

Abstract
NLRP3 inflammasome is a protein complex crucial to caspase-1 activation and IL-1β and IL-18 maturation. This receptor participates in innate immune responses to different pathogens, including the bacteria of genus Brucella. Our group recently demonstrated that Brucella abortus-induced IL-1β secretion involves NLRP3 inflammasome and it is partially dependent on mitochondrial ROS production. However, other factors could be involved, such as P2X7-dependent potassium efflux, membrane destabilization and cathepsin release. Moreover, there is increasing evidence that nitric oxide acts as a modulator of NLRP3 inflammasome. The aim of this study was to unravel the mechanism of NLRP3 inflammasome activation induced by B. abortus, as well as the involvement of bacterial nitric oxide (NO) as a modulator of this inflammasome pathway. We demonstrated that NO produced by B. abortus can be used by the bacteria to modulate IL-1β secretion in infected murine macrophages. Additionally, our results suggest that B. abortus-induced IL-1β secretion depends on a P2X7-independent potassium efflux, lysosomal acidification, cathepsin release, mechanisms clearly associated to NLRP3 inflammasome. In summary, our results help to elucidate the molecular mechanisms of NLRP3 activation and regulation during an intracellular bacterial infection.
Keywords: Brucella abortus, NLRP3 inflammasome, Nitric oxide, Immune evasion, ΔnarG mutant
INTRODUCTION
Over the past years, inflammasomes gained attention by their role on defense against pathogens and in the development of metabolic, neurodegenerative and autoinflammatory diseases as well as in cancer [1]. Inflammasomes are multiprotein platforms which control maturation of the proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 [2]. Several inflammasomes have been identified, and most of them include either receptors of the NOD-like receptor family of proteins (e.g. NLRP3, NLRP1 and NLRPC4) or AIM receptors (AIM2 inflammasome). However, the NLRP3 is by far the most studied inflammasome [3, 4]. NLRP3 pathway requires at least two signals: the first is provided by microbial molecules or endogenous cytokines and leads to the upregulation of NLRP3 and pro-IL-1β through the activation of the transcription factor NF-κB; the second signal is provided by diverse stimuli, such as pathogen or damage-associated molecular patterns (PAMPs or DAMPs, respectively), triggering the assembly of the NLRP3 inflammasome and multimerization of the adaptor molecule ASC [5]. However, the molecular interactions that engage the NLRP3 inflammasome in response to such distinct stimuli are still unclear. Recently, it has been shown that sodium (Na+) influx and most notably potassium (K+) efflux via purinergic receptor P2X7 are events related to NLRP3 inflammasome activation induced by bacterial toxins and particulate matter [6, 7]. A second model proposed that NLRP3 acts as a cell stress sensor, activated by reactive oxygen species (ROS) generated in spatial and temporal proximity to the inflammasome [8]. It has been also observed a correlation between ROS generation and K+ efflux. Several NLRP3 stimuli studied to date (e.g. ATP and membrane pore-forming proteins) triggered ROS production, frequently accompanied by K+ efflux and although the interplay between these events is not clear, it is possible that low intracellular [K+] triggers ROS production and vice-versa [9].
Increasing evidence has shown that NLRP3 inflammasome activation is important for host defense and effective pathogen clearance against microbial infections [10]. Previous reports have shown that the immune responses against Gram-positive bacteria such as Staphylococcus aureus or Listeria monocytogenes requires NLRP3 activation, the same is not true for Salmonella typhimurium or Francisella tularensis (Gram-negative bacteria) [11]. However, some Gram-negative enteropathogens, such as enterohemorrhagic Escherichia coli (EHEC) and Citrobacter rodentium, induce NLRP3 inflammasome activation in bone marrow-derived macrophages in a Toll-IL-1 receptor (TIR)-domain-containing adapter-inducing interferon-β (TRIF)-dependent pathway [12]. Furthermore, Neisseria gonorrhoeae induces a cathepsin B-dependent NLRP3 inflammasome activation and cell death [13]. Experiments with macrophages infected with Paracoccidioides brasiliensis also showed that endosomal-lysosomal acidification is a mechanism involved in NLRP3 inflammasome activation [14]. Brucella abortus is a facultative intracellular gram-negative cocobacillus, causative agent of brucellosis in humans and livestock. In humans, it causes undulant fever, endocarditis, arthritis and osteomyelitis; in livestock, it leads to abortion and infertility, resulting in significant economical losses [15, 16]. Our group published recently that IL-1β secretion in macrophages infected with Brucella abortus was partially dependent on mitochondrial ROS. Moreover, infected NLRP3 knockout (KO) macrophages secreted lower levels of IL-1β than wild-type cells and NLRP3 KO mice are more susceptible at four-weeks after infection when compared to wild-type animals [17]. Our previous study provided important insights into the role of NLRP3 inflammasome in response to B. abortus; however, the exact mechanism underlying the NLRP3-dependent host response needs to be better understood.
To deal with host immune response, some bacteria have evolved diverse strategies to manipulate inflammasome activation in host cells [18]. Legionella pneumophila, for example, controls ASC levels to manipulate inflammasome, apoptosome and NF-κB pathways, establishing the necessary environment for its replication within human monocytes [19]. The protein RipA from Francisella tularensis inhibits IL-1β, IL-18 and TNF-α secretion in macrophages to evade host immunity; mice infected with mutants for RipA produced higher levels of inflammatory cytokines when compared to the wild-type bacteria [20]. It was recently described that nitric oxide (NO) negatively regulates IL-1β processing, through inhibition of NLRP3 inflammasome assembly in cells infected with Mycobacterium tuberculosis [21]. In humans, Mycobacterium bacilli reside in granulomas, structures which may limit the availability of oxygen. Under these conditions, there is an increased expression of genes involved in denitrification, a energy-yelding metabolic process which converts nitrate (NO3−) to inert nitrogen gas (N2), with NO as a intermediate metabolite [22, 23]. However, the hypothesis of IL-1β production being modulated by Mycobacterium NO has not been tested yet. Recently, genomic analysis has revealed that members of the genus Brucella also possess denitrifying genes in their genomes [24, 25], and this raises the hypothesis of Brucella NO as a regulator of NLRP3-dependent IL-1β, since it has been shown that genes involved in denitrification regulate Brucella virulence in mice [26].
In the present study, we investigated the cellular and molecular mechanisms involved in NLRP3 activation during B. abortus infection. Most importantly, we demonstrated that NO produced by B. abortus is a modulator of NLRP3-dependent IL-1β production and acts as an evasion strategy used by this pathogen to avoid proper host innate immune responses.
RESULTS
The NLRP3-dependent IL-1β secretion in B. abortus-infected BMDMs requires potassium efflux
The NLRP3 inflammasome can be activated by a variety of stimuli, including extracellular ATP, microbial toxins (e.g., nigericin), and crystalline particles, all converging on potassium efflux [27]. Previous studies have reported that potassium efflux is important in caspase-1 activation, IL-1β processing, and cell death and NLRP3 activation is inhibited by high extracellular [K+] [6, 28]. To test whether potassium efflux is involved in B. abortus-induced NLRP3 inflammasome activation, two defined inhibitors were used to treat B. abortus-infected BMDMs, glibenclamide (a selective inhibitor for ATP-dependent potassium channels) and KCl (since high extracellular concentrations of K+ prevent NLRP3 activation). Addition of increasing concentrations of glibenclamide and KCl to macrophages prior to bacterial infection reduced IL-1β secretion in a dose-dependent manner (Fig 1A and 1B) without significant changes in TNF-α (Figs 1C and 1D), an inflammasome-independent cytokine. Moreover, Western blot analysis showed that treatment of BMDMs with glibenclamide and KCl prior to B. abortus infection does not significantly affect pro-IL-1 β and pro-caspase-1 levels but it results in a decrease in secreted IL-1β and caspase-1 mature forms (Fig 1E). These results suggest that potassium efflux induces a NLRP3-dependent caspase-1 activation and IL-1β secretion in B. abortus-infected BMDM.
Figure 1.

The effect of glibenclamide and KCl on IL-1β and TNF-α release in BMDMs infected with B. abortus. Cells were preincubated with glibenclamide (Glib) or KCl for 1h before infection with B. abortus strain 2308 (MOI of 100:1) and supernatants were harvested 17h after infection. IL-1β (A, B) and TNF-α (C, D) secretion were determined by ELISA. The graphs show results from a single experiment performed in triplicate, representative of three independent experiments with similar results and data are presented as mean ± SD. (E) Western blot analysis of lysates and supernatants of BMDMs infected with B. abortus, preincubated or not with glibenclamide (80 μM) or KCl (40 mM). Abbreviations: NI (non-infected), Ctrl (control), Glib (glibenclamide), KCl (potassium chloride), CL (cell lysate), SN (supernatant). Image is representative of two independent experiments. Significant differences between infected cells in the presence or absence of the inhibitor are denoted by one, two or three asterisks (for p<0.05, p<0.01 and p<0.001, respectively; one-way ANOVA + Bonferroni post-hoc test).
IL-1β secretion in B. abortus-infected BMDMs does not require the P2X7 receptor
The occurrence of extracellular ATP is considered a signal for the immune system, particularly during an inflammatory response. It is sensed by P2X receptors, whose activation by ATP opens a cation-specific channel and alters the ionic environment of the cell activating several pathways including the inflammasome [29]. Moreover, studies suggest that P2X7R opening causes a drastic change in K+ homeostasis which has an important role in caspase-1 activation and NLRP3-dependent IL-1β release [30, 31]. Therefore, we investigate the potential role of P2X7R in the IL-1β secretion induced by B. abortus in macrophages using two different approaches: first, by preincubating BMDMs with A740003 (a selective P2X7 purinoceptor antagonist) before infecting with B. abortus, and secondly, by quantifying IL-1β secretion in infected P2X7 knockout cells. As shown in the Fig 2A, IL-1β secretion in B. abortus-infected cells was not affected by the presence of the inhibitor, even when tested at higher concentrations (50 μM). As a control, cells were preincubated with A740003 at the highest concentration tested and then stimulated with LPS plus ATP. As expected, the presence of the inhibitor was sufficient to abolish IL-1β secretion. Moreover, no differences in cytokine secretion were observed between wild-type and P2X7R KO BMDMs, when infected with the bacteria (Fig 2B). The results suggest that P2X7R does not play an essential role in B. abortus-induced IL-1β secretion.
Figure 2.

The role of P2X7 receptor on IL-1β release in BMDMs infected with B. abortus. (A) BMDMs were preincubated with the P2X7 inhibitor A740003 for 30 min before stimulation with ATP (5 mM) or infection with B. abortus strain 2308 (MOI of 100:1). Ultrapure LPS (1 μg/mL) was added 4h before stimulation with 5 mM ATP. Supernatants were harvested 30 min after stimulation with ATP or 17h after infection with B. abortus. (B) wild type and P2X7R KO cells were infected with B. abortus strain S2308 (MOI of 100:1) or stimulated with LPS + ATP (as in A) and the supernatants collected 17h after infection. IL-1β secretion was determined by ELISA. C symbol denotes control, non-infected cells. The graphs show results from a single experiment performed in triplicate, representative of three independent experiments with similar results and data are presented as mean ± SD. Significant differences are denoted by three asterisks (for p<0.001; one-way ANOVA + Bonferroni post-hoc test).
IL-1β secretion in infected BMDMs was dependent on lysosomal acidification and cathepsin B release
Previous studies have shown that NLRP3 inflammasome can be activated by a number of signaling mechanisms other than potassium efflux, such as lysosomal acidification and cathepsin B release into the cytosol, resulting in IL-1β secretion [32–34]. The use of chloroquine, which inhibits endosomal-lysosomal system acidification and the chemical Ca074Me, which acts as a membrane-permeable inhibitor of the cathepsin B, can result in a reduction of inflammasome activation [35, 36]. Therefore, we examined whether B. abortus-induced IL-1β secretion in BMDMs is dependent on lysosomal acidification and cathepsin B release. Preincubation of BMDMs with increasing concentrations of chloroquine or Ca074Me resulted in a dose-response decrease of IL-1β secretion, as shown in the Figs 3A and 3B. TNF-α production, in turn, was unaffected (Figs 3C and 3D), except when cells were preincubated with the highest concentration of chloroquine used prior to infection with B. abortus (Fig 3C). Additionally, western blot analysis showed a similar pattern to those observed in glibenclamide and KCl, in which the treatment of BMDMs with chloroquine and Ca074Me prior to B. abortus infection does not significantly affect pro-IL-1 β and pro-Caspase-1 levels but it results in a decrease in secreted IL-1β and caspase-1 mature forms (Fig 3E). Together, these results suggest lysosomal acidification and cathepsin B release are important mechanisms to the activation of NLRP3 inflammasome and consequently caspase-1 activation and IL-1β release following B. abortus infection.
Figure 3.

The effect of chloroquine and Ca074Me on inflammasome activation in BMDMs infected with B. abortus. Cells were preincubated with Chloroquine (Chl) or Ca074Me for 1h before infection with B. abortus strain 2308 (MOI of 100:1) and supernatants were harvested 17h after infection. IL-1β (A, B) and TNF-α (C, D) secretion were determined by ELISA. The graphs show results from a single experiment performed in triplicate, representative of three independent experiments with similar results and data are presented as mean ± SD. (E) Western blot analysis of lysates and supernatants of BMDMs infected with B. abortus, preincubated or not with chloroquine (80 μM) or Ca074Me (12 μM). Abbreviations: NI (non-infected), Ctrl (control), Chl (chloroquine), Ca (Ca074Me), CL (cell lysate), SN (supernatant). Image is representative of two independent experiments. Significant differences between infected cells in the presence or absence of the inhibitor are denoted by three asterisks (for p<0.001, one-way ANOVA + Bonferroni post-hoc test).
NADPH oxidase-derived ROS is dispensable for IL-1β secretion in B. abortus-infected BMDMs
Several authors have proposed that NLRP3 inflammasome acts as a sensor of cellular stress when activated by reactive oxygen species (ROS) [9, 37, 38]. ROS generation is triggered by a plethora of stimuli, such as whole pathogens, pathogen-associated molecular patterns (PAMPs), asbestos, MSU (monosodium urate) crystals and damage-associated molecular patterns or DAMPs [9, 39, 40]. We described previously that IL-1β secretion induced by B. abortus is partially dependent on mitochondrial ROS, based on experiments with BMDMs preincubated with MitoTEMPO prior to bacterial infection [17]. However, it has been also proposed that NADPH oxidase-derived ROS could activate NLRP3 inflammasome, based on studies with chemical inhibitors and knockdown of NADPH subunits [41, 42]. We therefore investigated whether IL-1β secretion induced by B. abortus infection depends on NADPH oxidase-derived ROS. To perform this experiment, we infected BMDMs lacking gp91phox, defective in phagocyte ROS generation [43]. As observed in the Fig 4, gp91phox KO BMDMs showed no impairment of IL-1β secretion in response to B. abortus or LPS plus ATP, suggesting that NADPH oxidase-derived ROS does not play a role in NLRP3 inflammasome activation in BMDMs infected with B. abortus.
Figure 4.
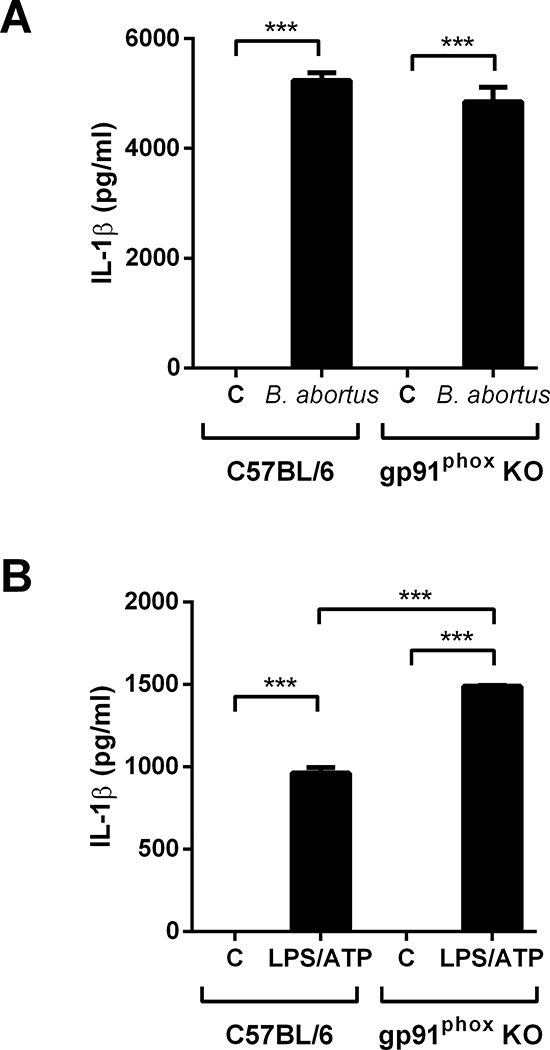
The role of NADPH oxidase on IL-1β release in BMDMs infected with B. abortus. (A) cells were infected with B. abortus strain 2308 (MOI of 100:1) and supernatants were harvested 17h after infection. As a control, cells were primed with ultrapure LPS (1 μg/mL) for 4 hours before stimulation with 5 mM ATP and supernatants were harvested 30 min after stimulation, as shown in (B). IL-1β secretion was determined by ELISA. The graphs show results from a single experiment performed in triplicate, representative of three independent experiments with similar results and data are presented as mean ± SD. Significant differences are denoted by three asterisks (for p<0.001, one-way ANOVA + Bonferroni post-hoc test).
Nitric oxide inhibits NLRP3 inflammasome activation and IL-1β secretion induced by B. abortus
The NLRP3 inflammasome activation is a process regulated by several mechanisms in order to prevent detrimental effects to the host. For instance, it was demonstrated that type I IFNs negatively regulate caspase-1 activation induced by NLRP3 agonists, but not that induced by AIM2 or NLRC4 ligands [44]. In addition, in vitro experiments suggested that an increase in extracellular osmolarity in macrophages inhibits hypotonicity-induced NLRP3 activation and IL-1β release [45]. It was also suggested that nitric oxide (NO) prevents IL-1β and IL-18 release by inhibiting caspase-1 activity, as well as preventing the assembly of the NLRP3 inflammasome via thiol nitrosylation [21, 46, 47]. Therefore, we assessed the potential role of NO in inhibiting NLRP3-dependent IL-1β secretion induced by B. abortus through incubation of BMDMs with L-NAME, (an analog of arginine that inhibits NO production) or SNAP (a nitrosothiol derivative used as NO donor), prior to infection. The results shown in the Figs 5A and 5B revealed opposite effects of L-NAME and SNAP, since the preincubation of cells with L-NAME prior to B. abortus infection increased IL-1β secretion in approximately 60%, while the preincubation with SNAP resulted in approximately 77% reduction in IL-1β secretion. NO production was assessed indirectly by measuring the concentration of nitrite (NO2−) by the Griess reaction. Interestingly, NO production was not diminished in Brucella-infected BMDMs in the presence of L-NAME (Fig 5C), although a 50% reduction was observed in cells stimulated with LPS. In addition, the incubation of BMDMs with SNAP prior to B. abortus infection resulted in a NO production 2.8 times higher than the infection alone (Fig 5D). Nevertheless, NO production in infected cells was reduced compared to stimulation with LPS in the presence of SNAP. The preincubation of cells with L-NAME or SNAP did not affect invasion or intracellular bacterial growth, as observed in the Fig 5E. To further investigate the phenomenon of inhibition of IL-1β secretion mediated by NO, we infected BMDMs with B. abortus in the presence or absence of bovine hemoglobin (Hg), a widely known NO scavenger [48–50]. These results strengthen our findings shown in the Fig 5, since the preincubation with Hg increased IL-1β secretion and strongly reduced NO2− detection in B. abortus-infected cells (Supporting Information Figs 1A and 1B), while not compromising intracellular bacterial replication (Supporting Information Figs 1C). In addition, as shown in Western blot analysis, the treatment of BMDMs with L-NAME and SNAP prior to B. abortus infection neither affect pro-IL-1β and pro-caspase-1 levels nor it has influenced NLRP3 expression. On the other hand, SNAP treatment resulted in a decrease in secreted IL-1 β and caspase-1 activation, a phenomenon not observed in BMDMs pretreated with L-NAME (Fig 5F).
Figure 5.

The effect of L-NAME and SNAP on IL-1β, nitrite production and bacterial replication in BMDMs infected with B. abortus. Cells were preincubated with L-NAME (2 mM) or SNAP (0.5 mM) for 30 min before infection with B. abortus strain 2308 (MOI of 100:1) or stimulation with ultrapure LPS (1 μg/mL) or nigericin (Nig, 20 μM). Prior to stimulation with nigericin, cells were primed with ultrapure LPS for 4 hours. Supernatants were harvested 17h after stimulation, except for LPS/Nig-stimulated cells, whose supernatants were harvested 30 min after stimulation. IL-1β (A and B) was determined by ELISA and nitrite (NO2−, C and D) by Griess reaction. The graphs show results from a single experiment representative of four independent experiments performed in triplicate, with similar results and data are presented as mean ± SD. (E) BMDMs were infected with B. abortus (MOI 100:1) and CFU counting were determined 2 and 24 hours post infection (hpi). The graph shows results from a single experiment representative of three independent experiments performed in quadruplicate, with similar results and data are presented as mean ± SD. (F) Western blot analysis of lysates and supernatants of BMDMs infected with B. abortus, preincubated or not with L-NAME (2 mM) or SNAP (0.5 mM). Abbreviations: NI (non-infected), Ctrl (control), LN (L-NAME), SN (SNAP), CL (cell lysate), SN (supernatant). Image is representative of two independent experiments. Significant differences between infected cells in the presence or absence of L-NAME or SNAP are denoted by two or three asterisks (for p<0.01 and p<0.001, respectively; one-way ANOVA + Bonferroni post-hoc test).
Bacterial nitric oxide modulates NLRP3-dependent IL-1β secretion in B. abortus-infected BMDMs
The results shown in the Fig. 5 (and Supporting Information Fig 1) led us to further investigate the role of NO on NLRP3-mediated IL-1β secretion in BMDMs infected with B. abortus, since infected macrophages preincubated or not with L-NAME produced NO at similar levels. We then infected wild-type and iNOS KO-derived BMDMs with B. abortus at the same conditions previously described. Herein, we did not observe significant differences in IL-1β secretion between both wild-type and iNOS KO cells (Fig 6A). However, although NO production was lower in iNOS defective cells than in wild-type macrophages (Fig 6B), it was still detected. This phenomenon was observed only in BMDMs infected with the bacteria, since iNOS KO cells stimulated with LPS did not produce NO in detectable levels as expected (Fig 6C). Quantitative real-time PCR analysis confirmed the lack of iNOS mRNA expression in B. abortus-infected iNOS KO cells (Fig 6D). These results raised the hypothesis that a NO metabolite produced by B. abortus could also modulate NLRP3-dependent IL-β secretion. Brucella is a member of the α-proteobacteria class and it is capable of reduction of nitrate to dinitrogen (N2) through a cascade of different enzymes that produces intermediate gaseous nitrogen oxide products, including NO [24–26]. Even though only a few pathogens have been shown to be denitrifiers, recent study suggested an important role of denitrification genes in host-pathogen interaction [51, 52]. Therefore, to assess the potential of bacterial NO in modulating NLRP3-dependent IL-1β secretion in macrophages, we generated bacterial mutants defective in the catalytic subunit α of enzyme nitrate reductase (narG gene), as described by Haine and coworkers [53]. The nitrate reductase is responsible for the first step in denitrification reaction, which reduces nitrate to nitrite, the substrate to the production of NO by nitrite reductase [54, 55]. The integrative disruption strategy used in this study, as well the genotypic and phenotypic characterization of B. abortus ΔnarG mutants are shown in the Supporting information Fig 2.
Figure 6.

The modulatory effect of bacterial NO in NLRP3-dependent IL-1β secretion in BMDMs infected with B. abortus. Cells were infected with B. abortus strain 2308 (MOI of 100:1) and supernatants were harvested 17h after infection. IL-1β secretion and NO production in BMDMs infected with B. abortus strain 2308 (MOI of 100:1) are shown in A and B, respectively. As a control, cells were stimulated with ultrapure LPS (1 μg/mL) for 17 hours (C). (D) Determination of iNOS mRNA expression in B. abortus-infected cells, normalized to 18S mRNA. Cells were also infected with B. abortus wild type (wt) or ΔnarG mutant and supernatants were harvested 17h after infection. The levels of NO production, IL-1β and TNF-α are shown in E, F and G, respectively. Cytokine secretion was determined by ELISA and NO production by Griess reagent. The graphs show results from a single experiment representative of four independent experiments performed in triplicate, with similar results and data are presented as mean ± SD. (H) Western blot analysis of lysates and supernatants of BMDMs infected with B. abortus (wild type or ΔnarG mutant). Image is representative of two independent experiments. (I) BMDMs were infected with B. abortus wild type (wt) or ΔnarG mutant (MOI 100:1) and CFU counting were determined 24 hours post infection. The graph shows results from a single experiment representative of three independent experiments performed in triplicate, with similar results and data are presented as mean ± SD. Abbreviations: NI (non-infected), wt (wild type bacteria), ΔnarG (nitrate reductase deficient bacteria), CL (cell lysate), SN (supernatant). Significant differences are denoted by three asterisks (for p<0.001, one-way ANOVA + Bonferroni post-hoc test).
As shown in the Fig 6E, NO detection was completely abrogated in iNOS KO cells infected with the ΔnarG mutant, suggesting that the NO detected in B. abortus-infected cells is, in part, produced by the bacteria. Moreover, IL-1β secretion was higher in ΔnarG-infected cells when compared to WT-infected cells (Fig 6F); although this difference was not observed in TNF-α production (Fig 6G). In addition, Western blot analysis showed an increase in secreted IL-1β and caspase-1 levels in BMDMs infected with the ΔnarG mutant when compared to those cells infected with the wild type bacteria (Fig 6H). Surprisingly, there were no significant differences in bacterial load in BMDMs infected either with ΔnarG mutant or the wild-type bacteria (Fig 6I), suggesting that B. abortus could trigger additional mechanisms to ensure its survival inside macrophages.
DISCUSSION
Several studies have shown that NLRP3 inflammasome activation is crucial for recognition of pathogenic microbes and allows the host to induce appropriate protective innate immune responses [3, 12, 56]. Our group recently demonstrated the importance of NLRP3 inflammasome for IL-1β secretion and resistance to B. abortus infection [17]; however, the molecular mechanisms underlying the inflammasome activation and regulation are not fully understood. The mechanisms of IL-1β secretion following NLRP3 activation were examined by treating BMDMs with specific inhibitors prior to infection with Brucella. The results demonstrated that K+ efflux, lysosomal-endosomal acidification and cathepsin B release are critical events to a NLRP3 inflammasome-dependent caspase-1 activation and IL-1β secretion in BMDMs infected with B. abortus. Additionally, recent findings from our group demonstrate that pyroptosis which is dependent on caspase-11 and GSDMD are central to potassium efflux and, consequently, to NLRP3 inflammasome activation in response to B. abortus [57]. In contrast, we show that the purinergic receptor P2X7 is not required to B. abortus-induced K+ efflux in BMDMs and this could be explained by different ways: i) this event is dependent on other potassium channels (e.g. pannexin- or a different P2X family member) or ii) K+ efflux in BMDMs is not restricted to the P2X7 receptor. It is important to emphasize that the reduction in caspase-1 activation and IL-1β secretion observed in BMDMs pretreated with all inhibitors does not correlate with a decline in cell viability, in intracellular bacterial load or in NLRP3 protein levels as observed in the Supporting information Fig 3.
Our previous study showed that IL-1β secretion induced by B. abortus is partially dependent on mitochondrial ROS. In addition, a recent study by others suggested that both mitochondrial ROS-dependent and -independent pathways are required for NLRP3 inflammasome activation [58], which raised the possibility of the NLRP3 inflammasome being also activated by other types of ROS in BMDMs infected with B. abortus. Therefore, we assessed whether phagocytic NADPH oxidase-derived ROS could also activate the NLRP3 inflammasome in infected BMDMs. However, the results showed that IL-1β secretion in gp91phox deficient BMDMs was not impaired in response to B. abortus infection. These results are in agreement with a previous report, in which it was suggested that NADPH oxidase is not required to control B. abortus replication in vivo [59]. This finding suggests that the superoxide (O2•−) produced by NADPH oxidase does not play a role in NLRP3 activation and IL-1β secretion, but it cannot rule out the possibility that other sources of O2•− could act in NLRP3-mediated immune responses to B. abortus, such as those produced by xanthine oxidase in peroxisomes [60].
Although the proinflammatory cytokine IL-1β is critical to host defense against many pathogens, exacerbated expression and secretion of this molecule can lead to tissue damage, and dysregulated inflammasome activation is related to the pathogenesis of a variety of inflammatory diseases [61, 62]. In this regard, NO has been pointed to as an important molecule produced by host cell to control NLRP3-dependent IL-1β secretion, given that it specifically inhibits the assembly of the NLRP3 inflammasome via thiol nitrosylation [21, 47]. Surprisingly, our results suggest that IL-1β secretion in B. abortus-infected BMDMs could be negatively modulated by bacterial NO, since NO production was unchanged by incubation of macrophages with L-NAME prior to infection but IL-1β secretion was increased. Moreover, uninfected cells incubated with a NO donor (SNAP) produced higher amounts of NO when compared to infected cells, and this could be explained by the fact that the NO donor itself is not able to induce ROS production. This is necessary to generate peroxynitrite (ONOO−), a product of the diffusion-controlled reaction of NO and O2•− and a highly reactive nitrogen species (RNS), a class of molecules produced in the respiratory burst [63, 64]. To test this hypothesis of bacterial NO regulating inflammasome activation, we constructed Brucella mutants for catalytic subunit of the nitrate reductase (ΔnarG), which were used to infect wild-type and iNOS KO BMDMs. The results are summarized in the Fig. 7 and they strongly suggest that NO detected in supernatants of infected cells are produced mainly by the bacteria, and it can regulate caspase-1 activation and IL-1β secretion. It was also shown that BMDMs infected with ΔnarG mutants produced similar amounts of TNF-α as well as their survival inside cells did not differ from that observed for wild-type bacteria. Our results are in line with those recently reported, showing that control of B. abortus replication in macrophages does not depend on host cell NO, given that infection of immortalized macrophages in the presence of NG-monomethyl-L-arginine, a nitric oxide synthase inhibitor, does not alter intracellular bacterial growth [59]. More importantly, we demonstrated that B. abortus produces NO as a mechanism of regulation of IL-1β secretion in macrophages, possibly via NLRP3 inflammasome, since it was previously showed that the assembly of this inflammasome is impaired by NO-dependent thiol nitrosylation [21]. Altogether, these results help to elucidate the molecular mechanisms of NLRP3 activation and regulation and suggest a potential evasion mechanism used by bacterial pathogens to modulate host innate immune responses.
Figure 7.

Mechanisms of activation of NLRP3 inflammasome and its regulation by nitric oxide in BMDMs infected with B. abortus. B. abortus infection induces lysosomal acidification, cathepsin B release and potassium (K+) efflux and leads to Caspase-1 activation and IL-1β secretion. These events can be inhibited by chloroquine, Ca074Me and Glibenclamide/KCl, respectively, without significant changes in pro-caspase-1 and pro-IL-1β expression. The NLRP3-dependent IL-1β production is also impaired by NO, produced either by macrophage iNOS or as an intermediate metabolite of the bacterial denitrification process. Chl: chloroquine; Glib: glibenclamide; ROS: reactive oxygen species.
MATERIAL AND METHODS
Reagents
Adenosine-5’-triphosphate disodium salt (ATP) and Escherichia coli ultrapure LPS were purchased from Invivogen (San Diego, CA, USA). DMEM, Fetal bovine serum (FBS), Penicillin/Streptomycin, Phosphate-Buffered Saline (PBS), Hepes, M-PER® Mammalian Protein Extraction Reagent and Bicinchoninic acid (BCA) protein assay kit was purchased from were purchased from Gibco/Thermo Fisher Scientific (Waltham, MA, USA). Potassium chloride (KCl) was purchased from Synth (Diadema, SP, Brazil). Sodium nitrate (NaNO3) and sodium nitrite (NaNO2) were purchased from Merck (Darmstadt, Germany). Nigericin sodium salt, Chloroquine diphosphate, Glibenclamide, A-740003, Ca074Me (methyl ester), S-Nitroso-N-acetyl-DL-penicillamine (SNAP), Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME), hemoglobin from bovine blood, Sulfanilamide, N-1-napthylethylenediamine dihydrochloride and protease inhibitor cocktail were purchased from Sigma/Aldrich (St. Louis, MO, USA). Mouse monoclonal anti-NLRP3 (AG-20B-0014-C100) and anti-Caspase-1 (AG-20B-0042-C100) antibodies were purchased from Adipogen (San Diego, CA, USA). Mouse monoclonal anti IL-1β (12242S), rabbit monoclonal anti-β-actin (4970S) and secondary HRP-linked anti-mouse IgG (7076S) and anti-rabbit IgG (7074S) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Clarity™ Western ECL Substrate was purchased from Bio-Rad (Philadelphia, PA, USA).
Ethics statement
All experiments involving animals were conducted in accordance with the Brazilian Federal Law number 11.794, which regulates the scientific use of animals in Brazil, the Institutional Animal Care and Use Committees (IACUC) guidelines and the Animal Welfare Act and Regulations guidelines established by the American Veterinary Medical Association Panel on Euthanasia. Animals were fed, housed and handled in strict agreement with these recommendations. All protocols were approved by the Committee for Ethics in Animal Experimentation (CETEA) at Universidade Federal de Minas Gerais UFMG under permit #128/2014.
Mice
The following strains were used in this work: C57BL/6 (wild type strain, obtained from the Federal University of Minas Gerais-UFMG, Brazil), gp91phox and iNOS KO (kindly provided by Dra. Leda Quércia Vieira, Department of Biochemistry and Immunology, Institute of Biological Sciences, UFMG, Brazil), P2X7 KO (kindly provided by Dr. José Carlos Alves Filho, Department of Pharmacology, Ribeirao Preto Medical School, University of Sao Paulo, Brazil). Genetically deficient and control mice were maintained at our facilities and used at 6 to 8 weeks of age. Mice were housed in filter-top cages and provided with sterile water and food ad libitum.
Bacteria
Brucella abortus smooth virulent strain 2308 was obtained from our laboratory collection. Frozen stocks were prepared from isolated colonies previously grown in Brucella broth medium (BB) + 1.5% agar for 3 days. One day prior to infection, B. abortus was grown in liquid BB (when necessary, 10 μg/mL kanamycin was added) and the optical density was measured in a spectrophotometer. In all experiments performed in this work 1 OD600 = 3 × 109 CFU/mL.
Generation of B. abortus ΔnarG mutants
We generated mutants for the catalytic subunit α of nitrate reductase (narG) using an integrative disruption strategy, as previously reported [53, 65]. Plasmid pSKoriT:narGKanR, bearing a kanamycin resistance gene (KanR) containing a internal fragment of the B. melitensis narG gene, was kindly provided by Dr. Xavier de Bolle (University of Namur, Namur, Belgium) and used to transform B. abortus, since alignment analysis revealed that a 99.7% identity between B. abortus and B. melitensis narG gene. All DNA manipulations performed to generate the plasmid are described elsewhere [53]. Two micrograms of the plasmid were introduced into B. abortus by electroporation, using the following parameters: 2.5 kV, 25 μF and 400 Ω and time constants of 10s. Electroporated bacteria were incubated in liquid BB for 6h without antibiotics. Integrative mutants were selected on BB agar 1.5% with 10 μg/mL kanamycin for 4–5 days. Disrupted mutants were selected by PCR, using the following primers: NarG Forward (5’-CTGCTCGTGGAAGATCTATGTC-3’), and NarG Reverse (5’-CTTGTAGCGGGCTTCAGTATAG-3’) (amplicon 649 pb), KanR Forward (5’-GGTATAAATGGGCTCGCGATAA-3’) and KanR Reverse (5’-CGACTGAATCCGGTGAGAATG-3’) (amplicon 507 pb). PCR was performed in a 20 μL reaction with 20 ng of genomic DNA, 0.5 μM each primer, 250 μM dNTPs, 2 mM MgCl2, 0.5 U GoTaq Flexi DNA polymerase (Promega, USA) and 5 μL of 5x colorless GoTaq Flexi Buffer. PCR cycling parameters were: initial DNA denaturation at 95 °C for 3 min and then cycled 35 times at 95 °C for 1 min, 54 °C for 1 min, and 72 °C for 1 min. We performed a final extension step of 7 min at 72 °C. The reaction products were detected by electrophoresis on 1% agarose gel and visualized with UV transilluminator after staining with Blue Green Loading dye I (LGC Biotecnologia, Brazil) to determine the size of amplified products.
Generation of bone marrow-derived macrophages (BMDMs)
Bone marrow cells were obtained from femur and tibiae of KO and wild type mice, and they were differentiated into BMDMs using a previously described protocol, with some modifications [66]. Briefly, cells were seeded on 24-well plates at 5 × 105 cell/mL (day 0) and maintained in DMEM medium containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 20% LCCM (L929-conditioned medium), at 37°C in a 5% CO2 atmosphere for 7 days. On day 4 of incubation, the medium was fully replaced. Four hours before stimulation or infection, BMDMs were maintained only in DMEM medium containing 1% FBS.
In vitro stimulation of BMDMs
In all experiments, BMDMs were maintained in DMEM medium containing 1% FBS. Cells were infected at a multiplicity of infection (MOI) of 100 in 0.5 mL DMEM plus 1% FBS, previously incubated with indicated concentrations of glibenclamide, KCl, A740003, chloroquine or Ca074Me for 1 hour, where applicable. Preincubation with L-NAME (2 mM), SNAP (0.5 mM) or hemoglobin (150 μM) was performed for 30 min before infection. As a control for nitrite production, cells were stimulated with ultrapure LPS (1 μg/ml) for 17 hours. As a control for IL-1β secretion, cells were primed with ultrapure LPS (1 μg/ml) for 4 h and stimulated with nigericin (20μM) or ATP (5 mM) for 30 min. Supernatants were collected at indicated times. Cells were then washed with PBS at room temperature, lysed with 25 μl/well M-PER® plus 10 mM NaF, 1 mM sodium orthovanadate, and 1:100 protease inhibitor cocktail and collected in 1.5 ml tubes. Both supernatants and cell lysates were stored at −80°C until use.
Western blot analysis
Protein concentrations from cell lysates were determined by BCA assay. Equal amounts of protein (15 μg) or 25 μl of supernatants collected from infected macrophages were were loaded onto 15% SDS-polyacrylamide gels and then transferred to nitrocellulose membranes, according to standard techniques. Membranes were blocked 1 h at room temperature with blocking buffer [TBS-T (TBS containing 0.1% Tween-20) plus 5% nonfat dry milk)] before incubation with anti-β-actin (1:4000), anti-IL-1β (1:1000), anti-caspase-1 (1:1000) or anti-NLRP3 (1:1000), overnight at 4°C. Subsequently, membranes were washed three times with TBS-T and incubated with a 1:1000 dilution of HRP-conjugated anti-mouse or anti-rabbit IgG antibody (1.5 h at room temperature). Blots were washed three times with TBS-T and developed with an ECL system according to the manufacturer’s protocols.
RNA isolation from BMDMs and quantitative real-time PCR
Total RNA from infected BMDMs was isolated using the TRIzol® reagent, accordingly to manufacturer instructions. Reverse transcription of 1 μg from total RNA was performed using illustra™ Ready-To-Go RT-PCR Beads (GE Healthcare, UK), following instructions of the manufacturer. Quantitative real-time PCR was conducted in a final volume of 10 μL containing the following: SYBR® Green PCR Master Mix (Thermo Fisher Scientific, USA), oligo-dT cDNA as the PCR template and 5 μM of primers. The PCR reaction was performed with QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific, USA), using the following cycling parameters: 60°C for 10 min, 95°C for 10 min, 40 cycles of 95°C for 15 sec and 60°C for 1 min, and a dissociation stage of 95°C for 15 sec, 60°C for 1 min, 95°C for 15 sec, 60°C for 15 sec. Primers were used to amplify a specific 100–120 base pairs fragment corresponding to specific gene targets as follows: iNOS Forward (5′-CAGCTGGGCTGTACAAACCTT-3′) and iNOS Reverse (5′-CATTGGAAGTGAAGCGTTTCG-3′); 18S Forward (5′-CGTTCCACCAACTAAGAACG-3′) and 18S Reverse (5′-CTCAACACGGGAAACCTCAC-3′). Data were analyzed using the threshold cycle (ΔΔCT) method and they were presented as relative expression units after normalization to the 18S gene. PCR measurements were conducted in triplicate.
Nitrite measurement by Griess reagent
The nitric oxide assay was performed as described previously [67]. The concentration of nitrite (NO2−), a stable metabolite of NO, was measured using Griess reagent (1% sulfanilamide and 0.1% naphthylethylenediamine dihydrochloride in 2.5% phosphoric acid). Briefly, 50 μL of cell culture supernatants was mixed with 50 μL of Griess reagent. Subsequently, the mixture was incubated protected from light at room temperature for 5 min and the absorbance at 550 nm was measured in a microplate reader. Fresh culture medium (DMEM + 1% FBS) was used as a blank in every experiment. The quantity of nitrite was determined from a sodium nitrite (NaNO2) standard curve.
Infection of BMDMs with B. abortus and determination of intracellular bacterial survival
BMDMs (5 × 105 cells/well) were infected with B. abortus (wild type and ΔnarG mutant) at a MOI of 100:1 in 0.5 mL DMEM plus 1% FBS. Bacteria were centrifuged onto macrophages at 400 × g for 10 min at 4°C then incubating the cells for 30 min at 37°C under 5% CO2. Cells were washed 3 times with PBS to remove extracellular bacteria and incubated for an additional 90 min in medium supplemented with 100 μg/mL gentamicin to kill extracellular bacteria. Thereafter, the antibiotic concentration was decreased to 10 μg/mL. Twenty-four hours post infection, cells were again with PBS and lysed with 0.1% (vol/vol) Triton X-100 in H2O and serial dilutions of lysates were rapidly plated onto BB + 1.5% agar. Three days after a incubation at 37°C, the number of colony forming units (CFU) was determined.
Determination of IL-1β and TNF-α in supernatants by enzyme-linked immunosorbent assay (ELISA)
Collected supernatants of stimulated BMDMs were thawed on the day of the assay and used for determination of IL-1β and TNF-α concentrations using Mouse DuoSet ELISA (R&D Systems), according to the manufacturer’s specifications.
Cell viability determination by MTS
BMDMs (5 × 105 cells/well) were seeded into 96-well plates and incubated with chemicals as previously indicated. The medium was completely removed and wells were filled with 100 μL DMEM medium containing 1% FBS + 20 μL 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt [MTS] (from CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega, USA). The plate was protected from exposure to light with aluminum foil and incubated at 37°C for 2 hours. Absorbance was recorded at 492 nm and the percentage of viable cells was determined in comparison to untreated cells.
Statistical analysis
All experiments were repeated at least three times with similar results. Graphs and data analysis were performed using GraphPad Prism 6 (GraphPad Software), using one-way ANOVA (Bonferroni post-hoc test) or student’s t test (Tukey’s post-hoc test).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Xavier de Bolle (University of Namur, Belgium) for providing the pSKoriT:narGKanR plasmid. We also thank to Dra. Leda Quércia Vieira, (Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Brazil) for providing the iNOS and gp9phox KO mice, and Dr. José Carlos Farias Alves Filho (Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, São Paulo, Brazil), for providing P2X7 KO mice.
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico, Grants 406883/2018-1, 402527/2013-5, and 302660/2015-1, Fundação de Amparo a Pesquisa do Estado de Minas Gerais, Grant APQ 837/15, APQ 1945/17, and Rede Mineira de Imunobiologicos Grant 00140-16 and National Institute of Health R01 AI116453.
Abbreviations
- ROS
reactive oxygen species
- P2X7
P2X purinoceptor 7
- ATP
adenosine triphosphate
- NO3-
nitrate
- NO2-
nitrite
- gp91phox
phagocyte NADPH oxidase, glycosylated subunit 91
- NADPH
Nicotinamide adenine dinucleotide phosphate
- iNOS
Inducible nitric oxide synthase
- narG
nitrate reductase 1, alpha subunit
- Kan
kanamycin
- MOI
multiplicity of infection
- Glib
glibenclamide
- Chl
chloroquine
- L-NAME
N(ω)-nitro-L-arginine methyl ester
- SNAP
S-Nitroso-N-Acetyl-D,L-Penicillamine
- Hg
hemoglobin
- Nig
nigericin
Footnotes
CONFLICT OF INTEREST DISCLOSURE
The authors have no commercial or financial conflicts of interest.
REFERENCES
- 1.Man SM and Kanneganti TD, Regulation of inflammasome activation. Immunol Rev 2015. 265: 6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Netea MG and Joosten LA, Inflammasome inhibition: putting out the fire. Cell Metab 2015. 21: 513–514. [DOI] [PubMed] [Google Scholar]
- 3.Franchi L, Munoz-Planillo R and Nunez G, Sensing and reacting to microbes through the inflammasomes. Nat Immunol 2012. 13: 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozaki E, Campbell M and Doyle SL, Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res 2015. 8: 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He Y, Hara H and Nunez G, Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci 2016. 41: 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM and Nunez G, K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013. 38: 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arlehamn CS, Petrilli V, Gross O, Tschopp J and Evans TJ, The role of potassium in inflammasome activation by bacteria. J Biol Chem 2010. 285: 10508–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T and Rouis M, NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol 2015. 4: 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tschopp J and Schroder K, NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 2010. 10: 210–215. [DOI] [PubMed] [Google Scholar]
- 10.Yang CS, Shin DM and Jo EK, The Role of NLR-related Protein 3 Inflammasome in Host Defense and Inflammatory Diseases. Int Neurourol J 2012. 16: 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP et al. , Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006. 440: 228–232. [DOI] [PubMed] [Google Scholar]
- 12.Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM et al. , TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 2012. 150: 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duncan JA, Gao X, Huang MT, O’Connor BP, Thomas CE, Willingham SB, Bergstralh DT et al. , Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol 2009. 182: 6460–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ketelut-Carneiro N, Silva GK, Rocha FA, Milanezi CM, Cavalcanti-Neto FF, Zamboni DS and Silva JS, IL-18 triggered by the Nlrp3 inflammasome induces host innate resistance in a pulmonary model of fungal infection. J Immunol 2015. 194: 4507–4517. [DOI] [PubMed] [Google Scholar]
- 15.Franco MP, Mulder M, Gilman RH and Smits HL, Human brucellosis. Lancet Infect Dis 2007. 7: 775–786. [DOI] [PubMed] [Google Scholar]
- 16.Oliveira SC, Giambartolomei GH and Cassataro J, Confronting the barriers to develop novel vaccines against brucellosis. Expert Rev Vaccines 2011. 10: 1291–1305. [DOI] [PubMed] [Google Scholar]
- 17.Gomes MT, Campos PC, Oliveira FS, Corsetti PP, Bortoluci KR, Cunha LD, Zamboni DS et al. , Critical role of ASC inflammasomes and bacterial type IV secretion system in caspase-1 activation and host innate resistance to Brucella abortus infection. J Immunol 2013. 190: 3629–3638. [DOI] [PubMed] [Google Scholar]
- 18.Rathinam VA, Vanaja SK and Fitzgerald KA, Regulation of inflammasome signaling. Nat Immunol 2012. 13: 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdelaziz DH, Gavrilin MA, Akhter A, Caution K, Kotrange S, Khweek AA, Abdulrahman BA et al. , Apoptosis-associated speck-like protein (ASC) controls Legionella pneumophila infection in human monocytes. J Biol Chem 2011. 286: 3203–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang MT, Mortensen BL, Taxman DJ, Craven RR, Taft-Benz S, Kijek TM, Fuller JR et al. , Deletion of ripA alleviates suppression of the inflammasome and MAPK by Francisella tularensis. J Immunol 2010. 185: 5476–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA and Sassetti CM, Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol 2013. 14: 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sohaskey CD, Regulation of nitrate reductase activity in Mycobacterium tuberculosis by oxygen and nitric oxide. Microbiology 2005. 151: 3803–3810. [DOI] [PubMed] [Google Scholar]
- 23.Sohaskey CD and Modesti L, Differences in nitrate reduction between Mycobacterium tuberculosis and Mycobacterium bovis are due to differential expression of both narGHJI and narK2. FEMS Microbiol Lett 2009. 290: 129–134. [DOI] [PubMed] [Google Scholar]
- 24.Paulsen IT, Seshadri R, Nelson KE, Eisen JA, Heidelberg JF, Read TD, Dodson RJ et al. , The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc Natl Acad Sci U S A 2002. 99: 13148–13153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DelVecchio VG, Kapatral V, Redkar RJ, Patra G, Mujer C, Los T, Ivanova N et al. , The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc Natl Acad Sci U S A 2002. 99: 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baek SH, Rajashekara G, Splitter GA and Shapleigh JP, Denitrification genes regulate Brucella virulence in mice. J Bacteriol 2004. 186: 6025–6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Latz E, Xiao TS and Stutz A, Activation and regulation of the inflammasomes. Nat Rev Immunol 2013. 13: 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F and Tschopp J, Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 2007. 14: 1583–1589. [DOI] [PubMed] [Google Scholar]
- 29.Miller CM, Boulter NR, Fuller SJ, Zakrzewski AM, Lees MP, Saunders BM, Wiley JS et al. , The role of the P2X(7) receptor in infectious diseases. PLoS Pathog 2011. 7: e1002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mortaz E, Adcock IM, Shafei H, Masjedi MR and Folkerts G, Role of P2X7 Receptors in Release of IL-1beta: A Possible Mediator of Pulmonary Inflammation. Tanaffos 2012. 11: 6–11. [PMC free article] [PubMed] [Google Scholar]
- 31.Franceschini A, Capece M, Chiozzi P, Falzoni S, Sanz JM, Sarti AC, Bonora M et al. , The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J 2015. 29: 2450–2461. [DOI] [PubMed] [Google Scholar]
- 32.Chu J, Thomas LM, Watkins SC, Franchi L, Nunez G and Salter RD, Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J Leukoc Biol 2009. 86: 1227–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA et al. , Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008. 9: 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen CC, Tsai SH, Lu CC, Hu ST, Wu TS, Huang TT, Said-Sadier N et al. , Activation of an NLRP3 inflammasome restricts Mycobacterium kansasii infection. PLoS One 2012. 7: e36292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Bari MA, Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother 2015. 70: 1608–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA and Rock KL, Multiple Cathepsins Promote Pro-IL-1beta Synthesis and NLRP3-Mediated IL-1beta Activation. J Immunol 2015. 195: 1685–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abais JM, Xia M, Zhang Y, Boini KM and Li PL, Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal 2015. 22: 1111–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim SR, Kim DI, Kim SH, Lee H, Lee KS, Cho SH and Lee YC, NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis 2014. 5: e1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF and Vercesi AE, Mitochondria and reactive oxygen species. Free Radic Biol Med 2009. 47: 333–343. [DOI] [PubMed] [Google Scholar]
- 40.Lamkanfi M and Dixit VM, A new lead to NLRP3 inhibition. J Exp Med 2017. 214: 3147–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM and Ojcius DM, ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem 2007. 282: 2871–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT and Tschopp J, Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008. 320: 674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH et al. , Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 1995. 9: 202–209. [DOI] [PubMed] [Google Scholar]
- 44.Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M et al. , Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011. 34: 213–223. [DOI] [PubMed] [Google Scholar]
- 45.Compan V, Baroja-Mazo A, Lopez-Castejon G, Gomez AI, Martinez CM, Angosto D, Montero MT et al. , Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 2012. 37: 487–500. [DOI] [PubMed] [Google Scholar]
- 46.Kim YM, Talanian RV, Li J and Billiar TR, Nitric oxide prevents IL-1beta and IFN-gamma-inducing factor (IL-18) release from macrophages by inhibiting caspase-1 (IL-1beta-converting enzyme). J Immunol 1998. 161: 4122–4128. [PubMed] [Google Scholar]
- 47.Hernandez-Cuellar E, Tsuchiya K, Hara H, Fang R, Sakai S, Kawamura I, Akira S et al. , Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol 2012. 189: 5113–5117. [DOI] [PubMed] [Google Scholar]
- 48.Reddix RA, Mullet D, Fertel R and Cooke HJ, Endogenous nitric oxide inhibits endothelin-1-induced chloride secretion in guinea pig colon. Nitric Oxide 1998. 2: 28–36. [DOI] [PubMed] [Google Scholar]
- 49.Privalle C, Talarico T, Keng T and DeAngelo J, Pyridoxalated hemoglobin polyoxyethylene: a nitric oxide scavenger with antioxidant activity for the treatment of nitric oxide-induced shock. Free Radic Biol Med 2000. 28: 1507–1517. [DOI] [PubMed] [Google Scholar]
- 50.Li Q, Chen Y, Li B, Luo C, Zuo S, Liu X, Zhang JH et al. , Hemoglobin induced NO/cGMP suppression Deteriorate Microcirculation via Pericyte Phenotype Transformation after Subarachnoid Hemorrhage in Rats. Sci Rep 2016. 6: 22070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stevanin TM, Laver JR, Poole RK, Moir JW and Read RC, Metabolism of nitric oxide by Neisseria meningitidis modifies release of NO-regulated cytokines and chemokines by human macrophages. Microbes Infect 2007. 9: 981–987. [DOI] [PubMed] [Google Scholar]
- 52.Kohler S, Foulongne V, Ouahrani-Bettache S, Bourg G, Teyssier J, Ramuz M and Liautard JP, The analysis of the intramacrophagic virulome of Brucella suis deciphers the environment encountered by the pathogen inside the macrophage host cell. Proc Natl Acad Sci U S A 2002. 99: 15711–15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haine V, Dozot M, Dornand J, Letesson JJ and De Bolle X, NnrA is required for full virulence and regulates several Brucella melitensis denitrification genes. J Bacteriol 2006. 188: 1615–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferguson SJ, Nitrogen cycle enzymology. Curr Opin Chem Biol 1998. 2: 182–193. [DOI] [PubMed] [Google Scholar]
- 55.Richardson DJ and Watmough NJ, Inorganic nitrogen metabolism in bacteria. Curr Opin Chem Biol 1999. 3: 207–219. [DOI] [PubMed] [Google Scholar]
- 56.Bauernfeind F and Hornung V, Of inflammasomes and pathogens--sensing of microbes by the inflammasome. EMBO Mol Med 2013. 5: 814–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cerqueira DM, Gomes MTR, Silva ALN, Rungue M, Assis NRG, Guimaraes ES, Morais SB et al. , Guanylate-binding protein 5 licenses caspase-11 for Gasdermin-D mediated host resistance to Brucella abortus infection. PLoS Pathog 2018. 14: e1007519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jabaut J, Ather JL, Taracanova A, Poynter ME and Ckless K, Mitochondria-targeted drugs enhance Nlrp3 inflammasome-dependent IL-1beta secretion in association with alterations in cellular redox and energy status. Free Radic Biol Med 2013. 60: 233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun YH, den Hartigh AB, Santos RL, Adams LG and Tsolis RM, virB-Mediated survival of Brucella abortus in mice and macrophages is independent of a functional inducible nitric oxide synthase or NADPH oxidase in macrophages. Infect Immun 2002. 70: 4826–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuppusamy P and Zweier JL, Characterization of free radical generation by xanthine oxidase. Evidence for hydroxyl radical generation. J Biol Chem 1989. 264: 9880–9884. [PubMed] [Google Scholar]
- 61.Dinarello CA, Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009. 27: 519–550. [DOI] [PubMed] [Google Scholar]
- 62.Franchi L, Eigenbrod T, Munoz-Planillo R and Nunez G, The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 2009. 10: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Radi R, Peroxynitrite, a stealthy biological oxidant. J Biol Chem 2013. 288: 26464–26472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Slauch JM, How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol Microbiol 2011. 80: 580–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haine V, Sinon A, Van Steen F, Rousseau S, Dozot M, Lestrate P, Lambert C et al. , Systematic targeted mutagenesis of Brucella melitensis 16M reveals a major role for GntR regulators in the control of virulence. Infect Immun 2005. 73: 5578–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weischenfeldt J and Porse B, Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc 2008. 2008: pdb prot5080. [DOI] [PubMed] [Google Scholar]
- 67.Macedo GC, Magnani DM, Carvalho NB, Bruna-Romero O, Gazzinelli RT and Oliveira SC, Central role of MyD88-dependent dendritic cell maturation and proinflammatory cytokine production to control Brucella abortus infection. J Immunol 2008. 180: 1080–1087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.