 10 nM was sufficient to significantly increase FAM220A mRNA levels (p=0.0001, Supplementary Fig. 9C). Most importantly, increased LNA concentrations (20 nM and 50 nM) did not further increase FAM220A mRNA levels, suggesting that an increased concentration of LNA does not indirectly effect FAM220A expression (Supplementary Fig. 9C).
Fig. 5.
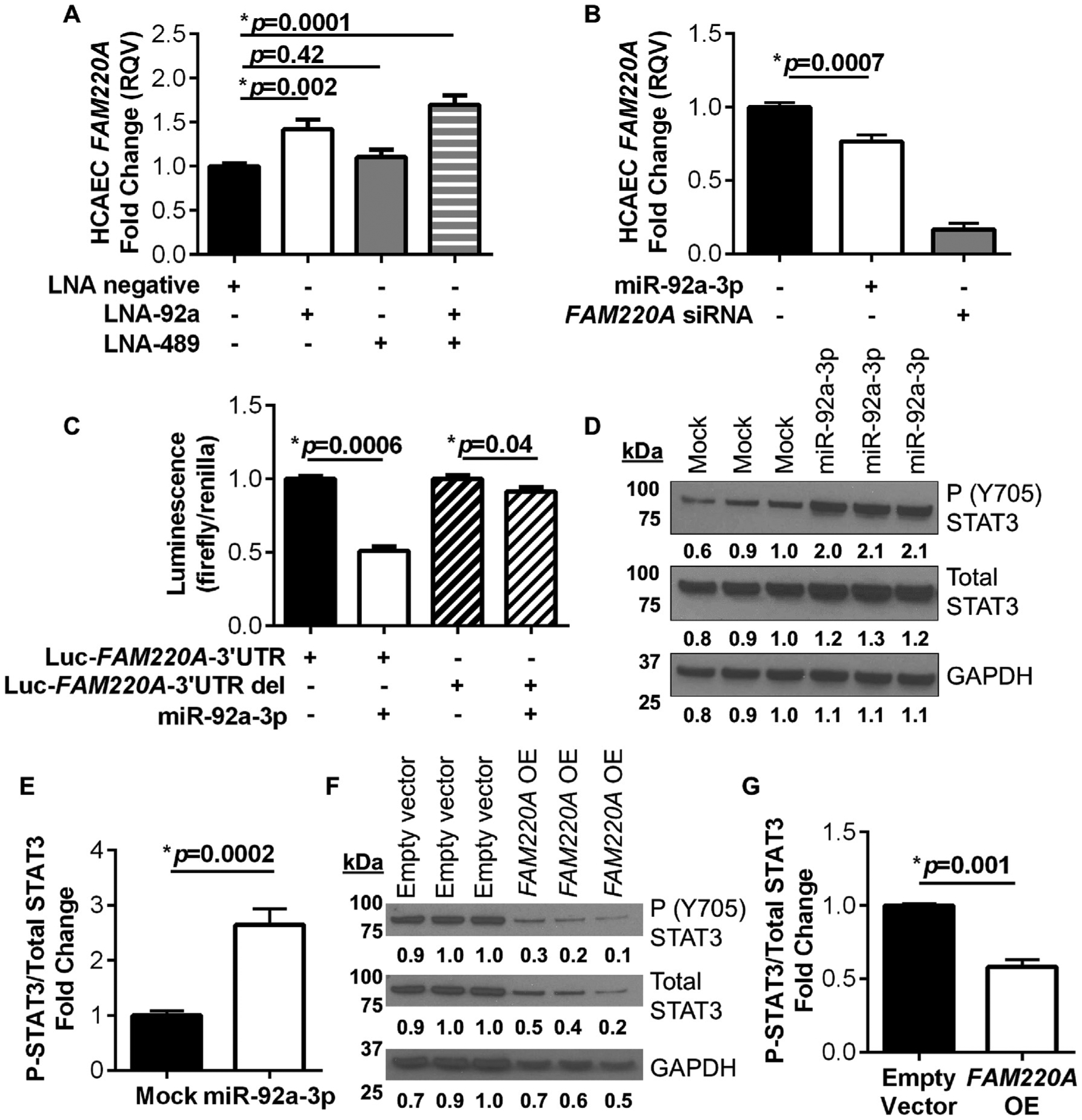
FAM220A is directly regulated by miR-92a-3p in human endothelial cells. (A) HCAEC FAM220A mRNA levels with miRNA inhibition; LNA-Scrambled (Scr), LNA-92a, or LNA-489. N = 9. (B) HCAEC FAM220A mRNA levels with miRNA over-expression; miR-92a-3p mimetics or siRNA FAM220A control. N = 8–12. (C) Gene reporter (luciferase) assays with dual transfection of miR-92a-3p mimics and luciferase reporter plasmids harboring the full-length human FAM220A 3′ UTR or mutated FAM220A with a 2 bp deletion in miR-92a-3p target site in HEK293 cells. N = 7. (D–G) Western blots and quantification for STAT3 Y705 phosphorylation, total STAT3, and GAPDH. (D–E) Over-expression of miR-92a-3p (mimetics) in HCAEC in growth factor-free culture media. N = 8. (F and G) Over-expression of FAM220A in HCAEC. N = 3. For comparisons between 2 groups Mann-Whitney non-parametric tests were used, and when comparing between > 2 groups One-way ANOVA tests were used with Bonferroni corrections, α = 0.025 for 2 comparisons and α = 0.017 for 3 comparisons.
To further demonstrate that miR-92a-3p regulates FAM220A, miR-92a-3p mimetics were transfected into HCAEC. We found that miR-92a-3p over-expression significantly reduced FAM220A mRNA levels, as quantified by real-time PCR (p=0.0007, Fig. 5B, Supplementary Fig. 9D). To determine if miR-92a-3p directly targets FAM220A at the predicted target site within the human FAM220A 3′ UTR, gene reporter (luciferase) assays with the full-length FAM220A 3′ UTR and site-directed mutagenesis were used. Upon dual transfection of the FAM220A luciferase reporter and miR-92a-3p mimetics in HEK293 cells, normalized luciferase activity (Firefly/Renilla) was significantly decreased compared to control cells without miR-92a-3p mimetics (p=0.0006, Fig. 5C). Most importantly, site-directed mutagenesis deleting 2 bases within the predicted miR-92a-3p target site in the FAM220A 3′ UTR blocked miR-92a-3p suppression of normalized luciferase activity (Fig. 5C). These results confirmed that miR-92a-3p directly regulates FAM220A at the predicted target site within the FAM220A 3’ UTR in humans. FAM220A has previously been reported as a negative regulator of STAT3 phosphorylation [25–27]. To determine if miR-92a-3p regulates STAT3 phosphorylation, HCAEC were transiently transfected with miR-92a-3p mimetics, and STAT3 Y705 phosphorylation levels were found to be significantly increased at 48 h by western blotting (p=0.0002, Fig. 5D and E, Supplementary Figs. 9E and F). To experimentally confirm that FAM220A negatively regulates STAT3 phosphorylation in human endothelial cells, FAM220A levels were increased in HCAEC by transient transfection of a FAM220A open reading frame plasmid. We found that FAM220A over-expression resulted in significantly reduced STAT3 Y705 phosphorylation by western blotting (p=0.001, Fig. 5F and G). These results support that miR-92a-3p directly regulates FAM220A in endothelial cells, which in turn regulates the phosphorylation of STAT3. These results support that miR-92a-3p inhibition in mice likely altered both the endothelial Fam220a expression and STAT3 activity that may have contributed to the observed reduction in atherosclerotic burden.
4. Discussion
Consistent with clinical observations and previous experimental studies, this study observed that renal injury potentiates hyperlipidemia and atherosclerosis [18,29–31]. The characteristics of the atherosclerotic lesion were also consistent with clinical and experimental observations of CKD-driven atherosclerosis including increased macrophage content (CD68 + staining) and necrotic area [18,29–31]. A potential link between CKD and CVD is endothelial dysfunction and miRNAs are key regulators of endothelial cell gene expression and function [5]. In this study, we report that miR-92a-3p is increased in aortic endothelium of Apoe−/− mice with renal impairment. Endothelial miRNAs offer great potential as therapeutic targets to treat atherosclerosis [6], and may provide an improved treatment strategy for CKD patients especially those with advanced CKD in whom lipid lowering therapies have reduced benefit [32]. In our study, dual LNA-92a and LNA-489 treatment with HDL delivery resulted in a dramatic reduction in atherosclerotic ORO lesion area without altering other lesion characteristics. Furthermore, dual miRNA inhibition altered the aortic endothelium gene expression profile. These results support a role for endothelial miR-92a-3p and miR-489–3p in CKD-associated atherogenesis that may have therapeutic potential.
Pathway analysis of mRNA sequencing results identified distinct pathways and biological processes, as well as transcriptional networks, which likely confer the mechanistic link(s) between endothelial miRNA activity and atherosclerosis in the setting of CKD. Thus, pathway analysis of changes in endothelium gene expression identified TGFβ and JAK/STAT pathways as being significantly altered by dual miRNA inhibition. Signaling molecules in the TGFβ pathway, namely SMAD3, were enriched within our sequencing analysis. Moreover, STAT3, a terminal transcription factor for the TGFβ signaling pathway was just out of significance for this analysis. The results support miRNA regulation of two critical cell signaling components, TGFβ and STAT3, which may be impacted by targeting endothelial miR-489–3p and miR-92a-3p. With mRNA sequencing and in silico prediction studies, the novel miRNA targets, Tgfb2 and Fam220a, were identified and confirmed by real-time PCR and gene reporter assays. First, miR-489–3p directly regulates Tgfb2 mRNA levels, which likely alters the TGFβ pathway influencing multiple anti-inflammatory processes that reduce atherogenesis [21–23,33]. Second, miR-92a-3p likely indirectly regulates pro-atherogenic STAT3 activity through direct regulation of FAM220A, a negative regulator of STAT3 [25–27]. Multiple studies have demonstrated that STAT3 is a pro-inflammatory transcription factor in vascular endothelial cells, and upon activation, promotes endothelial dysfunction and atherogenesis [24,28]. Therefore, dual inhibition of these miRNAs likely simultaneously activates an anti-inflammatory pathway (TGFβ) and inhibits a pro-inflammatory pathway (STAT3) to reduce atherosclerosis.
Identification of miR-489–3p and miR-92a-3p in this study suggests they may play a role in endothelial biology in the setting of CKD-associated atherosclerosis. To date, miR-489–3p has not been linked to endothelial biology, atherosclerosis, or CVD. However, miR-489–3p was induced in the kidney parenchyma by hypoxia-inducible factor 1 (HIF1) in a model of renal injury thereby demonstrating its relevance to kidney disease [17]. The potential regulation of miR-489–3p by HIF1 in endothelial cells should be further evaluated to determine if this mechanism contributes to miR-489–3p expression in vascular endothelium. In this study, 5/6Nx resulted in a 1.8-fold increase in endothelial miR-489–3p levels; however, these changes did not reach statistical significance due to variability between individual studies and within specific groups of mice. Nevertheless, miR-489–3p proved to be a viable target to reduce atherosclerosis. Our study defined a novel role for miR-489–3p in endothelial cells by demonstrating that i.) miR-489–3p directly targets Tgfb2 in endothelial cells and ii.) LNA inhibition of miR-489–3p in vivo significantly increased Tgfb2 expression in aortic endothelium. In contrast to the paucity of information on miR-489–3p, a pro-atherogenic role for miR-92a-3p in endothelial cells, similar to that found by the current study, has previously been reported [10,14]. Inhibition of miR-92a-3p in Ldlr−/− mice on high-fat diet resulted in increased expression of Klf2, Klf4, and nitric oxide synthase 3 (Nos3), and decreased phosphorylation of p65 in the aorta, which led to a reduction in endothelial dysfunction and atherogenesis [14]. Our study further defined a role for miR-92a-3p in a more severe hypercholesterolemic model, Apoe−/− mice, and in conjunction with renal injury. Our study identified and confirmed a novel direct target of miR-92a-3p, Fam220a, and further defined a role in regulating STAT3 activation by confirming the function of FAM220A within human endothelial cells. Together, these results reveal new roles for miR-489–3p and miR-92a-3p in atherosclerosis and renal injury/CKD. Although the link between renal injury and endothelial miRNA changes has not been extensively studied, CKD-associated oxidative stress may influence miR-92a-3p expression. A previous study by Shang et al. reported that accumulation of uremic toxins, e.g. indoxyl sulfate, in CKD increased oxidative stress and endothelium miR-92a-3p expression, which contributes to endothelial dysfunction [15]. This study also found a positive correlation between indoxyl sulfate and miR-92a-3p in serum of end-stage renal disease patients on hemodialysis [15]. Therefore, targeting circulating CKD-associated stimuli that induce endothelial miRNA changes (and endothelial dysfunction) holds potential therapeutic value to treat CKD-associated atherosclerosis.
Recent advances in RNA-based therapeutics have ushered in many new strategies to target both mRNAs and small RNAs for a variety of diseases. Currently, there are very few RNA-based drugs with F.D.A. approval. Very recently, patisiran, a siRNA-based drug to treat hereditary transthyretin amyloidosis, was the first siRNA drug approved by the F.D.A [34,35]. Mipomersen, an antisense oligonucleotide targeting apolipoprotein B, is the only F.D.A.-approved RNA-based drug to treat dyslipidemia or CVD, although multiple miRNA therapeutics are currently under clinical investigation [36–38]. However, none of these drugs are designed to directly treat CKD-associated atherosclerosis, an area devoid of clear understanding of the underlying pathophysiology in a population in urgent need of new therapeutic approaches. This study, therefore, greatly advances the field of miRNA therapeutics as it demonstrates the feasibility of a combinatorial inhibition approach using dual LNAs to target multiple miRNAs toward a desired outcome. Currently, there is a great need to better understand how multiple miRNAs cooperatively regulate biological pathways, and future studies may benefit from targeting a cassette of miRNAs within a regulatory network to illicit a greater physiological impact. Despite the advances in RNA-based therapeutics, limitations still exist in stability and delivery of RNA based therapeutics to specific tissues/cells [39]. One potential strategy for miRNA delivery is using lipoproteins, particularly HDL, since HDL has been demonstrated to stably transport small RNAs and transfer RNAs to endothelial cells [40,41]. In addition, utilizing HDL as the delivery vehicle may confer additional benefits including increased cholesterol efflux and reduced inflammation, which is supported by the clinical trials evaluating reconstituted HDL therapy for CVD. Two reconstituted HDL therapies, CSL112 and CER-001, have exhibited improved cholesterol efflux capacity, but the effect on atherosclerosis is unclear [42,43]. The CER-001 trials demonstrated a reduction in atheroma volume only for the subjects with the most severe level of atherosclerosis (> 30% atheroma volume) and lacked an effect on the entire cohort of acute coronary syndrome patients [44,45]. The additional benefit of HDL may have a larger impact for CKD patients with CVD since their HDL has reduced functionality [46]. Nevertheless, utilizing HDL as a delivery method for RNA-based therapies would add physiological benefit to the therapy and the combined therapeutic approach has greater clinical potential since it affects multiple aspects within the complex pathophysiology of CVD.
One potential limitation of this study is that we focused solely on gene expression changes in the aortic endothelium after LNA treatments in mice. These LNAs can readily be taken up by other non-endothelial cells and the LNA treatments have potential to inhibit miR-92a-3p and miR-489–3p in additional tissues and cell types, which may also contribute to the observed reduction in atherosclerotic lesion. To this point, we did observe that miR-92a-3p levels were reduced in the liver and the remaining non-endothelium part of the aorta after LNA-92a treatment. Nevertheless, the loss of miR-92a-3p was not found to affect plasma lipids levels as the total cholesterol in the plasma in treated mice was not significantly altered. At this time, we do not know the full consequence(s) of miRNA silencing in other tissues on the observed phenotype and further studies would be required to address this hypothesis. Furthermore, the dual LNA treatment significantly reduced the neutral lipid content without altering other components of the lesion, including CD68 + and MOMA-2 macrophage areas. This may represent loss of neutral lipid from non-macrophage areas of the lesion in addition to loss of neutral lipids from macrophages that remain in the lesion. Another potential explanation for no change to the macrophage area is reciprocal increase in new macrophages and decrease in cholesterol-loaded macrophages within the lesion. Nonetheless, further investigations are required to fully understand the mechanisms contributing to the movement of neutral lipids within this context.
Another limitation of the study is the potential that HDL, used as a carrier for the LNAs, contributed to the observed phenotype. The most important observation related to this point is that treatments with HDL complexed with both LNA-92a and LNA-489 in combination significantly decreased atherosclerosis in Apoe−/− 5/6Nx mice when compared to the proper control, i.e. HDL complexed with LNA scrambled control (LNA-Scr). Thus, these results suggest that the targets of the LNAs, not just the carrier HDL or the LNA chemistry, were responsible for a significant reduction in atherosclerosis. The reduction in lesion area is comparable to other miRNA atherosclerosis studies. In our study, we observed a 28.6% reduction in aortic sinus lesion area by Oil-Red-O staining (quantification) 7 days after a single injection of 40 mg/kg LNAs against miR-92a-3p and miR-489–3p (20 mg/kg each) in Apoe−/− mice fed a chow diet for 8 weeks after 5/6Nx surgery to induce renal injury. In 2014, Loyer et al. reported a 23% reduction of atherosclerosis in Ldlr−/− mice fed a high fat diet for 14 weeks after these mice received 2 treatments of miR-92a inhibitors at weeks 4 and 9; aortic sinus lesion area was quantified by Oil-Red-O at week 1414. Therefore, a single injection of LNAs against both miR-92a-3p and miR-489–3p was more effective than two injections of anti-miR-92a-3p alone, albeit in different models of atherosclerosis. In 2011, Rayner et al. reported a 35% reduction in atherosclerosis with inhibition of miR-33 at the end of a 20-week study in Ldlr−/− mice on a high fat diet, as quantified by H&E of aortic sinus lesion area [47]. In this study, Ldlr−/− mice were placed on a high fat diet for 14 weeks followed by 4 weeks of anti-miR-33 treatments (6 treatments total) [47]. For comparisons, targeting miR-33 with 6 injections was only slightly more effective than a single injection of LNAs against both miR-92a-3p and miR-489–3p; however, these were different models of atherosclerosis.
In summary, we identified elevated expression of aortic endothelial miRNAs associated with renal injury and hypercholesterolemia. Inhibition of miR-92a-3p and miR-489–3p through a dual LNA treatment strategy markedly reduced atherosclerosis and altered endothelial gene expression. Through in silico and pathway analyses, Tgfb2 and Fam220a were identified as novel target genes for miR-489–3p and miR-92a-3p, respectively. Further, we demonstrated that FAM220A is a direct target of miR-92a-3p in HCAEC and validated the regulation of STAT3 phosphorylation by FAM220A in endothelial cells. Therefore, these findings support the potential of a dual LNA-based miRNA inhibition strategy to treat atherosclerosis in the setting of CKD.
Supplementary Material
HIGHLIGHTS.
Aortic endothelial miR-92a-3p levels are increased in a mouse model of renal injury.
Dual inhibition of miR-92a-3p and miR-489–3p significantly reduced atherosclerotic lesion area (28.6%) after a week.
Dual miRNA inhibition in 5/6Nx;Apoe−/− mice significantly altered TGFβ signaling pathway and STAT3 transcriptional activity.
Tgfb2 and Fam220a are novel targets of miR-489–3p and miR-92a-3p, respectively.
Within human endothelial cells, miR-92a-3p directly regulates Fam220a, which negatively regulates STAT3 activity.
Acknowledgements
The authors would like to acknowledge Robert Taylor and Stuart Landstreet for human HDL purification through Lipoprotein and HDL Function Core at VUMC. The Center for Quantitative Sciences, specifically Shilin Zhao and Yan Guo, provided assistance with bioinformatics analyses. The authors would like to thank Leslie Sedgeman, Danielle Michell, and Ryan Allen for helpful discussion and critique of the project and manuscript.
Financial support
This work was supported by the National Institutes of Health [T32HL069765, P01HL116263 to MFL, R01HL128996 to KCV]; and National Science Foundation [1445197 to CBW].
Footnotes
Conflicts of interest
The authors declared they do not have anything to disclose regarding conflict of interest with respect to this manuscript.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.atherosclerosis.2019.01.023.
References
- [1].Go AS, Chertow GM, Fan DJ, McCulloch CE, Hsu CY, Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization, N. Engl. J. Med 351 (2004) 1296–1305. [DOI] [PubMed] [Google Scholar]
- [2].Liu H, Yan L, Ma GS, Zhang LP, Gao M, Wang YL, Wang SP, Liu BC, Association of chronic kidney disease and coronary artery disease in 1,010 consecutive patients undergoing coronary angiography, J. Nephrol 25 (2012) 219–224. [DOI] [PubMed] [Google Scholar]
- [3].Briasoulis A, Bakris GL, Chronic kidney disease as a coronary artery disease risk equivalent, Curr. Cardiol. Rep 15 (2013). [DOI] [PubMed] [Google Scholar]
- [4].Salmon AHJ, Ferguson JK, Burford JL, Gevorgyan H, Nakano D, Harper SJ, Bates DO, Peti-Peterdi J, Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction, J. Am. Soc. Nephrol 23 (2012) 1339–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schober A, Weber C, Mechanisms of MicroRNAs in atherosclerosis, Annu. Rev. Pathol. Mech 11 (2016) 583–616. [DOI] [PubMed] [Google Scholar]
- [6].Laffont B, Rayner KJ, MicroRNAs in the pathobiology and therapy of atherosclerosis, Can. J. Cardiol 33 (2017) 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kumar S, Kim CW, Simmons RD, Jo H, Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs, Arterioscler. Thromb. Vasc. Biol 34 (2014) 2206–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bartel DP, MicroRNAs: genomics, biogenesis, mechanism, and function, Cell 116 (2004) 281–297. [DOI] [PubMed] [Google Scholar]
- [9].Bartel DP, MicroRNAs: target recognition and regulatory functions, Cell 136 (2009) 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fang Y, Davies PF, Site-specific microRNA-92a regulation of Kruppel-like factors 4 and 2 in atherosusceptible endothelium, Arterioscler. Thromb. Vasc. Biol 32 (2012) 979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Boon RA, Hergenreider E, Dimmeler S, Atheroprotective mechanisms of shear stress-regulated microRNAs, Thromb. Haemostasis 108 (2012) 616–620. [DOI] [PubMed] [Google Scholar]
- [12].Liu H, Li G, Zhao W, Hu Y, Inhibition of MiR-92a may protect endothelial cells after acute myocardial infarction in rats: role of KLF2/4, Med. Sci. Mon 22 (2016) 2451–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, Urbich C, Zeiher AM, Dimmeler S, MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice, Science 324 (2009) 1710–1713. [DOI] [PubMed] [Google Scholar]
- [14].Loyer X, Potteaux S, Vion AC, Guerin CL, Boulkroun S, Rautou PE, Ramkhelawon B, Esposito B, Dalloz M, Paul JL, Julia P, Maccario J, Boulanger CM, Mallat Z, Tedgui A, Inhibition of microrna-92a prevents endothelial dysfunction and atherosclerosis in mice, Circ. Res 114 (2014) 434–443. [DOI] [PubMed] [Google Scholar]
- [15].Shang FQ, Wang SC, Hsu CY, Miao YF, Martin M, Yin YJ, Wu CC, Wang YT, Wu GH, Chien S, Huang HD, Tarng DC, Shiu YT, Cheung AK, Huang PH, Chen Z, Shyy JYJ, MicroRNA-92a mediates endothelial dysfunction in CKD, J. Am. Soc. Nephrol 28 (2017) 3250–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhou X, Qu Z, Zhu C, Lin Z, Huo Y, Wang X, Wang J, Li B, Identification of urinary microRNA biomarkers for detection of gentamicin-induced acute kidney injury in rats, Regul. Toxicol. Pharmacol 78 (2016) 78–84. [DOI] [PubMed] [Google Scholar]
- [17].Wei Q, Liu Y, Liu P, Hao J, Liang M, Mi QS, Chen JK, Dong Z, MicroRNA-489 induction by hypoxia-inducible factor-1 protects against ischemic kidney injury, J. Am. Soc. Nephrol 27 (2016) 2784–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yamamoto S, Zuo YQ, Ma J, Yancey PG, Hunley TE, Motojima M, Fogo AB, Linton MF, Fazio S, Ichikawa I, Kon V, Oral activated charcoal adsorbent (AST-120) ameliorates extent and instability of atherosclerosis accelerated by kidney disease in apolipoprotein E-deficient mice, Nephrol. Dial. Transplant 26 (2011) 2491–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison DG, Giddens DP, Jo H, A model of disturbed flow-induced atherosclerosis in mouse carotid artery by partial ligation and a simple method of RNA isolation from carotid endothelium, JoVE 40 (2010) 1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ponda MP, Barash I, Feig JE, Fisher EA, Skolnik EY, Moderate kidney disease inhibits atherosclerosis regression, Atherosclerosis 210 (2010) 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Robertson AKL, Rudling M, Zhou XH, Gorellk L, Flavell RA, Hansson GK, Disruption of TGF-beta signaling in T cells accelerates atherosclerosis, J. Clin. Invest 112 (2003) 1342–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mallat Z, Gojova A, Marchiol-Fournigault C, Esposito B, Kamate C, Merval R, Fradelizi D, Tedgui A, Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice, Circ. Res 89 (2001) 930–934. [DOI] [PubMed] [Google Scholar]
- [23].Toma I, McCaffrey TA, Transforming growth factor-beta and atherosclerosis: interwoven atherogenic and atheroprotective aspects, Cell Tissue Res. 347 (2012) 155–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dutzmann J, Daniel JM, Bauersachs J, Hilfiker-Kleiner D, Sedding DG, Emerging translational approaches to target STAT3 signalling and its impact on vascular disease, Cardiovasc. Res 106 (2015) 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ning HX, Rong Y, Zhang YJ, Ren FL, Chang ZJ, SIPAR interacts with STAT3 and negatively regulates its activities, Prog. Biochem. Biophys 32 (2005) 173–179. [Google Scholar]
- [26].Ren FL, Geng YT, Minami T, Qiu Y, Feng YR, Liu CX, Zhao J, Wang YY, Fan XZ, Wang YM, Li MD, Li J, Chang ZJ, Nuclear termination of STAT3 signaling through SIPAR (STAT3-Interacting Protein as a Repressor)-dependent recruitment of T cell tyrosine phosphatase TC-PTP, FEBS Lett. 589 (2015) 1890–1896. [DOI] [PubMed] [Google Scholar]
- [27].Ren FL, Su FQ, Ning HX, Wang YM, Geng YT, Feng YR, Wang YY, Zhang YQ, Jin Z, Li Y, Jia BQ, Chang ZJ, SIPAR negatively regulates STAT3 signaling and inhibits progression of melanoma, Cell. Signal 25 (2013) 2272–2280. [DOI] [PubMed] [Google Scholar]
- [28].Lim CP, Fu XY, Multiple roles of STAT3 in cardiovascular inflammatory responses, Prog. Mol. Biol. Transl 106 (2012) 63–73. [DOI] [PubMed] [Google Scholar]
- [29].Yamamoto S, Zhong J, Yancey PG, Zuo Y, Linton MF, Fazio S, Yang H, Narita I, Kon V, Atherosclerosis following renal injury is ameliorated by pioglitazone and losartan via macrophage phenotype, Atherosclerosis 242 (2015) 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bro S, Bentzon JF, Falk E, Andersen CB, Olgaard K, Nielsen LB, Chronic renal failure accelerates atherogenesis in apolipoprotein E-deficient mice, J. Am. Soc. Nephrol 14 (2003) 2466–2474. [DOI] [PubMed] [Google Scholar]
- [31].Bro S, Moeller F, Andersen CB, Olgaard K, Nielsen LB, Increased expression of adhesion molecules in uremic atherosclerosis in apolipoprotein-E-deficient mice, J. Am. Soc. Nephrol 15 (2004) 1495–1503. [DOI] [PubMed] [Google Scholar]
- [32].Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, Wanner C, Krane V, Cass A, Craig J, Neal B, Jiang LX, Hooi LS, Levin A, Agodoa L, Gaziano M, Kasiske B, Walker R, Massy ZA, Feldt-Rasmussen B, Krairittichai U, Ophascharoensuk V, Fellstrom B, Holdaas H, Tesar V, Wiecek A, Grobbee D, de Zeeuw D, Gronhagen-Riska C, Dasgupta T, Lewis D, Herrington W, Mafham M, Majoni W, Wallendszus K, Grimm R, Pedersen T, Tobert J, Armitage J, Baxter A, Bray C, Chen YP, Chen ZM, Hill M, Knott C, Parish S, Simpson D, Sleight P, Young A, Collins R, Investigators S, The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial, Lancet 377 (2011) 2181–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Frutkin AD, Otsuka G, Stempien-Otero A, Sesti C, Du L, Jaffe M, Dichek HL, Pennington CJ, Edwards DR, Nieves-Cintron M, Minter D, Preusch M, Hu JH, Marie JC, Dichek DA, TGF-beta 1 limits plaque growth, stabilizes plaque structure, and prevents aortic dilation in apolipoprotein E-null mice, Arterioscl. Throm. Vas 29 (2009) 1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wood H, FDA approves patisiran to treat hereditary transthyretin amyloidosis, Nat. Rev. Neurol 14 (2018) 570. [DOI] [PubMed] [Google Scholar]
- [35].Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S, Plante-Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH 3rd, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB, Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis, N. Engl. J. Med 379 (2018) 11–21. [DOI] [PubMed] [Google Scholar]
- [36].Michell DL, Vickers KC, HDL and microRNA therapeutics in cardiovascular disease, Pharmacol. Therapeut. 168 (2016) 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chakraborty C, Sharma AR, Sharma G, Doss CGP, Lee SS, Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine, Mol. Ther. Nucleic Acids 8 (2017) 132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Stein CA, Castanotto D, FDA-approved oligonucleotide therapies in 2017, Mol. Ther 25 (2017) 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kaczmarek JC, Kowalski PS, Anderson DG, Advances in the delivery of RNA therapeutics: from concept to clinical reality, Genome Med. 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT, MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins, Nat. Cell Biol 13 (2011) 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tabet F, Vickers KC, Cuesta Torres LF, Wiese CB, Shoucri BM, Lambert G, Catherinet C, Prado-Lourenco L, Levin MG, Thacker S, Sethupathy P, Barter PJ, Remaley AT, Rye KA, HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells, Nat. Commun 5 (2014) 3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gille A, D’Andrea D, Tortorici MA, Hartel G, Wright SD, CSL112 (apolipoprotein A-I [human]) enhances cholesterol efflux similarly in healthy individuals and stable Atherosclerotic disease patients, Arterioscl. Throm. Vas 38 (2018) 953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kootte RS, Smits LP, van der Valk FM, Dasseux JL, Keyserling CH, Barbaras R, Paolini JF, Santos RD, van Dijk TH, Dallinga-van Thie GM, Nederveen AJ, Mulder WJM, Hovingh GK, Kastelein JJP, Groen AK, Stroes ES, Effect of open-label infusion of an apoA-I-containing particle (CER-001) on RCT and artery wall thickness in patients with FHA, J. Lipid Res 56 (2015) 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tardif JC, Ballantyne CM, Barter P, Dasseux JL, Fayad ZA, Guertin MC, Kastelein JJP, Keyserling C, Klepp H, Koenig W, L’Allier PL, Lesperance J, Luscher TF, Paolini JF, Tawakol A, Waters DD, C.H.I. Significantly, Effects of the high-density lipoprotein mimetic agent CER-001 on coronary atherosclerosis in patients with acute coronary syndromes: a randomized trial, Eur. Heart J 35 (2014) 3277–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kataoka Y, Andrews J, Duong M, Nguyen T, Schwarz N, Fendler J, Puri R, Butters J, Keyserling C, Paolini JF, Dasseux JL, Nicholls SJ, Regression of coronary atherosclerosis with infusions of the high-density lipoprotein mimetic CER-001 in patients with more extensive plaque burden, Cardiovasc. Diagn. Ther 7 (2017) 252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yamamoto S, Kon V, Chronic kidney disease induced dysfunction of high density lipoprotein, Clin. Exp. Nephrol 18 (2014) 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ, Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis, J. Clin. Invest 121 (2011) 2921–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.