

Abstract
Small-molecule fluorescent probes are powerful tools for chemical biology; however, despite the large number of probes available, there is still a need for a simple fluorogenic scaffold which allows for the rational design of molecules with predictable photophysical properties and is amenable to concise synthesis for high throughput screening. Here, we introduce a highly modular quinoline-based probe containing three strategic domains that can be easily engineered and optimized for various applications. Such domains are allotted for 1) compound polarization, 2) tuning of photophysical properties, and 3) structural diversity. We successfully synthesized our probes in two steps from commercially available starting materials in overall yields of up to 95%. Facile probe synthesis was permitted by regioselective palladium-catalyzed cross coupling, which enables combinatorial development of structurally diverse quinoline-based fluorophores. We have further applied our probes to live-cell imaging, utilizing their unique two stage fluorescence response to intracellular pH. These studies provide a full demonstration of our strategy in rational design and stream-lined probe discovery to reveal the diverse potential of quinoline-based fluorescent compounds.
Graphical Abstract
■ INTRODUCTION
Fluorescence-based technologies have emerged as indispensable tools for both in vitro and in vivo biomedical studies, making them an essential facet of chemical biology.1–3 Fluorescent small molecules, in particular, have significant advantages over protein-based probes in optical imaging and analytical sensing due to their small, noninvasive size, high sensitivity, and fast response time.2–3 Because they have ubiquitous applications as cellular stains, environmental sensors, or biomolecular labels, a plethora of fluorescent probes have been developed over past decades.3 Although numerous fluorescent probes are known, the vast majority of these probes are developed through structural modification of a diminutive set of classical “core” dyes such as coumarin, fluorescein, BODIPY, or cyanine.3 This underscores the importance of intrinsic modularity of the “core” fluorophores in the elaboration and modification of dyes, but also reveals the limited number of available “core” scaffolds.3–4 Moreover, within the context of biological systems, “ideal” fluorescent probes are still in high demand, as properties such as cell permeability, cytotoxicity, selectivity/sensitivity, and dynamic range in the cellular environment are often difficult to predict based on rational design and theoretical calculations.5
Addressing these limitations, combinatorial approaches to the discovery of fluorescent probes have emerged as exciting alternatives for the exploration and optimization of fluorophores.6–7 As a result of successful implementation of combinatorial chemistry techniques – including major improvements in high-throughput screening and imaging methodologies – on fluorescent scaffolds, many rare and surprising probes such as tricarbocyanine-based photostable near-infrared (NIR) dyes8 or rosamine-based in vivo glutathione probes9 were developed.7 Yet, challenges still exist for both library design and the ease of synthetic methods, stressing the necessity of new tunable scaffolds that can be easily diversified.6
Underexplored Quinoline Scaffold.
Among many synthetic scaffolds, quinoline is a highly attractive structure for its relative synthetic versatility.4 Quinoline, being an important construction motif, is widely observed in natural products, drug discovery, as well as advanced functional materials.4, 10–12 Moreover, quinoline is the core structure of quinine, a first well-defined small molecule fluorophore,3, 13 and has been investigated as an intriguing molecular probe since the nitrogen can be used as the site for monitoring the interactions with target molecules through changes in fluorescence.4, 14–15 However, despite considerable efforts to develop novel synthetic scaffolds containing heterocyclic rings, quinoline has been under-explored as a fluorescent probe largely due to synthetic difficulties.
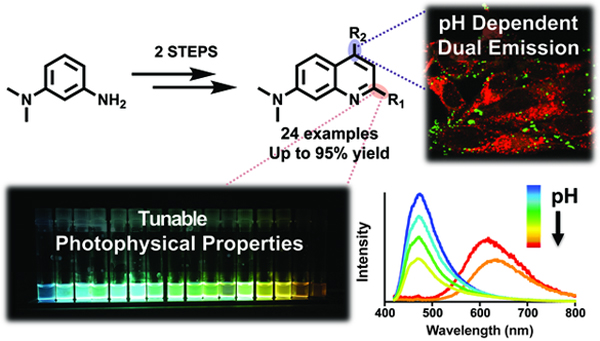
Here, we introduce a small, yet highly tunable quinoline-based “core” scaffold that is compatible with the generation of libraries of fluorescent molecules in two steps (Figure 1). Our scaffold includes three strategic domains that can be easily functionalized and optimized to present potential advances over classic and contemporary dyes. Multi-well plate analysis of the resulting chemical libraries allowed us to identify useful photophysical features in an efficient manner and our observations can serve as a guideline for next generation library design based on rational structure/photophysical relationships. We have further demonstrated the utility of one derivative with particularly unusual pH-sensitivity in multi-color live-cell imaging. We call this scaffold dimethylamino quinoline or DMAQ.
Figure 1.

(A) Modularity of dimethylamino quinoline (DMAQ) dyes consisting of a polarization domain and two tuning domains (B) Two-step synthesis of 2,4-disubstituted quinoline derivatives (15 monoarylated, 9 bisarylated DMAQ)
■ RESULTS AND DISCUSSION
Rational Design of DMAQ Scaffold.
In general, a simple and short synthetic route to generate a large number of target molecules is favored.16–17 However, most commonly used dyes require several steps for derivatization, resulting in low overall yields because they lack easy tunability or late-stage modification sites.6 In order to achieve desired step economy in obtaining dyes of interest, a highly efficient synthetic route to obtain a key precursor and its derivatives was investigated. Therefore, we designed our scaffold to contain three strategic domains: one for polarization and two tunable domains for structural diversity and/or late-stage chemical modification (Figure 1A). Polarization of the fluorophore is achieved by the electron-donating dimethylamino group at the 7-position of quinoline, which can push electron density into the core of DMAQ. The 2- and 4-positions of DMAQ are allotted for tuning domains, where electronically and structurally diverse chemical moieties can be installed to either push or pull the electron density. These substitutions were envisioned to orchestrate a versatile “push-pull” system of the fluorescent scaffold (Figure 1A). Generated molecules with a D(donor)-π-A(acceptor) system are anticipated to display varied photophysical (e.g. fluorescence, quantum yield, solvatochromism, Stokes shifts, etc.) and chemical properties (e.g. solubility, ion binding, acidity/basicity, etc.).
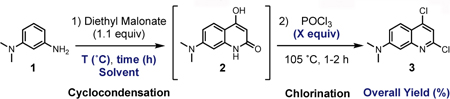
To accelerate the utilization of our scaffold, a synthetic route for rapid assembly of the DMAQ core was studied. Not surprisingly, over the past decades, several condensation and cascade reactions to efficiently access medicinally valuable multi-substituted quinolines have been reported with various mechanisms and conditions, including: Conard-Limpach-Knorr18–19, Skraub-Doebner-Von Miller20–22, Friedlaender23–24, halogen25 or copper26 mediated.4, 27–28However, many traditional condensation methods suffer from low yields, harsh reaction conditions, and operational complications, or require specialized starting materials or catalysts.4, 28 In this work, we report a significantly improved two-step synthesis of quinoline-based dyes from commercially available starting materials which provides good to excellent yields. The initial DMAQ library was generated by (1) a one-pot two-step cyclocondensation and chlorination procedure to afford key precursor 3, and (2) a regioselective Suzuki-Miyaura cross-coupling reaction at the 2-position (C-2) of DMAQ (Figure 1B). The second DMAQ library was achieved via a sequential cross-coupling reaction at C-2 and then at the 4-position (C-4).
Synthesis of the 2,4-dichloroquinoline Core.
The synthesis of the key precursor of the DMAQ probe, 2,4-dichloro-N,N-dimethylquinolin-7-amine 3, was first attempted following previous reports, which involved two steps, inefficient purifications such as filtration or recrystallization, and harsh conditions such as extremely high temperatures, up to 270 ˚C.29–30 Adapting the reported conditions, 3-(dimethylamino) aniline 1 was first cyclocondensed with neat diethyl malonate to form 7-(dimethylamino)-4-hydroxyquinolin-2(1H)-one 2. After filtration of solids, 2 was chlorinated using neat phosphorous oxychloride (POCl3) under reflux conditions to afford dichloroquinoline 3. The previously reported yield for compound 3 was estimated to be 54% (Figure S2);29–30however, we found that these conditions resulted in only 26 % yield on the gram scale (Table 1, entry 1), which is similar to the 25% yield of 2,4-dibromo-N,N-dimehtylquinolin-7-amine in a recent report (Figure S2).30
Table 1.
Conditions for Synthesis of Key Precursor 3
 | |||||
|---|---|---|---|---|---|
| entry | solvent | T (°C) | time (h) | POCl3(equiv.) | Yield (%) |
| 1 | None | 180 – 270 | 5 | 6 | < 26 |
| 2 | DPE | 160 – 200 | 15 | 10 | 96 |
| 3 | DPE | 160 – 200 | 15 | 10 | 72a |
| 4 | DPE | 160 – 190 | >20 | 10 | 80 |
| 5 | DPE | 160 | >24 | 3 | 75 |
All reactions in the table above were performed on the same scale (8.8 mmol) and monitored by thin layer chromatography (TLC) analysis for formation of 2.
Reported yield was obtained after intermediate 2 was isolated. DPE is diphenyl ether.
The poor reproducibility and low yield that we observed was presumed to be due to heterogeneity during the cyclocondensation reaction. Moreover, poor solubility of hydroxyquinolone 2 made column purification difficult and further affected the yield in the following halogenation reaction. We envisioned that an appropriate solvent with a high boiling point would keep 2 in solution and ensure that the reaction mixture remained homogenous. Gratifyingly, we found that diphenyl ether (DPE), which has boiling point of 258 ˚C, not only keeps the reaction mixture in a homogenous state, but also is inert to both cyclocondensation and halogenation reaction conditions, allowing one-pot two-step synthesis of 2 in an excellent yield on gram scale. Complete formation of 2 was monitored via thin layer chromatography (TLC) analysis (Figure S3B), which was then followed by addition of POCl3 to obtain 3 in 96% overall yield (Table 1, entry 2). The yield dropped to 72% when the chlorination reaction was performed after intermediate 2 was isolated by solid filtration (Table 1, entry 3). Cyclocondensation reactions were also attempted in milder conditions such as lower temperature (190 ˚C or 160 ˚C) or fewer equivalents of POCl3. Those conditions successfully afforded 2,but resulted in lower conversion and longer reaction times than that entry 2 conditions (Table 1, Entry 4–5). Thus, our one-pot, optimized conditions efficiently produce poly-halogenated quinoline 3, a core subunit which can be utilized in transition-metal-catalyzed cross coupling reactions to generate our fluorogenic probes as well as medicinally valuable bio-active compounds.31
Synthesis of Monoarylated DMAQ via Regioselective Cross-Coupling Reaction.
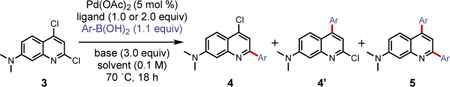
Palladium-catalyzed cross-coupling reactions of halogenated heteroarenes have played a pivotal role in the synthesis of important biological targets or their key precursors.32 We were interested in identifying a general catalyst system capable of C-2 selective arylation of dichloroquinoline 3, which would give us the flexibility for structure-photophysical property relationship studies of fluorophores. However, with heteroarenes bearing identical halogens, high selectivity is difficult to achieve as the final regioselectivity is determined by multiple factors including steric effects, electronic effects, and the presence of directing groups in the reactants.32 While many conditions have been investigated to achieve C-2 selective functionalization of 2,4-dichloroquinoline or 2,4-dichloropyridine, there was no reported condition for 3, which includes an electron donating substituent at the 7-position.32–33 Unlike 2,4-dichloroquinoline, which possess only one highly electrophilic position at C-2, 3 contains the dimethylamino group that could reduce the electrophilicity of C-2 and weaken selectivity. Additionally, the C-2 selectivity of the pyridine moiety seems to be highly depended on the choice of ligand or the presence of additives such as LiCl, which could alter the selectivity to C-4.32–33 Finally, consideration must be given to avoid bis-arylated products, as we would like to keep C-4 as an additional site for late-stage structural diversification or functionalization. In an effort to achieve highly regioselective and broadly compatible catalyst condition for electronically diverse aryl boronic acids, we screened several factors: electronically distinct boronic acids, ligands, bases, solvents, and temperature. With the support of the Penn/Merck High throughput experimentation center, a general and efficient condition for C-2 selective Suzuki Miyaura cross-coupling of 3 was investigated.
Screening was conducted with 5 mol % palladium acetate (Pd(OAc)2), 5 mol % (bidentate) or 10 mol % (monodentate) of eight different ligands, two different bases, cesium carbonate (Cs2CO3) or potassium carbonate (K2CO3) (3.0 equiv), and three different phenylboronic acids (1.1 equiv). We chose 4-(dimethylamino)phenyl-, phenyl-, and 4-(cyano)phenyl-boronic acids as three representative electron-rich, -neutral, and -deficient substrates, respectively. Eight ligands were chosen based on the previous report that showed high C-2 selectivity for dichloropyridine.16 Using 0.6 mg (2.5 µmol) of dichloroquinoline 3 per reaction, a total of 96 conditions were screened (Figure S4). Not surprisingly, mixtures of mono-coupling products (4, 4’), bis-coupling product (5), dechlorinated byproduct, and hydrolyzed byproducts were observed from these reactions (Tables S1–4 and Figures S5–7). As summarized in Table 2, the ligands exert a profound effect on the regioselectivity of the coupling as well as optimal reactivity to minimize bis product.32 Unlike previously reported conditions for C-2 selective Suzuki-Miyaura cross coupling of dichloropyridine using 1,1′-Ferrocenediyl-bis(diphenylphosphine) (DPPF), it was observed that, in general, triphenylphosphine (PPh3) and 1,3-bis(diphenylphosphino) propane (DPPP) highly favored coupling at C-2 over C-4 (Table 2, entries 1, 2, 5, 6, 9, and 10) while 1,1’-bis(di-i-propylphosphino)ferrocene (DIPPF) and tricyclohexylphosphine (PCy3) gave a low yield for 4 (Table 2, entries 3, 4, 7, 8, 11, and 12) due to poor C-2 selectivity or a high yield of 5. Nonetheless, our desired 4 was the major product in most screened conditions, presumably due to the intrinsic reactivity of C=N bond and the effect of the quinoline nitrogen coordinating to the Pd catalyst, confirming that the order of the relative ease of oxidative addition for 2,4-dichlorquinoline is C-2 > C-4.32,33–34
Table 2.
Selected results of regioselective cross-coupling reactions with three electronically distinct boronic acids
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | Ar-B(OH)2 | ligand | solvent | base | 4 (%) | 4’ (%) | 5 (%) | 3 (%) |
| 1 | PPh3 | THF | K2CO3 | 75 | 0 | 0 | 14 | |
| 2 | DPPP | Dioxane | Cs2CO3 | 76 | 2 | 3 | 12 | |
| 3 | DIPPF | THF | K2CO3 | 57 | 13 | 6 | 15 | |
| 4 | PCy3 | Dioxane | Cs2CO3 | 29 | 35 | 8 | 22 | |
| 5 | PPh3 | THF | K2CO3 | 75 | 6 | 10 | 0 | |
| 6 | DPPP | Dioxane | Cs2CO3 | 90 | 8 | 1 | 0 | |
| 7 | DIPPF | THF | K2CO3 | 60 | 5 | 16 | 17 | |
| 8 | PCy3 | Dioxane | Cs2CO3 | 36 | 3 | 38 | 22 | |
| 9 | PPh3 | THF | K2CO3 | 78 | 9 | 1 | 3 | |
| 10 | DPPP | Dioxane | Cs2CO3 | 59 | 4 | 17 | 17 | |
| 11 | DIPPF | THF | K2CO3 | 38 | 5 | 22 | 16 | |
| 12 | PCy3 | Dioxane | Cs2CO3 | 44 | 4 | 43 | 8 | |
Four selected catalyst conditions are shown for each boronic acid: triphenylphosphine (PPh3), 1,3-bis(diphenylphosphino) propane (DPPP), 1,1’-bis(di-i-propylphosphino)ferrocene (DIPPF), tricyclohexylphosphine (PCy3). The highlighted condition is the best condition for the corresponding boronic acid. The final solvent consists of 4:1 solvent:water. Percent composition of products was determined by chromatographic peak integration normalized with an internal standard.
After additional temperature screening at 60 ˚C, 70˚C, and 80 ˚C, we established that performing the reaction with 5 mol % of bis(triphenylphosphine) palladium chloride (Pd(PPh3)2Cl2) and 3 equiv of K2CO3 in 4:1 tetrahydrofuran (THF):water at 65 ˚C for 5 to 48 h afforded products in the highest yield with excellent C-2 regioselectivity for a broad scope of boronic acids. A range of aryl boronic acids, spanning electron donating to withdrawing para-substituted phenylboronic acids, sterically hindered arylboronic acids, and heteroaryl boronic acids, was subjected to the optimized reaction condition (Scheme 1). All of the electron-rich (4b), electron-neutral (4c-d), and electron poor (4e-f) phenylboronic acids afforded excellent isolated yields (>91%) of C-2 arylated products. Of sterically hindered phenylboronic acids, the presence of a methoxy functional group at the ortho- and para-position resulted in 92% (4g) and 94% (4i) yields, respectively, while at the meta-position gave a slightly lower isolated yield of 83% (4h). Though phenyl substituted 4a resulted in relatively low yield35 (39%), high yield for para-methoxy phenyl substituted 4i demonstrates protected phenol can be utilized for synthesis of 4a. To assess additional steric effects, 2,4-difluorophenylboronic acid and 9-anthraceneboronic acid were subjected to reaction with 3. While 4j was afforded in 87% yield, only a 20% yield was observed for 4k, although it was still the major arylated product. The low yield of 4k is most likely due to protodeboronation of the anthracene boronic acid, as significant amounts of anthracene product were observed (Figure S8).36 Furthermore, we examined C-2 arylation with heterocycles, 2-thienyl and 3-thienyl boronic acid, to afford 4m and 4n in 85% and 97% yields, respectively. The cross-coupling condition tolerated a variety of para-substituted functional groups, such as hydroxyl, methoxy, cyano, dimethylamino, and fluoro as well as a Boc-protected primary amine. Fluorine and primary amines are particularly important functional groups as fluorine is a frequently used substituent in both medicinal chemistry and organic material design37 and a primary amine is a convenient site for conjugation for biological studies. Interestingly, electron withdrawing aryl boronic acids reacted much more rapidly with starting material 3 than electron donating boronic acids. The overall scope of the study showed the robustness and generality of the reaction condition to electronic differences, steric hindrance, and varied functional groups with high yields and excellent regio-selectivity. This enabled the rapid construction of diverse arrays of monoaryl- or heteroarylquinolines.
Scheme 1.

Aryl/Heteroaryl Boronic Acid Substrate Scope
All reactions performed on 0.18 mmol scale. Reported yields are isolated yield.
Effect of the C-2 Substituents on Photophysical Properties of DMAQ.
Having accomplished the facile synthesis of a DMAQ library, we then turned our attention to studying the photophysical properties generated by variations in the push-pull system of DMAQ. Upon initial visual inspection, more electron withdrawing (hetero)aryl substitution on C-2 of DMAQ showed more red-shifted emission in DMSO, indicating the formation of a D-π-A system (Figure 2A). To provide a guideline for rational selection of substituents for future DMAQ derivatives, we analyzed 4b, 4a, 4i, 4c, 4d, 4e, and 4f in terms of the Hammett substituent constant for the para functional group on the C-2 phenyl ring (Figure 2B).38 Electronegativity and emission wavelength showed a positive correlation, as expected. This analysis is further supported by HOMO-LUMO calculations that show a movement of electron density from the C-7 dimethylamino group to the C-2 aryl group upon excitation from S0 to S1 (see full discussion in Supporting Information, Table S15–S16 amd Figure S31–S36.).
Figure 2.
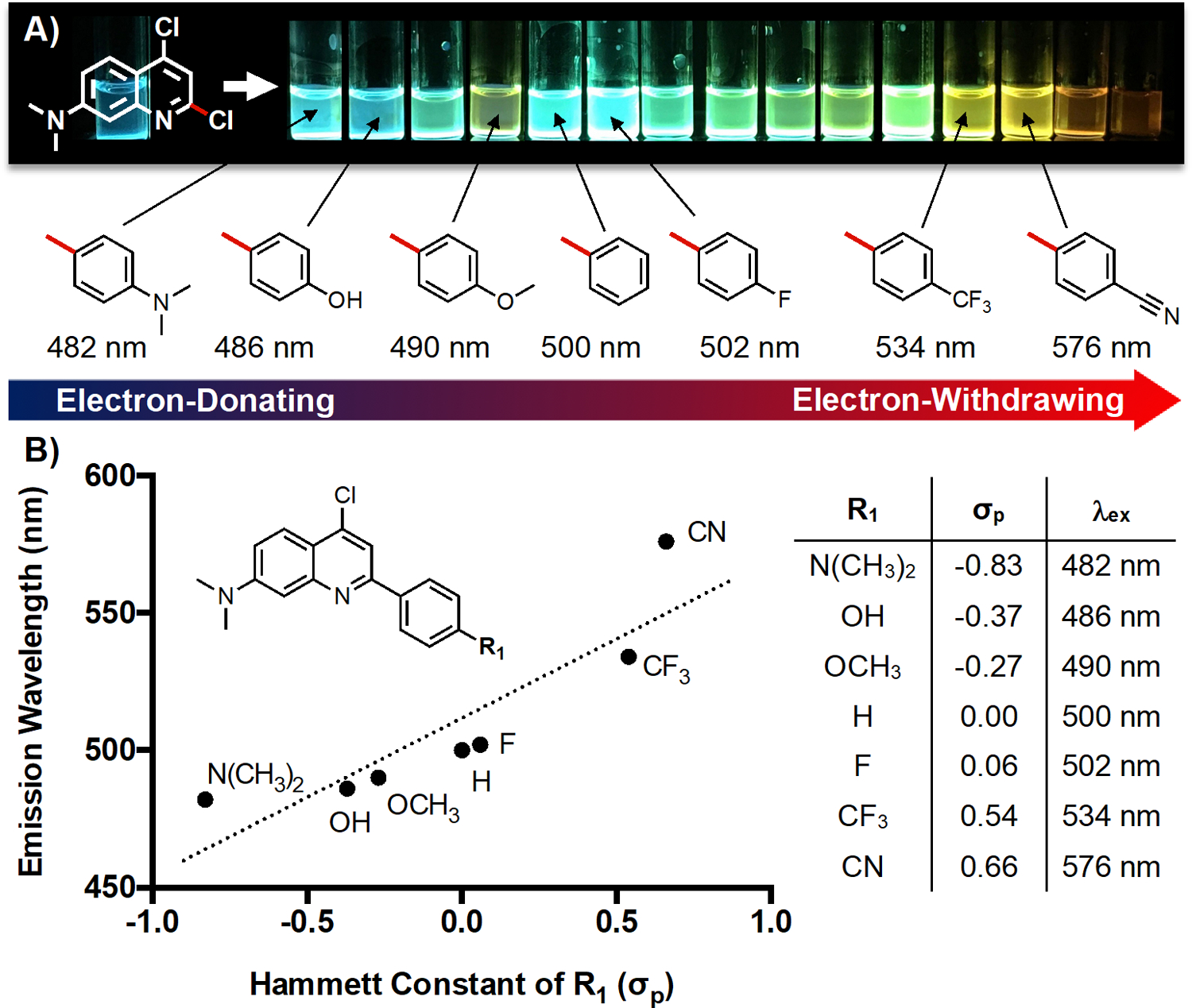
Structure-photophysical property relationship of DMAQ. A) A range of C-2 aryl substitution with emission maximum under 405 nm excitation is shown. The compound order is 4b, 4a, 4n, 4i, 4c, 4d, 4m, 4l, 4g, 4h, 4j, 4e, 4f, 4k, and 4l’ from left to right. Stock solutions of 4a-4n were made in DMSO (5 mM) and 4l’ in water with 0.1% TFA (5 mM). The image was taken using a handheld UV lamp (365 nm). B) Correlationbetween emission wavelength and Hammett constant (σp) of R1 substituents in monoaryl DMAQ. (R2 =0.79).
Photophysical Properties of Monoaryl DMAQ.
Strategies in fluorescent probe development consider not only the design and synthesis of fluorescent libraries, but also the evaluation and validation of the probes in a high-throughput manner. Thus, in order to obtain more quantitative information on the properties of our DMAQ library, we designed a multi-well assay amenable to assessing spectral properties such as fluorescence intensity and emission wavelength for each compound in the library (Figure 3).
Figure 3.

Plate reader analysis of photophysical properties of monoaryl DMAQ. Each compound (3,4a-4n) was prepared as a 5 mM stock solution in diemthyl sulfoxide (DMSO) except for 4l’ in water. Each well contains 1% DMSO in solution with a final 50 µM dye concentration. For polarity screening (left panel), toluene, basic alumina filtered dichloromethane (DCM), 0.1% trifluoroacetic acid in DCM (DCM+TFA), dimethylformamide (DMF), DMSO, and ethanol (EtOH) were used as solvents. For pH screening (right panel), 10 mM buffer solutions were prepared; pH 2 (hydrochloric acid), pH 4 (citric acid/phosphate buffer), pH 6.9 (citric acid/phosphate buffer), pH 7.3 (phosphate buffer), pH 10 (carbonate buffer), and pH 11 (sodium hydroxide). Emission spectra were measured at λex of 405 nm regardless of their optimal max absorption, and the image was taken under handheld UV lamp (365 nm).
The emission spectrum of each compound was measured under excitation at 405 nm, a common wavelength laser used in confocal microscopes for biological studies. Since the dimethylamino group and quinoline nitrogen in the DMAQ derivatives were expected to induce solvatochromism and pH sensitivity, six organic solvents with varying properties were chosen: nonpolar aprotic (toluene, DCM), nonpolar protic/acidic (DCM+TFA), polar aprotic (DMF, DMSO), and polar protic (EtOH). In addition, six different buffered aqueous solutions spanning pH 2 to pH 11 were used for screening. While our parent compound, 3, showed no significant environmental sensitivity, the rest (4a-4n) of the 2,4-substituted quinoline dyes showed solvent-dependent photophysical properties, confirming the design principle of tunability (spectra selected to illustrate certain observations are shown in Figure 3, complete sets of emission spectra for all dyes are shown in Figures S9–10). Furthermore, this tunability was generally predictable, with electron withdrawing para-substituted phenyl rings at the 2-position red-shifting the emission (e.g., 4f) as one would expect for the designed push-pull system.
Selected DMAQs with para-substituded aryl rings (4b,4c,4e,4f,4i,4l), (hetero)aryl rings (4m), and parent core DMAQ 3 showed emission spanning 453 nm to 516 nm in basic alumina filtered DCM (Figure 3B). Under nonpolar protic/acidic conditions (DCM+TFA), all compounds 4a-4n showed a significant bathochromic emission shift in comparison to emission in basic alumina-filtered DCM (Figure 3A). While 3 had a minimal bathochromic shift of 3 nm, (hetero)arylated DMAQs (4b,4c,4e,4f,4i,4l,4m) showed an 84 to 96 nm shift in the presence of 0.1% TFA. These results indicate the general pH sensitivity of the quinoline “core” scaffold. Moreover, many dyes showed a sensitivity to solvent polarity as well as pH. 4k, in particular, was highly solvatochromic, with an emission spanning 480 to 586 nm in various organic solvents (Figure 3C). This can be a very valubale property in which the dye could be used to sense its local environment on a protein surface or in a cellular compartment. Boc deprotection of 4l gave 4l’ with significantly increased water solubility and enhanced brightness (Figure 3D).
Dyes with explicitly pH-sensitive functional groups were also investigated, where the emission of phenol-functionalized 4a red-shifted in acidic environments, with an apparent pKa in the 4–6.9 range (Figure 3E). The pH range for fluorescence changes can be tuned through electronic effects; fluorine functionalized 4d exhibited a 118 nm red-shift in emission on going from pH 4 to pH 2 (Figure 3G), while the emission of ortho-methoxy functionalized 4g red-shifted 100 nm on going from pH 7 to pH 4 (Figure. 3H). The fluorescence of dimethylamino-substituted 4b showed an interesting three stage change with pH, where it was quenched at acidic pHs, and had two emission peaks at 480 and 600 nm at pH 7, with the second peak growing more prominent at basic pHs (Figure. 3F). Taken together, these data show the importance of the C-2 position in tuning the spectral peroperties of DMAQ dyes. The monoaryl DMAQ derivatives can easily be functionalized at C-4 for conjugation to other molecules or for further derivatization.
Synthesis of Bisaryl DMAQ via Sequential Cross-Coupling Reaction.
The ability to regio-selectively cross-couple aryl groups at the 2-C position of 3 provided us with the opportunity to further tune the properties of our dyes by substitution at the 4-C position. In screening conditions for sequential cross-coupling, we found that once the 2-arylated product was formed using our optimized regioselective cross coupling conditions, a second arylation cross-coupling reaction could be performed without additional Pd by addition of an excess of a second boronic acid and increasing the temperature to 85 ˚C (Figure 4A). Using three boronic acids used in Table 2, we investigated the efficiency of sequential cross coupling. In order to run sequential coupling reactions at 85 ˚C, dioxane was used instead of THF. Gratifyingly, we found that all reaction sequences gave good to excellent yields (HPLC yields shown in Figures S11–13).
Figure 4.

Sequential cross-coupling for bisaryl DMAQ dyes. A) Reaction scheme for sequential cross coupling. B-D) Emission spectra of 5a-5i in DMSO upon 405 nm excitation. They are categorized under same R1 where B) R1= N(CH3)2, C) R1= H, and D) R1= CN. Reported yield is isolated yield.
Effects of the C-4 Substituents on Photophysical Properties of DMAQ.
The nine compound doubly-subsituted DMAQ library was used to study the effect of the 4-C domain on DMAQ photophysical properties. Generally, electron withdrawing groups on the on 4-C phenyl ring red-shifted the emission of the corresponding parent DMAQ 4b, 4c, or 4f. 5c was particularly strongly affected by para-cyanophenyl modification at 4-C, resulting in a highly red shifted emission maximum of 593 nm compared to 482 nm for 4b (Figure 4B). Conversely, the emission maximum of 4f (576 nm) was strongly blue-shifted to 483 nm by addition of an electron donating amino group on 4-C to give 5g (Figure 4D). Interestingly, 5c and 5g both had the same set of substitutions, but in opposite positions, giving very different properties: 5c exhibited highly red-shifted emission while 5g was highly blue-shifted. This phenomenon implies that excitation involves the flow of electrons from the 2-C substituent to the 4-C substituent, so that matching this with appropriate electron donating (2-C) and electron withdrawing (4-C) groups lowers the energy for this transition and red-shifts the absorption (Table S5) and emission.
Photophysical Property of Bisaryl DMAQ.
5a-i were subjected to pH-dependent fluorescence spectral analysis by screening their emission in pH 2, 4, 6.9, 7.3, 10, and 11 as shown in Figure 3 for 4a – 4n (Figure S14). 5a, 5b, 5d, 5e, and 5f exhibited pH dependent dual emissive properties, where emission red-shifted up to 170 nm in solutions of pH 2 and/or pH 4 under single excitation at 405 nm. 5g and 5i showed pH dependent intensity changes; 5g had 20-fold higher emission at 525 nm in pH 11 buffer than pH 2 buffer. Lastly, 5c had quenched emission in buffers at or below pH 4, while 5h had quenched emission at pH 2 (Figure S20–21) under excitation at 405 nm.
Live Cell Imaging of DMAQ dyes.
The DMAQ compounds (4a-4n, 5a-5i) were all subjected to live HeLa cell imaging (Figure S15–17). All of them, except for 4f, were cell permeable and were suitable for live cell imaging. Yet it should be noted that modifying 4f to 5g-5i enabled good cell permeability, validating the benefit of tunable scaffold. Selected dyes were subjected to cytotoxicity assays (Table S6). Among them, an application of pH sensitive dye 5a in live cells was chosen as a proof-of-concept to illustrate the value of DMAQs. 5a demonstrated superior properties over many commercial pH sensitive probes (Figure 5). In a neutral to basic environment (pH >7), a broad absorption band with λex at 308 nm and 412 nm (green dotted line) and λem at 469 nm (green solid line) of 5a were observed (Figure 5A). However, at a pH of 4 or lower, absorbance and emission shifted to λ ex at 474 nm (red dotted line) and λem at 637 nm (red solid line), respectively. Having a bathochromic shift of 168 nm in emission from basic to acidic environment produced a large Stokes shift and gave 5a an unusual pH-dependent dual-emission characteristic. These properties allowed 5a to stain two different locations based in live HeLa cells. Green staining (Figure 5C) obtained by exciting at 405 nm and collecting the emission at 475±25 nm (Figure 5C) identified a subset of probe molecules presumably sequestered in neutral or basic compartments. Red staining was produced by exciting at 488 nm with the emission filter set at 650±25 nm (Figure 5D) and indicated a substantial fraction of the dye in a presumably acidic environment. A merged image did not show any yellow regions, indicating that 5a can orthogonally image two regions that differ in local environment (Figure 5F). This is in contrast to many pH-sensitive dyes which are based on a change in fluorescence quenching. For instance, the commercial dye, cell-permeant ratiometric pH indicator Seminaphthorhodafluor (SNARF) exhibits a pH-dependent emission shift from acidic to basic conditions. This pH dependence allows the ratio of the fluorescence intensity from the dye at two emission wavelengths – typically 580 nm and 640 nm – to be widely used for quantitative determination of pH. However, SNARF derivatives are only sensitive to pH changes near 7 and have a relatively small (60 nm) emission shift with changes in pH. Moreover, the published syntheses of SNARF variants are challenging (2–7 of steps and <18% yield).39–41 These limitations are addressed by 5a, which exhibits a 170 nm pH-dependent emission shift, and other DMAQ derivatives, all of which are easily synthesized, with a tunable pH sensor range that can be seen in (Figure S14)
Figure 5.

Fluorescence spectrum and live HeLa cell imaging after 3 h incubation of 5a. A) Normalized UV absorption spectra and emission spectra excited by 405 nm are shown in pH 7 and pH 2 buffer. 405 nm and 488 nm lasers for microscopy excitation are shown by arrows. B) Differential interference contrast (DIC) image of Hela cells after 3 h incubation of 5a C) λex = 405 nm, λem = 425–475 nm D) λex = 488 nm, λem = 650–700 nm E-G) Merged images of green (λex = 405 nm, λem = 425–475 nm) and red (λex = 488 nm, λem = 625–675 nm) in E), red (λex = 488 nm, λem = 650–700 nm) in F), and red (λex = 488 nm, λem = 700–750 nm) in G).
■ CONCLUSIONS
In conclusion, our work demonstrates a synthetic advance to develop the under-explored quinoline as a fluorescent scaffold “core” that allows both rational and combinatorial approach to create novel dyes. Our method has also served to simplify the synthesis of medicinally important substituted quinoline analogues. The power of the rationally designed DMAQ “core” scaffold in the discovery of new DMAQ dyes has been validated, although by no means fully explored. There is an exciting and immediate opportunity for the exploration and optimization of DMAQ probes for a wide range of applications. For instance, these DMAQ fluorophores exhibit solid state fluorescence in amorphous and/or crystalline solids, which have potential for application in organic materials (Figure S22). Combining a rationally designed tunable scaffold with high throughput analysis, we show more than 20 different 2,4-disubstituted DMAQ derivatives with a range of emission spectra spanning the visible region as well as interesting chemical properties. With this library, we were able to further understand the structure-photophysical relationship and intrinsic properties of the DMAQ scaffold. Emission of DMAQ could be rationally tuned by C-2 substituents and DMAQ dyes generally exhibited a large Stokes shift. Our study of 2,4-diarylated DMAQs revealed a probable photophysical mechanism for electron flow from the C-2 substituent to the C-4 substituent which can be explored in subsequent theoretical and experimental investigations. Building off of our results with 5c, further accentuating the electron donation of the C-2 position and electron withdrawal of the C-4 position could generate very small near IR dyes. Studies of the cellular localization of 5a and other dyes presented here will allow the microscopy community to take advantage of their unique properties. Based on these promising initial investigations, we expect to develop DMAQ as a multi-functional probe that can serve as both drug and sensor to answer challenging questions in complex biological processes. We envision that highly tunable and optimizable fluorescent probes can function in convenient, continuous assays, thereby providing useful tools for studying biological events in cellular environments.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by funding from the University of Pennsylvania, by the National Science Foundation (NSF CHE-1708759 to EJP), and by the National Institutes of Health (Grant NIH R01-GM118510 to DMC). Instrument support from the National Institutes of Health (Grant NIH RR-023444) includes HRMS. We thank Dr. Charles W. Ross III (University of Pennsylvania) for assistance in HRMS analysis, Dr. Patrick Carroll (University of Pennsylvania) for X-ray analysis, Dr. Jun Gu (University of Pennsylvania) for NMR assistance, and Dr. Simon Berritt (University of Pennsylvania) for helpful discussion for high throughput experimentation.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Experimental protocols, characterization data and NMR spectra of all new compounds.
References
- 1.Terai T; Nagano T, Fluorescent probes for bioimaging applications. Current Opinion in Chemical Biology 2008, 12 (5), 515–521. [DOI] [PubMed] [Google Scholar]
- 2.Jiang M; Gu X; Lam JWY; Zhang Y; Kwok RTK; Wong KS; Tang BZ, Two-photon AIE bio-probe with large Stokes shift for specific imaging of lipid droplets. Chemical Science 2017, 8 (8), 5440–5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavis LD; Raines RT, Bright Ideas for Chemical Biology. ACS Chemical Biology 2008, 3 (3), 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laras Y; Hugues V; Chandrasekaran Y; Blanchard-Desce M; Acher FC; Pietrancosta N, Synthesis of Quinoline Dicarboxylic Esters as Biocompatible Fluorescent Tags. The Journal of Organic Chemistry 2012, 77 (18), 8294–8302. [DOI] [PubMed] [Google Scholar]
- 5.Wang L; Zhang J; Kim B; Peng J; Berry SN; Ni Y; Su D; Lee J; Yuan L; Chang Y-T, Boronic Acid: A Bio-Inspired Strategy To Increase the Sensitivity and Selectivity of Fluorescent NADH Probe. Journal of the American Chemical Society 2016, 138 (33), 10394–10397. [DOI] [PubMed] [Google Scholar]
- 6.Finney NS, Combinatorial discovery of fluorophores and fluorescent probes. Current Opinion in Chemical Biology 2006, 10 (3), 238–245. [DOI] [PubMed] [Google Scholar]
- 7.Vendrell M; Zhai D; Er JC; Chang Y-T, Combinatorial Strategies in Fluorescent Probe Development. Chemical Reviews 2012, 112 (8), 4391–4420. [DOI] [PubMed] [Google Scholar]
- 8.Samanta A; Vendrell M; Das R; Chang Y-T, Development of photostable near-infrared cyanine dyes. Chemical Communications 2010, 46 (39), 7406–7408. [DOI] [PubMed] [Google Scholar]
- 9.Ahn Y-H; Lee J-S; Chang Y-T, Combinatorial Rosamine Library and Application to in Vivo Glutathione Probe. Journal of the American Chemical Society 2007, 129 (15), 4510–4511. [DOI] [PubMed] [Google Scholar]
- 10.Dhiman AK; Kumar R; Kumar R; Sharma U, Metal-Free Synthesis of 2-Substituted 3-(2-Hydroxyaryl)quinolines and 4-(2-Hydroxyaryl)acridines via Benzyne Chemistry. The Journal of Organic Chemistry 2017, 82 (23), 12307–12317. [DOI] [PubMed] [Google Scholar]
- 11.Michael JP, Quinoline, quinazoline and acridone alkaloids. Natural Product Reports 2008, 25 (1), 166–187. [DOI] [PubMed] [Google Scholar]
- 12.Foley M; Tilley L, Quinoline Antimalarials: Mechanisms of Action and Resistance and Prospects for New Agents. Pharmacology & Therapeutics 1998, 79 (1), 55–87. [DOI] [PubMed] [Google Scholar]
- 13.Kaufman TS; Rúveda EA, The Quest for Quinine: Those Who Won the Battles and Those Who Won the War. Angewandte Chemie International Edition 2005, 44 (6), 854–885. [DOI] [PubMed] [Google Scholar]
- 14.(a) Fahrni C; O’Halloran T, Aqueous Coordination Chemistry of Quinoline-Based Fluorescence Probes for the Biological Chemistry of Zinc J. Am. Chem. Soc 1999, 121, 11448–11458. [Google Scholar]; (b) Nowakowski A; Petering D, Reactions of the Fluorescent Sensor, Zinquin, with the Zinc-Proteome: Adduct Formation and Ligand Substitution. Inorg. Chem 2011, 50, 10124–10133. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jeffrey W Meeusen J; Tomasiewicz H; Nowakowski A; Petering D, TSQ (6-Methoxy-8-p Toluenesulfonamido-Quinoline), a Common Fluorescent Sensor for Cellular Zinc, Images Zinc Proteins, Inorg. Chem 2011, 50, 7563–7573. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hanaoka K; Kikuchi K; Kojima H; Urano Y; Nagano T, Development of a Zinc Ion-Selective Luminescent Lanthanide Chemosensor for Biological Applications. Journal of the American Chemical Society 2004, 126 (39), 12470–12476 [DOI] [PubMed] [Google Scholar]; (e) Li G; Zhu D; Xue L; Jiang H, Quinoline-Based Fluorescent Probe for Ratiometric Detection of Lysosomal pH. Organic Letters 2013, 15 (19), 5020–5023. [DOI] [PubMed] [Google Scholar]
- 15.Li W; Lin W; Wang J; Guan X, Phenanthro[9,10-d]imidazole-quinoline Boron Difluoride Dyes with Solid-State Red Fluorescence. Organic Letters 2013, 15 (7), 1768–1771. [DOI] [PubMed] [Google Scholar]
- 16.Wender PA; Jeffreys MS; Raub AG, Tetramethyleneethane Equivalents: Recursive Reagents for Serialized Cycloadditions. Journal of the American Chemical Society 2015, 137 (28), 9088–9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wender PA; Verma VA; Paxton TJ; Pillow TH, Function-Oriented Synthesis, Step Economy, and Drug Design. Accounts of Chemical Research 2008, 41 (1), 40–49. [DOI] [PubMed] [Google Scholar]
- 18.Curran ACW, 5,6,7,8-Tetrahydroquinolines. Part I. A novel synthesis of 7,8-dihydroquinolin-5(6H)-ones. Journal of the Chemical Society, Perkin Transactions 1 1976, (9), 975–977. [Google Scholar]
- 19.Misani F; Bogert MT, THE SEARCH FOR SUPERIOR DRUGS FOR TROPICAL DISEASES. II. SYNTHETIC STUDIES IN THE QUINOLINE AND PHENAN-THROLINE SERIES. SKRAUP AND CONRAD-LIMPACH-KNORR REACTIONS. The Journal of Organic Chemistry 1945, 10 (4), 347–365. [DOI] [PubMed] [Google Scholar]
- 20.Sakai N; Aoki D; Hamajima T; Konakahara T, Yb(OTf)3-catalyzed cyclization of an N-silylenamine with 2-methylene-1,3-cyclohexanedione to afford a 7,8-dihydroquinolin-5(6H)-one derivative and its application to the one-pot conversion to a 2,3,5-trisubstituted quinoline derivative. Tetrahedron Letters 2006, 47 (8), 1261–1265. [Google Scholar]
- 21.Eisch JJ; Dluzniewski T, Mechanism of the Skraup and Doebner-von Miller quinoline syntheses. Cyclization of .alpha.,.beta.-unsaturated N-aryliminium salts via 1,3-diazetidinium ion intermediates. The Journal of Organic Chemistry 1989, 54 (6), 1269–1274. [Google Scholar]
- 22.Corey EJ; Tramontano A, Total synthesis of the quinonoid alcohol dehydrogenase coenzyme (1) of methylotrophic bacteria. Journal of the American Chemical Society 1981, 103 (18), 5599–5600. [Google Scholar]
- 23.Ryabukhin SV; Naumchik VS; Plaskon AS; Grygorenko OO; Tolmachev AA, 3-Haloquinolines by Friedländer Reaction of α-Haloketones. The Journal of Organic Chemistry 2011, 76 (14), 5774–5781. [DOI] [PubMed] [Google Scholar]
- 24.Gladiali S; Chelucci G; Mudadu MS; Gastaut M-A; Thummel RP, Friedländer Synthesis of Chiral Alkyl-Substituted 1,10-Phenanthrolines. The Journal of Organic Chemistry 2001, 66 (2), 400–405. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X; Yao T; Campo MA; Larock RC, Synthesis of substituted quinolines by the electrophilic cyclization of n-(2-alkynyl)anilines. Tetrahedron 2010, 66 (6), 1177–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L; Chen D; Yao J; Zong Q; Wang J; Zhou H, CuX-Activated N-Halosuccinimide: Synthesis of 3-Haloquinolines via Electrophilic Cyclization of Alkynyl Imines. The Journal of Organic Chemistry 2017, 82 (9), 4625–4630. [DOI] [PubMed] [Google Scholar]
- 27.Li X; Mao Z; Wang Y; Chen W; Lin X, Molecular iodine-catalyzed and air-mediated tandem synthesis of quinolines via three-component reaction of amines, aldehydes, and alkynes. Tetrahedron 2011, 67 (21), 3858–3862. [Google Scholar]
- 28.Collet JW; Ackermans K; Lambregts J; Maes BUW; Orru RVA; Ruijter E, Modular Three-Component Synthesis of 4-Aminoquinolines via an Imidoylative Sonogashira/Cyclization Cascade. The Journal of Organic Chemistry 2018, 83 (2), 854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nasr M; Drach JC; Smith SH; Shipman C; Burckhalter JH, 7-Aminoquinolines. A novel class of agents active against herpes viruses. Journal of Medicinal Chemistry 1988, 31 (7), 1347–1351. [DOI] [PubMed] [Google Scholar]
- 30.Eisenhart TT; Howland WC; Dempsey JL, Proton-Coupled Electron Transfer Reactions with Photometric Bases Reveal Free Energy Relationships for Proton Transfer. The Journal of Physical Chemistry B 2016, 120 (32), 7896–7905. [DOI] [PubMed] [Google Scholar]
- 31.Jain S; Chandra V; Kumar Jain P; Pathak K; Pathak D; Vaidya A, Comprehensive review on current developments of quinoline-based anticancer agents. Arabian Journal of Chemistry 2016. [Google Scholar]
- 32.Dai X; Chen Y; Garrell S; Liu H; Zhang L-K; Palani A; Hughes G; Nargund R, Ligand-Dependent Site-Selective Suzuki Cross-Coupling of 3,5-Dichloropyridazines. The Journal of Organic Chemistry 2013, 78 (15), 7758–7763. [DOI] [PubMed] [Google Scholar]
- 33.Schröter S; Stock C; Bach T, Regioselective Cross-Coupling Reactions of Multiple Halogenated Nitrogen-, Oxygen-, and Sulfur-Containing Heterocycles. 2005; Vol. 61, p 2245–2267. [Google Scholar]
- 34.Reddy EA; Islam A; Mukkanti K; Bandameedi V; Bhowmik DR; Pal M, Regioselective alkynylation followed by Suzuki coupling of 2,4-dichloroquinoline: Synthesis of 2-alkynyl-4-arylquinolines. Beilstein Journal of Organic Chemistry 2009, 5, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee C-Y; Ahn S-J; Cheon C-H, Protodeboronation of ortho- and para-Phenol Boronic Acids and Application to ortho and meta Functionalization of Phenols Using Boronic Acids as Blocking and Directing Groups. The Journal of Organic Chemistry 2013, 78 (23), 12154–12160. [DOI] [PubMed] [Google Scholar]
- 36.Lennox AJJ; Lloyd-Jones GC, Selection of boron reagents for Suzuki-Miyaura coupling. Chemical Society Reviews 2014, 43 (1), 412–443. [DOI] [PubMed] [Google Scholar]
- 37.Guo P; Joo JM; Rakshit S; Sames D, C–H Arylation of Pyridines: High Regioselectivity as a Consequence of the Electronic Character of C–H Bonds and Heteroarene Ring. Journal of the American Chemical Society 2011, 133 (41), 16338–16341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hansch C; Leo A; Taft RW, A survey of Hammett substituent constants and resonance and field parameters. Chemical Reviews 1991, 91 (2), 165–195. [Google Scholar]
- 39.Marcotte N; Brouwer AM, Carboxy SNARF-4F as a Fluorescent pH Probe for Ensemble and Fluorescence Correlation Spectroscopies. The Journal of Physical Chemistry B 2005, 109 (23), 11819–11828. [DOI] [PubMed] [Google Scholar]
- 40.Nakata E; Nazumi Y; Yukimachi Y; Uto Y; Maezawa H; Hashimoto T; Okamoto Y; Hori H, Synthesis and photophysical properties of new SNARF derivatives as dual emission pH sensors. Bioorganic & Medicinal Chemistry Letters 2011, 21 (6), 1663–1666. [DOI] [PubMed] [Google Scholar]
- 41.Richter C; Ernsting NP; Mahrwald R, Operationally Simple and Selective One-Pot Synthesis of Hydroxyphenones: A Facile Access to SNARF Dyes. Synthesis 2016, 48 (08), 1217–1225. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.