

Intratumoral genetic heterogeneity is well known, as is the role of preexisting mutations in many documented cases of clinical relapse. But what is the extent of diversity and how does it evolve? To what degree are these mutations the product of selection like the consensus mutations that drive tumor growth, invasion, and metastasis? Is preexisting genetic resistance likely, or universal? And what are the implications for therapy?
Perspective
This Perspective focuses on two recent publications investigating intratumoral clonal and subclonal heterogeneity and implications for tumor evolution and therapy. These complementary works together present a strong case for intratumoral heterogeneity arising from neutral evolution after initial selection of driver mutations.
Reiter et al. 1 performed a comprehensive whole exome analysis of intratumoral heterogeneity within the primary tumor, between the primary and associated metastases, within individual metastases, and between metastases across multiple tumor types at diagnosis. To distinguish driver from passenger mutations, they utilized numerous bioinformatic approaches, and found good concordance in the driver mutations across these comparisons. A single biopsy from the primary was sufficient to capture nearly all of the driver genes found in associated metastases. Driver mutations were largely clonal (in all samples); the subclonal space contained very few new drivers. Results were consistent with a 91% concordance rate of qualitative response to targeted agents across lesions. The analysis was based on public data sources at low sequencing depth (approximately 10×) and variable accuracy. In fact, the authors attribute previous claims of driver heterogeneity to low tumor content, low depth, noise, and statistical issues inherent in taking a large number of samples.
In contrast to measurements obtained from literature databases, we investigated subclonal (< 10% allele frequency) heterogeneity in fresh diagnostic specimens of colorectal cancer 2 at exceptional depth (approximately 20,000×) and accuracy (< one technical error in 107 nucleotides sequenced) using duplex sequencing, which sequences both DNA strands and calls a mutation only if complementary changes are observed on both strands. 3 We concluded from the curve of new unique mutations observable as a function of depth that the “effective mutation rate” per base per new cancer cell added to the tumor is higher (approximately 6 × 10−7) than previously assumed (10−10 to 10−8). 2 The infinite sites assumption 4 states that a mutation in a particular base will occur uniquely in one cell at any instant, and is used in other current mathematical models of tumor evolution. 5 , 6 However, the product of the number of cells in a tumor at diagnosis (≥ 109) and the effective mutation rate we found is much greater than one, and this quantity is a reasonable estimate of the number cells in which mutations at a particular base would simultaneously occur. We therefore concluded that the “infinite sites assumption” would be violated, leading to a previously unanticipated accumulation of mutational diversity (Figure 1 ) beyond a critical tumor size (1/(effective mutation rate)) despite identical predictions to other current models early in tumor growth. We calculated that every conceivable mutation is present in at least one cell at diagnosis, and therefore any resistance mutation is preexisting. While previous clinical and molecular results implied that preexisting resistance would be frequent, we found it to be universal (probability of all cells wild type at a given locus at diagnosis ≈ 10−308).
Figure 1.
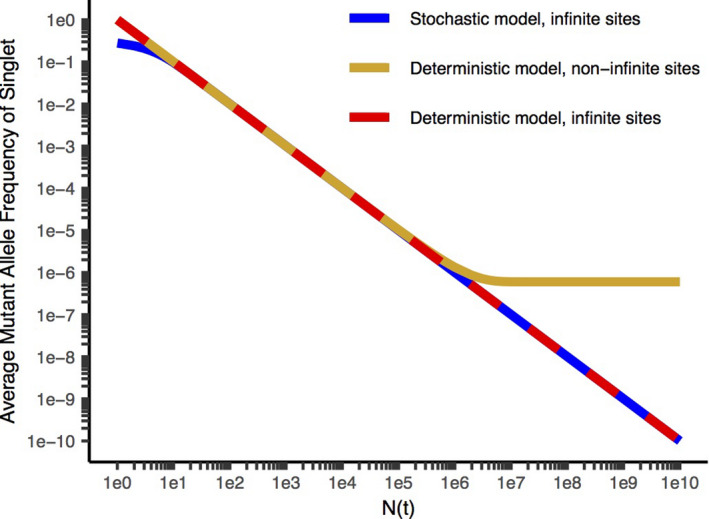
Average MAF (mutant allele frequency) for a given mutation vs. N(t) (the number of cells at the time it is formed), for the stochastic model with the infinite sites assumption 6 (blue), the deterministic model with the infinite sites assumption 5 (red), and without the infinite sites assumption 2 (gold). Sequencing to a depth D queries, on average, mutational events occurring when N(t) = D. Stochastic models consider random fluctuations from the expected average evolutionary trajectory and are more accurate at early times when the tumor is small compared with deterministic models that are based on the expected average evolutionary trajectory. The infinite sites assumption states that a mutation in a particular base will occur uniquely in one cell at any instant. Models without the infinite sites assumption are more accurate than those with it for larger tumor masses in which the number of cells approaches or exceeds the reciprocal of the effective mutation rate, since the expected number of instances of mutations at a particular base is equal to the product of the effective mutation rate and the number of cells dividing. The deterministic model with the infinite sites assumption leads to a reciprocal relationship between N(t) and the average MAF and a straight line with a slope of −1 on a log‐log plot. The deterministic model without infinite sites predicts that the MAF will approach a limiting value equal to the effective mutation rate for N(t) comparable to the reciprocal of the effective mutation rate and larger. This corresponds to a significant degree of additional diversity added late in tumor evolution, including during the clinical course, compared with the other models. Parameters are cell birth rate = 0.25/day, cell death rate = 0.18/day, effective mutation rate per base per new cell added to the tumor = 6.1 × 10−7. Reproduced from ref. 2 with permission.
Our study was limited to multiple exons of DNA polymerase genes totaling approximately 13 kilobase (kb). In contrast, Reiter et al. 1 covered the exome but with lower depth and accuracy of sequencing. Nonetheless, several important conclusions are common to both studies. Theoretical and experimental studies 7 , 8 suggested neutral evolution (random accumulation of mutations with minimal selection) after selection for driver genes, further concordant with an analysis of allele frequencies in The Cancer Genome Atlas (TCGA) database confirming neutral evolution across a variety of tumor types, 5 with studies of synonymous/nonsynonymous mutation ratios, 9 and with both studies under consideration here. Due to the branching nature of tumor evolution, deeper sequencing and rarer subclones probe later timepoints than before. Both studies see hope in regimens that match the mutational diversity within cancers with equally complex non–cross resistant treatment regimens. Regarding relapse, both studies conclude that not all driver mutations are resistance mutations, and not all resistance mutations are drivers. Both papers discuss the clinical importance of metastases rather than the primary, as diffuse metastases are generally the cause of cancer mortality. Both studies indicate that resistance can also arise via other genetic and epigenetic mechanisms, and by reversible phenotypic plasticity. Reiter et al. 1 stated that mutations in very rare cells are of minimal importance because such cells may have a very small probability of reaching fixation within the primary. In contrast, we argue that rare cells in numerous diffuse micrometastases can rapidly establish themselves as minor subclones. Micrometastases less than the angiogenic limit of 1–2 mm3 can receive oxygen and nutrients via diffusion, and only as they pass this size threshold do they undergo the “angiogenic switch” releasing proangiogenic factors to stimulate neovascularization. Competition for nutrients may be primarily against normal tissue cells that do not have a full complement of driver genes to compete effectively against transformed cells. We view rare cells as a major source of late relapse due to resistance mutations and have published a paradigm aimed at preventing any single cell from simultaneously possessing resistance to all the non–cross resistant components of a combination due to independent mutations. 10 In this regard, while the degree of concordance among drivers shown by Reiter et al. 1 is impressive, in our opinion there is legitimate concern about the role of minority nonconcordant drivers, as well as non–driver resistance mutations, including those in rare cells. While resistance to any single therapy is preexisting, simultaneous resistance to multiple non–cross resistant therapies in a single cell may, according to our results, often be acquired during treatment.
While both papers recommended non–cross resistant therapies as part of more complex therapeutic regimens, the question of whether this always implies simultaneous combination therapy remains open. Previous theoretical papers on this topic (reviewed in SI Appendix of ref. 2) have assumed treatment with the same therapy until relapse or progression. In contrast, if frequent adaptation of therapy is permitted in anticipation of probable undetected resistance events, optimal therapeutic sequences can contain both pulses of monotherapy and simultaneous combinations. 10 This paradigm is especially relevant when we consider the possible need for dose reductions due to toxicity in simultaneous combinations, and the frequent need for adequate dosages to penetrate tissue spaces and achieve antineoplastic activities.
Another significant unexplored area is the outgrowth of subclones with very high mutation rates in the presence of therapy. The genes controlling mutation rate are also randomly mutating in a cancer, and all possible mutations will be present. We term these hypermutator subclones, and believe based on simulation results 10 that they are particularly dangerous due to their ability to rapidly acquire independent mutational resistance mutations to multiple components of a complex therapeutic regimen.
To counter the extraordinary diversity of mutational resistance mechanisms in tumors, optimal therapy should:
Utilize (in simultaneous combination or in rapid monotherapy pulses) a sufficient number of non–cross resistant agents to minimize the likelihood that a single cell could harbor simultaneous corresponding resistance mutations to all available therapies relevant to a single cancer. 2 , 10 Cells with single resistance will already be present, but there is an opportunity to prevent multiple simultaneous resistance in single cells. 2 Up to four non–cross resistant therapies may be required, 2 and each of these therapies may itself contain several synergistic individual drugs in order to overcome network signaling plasticity unrelated to mutations, even within a single subclone. 10 Simultaneous administration of this number of agents in combination may not be feasible due to toxicity, but if therapy is changed frequently (rather than waiting for relapse) pulses of less complex combinations can achieve long‐term control of diverse disease by utilizing a large variety of therapies; 10 and:
If feasible, selectively target hypermutator subclones that drive intratumoral diversity and may be more likely to rapidly acquire multiple resistances in a single cell. 10 Based on simulation results, we believe multiply resistant hypermutator cells will often be present in end‐stage cancer patients. 2 , 10 We are developing methods to isolate and quantify hypermutator subclones.
The same principles apply to resistance driven by chromosomal rearrangements (including copy number variation) and stable epigenetic changes.
In summary, these results indicate that rare cells matter to cancer treatment, because they are a source of resistance mutations. While epigenetic mechanisms and phenotypic plasticity likely also contribute to primary resistance and early relapses, we believe medium‐term and late relapses will be increasingly caused by genetic changes. Treatment in the future should not be addressed to high prevalence consensus mutations only, but should also proactively consider rare singly resistant subclones (especially if they are hypermutators) and prevent their further evolution to multiple resistance by eliminating them early. Thus, later line therapies will become part of complex earlier therapeutic sequences, blurring the distinction between lines of therapy. Such a strategy must be guided by frequent high depth sequencing and mathematical models of evolutionary dynamics. 10 More sensitive and less costly noninvasive sequencing by liquid biopsy will be needed.
Funding
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Awards NCI P01‐CA077852 and NCI R01‐CA160674 (L.A.L.).
Conflict of Interest
L.A.L. and the University of Washington have a license agreement with TwinStrand Biosciences for the use and development of Duplex Sequencing technology. L.A.L. is a founding member of TwinStrand Biosciences. L.A.L. is a member of the Scientific Advisory Board of Stratos Genomics Inc. R.A.B. consults for AstraZeneca, Vertex, Zymeworks, and CStone, and is the Founder and Chief Scientific Officer of Onco‐Mind, LLC.
Supporting information
Supplementary Material
Acknowledgments
We thank Brendan Kohrn for critical analyses in ref. 2. We thank Robert Clarke, George Martin, Raymond Monnat, Daniel Promislow, and Louis Weiner for critical review of this manuscript.
References
- 1. Reiter, J.G. et al An analysis of genetic heterogeneity in untreated cancers. Nat. Rev. Cancer 19, 639–650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Loeb, L.A. et al Extensive subclonal mutational diversity in human colorectal cancer and its significance. Proc. Natl. Acad. Sci. U S A 116, 26863–26872 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schmitt, M.W. , Kennedy, S.R. , Salk, J.J. , Fox, E.J. , Hiatt, J.B. & Loeb, L.A. Detection of ultra‐rare mutations by next‐generation sequencing. Proc. Natl. Acad. Sci. U S A 109, 14508–14513 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kimura, M. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61, 893–903 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Williams, M.J. , Werner, B. , Barnes, C.P. , Graham, T.A. & Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 48, 238–244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bozic, I. , Gerold, J.M. & Nowak, M.A. Quantifying clonal and subclonal passenger mutations in cancer evolution. PLoS Comput. Biol. 12, e1004731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beckman, R.A. & Loeb, L.A. Efficiency of carcinogenesis with and without a mutator mutation. Proc. Natl. Acad. Sci. U S A 103, 14140–14145 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sottoriva, A. et al A Big Bang model of human colorectal tumor growth. Nat. Genet. 47, 209–216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martincorena, I. et al Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beckman, R.A. , Schemmann, G.S. & Yeang, C.‐H. Impact of genetic dynamics and single‐cell heterogeneity on development of nonstandard personalized medicine strategies for cancer. Proc. Natl. Acad. Sci. U S A 109, 14586–14591 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material