

Abstract
Direct functionalization of natural products is important for studying the structure–activity and structure–property relationships of these molecules. Recent advances in the transition-metal-catalyzed functionalization of C(sp3)–H bonds, the most abundant yet inert bonds in natural products, have allowed natural product derivatives to be created selectively. Strategies to achieve such transformation are reviewed.
Keywords: amination, catalysis, C—H bond functionalization, oxidation, regioselectivity
Graphical Abstract

1. Introduction
Chemists have explored the structure, synthesis, derivatization, and biological functions of natural products for decades, seeking new routes to their preparation, new classes of organic structures, and fundamental principles that govern their functions. These studies have been motivated by the wide use of such structures in the process of drug development.[1–6] For example, between 1940–2010, 41% of anti-cancer small-molecule drugs were either natural products or semi-synthetically derived from natural products.[1–3]
Natural products and their derivatives also assist in the discovery of new biological targets and provide tools to study the pharmacology of these targets.[7] Opioid drugs are a good example of this symbiosis (Figure 1). Although many cultures have used sap of the poppy seed for pain management for thousands of years, it was only in the 19th century that chemists crystallized the main active ingredient of opium, which is morphine (1).[8] Subsequently, Merck KGaA, Darmstadt, Germany scaled up the isolation process of morphine and started marketing morphine as a pain medication. The discovery of morphine has revolutionized pharmaceuticals to treat pain, and led to the discovery of opioid receptors, which are important for understanding and exploring conditions such as depression and drug addiction.[9] The unique polycyclic structure of this molecule has inspired the development of many synthetic strategies for the total synthesis of morphine.[10]
Figure 1.
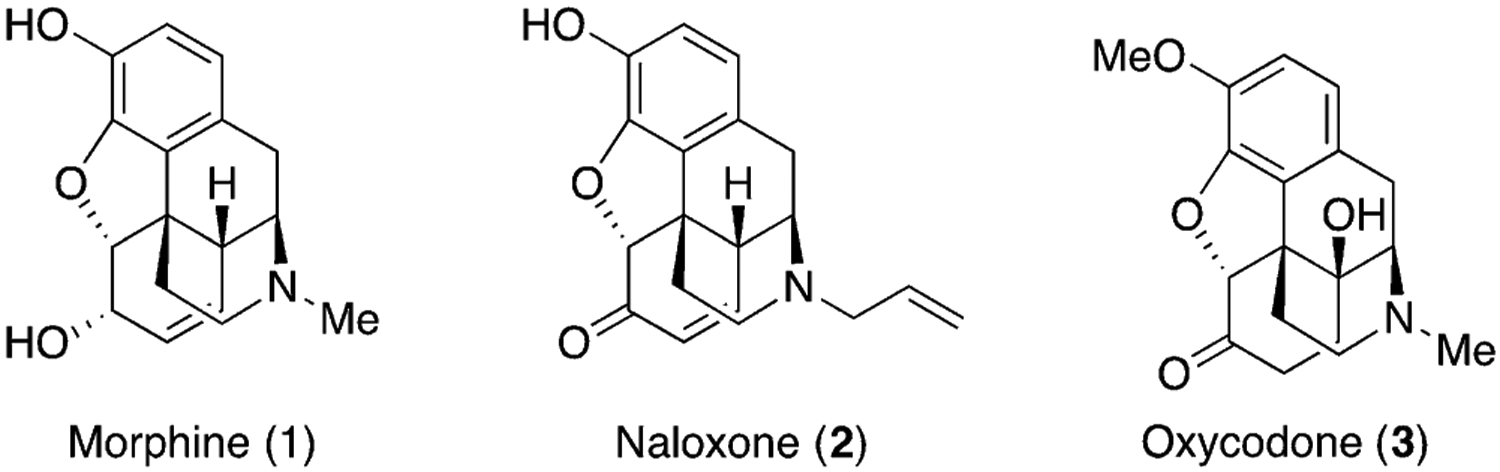
Structures of selected opioids.
Often, the full range of biological properties of a natural product are not understood until its derivatives are prepared. These derivatives help determine the structure–activity and structure–property relationships of a given natural product and ultimately help identify molecules that have improved pharmacological profiles. If a natural product is available in sufficient quantities through isolation or fermentation, direct functionalization (often termed late-stage functionalization) can rapidly provide analogues in a more cost-effective way than parallel resynthesis of each new analogue.[11] Chemists have used this strategy of direct functionalization extensively since the early days of natural product isolation. However, natural products are under-represented in modern screening libraries, in part because natural product isolation is slow and generates a limited number of molecules.[12]
The direct functionalization of abundant natural products holds great potential to create libraries of natural product derivatives that contain large numbers of molecules with deep-seated changes to the core structures, as well as changes to the peripheral functionality, while maintaining properties that are similar to those of natural products. The structural diversity of natural products makes them important starting points for the creation of complex, three-dimensional structures in modern screening libraries.[13]
In this Minireview, we discuss strategies for the functionalization of unactivated C–H bonds in natural products, summarize some of the challenges confronting the development of these reactions, and explain why the functionalization of C–H bonds can be a particularly valuable approach to the derivatization of natural products. In the 1990s, Bergman wrote that the discovery of selective reactions at C–H bonds in the presence of other functional groups was one of the “holy grails” of chemistry.[14] Since then, the chemistry of C–H bonds has undergone explosive growth, encompassing the development of new reagents and catalysts that allow selective cleavage of C–H bonds in the presence of a wide range of functional groups. At the same time, the selective functionalization of C–H bonds in both simple and complex structures remains challenging because these bonds are less reactive than functional groups toward many reagents and classes of catalysts.[15] Because many new strategies for the functionalization of natural products involve reactions catalyzed by transition-metal complexes,[16] this Minireview focuses on the state of the art in transition-metal-catalyzed functionalizations of C–H bonds in natural products.
2. Directed Functionalization of C–H Bonds in Natural Products.
In a “directed” C–H functionalization reaction, a Lewis basic functional group on the substrate binds to the catalyst and limits the reaction site to one of the many C–H bonds.[17–19] Such binding also dramatically accelerates the rate of C–H bond cleavage. Existing functional groups are typically converted into a functional group that can serve as a ligand to the transition-metal complex. For example, ketones have been converted into imines[20] and oximes,[21] alcohols have been converted into oximes,[22] and carboxylic acids have been converted into amides[23] to serve as directing groups in C–H bond functionalization reactions.
This type of directed C–H activation by transition-metal catalysts is a well-known organometallic reaction and is termed “cyclometallation”.[24–26] In 1985, Baldwin and co-workers reported an oxime-directed cyclometallation of dihydrolupenone derivative 4 (Figure 2A).[27] In this reaction, stoichiometric Na2PdCl4 was used to generate cyclometallated dimer 5, which was then oxidized with Pb(OAc)4 to give the C23-acetoxylated derivative of lupenone 6.
Figure 2.
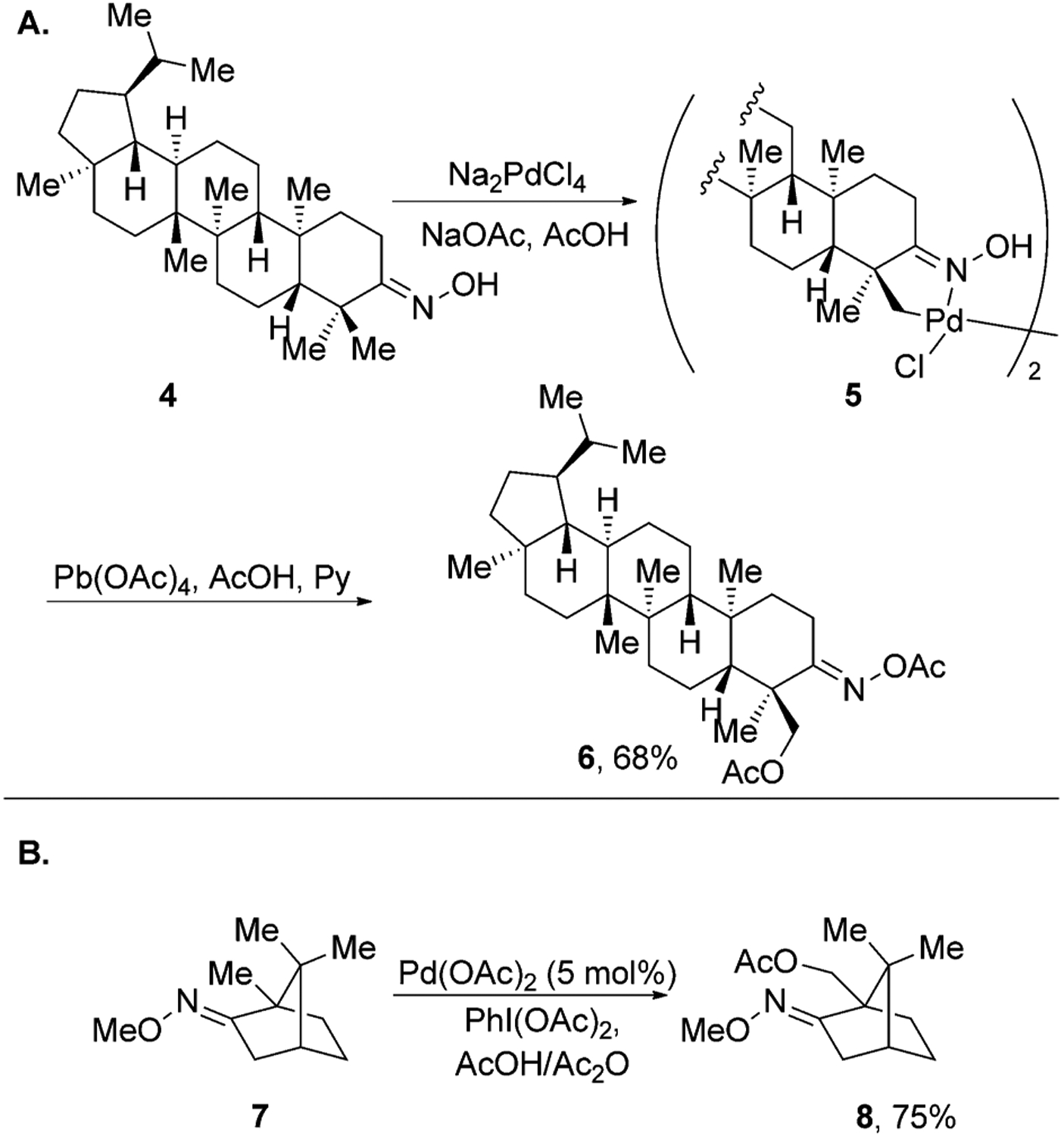
Oxime-directed C–H acetoxylation of natural products.
Almost two decades later, Sanford and colleagues reported an improved method that used catalytic palladium acetate and PhI(OAc)2 as the stoichiometric oxidant to achieve a similar transformation (Figure 2B).[21] This procedure was successfully applied to the acetoxylation of primary C–H bonds in camphor derivative 7.
C–H bond functionalization reactions require a stoichiometric oxidant, thus requiring that the reaction products be stable to further oxidation. Both of the procedures described above install an acetoxy group at the position of a C–H bond to form a product that is stable under the oxidative reaction conditions. However, installation of a hydroxy group is more challenging because the alcohol product can be further oxidized to the corresponding carbonyl compound.
In 2004, Schönecker and co-workers reported a copper-mediated procedure for the hydroxylation of steroids.[28] Recently, the Baran group improved the reaction conditions initially reported by Schönecker and demonstrated that this method could be applied to the synthesis of a variety of polyhydroxylated derivatives of natural steroids. (Figure 3).[29] For example, 3-methylestrone and dehydro-epi-androsterone were oxidized to their corresponding C12-hydroxylated analogues 9 and 10 in good yield. Based on the solid-state X-ray structure analysis of steroids and force field calculations, Schönecker proposed that this reaction proceeds through a seven-membered transition state in which the geometrically accessible γ-C–H bond is cleaved selectively. The imine directing group was cleaved under mild conditions using an aqueous solution of EDTA. Weaker C–H bonds, such as those at benzylic and allylic positions, as well as more sterically accessible primary C–H bonds, were not functionalized under these conditions.
Figure 3.

Directed C–H hydroxylation of steroids.
Although several groups have reported important advances in directing functionalization of unactivated C(sp3)–H bonds in natural products, the range of functional groups that have been introduced by this strategy is narrow. To overcome this limitation, Dong and co-workers developed a strategy to convert a C–H bond into a leaving group.[22] In this procedure, they first transformed the alcohol functional group of 11 into a bidentate directing group through the one-pot, two-step procedure shown in Figure 4. This directing group enabled tosylation of the C–H bonds β to the starting alcohol. The OTs group was then displaced by a variety of nucleophiles to generate azide, halogen, phenyl ether, phenyl thioether, and nitrile derivatives of 2H-pregnanediol in high yield.
Figure 4.
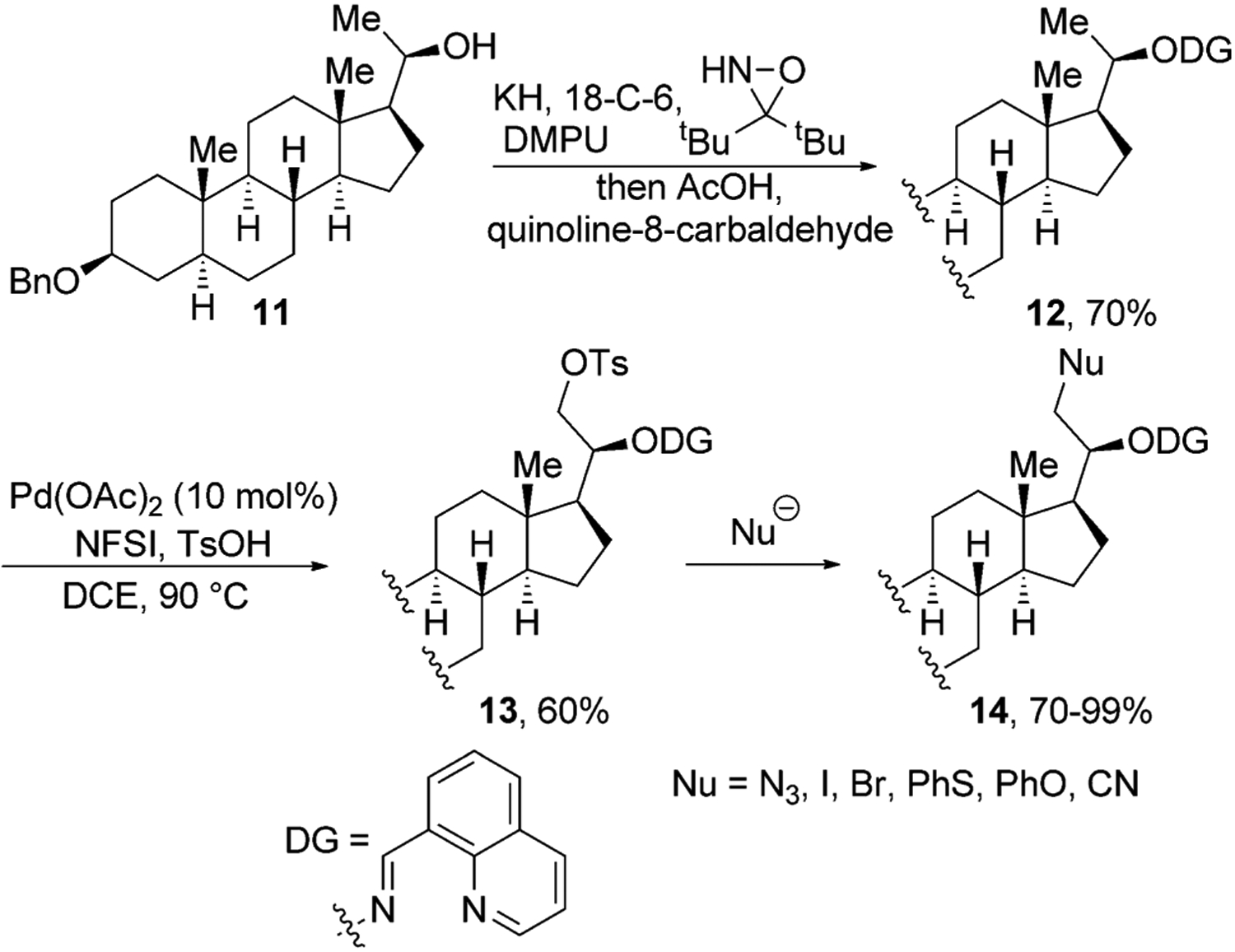
Directed tosylation of primary C–H bonds in 2H-pregnanediol. 18-C-6=18-crown-6, DCE=1,2-dichloroethane, DMPU=1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone, NFSI=N-fluorobenzenesulfonimide, Ts=tosyl.
Amides can also serve as directing groups. For example, Yu and co-workers reported an O-methylhydroxamide directing group that can be used in combination with catalytic Pd(OAc)2 to direct alkylation by alkylboronic acids to a nearby primary C–H bond.[23] Various derivatives of dehydroabietic acid 15 were prepared by using this strategy (Figure 5).
Figure 5.

Alkylation of primary C–H bonds of dehydroabietic acid using an amide directing group.
Directing groups can orient the catalyst toward a particular C–H bond through ionic or hydrogen-bonding interactions or through the reversible formation of covalent bonds. Enzymes use a similar strategy to present only specific C–H bonds of natural products to an active site for functionalization. Inspired by P450 enzymes that catalyze the hydroxylation of C–H bonds, Breslow and co-workers designed manganese porphyrin catalysts such as 19,[30] in these structures, cyclodextrin units are attached to the porphyrin ring to bind to the steroid. Such non-covalent binding strongly influences the regioselectivity of the oxidation reaction. For example, andostane-3,17-diol derivative 17 underwent hydroxylation only at the C6 position to give triol 18 in high yield after saponification (Figure 6)
Figure 6.
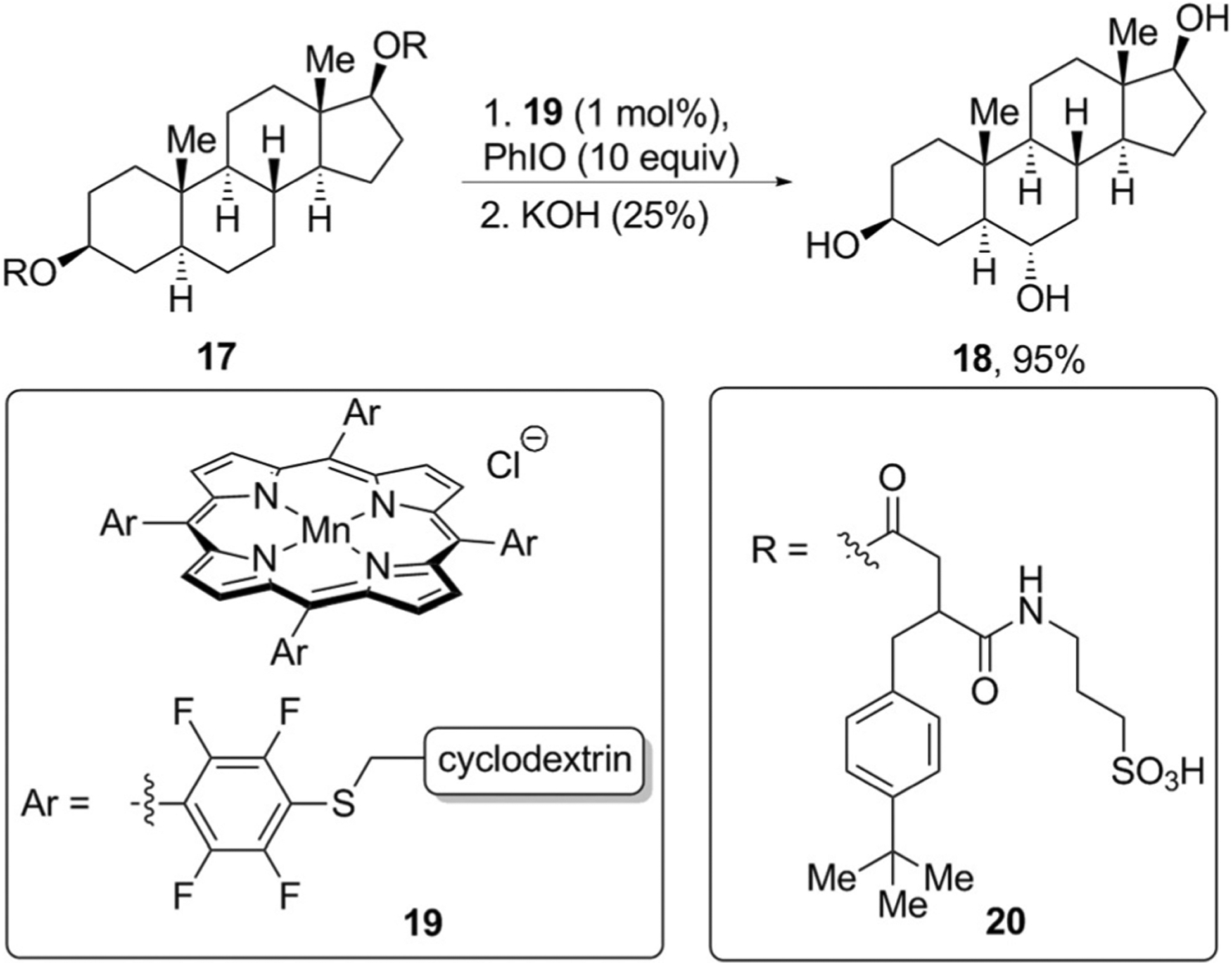
Templated C–H oxidation of natural products.
One of the drawbacks of directed functionalization is that the overall process requires installation and removal of the directing group before and after the reaction that functionalizes a C–H bond. This sequence often requires multiple steps and purifications. Hartwig and Simmons reported an alternative strategy in which a silyl unit is a reagent for functionalization that also attaches to an alcohol to control the site at which silylation of a C–H bond occurs.[31] This process involves a three-step, one-pot sequence encompassing the silylation of an alcohol, intramolecular silylation of the primary C–H bond, and Tamao–Fleming oxidation of the resulting oxasilole. Several natural products were hydroxylated with this sequence; in the most complex case, methyl oleanate was converted into methyl hederagenate in 61% overall yield (Figure 7). This procedure is a significant improvement upon Baldwin’s method,[27] which took ten overall steps, required the use of stoichiometric amounts of Pd(OAc)2, and gave methyl hederagenate in 36% overall yield.[32]H-Atom
Figure 7.
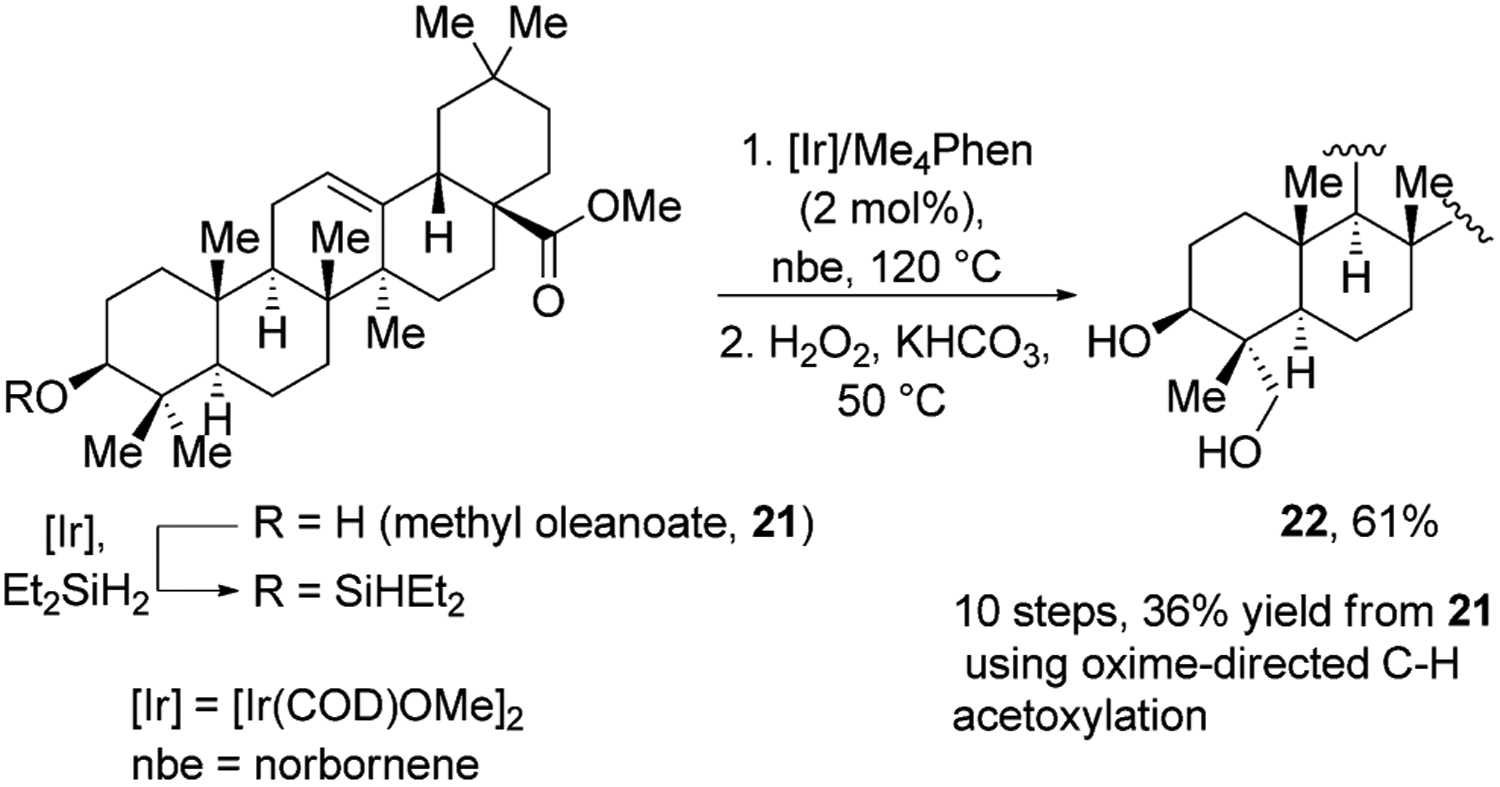
Silyl directed oxidation of oleanolic acid. Me4Phen=3,4,7,8-tetramethyl-1,10-phenanthroline, nbe=norbornene.
White and co-workers have used carboxylic acids in natural products to functionalize them with high site selectivity.[33] For example, taxane derivative 23 was converted into lactone 24 in moderate yield using iron catalyst 25. The authors found that it was crucial to add the reagents and catalyst in small portions over time. When the carboxylic acid was protected as a methyl ester, lactone 24 was not formed, thus indicating that formation of this product was directed by the carboxylic acid (Figure 8).
Figure 8.
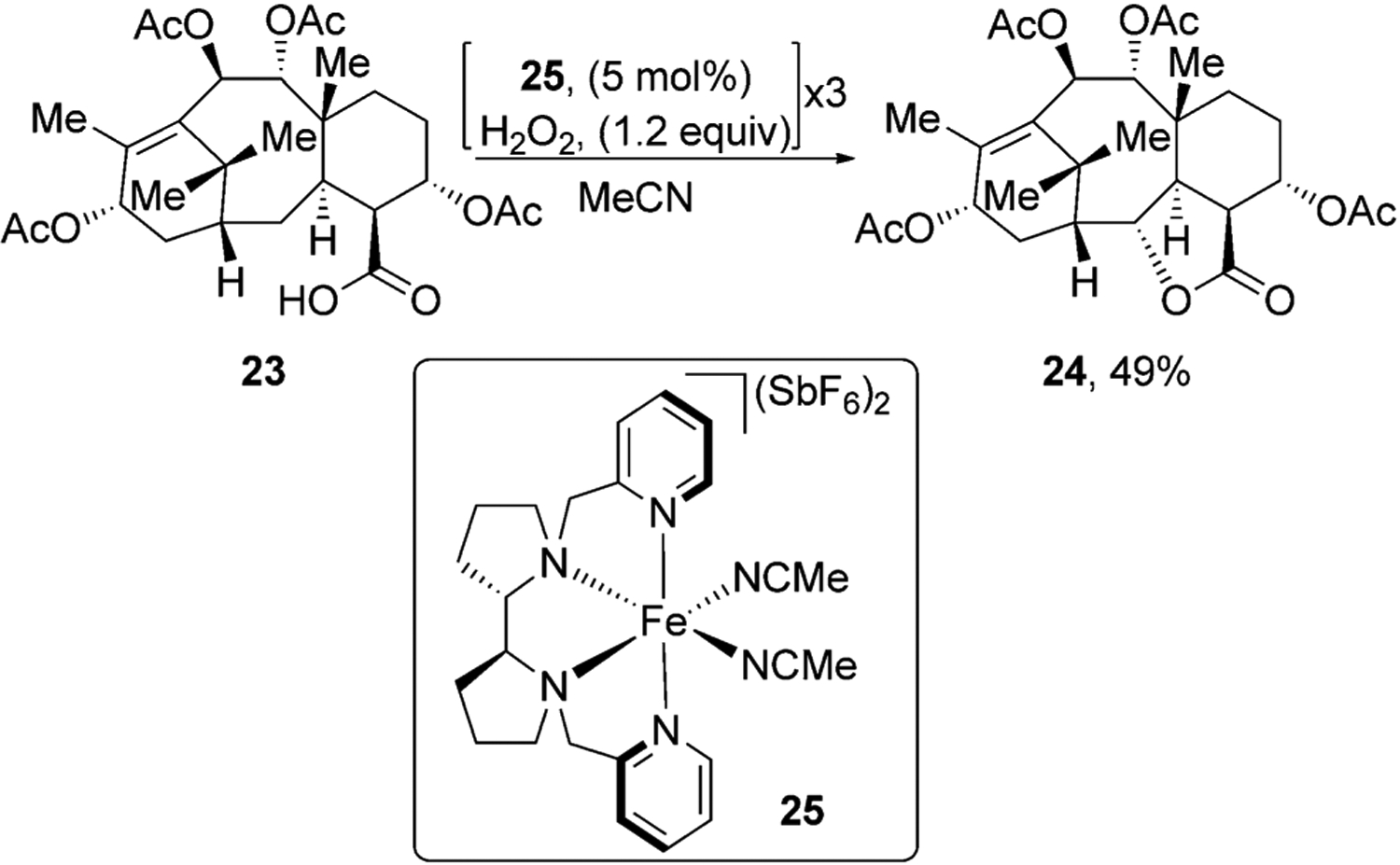
Carboxylic acid directed oxidation of a taxane derivative.
3. Undirected Functionalization of C–H Bonds in Natural Products.
Undirected C–H functionalization is more challenging than directed functionalization because such reactions occur without the strong binding of the substrate to the catalyst that precedes C–H bond cleavage in directed reactions and positions one of many C–H bonds close to the metal center. Two primary strategies have been used to cleave the C–H bonds in natural products without using a directing group. These are 1) homolytic cleavage of the C–H bonds to generate a carbon-centered radical, which is trapped by a reagent or transition-metal complex (Figure 9A), and 2) direct insertions of carbene, niterene, or oxene units of the corresponding metal complexes into a C–H bond (Figure 9B).
Figure 9.
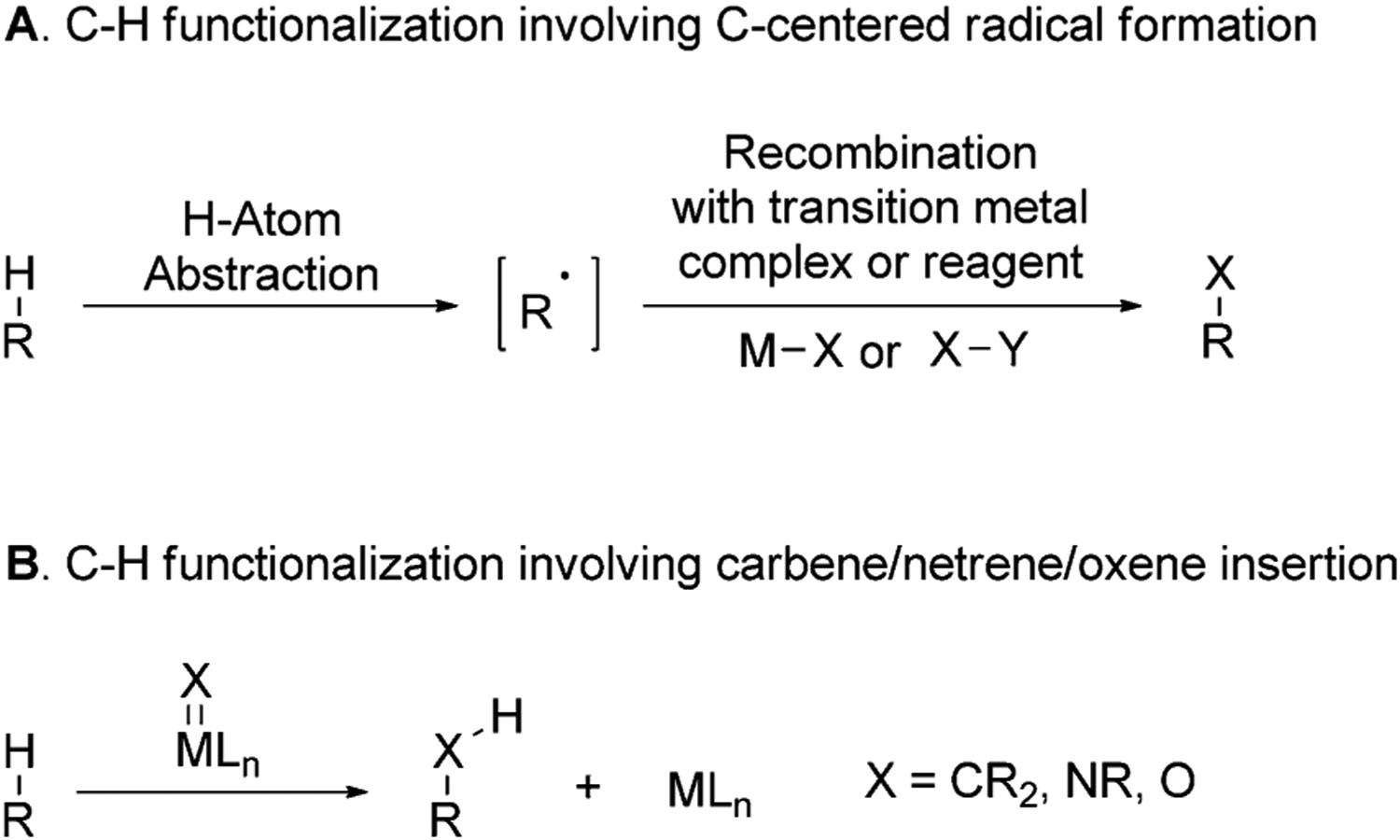
Strategies used for undirected cleavage of C–H bonds in natural products.
3.1. Undirected Functionalization of C–H bonds via Carbon-Centered Radicals
Chemists have long known that alkyl radicals can be formed through abstraction of the hydrogen atom in a C–H bond. In such reactions, abstraction of the H atom by a transition-metal complex, such as a metal oxo complex, or by an electrophilic radical species generated through homolytic dissociation of an oxidant, occurs. Both the metal complexes and electrophilic radical species capable of H-atom abstraction are electron deficient; thus, electron-rich C–H bonds are cleaved faster than electron-poor C–H bonds under the same conditions. In addition to these electronic effects on selectivity, the steric properties of the reagent reacting with the C–H bonds can affect the regioselectivity of the C–H bond functionalization.
C–H bond functionalization reactions that proceed via the formation of C-centered radicals offer some advantages over such reactions occurring through heterolytic C–H bond cleavage. In particular, functional groups that are highly reactive toward heterolytic bond cleavage, for example, O–H, N–H, and acidic C–H bonds, usually do not react under conditions that lead to homolytic C–H bond cleavage because these bonds are stronger than tertiary alkyl C–H bonds and are electronically deactivated. Thus, amides, carboxylic acids, ketones, nitriles, and tertiary alcohols are all well tolerated for homolytic cleavage of a C–H bond.
Compared to a simple hydrocarbon, such as n-decane (26), in which the properties of the secondary C–H bonds are similar to each other, natural products contain C–H bonds in a variety of steric and electronic environments. For example, in bile acid 27, there are a total of 45 different C–H bonds (Figure 10); 15 are primary, 20 are secondary, and 10 are tertiary C–H bonds. The presence of functional groups influences the electronic properties of the neighboring C–H bonds and further modulates the reactivity of these bonds toward electrophilic H-atom abstracting agents. For example, in certain bile acids such as 27, electron-withdrawing OAc groups deactivate the C–H bonds in the cyclic portion of the molecule, thereby leading to selective oxidation of 27, for example, at the C25 position, by a ruthenium catalyst.[34]
Figure 10.
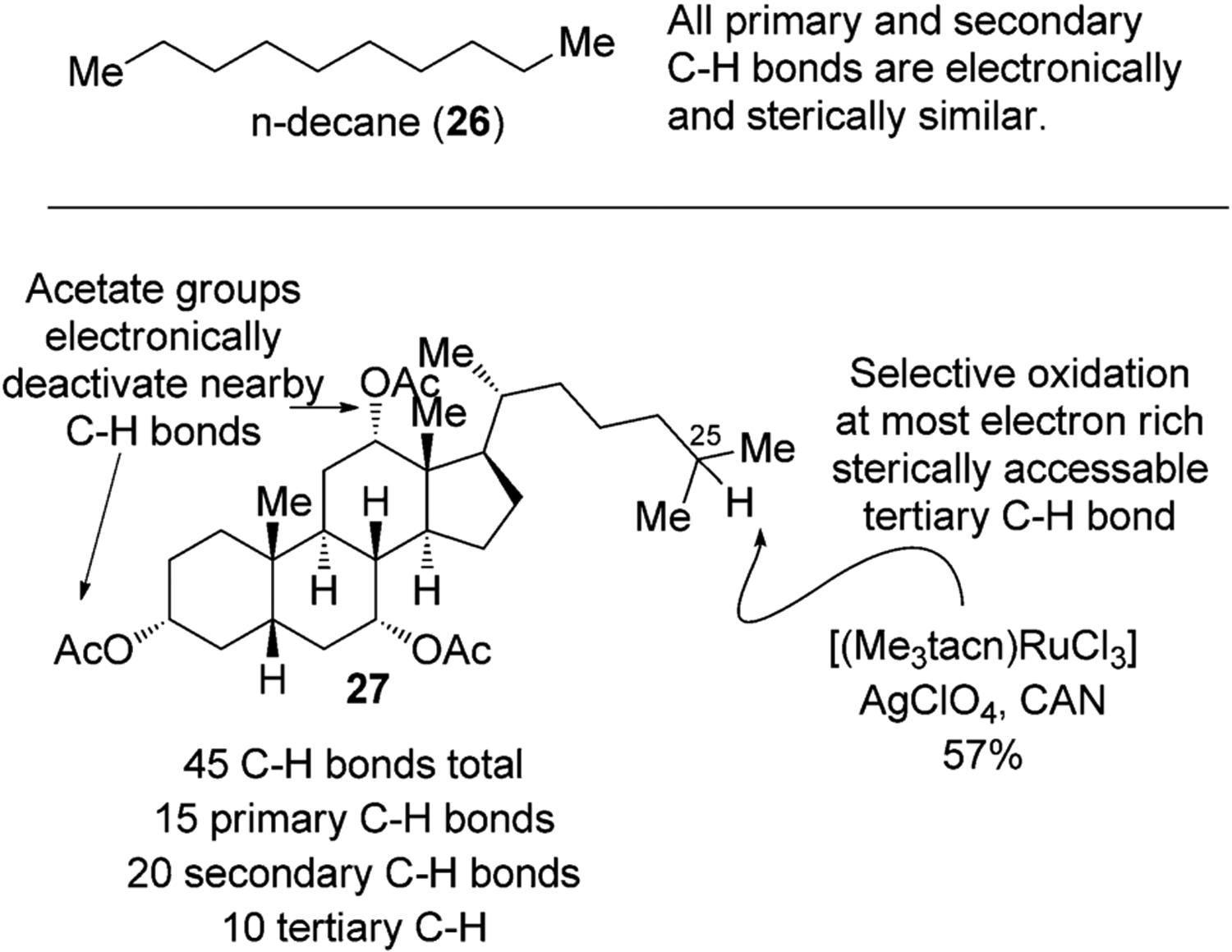
Different types of C–H bonds in natural products allow selective functionalization. CAN=ceric ammonium nitrate.
Hartwig and co-workers recently reported an iron-catalyzed C–H azidation method in which Zhdankin’s reagent 28 is the source of an azide, and iron(II) acetate with a pybox ligand L1 is the catalyst (Figure 11).[35,36] This reaction proceeds with excellent regioselectivity, favoring azidation at electron-rich, tertiary C–H bonds. For example, azidation of betulinic acid derivative 29a, which contains an electron-withdrawing alkoxycarbonyl group, did not give any azidation product, but a structurally similar betulin derivative 30a, which contains a less electron-withdrawing acetoxymethyl group at the position of the ester, gave azide 30b in good yield.
Figure 11.
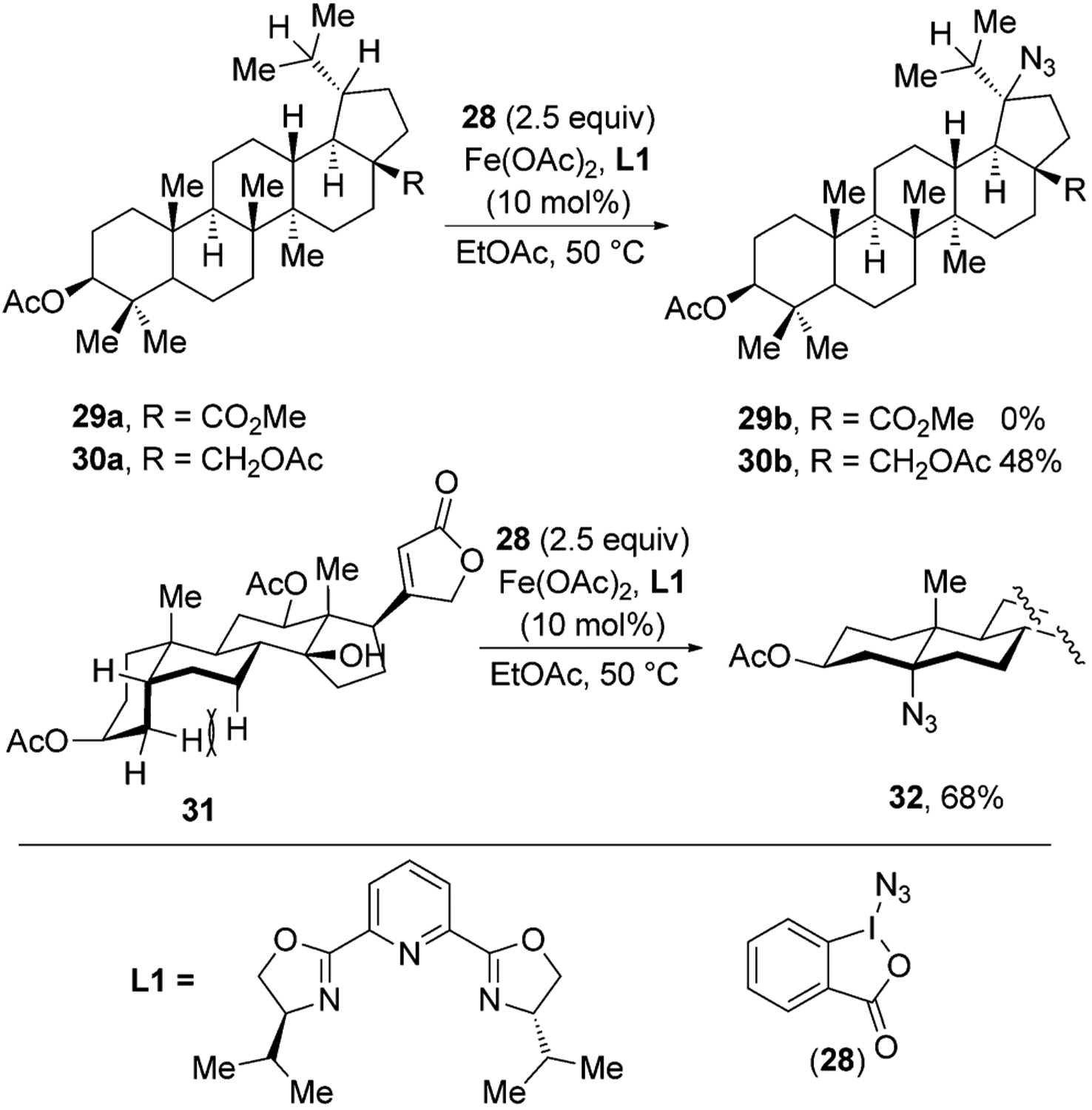
Fe-catalyzed C–H azidation of natural products.
C–H bonds at the junction of a cis-decalin ring system were particularly reactive toward the azidation process. For example, a derivative of digoxigenin gave the azide 32 in 68 % yield. This selectivity toward the cis-decalin feedstock occurs because C–H bonds at the cis-decalin ring junction are sterically more accessible and weaker than C–H bonds at the ring junction of the trans-decalin unit. C–H bonds at the ring junction of the cis-decalin motif are weaker than those at the trans junction because H-atom abstraction from both cis- and trans-decalin C–H bonds generate the same radical, but the cis-decalin is less stable than the trans isomer. In the case of the cis-decalin ring system, however, H-atom abstraction relieves the strain due to the unfavorable syn-pentane interactions in cis-decalin. The azidation process tolerates ketone, ester, amide, tertiary alcohol, and epoxide functional groups.
Consistent with this argument, transition-metal oxo complexes have been used for the oxidative functionalization of unactivated C–H bonds in natural products. The Groves group reported a series of functionalizations of C–H bonds in which a manganese-salen or manganese-porphyrin oxo complex abstracts a hydrogen atom from a C–H bond.[37–39] Trapping of the carbon-centered radical then occurs through halide or azide transfer from manganese complexes. For example, the manganese-salen complex 35 catalyzed azidation of artemisinin derivative 33 to form its azido analogue 34.[37] Similar to the azidation reaction reported by Hartwig and colleagues, this reaction selectively functionalized the C–H bond that is farthest from electron withdrawing functional groups. Manganese porphyrin catalyst 37 also catalyzes C–H functionalization reactions. For example, azido, chloro, and fluoro derivatives of 36 a have been prepared through direct C–H functionalization of C2–H bonds (Figure 12).
Figure 12.

Mn-catalyzed C–H azidation and halogenation of natural products. TBAF=tetrabutylammonium fluoride, TMP=tetramesityl porphyrin.
One of the attractive features of using metal oxo complexes for hydrogen-atom abstraction is that less-hindered secondary C–H bonds can be functionalized over weaker and more electron-rich tertiary C–H bonds by tuning the steric properties of the ligands. For example, the White group has shown that increasing the steric bulk of the ligand in the iron catalyst 25 through the formal addition of trifluoroaryl substituents on the ligand changes the regioselectivity of C–H bond oxidation in artemisinin from formation of a tertiary alcohol through oxidation of a tertiary C–H bond to formation of a ketone through oxidation of a secondary C–H bond (Figure 13).[40]
Figure 13.
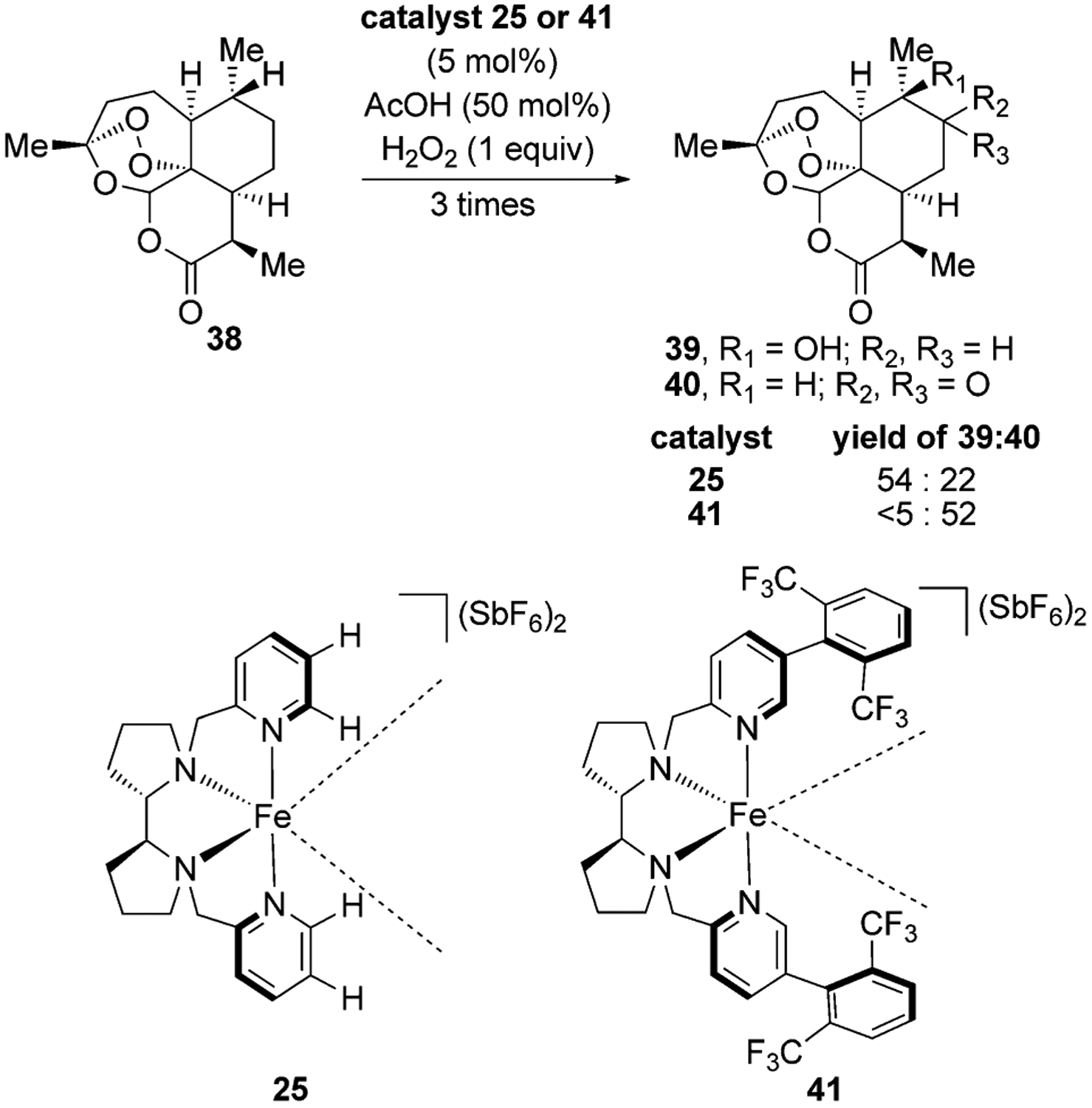
Catalyst-controlled C–H oxidation of artemisinin.
A distinct class of metal oxo complexes that catalyze oxidative functionalization of unactivated C–H bonds are polyoxometallates. These complexes absorb visible light to produce the active form of the catalyst for hydrogen-atom abstraction.[41] For example, the decatungstate anion (W10O324−) absorbs light between 35–400 nm and cleaves C–H bonds to generate C-centered radicals.42–43 In one recent example, C-centered radicals generated in this fashion were trapped by N-fluorobenzenesulfonamide (NFSI) to obtain flourinated derivatives of several terpenes (Figure 14).43
Figure 14.
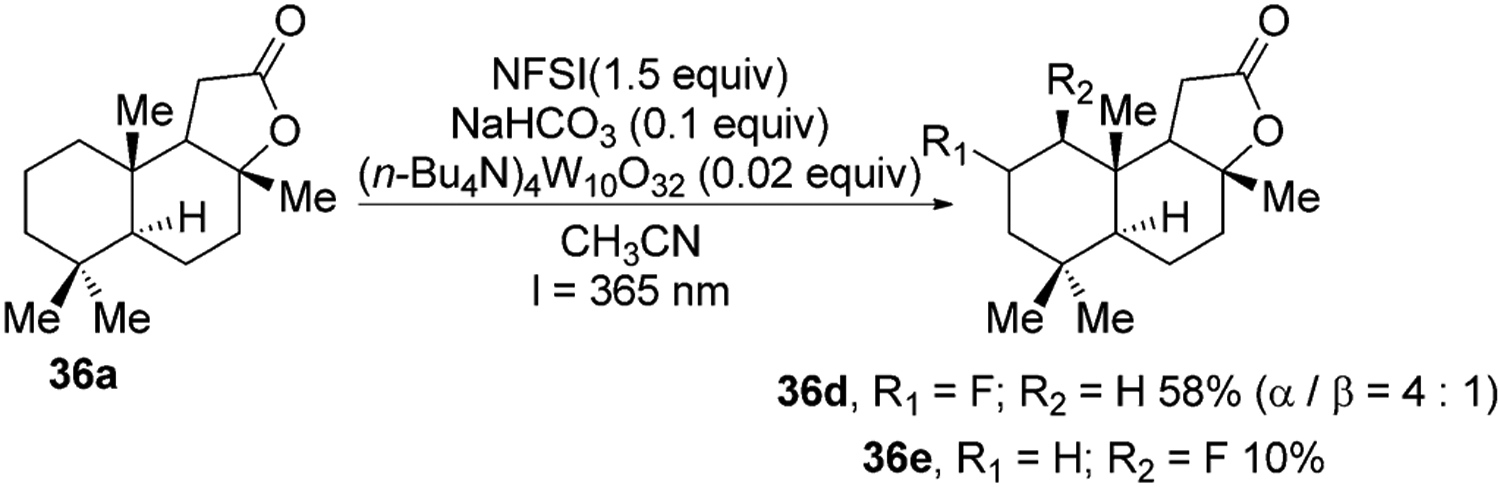
Decatungstate anion mediated C–H fluorination of sclareolide.
3.2. Insertion Reactions
Insertions of carbene and nitrene groups catalyzed by transition-metal complexes also have led to the functionalization of natural products. An important feature of many of these reactions is the retention of configuration that occurs at the C–H bond into which the carbene or nitrenes inserts. This retention contrasts with the formation of racemic product from reactions occurring via free radicals. For example, Du Bois and co-workers have shown that Rh2(esp)2 catalyzes the selective insertion of a nitrenes derived from DsfNH2 (Dsf = 2,6-difluorophenylsulfonyl) into the electron-rich tertiary C–H bonds of natural products.[44] Nitrene insertion occurred selectively into the tertiary C–H bond in a cyclohexamide derivative that is farthest from the electron-withdrawing ketone and glutarimide groups to give sulfonamide 43 as single stereoisomer (Figure 15).
Figure 15.
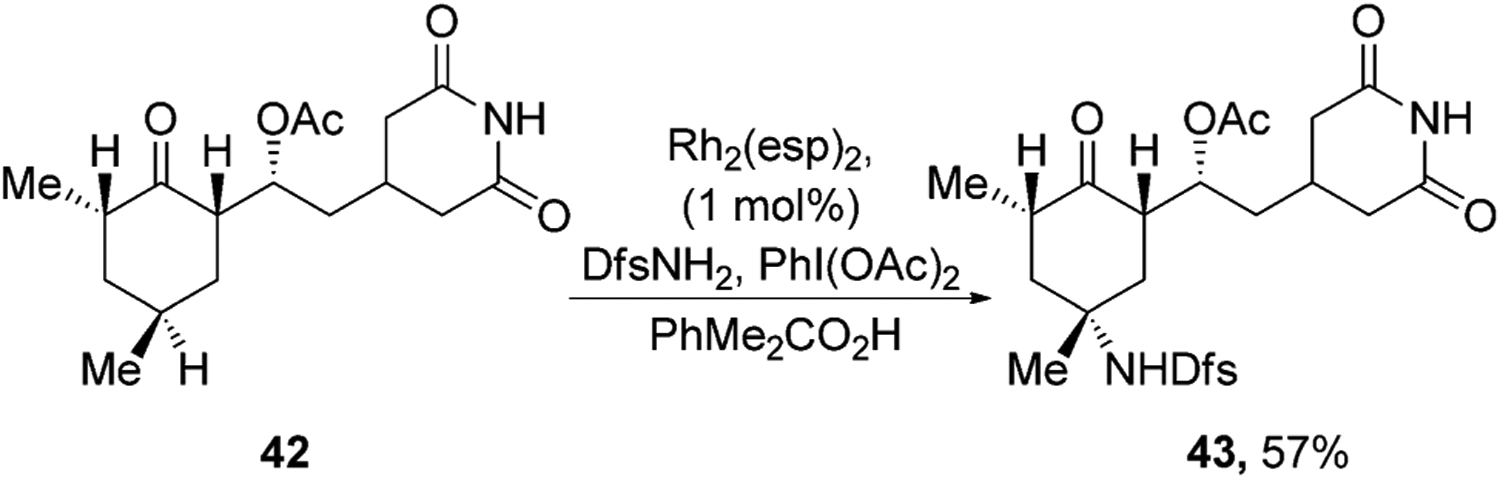
Rh-catalyzed nitrene insertion. Dsf=2,6-difluorophenylsulfonyl, Rh2(esp)2=Bis[rhodium(α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid)].
4. Concluding Remarks and Future Directions
Although limitations arising from the selectivity and reactivity of C–H bonds make synthetic chemistry occurring at these positions challenging, the benefits of learning how to control the functionalization of unactivated C–H bonds are high. In this Minireview, we have discussed the principal strategies used to conduct the functionalization of C–H bonds in natural products with transition-metal catalysts. As demonstrated by the examples discussed here, regioselective functionalization of primary, secondary, and tertiary C–H bonds has been achieved in several remarkable cases by using directing groups and the innate reactivity of particular C–H bonds (e.g., electron-rich or sterically hindered C–H bonds) in natural products. However, significant challenges remain, despite major advances in controlling the regioselectivity of C–H bond functionalization with transition-metal catalysts. For example, no systems are currently available for functionalizing primary C–H bonds in complex molecules without the aid of directing groups or for functionalizing sterically hindered C–H bonds over sterically accessible ones. Likewise, control of stereochemistry during C–H bond functionalization, particularly in complex molecules, is an unsolved problem. Most natural products have at least one stereogenic center. Thus it is necessary to develop diastereoselective reactions to obtain single isomers of natural product derivatives. Insertion reactions of nitrenes is one example in which C–H bond functionalization proceeds with high stereoselectively; however, this reaction yields only the stereoisomer resulting from retention of configuration.
In addition to the issues of selectivity, the simple availability of natural products often limits the derivatization of natural products. Highly functionalized natural products are often isolated in small quantities from their natural sources and are too expensive for academic laboratories to obtain in sufficient quantities from commercial sources to explore their reactivities. Thus, close collaboration between groups working on biosynthesis and C–H bond functionalization, as well as between industrial and academic research laboratories, would be valuable for producing new classes of molecules.
Acknowledgements
We thank the Department of NIH (GM-58108 and GM 11581201) for current support of our studies on the functionalization of C-H bonds.
Biographies

Rashad Karimov is an Assistant Professor of Chemistry at Auburn University. He received his B.S. degree from Lomonosov Moscow State University and his MS degree from University of Minnesota, Duluth. He completed his Ph.D. in 2014 at the Tri-Institutional Chemical Biology Program under the direction of Prof. David Gin and Prof. Derek Tan, studying the chemical synthesis of immunoadjuvant natural products. Prior to his current position, he was a postdoctoral fellow in the laboratory of Prof. John Hartwig, where he worked on the C–H functionalization of natural products.

Prof. Hartwig is the Henry Rapoport Professor at the University of California, Berkeley. His group seeks to discover new reactions catalyzed by transition-metal complexes and to reveal new reaction mechanisms. He recently authored the textbook Organotransition Metal Chemistry: From Bonding to Catalysis. He is the 2006 recipient of the ACS Award in Organometallic Chemistry and the 2013 ACS H.C. Brown Award in Synthetic Methods. He was elected to the National Academy of Sciences in 2012.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
The ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201710330.
This article is part of the Special Issue to commemorate the 350th anniversary of Merck KGaA, Darmstadt, Germany. More articles can be found at http://doi.wiley.com/10.1002/anie.v57.16.
References
- 1.Newman DJ, Cragg GM, J. Nat. Prod 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ, Cragg GM, J. Nat. Prod 2012, 75, 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bioactive Compounds from Natural Sources: Natural Products as Lead Compounds in Drug Discovery, 2. Aufl. (Hrsg.: C. Tringali), CRC Press, Boca Raton, 2012, S. 299–451. [Google Scholar]
- 4.Ganesan A, Curr. Opin. Chem. Biol 2008, 12, 306–317. [DOI] [PubMed] [Google Scholar]
- 5.Koehn FE, Carter GT, Nat. Rev. Drug Discovery 2005, 4, 206–220. [DOI] [PubMed] [Google Scholar]
- 6.Butler MS, J. Nat. Prod 2004, 67, 2141–2153. [DOI] [PubMed] [Google Scholar]
- 7.Carlson EE, ACS Chem. Biol 2010, 5, 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luch A, Molecular, clinical, and environmental toxicology, Birkhäuser, Boston, 2009. [PubMed] [Google Scholar]
- 9.Pasternak GW, Pan YX, Pharmacol. Rev 2013, 65, 1257–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hudlicky T, Butora G, Feamley SP, Gum AG, Stabile MR, A Historical Perspective of Morphine Syntheses (Studies in Natural Products Chemistry, Vol. 18) (Ed.: Atta-ur-Rahman), Elsevier, Amsterdam, 1996, pp. 43–154. [Google Scholar]
- 11.Morrison KC, Hergenrother PJ, Nat. Prod. Rep 2014, 31, 6–14. [DOI] [PubMed] [Google Scholar]
- 12.Li JWH, Vederas JC, Science 2009, 325, 161–165. [DOI] [PubMed] [Google Scholar]
- 13.Bauer RA, Wurst JM, Tan DS, Curr. Opin. Chem. Biol 2010, 14, 308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arndtsen BA, Bergman RG, Mobley TA, Peterson TH, Acc. Chem. Res 1995, 28, 154–162. [Google Scholar]
- 15.Hartwig JF, J. Am. Chem. Soc 2016, 138, 2–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. A recent comprehensive review of non-enzymatic derivatization of natural products:; Shugrue CR, Miller SJ, Chem. Rev 2017, 117, 11894–11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartwig JF, Larsen MA, ACS Cent. Sci 2016, 2, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyons TW, Sanford MS, Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang M, Zhang YF, Jie XM, Zhao HQ, Li G, Su WP, Org. Chem. Front 2014, 1, 843–895. [Google Scholar]
- 20.Jun CH, Hong JB, Kim YH, Chung KY, Angew. Chem. Int. Ed 2000, 39, 3440–3442; Angew. Chem. 2000, 112, 3582–3584. [DOI] [PubMed] [Google Scholar]
- 21.Desai LV, Hull KL, Sanford MS, J. Am. Chem. Soc 2004, 126, 9542–9543. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Yan GB, Ren Z, Dong GB, Nat. Chem 2015, 7, 829–834. [DOI] [PubMed] [Google Scholar]
- 23.Wang DH, Wasa M, Giri R, Yu JQ, J. Am. Chem. Soc 2008, 130, 7190–7191. [DOI] [PubMed] [Google Scholar]
- 24.Kleiman JP, Dubeck M, J. Am. Chem. Soc 1963, 85, 1544–1545. [Google Scholar]
- 25.Cope AC, Siekman RW, J. Am. Chem. Soc 1965, 87, 3272–3273. [Google Scholar]
- 26.Fahey DR, J. Organomet. Chem 1971, 27, 283–292. [Google Scholar]
- 27.Baldwin JE, Jones RH, Najera C, Yus M, Tetrahedron 1985, 41, 699–711. [Google Scholar]
- 28.Schönecker B, Zheldakova T, Lange C, Günther W, Görls H, Bohl M, Chem. Eur. J 2004, 10, 6029–6042. [DOI] [PubMed] [Google Scholar]
- 29.See YY, Herrmann AT, Aihara Y, Baran PS, J. Am. Chem. Soc 2015, 137, 13776–13779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Breslow R, Angew. Chem. Int. Ed 2000, 39, 2692–2694; Angew. Chem. 2000, 112, 2804–2806. [DOI] [PubMed] [Google Scholar]
- 31.Simmons EM, Hartwig JF, Nature 2012, 483, 70–73. [DOI] [PubMed] [Google Scholar]
- 32.García-Granados A, López PE, Melguizo E, Parra A, Simeó Y, J. Org. Chem 2007, 72, 3500–3509. [DOI] [PubMed] [Google Scholar]
- 33.Bigi MA, Reed SA, White MC, J. Am. Chem. Soc 2012, 134, 9721–9726. [DOI] [PubMed] [Google Scholar]
- 34.McNeill E, Du Bois J, Chem. Sci 2012, 3, 1810–1813. [Google Scholar]
- 35.Karimov RR, Sharma A, Hartwig JF, ACS Cent. Sci 2016, 2, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma A, Hartwig JF, Nature 2015, 517, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang XY, Bergsten TM, Groves JT, J. Am. Chem. Soc 2015, 137, 5300–5303. [DOI] [PubMed] [Google Scholar]
- 38.Liu W, Groves JT, J. Am. Chem. Soc 2010, 132, 12847–12849. [DOI] [PubMed] [Google Scholar]
- 39.Liu W, Huang XY, Cheng MJ, Nielsen RJ, Goddard WA, Groves JT, Science 2012, 337, 1322–1325. [DOI] [PubMed] [Google Scholar]
- 40.Gormisky PE, White MC, J. Am. Chem. Soc 2013, 135, 14052–14055. [DOI] [PubMed] [Google Scholar]
- 41.Hill CL, J. Mol. Catal. A 2007, 262, 2–6. [Google Scholar]
- 42.Tzirakis MD, Lykakis IN, Orfanopoulos M, Chem. Soc. Rev 2009, 38, 2609–2621. [DOI] [PubMed] [Google Scholar]
- 43.Halperin SD, Fan H, Chang S, Martin RE, Britton R, Angew. Chem. Int. Ed 2014, 53, 4690–4693; Angew. Chem. 2014, 126, 4778–4781. [DOI] [PubMed] [Google Scholar]
- 44.Roizen JL, Zalatan DN, Du Bois J, Angew. Chem. Int. Ed 2013, 52, 11343–11346; Angew. Chem. 2013, 125, 11553–11556. [DOI] [PMC free article] [PubMed] [Google Scholar]