

This review provides a state-of-the-art description of the performance of Sanger cycle sequencing of the 16S rRNA gene for routine identification of bacteria in the clinical microbiology laboratory. A detailed description of the technology and current methodology is outlined with a major focus on proper data analyses and interpretation of sequences. The remainder of the article is focused on a comprehensive evaluation of the application of this method for identification of bacterial pathogens based on analyses of 16S multialignment sequences.
KEYWORDS: 16S rRNA, bacteria, cycle sequencing, identification
SUMMARY
This review provides a state-of-the-art description of the performance of Sanger cycle sequencing of the 16S rRNA gene for routine identification of bacteria in the clinical microbiology laboratory. A detailed description of the technology and current methodology is outlined with a major focus on proper data analyses and interpretation of sequences. The remainder of the article is focused on a comprehensive evaluation of the application of this method for identification of bacterial pathogens based on analyses of 16S multialignment sequences. In particular, the existing limitations of similarity within 16S for genus- and species-level differentiation of clinically relevant pathogens and the lack of sequence data currently available in public databases is highlighted. A multiyear experience is described of a large regional clinical microbiology service with direct 16S broad-range PCR followed by cycle sequencing for direct detection of pathogens in appropriate clinical samples. The ability of proteomics (matrix-assisted desorption ionization-time of flight) versus 16S sequencing for bacterial identification and genotyping is compared. Finally, the potential for whole-genome analysis by next-generation sequencing (NGS) to replace 16S sequencing for routine diagnostic use is presented for several applications, including the barriers that must be overcome to fully implement newer genomic methods in clinical microbiology. A future challenge for large clinical, reference, and research laboratories, as well as for industry, will be the translation of vast amounts of accrued NGS microbial data into convenient algorithm testing schemes for various applications (i.e., microbial identification, genotyping, and metagenomics and microbiome analyses) so that clinically relevant information can be reported to physicians in a format that is understood and actionable. These challenges will not be faced by clinical microbiologists alone but by every scientist involved in a domain where natural diversity of genes and gene sequences plays a critical role in disease, health, pathogenicity, epidemiology, and other aspects of life-forms. Overcoming these challenges will require global multidisciplinary efforts across fields that do not normally interact with the clinical arena to make vast amounts of sequencing data clinically interpretable and actionable at the bedside.
INTRODUCTION
Nucleic acid sequencing of the bacterial 16S rRNA gene (here designated 16S) has been used for several decades to identify clinical and environmental isolates and to assign phylogenetic relationships. Carl Woese and George Fox pioneered comparisons of 16S sequence data prior to the development of DNA sequencing methods to perform complex phylogenetic studies, initially using it to classify methanogenic bacteria and to describe the Archaebacterium Halobacterium volcanii (1–3). Subsequent accumulation of large amounts of small-subunit rRNA gene sequence data (16S of bacteria and 18S rRNA of eukaryotes) allowed other phylogenetic reconstruction studies, which established the three fundamental domains (Archaea, Bacteria, and Eucarya) within the universal tree of life (4). 16S remains the most widely used stable target for bacterial identification and genetic evolutionary studies, because other highly conserved genes have not been as thoroughly studied. However, various 16S regions and/or a longer gene sequence (i.e., up to ∼1,060 bp) are required for definitive identification of many bacterial genera and/or species as outlined here and in the recently published revision of CLSI MM-18-A2 (5).
Definitive identification of human bacterial pathogens using targeted partial 16S cycle sequencing has been used in clinical microbiology laboratories for the past ∼30 years (6–15). The advent of commercial capillary gel genetic analyzers and the availability of public database repositories containing a large amount of 16S sequence data made this feasible. GenBank (NCBI) currently contains >29,000,000 entries for 16S sequences of various lengths and quality derived from a diverse range of bacteria recovered from various clinical/environmental sources (https://www.ncbi.nlm.nih.gov/genbank); many of these entries contain just 16S sequences, but increasingly partial or entire genomes containing complete 16S sequences are being deposited. Several excellent reviews of the impact and utility of this method on clinical microbiology practices were published more than a decade ago and outlined the labor intensity, expense, and technological constraints of Sanger sequencing methods available at that time (6, 11, 16). Substantial advances have subsequently occurred in the efficiency of cycle sequencing and the standardized interpretation of 16S sequence data for assigning a definitive bacterial genus- and/or species-level identification. Whereas diagnostic laboratories may have previously referred a clinical isolate to an academic core facility for partial targeted 16S sequencing analysis, advances in the efficiency of PCR technology, sequencing instrumentation, and 16S sequence interpretation made its performance in-house possible (17). Therefore, genomic identification using variable regions within the 16S gene for species-specific differentiation has been widely used in the pre-matrix-assisted desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) era for precise identification of a wide range of clinically relevant bacterial pathogens where phenotypic methods could not provide an adequate level of discrimination or gave discrepant results (18–24). Many larger facilities also routinely use 16S universal PCR or broad-range PCR and cycle sequencing to identify amplified bacterial DNA directly from clinical isolates or samples (25–27).
MALDI-TOF MS has recently supplanted routine phenotypic tests to a large degree as the routine identification method used for pathogen identification and revolutionized the ability of clinical microbiology laboratories to rapidly identify a much wider diversity of microorganisms (23, 28, 29). A combination of phenotypic and genotypic tests is commonly performed to arrive at specific bacterial identification, including a Gram stain, rapid biochemical tests, MALDI-TOF MS, and, where necessary, genetic analysis of 16S or other gene targets. Ready access to isolate biorepositories storing clinical strains characterized by phenotypic tests and 16S sequencing are essential for the ongoing expansion of existing MALDI-TOF MS databases to include more unusual pathogens (18, 19, 21–24, 29–32). Genetic analyses of 16S sequences either through Sanger cycle or next-generation sequencing methods will continue to be an important diagnostic technology alongside current proteomic methods, which will be addressed (11, 25, 33).
This review describes the state-of-the-art approach for performing fast 16S cycle sequencing (Sanger) for routine identification of bacterial pathogens in the clinical microbiology laboratory, building upon a comprehensive previous Clinical Microbiology Reviews article by Clarridge (16). Because of the widespread use of 16S for isolate identification and its accelerating use for metagenomics and microbiome studies, we provide a detailed analysis of the identified limitations of using this target for these various applications. Gaps remain in the currently available sequence database(s) that limits analysis and interpretation of 16S sequence data. GenBank (NCBI) holds the most sequences but has no curation in place to ensure correct sequence and annotation content. Other more curated databases are often not representative of the microbial diversity, because they mainly focus on some type and reference strains. Longitudinal clinical experience from a large integrated regional clinical microbiology is described that routinely used both 16S sequencing along with MALDI-TOF MS for the identification of bacterial pathogens. A detailed analysis of the current utility of using the 16S target for bacterial identification is presented based on analysis of sequence alignments done to revise CLSI MM-18 A2 (5), and the ability of 16S to discriminate clinically relevant genus/species is outlined in Tables 3 to 12. Aside from primer selection, the main factors limiting 16S discriminatory ability for clinical/environmental bacterial isolates is the current lack of available sequence data and a high degree of 16S homology between several related genera and/or species. The current and future use of proteomics and next-generation sequencing is discussed as a replacement for targeted 16S rRNA gene capillary cycle sequencing.
THE 16S rRNA GENE AND PRIMER SELECTION
The prokaryotic rRNA genes specifically include 5S, 16S, and 23S and intergenic regions (34–36). The 16S rRNA gene is about ∼1,500 nucleotides long (∼1.5 kb, although this is an average and some organisms can have 16S sequences that are shorter or longer) and is part of the 30S small subunit of prokaryotic ribosomes that binds to the Shine-Dalgarno sequence at the 3′ end (36–40). 16S rRNA has several functions, including a structural role as well as being crucial to protein synthesis. Along with 23S, it provides a scaffold to assist with the binding of the 50S and 30S ribosomal subunits, as well as defining the ribosomal protein positions (34–36). The 3′ end of 16S RNA also binds to the S1 and S21 proteins, known to be involved in initiation of protein synthesis by RNA-protein cross-linking (41). All microorganisms have at least one copy of 16S, making it ubiquitous, and as it is highly conserved and evolves slowly, it is the most widely used single target for phylogenetic studies of bacteria and archaea (3, 4, 42). Multiple sequences of the 16S can exist with a single bacterium, and some copies may differ (43, 44). Genomic sequencing studies also show that many bacterial species have intragenic heterogeneity (i.e., harbor multiple 16S gene copies and polymorphisms between these copies) that allow interspecies subtyping via partial or full sequencing of 16S (45, 46). Interspecies discrimination based on intragenic heterogeneity has been demonstrated for a variety of human pathogens, including Neisseria (47, 48), Haemophilus (49), Salmonella (50), and Listeria (51) species. Horizontal transfer of 16S also occurs, albeit infrequently and only at the intragenus or intraspecies level (52). Although this is much more restricted than bacterial horizontal transfer of operational genes (i.e., enzyme-encoding genes), some investigators question whether 16S should be the only target used for identification or phylogenetic purposes. However, 16S has been extensively studied and applied to establish a species description, species taxonomy, and phylogenetic relationships. Therefore, 16S is the molecular target of choice for genus- or species-level identification in the clinical laboratory because of its ubiquitous nature amongst bacteria and archaea (∼10% to 15%) and the abundance of sequence data compared to that for other targets (5).
The 16S gene contains mosaics of sequence that range from highly conserved, variable, and hypervariable regions, as illustrated previously by Baker and colleagues in their schematic of the Escherichia coli one (Fig. 1) (39). Within certain stretches, 16S provides genus- and/or species-specific signatures that enable accurate identification depending on the targeted gene regions for a particular bacterium/microorganism group(s). Universal 16S primers can be designed to target the conserved regions of 16S, of which some motifs are shared across the entire kingdom (“eubacterial primers”). In laboratory practice, these primers most often target the first ∼500 bp of the small ribosomal subunit gene, because analysis of the V1-V3 regions is considered sufficient to allow accurate identification of most specific genera/species; hence, most 16S sequences currently deposited in public databases correspond to this part of the gene. Inaccurate biological conclusions, however, are derived from experiments using suboptimal 16S primer design because of nonamplification and/or detection of some critical genera and/or species (i.e., some species or groups are missed entirely or proportionally misrepresented within the population), and there will be a significant loss of taxonomic classification by shorter 16S sequences (53). It is important to remember that forward (F; defined as oligonucleotide sequence that is complementary to the antisense strand of double-stranded DNA) and reverse (R; defined as oligonucleotide sequence that is complementary to the sense strand of double-stranded DNA) primer sets targeting the 16S V1-V3 regions were historically designed for environmental microbiome community analysis (54–56) and not for clinical isolates (Fig. 1). More rigorous identification of human pathogens requires the use of other 16S specific F/R primer pairs, as previously reported (5, 57). Although previous analyses show it is difficult to design primers to universally detect all prokaryotic 16S rRNA gene sequences (39, 58, 59), this has been achieved (SmartGene patent number EP1863922B1). Optimal primer design can also mitigate the specificity issues of using standard 16S primers for broad-range PCR/sequencing or microbiome analyses, which decrease amplification of potential contaminants and cross-reactivity with common human host DNA sequences (60, 61).
FIG 1.

E. coli 16S rRNA gene and locations of conserved and variable regions.
Partial targeted sequencing of the 16S V1-V3 region (i.e., first ∼500 bp), however, may not provide enough coverage of variable regions to allow unambiguous species-level identification of a number of important human bacterial pathogens, as is outlined in detail by the recently revised Clinical and Laboratory Standards Institute (CLSI) guideline MM-18-A2 (5). Initial interpretive criteria published by the Clinical and Laboratory Standards Institute (CLSI MM-18) for the identification of a human bacterial pathogen by partial 16S DNA target sequencing recommended the use of specific bacterial primer pairs (i.e., 4F, 27F, 534R, and 801R) that target the first ∼500 bp of the gene (i.e., V1-V3 region) (62). The recently published CLSI MM-18-A2 update also indicates a longer section of 16S sequence across several gene regions (V1-V6) (∼1,060 bp, covering the V5 and V6 variable regions), or even the entire gene (1,540 bp) may need to be analyzed within many genera to achieve a species-level identification (5). 16S multisequence alignments within all major bacterial pathogen genera/species were analyzed across the various gene regions to make the recommendations outlined by CLSI MM-18-A2 for bacterial identification using a shorter (V1-V3) or longer sequence (5). This work highlighted that an international standard should be developed for the use of 16S primers for various clinical applications, particularly for the accurate identification of human pathogens, or human microbiome analyses that should include the precise limitations of particular published 16S primer pairs for this purpose.
Use of the 16S target alone may not be sufficient to reliably identity many human clinical pathogens for several reasons, such as (i) high genetic similarity within specific microorganisms or groups, (ii) the presence of variable copy numbers of 16S rRNA genes with sequence variation in their genomes (45, 46, 63), and (iii) the lack of 16S sequence information currently available in published data repositories. This is not surprising, since the vast majority of microorganism/microorganism groups have yet to be identified or classified (i.e., only an estimated 1% of all microbes have been discovered) (64, 65). In any case, 16S sequencing will yield a result allowing an approximate organism classification against those present in the database.
Several other genetic targets have been used for research purposes for improved identification of microorganism/microorganism groups and most commonly include the conserved genes in the ribosomal region (rpoA, rpoB, rpoC, and rpoD) (66–68), the spacer region 16S-23S (69), DNA metabolic enzymes (gyrA and gyrB) (70–72), DNA repair genes (recA and recN) (73, 74), the elongation factor Tu gene (tuf) (75–78), superoxide dismutase (sodA) (75, 79), and the chaperonin family of proteins (cpn60) (75, 80). Other, more rarely reported, genetic targets, such as dnaJ, may also be efficient microbial identification targets (81–84). These alternative gene targets, like 16S, have functionally conserved regions with flanking regions of variability, making them ideal for potentially closer separation of related species. Primer selection for alternate targets will often not be universal or “eubacterial” and must be carefully designed to amplify and sequence the intended microorganism/microorganism group. As such, alternate target analysis aside from the rpoB gene and the spacer region 16S-23S has been used mainly for research studies to distinguish specific genera/species (85–87). Because of the rather limited amount of data available for most alternate targets with a number of species not adequately covered (compared to 16S), one would have to make the necessary efforts to build, populate, and validate an in-house database before clinical use. Clinical laboratories should not rely on alternate target analysis alone for reporting identification on clinical isolates unless they have developed a comprehensive gene target database and subsequently done extensive preclinical validation of this bioinformatics tool.
PRINCIPLES OF SANGER AND PYROSEQUENCING METHODS
Partial or complete target sequencing of 16S is commonly performed in the clinical laboratory using either chain termination (Sanger) or pyrosequencing chemical analysis. Figure 2 gives a schematic outline of a chain termination and a pyrosequencing reaction (88, 89).
FIG 2.
Schematic outline of a chain termination and pyrosequencing reaction.
Chain Termination Sequencing
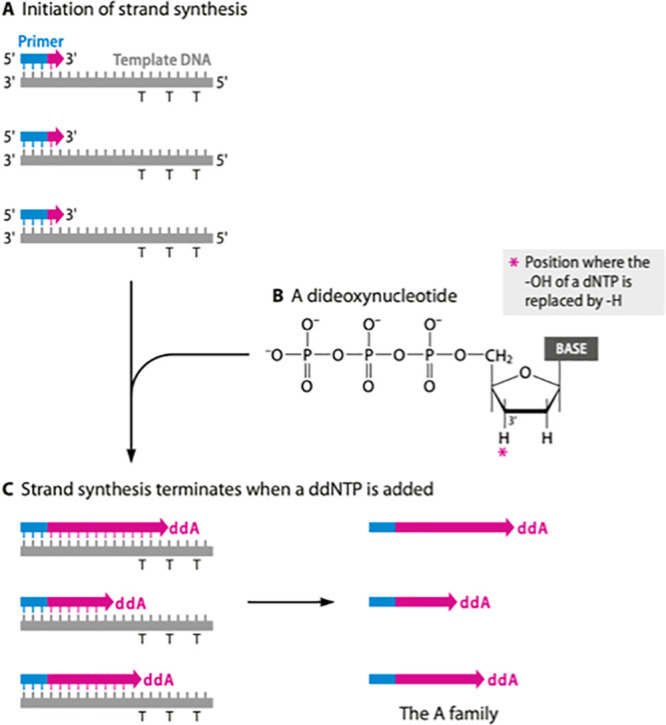
During a Sanger procedure, PCR is initially performed using short oligonucleotide 16S primers to synthesize complementary amplicons to the template (90, 91). Secondary cycle sequencing of the amplicon involves a thermostable DNA polymerase, a primer designed to anneal to the template nucleic acid, and small amounts of the required double-stranded DNA template. Four chain-terminating dideoxynucleoside triphosphates (ddNTPS; ddATP, ddTTP, ddGTP, and ddCTP) labeled with individual fluorescent markers of different spectra are also added to the reaction mix at a lower concentration than the deoxyribonucleotide triphosphates (DNTPS; dATP, dTTP, dGTP, and dCTP). Synthesis of DNA by DNA polymerase incorporates ddNTPs, causing termination of sequence elongation, as these bases lack a 3′-hydroxyl group needed to polymerize to the next nucleotide normally provided by a dNTP base. Each incorporated ddNTP is in a chain-terminated fragment at the same position as the dNTP base in the DNA template.
The cycle sequencing reaction successively builds up DNA strands of different lengths that have a different fluorescently labeled ddNTP (A, T, C, or G) (i.e., dye terminators) at the 3′ end due to many chain termination events (88). BigDye (Applied Biosystems Inc., Thermo Fisher Scientific, Foster City, CA) terminators use single energy transfer molecules, which include an energy donor and acceptor (i.e., dichlororhodamine or rhodamine) dye connected by a highly efficient energy transfer linker (91). These dyes have significantly less overlap at their maximum excitation wavelength than conventional rhodamine dyes, so that sequencing products are produced with a cleaner fluorescent signal and improved base-calling accuracy, particularly at longer read lengths (91).
The single-stranded DNA (ssDNA) fragment mixture generated by the fluorescence cycle sequencing reaction is loaded by electrokinetics into a polyacrylamide (PA; acrylamide monomers [CH2=CH-CO-CH2] cross-linked with N,N′-methylenebisacrylamide or bis unit [CHS=CH-CO-NH-CH2-NH-CO-CH=CH2]) gel capillary housed in an automated genetic analyzer, where the fragments are separated by electrophoresis and sequentially read by a fluorometric detector to generate an electropherogram trace of the derived DNA sequence (92, 93). Sequences between 100 and ∼1,300 nucleotides long can be resolved into a series of bands on a PA gel even when ssDNA fragments differ by only one nucleotide (94). Automated capillary sequencing genetic analyzers typically house four or more thin-column capillaries (0.1-mm diameter, 50 to 80 cm long) filled with PA; the optimal PA concentration in the 6% to 7% gel matrix is an acrylamide/bis ratio of 19:1 for resolution of ssDNA fragments between 100 and 750 nucleotides long (95). Longer sequence reads may be obtained by altering the pore size of the gel by using a different PA concentration and PA/bis ratio (92).
Pyrosequencing
Pyrosequencing determines the order of nucleotides in template DNA by synthesis and detection of released pyrophosphate (PPi) upon nucleotide base (A/T/C or G) incorporation (89). This method is mainly used for fast and accurate short reads of DNA templates that do not contain repetitive homopolymer regions (if known) (95). The ssDNA template is hybridized to a sequencing primer and incubated with several enzymes. DNA polymerase synthesizes the complementary sequence, while ATP sulfurylase converts PPi to ATP in the presence of adenosine 5′ phosphosulfate. ATP then acts as a substrate for the luciferase-mediated conversion of luciferin to oxyluciferin that generates visible light proportional to the amount of ATP produced. The light produced by this enzymatic reaction is detected by a charge-coupled device camera and analyzed in a pyrogram. The intensity of light measured by a pyrosequencer, such as the Pyromark (Qiagen, Germantown, MD), determines if there are multiple bases in a row in the sequence (i.e., single G versus GGG). The previous nucleotide is degraded by apyrase before the next one is added for synthesis. No light is emitted if the added base is not complementary to the first unpaired base of the template and dNTPs are incorporated until the entire strand is synthesized. Pyrosequencing can only sequence lengths of DNA that are ∼300 to 500 nucleotides long, which is much shorter than that obtained by the Sanger method (89).
OVERVIEW OF PCR/CYCLE SEQUENCING USING AN AUTOMATED GENETIC ANALYZER
This section briefly outlines the principles and procedures for performing sequential 16S PCR/cycle sequencing in the clinical microbiology laboratory. The sequential steps performed in a PCR/cycle sequencing procedure using a fast protocol in the clinical laboratory has previously been reported and are summarized in Fig. 3 (17, 96, 97). However, the individual steps are similar throughout the procedure if standard PCR protocols are used. Comprehensive detailed procedures for each step performed in a cycle sequencing analysis have been published elsewhere (92, 95). In-house sequencing procedures should be performed in a facility that has strict separation between pre- and post-PCR environments either by physically separating these work areas or using self-contained hoods with dedicated equipment in each area (96, 97). The reader is referred to previously published reports for a detailed discussion of the front-end handling and storage of isolates for molecular analyses as well as nucleic acid sequencing (95, 98). Freshly collected clinical isolates should be obtained that are free from preservatives that can interfere with PCR/cycle sequencing reactions.
FIG 3.

Summary of steps of a fast 16S PCR and cycle sequencing procedure.
DNA Extraction/Purification and the Use of Controls
Accurate high-quality sequencing data are highly reliant on efficient extraction and purification of nucleic acid, so the template is free of contaminants. The use of “DNA-free” reagents is highly recommended to minimize detection of contaminating bacterial DNA from commercial products (60, 95). Several methods are available for preparing microbial DNA for sequence analysis, and different protocols need to be verified for different pathogen types as well as clinical isolates (i.e., Gram-negative versus Gram-positive, etc.). Some bacteria, such as Mycobacterium spp., are more difficult to lyse, and special extraction protocols are required for sequencing these genera (99). Clinical laboratories must verify the isolate/isolate extraction method used with manufacturer cycle sequencing protocols as part of the overall method validation process. Although other manual DNA extraction methods, such as proteinase K lysis, bead beating, or direct boiling, may be used (100–103), clinical laboratories currently rely on commercial manual or automated extraction methods for in-house cycle sequencing depending on the number of isolates to be tested and the type of downstream nucleic acid to be sequenced (17). The DNAzol reagent (contains guanidine isothiocyanate) protocol (Thermo Fisher Scientific) is a cost-effective method for recovery of bacterial genomic DNA from a wide variety of liquid and solid isolates. A commercial manual spin-column filtration method efficiently extracts a small number of isolates (i.e., ≤24 isolates), whereby the isolate is processed through several manual steps (101, 102). For example, high-purity yields of DNA can be routinely obtained from bacteria and human blood or tissues using the PureLink genomic DNA kit (Invitrogen, Thermo Fisher Systems) or the DNeasy blood and tissue kit (Qiagen), but there are many other commercial suppliers of similar kits on the market (produced by EdgeBio, Qiagen/MoBio, etc.). A simple DNA extraction for cycle sequencing of bacteria, fungi, and food types can also be achieved using the PrepMan Ultra isolate preparation reagent (Applied Biosystems, Thermo Fisher Scientific). Commercial automated extraction instruments allow more efficient isolation of nucleic acid from a large number of isolates/isolates (i.e., >24 per run), and many of these platforms deploy magnetic bead particle technology to separate and purify nucleic acids (104). Specialized procedures are also required for efficient extraction of nucleic acid from formaldehyde or formaldehyde-fixed, paraffin-embedded tissues, molds, or difficult clinical isolates, such as stools, that contain lots of particulate matter that may inhibit PCR (105–109).
The NanoDrop (Thermo-Fisher) instrument provides an efficient, reliable means to check DNA purity using spectrophotometric optical density measurements by fluorescence at 260 and 280 nm; the A260/A280 ratio should be 1.8 to 2.0. Lower ratios indicate protein contamination, but nucleic acid contamination will not be detected (108). Other methods may be used but are more laborious, including total phosphorous content, dye intercalations, and limiting dilution (92, 95). A more exact measurement of DNA concentration is required for NGS applications, and this is currently achieved in clinical operations by using either an Agilent Bioanalyzer or a Qubit fluorometer (Life Technologies, Thermo Fisher Scientific), whereby intercalated dyes provide a precise quantitation of the amount of DNA present in the isolate down to the picogram level (109, 110). However, DNA purity still must be measured using the above A260/A280 ratio prior to proceeding with downstream NGS procedures.
Optimization of the quantity of DNA template added to a cycle sequencing reaction is done according to the PCR product size. Sequencing PCR products between ∼100 to 200 bp requires 1 to 3 ng of template, but larger PCR products between ∼1,000 and 2,000 bp will require 10 to 40 ng (92, 98). A Beer-Lambert Law calculator can be used to determine the DNA concentration in the PCR product by multiplying the UV absorbance of the isolate at 260 nm by either 33 or 50 μg/ml for ssDNA versus dsDNA templates, respectively (111). Too much DNA template added to a sequencing reaction rapidly depletes the reagents and the dye label in the reaction mixture, whereas too little DNA results in a poor electropherogram trace (i.e., reduced peak height and strength), which makes data analysis difficult or uninterpretable.
Fast PCR/Cycle Sequencing Processes
Clinical laboratories routinely perform in-house PCR/cycle sequencing protocols for several applications. Although standard PCR can be used to perform these procedures, use of a fast protocol allows shortened PCR cycling times, which reduces the time to reporting and increases overall testing throughput. Potential contamination can be minimized by using DNA-free reagents and enzymes from commercial products, and purified primers avoid misannealing problems (60, 112). Fast PCR decreases the overall procedural cycle time by using “fast” primers (i.e., higher thermodynamic melting temperature [Tm] from 64°C to 77°C), DNA polymerases that are thermodynamically stable at higher temperature with a higher extension rate, typically 2 to 4 kb/min. Fast-ramping thermal cyclers are used that perform the reaction at higher temperature and speed with greater thermal uniformity, because the temperature differential between PCR cycle steps is reduced (96). Some commercial thermal cyclers allow both fast-ramping and standard protocols (i.e., Veriti thermal cycler [Applied Biosystems] and C1000 Touch thermal cycler with dual 48/48 fast reaction module [Bio-Rad]). Fast PCR protocol changes also include combining the annealing and extension steps and eliminating the final extension step for short (<250-bp) amplicons (96, 97). A fast PCR/cycle sequencing protocol decrease the PCR time to only 30 to 40 min. The total procedure takes less than half (∼4 to 5 h) the time required to complete a conventional PCR/cycle sequencing run, so it can be completed within a day provided data interpretation is completed using a commercial system that interprets the electropherogram data as it is generated (see “Sequence Data Analysis and Interpretation,” below).
Enzymatic purification of PCR amplicons prior to cycle sequencing using exonuclease 1/shrimp alkaline phosphatase (Exo1/SAP-IT) treatment is preferred, as it is a simple, reliable method that effectively cleans up a large number of isolates with minimal manual manipulation (113). ExoSAP-IT (P/N 78200; USR Corporation, Thermo Fisher Scientific) cleanup reagent is active in commonly used buffers, so it may be added directly to the PCR product: Exo1 degrades single-stranded DNA, such as unused primers, and recombinant SAP dephosphorylates unused primers and dNTPs. Enzymatic cleanup includes an initial treatment (15 min at 37°C) followed by heat incubation (80°C for 15 min) that allows enzymatic deactivation. Other more laborious methods that may be used for this step include serial dilution, ethanol precipitation, column ultrafiltration, and gel purification (95, 114).
Clinical laboratories should perform cycle sequencing using commercial protocols and reagents, so that reliable results are obtained with minimal assay optimization (i.e., commercial primers, DNA enzymes, and fast PCR/cycle sequencing mixtures are already optimized to work together for a range of DNA templates). For example, the BigDye direct sequencing kit (Applied Biosystems, ThermoFisher) includes a set of universal primers (M13 forward and reverse) for 16S sequencing and eliminates the need to perform another post-PCR purification step prior to cycle sequencing. These primers can also be used for the subsequent cycle sequencing step. By performing PCR to cycle sequencing steps in a single tube, the BigDye kit not only decreases manual manipulation but also makes obtaining sequence data much faster (91). However, difficult DNA template sequences (i.e., homopolymer G-C-rich regions) may require the use of specific commercial master mixes and reagents that have been optimized to work with a DNA polymerase with high processivity and fidelity to ensure sequencing efficiency and accuracy (115).
Troubleshooting Sequencing Problems
DNA extraction and amplification controls are necessary to ensure that subsequent sequencing reactions are performed according to regulatory and accreditation requirements (95). Positive and negative extraction, amplification, and sequencing controls are important for monitoring assay integrity as well as troubleshooting. The same solution used as a starting matrix for isolates in a sequencing run (e.g., distilled water free of either reagents or template DNA) can be used as the DNA extraction negative control, which is run through the entire procedure to detect possible environmental or reagent contamination. This negative control should produce no more than baseline traces in the electropherogram without sequence data. If amplicon is produced, sources of contamination are found most often in the DNA extraction reagents and/or the isolate handling protocol. The DNA extraction positive control should be a unique organism that is not a human pathogen but is representative of the expected bacteria isolate(s) that produces a unique, easily distinguished sequence pattern. To further reduce the risk of amplicon contamination, the positive-control strains should be rotated and not consist of pathogens expected to be contained in the samples being tested. Uracil DNA N-glycosylase enzyme and deoxynucleotide triphosphate mixes also should be supplemented with 2′-deoxyuridine 5′-triphosphate (dUTP) in the PCR mix to prevent carryover contamination of dUTP-containing amplicons (95, 98). Long periods of cold storage of purified PCR amplicons should also be avoided prior to template cycle sequencing as another measure to prevent degradation and contamination.
Amplification reactions should also include a negative and positive reaction tube. The negative control monitors the integrity of the amplification reagents, and the positive control should be like the one being used to control the extraction. Most commercial PCR kits already contain a positive internal control (i.e., genomic DNA extracted from a microorganism whose sequence is known), and this template can also serve as a control for the cycle sequencing reactions. HPLC-grade water should be used as the negative control for the sequencing procedure, and it should not produce sequencing data. Alternatively, a known sterile isolate matrix may be used in broad-range PCR/cycle sequencing assays to mimic the specific clinical isolate material being analyzed (98). Possible contamination is indicated by obtaining sequencing data from the negative control, and the microorganism’s identity may indicate the source of contamination (i.e., introduced during the procedure or within reagents).
Troubleshooting of poor-quality sequencing data involves investigating various causes, as outlined in previously published guidelines (92, 95). The automated sequencer trace most often shows no recognizable signal or signal loss after the start of base calling, unexpected gaps or termination, mixed signal with multiple overlapping peaks, or misshaped peaks or background noise resulting in missed or incorrect base calls (92, 93). Common reasons for low-quality data include (i) poor-quality DNA template (i.e., inefficient DNA isolation or low concentration), (ii) inadequate cleanup of the template, (iii) poor primer design (i.e., disparate Tm of F/R primers or primer Tm too low for fast PCR) or impurity (e.g., primer fragments) resulting in poor annealing, (iv) multiple annealing sites or failed PCR amplification, (v) inadequate cleanup of the PCR products, (vi) PCR and/or cycle sequencing reactions not optimized for DNA template/primers, (vii) wrong software mobility file used to interpret the dyes used, and (viii) overall low signal strength so that the software cannot interpret the raw data (92, 94, 95). Gaps or sequence termination may also occur when a low-fidelity DNA polymerase is used that cannot read through difficult template regions (i.e., homopolymer that is highly GC-rich) (116). Use of an automated sequencing quality control analysis program can assist troubleshooting DNA sequencing problems that limit sequence read length (e.g., QualTrace II [Nucleics]). Consultation with the manufacturer of the reagent kits and automated genetic analyzer often assist with identification of the problem(s) if a solution is not immediately evident.
Sequence Data Analysis and Interpretation
Quality checks of sequence data are important, because base-calling algorithms that determine the nucleic acid from the signal peak may produce wrong or incomplete base calls, particularly if the amount of input DNA is low and/or the template harbors insertions of deletions or conformational complexity (115, 117). Clinical laboratories that use an external sequencing service (i.e., university core facility) must ensure that the referral laboratory meets the appropriate regulatory and accreditation requirements for diagnostic testing, which includes employment of highly trained, knowledgeable personnel capable of communicating about encountered technical and organizational issues (94). The referral laboratory should routinely provide the individual electropherogram results as a DNA sequence chromatogram file (e.g., *.scf or *.ab1) for each sequenced isolate, and these files should include associated quality score metrics, such as phred scores. This is essential for ensuring the quality and accuracy of the clinical isolate’s identification and antibiotic susceptibility profile.
Sequencing should be initiated far enough upstream to ensure that the region of interest lies within the clear range to be analyzed. This range should cover as many variable regions as possible to optimize species differentiation (while being aware that the positions of variable regions differ between different bacterial families). Most capillary sequencing instruments produce good-quality sequences of ∼600 to 800 bp in length, but some well-tuned instruments may even exceed ∼1,000 bp in length for cultured isolates (92, 93). A sequence commonly has a few bases of poor quality at the beginning and end of the trace with a high-quality region in the middle of variable length; a consistent procedure should be used to review and trim poor-quality sequence data from the 3′ and 5′ ends before proceeding with further analyses (95). Manual sequence editing of base calls for nucleotides other than the one initially reported by the base caller must be documented and should be recorded.
Quality checks of sequence data should start with aligning and assembling all sequence fragments from an isolate to generate a contig and a consensus sequence; this can be achieved either by using a generic assembler software or by using application-specific software, which will perform the alignment against an automatically or manually selected reference sequence; reference-driven alignments are generally easier to interpret and may be more precise (Fig. 4) (5). Initial review of raw sequence data should primarily focus on alignment accuracy; all contig fragments should align within the boundaries of the expected target gene sequence. After trimming the 5′ and 3′ ends of the fragments to contain only valid calls, the resulting consensus sequence should span the expected read length. Bidirectional sequencing and alignment with a known reference sequence is essential for interpreting mixed or unclear base calls, and adequate coverage by other sequencing fragments can be very helpful in that regard (5). Once trimming has been performed, one should systematically verify the contig for read accuracy; it is recommended that users align the chromatogram fragments against a reference sequence that is close to the species expected in the isolate to obtain meaningful events to check while saving time (5).
FIG 4.

Multisequence alignment compared to a reference sequence ensures proper data interpretation.
The following events should be verified and edited where necessary. (i) Mismatches with the reference sequence may be misread bases (to be edited) or real mismatches, which may reflect a different species or intraspecies diversity. The reverse-complementary strand may help to differentiate between artifacts and real mismatches. (ii) Insertions and deletions can occur as artifacts when base-calling software shifts a chromatogram peak or double reads multiple chromatogram peaks for the same nucleotide (e.g., homopolymer stretches that cause polymerase stutter). Given the highly conserved nature of the 16S rRNA gene, insertions and deletions are less frequently encountered in the context of intraspecies or intraoperon diversity. (iii) An ambiguous base call is assigned by the instrument software whenever it cannot accurately determine a base at a particular position; either the International Union of Biochemistry (IUB) code for a mixture or an “N” (nucleotide) is inserted (118). Base pair differences will be detected by the sequencer’s base caller software if it has been configured to detect these ambiguities as IUB codes (recommended setting). Ambiguous base calls occur for many reasons, including (i) high noise levels (usually most prominent at the ends of a chromatogram or in the case of technical problems), (ii) the presence of multiple isolates in the sample, (iii) shifts downstream of insertions/deletions, or (iv) 16S intraoperon diversity in a species (119). To distinguish technical from biological ambiguities, one should verify if chromatogram peaks for the nucleotide concerned are all overlaid with the same noise, which indicates problems with signal detection in the sequencing reaction (Note that chromatogram peak height generally does not reflect quantitative relations of nucleotides well [and, thus, of subpopulations] due to signal normalization by the base-calling software.) In cases where a chromatogram peak cannot be assigned to a single nucleotide, one should apply the appropriate IUB code instead of assigning a less meaningful N. (Note that some base-calling software systems propose parametrizations that allow automated assignment of IUB codes.)
Given the potential impact of sequence edits on the resulting species identification, it is important to make these edits traceable, ideally automatically via the editing software used. Edited consensus 16S sequences are usually searched against one or several reference databases, using rapid search algorithms such as the basic local alignment search tool, or BLAST (https://blast.ncbi.nlm.nih.gov/), which screens large data sets to obtain a ranked list of the closest matching sequences as well as pairwise alignments of the isolate sequence with selected reference sequences (120, 121). One needs to be aware that matches are ordered by match score in BLAST; the best-matching sequence for a species identification does not always show on top. Please be aware that optimal similarity in BLAST searches against a reference database relies on keeping the number of ambiguous bases, and especially of undetermined positions (N), to a minimum. Microorganisms may also be misidentified using the BLAST algorithm for sequence interpretation, so the match list should be reviewed carefully for the important parameters outlined in Table 1 (120, 121); a matching reference sequence should be retained as possible identification, provided that the following criteria are fulfilled.
TABLE 1.
Important parameters reviewed after a BLAST search for 16S sequencesa
| Parameter | Definition |
|---|---|
| Match accuracy | Best matching reference sequences should show the lowest no. of mismatches (not necessarily reflected by default sorting by BLAST score) |
| Match length | Matching reference sequences should cover an isolate sequence entirely; shorter matching sequences by BLAST should be verified by alignment with the isolate sequence for missing mismatches on the edges |
| Match consistency | An isolate sequence that matches a no. of reference sequences within the same species and genus annotation increases the confidence for such species and genus identification |
| Match differentiation | To be able to estimate the degree of an isolate’s differentiation to the next closest species, sequences derived from closely related but different species should appear on the list of matching reference sequences, to be evaluated in this context |
See reference 5.
Match accuracy.
Match accuracy is the degree of similarity between the isolate sequence and the matching reference sequence. The higher the similarity, the better the match. However, one needs to be aware of the following issues, as outlined in CLSI MM18-A2 (5): ambiguity codes (IUB, IUPAC) are interpreted by the BLAST search algorithm as full, instead of partial, mismatches, so a sequence containing a number of ambiguous bases will rank lower on the list despite the presence of partially matching bases. In addition, BLAST scores rank matching reference sequences according to the sequence length above the mismatch number. Thus, a retrieved reference sequence may appear in a higher-ranked position according to the BLAST score based on its overall match length, even though it has more mismatches than another lower-ranked sequence based on its shorter match length. In the past, a similarity threshold of >98.5% has been recommended as a rule of thumb for assigning a 16S sample sequence to a certain species (16, 122). While this simple cutoff makes interpretation easier in diagnostic laboratories, it can easily be misleading for several reasons that have been previously reported (16, 122).
Match and database coverage.
The amount of 16S sequence coverage, as defined by the adequate representation of genus-relevant variable and conserved regions, affects the similarity of a sample to a reference sequence. If conserved stretches are predominant in the sample sequence, the match similarity to the best-matching sequence will exceed the cutoff without yielding an unambiguous identification. Therefore, a similarity score would require a minimum coverage of variable regions for the genus or genera involved. Genus diversity also plays a role, because some genera are highly diverse whereas others are not (e.g., Mycobacterium). Slow-growing mycobacteria, such as M. genavense, would not separate from other atypical mycobacteria using 16S sequencing with a cutoff of 98.5% (123). In the case of a standard sequencing of the first 500 bp, M. genavense and M. triplex are genetically different by only 4 mismatches or <1% of the V1-V3 sequence, but they can be clearly differentiated on this basis as outlined below. Species diversity also plays a role because highly diverse species, such as Fusobacterium nucleatum, which includes a number of at least 5 subspecies (https://lpsn.dsmz.de/), exhibit an intraspecies diversity of 10 to 12 mismatches (>2%) within the first 500 bp between variants. If the reference database used does not cover explicitly the relevant subspecies and variants as outlined below, the sample sequence will not be matched with a high enough score for species identification. Achievable match similarity also depends on the adequate coverage of species, subspecies, and variants in the reference database used. If the respective variant is not present in the database, a sample sequence may match only references below the cutoff, yielding an inconclusive result. Missing reference sequences can also lead to an unambiguous match above the cutoff with a reference sequence present, whereas the correct result should have been ambiguous and not definitive. (Note that BLAST match lists should always be reviewed for other possible matches, and in a multialignment with the sample sequence, the relevance of mismatches will become transparent with regard to the variable regions relevant to the genus involved [see “Match differentiation,” below]).
Match length.
Matching reference sequences should span the longest alignment possible or ideally align with the full isolate sequence. At an equal number of mismatches, longer matches should be given preference for identification, thereby ensuring better reliability. (Note that BLAST tends to truncate sequence matches when alignment become uncertain at the 5′ and 3′ end due to mismatches or insertions/deletions.) This can lead to matching references ranking high on the list despite the presence of mismatches on the edges, which then are not accounted for or displayed in the pairwise alignments. Therefore, one should always verify the match length of an isolate sequence with a reference before calling an identification. In cases of the doubt of mismatches being present at the edges of references, one should perform a pairwise or multiple alignment that includes the isolate sequence.
Match consistency.
The list of matching reference sequences (e.g., species and genus names) should be reviewed for naming consistency within the species and within the genus. (Note that ongoing taxonomic name changes have created confusion for clinical laboratorians and clinicians for clinically relevant microorganisms.) Thus, the best-matching reference sequences on the ranking list ideally should be consistently annotated with the same species name (provided that the reference database contains multiple entries of this species) or with the same genus (provided that the reference database contains only one or a few entries per species); mismatches can reflect the natural intraspecies variability. If the best-matching references contain other species at an equal number of mismatches, a species call for identification may not be possible on this basis; in such cases, a genus identification call could be made. If the best-matching reference sequences come from different genera at equal mismatches and scores, only an identification on the family level should be envisioned (e.g., see E. coli and Shigella spp. to be interpreted as “Enterobacterales”). Match consistency can be indicative of problems with inconsistent coverage of a species or genus in a database, or of a poor choice of the sequenced region, to be variable enough for differentiation between certain species and genera.
Match differentiation.
Match differentiation is the ability to call a species identification while making sure that no other species would match as well. The number of matches and mismatches between the isolate sequence and the best-matching references of the closest species are considered. The match list should show representative sequences of more than one species to enable differentiation from the next closest one. Match differentiation refers to intraspecies variability (thus, tolerable mismatches) and interspecies mismatches (thereby allowing species differentiation); therefore, it is important that the match list also includes the next closest species. A multiple alignment of the isolate sequence with the best-matching reference sequences of the closest species (two or three) is often helpful in assessing the differentiation concerning the position of mismatches; mismatches of an isolate sequence occurring in variable regions, where species of this genus usually differ, are indicative of a nonmatch to a species and should be documented. In these cases, one may report “close to” the closest matching species.
Reference databases.
Clinical laboratories should use a reference database for microbial identification that contains representative sequences of good quality for all species and genera that a user expects to detect. Thus, an isolate sequence is likely to match a relevant reference sequence, either of the species searched or of another closely related species. To achieve best possible match accuracy (Table 1), a reference database should include naturally occurring variants of all species and subspecies so that more than one good-quality reference sequence is available for each species (124). Sequences submitted for species that are rare or not previously described can be useful if such a case is detected in the laboratory; such a match may give hints to observations made by other investigators. However, such sequences may also confound the match results with regard to established species; thus, one should be able to blind them out. In any case, sequences from the public domain should be represented with key characteristics (i.e., the original annotation, referring to author, submission, source, etc., and original repository where the sequence comes from). After having performed a BLAST search, a multialignment (e.g., by CLUSTAL or by an equivalent method) of the best-matching species and their variants can help to accurately detect and assess the following problems: (i) mismatches on the edges of a sequence, which were not considered by the BLAST algorithm due to alignment break-off; (ii) match and mismatch consistency between the isolate and the best-matching sequences but still diverse reference sequences (note that the alignment in these cases can show if mismatches are located in hypervariable regions for this genus [indicating a different species] or in regions where mismatches are balanced and, thus, not significant [indicative of a species variant]); and (iii) match of reference sequences is not complete enough to see if essential information is missing for species-level identification (multialignments are invaluable in showing where there are hidden mismatches at the edges of an alignment and in defining areas of insertion or deletion that may affect alignment accuracy, and they may also indicate that the mismatches occur within the regions where interspecies variability is observed [5]); (iv) reduced variability between closely related species (i.e., near-complete sequence similarity across the entire 16S rRNA target gene) (5).
A multialignment is also necessary to subsequently construct a dendrogram, which is a useful graphical display for understanding the phylogenetic relationships between the query and reference sequences (125). Methods commonly used for generating dendrograms are the neighbor-joining (NJ) method, the unweighted pair group method with arithmetic averages (UPGMA), and the weighted pair group method with arithmetic averages (126). If bacterial isolates are closely related, these phylogenetic methods have equivalent performance, but if isolate sequences are not closely related, then the choice of phylogenetic method may affect dendrogram relationships, as previously illustrated (16, 126). To build significant dendrograms, one should use sequences of maximum length and maximum overlap; in the case of similar sequences within genera, mismatches within the 16S gene sequence within the first ∼500 bp or the last ∼1,000 bp, depending on the length of sequence analyzed and the alignment tool, can also affect the comparison of sequences (i.e., percentage dissimilarity) and, thus, the dendrogram (16, 127). In addition, naturally occurring insertions or deletions are likely not reflected by dendrogram matrices, which only account for positions covered by all sequences. Taxonomists must consider these potential pitfalls in their analyses and assignment of exact relationships between the higher bacterial taxa (128). Generation of a dendrogram may better show relatedness between isolates than either percent dissimilarity or concise sequence alignment comparison (6). Although strains may seem similar to each other based on their percent dissimilarity (i.e., ≤1%), based on the positions of the mismatches within 16S, a dendrogram may show this not to be the case (127). Rooting a phylogenetic tree using a somewhat distantly related sequence of a different genus can help to build more stable clusters of very similar sequences; bootstrapping will indicate the robustness of a branch but is generally low for highly similar sequences from genes, such as the 16S (129). Dendrogram analyses may be helpful when analyzing an unknown sequence, particularly an isolate’s relationship to other closely and distantly related major genera. A phylogenetic analysis of the unknown isolate can indicate where a species groups, even when there is not a closely related sequence to compare within available databases (16, 126, 130).
The final sequence is analyzed by comparing it to similar sequences available in a public and/or commercial database. Accurate identification of clinical isolates using 16S is highly dependent upon access to accurate databases that contain a sufficient number of high-quality sequences for a particular genus/species that have the correct taxonomic nomenclature assigned (16). DNA sequence databases commonly used for diagnostic bacterial identification are outlined in Table 2. Reference databases are powerful tools for sequence analysis, but their strengths and limitations should be specifically outlined by the clinical laboratory in their standard operating procedure. Some resource databases are freely available on the Internet, but many are unverified and depend on ongoing funding from public or private sources to maintain their content. Clinical laboratories must ensure that the database(s) used is clinically relevant and meets the diagnostic rigor required for diagnostic coverage, quality, and maintenance, as previously outlined above and in CLSI MM-18 (5). The most current database version should be used, and the derived sequence interpretation also should be cross-checked using one or more of these sources. Due to the rapid changes occurring in taxonomy and nomenclature of many clinically relevant bacterial pathogens (131), clinical laboratories should only use databases that are kept current by regular updates.
TABLE 2.
Overview of some publicly and commercially available reference databases for bacterial identification using 16S rRNA gene sequence interpretationa
| Database | DNA target(s) | No. of sequences | Curation | Alignment of clustered sequences | Link | Comment |
|---|---|---|---|---|---|---|
| NCBI nt (Genbank NCBI) | All | ≈21,000,000 | Limited | No | https://blast.ncbi.nlm.nih.gov/Blast.cgi (select appropriate dataset in the menu in order to restrict and accelerate the search); for downloads, ftp://ftp.ncbi.nlm.nih.gov/blast/db/ | Hosts all published sequences; excellent coverage; frequent updates; many redundant entries; frequent erroneous entries; use for unusual or new species |
| Greengenes (consortium comprised of Second Genome Inc., University of Colorado, and University of Queensland) | 16S rRNA genes | ≈1,200,000 | Yes; manual sequences >12,000 bp; taxonomy curation | Yes: sequence clusters at various similarity percentages | Searches, http://greengenes.lbl.gov/Download/Tutorial/Tutorial_19Dec05.pdf; downloads, https://greengenes.secondgenome.com/ | Includes several tools from chromatogram analysis to alignments; latest version from 2013; unclear updates, some taxonomy information may be outdated |
| RDP (Michigan State University) | 16S rRNA | ≈3,200,000 | Yes; manual sequences >12,000 bp; taxonomy curation | Yes; Aligned | Searches, https://rdp.cme.msu.edu/seqmatch/seqmatch_intro.jsp and https://rdp.cme.msu.edu/index.jsp | Manually and not regularly updated; last update was May 2015; various tools available to analyze user data further |
| SILVA (Max Plank Institute for Marine Microbiology) | ≈5,000,000, includes small ribosomal subunit for eukaryotes | Yes; manual sequence quality; taxonomy curation | Yes; multiple cluster sets available | Search, http://www.arb-silva.de/aligner/; downloads, http://www.arb-silva.de/download/arb-files/ | Continually updated; tools available to analyze user data; genes other than 16S rRNA | |
| Molzym SepsiTest | ≈7,043 | Manual | No | CE-IVD database works with kit but also with sequences generated otherwise | ||
| SmartGene IDNS Bacteria Module 3.9.x | 16S rRNA and rpoB | ≈800,000 16S, 358,000 centroid annotated | Yes; quality filters for sequence quality, centroid annotation for annotation qualification | Centroid annotation for most representative sequence per species | CE-IVD; proprietary centroid annotation; quality filtered, continually updated; tools available to analyze user data; genes other than 16S rRNA | |
| MicroSEQ 3.1 | 16S rRNA | 2,300 | Sequences of collection and type strains | No | Compatible with the MicroSEQ sequencing kit of ThermoFisher; mainly for 500-bp sequencing |
Entries with gray shading represent publicly available databases, and those without gray shading represent commercially available databases. See reference 5.
Manually copying and pasting an isolate’s sequence into a website’s search window to perform an interpretation search may also produce errors. Users should verify that the entire sequence being interrogated is accurately copied from the 5′ to the 3′ end (or that the software takes care of resolving this), and that no older sequence is accidentally pasted from the computer’s cache. If BLAST is being used as the search algorithm (120), the users should record and understand its settings, or a standardized parametrization is used, which has proven its adequacy for targets such as 16S. Analysis software that contains preparameterized BLAST search tools, easy-to-use, multiple alignment tools, and other functionalities, along with valid reference sequences, can avoid these pitfalls and streamline interpretation. All isolate sequence results should record the interpretation database(s) used along with its version to troubleshoot isolate result traceability.
Sequence databases also vary widely in terms of the target gene data available. One can distinguish curated and noncurated databases, and within the curated ones, those where manual curation is performed and where the curation is achieved via algorithm-based methods. All these databases have their advantages and disadvantages, but for diagnostic purposes, they should use the most current nomenclature and taxonomic organization and contain only curated sequences that are quality assured for accuracy, completeness, and annotations. Most of the bacterial sequence data deposited in public databases, such as GenBank (the world’s largest noncurated repository; https://www.ncbi.nlm.nih.gov/genbank), correspond to the 5′ region of 16S, but linked gene name/sequence can be uploaded, so the database is largely unverified. GenBank also contains both pathogenic and nonpathogenic human, animal, and environmental data that can generate some unusual matches against an isolate’s 16S sequence in BLAST (124). Furthermore, the presence in GenBank of many redundant entries (i.e., identical sequences with the same species annotation) can even mask relevant matches to sequences of other species. The same criteria outlined above should be applied when using a commercial database in the clinical laboratory for isolate sequence interpretation. Applied Biosystems (Life Technologies, Thermo Fisher Scientific) has software for bacterial 16S rRNA gene and fungal D1/D2 regions of the 26S rRNA gene (MicroSEQ ID Analysis); this software package and its manually curated reference database, however, is accessible only to users of the respective kits and often favors partial sequencing. Molzym (Bremen, Germany) also provides its own manually curated reference databases in the context of sepsis diagnostics. SmartGene IDNS is a commercially available software package that supports automated or semiautomated sequence analysis from raw data to the report for all current sequencing platforms; it also comes with its own reference database (21, 130, 132–135). The SmartGene IDNS (Zug, Switzerland) provides comprehensive curated databases for bacterial and fungal sequences using automated algorithm-based methods, which houses nonredundant and representative sequences for each species, full-length sequences, or sequences from collection strains, etc., within different containers.
Overall laboratory resource availability, technologist expertise, and the required operational efficiency should be considered when selecting a reference sequencing interpretation database(s). Databases developed for 16S sequence analyses of specific genera or groups of microorganisms should also be used where verification studies show improved quality and accuracy of results. Turenne et al. compared the identification of 79 mycobacterial type strain sequences by analyses using either GenBank, the Ribosomal Database Project (RDP-II), or the 16S database of RIDOM (136). The RIDOM database contained an identical matching sequence for each submitted type strain, but only about a quarter of them could be accurately identified using BLAST either on GenBank or RDP-II (the open-access 16S RIDOM database has since been closed). Sequence-based identification within Nocardia spp. may also be problematic due to this genus’s high degree of intra- and interspecies genomic variability within this genus (137). Helal et al. compared clustering and classification algorithms within GenBank to identify 364 known and yet-to-be-identified Nocardia 16S sequences (138). These investigators found that the identification of centroids of 16S rRNA gene sequence clusters using novel distance matrix clustering enabled the identification of the most representative sequences for individual Nocardia species and allowed the quantitation of inter- and intraspecies variability.
GenBank/NCBI makes available a type strain match filter via its type material annotation (139). Using this resource, one can select matches from type strains, of which there are currently only ∼20,300. However, one should be aware of missing species and variants and linked 16S sequences that are sometimes partial or even fragmented, leading to coverage issues with BLAST (see “Match and database coverage,” above).
IDENTIFICATION OF CLINICALLY RELEVANT BACTERIAL PATHOGENS USING 16S rRNA GENE SEQUENCING
This section provides the readers with a comprehensive assessment of the use of this method for identification of bacteria within various taxonomic groups that cause human disease according to how things currently stand. Because NGS studies are rapidly changing our understanding of the classification and taxonomy of important groups of pathogens, it is important to consult online databases to verify that one is accessing the most up-to-date information for specific microorganisms and groups. Some recommended sites include the International Journal of Systematic and Evolutionary Microbiology (IJSEM), The Taxonomy Database of the National Center of Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/taxonomy), the List of Prokaryotic Names with Standing in Nomenclature (LSPN) (https://lpsn.dsmz.de/), and Deutsche Sammlung von Mikroorganismen und Zellkulturen (https://www.dsmz.de).
Overview of Pathogen Identification
Clinical microbiology laboratories must be able to rapidly and accurately identify a diverse range of bacterial isolates in order to diagnose the etiology of infection and provide guidance about appropriate antibiotic treatment. Partial or complete sequencing of 16S has proven to be an invaluable tool for providing a reliable identification of infections caused by unusual or rarely encountered bacteria, particularly in the pre-MALDI-TOF MS era (16). A genus- and species-level identification of bacterial isolates was obtained by 16S rRNA gene sequencing in >90% and 65% to 83%, respectively, depending on the group of bacteria and the criteria used for species definition in cases where conventional phenotypic methods had failed (11, 16). With the emergence of 16S rRNA gene sequencing as an identification tool in the last 20 years, the usefulness of commercial databases has also undergone limited clinical evaluation. The MicroSeq 500 16S rDNA-based identification system can reliably identify >80% of clinically relevant bacterial isolates with atypical phenotypic profiles and 89.2% of unusual aerobic Gram-negative bacilli (8, 10, 135, 140, 141). It has also proven useful for the identification of some slow-growing bacteria, such as Mycobacterium species, notwithstanding the limitations of using this target (i.e., 16S sequences cannot differentiate species within the M. tuberculosis complex, the M. avium intracellulare complex, or the M. chelonae/M. abscessus complex) (136, 142, 143). Simmons et al. compared the identification by conventional methods of a diverse group of bacterial clinical isolates with gene sequences interrogated by the SmartGene and MicroSeq databases (135). Of 300 isolates, SmartGene identified 295 (98%) to the genus level and 262 (87%) to the species level, with 5 (2%) being inconclusive. MicroSeq identified 271 (90%) to the genus level and 223 (74%) to the species level, with 29 (10%) being inconclusive. SmartGene and MicroSeq agreed on the genus for 233 (78%) isolates and the species for 212 (71%) isolates. Conventional methods identified 291 (97%) isolates to the genus level and 208 (69%) to the species level, with 9 (3%) being inconclusive. SmartGene, MicroSeq, and conventional identifications agreed for 193 (64%) of the results.
Utilization of 16S PCR/sequencing to identify clinically relevant bacteria that previously would have been mis- or unidentified from clinical specimens has also provided insight into the epidemiological and pathogenic potential of rare or unusual bacteria in human infections. Woo and colleagues summarized the novel bacterial species discovered from human specimens in just 7 years, from 2001 to 2007 (11); a total of 215 novel species, 29 belonging to novel genera, were reported. In addition, 100 (15 novel genera) novel species were found in 4 or more patients, and the largest numbers were of the genera Mycobacterium and Nocardia. Then and now, the oral cavity/dental-related specimens and the gastrointestinal tract were the most important reservoirs for discovery of novel species (11). This agrees with the huge diversity of microbiota identified at these important body sites by the human microbiome project (144, 145). Since their discovery, Streptococcus sinensis, Laribacter hongkonensis, Clostridium hathewayi, and Borrelia spielmanii have been more fully characterized, including their epidemiology and routes of transmission (11). Prospective local experience with 16S sequencing can also help define regional epidemiology of novel opportunistic pathogens. Performance of 16S sequencing on a large number of clinically relevant pathogens over the past decade in our laboratory revealed the epidemiology of invasive infections, such as bacteremia, due to several unusual bacteria, including Eggerthella lenta (146) and Peptoniphilus (147) and Actinomyces (21) species.
Current Limitations of the 16S rRNA Gene Target for Pathogen Identification
Our group recently collaborated on updating the Clinical and Laboratory Standards Institute (CLSI MM-18-A2) document entitled Interpretive Criteria for Identification of Bacteria and Fungi by DNA Target Sequencing; Approved Guideline (5). This important clinical laboratory guideline provides interpretive criteria for identification of a wide range of clinically relevant bacteria and fungi to the genus and species levels using partial or complete 16S sequencing. To revise this document, we performed comprehensive multialignments to analyze relevant 16S sequences for most clinically relevant pathogens and closely related environmental species. Although more 16S sequence data are available for human pathogens within public/private databases than other gene targets, one must recognize that few to no sequences (i.e., defined as ≤5 individual 16S sequences/species currently deposited in GenBank [NCBI]) have been published for a wide variety of the pathogenic organism/microorganism groups outlined here. The statistics about genus homology, shown in Tables 3 to 12, were generated by using the best representative sequences for each species of optimal length (where available) from GenBank/NCBI (where available), grouping them by genus and then aligning them to cover at least ∼50 to 1,200 bp of the 16S gene using MAFFT V7 (148), excluding sequences of species not covering these positions. Each alignment was analyzed by column/position: a column where all the species-sequences have the same nucleotide was counted as an identical position, and a column where at least one species-sequence had a gap or a different nucleotide was counted as a divergent position. The counting started at the first common position of all sequences and stopped at the last common position to avoid recording diversity where sequences were shorter. The percentages give an idea about the homology of a genus; in general, genera with few species tend to display a higher degree of homology.
TABLE 3.
Summary of ability of 16S rRNA gene target to identify clinically relevant Staphylococcaceae, Micrococcaceae, and Dermacoccaceae
| Genus | No. of sequences in the genus MSAa | Total no. of tested positions | No. of identical positions | No. of divergent positions | % Identity |
|---|---|---|---|---|---|
| Staphylococcus | 46 | 1,387 | 1,237 | 150 | 89.19 |
| Micrococcus | 9 | 1,386 | 1,314 | 72 | 94.81 |
| Citricoccus | 3 | 1,457 | 1,435 | 22 | 98.49 |
| Kytococcus | 2 | 1,445 | 1,419 | 26 | 98.20 |
| Dermacoccus | 4 | 1,471 | 1,438 | 33 | 97.76 |
| Kocuria | 20 | 1,429 | 1,212 | 217 | 84.81 |
| Rothia | 8 | 1,394 | 1,273 | 121 | 91.32 |
| Luteipulveratus | 0 | 0 | |||
| Auritidibacter | 0 | 0 |
MSA, multisequence alignment.
TABLE 4.
Summary of ability of 16S rRNA gene target to identify clinically relevant Streptococcaceae, Lactobacillaceae, Leuconostocaceae and Enterococcaceae
| Genus | No. of sequences in the genus MSAa | Total no. of tested positions | No. of identical positions | No. of divergent positions | % Identity |
|---|---|---|---|---|---|
| Streptococcus | 87 | 1,360 | 1,069 | 291 | 78.60 |
| Enterococcus | 44 | 1,470 | 1,270 | 200 | 86.39 |
| Aerococcus | 7 | 1,473 | 1,277 | 196 | 86.69 |
| Abiotrophia-Granulicatella | 0 | 0 | |||
| Dolosigranulum | 0 | 0 | |||
| Helcococcus | 2 | 1,346 | 1,262 | 84 | 93.76 |
| Facklamia | 6 | 1,400 | 1,203 | 197 | 85.93 |
| Gemella | 6 | 1,372 | 1,237 | 135 | 90.16 |
| Lactococcus | 11 | 1,454 | 1,185 | 269 | 81.50 |
| Leuconostoc | 13 | 1,491 | 1,334 | 157 | 89.47 |
| Pediococcus | 11 | 1,488 | 1,309 | 179 | 87.97 |
| Vagococcus | 10 | 1,383 | 1,218 | 165 | 88.07 |
| Globicatella | 0 | 0 |
MSA, multisequence alignment.
TABLE 5.
Summary of ability of 16S rRNA gene target to identify clinically relevant Actinomycetaceae, Corynebacteriaceae, Micrococcaceae, Microbacteriaceae, Paenibacillaceae, Cellulomonadaceae, Listeriaceae, Intrasporangiaceae (Dermatobactereaceae and Dermatophilaceae), Pseudonocardiaceae, Bacillaceae, Erysipelothrichaceae, Promicromonosporaceae, Dermabacteriaceae, and Brevibacteriaceae
| Genus | No. of sequences in the genus MSAa | Total no. of tested positions | No. of identical positions | No. of divergent positions | % Identity |
|---|---|---|---|---|---|
| Arcanobacterium | 8 | 1,356 | 1,235 | 121 | 91.08 |
| Arthrobacter | 41 | 1,362 | 1,079 | 283 | 79.22 |
| Bacillus | 217 | 1,419 | 710 | 709 | 50.04 |
| Geobacillus | 11 | 1,500 | 1,404 | 96 | 93.60 |
| Brachybacterium | 19 | 1,397 | 1,247 | 150 | 89.26 |
| Brevibacterium | 28 | 1,406 | 978 | 428 | 69.56 |
| Corynebacterium | 93 | 1,399 | 944 | 455 | 67.48 |
| Cellulosimicrobium | 5 | 1,442 | 1,377 | 65 | 95.49 |
| Curtobacterium | 7 | 1,484 | 1,094 | 390 | 73.72 |
| Erysipelothrix | 4 | 1,512 | 1,386 | 126 | 91.67 |
| Exiguobacterium | 16 | 1,370 | 1,217 | 153 | 88.83 |
| Geobacillus | 11 | 1,500 | 1,404 | 96 | 93.60 |
| Knoellia | 5 | 1,447 | 1,395 | 52 | 96.41 |
| Janibacter | 8 | 1,381 | 1,306 | 75 | 94.57 |
| Leifsonia | 11 | 1,409 | 1,277 | 132 | 90.63 |
| Listeria | 12 | 1,170 | 1,070 | 100 | 91.45 |
| Microbacterium | 91 | 1,277 | 1,004 | 273 | 78.62 |
| Oerskovia | 4 | 1,467 | 1,460 | 7 | 99.52 |
| Paraoerskovia | 2 | 1,477 | 1,452 | 25 | 98.31 |
| Paenibacillus | 187 | 1,310 | 814 | 496 | 62.14 |
| Pseudoclavibacter | 5 | 1,418 | 1,274 | 144 | 89.84 |
| Kocuria | 20 | 1,429 | 1,212 | 217 | 84.81 |
| Trueperella | 4 | 1,446 | 1,361 | 85 | 94.12 |
MSA, multisequence alignment.
TABLE 6.
Summary of ability of 16S rRNA gene target to identify clinically relevant Enterobacterales (formerly Enterobacteriaceae)
| Genus | No. of sequences in the genus MSAa | Total no. of tested positions | No. of identical positions | No. of divergent positions | % Identity |
|---|---|---|---|---|---|
| Escherichia | 4 | 1,463 | 1,435 | 28 | 98.09 |
| Shigella | 4 | 1,539 | 1,530 | 9 | 99.42 |
| Pantoea | 13 | 1,424 | 1,332 | 92 | 93.54 |
| Klebsiella | 7 | 1,379 | 1,322 | 57 | 95.87 |
| Raoultella | 4 | 1,453 | 1,426 | 27 | 98.14 |
| Cronobacter | 7 | 1,548 | 1,499 | 49 | 96.83 |
| Enterobacter | 11 | 1,428 | 1,340 | 88 | 93.84 |
| Proteus | 5 | 1,466 | 1,448 | 18 | 98.77 |
| Citrobacter | 13 | 1,456 | 1,376 | 80 | 94.51 |
| Salmonella | 2 | 1,505 | 1,480 | 25 | 98.34 |
| Morganella | 0 | 0 | |||
| Providencia | 9 | 1,436 | 1,370 | 66 | 95.40 |
| Cedecea | 3 | 1,466 | 1,446 | 20 | 98.64 |
| Edwardsiella | 5 | 1,549 | 1,537 | 12 | 99.23 |
| Hafnia | 3 | 1,415 | 1,371 | 44 | 96.89 |
| Serratia | 18 | 1,379 | 1,265 | 114 | 91.73 |
| Yersinia | 18 | 1,449 | 1,395 | 54 | 96.27 |
MSA, multisequence alignment.
TABLE 7.
Summary of ability of 16S rRNA gene target to identify clinically relevant Pseudomonaceae, Burkholderiaceae, Ralstoniaceae, Moraxellaceae, Xanthomonadaceae, Comamonadaceae, Alcaligenaceae, Caulobacteraceae, Acetobacteraceae, Methylobacteriaceae, Neisseriaceae, Flavobacterium, Rhizobiaceae, Rhodobacteriaceae, Flavobactereaceae, Rhodospirillaceae, Brucellaceae, Sphingobacteriaceae, and Legionellaceae
| Genus | No. of sequences in the genus MSAa | Total no. of tested positions | No. of identical positions | No. of divergent positions | % Identity |
|---|---|---|---|---|---|
| Pseudomonas | 171 | 1,394 | 922 | 472 | 66.14 |
| Ralstonia | 6 | 1,546 | 1,472 | 74 | 95.21 |
| Burkholderia | 27 | 1,484 | 1,427 | 57 | 96.16 |
| Acinetobacter | 47 | 1,450 | 1,254 | 196 | 86.48 |
| Stenotrophomonas | 13 | 1,446 | 1,340 | 106 | 92.67 |
| Acidovorax | 15 | 1,434 | 1,341 | 93 | 93.51 |
| Achromobacter | 19 | 1,405 | 1,345 | 60 | 95.73 |
| Alcaligenes | 4 | 1,470 | 1,429 | 41 | 97.21 |
| Advenella | 5 | 1,431 | 1,359 | 72 | 94.97 |
| Paenalcaligenes | 2 | 1,514 | 1,375 | 139 | 90.82 |
| Kerstersia | 2 | 1,460 | 1,435 | 25 | 98.29 |
| Brevundimonas | 26 | 1,373 | 1,181 | 192 | 86.02 |
| Comamonas | 19 | 1,436 | 1,229 | 207 | 85.58 |
| Cupriavidus | 17 | 1,441 | 1,339 | 102 | 92.92 |
| Delftia | 5 | 1,496 | 1,340 | 156 | 89.57 |
| Asaia | 7 | 1,397 | 1,378 | 19 | 98.64 |
| Methylobacterium | 33 | 1,394 | 1,173 | 221 | 84.15 |
| Roseomonas | 26 | 1,390 | 1,104 | 286 | 79.42 |
| Neisseria | 20 | 1,325 | 1,168 | 157 | 88.15 |
| Bergeyella | 0 | 0 | |||
| Weeksella | 2 | 1,482 | 1,457 | 25 | 98.31 |
| Myroides | 8 | 1,420 | 1,245 | 175 | 87.68 |
| Legionella | 51 | 1,315 | 969 | 346 | 73.69 |
| Chryseobacterium | 93 | 1,379 | 1,025 | 354 | 74.33 |
| Elizabethkingia | 3 | 1,521 | 1,492 | 29 | 98.09 |
| Empedobacter | 2 | 1,470 | 1,435 | 35 | 97.62 |
| Rhizobium | 66 | 1,504 | 966 | 538 | 64.23 |
| Bordetella | 14 | 1,456 | 1,395 | 61 | 95.81 |
| Oligella | 2 | 1,488 | 1,441 | 47 | 96.84 |
| Haematobacter | 2 | 1,388 | 1,387 | 1 | 99.93 |
| Agrobacterium | 6 | 1,377 | 1,319 | 58 | 95.79 |
| Moraxella | 15 | 1,431 | 1,193 | 238 | 83.37 |
| Paracoccus | 49 | 1,347 | 1,064 | 283 | 78.99 |
| Psychrobacter | 34 | 1,401 | 1,239 | 162 | 88.44 |
| Ochrobactrum | 17 | 1,393 | 1,219 | 174 | 87.51 |
| Sphingobacterium | 37 | 1,418 | 991 | 427 | 69.89 |
| Pannonibacter | 2 | 1,406 | 1,378 | 28 | 98.01 |
| Brucella | 7 | 1,406 | 1,397 | 9 | 99.36 |
| Pseudochrobactrum | 4 | 1,386 | 1,376 | 10 | 99.28 |
MSA, multisequence alignment.
TABLE 8.
Summary of ability of 16S rRNA gene target to identify clinically relevant Pasteurellaceae, Francisellaceae, Bartonellaceae, Flavibacteriaceae, Neisseriaceae, Porphyromonadaceae, Sutterellaceae, Cardiobacteriaceae, and Leptotrichiaceae
| Genus | No. of sequences in the genus MSAa | Total no. of tested positions | No. of identical positions | No. of divergent positions | % Identity |
|---|---|---|---|---|---|
| Actinobacillus | 17 | 1,350 | 1,121 | 229 | 83.04 |
| Aggregatibacter | 3 | 1,460 | 1,351 | 109 | 92.53 |
| Bartonella | 26 | 1,382 | 1,256 | 126 | 90.88 |
| Cardiobacterium | 2 | 1,508 | 1,459 | 49 | 96.75 |
| Capnocytophaga | 8 | 1,460 | 1,219 | 241 | 83.49 |
| Haemophilus | 13 | 1,367 | 1,084 | 283 | 79.30 |
| Kingella | 5 | 1,410 | 1,286 | 124 | 91.21 |
| Eikenella | 0 | 0 | |||
| Pasteurella | 12 | 1,377 | 1,128 | 249 | 81.92 |
| Dysgonomonas | 7 | 1,414 | 1,203 | 211 | 85.08 |
| Paludibacter | 2 | 1,470 | 1,346 | 124 | 91.56 |
| Streptobacillus | 5 | 1,416 | 1,299 | 117 | 91.74 |
MSA, multisequence alignment.
TABLE 9.
Summary of ability of 16S rRNA gene target to identify clinically relevant Campylobacteraceae, Helicobacteraceae, and Leptospiraceae
| Genus | No. of sequences in the genus MSAa | Total no. of tested positions | No. of identical positions | No. of divergent positions | % Identity |
|---|---|---|---|---|---|
| Campylobacter | 27 | 1,672 | 1,108 | 564 | 66.27 |
| Helicobacter | 38 | 1,828 | 1,079 | 749 | 59.03 |
| Arcobacter | 21 | 1,403 | 1,182 | 221 | 84.25 |
| Leptospira | 21 | 1,319 | 1,113 | 206 | 84.38 |
MSA, multisequence alignment.
TABLE 10.
Summary of ability of 16S rRNA gene target to identify clinically relevant Clostridiaceae, Actinomycetaceae, Atopobaceae, Bifidobactereaceae, Rumminococcaceae, Eggerthellaceae, Eubacteriaceae, Lactobacillaceae, Coriobacteriaceae, Peptoniphilaceae, Peptostreptococcaceae, Propionibacteriaceae, and Halbacteriaceae
| Genus | No. of sequences in the genus MSAa | Total no. of tested positions | No. of identical positions | No. of divergent positions | % Identity |
|---|---|---|---|---|---|
| Actinobaculum | 2 | 1,474 | 1,405 | 69 | 95.32 |
| Actinotignum | 3 | 1,480 | 1,370 | 110 | 92.57 |
| Actinomyces | 44 | 1,427 | 932 | 495 | 65.31 |
| Anaerosphaera | 0 | 0 | |||
| Atopobium | 5 | 1,442 | 1,276 | 166 | 88.49 |
| Olsenella | 4 | 1,452 | 1,329 | 123 | 91.53 |
| Bifidobacterium | 46 | 1,407 | 1,086 | 321 | 77.19 |
| Blautia | 8 | 1,460 | 1,259 | 201 | 86.23 |
| Clostridium | 135 | 1,557 | 723 | 834 | 46.44 |
| Hungatella | 0 | 0 | |||
| Robinsoniella | 0 | 0 | |||
| Eggerthella | 2 | 1,428 | 1,372 | 56 | 96.08 |
| Paraeggerthella | 0 | 0 | |||
| Eubacterium | 22 | 1,394 | 772 | 622 | 55.38 |
| Filifactor | 2 | 1,526 | 1,402 | 124 | 91.87 |
| Lactobacillus | 171 | 1,431 | 828 | 603 | 57.86 |
| Megasphaera | 8 | 1,532 | 1,346 | 186 | 87.86 |
| Peptoniphilus | 9 | 1,427 | 1,095 | 332 | 76.73 |
| Anaerosphaera | 0 | 0 | |||
| Peptococcus | 2 | 1,488 | 1,431 | 57 | 96.17 |
| Finegoldia | 0 | 0 | |||
| Parvimonas | 0 | 0 | |||
| Propionibacterium | 5 | 1,444 | 1,277 | 167 | 88.43 |
| Ruminococcus | 10 | 1,412 | 997 | 415 | 70.61 |
| Slackia | 6 | 1,338 | 1,109 | 229 | 82.88 |
| Solobacterium | 0 | 0 | |||
| Turicibacter | 0 | 0 |
MSA, multisequence alignment.
TABLE 11.
Summary of ability of 16S rRNA gene target to identify clinically relevant Bacteroideaceae, Desulfovibrionaceae, Veillonellaceae, Fusobacteriaceae, Leptotrichiaceae, Acidaminococcaceae, Porphyromonaceae, Prevotellaceae, Selenomonadaceae, and Sutterellaceae
| Genus | No. of sequences in the genus MSAa | Total no. of tested positions | No. of identical positions | No. of divergent positions | % Identity |
|---|---|---|---|---|---|
| Bacteroides | 44 | 1,466 | 783 | 683 | 53.41 |
| Parabacteroides | 8 | 1,473 | 1,271 | 202 | 86.29 |
| Macellibacteroides | 0 | 0 | |||
| Alistipes | 6 | 1,492 | 1,291 | 201 | 86.53 |
| Dialister | 4 | 1,511 | 1,326 | 185 | 87.76 |
| Veillonella | 9 | 1,468 | 1,317 | 151 | 89.71 |
| Bilophila | 0 | 0 | |||
| Desulfovibrio | 49 | 1,381 | 820 | 561 | 59.38 |
| Fusobacterium | 11 | 1,450 | 1,273 | 177 | 87.79 |
| Acidaminococcus | 2 | 1,564 | 1,501 | 63 | 95.97 |
| Anaerobiospirillum | 2 | 1,475 | 1,367 | 108 | 92.68 |
| Porphyromonas | 18 | 1,387 | 946 | 441 | 68.20 |
| Prevotella | 49 | 1,445 | 958 | 487 | 66.30 |
| Selenomonas | 9 | 1,410 | 1,078 | 332 | 76.45 |
| Mobiluncus | 2 | 1,498 | 1,460 | 38 | 97.46 |
| Odoribacter | 3 | 1,450 | 1,179 | 271 | 81.31 |
| Butyricimonas | 4 | 1,485 | 1,375 | 110 | 92.59 |
| Sutterella | 3 | 1,456 | 1,343 | 113 | 92.24 |
MSA, multisequence alignment.
TABLE 12.
Summary of ability of 16S rRNA gene target to identify clinically relevant aerobic actinomycetes and Mycobacterium
| Genus | No. of sequences in the genus MSAa | Total no. of tested positions | No. of identical positions | No. of divergent positions | % Identity |
|---|---|---|---|---|---|
| Actinomadura | 55 | 1,481 | 1,090 | 391 | 73.60 |
| Gordonia | 33 | 1,369 | 1,194 | 175 | 87.22 |
| Nocardia | 102 | 1,335 | 1,093 | 242 | 81.87 |
| Nocardioides | 88 | 1,396 | 1,052 | 344 | 75.36 |
| Nocardiopsis | 40 | 1,365 | 1,138 | 227 | 83.37 |
| Rhodococcus | 42 | 1,353 | 1,076 | 277 | 79.53 |
| Segniliparus | 2 | 1,457 | 1,441 | 16 | 98.90 |
| Streptomyces | 559 | 1,467 | 936 | 531 | 63.80 |
| Tsukamurella | 7 | 1,475 | 1,451 | 24 | 98.37 |
| Mycobacterium | 180 | 1,370 | 1,038 | 332 | 75.77 |
MSA, multisequence alignment.
The lack of currently available 16S sequence data is a serious limitation to comprehensive clinical pathogen identification (it is an even bigger problem with alternative targets, such as rpoB) but also has broader implications for reliance on this target for metagenomics and microbiome studies. One should be careful when a species is represented by only one or a few sequences, especially if these few sequences differ a lot. One solution to this problem is to apply high-precision sequencing of nearly full-length 16S, either by Sanger sequencing using appropriate primers or using a next-generation sequencing protocol (149), while focusing on sequence quality. There is also a high degree of complete similarity across the length of 16S for many other organism/microorganism groups that does not allow identification to the species level for some or all species within certain genera. The detailed analyses of 16S sequences for clinically relevant bacteria are outlined in the following sections.
Staphylococcus and related aerobic Gram-positive cocci.
Table 3 outlines the 16S sequence diversity for clinically relevant genera within the Staphylococcaceae, Micrococcaceae, and Dermacoccaceae families. Comparison of the number of identical versus divergent 16S positions for various genera shows a wide range of percent identity, with Kocuria and Micrococcus being the most divergent genera. However, within each of these clinically important genera, several species cannot be reliably identified based on 16S analysis. In aerobic GPC groups, the most variability occurs in the V6 region and beyond, so that species-level differentiation often requires sequencing longer stretches of 16S sequences to include these regions. An alternative target such as rpoB is needed to obtain a reliable species-level identification for all organisms and groups within the Micrococcaceae and Dermacoccaceae; however, there are only a few full-length rpoB sequences available for most genera, which is currently insufficient for implementing a differentiation scheme.
Staphylococcus is a very homogeneous genus, and sequencing of a longer stretch (up to 1,060 bp) of the 16S is recommended to differentiate species with enough base pair mismatches to increase certainty (5). S. aureus and S. lugdunensis can, however, be differentiated by 16S sequence variability in the first 500 bp of 16S. Many coagulase-negative staphylococci (CoNS) are closely related and, due to genetic similarity across the 16S gene, cannot be differentiated with certainty, even with a longer 16S sequence (5, 150). Some species, such as S. capitis/S. caprae or S. agnetis/S. hyicus, have identical 16S sequences, except for some facultative base pair mismatches in regions V6 and V7. Others, such as S. pasteuri/S. warneri or S. carnosus/S. piscifermentans, have either identical or nearly identical 16S sequences, so this target gene cannot be used for differentiation. Limited 16S sequence data are currently available for several Staphylococcus species isolated from human (S. massiliensis [151, 152]), animal, and/or environmental sources (S. felis [153], S. fleuretti [154], S. lutrae [155], S. microti [156], S. muscae [157], S. rostri [603], S. simiae [158] and S. stepanovicii [159]). It should also be noted that S. massiliensis is closely related to S. piscifermentans, S. condimenti, S. carnosus subsp. carnosus, S. carnosus subsp. utilis, and S. simulans (151).
Micrococcus and Citricoccus are closely related genera, but they can be distinguished based on 16S sequence variability within the first ∼500 bp of 16S (5). Hypervariable regions within V2 and V6 allow differentiation of Micrococcus spp. and most Citricoccus spp. C. muralis and C. nitriphenolicus cannot be distinguished by 16S, as their sequences only differ by a single base pair mismatch in V2. Limited 16S sequence data are currently available for the environmental organism Micrococcus lactis (160), which has recently been moved to a new genus, Neomicrococcus, which also includes N. aestuarii (formerly known as Zhihengliuella aerstuarii) (161). Limited 16S sequence data are currently available for most environmental Citricoccus spp. (i.e., C. muralis, C. nitrophenolicus, C. parietis, and C. zhacaiensis) except for C. alkalitolerans. “C. massiliensis” is a new bacterial species recently isolated from human skin by culturomics whose 16S sequence has a high degree of identity (98.61%) with C. nitrophenolicus (162).
Dermacoccus spp. have high-level identity (“highly identical”) with regard to 16S, with only a few facultative base pair mismatches in region V6. Dermacoccus spp. cannot be distinguished based on 16S sequence variability within the first ∼500 bp (5). D. nishihomiyaensis is an important part of the skin microbiome whose depletion may play a role in atopic dermatitis (163). Limited 16S sequence data are currently available for the environmental species D. abyssi, D. barathri, and D. profundi, but the former two species have identical 16S sequences (164, 165). D. barathri, however, can cause rare opportunistic infections in humans (166). The Dermacoccus genus is also closely related to the environmental organism Luteipulveratus mongoliensis (167) according to the limited 16S sequence data available for the latter species. Kytococcus is a highly identical genus including animal and environmental species, such as K. aerolatus, K. sendentarius, and K. schroeteri (168). Although Kytococcus species have mainly been isolated from the environment, K. schroeteri causes human infection, including endocarditis and osteomyelitis (169, 170). Kytococcus aerolatus and K. schroeteri are identical, with some facultative mismatches in region V6 (5).
Kocuria and Rothia genera can be differentiated using a longer 16S sequence (1,060 bp). Kocuria species are best differentiated by variability in regions V1, V2, and V6. K. rhizophila and K. arsenatis have identical 16S sequences. Limited 16S sequence data are also currently available for K. arsenatis (171) and several other environmental Kocuria spp., including K. aegyptia (172), K. atrinae (173), K. carniphila (formerly K. varians) (174), K. gwangalliensis (175), K. halotolerans (176), K. himachalensis (177), K. koreensis (178), and K. salsicia (179). Several Kocuria spp. inhabit the skin microbiome of animals and humans (180), and some have recently been identified as causes of human infection, including K. rosea, K. carniphila, and K. massiliensis, but limited 16S sequence data are currently available for these species (181). Rothia aeria and R. dentocariosa can be differentiated by variability in regions V4 and V6 (5). Limited 16S sequence data are also available for Rothia endophytica, found in plants (182).
Limited 16S sequence data are currently available for Auritidibacter ignavus (i.e., ear swab from a man with otitis externa) (183). Auritidibacter is closely related based on 16S sequences to several Kocuria spp., including K. atrinae, K. rosea, K. polaris, and K. palustris. Other closely related species include Yaniella soli (184), Y. flava (185), Arthrobacter cumminsii (186), and Calidifontibacter indicus (187). However, except for A. cumminsii, only one 16S sequence is available for all these organisms. A longer 16S sequence (1,060 bp) allows identification of A. ignavus based on variability across the entire gene, but identification cannot be reliably made from shorter 16S sequences due to similarity in the first ∼500 bp of the gene and the limited availability of sequence data (5).
Streptococcus, Streptococcus-like organisms, and Enterococcus.
Table 4 outlines 16S sequence diversity for clinically relevant genera within the Streptococcaceae, Lactobacillaceae, Leuconostocaceae, and Enterococcaceae families. Comparison of the number of identical versus divergent 16S positions for various genera shows a wide range of percent identity, with Streptococcus and Lactococcus being the most divergent genera. However, within each of these clinically important genera are several species that cannot be reliably identified based on 16S analysis. Approximately 20% of Streptococcus species cannot be distinguished using 16S. Hypervariable regions in Streptococcus species 16S sequences occur within V1-3 and V6, so that many species can only be differentiated by distinct base pair mismatches across these variable regions. Therefore, a long 16S sequence (i.e., ∼1,060 bp) is recommended for a reliable species-level identification (5). Clinically important pathogens S. pneumoniae and S. pseudopneumoniae are very closely related but can be differentiated from each other and from S. mitis via one or two mismatches in the region between ∼600 and 900 bp (5). Differentiation by rpoB is also compromised by high genetic similarity among these closely related Streptocccus spp. (66). Most S. viridans groups (S. mitis, S. salivarius, S. bovis, and S. mutans), with the exception of the S. anginosus group, are closely related, and, due to similarity across 16S, they cannot be differentiated, even with a longer 16S sequence (5). Limited 16S sequence data are currently available for S. acidominimus (188), S. devriesei (189), and S. massiliensis (190). Beta-hemolytic streptococci within the Lancefield typing scheme can be identified by sequence variability within the first ∼500 bp of 16S (5). Aerococcus spp. can be differentiated by 16S variability in regions V2 and V7, but the clinical pathogens A. viridans and A. urinae have identical 16S sequences (5). Limited 16S sequence data are also currently available for A. sanguinicola (191), A. suis (192), and A. urinaehominis (193).
Abiotrophia and Granulicatella can be differentiated due to 16S sequence variability within regions V1-3 and V6 (5). Abiotrophia-Granulicatella genera are, however, closely related to Facklamia based on 16S sequence data. Limited 16S sequence data are currently available for G. balaenopterae (194) and most Facklamia spp., including human isolates (i.e., F. hominis, F. ignava, F. languida, F. sourekii, and F. tabacinasalis) (195) and F. miroungae (196). Facklamia spp. are highly identical, but variability in 16S V1-3 and V6 allows differentiation; however, analyses across these regions are required to ensure accuracy. Dolosigranulum pigrum (197, 198) is closely related to D. paucivorans (199), and the Facklamia, Globicatella, Helcococcus, and Ignavigranum genera are all reported to cause human infections (195, 198, 200–202). Globicatella sanguinis and G. sulfidifaciens share identical 16S sequences and, thus, cannot be differentiated (5). Limited 16S sequence data are currently available for all of these genera/species, with the exception of Helcococcus kunzii (202) and H. ovis (203). A single 16S sequence is available for Ignavigranum ruoffiae (200).
Alloiococcus otitis (204) is closely related to Alkalibacterium spp. based on 16S sequence analyses (5). Limited 16S sequence data are currently available for A. otitis and environmental species, including Alkalibacterium pelagium and A. thallasium (205).
Gemella spp. can be differentiated by 16S sequence variability within regions V2, V3, and V6 (5). Limited 16S sequence data are currently available for G. asaccharolytica, G. bergeri, and G. cuniculi (206, 207).
Lactococcus spp. can be differentiated by 16S sequence variability within regions V1-3 and V6 (5). Closely related species, such as L. fujiensis and L. chungangensis, differentiate in region V6 alone (5, 208). Others, such as L. garvieae and L. formosensis, are almost identical within 16S. Lactococcus spp. are closely related to several Streptococcus spp. Limited 16S sequence data are currently available for several human/animal species (L. plantarum [209], S. caballi and S. henryi [210], S. danieliae [211], S. merionis [212], S. porcorum [213], S. saliviloxodontae [214], S. entericus [215], and S. lactarius [216]) and environmental species (L. taiwanensis [217], L. hircilactis and L. laudensis [218], L. fujiensis, L. chungangensis [219], and L. formosensis [220]).
Leuconostoc is a very homogeneous genus within 16S, but long sequence stretches spanning V1-V7 (∼1,060 bp) allow accurate species-level identification (5). L. fallax shows an insertion in region V1. L. citreum and L. holzapfeli are homologous and cannot be differentiated by 16S. L. mesenteroides and L. pseudomesenteroides have highly identical 16S sequences and are identical within the first ∼500 bp of region V1-V3. Limited 16S sequence data are currently available for L. kimchi (221), L. lactis (222), L. miyukkimchii (223), and L. palmae (224).
Pediococcus is highly identical within 16S, but distinct base pair mismatches distributed over regions V1-V7 (∼1,060 bp) allow differentiation, whereas region V3 is less helpful for some species due to identical or almost identical sequence similarity (i.e., P. damnosus, P. ethanolidurans, P. inopinatus, and P. parvulus) (5). Limited 16S sequence data are currently available for several environmental species, including P. argentinicus (225), P. cellicola (226), and P. siamensis (227).
Vagococcus is best differentiated by 16S variability within regions V1 and V2 (5). V. carniphilus and V. fluvialis are closely related, and differentiation within the first ∼500 bp requires good-quality sequencing data. Limited 16S sequence data are currently available for several environmental species, including V. acidifermentans (228), V. elongatus (229), V. entomophilus (230), V. fessus (231), and V. penaei (232).
Weisella viridescens, W. cibaria, and W. confuse are known as opportunistic pathogens involved in human infections (233), and Weisella spp. can be differentiated by 16S variability within regions V1, V2, and V7 (5). W. fabalis, W. fabaria, W. ghanensis are closely related and exhibit genetic similarity over the entire 16S. Limited 16S sequence data are currently available for many environmental species, including W. ceti (234), W. beninensis (233), W. diestrammenae (235), W. fabalis (236), W. fabaria, W. ghanensis, W. jogaejeotgali, W. kandleri (237), W. oryzae, W. paramesenteroides, W. thailandensis, and W. uvarum (238).
Enterococcus spp. can be differentiated by 16S variability within regions V1-3, whereas region V6 is highly genetically identical among species (5, 239). In general, 16S sequences are highly identical among Enterococcus spp., and quite a number of species cannot be differentiated, including E. haemoperoxindus/E. moraviensis, E. devriesei/pseudoavium/viikkiensis/xiangfangensis, E. avium/gilvus/raffinosus/malodoratus, E. casseliflavus/gallinarum, and E. durans/hirae/lactis (5). Sequencing of an alternative target, such as rpoB, is promising (66), with the caveat that for many species, appropriate sequences are still missing in the public domain. Limited 16S sequence data are currently available for many animal, human, and environmental Enterococcus spp., including E. alcedinis, E. asini, E. caccae (240), E. camelliae, E. canintestini, E. eurekensis, E. haemoperosidus, E. lemanii, E. moraviensis, E. olivae, E. pallens (241), E. phoeniculicola, E. plantarum, E. quebecensis, E. ratti, E. rivorum, E. rotai, E. saccharolyticus, E. termitis, E. ureasiticus, E. ureilyticus, and E. villorum (242).
Aerobic Gram-positive bacilli.
Table 5 outlines 16S sequence diversity for clinically relevant genera within the Actinomycetaceae, Corynebacteriaceae, Micrococcaceae, Microbacteriaceae, Paenibacillaceae, Cellulomonadaceae, Listeriaceae, Intrasporangiaceae, Pseudonocardiaceae, Bacillaceae, Erysipelothrichaceae, Promicromonosporaceae, Dermabacteriaceae, and Brevibacteriaceae families. Comparison of the number of identical versus divergent 16S positions for various genera shows a wide range of percent identity, with Bacillus and Corynebacterium being the most divergent genera. However, within each of these clinically important genera, several species cannot be reliably identified based on 16S analysis. Approximately ∼35% of Corynebacterium species cannot be distinguished using 16S.
Arcanobacterium spp. can be differentiated by variability within 16S regions V1-3 and V6 (while A. phocae and A. phocisimile differentiate only by a few mismatches) and for Trueperella spp. in regions V1-3 (5). The 4 species of Trueperella (T. abortisuis, T. bernardiae, T. bonsai, and T. pyogenes) can be differentiated in 16S regions V1-3 by only a few nucleotide insertions/deletions. A 16S sequence should be analyzed across these regions for differentiation of these species; the rest of the 16S is highly identical and not helpful for differentiation. Limited 16S sequence data are currently available for several animal species, including A. canis, A. hippocoleae, A. phocisimile, and T. bonsai (243). Arthrobacter spp. can be differentiated by variability within 16S regions V1, V2, V3, and V6, except for A. koreensis and A. luteolus. Arthrobacter spp. frequently show insertions/deletions in region V3 (5). Limited 16S sequence data are available for several clinical and environmental species, including A. albus (244), A. sanguinis and A. soli (245), A. halodurans (246), and A. tecti (247). A. pascens and Pseudarthrobacter oxydans 16S sequences are similar, but they can be differentiated in 16S region V6.
Bacillus can generally be differentiated by variability in 16S within regions V1-3 and V6 (5). Many Bacillus spp. are highly related, and long stretches of 16S (∼1,060 bp) should be analyzed for a definitive identification. B. anthracis, B. cereus, B. wiedmannii, and B. thuringensis have almost identical 16S sequences and cannot be differentiated using 16S (248). Geobacillus is rarely isolated from clinical specimens and is a highly genetically identical genus, with many closely related species (249). Species-level differentiation requires analyses of a longer 16S sequence spanning variable regions V1-3, V6, and V7 but is limited to very few mismatches over several variable regions (5). Limited 16S sequence data are currently available for several environmental species, including G. galactosidasium, G. jurassicus, G. thermantarcticus, and G. vulcani (250). Several Paenibacillus spp. have been reported to cause human infection, although others are recognized as common contaminants of clinical specimens (251). Paenibacillus spp. can be differentiated by 16S variability within regions V1, V2, V3, and V6, but closely related species may require analysis of full-length 16S sequences (5). Limited 16S sequence data are currently available for several environmental species, including P. brasilensis (252).
Brachybacterium can be differentiated by 16S variability within regions V1, V2, and V6 (5). Brachybacterium spp. rarely cause human infection (253). Limited 16S sequence data are currently available for several environmental species, including B. alimentarium, B. fresconis, B. saurashtrense, B. squillarum, B. tyrofermentans, and B. zhongshanense (254, 255).
Brevibacterium can be differentiated by 16S variability within regions V1, V2, and V6/V7, with some species, such as B. casei or B. paucivorans, showing several deletions in V3 (5). B. casei is most frequently isolated from clinical isolates (256). B. frigoritolerans and B. halotolerans show distinctly different 16S sequences compared to other species within the Brevibacterium genus and, thus, may represent a subspecies or another genus. Limited 16S sequence data are currently available for several human and environmental species, including B. massliense (257), B. ravenspurgense (245), B. sanguinis and B. paucivorans (258), B. album, B. ammoniilyticum, B. antiquum, B. celere, B. daeguense, B. jeotgali, B. marinum, B. oceani, B. picturae, B. pityocampae, B. salitolerans, B. samyangense, B. sandarakinum, B. senegalense, B. siliguriense, and B. yomogidense (245).
Corynebacterium is a large genus that contains many species that are pathogenic and nonpathogenic to humans (259, 260). Many Corynebacterium spp., such as C. durum, show multinucleotide insertions in regions V1 and V3. Corynebacterium can be differentiated by 16S variability within regions V1-3 and regions V6-8 (5). Limited 16S sequence data are currently available for several human Corynebacterium spp., including C. massilense and C. mycetoides (261). Genetic analyses of 168 Corynebacterium spp. show that the rpoB target may provide additional diversity for separating some closely related species (67). Turicella otitidis (only species of this genus) is closely related to some Corynebacterium spp., and recent large-scale phylogenetic studies indicate that this organism should be moved back into the Corynebacterium genus (262). T. otitidis can be easily differentiated via 16S variability within regions V1-3 (5). This organism primarily causes acute and chronic otitis media in humans (263). Limited 16S sequence data are currently available for Turicella otitidis and closely related clinical and environmental Corynebacterium spp., including human isolates (i.e., C. freiburgense, C. hansenii, C. lipophiloflavum, C. mycetoides, C. pilbarense, C. lactis, and C. massiliense) (259, 260) and animal and environmental isolates (i.e., C. spheniscorum, C. terpenotabidum, C. nuruki, C. halotolerens, and C. deserti) (260, 264–266).
Cellulosimicrobium-Luteimicrobium-Promicromonospora genera are closely related and rarely isolated from clinical specimens but can be differentiated by 16S variability within regions V1-2 and region V6 (5). Several isolates previously identified as Oerskovia turbata are more closely related to Cellulosimicrobium, and a new species name of C. funkei was proposed (267). Limited 16S sequence data are currently available for several environmental species, including C. terreum (268), Luteimicrobium xylanilyticum, L. subarcticum, L. album, and Promicromonospora flava (269).
Cellumonas can be differentiated by 16S variability within regions V1-2 and region V6 (5). Limited 16S sequence data are currently available for several environmental species, including C. soli, C. aerilata, C. biazotea, C. bogoiensis, C. carbonis, C. shitinilytica, C. composti, C. gelida, C. humilata, C. iranensis, C. marina, C. oligotrophica, C. pakistanensis, C. persica, C. phragmiteti, C. terrae, C. uda, and C. xylanilytica (270).
Curtobacterium spp. are rarely isolated from clinical isolates but can be differentiated by sequencing a long stretch of 16S to include variability within regions V2, V3, V6, and V7 (5). Limited 16S sequence data are currently available for several species, including C. albidum (271).
Dermabacter hominis, Brachybacteria spp., Helcobacillus massiliensis, and Devriesea agamarum are closely related genera with highly identical 16S sequences, but differentiation is possible by variability in 16S regions V1, V2, V4, V6, and V7 (5, 272). B. conglomeratum and B. paraconglomeratum cannot be differentiated with certainty by 16S sequencing. Limited 16S sequence data are currently available for several human and environmental species, including H. massiliensis (273), Devriesea agamarum (274), B. squillarum, B. zhongshanense, B. fesonis, B. saurashtrense, and B. tyrofermentans (254, 255, 275).
Erysipelothrix is a genetically identical genus with a few distinct base pair mismatches across 16S within all variable regions that allow differentiation (5). E. tonsillarum and E. rhusiopathiae have very similar sequences and cannot be reliably differentiated using 16S (276). Limited 16S sequence data are currently available for E. inopinata (277).
Exiguobacterium is rarely isolated from clinical isolates, but it is a highly genetically identical genus with limited variability within 16S (278). Many closely related species should be differentiated by analyses of a longer 16S sequence spanning variable regions V1-3 and V6 (5). Limited 16S sequence data are currently available for several environmental species, including E. alkaliphilum, E. aquaticum, E. artemiae, and E. soli (279).
Knoellia spp. are rarely isolated from clinical specimens, but differentiation is allowed by variability in 16S regions V1 and V2 (5). Limited 16S sequence data are currently available for several environmental species, including K. aerolata, K. flava, and K. subterranean (280–282). Janibacter spp. are rarely isolated from clinical specimens (283, 284). Janibacter is a very homogeneous genus, but differentiation occurs by analyses of a long 16S sequence across variable regions V1-3, V6, and V7 (5, 285). Limited 16S sequence data are currently available for several environmental species, including J. alkaliphilus, J. corallicola, J. cremeus, and J. hoylei (286–288). Leifsonia spp. rarely cause human infections (289, 290). Leifsonia is another highly identical genus, but differentiation occurs by analyses of a long 16S sequence across variable regions V1-2, V3, and V6 (5). Limited 16S sequence data are currently available for several environmental Leifsonia spp. and Lysinimonas kribbensis (291), including L. antarctica, L. bigeumensis, L. lichenia, L. naganoensis, L. pindariensis, and L. psychrotolerans (292, 293).
Listeria spp. differentiation should be performed by analyses of 16S variability within regions V1, V2, V6, and V8. Highly related species, such as L. monocytogenes and L. innocua, differ by only a single distinct base pair mismatch within regions V2 and V8 (5). Limited 16S sequence data are currently available for several environmental (agricultural and natural environments) species, including L. aquatic, L. booriae, L. cornelliensis, L. fleischmanii, L. floridensis, L. grandensis, L. marthii, L. newyorkensis, L. riparia, L. rocourtiae, and L. weihenstephanensis (51, 294).
Several Microbacterium spp. cause human infections, including bacteremia and endophthalmitis (295). Microbacterium spp. differentiation should be performed by analyses of variability within a long 16S sequence covering regions V1, V2, V4, and V6, because single base pair mismatches are spread across the entire gene (5). Limited 16S sequence data are currently available for several environmental (agricultural and natural environments) species, including M. arthrosphaerae, M. marinum, M. mitrae, M. neimengense, M. pseudoresistens, M. saperdae, and M. soli (296).
Oerskovia spp. rarely cause human infections (297–299). Oerskovia-Paraoerskovia are both highly identical genera, but a few base pair mismatches allow species differentiation by variability in a longer 16S sequence across regions V1, V2, and V7 (5). O. jenensis and O. paurometabola cannot be differentiated because of complete 16S identity. Limited 16S sequence data are currently available for several environmental species, including O. jenensis, P. marina, and P. sediminicola (300–302). Pseudoclavibacter spp. is a rare cause of human infections (303–305). Pseudoclavibacter can be differentiated by 16S variability within regions V1 and V2 (5). Limited 16S sequence data are currently available for several environmental species, including P. caeni (306), P. chungangensis (307), and P. soli (308).
Rothia-Kocuria are closely related genera, and several species have been increasingly reported to cause human infections (309, 310). Differentiation can be achieved by 16S variability within regions V1, V2, and V6 (5). Limited 16S sequence data are currently available for several environmental (seawater and soil) species, including R. endophytica, K. aergyptia, K. atrinae, K. gwangalliensis, K. halotolerans, K. himachalensis, K. koreensis, and K. salsicia (173, 178, 179, 182).
Enterobacterales (formerly Enterobacteriaceae).
Enterobacteriaceae is a large, complex family that currently contains more than 30 genera and over 100 species. Although the systematic classification of Enterobacteriaceae is still being debated, a new taxonomic classification has recently been proposed for this large, complex organism group, which contains many major Gram-negative enteric pathogens. Alnajar et al. have recently proposed placing Enterobacteriaceae in the order Enterobacterales, within the class Gammaproteobacteria, based on phylogenetic analysis of the many diverse species (311). Their work supports the existence of seven distinct monophyletic clades of genera within the order, making it taxonomically relevant to divide the former Enterobacteriaceae family into seven families, including Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaveae fam. nov. In addition, this classification system would separate and distribute many clinically relevant pathogens among these families. For example, the Enterobacter-Escherichia clade is the largest group within the order Enterobacterales and consists of genera “Atlantibacter,” Buttiauxella, Cedecea, Citrobacter, Cronobacter, Enterobacter, Escherichia, Franconibacter, Klebsiella, Kluyvera, Kosakonia, Leclercia, Lelliottia, Mangrovibacter, Pluralibacter, Raoultella, Salmonella, Shigella, Shimwellia, Siccibacter, Trabulsiella, and Yokenella. The Erwinia-Pantoea clade, which is present in a monophyletic grouping with the Enterobacter-Escherichia clade, consists of the genera Erwinia, Pantoea, Phaseolibacter, and Tatumella. The Pectobacterium-Dickeya clade constis of the genera Brenneria, Dickeya, Lonsdalea, Pectobacterium, and Sodalis. The Yersinia-Serratia clade consists of the genera Chania, Ewingella, Rahnella, Rouxiella, Serratia, and Yersinia, the Hafnia-Edwardsiella clade consists of the genera Edwardsiella, Hafnia, and Obesumbacterium, the Proteus-Zenorhabdus clade consists of the genera Arseophonus, Moellerella, Morganella, Photorhabdus, Proteus, Providencia, and Xenorabdus, and the Budvicia clade consists of the genera Budvicia, Leminorella, and Pragia (311).
Although this reclassification may make taxonomic sense from a genetic perspective, it has not currently been widely adopted by clinical microbiology laboratories or the diagnostic industry that provides instrumentation, software, and databases to the diagnostic sector (312). Widespread approval of this scheme will be required by clinical microbiologists, industry partners, and their regulatory authorities before these taxonomic changes are translated into clinical practice. Clinicians will also need to be educated about taxonomy changes being reported to avoid confusion regarding antimicrobial therapy and the epidemiological significance of an organism-infection combination. Finally, the taxonomic and nomenclature changes outlined by Alnajar and colleagues do not solve the clinical problem of not being able to clearly separate E. coli from Shigella (313). This section, therefore, outlines the historically accepted genus and species names for the important human pathogens within this important organism group, because this is the nomenclature that will be in use in most clinical laboratories. Table 6 outlines the 16S sequence diversity for genera within the Enterobacterales family that commonly cause human infections (78, 314). Due to similarity within this target or a lack of available sequence data, ∼10% of these organisms cannot be identified to the species level using 16S. Escherichia-Shigella-Pantoea-Klebsiella-Raoultella-Cronobacter genera are highly identical genera (78), with only single mismatches across all 16S variable regions (5). Within the Escherichia genus, E. coli and E. fergusonii have only a few mismatches in region V1. Klebsiella spp. and Raoultella spp. can best be differentiated by variability in regions V3-V6, whereas Pantoea spp. only show variability in regions V1 and V2 (5). Several important human pathogens and nonpathogens (depending on the sample), including Escherichia coli, Shigella dysenteriae, Escherichia fergusonii, and Shigella flexneri, have highly identical 16S sequences and cannot be distinguished by sequencing or routine proteomics (MALDI-TOF) methods (5, 29, 315, 316). Alternate target sequencing using rpoB improves separation but cannot completely differentiate Escherichia coli/Shigella species (317). Thus, results obtained by 16S sequencing for one of these organisms should always be clinically correlated before reporting its presence as pathogenic; assessment of the presence of plasmids carrying toxins (e.g., by direct PCR is also helpful in this regard). Limited 16S sequence data are currently available for several human and environmental species, including Raoultella electrica (318), Cronobacter condimenti (319), Cronobacter universalis (320), Pantoea deleyi, and Pantoea wallisi (321).
Many other genera within the Enterobacteriaceae are also highly identical within 16S. Enterobacter species differentiation occurs by a few base pair mismatches across 16S variable regions V1, V3, and V6-7, whereas region V2 is not helpful (5, 322). A longer 16S sequence (∼1,060 bp) is needed to differentiate closely related Enterobacter species. Proteus 16S variability is restricted to single distinct mismatches with V2 and V5 regions (5, 323, 324). P. hauseri and P. vulgaris cannot be differentiated by 16S sequencing (325). Differentiation of Citrobacter species relies on limited genetic variability in both regions V3 and V6 (5). Limited sequence data are currently available for C. rodentium (326). Salmonella contains many serovars that are known as S. enterica (327) that do not correlate or differentiate according to specific 16S sequences (328); S. enterica, S. bongori, and S. subterranea differentiate in regions V3 and V6 by only a few base pair mismatches (5). Limited sequence data are currently available for S. subterranea (5). Morganella spp. can be differentiated by 16S variability within region V3 and by a few base pair mismatches in V2 (5). Serratia spp. can mostly be differentiated by 16S variability within regions V1, V2, V3, and V7, but S. grimesii and S. liquefaciens can only be differentiated by variability within region V7 (5, 329). Limited sequence data are currently available for S. glossinae (330, 331). Cedecea-Hafnia-Edwardsiella-Providencia genera are genetically similar, but enough variability occurs within 16S for differentiation (78, 314). Cedecea spp. can be differentiated within regions V1 and V3, Hafnia alvei and H. paralvei within regions V3 and V7, Edwardsiella spp. within regions V3 and V7 (with the exception of E. piscida and E. tarda, which have identical 16S sequences), and Providencia spp. within the combined regions V2, V3, V6, and V7 (5). Limited 16S sequence is currently available for H. paralvei (331), E. hoshinae (332), P. sneebia (333), and P. thailandensis (334). Yersinia spp. can be differentiated by 16S variability in regions V1, V2, and V6 (5, 335). Some species, such as Y. frederiksenii and Y. nurmii, are almost identical across 16S. Limited 16S sequence data are currently available for Y. entomophaga (336) and Y. pekkanenii (337).
Glucose-nonfermenting Gram-negative bacilli.
Table 7 outlines 16S sequence diversity for several clinically relevant and environmental genera of Gram-negative nonfermenters within the listed families. Comparison of the number of identical versus divergent 16S positions for various genera shows a wide range of percent identity. The most 16S sequence data are available for Pseudomonas species, and it is also one of the most divergent genera. However, within each of these clinically important nonfermenter genera are several species that cannot be reliably identified based on 16S analysis.
Many nonfermenter genera are highly identical within 16S. Pseudomonas is one of the most complex nonfermenter genera with the largest number of species (338, 339). Many Pseudomonas spp. can be differentiated by variability within the first ∼500 bp (regions V1-3), except for some rather rare environmental species. Many Pseudomonas spp. can only be differentiated by a few base pair mismatches within 16S; P. toyotomiensis (340) and P. chengduensis (341) are highly identical over the entire 16S, while P. punonensis (342), P. straminea (343), and P. argentinensis (344) are highly identical within the first ∼500 bp of 16S (5). Ralstonia is a highly identical genus that was previously classified within Pseudomonas (345). R. solanacearum and R. pseudosolanacearum cannot be differentiated by 16S sequencing (346), and R. syzygii is also closely related to these species; differentiation relies on only a few base pair mismatches (5). Limited sequence data are currently available for R. pseudosolanacearum (346). Pandoraea contains several clinical species that are emerging as important pulmonary pathogens in susceptible patients, particularly cystic fibrosis (347). Attempts to differentiate Pandoraea species requires analysis of a long stretch of the 16S gene that includes region V6. Limited 16S sequence data are currently available for several human and environmental species, including P. apista and P. pulmonicola (348), P. faecigallinarum, P. oxalativorans (347), P. thiooxydans (349), and P. vervacti (350).
Brevundimonas spp. rarely cause human infection, but B. vesticularis has increasingly been reported as a cause of bacteremia (351). Brevundimonas diminuta and B. faecalis cannot be differentiated by 16S (5). Other Brevundimonas spp. can be differentiated by precise sequencing of the first ∼650 bp via only a few base pair mismatches; B. abyssalis and B. aveniformis show a multinucleotide insertion in region V6 (5). Limited 16S sequence data are currently available for B. faecalis and B. vancanneytii (351, 352) and several environmental isolates, including B. abyssalis, B. aveniformis, B. bacteroides, B. basaltis, B. denitrificans, B. halotolerans, B. lenta, B. poindeterae, B. staleyi, B. variabilis, and B. viscosa (353).
Comamonas spp. are environmental organisms that infrequently cause human infections (354, 355); species can be differentiated within the 16S V1-3 regions (5). Although analyses of 16S sequences support the separation of Delftia from Comamonas, this phylogeny was not supported in the gyrB tree (356). Limited 16S sequence data are currently available for Comamonas spp., including C. testosterone (357), and several environmental species, C. badia, C. composti, C. granuli, C. guangdongensis, C. humi, C. kersterii, C. koreenensis, C. nitrativorans, C. terrae, C. thiooxydans, C. zonglianii, and C. odontotermitis (356).
Cupriavidus spp. are environmental organisms that have low human pathogenicity (358–360). Although analyses of 16S sequences support separation of Cupriavidus from Ralstonia, this phylogeny was not supported in the rpoB tree (356). Cupriavidus spp. can be differentiated by variability in the first ∼500 bp up to ∼650 bp (V1-4) (5). Limited 16S sequence data are currently available for several environmental species, including C. alkaliphilus, C. laharis, C. numazuensis, and C. pampae (360). Delftia is another highly identical genus that infrequently causes human infections (356, 361). There is no variability before position ∼450 bp (V3), and there are only a few base pair mismatches across the rest of 16S. D. lacustris and D. tsuruhatensis cannot be differentiated by 16S sequencing (5).
The Acinetobacter genus is very ancient and extremely diverse genus, and a recent whole-genome phylogenetic study showed that highly divergent species share more orthologues than certain strains within a species (362). Acinetobacter spp. can be differentiated by 16S variability within the first ∼500 bp up to ∼750 bp (V1-V4) (5). Limited 16S sequence data are currently available for several environmental Acinetobacter spp., including A. bohemicus, A. brisouii, A. harbinensis, A. indicus, A, kookii, A. pakisstanensis, A. puyangensis, A. gingfengensis, A. rudis, and A. variabilis (363). Stenotrophomonas maltophilia is an important opportunistic human pathogen that can be differentiated from the environmental organism S. daejeonensis by sequencing the first ∼750 bp (5, 364). Limited 16S sequence data are currently available for several environmental species, including S. daejeonensis, S. ginsengisoli, S. pavanii, and S. terrae (365).
The Burkholderia genus was recently separated into two distinct genera based on phylogenetic clustering; most animal and plant pathogens were retained in Burkholderia, and the environmental species found in soil, water, and the rhizospheres of plants were moved into a new genus, Paraburkholderia (366). Burkholderia species are important human opportunistic pathogens that cause respiratory infections. Members of the B. cepacia complex (e.g., nine genomic species, including B. cepacia, B. multivorans, B. cenocepacia, B. stabilis, B. vietnamiensis, B. dolosa, B. ambifaria, B. anthina, and B. pyrrocinia) in particular play a role in cystic fibrosis (367). B. pseudomultivorans causes human respiratory infection and was recently added to the B. cepacia complex (368). Burkolderia species are genetically identical, and many of them cannot be differentiated by 16S sequencing of regions V1-2, particularly within closely related complexes (i.e., B. cepacia complex and select agents, B. mallei/B. pseudomallei) (5); analyzing a longer 16S sequence is helpful for differentiation of some, but not all, species. B. vietnamensis, e.g., can be differentiated within the B. cepacia complex due to some 16S variability in the V6 region (5). B. metallica is also closely related to B. cepacia. Limited 16S sequence data are available for some environmental species, including P. ginsengisoli and B. pseudomultivorans (368, 369). Acidovorax is a highly homogeneous genus comprised of environmental organisms found in soil and water that are important plant pathogens (370). The genus is closely related to Burkholderia and includes several species that have been reported to cause rare human infections, including A. orzae, A. temperans, and A. avenae (371–373). Sequencing of more than the first ∼500 bp is recommended for species-level differentiation, especially between A. facilis and A. radicis (5). A. avenae, A. citrulli, A. cattleyae, and A. oryzae are highly identical and cannot be differentiated with certainty by 16S. Limited 16S sequence data are currently available for several environmental species, including A. anthurii, A. konjaci, A. radicis, A. soli, and A. wautersii.
Achromobacter is another highly identical genus comprised of clinical and environmental organisms that cause opportunistic infections in humans, particularly pulmonary infections in susceptible populations, such as cystic fibrosis (374–376). Achromobacter spp. cannot be differentiated with certainty by 16S sequencing, because there are only a few base pair mismatches around positions ∼450 (V3) and ∼1,010 (V6) (5). Therefore, if species differentiation is attempted, a long fragment (∼1,060 bp) should be sequenced across both 16S regions. Limited 16S sequence data are currently available for A. marplatensis and A. animicus (5, 374) and several environmental isolates, including A. aegrifaciens, A. anixfer, A. dolens, and A. insuavis.
Limited 16S sequence data are currently available for several clinical and environmental species in the highly related Alcaligenes-Advenella-Kerstersia-Paenalcaligenes genera (377, 378), including A. faeciporci (379), K. similis (380), P. hermetiae (381), P. hominis (382), and P. suwonensis (383).
Asaia are environmental organisms that infrequently cause infections (384, 385). Limited 16S sequence data are currently available for several Asaia spp. for several environmental species, including A. astilbis, A. platycodi, A. prunellae, and A. spathodeae (386).
Methylobacterium spp. have almost no interspecies variability in the 16S V3 region, but the V1, V2, V4, and V6 regions can be used for differentiation (5). However, some species, such as M. gregans and M. hispanicum, cannot be differentiated by 16S sequencing, and others, such as M. phyllostachyos, M. longum, and M. tardum, only differentiate by a few base pair mismatches in region V6 (around position ∼1,060). Limited 16S sequence data are currently available for several environmental Methylobacterium spp., including M. aerolatum, M. bullatum, M. cerastii, M. dankookense, M. gnaphali, M. goesingense, M. gossipiicola, M. iners, M. isbiliense, M. joetgali, M. longum, M. oxalidis, M. persicinum, M. phyllostachyos), M. pseudosasicola, M. soli, M. suomiense, M. tarhaniae, M. thuringiense, M. trifolii, and M. variabile (387).
Some Neisseria spp. are major human pathogens (388). Neisseria spp. can be differentiated within regions V2 and V3. Neisseria meningitidis and N. polysaccharea have only a few mismatches around position ∼150 bp (V2), whereas N. perflava, N. subflava, and N. flavescens have almost identical 16S sequences (5). Limited 16S sequence data are currently available for several clinical and environmental Neisseria spp., including N. animalis, N. dentiae, N. iguana, and N. wadsworthii (389).
Limited 16S sequence data are currently available for Bergeyella zoohelcum and closely related Chryseobacterium spp. (5).
Limited 16S sequence data are currently available for Weeksella virosa and closely related Empedobacter spp. (5).
Elizabethkingia meningoseptica and E. anophelis cannot be differentiated by 16S sequencing (5). Limited 16S sequence data are currently available for E. miricola (390).
Rhizobium-Agrobacterium are highly identical genera, and there is variability in the 16S regions V2, V4, and V6 (around position ∼1,050 bp) that allows species differentiation, whereas region V3 is not useful (5). Limited 16S sequence data are currently available for several plant Rhizobium-Agrobacterium spp., including R. aggregatum, R. calliandrae, R. cauense. R. endophyticum, R. fabae, R. freirei, R. halophytocola, R. jauaris, R. laguerreae, R. poessense, R. lupine, R. oryzae, R. petrolearium, R. pseudoryzae, R. selenitrireducens, R. skierniewicense, R. smilacinae, R. spaerophysae, R. straminoryzae, R. subbaronis, and R. soli (391).
Bordetella spp. cause serious human infection (392, 393). Most species share highly identical 16S sequences, and differentiation is only possible due to some variability in only regions V1 and V3. Bordetella pertussis, B. bronchiseptica, B. holmesii, and B. parapertussis have almost identical sequences and cannot be differentiated by 16S (5).
Limited 16S sequences are currently available for Oligella ureolytica (5).
Moraxella catarrhalis is the most common species isolated from clinical specimens (394). Moraxella has genetic variability over long 16S stretches, mostly beyond position ∼1,050 bp (V6) (5). Limited 16S sequence data are currently available for several clinical and environmental species, including M. lincolnii (395), M. boevrei, M. caviae, M. equi, M. oblonga, M. ovis, M. pluranimalium, and M. porci (396).
Paracoccus has 16S variability around positions ∼550 to 650 bp and position ∼1,000 bp, which allows species differentiation (5). Limited 16S sequence data are currently available for several environmental species, including P. aerstuarii, P. alkenifer, P. bengalensis, P. aeni, P. chinensis, P. fistulariae, P. haeundaensis, P. halophilus, P. huijuniae, P. isoporae, P. kocurii, P. kondraievae, P. koreensis, P. limosus, P. methylutens, P. niistensis, P. pacificus, P. rhizosphaerae, P. saliphilus, P. seriniphilus, P. solventivorans, P. sphaerophysae, P. stylophorae, P. sulfuroxidans, P. thiocyanatus, and P. tibetensis (397).
Psychrobacter-Geopsychrobacter intraspecies 16S variability is observed around base pair positions ∼100, ∼150 to 300, and ∼480 (5). Limited 16S sequence data are currently available for several environmental marine Psychrobacter/Geopsychrobacter spp., including G. electrodiphilus, P. aestuarii, P. arenosus, P. fulvigenes, P. jeotgali, P. luti, P. lutiphocae, P. proteolyticus, P. salsus, P. urativorans, and P. vallis (398).
Haematobacter spp. cannot be differentiated by 16S (5). Limited 16S sequences are currently available for H. missouriensis and H. massiliensis (399).
Myroides spp. are rare opportunistic pathogens, and infections have been reported mainly in China (400, 401). Limited 16S sequence data are currently available for several environmental marine Myroides spp., including M. guanonis, M. pelagicus, M. profundi, and M. phaeus.
Inguilinus limosus was initially isolated from patients with cystic fibrosis (402). Limited 16S sequence data are currently available for Inguilinus ginsengisoli (403).
Ochrobactrum spp. infrequently cause human infections, and misidentification can occur in the clinical laboratory (404, 405). Limited 16S sequence data are currently available for several Ochrobactrum-Paenochrobactrum-Pseudochrobactrum spp. (73).
Sphingobacterium spp. infrequently cause human infections (406). Sphingobacterium has additional genetic variability between 16S positions ∼600 and 750 bp (V4) that allow species differentiation (5). Limited 16S sequence data are currently available for several plant environmental species, including S. anhuiense, S. arenae, S. bambusae, S. caeni, S. cladoniae, S. composti, S. detergens, S. hotanense, S. kyonggiense, S. nematocida, S. pakistanense, S. pyschroaquaticum, S. shayense, S. thermophilum, S. wenxiniae, S. alimentarium, and S. lactis (407).
Pannonibacter phragmeitetus has caused bloodstream infections (408). Limited 16S sequence data are currently available for Pannonibacter indicus, an environmental organism isolated from a hot spring (409).
Brucella species are major zoonotic pathogens that cause human infections. Recent phylogenetic studies show that all Brucella species are monophyletic and closely related to the Ochrobactrum genus (410). Brucella is a highly identical genus, except for B. ceti and B. inopinata (5, 411). Limited 16S sequence data are currently available for several clinical and environmental species, including B. inopinata (412), B. microti (413), and B. pinnipedialis (411). B. melitensis cannot be differentiated by 16S (5).
Most cases of human Legionella infection (97.8%) are caused by L. pneumophila, L. longbeachae, Legionella bozemanii, and L. dumoffii (414). Legionella spp. can be differentiated by a longer 16S sequence that includes variable regions V1-6 (5). Limited 16S sequence data are currently available for several clinical (L. cardiac, L. steelei, L. tucsonensis, L. wadsworthii, L. lansingensis, and L. jordanis) (414) and water environmental species, including L. adelaidensis, L. beliardensis, L. birminhanensis, L. brunensis, L. cherrii, L. cincinnatiensis, L. drancourtii, L. dresdenensis, L. erythra, L. fairfieldensis, L. fallonii, L. geestiana, L. gratiana, L. hackeliae, L. impletisoli, L. isrealensis, L. jamestownensis, L. massiliensis, L. moravica, L. nagasackiensis, L. norrlandica, L. parisiensis, L. quateirensis, L. quinlivani, L. santicrucis, L. shakespeari, L. spiritensis, L. steigerwaltii, L. tunisiensis, L. waltersii, L. worsleiensis, and L. yabuuchiae (415).
Fastidious Gram-negative coccobacilli.
Table 8 outlines 16S sequence diversity within clinically relevant genera within the Pasteurellaceae, Bartonellaceae, Cardiobacteriaceae, Neisseriaceae, and Francisellaceae families across 108 species within 13 genera. The HACEK group of bacteria (Haemophilus spp., Aggregatibacter spp., Cardiobacterium hominis, Eikenella corrodens, and Kingella spp.) and Bartonella spp. have long been recognized as causing infective endocarditis and other human infections (416–418). Comparison of the number of identical versus divergent 16S positions for various genera shows a wide range of percent identity. The most 16S sequence data are available for Bartonella, while Haemophilus is the most divergent genus. However, within each of these clinically important genera are several species that cannot be reliably identified based on 16S analysis. Haemophilus is highly identical throughout much of 16S. H. aegyptius and H. influenzae are closely related and can only be differentiated by a few base pair mismatches within regions V2 and V4 (5). H. influenzae can only be differentiated from H. haemolyticus by a few base pair mismatches within regions V2, V5, and V6 (5). Limited 16S sequence data are currently available for several clinical and animal species, including H. haemoglobinophilus, H. parahaemolyticus (417), H. felis, and H. piscium. Aggregatibacter spp. can be differentiated by a 16S sequence covering the variable regions V2, V3, and V5. A. aprophilus and A. segnis are highly related, and 16S differentiation occurs by only a few distinct base pair mismatches within regions V2 and V3 (5, 417). Limited 16S sequence data are currently available for several Suttonella-Cardiobacterium spp., including S. indologenes (formerly Kingella indologenes) (419). Limited 16S sequence data are currently available for several Kingella-Eikenella spp., including K. potus (420). Bartonella is a highly identical genus throughout much of 16S (421). Species-level differentiation can be achieved, but only a few distinct base pair mismatches are spread over a longer 16S sequence encompassing regions V1-4 (5). Some Bartonella spp. share almost identical sequences, but they can be differentiated by only a few base pair mismatches in 16S, such as B. henselae and B. koehlerae in region V6 or B. rochalimae and B. clarridgeiae in region V2. Limited 16S sequence data are currently available for several clinical and environmental species, including B. taylorii, B. doshiae, B. elizabethae, B. rochalimae, B. acomydis, B. alsatica, B. birtlesii, B. bovis, B. callosciur, B. capreki, B. chomelii, B. clarridgeiae, B. coopersplainsensis, B. jaculi, B. japonica, B. koehlerae, B. pachyuromydia, B. queenslandensis, B. rattaustraliani, B. senagalens, B. sylvatica, and B. tribocorum (422, 423).
Actinobacillus is a highly identical genus according to 16S (424). A. equuli and A. hominis can be differentiated by 16S variability in region V3 (5). A. suis and A. ureae have highly similar 16S, but a longer sequence beyond ∼450 bp covering regions V1-V3 allows differentiation. Limited 16S sequence data are currently available for several clinical and environmental species, including A. ureae (425), A. seminis, A. anserigormium, and A. scotiae (424).
Capnocytophaga is an identical genus with regard to 16S. C. canimorsus and C. cynodegmi share similar 16S sequences but can be differentiated by a few base pair mismatches within the variable regions of V2-4 (5). Limited 16S sequence data are currently available for several species, including C. haemolytica and C. leadbetteri (426).
Dysgonomonas-Paludibacter-Parabacteroides are highly related genera according to 16S. Dysgonomonas gadei and D. termitidis can be differentiated by 16S variable regions V3-6 (5). Limited 16S sequence data are currently available for several clinical and environmental species, including D. gadei, D. mossii, D. hofstadii, D. oryzarvi, D. termitidis, Parabacteroides johnsonii, and Paludibacter propionicigenes (427, 428).
Fransicella is an identical genus throughout much of 16S. F. tularensis and F. hispaniensis are closely related but can be differentiated by a few base pair mismatches in 16S within regions V3 and V4 (5). Limited 16S sequence data are currently available for several species, including F. hispaniensis, F. guangzhouensis, and F. halioticida (429).
Pasteurella spp. can be differentiated by variability in 16S within regions V1, V2, and V3 (5). Pasteurella canis and P. dagmatis are, however, very closely related, with only a few distinct base pair mismatches in 16S within region V6. Limited 16S sequence data are currently available for several species, including P. stomatis, P. oralis, P. skyensis, P. langaaensis, and P. testudinis (430, 431).
Limited 16S sequence data are currently available for several Streptobacillus spp., including S. hongkongensis (432).
Campylobacterales.
Table 9 outlines 16S sequence diversity for clinically relevant genera within the Helicobacteraceae, Campylobacteraceae, and Leptospiraceae families. Comparison of the number of identical versus divergent 16S positions for various genera shows a wide range of percent identity. The most 16S sequence data are available for Helicobacter, and it, as well as Campylobacter, are highly divergent genera. However, within each of these clinically important genera are several species that cannot be reliably identified based on 16S analysis. Some Campylobacter species are important animal and human pathogens (433). C. coli and C. jejuni are almost identical across 16S, with very few, maybe inconsistent, base pair mismatches within region V5, whereas C. jejuni shows some distinct intraspecies variability in regions V1 and V4. Longer insertions (roughly 30 to 40 bp) were observed for C. curvus, C. sputorum, and C. rectus beyond region V2 (5). Limited 16S sequence data are currently available for several agricultural and environmental species, including C. avium, C. insulaenigrae, C. peloridis, and C. volucris (434–436). Helicobacter pylori causes peptic ulcer disease (437). H. acinonychis and H. pylori are closely related but can be differentiated by sequencing a longer stretch of 16S including regions V2 and V4-V6. Some species, such as H. bilis, H. canis, H. fenneliae, H. macacae, H. marmotae, H. mastomyrinus, and H. typhlonius, show an important insertion of sometimes >150 bp following region V2 (5). Limited 16S sequence data are currently available for several animal species, including H. acinonychis, H. anseris, H. aurati, H. baculiformis, H. branta, H. cholecystus, H. cynogastricus, H. equorum, H. marmotae, H. mastomyrinus, H. mesocricetorum, H. muridarum, H. pametensis, H. rodentium, H. salmononis, and H. typhlonius (438, 439).
Some Arcobacter species cause human infection (440), and they can be differentiated by 16S variability within regions V2, V4, and V5 (5). Limited 16S sequence data are currently available for several animal and environmental species, including A. anaerophilus, A. bivalviorum, A. cloacae, A. defluvii, A. ellisi, A. halophilus, A. marinus, A. molluscorum, A. mytili, A. nitrofigilis, A. skirrowii, A. suis, A. thereius, A. trophiarum, and A. venerupis (440).
Seven Leptospira species have been established as pathogenic, including L. interrogans, L. borgpetersenii, L. kirschneri, L. noguchii, L. santarosai, L. weilii, and L. alexanderi (441). Phylogenetic analysis of 16S sequences showed that L. alstonii and L. kmetyi clustered with the pathogenic Leptospira species, but they have not yet been isolated from humans (442, 443). Leptospira spp. can be differentiated by 16S variability within regions V2, V5, and V6 (5). Some species, such as L. licerasiae and L. wolffii, require a long 16S sequence (∼1,060 bp) for differentiation (5). Limited 16S sequence data are currently available for several human species, including L. alstonii, L. broomii, L. inadei, L. terpstrae, L. vanthielii, L. wolbachii, L. wolfii, and L. yangagawae, and environmental species, including L. idonii and L. kmetyi.
Gram-positive anaerobes.
Table 10 outlines 16S sequence diversity for clinically relevant genera within the Clostridiaceae, Actinomycetaceae, Atopobaceae, Bifidobactereaceae, Rumminococaceae, Eggerthellaceae, Eubacteriaceae, Lactobacillaceae, Coriobacteriaceae, Peptoniphilaceae, Peptostreptococcaceae, Propionibacteriaceae, and Halobacteriaceae families. Comparison of the number of identical versus divergent 16S positions for various genera shows a wide range of percent identity. The most 16S sequence data are available for Lactobacillus and Clostridium, while a high number of divergent positions are found in many of these anaerobic genera (Table 10). However, within each of these clinically important anaerobic genera, several species cannot be reliably identified based on 16S analysis. Actinotignum is closely related to some species within the Actinomyces, Trueperella, Actinobaculum, and Arcanabacterium genera within the family Actinomycetaceae (444). The genus Actinotignum recently was split off the genus Actinobaculum and contains three species, A. sanguinis, A. schaalii, and A. urinale. All three species can be differentiated by variability within 16S regions V1-3 and separated from the genus Actinobaculum (described above), although limited 16S sequence data are currently available for Actinotignum spp. The genus Actinobaculum consists of A. suis and A. massiliense, which have been split off the genus Actinomyces (445). Actinobaculum suis and A. massiliense can be differentiated by a few variable nucleotides in 16S V2 and V3 (5).
Actinomyces is a genus that currently contains 44 species (446). Actinomyces spp. can be involved in invasive infection of many tissues, including bone, that can cause sepsis, creating the need for species identification. A. israelii historically has been recognized as the main pathogen of human actinomycosis, but there is increased isolation of other potentially pathogenic species (e.g., Actinomyces naeslundii, Actinomyces meyeri, Actinomyces neuii, Actinomyces timonensis, and Actinomyces turicensis) (21, 446). Actinomyces spp. can be differentiated by 16S variability within the first ∼500 bp, covering regions V1-V3, although insertions/deletions of multiple nucleotides observed in these regions makes multialignments challenging (5). Thermactinomyces spp. are closely related to Actinomyces spp., but sequencing of a longer 16S stretch (∼1,060 bp) is recommended for differentiation (5). Limited 16S sequence data are currently available for many Actinomyces species, including several isolated from animals (A. bovis, A. bowdenii, A. catuli, A. coleocanis, A. denticolens, A. vaccimaxillae, A. weissii, and A. marimammalium) as well as several human (A. georgiae, A. graevenitzii, A. hominis, A. hongkongensis, A. nasicola, A. neuii, A. oricola, A. radicidentis, A. ruminicola, A. slackia, A. suimastitidis, A. timonensis, and Thermoactinomyces daqus) and environmental (A. naturae and T. intermedius) species (446, 447).
Atopobium is closely related to Olsenella and Coriobacterium glomerans (448, 449). Atopobium and Olsenella genera are well differentiated within 16S region V2 but are identical in other regions (5). Some species within these genera can also be differentiated by 16S variability within regions V2 and V5. Limited 16S sequence data are currently available for several human and animal species, including A. deltae and A. fossor, as well as O. umbonata and several novel Olsenella spp. recently isolated from the human colon (449).
Bifidobacterium spp. can be differentiated by 16S variability within regions V2 and V6 (5). Region V3 shows multinucleotide insertions and deletions that also may be helpful, and a longer 16S sequence (∼1,060 bp) is recommended that spans the V1-V3 regions. Limited 16S sequence data are currently available for several human, animal, and environmental species, including B. gallinarum, B. actinocoloniiforme, B. aesculapii, B. bivatii, B. bohemicum, B. bombi, B. callitrichos, B. cuniculi, B. mongoliense, B. moukalabense, B. pullorum, B. reuteri, B. saguini, B. stellenboschense, and B. tsurumiense (450, 451).
Blautia is a highly identical genus within 16S, with species-specific base pair mismatches in regions V1, 2, 4, 5, and 6. Limited 16S sequence data are currently available for several human species, including B. faecis, B. glucerasea, B. hansenii, B. luti, B. stercoris, B. wexlerae, and several novel species recently isolated from the human colon (452, 453).
Clostridium-Clostridioides-Hungatella species differentiation is feasible over all variable regions including V2 (which shows insertions/deletions), up to region V6 (5). The genus Clostridium has recently been reorganized, and the clinically relevant species C. difficile and C. mangenotii are now part of a new genus called Clostridioides (454). Limited 16S sequence data are currently available for several species, including H. hathewayi, C. difficile, C. amylolyticum, C. carboxidivorans, C. kluyveri, C. ljungdahlii, C. mangenotii, and C. sulfidigenes (455).
Eggerthella-Paraeggerthella spp. can be differentiated by 16S variability within regions V1, 2, and 5 (5). Eggerthella lenta was previously classified as Eubacterium lentum. It is the most common anaerobic Gram-positive cause of bloodstream infections and is associated with polymicrobial intra-abdominal infections (146, 456). Limited 16S sequence data are currently available for several human species, including E. sinensis and P. hongkongensis. Eubacterium-Filifactor spp. can be differentiated by 16S variability within regions V1, 2, 3, 5, and 6 (5). E. dolichum and E. tortuosum show insertions in V2 and V3. Differentiation of some species may, however, require analysis of longer 16S sequences (∼1,060 bp). Limited 16S sequence data are currently available for several species isolated from animals (E. fissicatena, E. ruminantium, and E. uniforme), the environment (E. acidaminophilum, E. aggregans, E. callanderi, and E. tarantellae), and humans, including E. barkeri, E. budayi, E. cellulosolvens, E. combesii, E. contortum, E. coprotanoligenes, E. dolichum, E. eiligens, E. hallii, E. moniliforme, E. multiforme, E. nitritogenes, E. oxidoreducens, E. plexicaudatum, E. pyruvatinorans, E. ramulus, E. siraeum, E. tortuosum, and E. ventriosum (457).
Lactobacillus species are mainly found in dairy products (e.g., Lactobacillus delbrueckii subsp. bulgaricus and L. helveticus) or in human and animal gastrointestinal tracts (e.g., Lactobacillus acidophilus and Lactobacillus gasseri), but many species demonstrate remarkable adaptability to diverse habitats (e.g., Lactobacillus plantarum, L. pentosus, L. brevis, and L. paracasei) (458). Lactobacillus are closely related and, due to currently unclear taxonomy for some subspecies (e.g., L. paracasei), a longer 16S sequence that includes V2-7 is recommended to ensure species identity (5). A recent study shows that elongation factor Tu (tuf gene), 60-kDa heat shock protein (hsp60 gene), and phenylalanyl-tRNA synthase (pheS gene) targets provide better discrimination of closely related species in the Lactobacillus acidophilus, L. casei, and L. plantarum groups (459).
Oribacterium asaccharolyticum and O. parvum are almost identical, with only a few or single base pair mismatches within 16S regions V4, V5, and V6, so that sequencing at least ∼1,060 bp is recommended (5). Limited 16S sequence data are currently available for several Oribacterium spp. from the human oral cavity, including O. asaccharolyticum and O. parvum (460).
Several species within the Peptostreptococcus-Finegoldia-Peptococcus genera cause human infections, including bloodstream infections (147, 461). Limited 16S sequence data are currently available for the canine pathogen P. canis and several human species, including Peptococcus niger, Peptoniphilus duerdenii, and P. koenoeneniae, two recently described species isolated from human wound infections (462, 463).
Several human skin species previously classified as Propionibacterium have recently been moved into a new genus, “Cutibacterium,” including C. acnes and C. avidum, both of which are opportunistic human pathogens (464). Limited 16S sequence data are currently available for P. propionicum isolated from humans (5) and several animal and environmental Propionibacterium spp., including P. australiense, P. cyclohexanicum, P. jensenii, P. microaerophilum, and P. thoenii (465).
Ruminococcus-Blautia are part of the human gut microbiome, and several species that were previously classified within the Ruminococcus genus have recently been moved to the Blautia genus (466). Limited 16S sequence data are currently available for several human Ruminococcus-Blautia spp., including B. obeum, R. bromii, R. callidus, R. champanellensis, R. faecis, R. gauvreauii, R. lactaris, and R. torques.
Slackia species are part of the human and animal gut microbiome (467). S. exigua has been reported to cause human wound and intraabdominal infections (468). Limited 16S sequence data are currently available for several Slackia species isolated from humans and animals, including S. equolifaciens, S. heliotrinireducens, S. isoflavoniconvertens, S. piriformis, and S. faecicanis (469).
Robinsoniella peoriensis was originally isolated from a swine manure storage pit but has subsequently been reported as a cause of human infection (469, 470). Limited 16S sequence data are currently available for this organism and closely related animal and environmental Clostridium spp. (C. jejuense, C. aminovalericum, and C. xylanovorans), as well as Blautia faecis and C. aldenense, isolated from humans (471, 472).
Solobacterium moorei was first described in 2000 and has since been reported to cause a variety of human infections, including mixed surgical wound infections (473). Limited 16S sequence data are currently available for this organism.
Limited 16S sequence data are currently available for Turicibacter sanguinis (474) and closely related species from soil/water, including Lysinibacillus sinduriensis, L. contaminans, L. mangiferihumi, Bacillus endoradicis, and Tepidibacillus fermentans.
Gram-negative anaerobes.
Table 11 outlines the 16S sequence diversity of clinically relevant genera within the Bacteroideaceae, Desulfovirionaceae, Veillonellaceae, Fusobacteriaceae, Leptotrichiaceae, Acidaminococcaceae, Porphyromonaceae, Prevotellaceae, Selenomonadaceae, and Sutterellaceae families. Comparison of the number of identical versus divergent 16S positions for various genera shows a wide range of percent identity. The most 16S sequence data are available for Bacteroides, Desulfovibrio, and Prevotella genera, and a high number of divergent positions is found in many of these anaerobic genera (Table 11). However, within each of these clinically important anaerobic genera, there are several species that cannot be reliably identified based on 16S analysis.
Bacteroides spp. can be differentiated by 16S variability within regions V1-3 (5). Some Bacteroides spp., such as B. coagulans, B. galacturonicus, and B. xylanolyticus, show deletions within region V3. Limited 16S sequence data are currently available for several human, animal (B. gallinarum, B. faecichinchillae, B. paurosaccharolyticus, B. propioninifaciens, B. coprosuis, B. stercorirosoris, and B. xylanolyticus), and environmental (B. reticulotermitis and Pseudobacteroides cellulosolvens) species, including several that are closely related (B. barnesiae, B. cellulosilyticus, B. clarus, B. coagulans, B. eggerthii, B. faecis, B. fluxus, B. galacturonicus, B. massiliensis, B. nordii, B. oleiciplenus, B. pectinophilus, B. rodentium, B. salanitronis, B. salyersiae, and Prevotella zoogleoformans) (475). A recent whole-genome phylogenetic analysis showed little difference between the Parabacteroides and Bacteroides genera (475). Limited 16S sequence data are currently available for several human and environmental Parabacteroides-Macellibacteroides spp., including P. gordonii, P. johnsonii, P. chinchilla, M. fermentans, and P. chartae.
Veillonella are strict anaerobes, currently classified in the Negativicutes phylum, that are among the most abundant organisms of the oral and intestinal microflora of animals and humans (476). Veillonella are Gram-negative organisms, but recent whole-genome and 16S sequencing studies show that this genus is more closely related to the Firmicutes phylum (476). Although Veillonella spp. are highly identical, differentiation requires a longer 16S sequence due to the limited variability across regions V1-3 (5). V. rodentium, V. rogosae, and V. tobetsuensis are very closely related, and V. denticariosi, V. dispar, and V. parvula are also highly identical within 16S. Limited 16S sequence data are currently available for Dialister-Veillonella spp., including several that have been isolated from animals (D. succinatiphilus, V. caviae, V. criceti, V. magna, V. ratti, V. rodentium, and V. montpellierensis) and humans (D. propionicifaciens, V. denticariosi, and V. tobetsuensis) (476, 477). Veillonella magna and Megasphaera spp. have highly similar 16S sequences. The Selenomonas-Megasphaera-Sporomusa branch includes members of the Firmicutes phylum with Gram-negative-type cell envelopes that were recently moved to the Negativicutes class, but recent whole-genome sequence analyses show these organisms are closely related to Clostridia (455). Megasphaera-Veillonella-Anaeroglobus geminatus are closely related but can be differentiated by 16S variability in regions V2, V3, V6, and V7 (5). Megasphaera sueciensis and M. paucivorans are highly identical, except for a few base pair mismatches in 16S within region V1 (5). Limited 16S sequence data are currently available for several species, including M. cerevisiae, M. sueciensis, M. paucivorans, and V. magna (478). Limited 16S sequence data are also currently available for human Negativicoccus and closely related species, including N. succinicivorans, V. magna, and D. propionicifaciens (130).
Selenomonas flueggi and S. infelix are closely related but can be differentiated by 16S variability within region V3 (5). Limited 16S sequence data are currently available for several environmental Selenomonas spp., including S. bovis, S. lacticifex, and S. lipolytica (455).
Thermodesulvovibrio is easily separated from Desulfovibrio, which is a highly identical genus, where some species, such as D. indonesiensis and D. marinus, differ across 16S in only a few base pair positions. A longer 16S sequence (∼1,060 bp) that includes variable regions within V2-6 allows differentiation (5). Limited 16S sequence data are currently available for several Desulfovibrio-Thermodesulfovibrio-Bilophila environmental species from groundwater, including D. aespoeensus, D. africanus, D. alcoholivorans, D. alkalitolerans, D. bastinii, D. butyratiphilus, D. frigidus, D. fructosivorans, D. gigas, D. halophilus, D. indonesiensis, D. intestinalis, D. magneticus, D. marinisediminis, D. marinus, D. marrakechensis, D. oceani, D. oxamicus, D. piger, D. profundus, D. psychrotolerans, D. salexigens, D. senezii, D. simplex, D. zosterae, T. aggregans, T. islandicus, and T. yellowstonii (479). Limited 16S sequence data are also available for the human species Bilophila wadsworthia (480).
Fusobacterium is a highly identical genus, and some species show deletions within 16S region V1 (5). Sequencing of a long 16S stretch that includes region V7 helps with species-level differentiation. Limited 16S sequence data are currently available for several Fusobacterium species isolated from animals (F. equinum and F. simiae) and humans (F. mortiferum and F. ulcerans) (481).
Leptotrichia are an important part of the human oral flora. Leptotrichia spp. can be differentiated by 16S variability within regions V1-3 and V6 (5). Limited 16S sequence data are currently available for several human Leptotrichia spp., including L. goddfellowii and L. hongkongensis (482).
Acidaminococcus-Phascolarctobacterium-Anaerovibrio are part of the gut microbiome. Limited 16S sequence data are currently available for several animal and human species, including P. faecium, P. succinatutens, and Anaerovibrio lipolyticus (483–485).
Alistipes spp. can be differentiated by 16S variability across regions V1-3, provided the entire sequences are available (5). Limited 16S sequence data are currently available for several human species, including A. indistinctus, A. onderdonkii, A. putredinis, A. shahii, and A. timonensis (486, 487).
Limited 16S sequence data are currently available for several human Anaerobiospirillum spp. and closely related environmental (Aeromonas sharmana and Tolumonas osonensis) and human species, including A. succiniciproducens, A. thomasii, Ruminobacter anylophilus, Helicobacter oris, Succinatimonas hippie, and Aeromonas diversa (488).
Peptoniphilus-Anaerosphaera-Parvimonas are closely related, and several species within the genera are identical, including Peptoniphilus asaccharolyticus and P. olsenii (5). Limited 16S sequence data are currently available for several human (P. methioninivorax, P. stercorisuis, and A. aminiphila) and environmental species, including P. coxii, P. duerdenii, P. gorbachii, P. koenoeneniae, P. lacrimalis, P. olsenii, and P. tyrrelliae (461, 463, 489).
Porphyromonas endodontalis and P. gingivicanis are identical and very closely related, so that analysis of a longer 16S stretch up to region V4 is required for differentiation (5). Limited 16S sequence data are currently available for several human species, including P. bennonis, P. circumdentaria, P. somerae, and P. uenonis (490). Prevotella is a highly identical genus with many very closely related species, so a long 16S sequence that includes regions V6 and V7 is required for differentiation (5). Limited 16S sequence data are currently available for the environmental isolate P. paludivivens and several human species, including P. clara, P. xylaniphila, P. albensis, P. amnii, P. aurantiaca, P. brevis, B. bergensis, B. bryantii, P. corporis, P. dentasini, P. enoeca, P. fusca, P. jejuni, P. maculosa, P. oryzae, P. pleuritidis, P. saccharolytica, P. scopos, P. shahii, and P. stercorea (491).
Mobiluncus is an important part of the vaginal bacterial flora (492). M. curtisii and M. mulieris are closely related, although limited 16S data are available for both species. However, a long 16S sequence that includes variable regions within V2-6 allows differentiation (5).
Limited 16S sequence data are currently available for several human and animal Odoribacter-Butyricimonas spp., including O. splanchnicus, O. laneus, B. faecihominis, B. paravirosa, B. synergistica, and B. virosa (493, 494).
Sutterella-Parasutterella spp. can be differentiated by 16S hypervariability within region V3 (5). Limited 16S sequence data are currently available for several closely related human, animal, and environmental Sutterella-Parasutterella species, including S. parvirubra, S. stercoricanis, P. secunda, P. excrementihominis, and Glaciimonas immobilis (495).
Aerobic actinomycetes (Mycobacterium, Nocardia, and related genera).
The aerobic actinomycetes are a large Gram-positive bacillary organism group that consists of heterogenous and taxonomically divergent genera (496). Human infection is acquired from environmental sources. Mycobacterium and Nocardia species are the most common isolates in the clinical laboratory. Aerobic actinomycete taxonomy has evolved significantly, with new species being identified (for example, for Mycobacterium spp. [497, 498] and for Nocardia spp. [137, 499]). For the genus Nocardia, sequencing 16S rRNA, secA, and other loci has led to improved complex and species differentiation, with better correlation to human-pathogenic potential and antimicrobial susceptibility profiles (500–502). Additional description and analysis of phylogenetic relationships for Gordonia, Rhodococcus, Nocardia, Skermania, Tsukamurella, and Turicella are available (137, 503, 504). This section outlines the ability of 16S to differentiate aerobic actinomycetes in the clinical laboratory.
(i) Mycobacterium.
The genus Mycobacterium consists of more than 180 species; recently, a subdivision of this genus has been proposed, with the creation of four new additional genera, Mycolicibacterium (encompassing M. fortuitum-vaccae like species), Mycolicibacter (M. terrae-like species), Mycolicibacillus (M. triviale-like species), and Mycobacteroides (M. abscessus-chelonae-like species) (505). However, Nouioui and colleagues (506) argue the Mycobacterium genus should not be split. While this issue remains under discussion, we will consider members of the genus Mycobacterium as one genus.
Table 12 outlines the 16S sequence diversity within Mycobacterium prior to its recent separation into 4 distinct genera, as outlined above. Comparison of the number of identical versus divergent 16S positions for Mycobacterium spp. shows a wide range of divergence, resulting in a lower percent identity (∼76%). Mycobacteria can be divided into three major groups: M. tuberculosis complex, nontuberculous mycobacteria (NTM), and M. leprae. Most mycobacterial species can be differentiated within the regions V1-3; however, for efficient identification, a stretch of about ∼500 bp covering these regions should be sequenced (5). Many rapidly growing species, such as M. abscessus, M. mucogenicum, M. fortuitum, and many others, show a multinucleotide (about 10- to 14-bp) deletion in region V3, which should be considered for identification; however, this feature can also be observed with some rather slow-growing species, such as M. genavense, M. interjectum, and others (507).
The Mycobacterium tuberculosis complex is composed of M. tuberculosis, M. africanum, M. canettii, M. bovis, M. bovis BCG, M. microti, M. orygis, M. caprae, M. pinnipedii, M. suricattae, and M. mungi (508). Within the M. tuberculosis complex, M. tuberculosis, M. bovis, M. bovis BCG, and M. africanum most commonly cause human infections (509). Because of high genomic similarity, they cannot be differentiated by 16S sequencing or by sequencing of the rpoB gene (5).
Nontuberculous mycobacteria (sometimes referred to as atypical mycobacteria) include many diverse mycobacterial species naturally found in environmental sources, but some play an important role as human pathogens in patients with underlying lung disease, those who are immunocompromised, and otherwise healthy individuals. NTM are broadly divided by their growth rate into slowly and rapidly growing mycobacteria but should be differentiated to the species (or complex) level to be able to recognize the isolate as a pathogen and guide antimicrobial therapy. Species- or complex-level differentiation can be achieved by 16S rRNA gene sequencing; however, since many NTM differ by only a few positions over the V1-3 stretch, accurate base calling by the sequencer is crucial to rule out artefactual mismatches (5).
The most frequently isolated NTM, the Mycobacterium avium complex (MAC), can be broadly divided into M. avium-related (sub)species and M. intracellulare-related species, which can be differentiated from each other by a few mismatches over the region V1-V3 (5). Species differentiation within MAC requires sequencing of other gene targets, such as internal transcribed sequence rpoB, but may not be needed routinely. However, it is important for epidemiological studies, source tracking, and outbreak investigations (i.e., M. chimera outbreak associated with heater-cooler devices) (510, 511).
Within rapidly growing NTM, the so-called Mycobacterium chelonae-abscessus complex contains two genetically closely related species, M. abscessus and M. chelonae, with different clinical presentations and antimicrobial susceptibilities; thus, they should be differentiated from each other. M. abscessus taxonomy has been under debate, but it is currently considered one species (M. abscessus) with 3 subspecies, subsp. abscessus, subsp. massiliense, and subsp. bolletti (512). These subspecies cannot be differentiated by sequencing of the 16S rRNA gene; a multilocus approach including several targets (i.e., rpoB, secA, and hsp65) provides higher discriminatory power. Sequencing of the erm41 gene (which is truncated in most M. massiliense) can aid subspecies differentiation and, most importantly, in the assessment of inducible antibiotic (macrolide) resistance. Additional species in the Mycobacterium chelonae-abscessus complex (besides M. chelonae and M. abscessus) include M. saopaulense, M. franklinii, M. salmoniphilum, and M. immunogenum, which cannot be differentiated by sequencing of the 16S rRNA gene and require additional targets (i.e., rpoB and hsp65) (513).
M. celatum is one of the rare mycobacterial species for which multiple 16S rRNA operons have been described. The 16S sequence within V2 often shows a shift of a few base pairs, yielding ambiguous Sanger sequence reads downstream; sequencing into V2 from both sides solves the problem and shows an insertion of a few base pairs in one of the operons, which explains this unique shift (119).
Some NTM, such as M. kansasii, M. gordonae, and M. flavescens, were described with >1 sequevar; in these cases it is useful to check whether the reference database used for the sequence searches actually contains these sequevars to achieve an accurate match (514).
Finally, several clinically important mycobacterial species show high sequence similarity to other species of less clinical importance, such as M. leprae and M. lepraemurium. Careful review of the sequences (here in regions V2 and V3) reveal a few, stable mismatches that allow differentiation. In cases where such differentiation cannot be achieved, one should report “close to” the closest pathogenic species, which gives an idea about the potential pathology in this case, and try to integrate this result into the clinical context.
(ii) Nocardia and other aerobic actinomycetes.
This complex group of organisms contains many species that cause serious human infection, especially in immunocompromised patients. Table 12 outlines the 16S sequence diversity within clinically relevant genera of aerobic actinomycetes. Comparison of the number of identical versus divergent 16S positions for various genera shows a wide range of percent identity. Most 16S sequence data are available for Nocardia and Streptomyces genera, but a high number of divergent positions are found in many genera within this group of microorganisms (Table 12). Several species within each of these clinically important aerobic actinomycetes genera cannot be reliably identified based on 16S analysis.
Of the genera listed in Table 12, Nocardia species are the most implicated in human infections. The genus Nocardia has a complicated taxonomic history that was recently reviewed by Conville and colleagues (137). Nocardia asteroides, the type species of the genus, used to be the most frequently reported nocardial taxon from human specimens. In 1988, Wallace and colleagues (515) reported six drug pattern types among a study of 78 clinical isolates previously identified as Nocardia asteroides, with the type strain of N. asteroides placed within a “miscellaneous” group and showing a unique susceptibility pattern. It is now clear that organisms previously identified in patient specimens as N. asteroides were likely misidentified by today's standards, and most appear to be members of these differentiated species (137).
While 16S rRNA sequencing (particularly gene regions 160 to 220 and 580 to 650) can aid in species-level identification of some Nocardia species (i.e., N. farcinica), a major difficulty with the use of this target is the high level of sequence similarity among species. For example, N. brevicatena/N. paucivorans and N. abscessus/N. asiatica/N. arthritidis have identical or nearly identical 16S rRNA gene sequence similarities. However, a longer 16S sequence of up to ∼1,200 bp (V4-V6) should be analyzed for optimal species resolution (5). Laboratories using 16S rRNA for identification of Nocardia species should consider different reporting levels, species, complex, or group, as appropriate. As described for Mycobacterium celatum, Sanger sequencing chromatograms should be carefully evaluated for evidence of multiple copies of the 16S rRNA gene with dissimilar nucleotide sequences (516).
Multilocus sequence analysis (MLSA) using concatenated sequences of 4 to 5 housekeeping genes (16S rRNA, gyrB, secA, hsp65, and/or rpoB) can provide higher accuracy and discriminatory power in the molecular identification of Nocardia spp. (500). Since sequencing 4 to 5 targets may be prohibitive for many clinical laboratories, using 3 (or even 2) targets has been proposed (i.e., 16S rRNA, gyrB, and secA), which provide species (or complex) assignment for the majority of isolates or can raise suspicion on the occurrence of a novel species.
Species within the N. abscessus complex (N. abscessus, N. athritidis, N. asiatica, N. beijingensis, and N. pneumoniae) are highly related and cannot be differentiated within the first ∼500 bp of 16S. N. abscessus, N. asiatica, and N. arthritidis are in fact almost identical over the entire 16S gene and may not separate using this target. N. beijingensis is highly identical to N. arthritidis and N. araoensis in 16S, but differentiation may occur by a few variable base pair positions within V1-V2 (5). N. exalbida cannot be separated from N. gamkensis because they share identical 16S sequences; limited 16S sequence data are also available for the latter species (5). N. brasiliensis is closely related to N. vulneris, and they can be differentiated by a few base pair mismatches in regions V1-3 and V4-6, or a longer sequence of up to ∼1,200 bp may be needed to separate these species. Limited 16S sequence data are available for other Nocardia spp. that are closely related to N. brasiliensis, including N. iowensis, N. altamirensis, N. jiangsuensis, N. kroppenstedtii. N. farcinica, and N. kroppenstedtii, and are closely related to each other and to N. cyriacigeorgica; differentiation of these clinically relevant species relies on a few mismatches in regions V1-V2 and V4, but a longer 16S sequence of up to ∼1,200 bp may be required (5). N. brevicatana is closely related to the N. paucivorans complex; differentiation of these clinically relevant species relies on a few mismatches in regions V1-V2, but a longer 16S sequence of up to ∼1,200 bp may be required (5). Several species within the N. nova complex (N. africana, N. aobensis, N. cerradoensis, N. elegans, N. kruczakiae, N. mikamii, N. nova, N. vermiculata, and N. veterana) are closely related and cannot be differentiated by 16S (5). N. nova cannot be differentiated from N. vermiculata, and N. cerradoensis and N. africana cannot be separated. Differentiation may be attempted for other species of this complex by a few base pair mismatches in 16S regions V1-V2 and V4-V5 (5). N. otitidiscaviarum can be identified by mismatches within 16S regions V1-2 and V4-5 (5). N. pseudobrasiliensis cannot be differentiated from N. rayongensis within the first ∼500 bp of 16S, but a few base pair mismatches in regions V4-5 may allow separation (5). Limited 16S sequence data are available for N. rayongensis, N. vermiculata, N. mikamii, and N. miyunensis. Species within the N. transvalensis complex (N. blacklockiae, N. transvalensis, and N. wallacei) are highly identical, and the last two species cannot be differentiated by 16S. N. blacklockiae may be differentiated by a few mismatches in regions V2 and V6 (5).
Clinically relevant aerobic actinomycetes (besides Mycobacterium and Nocardia) are shown in Table 12. For the most part, 16S rRNA sequencing can provide reliable genus-level identification that is sufficient in most cases. Species-level identification requires additional targets, such as choE for R. hoagii (equi) (504), groEL, rpoB, secA1, and ssrA genes for Tsukamurella spp. (517), and hsp65 and gyrB for Gordonia spp. (503).
Actinomadura is a highly identical genus that contains many environmental species. A. madurae and A. pelletieri are the most common species that cause mycetoma (518, 519). While resolution to genus occurs by mismatches with the V2, V3, and V4 16S regions, a full-length sequence is required to differentiate many Actinomadura spp. A. madurae is closely related to A. bangladeshensis but can be separated by mismatches in the V2 region (5).
Gordonia is a highly identical genus that contains many environmental species. G. terrae, G. bronchialis, G. sputi, and G. otitidis are mainly recovered from respiratory samples (503). Gordonia species may also cause acute peritonitis in patients on peritoneal dialysis (20, 520). G. terrae is closely related to G. lacunae, G. hongkongensis, and G. didemni, as there are only a few mismatches within the V2 and V4 16S regions, while G. bronchialis, G. sputi, G. aichiensis, G. otitidis, and G. polyisoprenivorans can be differentiated by mismatches in the V1-V3 regions (5). A longer 16S sequence of up to ∼1,200 bp is required to separate some other species. Limited 16S sequence data are available for several species, including G. alkaliphila, G. caeni, G. cholesterolivorans, G. defluvii, G. desulfuricans, G. didemni, G. effusa, G. hankookensis, G. hirsuta, G. humi, G. iterans, G. jinhuaensis, G. kroppenstedtii, G. namibiensis, G. neofelifaecis, G. otitidis, G. phosphorivorans, G. rhizosphera, G. shandongensis, G. sinesedis, G. soli, and G. westfalica.
Rhodococcus is a highly identical genus that contains many environmental species. R. hoagii (equi), R. erythropolis, and R. globerulus are most commonly implicated in human infections (504). Although a genus-level identification occurs by mismatches in regions V1, V4, and V6, a full-length 16S sequence is required for resolution of many Rhodococcus species (5). R. hoagii is closely related to R. soli, with only a few mismatches in the 16S V1 region (5). Some Rhodococcus species are closely related to Nocardia species; R. globerulus and N. globerula cannot be differentiated by 16S, while R. erythropolis is also like N. coeliaca. Other closely related species include R. baionurensis, R. degradans, and R. gingshengii, and the last two species share identical 16S sequences (5). Limited 16S sequence data are available for many environmental species, including R. aerolatus, R. agglutinans, R. antrifimi, R. artemisiae, R. biphenylovorans, R. defluvii, R. degradans, R. enclensis, R. humicola, R. imtechensis, R. kunmingensis, R. lactis, R. marinonascens, R. nanhaiensis, R. pedocola, R. percolatus, R. soli, R. sovatensis, R. trifolii, and R. tukisamuensis. Segniliparus rugosus can be identified and separated from Rhodococcus by mismatches within the 16S V1-V2 regions (5). S. rugosus is closely related to S. rotundus. Limited 16S sequence data are available for Segniliparus species.
Streptomyces is a large genus that contains more than 600 environmental species that may rarely cause human infections (521, 522). Limited 16S sequence data are available in reference databases, but many Streptomyces species are highly identical within 16S (5). S. somaliensis is identical to S. flavofungini and cannot be differentiated, while S. albidoflavus and S. violascens are also closely related.
Tsukamurella is a highly identical genus that contains many environmental species that rarely cause infection in immunocompromised patients (517, 523). A full-length 16S sequence is required to differentiate many Tsukamurella species, as only a few mismatches occur in V2, V3, V4, V6, and V7 regions (5). T. paurometabola is closely related to T. strandjordii and T. inchonensis, as there is only a single mismatch in the V3 and V6 regions. T. pulmonis is also closely related to T. tyrosinosolvens, T. sinensis, and T. strandjordii, with only a few mismatches throughout 16S. Limited 16S sequence data are available for T. serpentis, T. sinensis, and T. soli.
PATHOGEN DISCOVERY AND CHARACTERIZATION DIRECTLY FROM CLINICAL SPECIMENS
Broad-range 16S PCR and sequencing enable analysis of important culture-negative isolates by detecting bacterial nucleic acid through targeting conserved sequences (described in the introduction) (25–27). Cycle sequencing of the amplicon generated by broad-range PCR enables identification of the organism(s) (7, 57). This approach has been successfully applied to clinical isolates from normally sterile sites to diagnose invasive bacterial or fungal infections, including infective endocarditis (7, 524), and bone and joint infections, including prosthetic joint infection (525–527) and meningitis (528). Analysis of specimens with contaminating body flora should be avoided. Broad-range PCR/sequencing is best used in situations where infection is strongly suspected but routine bacterial cultures are negative because the organism is fastidious or uncultivable or antibiotics were administered prior to specimen collection (529). Novel pathogens also have been discovered using universal broad-range 16S primers/probes due to the high sensitivity of the procedure for detecting low-copy-number targets (7, 12, 530). Tropheryma whipplei was identified as the cause of Whipple’s disease using a broad-range PCR approach (531, 532). The causative agent of peptic ulcer diseases, Helicobacter pylori, was also found to be the dominant microbiota in the human stomach using broad-range PCR testing (533). Broad-range 16S PCR has also been used to detect clinically relevant Chlamydia spp. infecting humans and animals (534).
The overall sensitivity and detection limits of broad-range PCR are influenced by several factors that have previously been outlined, including prior culture enrichment of the isolate, the use of optimal molecular laboratory practices and decontamination procedures, the nucleic acid extraction method, the choice of primers/probes, the assay conditions used, the concentration of the amplified products prior to sequencing, and the appropriate use of procedural controls (25). Sterile collection of the clinical sample and isolate selection for broad-range PCR testing are also critically important to preventing ambiguous results by the inadvertent amplification of one or more commensal contaminant organisms. Interpretation of broad-range PCR data can be challenging, as the procedure is prone to contamination with not only bacterial but also host human DNA so that false-positive results occur (525, 535). Investigators have previously outlined the sources of possible contamination that happen during isolate collection, nucleic acid extraction, or PCR analyses (536). Ideally, a separate dedicated isolate should be collected from acceptable sites (i.e., sterile tissues and fluids) whenever this test is clinically requested, but that is often not feasible. Clinical isolates obtained from body sites/sources that are known to be contaminated with commensal flora are unacceptable for broad-range PCR testing, since the genetic material from the flora will generate too much noise and, thus, render an uninterpretable result.
A human DNA amplification control as well as appropriate negative and positive controls should be used for DNA extraction and carried through the entire procedure (524, 525, 535). A negative isolate control is also useful and should be made up of an aliquot of the negative clinical isolate matrix being tested (i.e., cerebrospinal fluid [CSF] that is culture negative). Commercial reagents should be sterilized using filtration or other methods prior to use to ensure sterility but avoid contamination with exogenous DNA (536). Various approaches have also been taken to mitigate external bacterial DNA contamination, including DNase treatment, restriction endonuclease digestion, UV irradiation, and 8-methoxypsoralen in combination with long-wave UV light to intercalate contaminating DNA into double-stranded DNA (537). Alternatively, DNA decontamination procedures are not necessary when employing a broad-range primer extension-PCR (PE-PCR) strategy (538). Interpretation of broad-range PCR data can also be challenging, as contaminating human DNA in the isolate may also be recognized by universal 16S primers/probes, resulting in a false-positive broad-range result (539). The recent use of dual priming oligonucleotide (DPO) primers has documented improved accuracy and specificity of 16S ribosomal PCR/sequencing reactions not only for isolate identification but also for universal broad-range detection from clinical isolates (535, 538, 540).
Despite the proven utility of broad-range PCR/sequencing for aiding the diagnosis of specific infection types outlined, there have been few evidence-based prospective studies evaluating its diagnostic impact in patients suspected to have infectious disease but not limited to a particular type of infection. Rampini et al. (529) showed a high concordance of >90% for their molecular 16S broad-range PCR assay compared to the gold standard of routine bacterial culture for 394 clinical specimens. Another 231 specimens of various types (i.e., aspirates and biopsy specimens, CSFs, tissues, heart valves, wound swabs, abscesses materials, and ascites) were also tested retrospectively using a molecular assay that showed sensitivity, specificity, and positive and negative predictive values of 42.9%, 100%, 100%, and 80.2% for culture-negative bacterial infections and improved patient care in patients pretreated with antibiotics (529). In 2016, our laboratory developed and implemented an in-house broad-range PCR/sequencing assay using DPO primers/probes that were designed to detect the widest range of known bacterial pathogens (524). We have previously described our experience with the procedure for the diagnosis of bacterial endocarditis and found it to be more sensitive than valve tissue Gram stain and culture and that sequence data were valuable even when blood cultures were positive (524).
Since 2016, we have performed this test on 602 specimens. Molecular testing is done routinely on explanted heart valves where infective endocarditis is suspected, and otherwise the test is restricted to the infectious diseases service following consultation with the microbiologist on call (i.e., adequate amount of a specimen from a sterile site that has not been used for prior testing). The most common specimens tested were tissue (50.0% of specimens) including heart valves or other cardiac tissue (23.4%) and musculoskeletal tissue (19.6%). Cerebrospinal fluid (15.7%) and other sterile fluids (34.3%), such as synovial (13.1%), pleural (5.9%), and aspirates (10.2%), made up the remainder of specimens. Organisms were identified in 37.9% of specimens, including 42.2% of tissues, 25.3% of CSF, and 40.1% of sterile fluids. Positive specimen types were most commonly either pleural fluid (69.4%) or sterile aspirates (41.9%). Specimens with organisms seen on Gram stain were more often positive (80.6%) than those with negative Gram stain (33.9%). Streptococcus species comprised 40.4% of all positives, including S. pneumoniae, which was the single most commonly detected organism (10.0% of positives, the majority from pleural fluid). Other identified organisms included Staphylococcus species (19.1%), a variety of Gram-negative bacilli (15.2%), anaerobes (11.3%), and one case of Tropheryma whipplei in heart valve tissue. Overall, it is worth noting that, in our experience, a positive 16S broad-range PCR/sequencing result correlates with the specimen Gram stain result in only 85% of cases. Correlation with the patient’s clinical condition is also critical for accurate interpretation of the molecular result, and, due to the restricted ordering of broad-range 16S tests in our jurisdiction, the test results were discussed with the infectious diseases service directly involved in the patient’s care.
COMPARISON OF MALDI-TOF MS AND NEXT-GENERATION SEQUENCING VERSUS SANGER 16S SEQUENCING FOR PATHOGEN IDENTIFICATION
Clinical microbiology laboratories rely on a variety of methods for pathogen identification. This section provides a brief synopsis of the current utility of both proteomics and advanced next-generation sequencing methods for this purpose. Table 13 provides a comparison of the advantages and disadvantages of each of these approaches. However, it is important for clinical laboratories to recognize that none of these methods provides a universal solution for accurately identifying all human pathogens or for separation from highly similar environmental organisms. Rather, a comparison and correlation of both phenotypic methods with proteomic and molecular analyses is necessary for the widest capability for broad pathogen identification. Essential correlations between the Gram stain, colony morphology on culture plates, biochemical profiles, and proteomics and molecular analyses will be an essential part of the clinical laboratory’s pathogen characterization toolbox for the foreseeable future.
TABLE 13.
Comparison of MALDI-TOF and nucleic acid sequencing methods for pathogen identification
| Comparison | MALDI-TOF | Sanger 16S sequencing | Next-generation sequencing |
|---|---|---|---|
| Availability | Widespread adoption | Limited in clinical labs | Limited in clinical labs |
| Application specific | Universal use possible for bacteria/fungi | Sent to a core facility with shared instrumentation | |
| Suitable for typing/molecular resistance | Universal use for other microorganisms | ||
| Expansion to typing/molecular resistance | |||
| Procedure complexity | Low | High; little or no automation | High-limited (application-specific) automation |
| Lower when using a commercial kit | Kits available for library preparation | ||
| CLSI MM18-A2 guides analyses | |||
| Accuracy | High depending on microorganism/group being interrogated | High if several regions covered (long reads with excellent coverage of 16S feasible | Whole-genome sequencing provides full coverage of 16S operons |
| Applicable to pure cultures | Sequences must be edited and trimmed | Application to pure cultures | |
| Application to pure cultures or normally sterile clinical samples with a single pathogen | Usually less sensitive depending on assembly processes | ||
| Intraoperon diversity problematic | |||
| All pipelines need to be thoroughly developed for various clinical application | |||
| Applicable to pure cultures | |||
| Databases | Ongoing development limited for various microorganisms/groups | Covers all microorganisms/groups | Rapidly expanding deposit of WGS data in public databases without curation |
| Requires ongoing validation against phenotypic and sequencing results | Several public databases are outlined in Table 2, but coverage for bacterial species of interest and reliability of annotation and sequences must be assessed | Some freely accessible target-specific 16S databases outlined in Table 2 can be used (i.e., in packages such as Qiime, www.qiime.org), but coverage for bacterial species of interest and reliability of annotation and sequences must be assessed | |
| Commercial databases also available | Whole genomes are available under the genome section of NCBI/GenBank and can be searched using BLAST | ||
| Commercial databases are also available as part of an analysis package | |||
| Cost/test | Low, but MS instrument cost, maintenance, and database use need to be considered | High when using a commercial kit linked to specific instruments and reagents | High, but costs decrease when pooling samples |
| Cost/sample is rapidly decreasing with increased throughput and read length and depends on read length | |||
| Instrumentation | MALDI-TOF MS instruments expensive but supplied by Becton-Dickinson (Bruker) or bioMérieux (Shimatzu) | Requires purchase of an automated genetic analyzer | Requires purchase of one or more NGS instruments, maintenance contracts, and reagent-rental agreements |
| Capillary electrophoresis columns must be regularly maintained and replaced | Shared core facility to minimize costs | ||
| Separate installation to avoid contamination | Separate installation to avoid contamination | ||
| Quality assurance | Laser must be regularly calibrated | Appropriate controls must be included with each run | Appropriate controls much be included with each run |
| QA organisms should be regularly run to verify performance | Appropriate controls must be used for each step of the procedure | Appropriate controls must be included with each step of the procedure | |
| Sequence trimming/editing allows identification of contamination problems | Checks on read generation, read filtering (elimination of nonspecific reads), mean read length, phred scores, concatenation/assembly efficiency must be done to ensure quality results | ||
| Testing capacity and throughput | Hundreds of individual isolates per day can be analyzed depending on the no. of instruments used | Suitable for single and few samples | Higher throughput than Sanger depending on the method and instrument being used |
| No more than 8–12 isolates can be run in a day | Pooling of samples is customary to reduce the per-sample cost | ||
| Throughput has never been automated | |||
| Data analysis | MALDI-TOF immediately provide an answer by analysis of an isolates spectral profile against the onboard database | Complex and general understanding of BLAST and alignments | Complex and a major barrier to clinical implementation |
| Requires sequence editing and analysis against a reference sequence | Requires appropriate storage of large amounts of sequence data | ||
| A multialignment against a close reference sequence should be performed | Requires knowledge in bioinformatics and general informatics | ||
| Reduced errors using curated commercial or online reference database | Delayed results often taking days to complete | ||
| Reliable results analysis using CLSI MM18-A2 guideline |
Will Proteomics Replace 16S Cycle Sequencing for Bacterial Identification?
The ability of clinical microbiology laboratories to rapidly and accurately identify a wide range of human bacterial pathogens to the genus and species level has been revolutionized by the widespread implementation of proteomics analysis using MALDI-TOF MS (541–545). While MALDI-TOF MS has only recently been adopted widely for diagnostics, Anhalt and Fenselau (546) first showed in 1975 that mass spectrometry could be used for bacterial identification. Extraction of basic cytoplasmic proteins, including ribosomal and mitochondrial proteins, heat shock proteins, DNA binding proteins, and RNA chaperone proteins, requires initial lysis of the organisms with organic solvents under acidic conditions (i.e., ethanol, formic acid, and acetonitrile) prior to MALDI-TOF MS instrument analyses (542). Proteomics biomarkers detected by MALDI-TOF MS spectra are largely intracellular proteins that range in size from 4 to 15 kDa and are mainly highly conserved ribosomal housekeeping proteins (i.e., 16S) (542).
In less than a decade of widespread use, MALDI-TOF MS has revolutionized the time it takes clinical microbiology laboratories to identify pathogens (i.e., MALDI-TOF MS identification is at least 24 h faster than routine phenotypic methods) and in many cases eliminated the need to routinely perform other types of complex analyses (541, 547, 548). Many published studies have demonstrated the accuracy of MALDI-TOF for the identification of a broad spectrum of bacterial pathogens (28, 29, 549, 550). In addition, MALDI-TOF MS now is also able to identify many different types of yeast, some fungi, Nocardia, and Mycobacterium, with the development of spectral profiles for these complex organisms (31, 32, 551–553). Overall, 98% of routine clinical isolates are identified to the genus level and >90% to the species level, and <1% are incorrectly identified (549). CLSI M18 A2 and M52 provide clinical laboratories with a detailed summary of the current diagnostic utility and pitfalls of using MALDI-TOF MS for identification of a wide variety of microorganisms/groups (5, 29).
However, MALDI-TOF MS has not eliminated the need to perform 16S sequencing in larger, more complex clinical microbiology laboratories where more difficult-to-identify fastidious, atypical, or unusual bacterial strains are encountered. Bizzini and colleagues performed one of the largest studies of difficult-to-identify bacterial strains and compared the ability of MALDI-TOF MS as an alternative method to 16S rRNA gene sequencing (545). Among 410 clinical isolates from 207 different difficult-to-identify species that previously had required 16S rRNA gene sequencing, the Microflex LT instrument (Bruker) and Biotyper automation 3.1 software, using a library of 3,740 spectra and criteria proposed by the manufacturer, gave a valid species-level identification score in only 45.9% of the strains. However, no misidentifications at the genus level occurred. Overall, MALDI-TOF MS yielded a score of x ≥ 2.0 for 204/410 (49.8%) of isolates and an x score between 1.7 and 2 for 73/410 (17.8%) isolates. Among the 73 isolates giving a score of <2.0, as recommended by clinical microbiology for accurate species-level identification, 66/73 (90.4%) were concordant at the species level and 7/73 (9.6%) at the genus level. Hence, 254/410 (62%) strains were concordant at the species level between 16S sequencing and MALDI-TOF MS. However, when only a score of ≥2.0 is considered, only 188 (45.9%) of these isolates would have achieved a reliable identification score on MALDI-TOF and would not have to be secondarily sequenced
Our large regional clinical microbiology has had a similar experience throughout implementation of MALDI-TOF MS. Although proteomics analyses have reduced the need for 16S rRNA gene sequencing to definitively identify many genera/species, the same numbers of difficult-to-identify isolates have been sent for molecular analysis despite using MALDI-TOF as the main identification method since 2014. Between 2010 and 2016, the types of clinically relevant organisms that required sequencing in our laboratory were related to the ability of the primary routine testing over this period to adequately provide an identification, as conventional biochemical testing is much less capable than MS. Overall, the largest group of organisms that required sequencing was Gram-positive bacilli, which are typically difficult to definitively identify biochemically: Gram-positive bacilli comprised 48.5% of all sequenced isolates from 2010 to 2016, including aerobic (18.2%), anaerobic (25.2%), and aerobic (5.1%) actinomycetes. Anaerobes collectively made up 40.1% of all isolates, disproportionate to the frequency at which they were cultured and again attributable to the difficulty identifying them biochemically (554–556). While anaerobes comprised almost half of all the isolates our laboratory sequenced in 2011, only 23.6% of sequenced isolates were anaerobes by 2016. This shows the dramatic impact MS has had on our reliance on sequencing and the ongoing limitations of current routine MALDI-TOF MS databases for anaerobe identification, although the Bruker Microflex LT system with an expanded database had improved performance (555). Even though our laboratory continues to perform 16S sequencing on a steady number of isolates, we increasingly use it as a gold standard reference test for verification of MALDI-TOF MS identification of unusual or rare organisms.
MALDI-TOF MS’s major advantage over other microbiological identification methods is its ability to rapidly and reliably identify a wide variety of microorganisms directly from the primary selective isolation medium. Despite the tremendous diagnostic advances realized by routine use of MALDI-TOF MS, the results of the colony morphology, Gram stain reaction, and rapid spot biochemical tests may still be required for confirmation of a proteomics bacterial pathogen identification. Although MALDI-TOF MS isolate analysis has a significantly lower cost than conventional phenotypic testing, significant capital is required to purchase and maintain a sophisticated mass spectrometry instrument (548). Although MALDI-TOF has allowed improved performance and increased capability compared to phenotypical analyses in clinical microbiology laboratories, it has not entirely replaced 16S sequencing (543, 545). However, since commercial MALDI-TOF MS systems (i.e., Vitek MS [bioMérieux] and MALDI Biotyper and Microflex LT [Bruker]) databases are based on ribosomal proteomic marker spectral profiles within 16S, it should not be surprising that its performance for accurate genus/species identification is not better in cases where 16S sequence analysis is known to be challenging; molecular analysis has advantages for resolution in these cases. Therefore, differentiating and identifying species with identical or almost identical 16S sequences is also problematic for MALDI-TOF MS, so the main advantages of proteomics compared to molecular analyses are MALDI-TOF MS’s ease of use, short hands-on time, and expense compared to 16S aside from the capital expenditure for an MS instrument (Table 13). Compared to MALDI-TOF MS, 16S cycle sequencing is still an expensive and rather complex procedure that takes at least a day to perform, analyze, and report. Unfortunately, 16S sequencing has not had the benefit of a similar effort in commercial automation, simplification, and curated database development, which has affected its broad routine use in clinical laboratories.
MALDI-TOF MS technology’s main limitation is that identification of new isolates is possible only if the spectral database contains peptide mass fingerprints of the type strains of specific genera/species/subspecies/strains (28, 29). Although MALDI-TOF MS databases are in continuous development, clinical isolates may not be identified by either commercial system (i.e., MALDI Biotyper and Vitek MS, bioMérieux), because the organism is not included in their databases or because the ribosomal protein spectrum is too similar to that of another species (29). Currently, clinical microbiology laboratories are largely reliant on industry for MALDI-TOF MS database updates. Individual researchers as well as clinical laboratories have limited accessibility on either of these platforms for in-house development because of the proprietary nature of their software and proteomic databases. User development of their own spectral profiles or assays has also been limited by the specialized training and expense of accessing expanded databases within these commercial systems. Several research groups have developed open-source software and databases to get around this issue, including MALDIquant, SpectraBank, mMass, Mass-Up, and pkDACLASS, but this is not a comprehensive list (www.mmass.org and www.sing-group.org/mass-up) (557–559). What would be clinically helpful is an apparatus-independent open-access database of MALDI-TOF spectral profiles that is linked to 16S sequences for validation; such a database should be linked to an archived clinical strain repository for each institute, against which external variants of the encountered species could be searched.
Another current disadvantage of MALDI-TOF MS is the need for a pure culture/colony to be able to perform reliable identification. This means that the organism must grow to some extent on culture media in a pure culture that is free from contamination by other bacteria. The organisms must also be alive when sampled in order to generate enough protein to be measured in the MALDI-TOF instrument. As outlined in more detail in the recently published CLSI M58 guidelines and the updated CLSI MM-18 A2 guidelines, MALDI-TOF MS misidentifications commonly occur with taxonomically closely related bacteria, such as Escherichia coli and Shigella, coagulase-negative staphylococci, viridans streptococci, some Gram-negative nonfermenters, and Bacillus cereus group species (29). Each of the two commercially available systems has specific limitations, as documented by previous studies (68, 556, 560–563). Jamal and colleagues performed a large comparative evaluation of the Vitek MS and Microflex LT (Bruker) platforms using a collection of 827 clinically important Gram-positive cocci and found that these systems correctly identified 97.2% and 94.7%, respectively (560). Although both systems reliably identified Staphylococcus aureus, beta-hemolytic streptococci, and enterococci, their databases for coagulase-negative staphylococci and viridans streptococcal species spectral profiles need to be expanded to improve performance. Seng and colleagues (563) showed that only 86 (22.3%) of 385 CoNS isolates were identified to the genus level using the Bruker Biotyper database. The Microflex LT system also misidentified several S. pneumoniae isolates as S. mitis (564). Carbonnelle and colleagues showed that 23 reference strains representative of clinically relevant species and subspecies of Micrococcaceae could be used as a database for the rapid identification of clinical CoNS isolates, which allowed accurate species-level identification for 97.4% of their CoNS isolates with MALDI-TOF MS (562). Spanu and colleagues also used MALDI-TOF MS analysis (Bruker Biotyper software, V2.0, using default parameter settings by the standard pattern-matching algorithm against the spectra of a reference database encompassing 46 Staphylococcus species and subspecies) to characterize 450 CoNS Staphylococcus species isolated from blood cultures and compared its performance to that of reference identification using rpoB sequence analysis (68). MALDI-TOF MS gave a correct species and subspecies identification for 447/450, 99.3% of the isolates, with only 3 being misidentified.
MALDI-TOF or 16S sequencing also should not currently be used for the routine identification of biopathogens that are categorized as potential risk level 3 bioterrorism agents (i.e., B. anthracis, Brucella spp., Yersinia pestis, and Fransicella tularensis), as well as several other pathogens that require increased containment measures (i.e., Burkholderia pseudomallei/mallei) (29, 565, 566). Clinical laboratories should continue to follow the recommended CDC algorithms, which rely on phenotypic testing for preliminary identification of a bioterrorism agent and immediately send the isolate to their public health reference laboratory for confirmatory identification according to previously published guidelines (https://www.selectagents.gov, https://clinmicro.asm.org/index.php/science-skills/guidelines/sentinel-guidelines).
As outlined in “Current Limitations of 16S rRNA Gene Target for Pathogen Identification,” above, broad-range 16S analyses can be used for discovery of novel species or undescribed variants, a major advantage compared to MALDI-TOF MS. Even if the isolate’s sequence is not exactly represented in the database, comparison of a 16S multisequence alignment to close but not identical matching references allows, with or without the help of phylogenetic analysis, clinical assignment of the organism to a known species/microorganism group with a high level of confidence. This information provides valuable information to the clinician trying to diagnose and treat patients with rare infections. Since most of the bacterial kingdom has not been discovered (64) and evolution continuously produces new variants of established species, the next section outlines how 16S or whole-genome sequencing will allow clinical laboratories to cope with the vast unknown. Ideally, genome-based DNA sequencing would be the gold-standard basis for bacterial organism identification and virulence profiling (e.g., structural genes provide identification, while others indicate potential for resistance or pathogenicity), and protein profiles would show what the organism actually does, what metabolism it has, and what proteins are expressed to render its specific proteomic classification (e.g., pathogenic, resistant, small colony variant, etc.). Therefore, genomic analyses will continue to complement and assist with enhancements to diagnostic proteomic analyses.
Will NGS Replace Single Targeted 16S Sequencing for Bacterial Identification?
Analysis of the whole genome of numerous pathogens can be done in one next-generation sequence (NGS) run, either from clinically recovered bacterial isolates or from metagenomics analyses (i.e., multiple species present in patient material from one individual). In contrast to Sanger sequencing, a major advantage of NGS is that a single protocol can be used for all pathogens for both identification and typing applications. Therefore, clinical microbiology laboratories are using this technology for a variety of applications, because both the investment and the running costs of NGS have substantially decreased during the last decade (567, 568). Next-generation sequencing holds unprecedented promise as a method for definitive pathogen identification, detection, and tracking of antimicrobial resistance, metagenomic analyses, and molecular epidemiological surveillance of outbreaks (25, 568–572). This section outlines not only the ability of NGS to revolutionize the identification of known and previously uncharacterized pathogens but also the obstacles preventing its widespread adoption in clinical laboratories.
Haemophilus influenzae was the first bacterium to be wholly sequenced in 1995 using the Sanger method, and it took more than a year to complete (573). Although the amount of available whole-genome sequencing data in public databases has rapidly increased since the introduction of NGS, according to the Microbial Genomes Resource database in NCBI, there have been ∼150,000 complete bacterial genome sequences deposited as of 2018, but most of these data have been obtained using Sanger cycle sequencing (https://www.ncbi.nlm.nih.gov/genome/microbes/). Many of our comments about Sanger cycle sequencing are also relevant to the performance of NGS in a diagnostic setting, in that standardized procedures will need to be put in place that include appropriate controls and quality checks across the sample test cycle (i.e., sample acquisition, NGS analysis, and data interpretation and analysis) (98, 574). Today’s NGS platforms provide rapid analyses of full genomes of clinical isolates (e.g., for strain characterization, assessment of resistance or pathogenicity, and epidemiological typing). Another application is the quantitative determination of the composition of a bacterial population (e.g., nonsterile clinical samples) to determine the 16S microbiome present by sequencing all 16S operons. Both approaches for performing high-throughput NGS testing can be streamlined through automation so that this method will progressively become more rapid and less expensive. In terms of efficacy and user-independent accuracy, one could imagine microbiome-like NGS approaches eventually replacing (to some extent) current culture-based testing of nonsterile materials in the clinical laboratory. However, NGS-based approaches (e.g., 16S microbiome analysis) require data analysis processes and infrastructure similar to those of Sanger cycle sequencing, such as matching reads to meaningful reference databases and classifying reads with regard to match accuracy, match length, match consistency, and match differentiation, which all have substantial impact on the accuracy of results for both species-level identification and microbiome analyses.
Widespread adoption of NGS in clinical microbiology depends on standardized and simplified/automated procedures that can be efficiently executed for various specimen volume ranges (i.e., from testing only a few to a high number of samples) while ensuring a timely result. Access to curated and regularly updated reference databases for species identification is important, as is the availability of quantification standards and an archived strain repository of inter- and intrapatient case comparisons or case follow-up. The NGS analysis of bacterial genomes would benefit from the availability of curated reference genomes for various clinically relevant species, which would not only standardize analysis procedures but also facilitate the assembly and mapping of genes. Genomes from clinical isolates could be searched against a repository of reference genomes that are linked to a strain repository where information about phenotype, pathogenicity, and antibiotic resistance are also housed. Several groups have begun to develop such comprehensive databases, including Advanced Molecular Detection (AMD), Centers for Diseases Control and Prevention (CDC, Atlanta, GA [https://www.cdc.gov/amd]) (no genomes uploaded yet), and NCBI (https://www.ncbi.nlm.nih.gov/refseq/about/prokaryotes/), but so far none of them covers all of the isolate characteristics listed above. So far, there is limited availability and clinical access to annotated pathogen-based whole-genome sequencing data that has been curated (i.e., quality assured), and most of this resides in proprietary commercial entities (e.g., CosmosID, Aperiomics, etc.). In addition, clinical epidemiologic and outcome studies will be required to interpret vast amounts of NGS pathogen data into clinically relevant, actionable information. Several excellent recent reviews have been published that provide a much more detailed description of the current status of NGS than can be provided here (567, 568, 570, 571).
NGS requires no specific target primers, unlike Sanger sequencing, because the whole genome of a pathogen is sequenced at random. Prior to sequencing, fragmentation of the genome is performed, since the maximum length sequenced by a benchtop NGS sequencer varies between 100 and 1,000 bases, whereas the size of common human pathogens ranges somewhere between 2 and 5 Mb, so the genomes cannot be sequenced in one part (575, 576). Therefore, NGS starts with the robust preparation of libraries that contains a representative source of the DNA or RNA of the genome under investigation, in which fragments of DNA and RNA are fused to adapters and barcodes that distinguish the DNA of the sequenced isolates, followed by clonal amplification, normalization, and sequencing (575–577). NGS is a much higher-throughput technology that provides a comprehensive analysis of a microbial genome at a sequence data rate that is substantially quicker (i.e., hundreds to thousands of times faster than Sanger). However, current NGS technologies are potentially less accurate than Sanger sequencing, and the shorter sequence read lengths can lead to difficulties with subsequent secondary assembly (575). Genome assembly from the multitude of short reads obtained by NGS is also more laborious if a reference sequence is not available for comparison, so assembly must be done de novo.
Clinical laboratories also need to be aware of the limitations of the NGS method/platform being used, because there are specific types of sequencing, reading, and analysis errors that have been reported (575, 578). Extensive information on currently available NGS platforms is available on the commercial supplier’s websites, and an overview of the properties of current NGS platforms is outlined in Table 14. It is clear that different vendors are producing high-throughput DNA sequencing machines that operate with significantly different underlying technologies that produce dissimilar types and quantities of sequence information (575, 577). In general, currently available NGS systems simultaneously determine the sequence of DNA bases from many thousands or even millions of short DNA templates (i.e., massively parallel sequencing) in a single biochemical reaction volume. Each template molecule may be affixed to a separate solid surface and then clonally amplified to increase signal strength using various sequencing methods, such as semiconductor sequencing, which measures a pH change with nucleotide incorporation (Ion Torrent PGM, Life Technologies, Thermo Fisher Scientific), and sequencing by synthesis of fluorescent reversible terminators (MiSeq; Illumina). Overall, these shorter read systems function well when there is a reference sequence available for comparison and mapping against the test isolate’s sequence (568, 575, 578). Currently, most clinical laboratories have used either the Illumina MiSeq or Ion Torrent PGM platforms for either targeted or whole-genome sequencing of bacterial isolates for various applications because of their overall read length and accuracy (576, 579). Other technologies are designed to rapidly sequence single long DNA templates in an approach called single molecular real-time (SMRT) sequencing, where fluorescent nucleotides are incorporated (Pacific Biosciences), which supports different applications and complements the information derived from shorter DNA sequence analyses. This platform may be used in research settings for shotgun sequencing of unknown isolates where no reference sequence is available, because its longer read lengths and advanced analytics improve and simplify de novo genome assembly (580, 581). An exception to this is the third generation of sequencers, such as MinION (Oxford Nanopore) and Sequel (Pacific Biosciences), which can generate larger fragments (more than 200 kb) (580, 582). These sequencers, however, are not yet in widespread use in the clinical microbiology laboratory because of their lack of affordability, the lower quality of the sequences, and the low throughput. Oxford Nanopore platforms use ionic current sensing as DNA is passed through nanopores, causing a current change that is specific for the type of nucleotide present, and different platforms are available for large-scale analyses or on a miniaturized scale using the portable MinION system (582, 583). Nanopore technology allows direct, electronic analysis of a variety of analytes regardless of length, including DNA, RNA, or small proteins. Although the overall error rate of nanopore technology initially did not meet the accuracy requirements for routine diagnostic testing due to lack of nucleotide specificity, this approach has shown steady improvement with the use of new sensing modalities and device pore architecture (582, 584). Combining NGS approaches using a system with higher read coverage and accuracy (i.e., Illumina), as well as a system with longer read coverage and lower accuracy (i.e., Nanopore), also may provide improved performance for some applications in the clinical laboratory setting (568, 583, 584).
TABLE 14.
Comparison of available next-generation sequencing platformsa
| Company | Instrument | Output/run (Gb) | Maximum read length (bp) | No. of reads (×106) | Running time |
|---|---|---|---|---|---|
| Illumina | MiniSeq | 0.6–7.5 | 2 × 150 | 25 | 4–24 h |
| MiSeq | 0.3–15 | 2 × 300 | 25 | 5–55 h | |
| NextSeq | 20–120 | 2 × 150 | 130/400 | 12–30 h | |
| HiSeq 3,000 | 125–700 | 2 × 150 | 2,500 | <1–3.5 days | |
| ThermoFisher | Ion PGM | 0.03–2 | 200–400 | 0.4–5.5 | 2–7 h |
| Ion 5S | 0.6–15 | 200–400 | 3–80 | 2.4–4 h | |
| Ion 5S XL | 0.6–15 | 200–400 | 3–80 | <24 h | |
| Oxford Nanopore | MinION | 21–42 | 230,000–300,000 | 2.2–4.4 | 1 min–48 h |
| Pacific Biosciencesb | Sequel | 0.75–1.25 | >20,000 | 370,000 | 30 min–6 h |
| Pacific Biosciencesb | RSII | 0.5–1 | >20,000 | 55,000 | 30 min–4 h |
Republished from the Journal of Biotechnology (568).
The Pacific Biosciences data are per smart cell; both the Sequel and the RSII can run 1 to 16 smart cells in one run.
Many bioinformatics, database, and clinical interpretation challenges remain in routinely applying one or more of these technologies to the daily clinical workflow of a diagnostic setting in order to produce a timely, reportable result that is clinically relevant (585). The lack of automated interpretation software that translates sequence data directly into actionable clinical information is one of the biggest barriers to NGS implementation into routine clinical practice (586). For NGS to be widely adopted, clinical microbiologists need to be able to provide a clinically relevant result in a time frame that is impactful for patient care. As outlined earlier (see “The 16S rRNA Gene and Primer Selection,” above), a definitive bacterial identification can be reported the same day using partial targeted Sanger sequencing of the 16S rRNA gene and a fast-cycle sequencing method, particularly if sequence analysis and interpretation are done in real time using a commercial curated database. To date, the fastest NGS protocol reported using targeted 16S-23S rRNA gene analyses to accomplish bacterial identification cost ~$90 (i.e., conversion from 70 pounds sterling) and took ≤4 days, the rate-limiting step being the complex analyses of NGS data (588).
Key bioinformatics challenges created by NGS data include, but are not limited to, aligning (mapping) large numbers of reads to a reference genome or de novo assembly of novel genomes, multiple alignment of huge numbers of reads and rare variant detection for amplicon sequencing projects, and file formats and computational tools for efficient storage and manipulation of multigigabyte sequence data files (577, 585, 587). Deurenberg and colleagues recently outlined the multitude of software packages frequently used for NGS data analyses in their clinical laboratory for various applications, including annotation, assembly, data quality checks, identification, detection and tracking of antimicrobial resistance, metagenomics and phylogeny, resistance and single-nucleotide polymorphism calling, genotyping, virulence, and visualization and comparison studies (567, 568, 570, 577).
Another rate-limiting step to the routine adoption of NGS sequencing in clinical microbiology laboratories is the availability of an open-access, comprehensive bacterial genome database that would readily enable comparison of curated sequence data that are fully annotated from an isolate(s) with a large number of existing strains. Given the current significant knowledge gaps outlined for the 16S rRNA gene (i.e., the most interrogated single-target gene for bacteria from clinical and environmental sources) within large public databases (i.e., GenBank and NCBI), it will clearly take some time to achieve this objective (see “Overview of fast PCR/Cycle Sequencing Using an Automated Genetic Analyzer,” above). However, the publicly available database outlined in Table 2 can be used (i.e., in packages such as Quantitative Insights into Microbial Ecology [Qiime] [www.Qiime.org]), but one has to assess the coverage of the bacterial species of interest and the reliability of sequences and their annotation. This is also a problem for interrogation of antimicrobial resistance. Microbial genome sequencing for resistance prediction and direct patient care requires widespread access to databases that have been curated and thoroughly evaluated as part of clinical trials or regulatory submission studies (570, 589). Current databases are maturing, but many resistance mechanisms need to be studied (590). The Canadian Comprehensive Antibiotic Resistance Database (CARD) is an example of a genomic antimicrobial database that combines molecular targets with exact sequence data and allows quick searches in new as well as nonannotated genomes for resistance mechanisms, determinants, and targets for individual drugs (591). To reliably identify drug-specific resistance markers using NGS, standards for quality criteria (i.e., reproducibility, sensitivity, specificity, and robustness) also must be established (592). Many bioinformatics tools are also rapidly emerging to facilitate the identification of antibiotic resistance markers in metagenomics data, and several of these have been previously described (570, 586). Global development of an open-access database of whole-genome bacterial sequence for bacterial pathogens known to cause human and animal diseases must be a priority for enabling the accelerated medical and veterinary implementation of this important technology.
Larger clinical laboratories implementing in-house NGS for one or more applications will need to validate each test offered and develop a rigorous quality control/assurance program that includes proficiency testing (574, 577, 593). Because NGS cannot be validated for all microbial species, several indicator species will likely be chosen to universally validate the entire procedure, including not only the laboratory but also bioinformatics components (568). An internal control, such as the commercial PhiX Control, provided and used for Illumina sequencing runs, or a housekeeping gene may also become standard practice (594). Overall, NGS data must be consistent when applied to diagnostic applications so that the repeatability and reproducibility of the procedure can be determined prior to implementation. However, a recent survey highlights that many clinical laboratories already performing NGS are using diverse sequencing and bioinformatics approaches across institutions, so that the results obtained at one facility may not be obtained in entirety by another laboratory, even within the same jurisdiction (593). This highlights the need for external quality assurance and proficiency testing standards development for clinical microbiology NGS tests. However, it will be complicated to control for all of the steps (high-quality DNA extraction, library preparation steps, sequencing reactions on different platforms, and the bioinformatics analyses) (587, 593). The development of international standards for not only validation but also external quality assurance of NGS procedures and testing is, however, critically important to the wide adoption of this technology by clinical microbiology laboratories.
Commercial suppliers are constantly improving available systems, and there are several other NGS platforms that will be marketed in the near future, ensuring that the capabilities will be available for diagnostic applications, including targeted sequencing of one or more bacterial genes or whole-genome sequencing. However, even if the price point for NGS instruments or contract sequencing companies makes NGS services easily affordable for smaller clinical laboratories, the preparation of isolates/clinical isolates for NGS and the subsequent analyses of the large, complex data set(s) generated will require ready access to technical and bioinformatics skills and computational infrastructure that may be beyond the capabilities of these facilities. Therefore, many clinical laboratories will elect to refer isolates/clinical isolates for NGS to larger clinical or research facilities or commercial suppliers rather than developing this technology in-house.
Migration toward routine NGS for a diverse number of clinical applications will accelerate over the next decade as the current barriers that limit its widespread adoption are addressed (587). Improvements in future generations of sequencing platforms, along with the development of rapid bioinformatics analytics, will greatly increase capacity and read length while reducing cost to a point where it is clinically feasible to perform whole-genome analyses on a routine basis. Bacterial whole-genome sequencing could become a routine tool in species identification, detection, and tracking of antimicrobial resistance, strain typing, microbiome characterization in nonsterile sites, and pathogen identification in hard-to-culture specimens, like prosthetic device infections (25, 567, 568, 577, 595). These applications will have a considerable impact on clinical diagnostics, epidemiology, and infection control. Sequencing a bacterial genome also will no longer be prohibited by cost, as it was estimated several years ago that it took as little as $100 to determine 150-fold coverage of an S. pneumoniae genome (596). Tuite and colleagues have also demonstrated that prediction of antimicrobial resistance by genome sequencing is possible for Gram-negative bacilli, but markers of in vitro resistance did not necessarily result in phenotypic expression (597). Genomic antimicrobial resistance databases will require continuous updating, quality control, and validation against standard phenotypic testing. Recently, the genome of the E. coli strain implicated in the outbreak in Europe was completely sequenced in a few hours using the Ion Torrent platform (598). Whole-genome sequencing was also shown to be superior to the gold standard typing method, pulsed-field gel electrophoresis, during a recent Canadian Listeria monocytogenes outbreak (599). Nosocomial methicillin-resistant Staphylococcus aureus outbreaks have also been efficiently investigated using whole-genome sequencing of the isolates (600, 601). Deep sequencing may also have a broader future application in diagnostic metagenomics and patient-specific microbial community analysis (25, 602).
FUTURE DIRECTIONS
Identifying bacteria isolated in the clinical laboratory by proteomics and sequence rather than phenotype has dramatically improved the diagnostic and epidemiological capabilities of clinical microbiology laboratories and allowed biochemically ambiguous, rare, and novel isolates to be described. Both proteomic and molecular identification methods allow bacterial identification that is more accurate and reproducible than that previously obtained using biochemical tests alone. However, with the steady adoption of next-generation sequencing technologies, the clinical microbiology laboratory is poised to be able to provide clinicians, infection control programs, and public health with a level of strain discrimination that was not previously possible.
One of the important improvements needed for sequence-based identification is related to data analysis. Current sequence analysis generally relies on BLAST searches against one or several reference databases, generating match lists ordered by score. When looking at the results, one may find a certain number of mismatches for the first match as well as the second and further matches. However, one needs to know whether these matches and mismatches still allow identification to the species by allowing differentiation of the second-best matching species and subsequent further matches. To determine this, there is a need for pairwise, or, even better, multiple alignments of the sample sequence to determine where the best matches occur and to visualize if the mismatches occur within a variable region of the genus where all species can be accurately differentiated. Such a process, however, is time-consuming and requires the ability to easily generate and analyze such multialignments.
Ideally, a search of a sample sequence against a database would consider the base pair position(s) of any mismatches and, thus, weigh them differently if they seem to be relevant for species differentiation. In our opinion, such technology would be rather helpful, not only for 16S but also for many other sequence-based assays, where positions of matches and mismatches make a difference in terms of clinical outcome, unlike a BLAST result, where all mismatches are weighted the same. Along with the weighting of matches and mismatches, the data analysis system also should consider the known diversity of each species to reliably recognize or rule out a variant of a species. Finally, all this should be automated to yield timely clinical results in as easy and routine a manner as possible while providing enough insight to troubleshoot errors for a more detailed investigation. Such an automated sequencing analysis system will also be essential for NGS read matching for a variety of clinical applications (e.g., isolate identification and virulence detection, metagenomics, population analysis, etc.).
Reflecting on the previous conclusions of Clarridge et al., it is remarkable that the listed barriers to widespread clinical laboratory adoption of 16S Sanger sequencing in 2004 for microbial identification are the same as those that will need to be overcome in the next decade to allow routine use of NGS for various applications (6). One major difficulty independent of technology is the definition of microbial taxonomy by humans and how this is reflected by the microorganism’s genetic and proteomic markers; in this regard, new sequencing technologies such as NGS will allow assessment of several genetic markers or whole genomes and, thus, hold the promise of better resolution. However, potential species differentiation greatly depends on the assessment of markers that represent the evolutionary clock of the microorganism involved, which is rather difficult to assess and establish; such assessment should also provide insights about the tolerance of variation within a species and about the boundaries separating it from the next closest one.
The new technologies include, but are not limited to, requirements for great technical skills, high cost of equipment, and the need for user-friendly comparative sequencing analysis software and validated databases. An additional barrier to the widespread uptake of NGS by clinical microbiology laboratories is the lack of bioinformatics expertise to assist with the development of user-friendly data analysis pipelines; standardization and validation of analysis methods, including updates and ongoing quality assurance, will be essential for case comparison of results generated by different facilities or for case follow-ups.
A future challenge for large clinical, reference, and research laboratories, as well as for industry, will be the translation of vast amounts of accrued NGS microbial data into convenient algorithm testing schemes for microbial identification, genotyping, and metagenomics and for microbiome analyses into meaningful, actionable information that clinicians can readily understand, as well as making this technology widely available to all patients served by small- or medium-sized laboratories. These challenges will not be faced by clinical microbiologists alone but by every scientist involved in a domain where the natural diversity of genes and gene sequences plays a critical role with regard to disease, health, pathogenicity, epidemiology, and other aspects of life-forms. Overcoming these challenges will require global multidisciplinary efforts across fields that would not normally interact with the clinical arena to make vast amounts of sequencing data clinically interpretable and actionable at the bedside.
ACKNOWLEDGMENTS
We thank Patrick Lane (Sceyence Studios) for his art enhancement of the illustrations. Lori Burnie-Watson assisted with the compilation of the manuscript.
S. Emler, L. Cerutti, and A. Gürtler are directors at SmartGene, a company providing services in the field of bioinfomatics, which should be seen as a potential conflict of interest.
Biographies

Deirdre L. Church [M.D., Ph.D., FRCPC D(ABMM)] is a dually trained subspecialist in infectious diseases and clinical microbiology. She is Professor, Departments of Pathology & Laboratory Medicine and Medicine, Cumming School of Medicine, University of Calgary. She is a member of several professional organizations, including AMMI, ASM, and ESCMID, and served on the Microbiology Resource Committee, College of American Pathologists. Her research has been extensively published in peer-reviewed journals. She was the Section Editor/author (Aerobic Bacteriology) of the 3rd and 4th editions of the Clinical Microbiology Procedures Handbook, ASM Press, and a coauthor for the 7th edition of Koneman’s Color Atlas and Textbook of Diagnostic Microbiology, Wolters Kluwer. Her current research focuses on the characterization and spread of antibiotic resistance, bloodstream infections, and the use of advanced technology.

Lorenzo Cerutti (Ph.D.) studied life sciences at the University of Lausanne, where he obtained a Ph.D. in biology at the Swiss Institute of Cancer Research. He acquired his experience in bioinformatics at the Wellcome Trust Sanger Center, where he worked for the first draft of the human genome. He spent several years at the Swiss Institute of Bioinformatics doing research on sequence analysis methods. Later, he worked as a bioinformatician at SmartGene, a privately owned company, where he developed NGS analysis pipelines and automated sequence annotation methods. He is now working on large NGS projects at the Health 2030 Genome Center in Geneva.

Antoine Gürtler is a bioinformatician working for SmartGene in Switzerland. Born in Geneva, he obtained his biology bachelor at the University of Geneva in 2018. Interested in working for an application service provider in life sciences, he took the opportunity to accomplish a master’s in molecular life sciences, mention bioinformatics, in collaboration between SmartGene and the University of Lausanne in 2019. During his master’s thesis, which focused on the classification of bacterial 16S sequences at the species level, he categorized the intra- and interspecies diversity with the goal of developing useful algorithms for species identification. During his spare time, Antoine likes to enjoy the fresh air of the Swiss mountains with his family and friends.

Thomas Griener (M.D., Ph.D.) is a Medical Microbiologist at Alberta Precision Laboratories and Clinical Assistant Professor in the Department of Pathology and Laboratory Medicine at the University of Calgary. He previously studied disease mechanisms and novel therapeutics for enterohemorrhagic E. coli infection and received his Ph.D. in microbiology and infectious diseases from the University of Calgary. His research currently focuses on molecular diagnostic microbiology and test utilization.

Adrian Zelazny [Ph.D., D(ABMM)] is the Chief of the Microbiology Service, Director of the Mycobacteriology section, and Director of the Microbiology Fellowship at the Microbiology Service, Department of Laboratory Medicine, Clinical Center, National Institutes of Health, in Bethesda, Maryland. He has published more than 100 peer-reviewed manuscripts and review articles. His research has focused on chronic infections and outbreaks by mycobacteria and fungi, microbiology and host-pathogen interactions in immunocompromised patients, and novel proteomic- and genomic-based diagnostic approaches. Dr. Zelazny serves on the editorial board of the Journal of Clinical Microbiology and in several Committees of the Clinical and Laboratory Standards Institute (CLSI).

Stefan Emler did his medical school at the Ludwig-Maximilian-University in Munich/Germany. He then worked at the Geneva University Hospital in Switzerland, where he got his training in Internal Medicine and Infectious Diseases and developed his research activity in molecular microbiology. He then worked as a director of clinical microbiology and molecular diagnostics for a privately owned laboratory and as consultant and medical director for Global Business Development at Roche Diagnostics. He cofounded and works as CEO for SmartGene, a Swiss-based application service provider, active globally in developing and marketing integrated cloud-based solutions for molecular diagnostics based on gene and genome sequencing. He is an active member with several professional associations, including ASM, and has contributed work as an expert on a number of scientific boards, including the European Commission for Horizon projects and CLSI. His scientific interests are in the comprehension of the natural diversity of microorganisms and viruses in the context of evolution, epidemics, and clinical relevance.
REFERENCES
- 1.Gupta R, Lanter JM, Woese CR. 1983. Sequence of the 16S ribosomal RNA from Halobacterium volcanii, an archaebacterium. Science 221:656–659. doi: 10.1126/science.221.4611.656. [DOI] [PubMed] [Google Scholar]
- 2.Olsen GJ, Pace NR, Nuell M, Kaine BP, Gupta R, Woese CR. 1985. Sequence of the 16S rRNA gene from the thermoacidophilic archaebacterium Sulfolobus solfataricus and its evolutionary implications. J Mol Evol 22:301–307. doi: 10.1007/BF02115685. [DOI] [PubMed] [Google Scholar]
- 3.Fox GE, Magrum LJ, Balch WE, Wolfe RS, Woese CR. 1977. Classification of methanogenic bacteria by 16S ribosomal RNA characterization. Proc Natl Acad Sci U S A 74:4537–4541. doi: 10.1073/pnas.74.10.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woese CR, Kandler O, Wheelis ML. 1990. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A 87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clinical Laboratory Standards Institute (CLSI). 2018. Interpretive criteria for identification of bacteria and fungi by DNA target sequencing; MM18-A2 approved guideline. CLSI, Wayne, PA. [Google Scholar]
- 6.Clarridge JE III, Attorri SM, Zhang Q, Bartell J. 2001. 16S ribosomal DNA sequence analysis distinguishes biotypes of Streptococcus bovis: streptococcus bovis biotype II/2 is a separate genospecies and the predominant clinical isolate in adult males. J Clin Microbiol 39:1549–1552. doi: 10.1128/JCM.39.4.1549-1552.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drancourt M, Bollet C, Carlioz A, Martelin R, Gayral JP, Raoult D. 2000. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol 38:3623–3630. doi: 10.1128/JCM.38.10.3623-3630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang YW, Ellis NM, Hopkins MK, Smith DH, Dodge DE, Persing DH. 1998. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol 36:3674–3679. doi: 10.1128/JCM.36.12.3674-3679.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mignard S, Flandrois JP. 2006. 16S rRNA sequencing in routine bacterial identification: a 30-month experiment. J Microbiol Methods 67:574–581. doi: 10.1016/j.mimet.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Woo PC, Ng KH, Lau SK, Yip KT, Fung AM, Leung KW, Tam DM, Que TL, Yuen KY. 2003. Usefulness of the MicroSeq 500 16S ribosomal DNA-based bacterial identification system for identification of clinically significant bacterial isolates with ambiguous biochemical profiles. J Clin Microbiol 41:1996–2001. doi: 10.1128/jcm.41.5.1996-2001.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woo PC, Lau SK, Teng JL, Tse H, Yuen KY. 2008. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect 14:908–934. doi: 10.1111/j.1469-0691.2008.02070.x. [DOI] [PubMed] [Google Scholar]
- 12.Bottger EC. 1989. Rapid determination of bacterial ribosomal RNA sequences by direct sequencing of enzymatically amplified DNA. FEMS Microbiol Lett 53:171–176. [DOI] [PubMed] [Google Scholar]
- 13.Becker K, Harmsen D, Mellmann A, Meier C, Schumann P, Peters G, von Eiff C. 2004. Development and evaluation of a quality-controlled ribosomal sequence database for 16S ribosomal DNA-based identification of Staphylococcus species. J Clin Microbiol 42:4988–4995. doi: 10.1128/JCM.42.11.4988-4995.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cloud JL, Conville PS, Croft A, Harmsen D, Witebsky FG, Carroll KC. 2004. Evaluation of partial 16S ribosomal DNA sequencing for identification of Nocardia species by using the MicroSeq 500 system with an expanded database. J Clin Microbiol 42:578–584. doi: 10.1128/jcm.42.2.578-584.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolbert CP, Persing DH. 1999. Ribosomal DNA sequencing as a tool for identification of bacterial pathogens. Curr Opin Microbiol 2:299–305. doi: 10.1016/S1369-5274(99)80052-6. [DOI] [PubMed] [Google Scholar]
- 16.Clarridge JE., III 2004. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev 17:840–862. doi: 10.1128/CMR.17.4.840-862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Church D. 2013. Principles of capillary based sequencing for clinical microbiologists. Clin Microbiol Newsl 35:11–18. doi: 10.1016/j.clinmicnews.2012.12.003. [DOI] [Google Scholar]
- 18.Cordovana M, Kostrzewa M, Soki J, Witt E, Ambretti S, Pranada AB. 2018. Bacteroides fragilis: a whole MALDI-based workflow from identification to confirmation of carbapenemase production for routine laboratories. Anaerobe 54:246–253. doi: 10.1016/j.anaerobe.2018.04.004. [DOI] [PubMed] [Google Scholar]
- 19.De Carolis E, Posteraro B, Lass-Florl C, Vella A, Florio AR, Torelli R, Girmenia C, Colozza C, Tortorano AM, Sanguinetti M, Fadda G. 2012. Species identification of Aspergillus, Fusarium and Mucorales with direct surface analysis by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin Microbiol Infect 18:475–484. doi: 10.1111/j.1469-0691.2011.03599.x. [DOI] [PubMed] [Google Scholar]
- 20.Lam JY, Wu AK, Leung WS, Cheung I, Tsang CC, Chen JH, Chan JF, Tse CW, Lee RA, Lau SK, Woo PC. 2015. Gordonia species as emerging causes of continuous-ambulatory-peritoneal-dialysis-related peritonitis identified by 16S rRNA and secA1 gene sequencing and matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS). J Clin Microbiol 53:671–676. doi: 10.1128/JCM.02971-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynch T, Gregson D, Church DL. 2016. Species-level identification of Actinomyces isolates causing invasive infections: multiyear comparison of Vitek MS (Matrix-assisted laser desorption ionization-time of flight mass spectrometry) to partial sequencing of the 16S rRNA gene. J Clin Microbiol 54:712–717. doi: 10.1128/JCM.02872-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanguinetti M, Posteraro B. 2014. MALDI-TOF mass spectrometry: any use for aspergilli? Mycopathologia 178:417–426. doi: 10.1007/s11046-014-9757-1. [DOI] [PubMed] [Google Scholar]
- 23.Sanguinetti M, Posteraro B. 2016. Mass spectrometry applications in microbiology beyond microbe identification: progress and potential. Expert Rev Proteomics 13:965–977. doi: 10.1080/14789450.2016.1231578. [DOI] [PubMed] [Google Scholar]
- 24.Veloo ACM, Jean-Pierre H, Justesen US, Morris T, Urban E, Wybo I, Kostrzewa M, Friedrich AW, Morris T, Shah H, Jean-Pierre H, Justesen US, Wybo I, Nagy E, Urban E, Kostrzewa M, Veloo A, Friedrich AW. 2018. Validation for a anaerobic bacteria optimized MALDI-TOF MS biotyper database: the ENRIA project. Anaerobe 54:224–230. doi: 10.1016/j.anaerobe.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 25.Sibley CD, Peirano G, Church DL. 2012. Molecular methods for pathogen and microbial community detection and characterization: current and potential application in diagnostic microbiology. Infect Genet Evol 12:505–521. doi: 10.1016/j.meegid.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sontakke S, Cadenas MB, Maggi RG, Diniz PP, Breitschwerdt EB. 2009. Use of broad range16S rDNA PCR in clinical microbiology. J Microbiol Methods 76:217–225. doi: 10.1016/j.mimet.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Harris KA, Hartley JC. 2003. Development of broad-range 16S rDNA PCR for use in the routine diagnostic clinical microbiology service. J Med Microbiol 52:685–691. doi: 10.1099/jmm.0.05213-0. [DOI] [PubMed] [Google Scholar]
- 28.Doern CD, Butler-Wu SM. 2016. Emerging and future applications of matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry in the clinical microbiology laboratory: a report of the Association for Molecular Pathology. J Mol Diagn 18:789–802. doi: 10.1016/j.jmoldx.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Clinical Laboratory Standards Institute (CLSI). 2016. Methods for the identification of cultured microorganisms using matrix-assisted laser desroption ionization time-of-flight mass spectrometry. M52 approved guideline. CLSI, Wayne, PA. [Google Scholar]
- 30.Erler R, Wichels A, Heinemeyer EA, Hauk G, Hippelein M, Reyes NT, Gerdts G. 2015. VibrioBase: a MALDI-TOF MS database for fast identification of Vibrio spp. that are potentially pathogenic in humans. Syst Appl Microbiol 38:16–25. doi: 10.1016/j.syapm.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Fraser M, Brown Z, Houldsworth M, Borman AM, Johnson EM. 2016. Rapid identification of 6328 isolates of pathogenic yeasts using MALDI-ToF MS and a simplified, rapid extraction procedure that is compatible with the Bruker Biotyper platform and database. Med Mycol 54:80–88. doi: 10.1093/mmy/myv085. [DOI] [PubMed] [Google Scholar]
- 32.Sanguinetti M, Posteraro B. 2017. Identification of molds by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol 55:369–379. doi: 10.1128/JCM.01640-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Angeletti S, Dicuonzo G, Avola A, Crea F, Dedej E, Vailati F, Farina C, De Florio L. 2015. Viridans group streptococci clinical isolates: MALDI-TOF mass spectrometry versus gene sequence-based identification. PLoS One 10:e0120502. doi: 10.1371/journal.pone.0120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutell RR, Larsen N, Woese CR. 1994. Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol Rev 58:10–26. doi: 10.1128/MMBR.58.1.10-26.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noller HF, Woese CR. 1981. Secondary structure of 16S ribosomal RNA. Science 212:403–411. doi: 10.1126/science.6163215. [DOI] [PubMed] [Google Scholar]
- 36.Woese CR, Gutell R, Gupta R, Noller HF. 1983. Detailed analysis of the higher-order structure of 16S-like ribosomal ribonucleic acids. Microbiol Rev 47:621–669. doi: 10.1128/MMBR.47.4.621-669.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. 1985. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci U S A 82:6955–6959. doi: 10.1073/pnas.82.20.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shine J, Dalgarno L. 1975. Growth-dependent changes in terminal heterogeneity involving 3′-adenylate of bacterial 16S ribosomal RNA. Nature 256:232–233. doi: 10.1038/256232a0. [DOI] [PubMed] [Google Scholar]
- 39.Baker GC, Smith JJ, Cowan DA. 2003. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55:541–555. doi: 10.1016/j.mimet.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 40.Woese CR, Fox GE, Zablen L, Uchida T, Bonen L, Pechman K, Lewis BJ, Stahl D. 1975. Conservation of primary structure in 16S ribosomal RNA. Nature 254:83–86. doi: 10.1038/254083a0. [DOI] [PubMed] [Google Scholar]
- 41.Odom OW, Deng HY, Dabbs ER, Hardesty B. 1984. Binding of S21 to the 50S subunit and the effect of the 50S subunit on nonradiative energy transfer between the 3′ end of 16S RNA and S21. Biochemistry 23:5069–5076. doi: 10.1021/bi00316a037. [DOI] [PubMed] [Google Scholar]
- 42.Doolittle WF. 1999. Phylogenetic classification and the universal tree. Science 284:2124–2129. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- 43.Louca S, Doebeli M, Parfrey LW. 2018. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 6:41. doi: 10.1186/s40168-018-0420-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Větrovský T, Baldrian P. 2013. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:e57923. doi: 10.1371/journal.pone.0057923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun DL, Jiang X, Wu QL, Zhou NY. 2013. Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl Environ Microbiol 79:5962–5969. doi: 10.1128/AEM.01282-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marchandin H, Teyssier C, Simeon De Buochberg M, Jean-Pierre H, Carriere C, Jumas-Bilak E. 2003. Intra-chromosomal heterogeneity between the four 16S rRNA gene copies in the genus Veillonella: implications for phylogeny and taxonomy. Microbiology 149:1493–1501. doi: 10.1099/mic.0.26132-0. [DOI] [PubMed] [Google Scholar]
- 47.Mechergui A, Achour W, Hassen AB. 2014. Intraspecific 16S rRNA gene diversity among clinical isolates of Neisseria species. APMIS 122:437–442. doi: 10.1111/apm.12164. [DOI] [PubMed] [Google Scholar]
- 48.Sacchi CT, Whitney AM, Reeves MW, Mayer LW, Popovic T. 2002. Sequence diversity of Neisseria meningitidis 16S rRNA genes and use of 16S rRNA gene sequencing as a molecular subtyping tool. J Clin Microbiol 40:4520–4527. doi: 10.1128/jcm.40.12.4520-4527.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sacchi CT, Alber D, Dull P, Mothershed EA, Whitney AM, Barnett GA, Popovic T, Mayer LW. 2005. High level of sequence diversity in the 16S rRNA genes of Haemophilus influenzae isolates is useful for molecular subtyping. J Clin Microbiol 43:3734–3742. doi: 10.1128/JCM.43.8.3734-3742.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hellberg RS, Haney CJ, Shen Y, Cheng CM, Williams-Hill DM, Martin WB. 2012. Development of a custom 16S rRNA gene library for the identification and molecular subtyping of Salmonella enterica. J Microbiol Methods 91:448–458. doi: 10.1016/j.mimet.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 51.Hellberg RS, Martin KG, Keys AL, Haney CJ, Shen Y, Smiley RD. 2013. 16S rRNA partial gene sequencing for the differentiation and molecular subtyping of Listeria species. Food Microbiol 36:231–240. doi: 10.1016/j.fm.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 52.Kitahara K, Yasutake Y, Miyazaki K. 2012. Mutational robustness of 16S ribosomal RNA, shown by experimental horizontal gene transfer in Escherichia coli. Proc Natl Acad Sci U S A 109:19220–19225. doi: 10.1073/pnas.1213609109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martinez-Porchas M, Villalpando-Canchola E, Vargas-Albores F. 2016. Significant loss of sensitivity and specificity in the taxonomic classification occurs when short 16S rRNA gene sequences are used. Heliyon 2:e00170. doi: 10.1016/j.heliyon.2016.e00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watanabe K, Kodama Y, Harayama S. 2001. Design and evaluation of PCR primers to amplify bacterial 16S ribosomal DNA fragments used for community fingerprinting. J Microbiol Methods 44:253–262. doi: 10.1016/s0167-7012(01)00220-2. [DOI] [PubMed] [Google Scholar]
- 55.Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 1991. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703. doi: 10.1128/jb.173.2.697-703.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sipos R, Szekely AJ, Palatinszky M, Revesz S, Marialigeti K, Nikolausz M. 2007. Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol Ecol 60:341–350. doi: 10.1111/j.1574-6941.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 57.Petti CA. 2007. Detection and identification of microorganisms by gene amplification and sequencing. Clin Infect Dis 44:1108–1114. doi: 10.1086/512818. [DOI] [PubMed] [Google Scholar]
- 58.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glockner FO. 2013. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ. 2008. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl Environ Microbiol 74:2461–2470. doi: 10.1128/AEM.02272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW. 2014. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol 12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Handschur M, Karlic H, Hertel C, Pfeilstocker M, Haslberger AG. 2009. Preanalytic removal of human DNA eliminates false signals in general 16S rDNA PCR monitoring of bacterial pathogens in blood. Comp Immunol Microbiol Infect Dis 32:207–219. doi: 10.1016/j.cimid.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 62.Clinical Laboratory Standards Institute (CLSI). 2008. Interpretive criteria for identification of bacteria and fungi by DNA target sequencing; M18 approved guideline. CLSI, Wayne, PA. [Google Scholar]
- 63.Ibal JC, Pham HQ, Park CE, Shin JH. 2019. Information about variations in multiple copies of bacterial 16S rRNA genes may aid in species identification. PLoS One 14:e0212090. doi: 10.1371/journal.pone.0212090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lennon JT, Locey KJ. 2016. The underestimation of global microbial diversity. mBio 7:e01298-16. doi: 10.1128/mBio.01298-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Locey KJ, Lennon JT. 2016. Scaling laws predict global microbial diversity. Proc Natl Acad Sci U S A 113:5970–5975. doi: 10.1073/pnas.1521291113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Drancourt M, Roux V, Fournier PE, Raoult D. 2004. rpoB gene sequence-based identification of aerobic Gram-positive cocci of the genera Streptococcus, Enterococcus, Gemella, Abiotrophia, and Granulicatella. J Clin Microbiol 42:497–504. doi: 10.1128/jcm.42.2.497-504.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khamis A, Raoult D, La Scola B. 2005. Comparison between rpoB and 16S rRNA gene sequencing for molecular identification of 168 clinical isolates of Corynebacterium. J Clin Microbiol 43:1934–1936. doi: 10.1128/JCM.43.4.1934-1936.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Spanu T, De Carolis E, Fiori B, Sanguinetti M, D'Inzeo T, Fadda G, Posteraro B. 2011. Evaluation of matrix-assisted laser desorption ionization-time-of-flight mass spectrometry in comparison to rpoB gene sequencing for species identification of bloodstream infection staphylococcal isolates. Clin Microbiol Infect 17:44–49. doi: 10.1111/j.1469-0691.2010.03181.x. [DOI] [PubMed] [Google Scholar]
- 69.Peker N, Garcia-Croes S, Dijkhuizen B, Wiersma HH, van Zanten E, Wisselink G, Friedrich AW, Kooistra-Smid M, Sinha B, Rossen JWA, Couto N. 2019. A comparison of three different bioinformatics analyses of the 16S-23S rRNA encoding region for bacterial identification. Front Microbiol 10:620. doi: 10.3389/fmicb.2019.00620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shin HB, Yoon J, Lee Y, Kim MS, Lee K. 2015. Comparison of MALDI-TOF MS, housekeeping gene sequencing, and 16S rRNA gene sequencing for identification of Aeromonas clinical isolates. Yonsei Med J 56:550–555. doi: 10.3349/ymj.2015.56.2.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou M, Yang Q, Kudinha T, Zhang L, Xiao M, Kong F, Zhao Y, Xu YC. 2016. Using matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) complemented with selected 16S rRNA and gyrB genes sequencing to practically identify clinical important Viridans group streptococci (VGS). Front Microbiol 7:1328. doi: 10.3389/fmicb.2016.01328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen ML, Tsen HY. 2002. Discrimination of Bacillus cereus and Bacillus thuringiensis with 16S rRNA and gyrB gene based PCR primers and sequencing of their annealing sites. J Appl Microbiol 92:912–919. doi: 10.1046/j.1365-2672.2002.01606.x. [DOI] [PubMed] [Google Scholar]
- 73.Scholz HC, Al Dahouk S, Tomaso H, Neubauer H, Witte A, Schloter M, Kampfer P, Falsen E, Pfeffer M, Engel M. 2008. Genetic diversity and phylogenetic relationships of bacteria belonging to the Ochrobactrum-Brucella group by recA and 16S rRNA gene-based comparative sequence analysis. Syst Appl Microbiol 31:1–16. doi: 10.1016/j.syapm.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 74.Glazunova OO, Raoult D, Roux V. 2010. Partial recN gene sequencing: a new tool for identification and phylogeny within the genus Streptococcus. Int J Syst Evol Microbiol 60:2140–2148. doi: 10.1099/ijs.0.018176-0. [DOI] [PubMed] [Google Scholar]
- 75.Ghebremedhin B, Layer F, Konig W, Konig B. 2008. Genetic classification and distinguishing of Staphylococcus species based on different partial gap, 16S rRNA, hsp60, rpoB, sodA, and tuf gene sequences. J Clin Microbiol 46:1019–1025. doi: 10.1128/JCM.02058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heikens E, Fleer A, Paauw A, Florijn A, Fluit AC. 2005. Comparison of genotypic and phenotypic methods for species-level identification of clinical isolates of coagulase-negative staphylococci. J Clin Microbiol 43:2286–2290. doi: 10.1128/JCM.43.5.2286-2290.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hwang SM, Kim MS, Park KU, Song J, Kim EC. 2011. Tuf gene sequence analysis has greater discriminatory power than 16S rRNA sequence analysis in identification of clinical isolates of coagulase-negative staphylococci. J Clin Microbiol 49:4142–4149. doi: 10.1128/JCM.05213-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Paradis S, Boissinot M, Paquette N, Belanger SD, Martel EA, Boudreau DK, Picard FJ, Ouellette M, Roy PH, Bergeron MG. 2005. Phylogeny of the Enterobacteriaceae based on genes encoding elongation factor Tu and F-ATPase beta-subunit. Int J Syst Evol Microbiol 55:2013–2025. doi: 10.1099/ijs.0.63539-0. [DOI] [PubMed] [Google Scholar]
- 79.Poyart C, Quesne G, Boumaila C, Trieu-Cuot P. 2001. Rapid and accurate species-level identification of coagulase-negative staphylococci by using the sodA gene as a target. J Clin Microbiol 39:4296–4301. doi: 10.1128/JCM.39.12.4296-4301.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kwok AY, Su SC, Reynolds RP, Bay SJ, Av-Gay Y, Dovichi NJ, Chow AW. 1999. Species identification and phylogenetic relationships based on partial HSP60 gene sequences within the genus Staphylococcus. Int J Syst Bacteriol 49(Part 3):1181–1192. doi: 10.1099/00207713-49-3-1181. [DOI] [PubMed] [Google Scholar]
- 81.Shah MM, Iihara H, Noda M, Song SX, Nhung PH, Ohkusu K, Kawamura Y, Ezaki T. 2007. dnaJ gene sequence-based assay for species identification and phylogenetic grouping in the genus Staphylococcus. Int J Syst Evol Microbiol 57:25–30. doi: 10.1099/ijs.0.64205-0. [DOI] [PubMed] [Google Scholar]
- 82.Nhung PH, Shah MM, Ohkusu K, Noda M, Hata H, Sun XS, Iihara H, Goto K, Masaki T, Miyasaka J, Ezaki T. 2007. The dnaJ gene as a novel phylogenetic marker for identification of Vibrio species. Syst Appl Microbiol 30:309–315. doi: 10.1016/j.syapm.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 83.Liu H, Li Y, Huang X, Kawamura Y, Ezaki T. 2003. Use of the dnaJ gene for the detection and identification of all Legionella pneumophila serogroups and description of the primers used to detect 16S rDNA gene sequences of major members of the genus Legionella. Microbiol Immunol 47:859–869. doi: 10.1111/j.1348-0421.2003.tb03452.x. [DOI] [PubMed] [Google Scholar]
- 84.Yamada-Noda M, Ohkusu K, Hata H, Shah MM, Nhung PH, Sun XS, Hayashi M, Ezaki T. 2007. Mycobacterium species identification–a new approach via dnaJ gene sequencing. Syst Appl Microbiol 30:453–462. doi: 10.1016/j.syapm.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 85.Gundi V, Dijkshoorn L, Burignat S, Raoult D, La Scola B. 2009. Validation of partial rpoB gene sequence analysis for the identification of clinically important and emerging Acinetobacter species. Microbiology 155:2333–2341. doi: 10.1099/mic.0.026054-0. [DOI] [PubMed] [Google Scholar]
- 86.Renesto P, Gouvernet J, Drancourt M, Roux V, Raoult D. 2001. Use of rpoB gene analysis for detection and identification of Bartonella species. J Clin Microbiol 39:430–437. doi: 10.1128/JCM.39.2.430-437.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Drancourt M, Raoult D. 2002. rpoB gene sequence-based identification of Staphylococcus species. J Clin Microbiol 40:1333–1338. doi: 10.1128/jcm.40.4.1333-1338.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qiagen. 2018. Pyrosequencing technology and platform review. Qiagen, Hilden, Germany: https://www.qiagen.com/no/resources/technologies/pyrosequencing-resource-center/technology-overview/. [Google Scholar]
- 90.Sanger F, Coulson AR. 1978. The use of thin acrylamide gels for DNA sequencing. FEBS Lett 87:107–110. doi: 10.1016/0014-5793(78)80145-8. [DOI] [PubMed] [Google Scholar]
- 91.Applied Biosystems (ABI). 2010. BigDye terminator v3.1 cycle sequencing kit: protocol. ABI ThermoFisher Scientific, Foster City, CA. [Google Scholar]
- 92.Applied Biosystems (ABI). 2009. DNA sequencing by capillary electrophoresis. Chemistry guide. ABI, Thermo Fisher Scientific, Foster City, CA. [Google Scholar]
- 93.Wallis Y, Morrell N. 2011. Automated DNA sequencing. Methods Mol Biol 688:173–185. doi: 10.1007/978-1-60761-947-5_12. [DOI] [PubMed] [Google Scholar]
- 94.Slatko BE, Kieleczawa J, Ju J, Gardner AF, Hendrickson CL, Ausubel FM. 2011. First generation automated DNA sequencing technology. Curr Protoc Mol Biol Chapter 7:Unit7 2. doi: 10.1002/0471142727.mb0702s96. [DOI] [PubMed] [Google Scholar]
- 95.Clinical Laboratory Standards Institute (CLSI). 2014. Nucleic acid sequencing methods in diagnostic laboratory medicine. MM09-A2 approved guideline. CLSI, Wayne, PA. [Google Scholar]
- 96.Applied Biosystems (ABI). 2008. Advances in fast PCR contribute to a fast resequencing workflow. ABI application note ThermoFisher Scientific, Foster City, CA. [Google Scholar]
- 97.Platt AR, Woodhall RW, George AL Jr. 2007. Improved DNA sequencing quality and efficiency using an optimized fast cycle sequencing protocol. Biotechniques 43:58–62. doi: 10.2144/000112499. [DOI] [PubMed] [Google Scholar]
- 98.Clinical Laboratory Standards Institute (CLSI). 2011. Establishing molecular testing in clinical laboratory environments: M19-A approved guideline. CLSI, Wayne, PA. [Google Scholar]
- 99.Belisle JT, Mahaffey SB, Hill PJ. 2009. Isolation of Mycobacterium species genomic DNA. Methods Mol Biol 465:1–12. doi: 10.1007/978-1-59745-207-6_1. [DOI] [PubMed] [Google Scholar]
- 100.Millar BC, Jiru X, Moore JE, Earle JA. 2000. A simple and sensitive method to extract bacterial, yeast and fungal DNA from blood culture material. J Microbiol Methods 42:139–147. doi: 10.1016/s0167-7012(00)00174-3. [DOI] [PubMed] [Google Scholar]
- 101.Lorenz MG, Muhl H, Disque C. 2015. Bacterial and fungal DNA extraction from blood samples: manual protocols. Methods Mol Biol 1237:109–119. doi: 10.1007/978-1-4939-1776-1_11. [DOI] [PubMed] [Google Scholar]
- 102.Lazarevic V, Gaia N, Girard M, Francois P, Schrenzel J. 2013. Comparison of DNA extraction methods in analysis of salivary bacterial communities. PLoS One 8:e67699. doi: 10.1371/journal.pone.0067699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goldenberger D, Perschil I, Ritzler M, Altwegg M. 1995. A simple “universal” DNA extraction procedure using SDS and proteinase K is compatible with direct PCR amplification. PCR Methods Appl 4:368–370. doi: 10.1101/gr.4.6.368. [DOI] [PubMed] [Google Scholar]
- 104.Berensmeier S. 2006. Magnetic particles for the separation and purification of nucleic acids. Appl Microbiol Biotechnol 73:495–504. doi: 10.1007/s00253-006-0675-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karakousis A, Tan L, Ellis D, Alexiou H, Wormald PJ. 2006. An assessment of the efficiency of fungal DNA extraction methods for maximizing the detection of medically important fungi using PCR. J Microbiol Methods 65:38–48. doi: 10.1016/j.mimet.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 106.Verheyen J, Kaiser R, Bozic M, Timmen-Wego M, Maier BK, Kessler HH. 2012. Extraction of viral nucleic acids: comparison of five automated nucleic acid extraction platforms. J Clin Virol 54:255–259. doi: 10.1016/j.jcv.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 107.Imyanitov EN. 2006. Isolation of nucleic acids from paraffin embedded archival tissues and other diffciult sources In Kieleczawa J. (ed), DNA sequencing II: optimizing preparation and cleanup. Jones and Bartlett Publishers Inc, Sudbury, MA. [Google Scholar]
- 108.Desjardins P, Conklin D. 2010. NanoDrop microvolume quantitation of nucleic acids. J Vis Exp 45:2565. doi: 10.3791/2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nakayama Y, Yamaguchi H, Einaga N, Esumi M. 2016. Pitfalls of DNA quantification using DNA-binding fluorescent dyes and suggested solutions. PLoS One 11:e0150528. doi: 10.1371/journal.pone.0150528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Davies J, Denyer T, Hadfield J. 2016. Bioanalyzer chips can be used interchangeably for many analyses of DNA or RNA. Biotechniques 60:197–199. doi: 10.2144/000114403. [DOI] [PubMed] [Google Scholar]
- 111.Tataurov AV, You Y, Owczarzy R. 2008. Predicting ultraviolet spectrum of single stranded and double stranded deoxyribonucleic acids. Biophys Chem 133:66–70. doi: 10.1016/j.bpc.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 112.Stinson LF, Keelan JA, Payne MS. 2019. Identification and removal of contaminating microbial DNA from PCR reagents: impact on low-biomass microbiome analyses. Lett Appl Microbiol 68:2–8. doi: 10.1111/lam.13091. [DOI] [PubMed] [Google Scholar]
- 113.Mardis E, McCombie WR. 2017. Preparing polymerase chain reaction (PCR) products for capillary sequencing. Cold Spring Harb Protoc 2017:pdb.prot094599. doi: 10.1101/pdb.prot094599. [DOI] [PubMed] [Google Scholar]
- 114.Kieleczawa J. 2006. Evaluation of methods for cleanup of DNA sequencing reactions, p 219–240. In Kieleczawa J. (ed), DNA sequencing II: optimizing preparation and cleanup. Jones and Bartlett Publishers Inc, Sudbury, MA. [Google Scholar]
- 115.Ewing B, Hillier L, Wendl MC, Green P. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 116.Kieleczawa J. 2006. Fundamentals of sequencing of difficult templates–an overview. J Biomol Tech 17:207–217. [PMC free article] [PubMed] [Google Scholar]
- 117.Ewing B, Green P. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194. doi: 10.1101/gr.8.3.186. [DOI] [PubMed] [Google Scholar]
- 118.Anonymous. 1986. Nomenclature committee of the International Union of Biochemistry (NC-IUB). Nomenclature for incompletely specified bases in nucleic acid sequences. Recommendations 1984. J Biol Chem 261:13–17. [PubMed] [Google Scholar]
- 119.Reischl U, Feldmann K, Naumann L, Gaugler BJ, Ninet B, Hirschel B, Emler S. 1998. 16S rRNA sequence diversity in Mycobacterium celatum strains caused by presence of two different copies of 16S rRNA gene. J Clin Microbiol 36:1761–1764. doi: 10.1128/JCM.36.6.1761-1764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 121.Karlin S, Altschul SF. 1990. Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc Natl Acad Sci U S A 87:2264–2268. doi: 10.1073/pnas.87.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fox GE, Wisotzkey JD, Jurtshuk P Jr. 1992. How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Bacteriol 42:166–170. doi: 10.1099/00207713-42-1-166. [DOI] [PubMed] [Google Scholar]
- 123.Bottger EC, Teske A, Kirschner P, Bost S, Chang HR, Beer V, Hirschel B. 1992. Disseminated “Mycobacterium genavense” infection in patients with AIDS. Lancet 340:76–80. doi: 10.1016/0140-6736(92)90397-l. [DOI] [PubMed] [Google Scholar]
- 124.Clayton RA, Sutton G, Hinkle PS Jr, Bult C, Fields C. 1995. Intraspecific variation in small-subunit rRNA sequences in GenBank: why single sequences may not adequately represent prokaryotic taxa. Int J Syst Bacteriol 45:595–599. doi: 10.1099/00207713-45-3-595. [DOI] [PubMed] [Google Scholar]
- 125.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hall BE. 2017. Phylogenetic trees made easy, 5th ed Oxford University Press, New York, NY. [Google Scholar]
- 127.Kattar MM, Chavez JF, Limaye AP, Rassoulian-Barrett SL, Yarfitz SL, Carlson LC, Houze Y, Swanzy S, Wood BL, Cookson BT. 2000. Application of 16S rRNA gene sequencing to identify Bordetella hinzii as the causative agent of fatal septicemia. J Clin Microbiol 38:789–794. doi: 10.1128/JCM.38.2.789-794.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Garrity MG. 2001. The road map to the manual, p 119–166. In Garrity GM. (ed), Bergey's manual of systematic bacteriology. Springer-Verlag, New York, NY. [Google Scholar]
- 129.Williams TA, Heaps SE, Cherlin S, Nye TM, Boys RJ, Embley TM. 2015. New substitution models for rooting phylogenetic trees. Philos Trans R Soc Lond B Biol Sci 370:20140336. doi: 10.1098/rstb.2014.0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Church DL, Simmon KE, Sporina J, Lloyd T, Gregson DB. 2011. Identification by 16S rRNA gene sequencing of Negativicoccus succinicivorans recovered from the blood of a patient with hemochromatosis and pancreatitis. J Clin Microbiol 49:3082–3084. doi: 10.1128/JCM.01913-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Munson E, Carroll KC. 2017. What's in a name? New bacterial species and changes to taxonomic status from 2012 through 2015. J Clin Microbiol 55:24–42. doi: 10.1128/JCM.01379-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kwiatkowski NP, Babiker WM, Merz WG, Carroll KC, Zhang SX. 2012. Evaluation of nucleic acid sequencing of the D1/D2 region of the large subunit of the 28S rDNA and the internal transcribed spacer region using SmartGene IDNS software for identification of filamentous fungi in a clinical laboratory. J Mol Diagn 14:393–401. doi: 10.1016/j.jmoldx.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 133.Slechta ES, Hohmann SL, Simmon K, Hanson KE. 2012. Internal transcribed spacer region sequence analysis using SmartGene IDNS software for the identification of unusual clinical yeast isolates. Med Mycol 50:458–466. doi: 10.3109/13693786.2011.630683. [DOI] [PubMed] [Google Scholar]
- 134.Conville PS, Murray PR, Zelazny AM. 2010. Evaluation of the integrated database network system (IDNS) SmartGene software for analysis of 16S rRNA gene sequences for identification of Nocardia species. J Clin Microbiol 48:2995–2998. doi: 10.1128/JCM.00681-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Simmon KE, Croft AC, Petti CA. 2006. Application of SmartGene IDNS software to partial 16S rRNA gene sequences for a diverse group of bacteria in a clinical laboratory. J Clin Microbiol 44:4400–4406. doi: 10.1128/JCM.01364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Turenne CY, Tschetter L, Wolfe J, Kabani A. 2001. Necessity of quality-controlled 16S rRNA gene sequence databases: identifying nontuberculous Mycobacterium species. J Clin Microbiol 39:3637–3648. doi: 10.1128/JCM.39.10.3638-3648.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Conville PS, Brown-Elliott BA, Smith T, Zelazny AM. 2017. The complexities of Nocardia taxonomy and identification. J Clin Microbiol 56:e01419-17. doi: 10.1128/JCM.01419-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Helal M, Kong F, Chen SC, Bain M, Christen R, Sintchenko V. 2011. Defining reference sequences for Nocardia species by similarity and clustering analyses of 16S rRNA gene sequence data. PLoS One 6:e19517. doi: 10.1371/journal.pone.0019517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Federhen S. 2015. Type material in the NCBI Taxonomy Database. Nucleic Acids Res 43:D1086–D1098. doi: 10.1093/nar/gku1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Teng JL, Yeung MY, Yue G, Au-Yeung RK, Yeung EY, Fung AM, Tse H, Yuen KY, Lau SK, Woo PC. 2011. In silico analysis of 16S rRNA gene sequencing based methods for identification of medically important aerobic Gram-negative bacteria. J Med Microbiol 60:1281–1286. doi: 10.1099/jmm.0.027805-0. [DOI] [PubMed] [Google Scholar]
- 141.Lau SK, Ng KH, Woo PC, Yip KT, Fung AM, Woo GK, Chan KM, Que TL, Yuen KY. 2006. Usefulness of the MicroSeq 500 16S rDNA bacterial identification system for identification of anaerobic Gram positive bacilli isolated from blood cultures. J Clin Pathol 59:219–222. doi: 10.1136/jcp.2004.025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hall L, Doerr KA, Wohlfiel SL, Roberts GD. 2003. Evaluation of the MicroSeq system for identification of mycobacteria by 16S ribosomal DNA sequencing and its integration into a routine clinical mycobacteriology laboratory. J Clin Microbiol 41:1447–1453. doi: 10.1128/JCM.41.4.1447-1453.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gillman LM, Gunton J, Turenne CY, Wolfe J, Kabani AM. 2001. Identification of Mycobacterium species by multiple-fluorescence PCR-single-strand conformation polymorphism analysis of the 16S rRNA gene. J Clin Microbiol 39:3085–3091. doi: 10.1128/jcm.39.9.3085-3091.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. 2007. The human microbiome project. Nature 449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Group NHW, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J, Little AR, Peavy H, Pontzer C, Portnoy M, Sayre MH, Starke-Reed P, Zakhari S, Read J, Watson B, Guyer M, NIH HMP Working Group . 2009. The NIH Human Microbiome Project. Genome Res 19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ugarte-Torres A, Gillrie MR, Griener TP, Church DL. 2018. Eggerthella lenta bloodstream infections are associated with increased mortality following empiric piperacillin-tazobactam (TZP) monotherapy: a population-based cohort study. Clin Infect Dis 67:221–228. doi: 10.1093/cid/ciy057. [DOI] [PubMed] [Google Scholar]
- 147.Brown K, Church D, Lynch T, Gregson D. 2014. Bloodstream infections due to Peptoniphilus spp.: report of 15 cases. Clin Microbiol Infect 20:857–860. doi: 10.1111/1469-0691.12657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Katoh K, Rozewicki J, Yamada KD. 2017. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20:1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Burke CM, Darling AE. 2016. A method for high precision sequencing of near full-length 16S rRNA genes on an Illumina MiSeq. PeerJ 4:e2492. doi: 10.7717/peerj.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shin JH, Kim SH, Jeong HS, Oh SH, Kim HR, Lee JN, Yoon YC, Kim YW, Kim YH. 2011. Identification of coagulase-negative staphylococci isolated from continuous ambulatory peritoneal dialysis fluid using 16S ribosomal RNA, tuf, and sodA gene sequencing. Perit Dial Int 31:340–346. doi: 10.3747/pdi.2010.00073. [DOI] [PubMed] [Google Scholar]
- 151.Al Masalma M, Raoult D, Roux V. 2010. Staphylococcus massiliensis sp. nov., isolated from a human brain abscess. Int J Syst Evol Microbiol 60:1066–1072. doi: 10.1099/ijs.0.006486-0. [DOI] [PubMed] [Google Scholar]
- 152.Zong Z. 2012. The newly-recognized species Staphylococcus massiliensis is likely to be part of the human skin microflora. Antonie Van Leeuwenhoek 101:449–451. doi: 10.1007/s10482-011-9635-5. [DOI] [PubMed] [Google Scholar]
- 153.Kwaszewska A, Lisiecki P, Szemraj M, Szewczyk EM. 2015. Animal Staphylococcus felis with the potential to infect human skin. Med Dosw Mikrobiol 67:69–78. [PubMed] [Google Scholar]
- 154.Moser A, Stephan R, Ziegler D, Johler S. 2013. Species distribution and resistance profiles of coagulase-negative staphylococci isolated from bovine mastitis in Switzerland. Schweiz Arch Tierheilkd 155:333–338. doi: 10.1024/0036-7281/a000468. [DOI] [PubMed] [Google Scholar]
- 155.Foster G, Ross HM, Hutson RA, Collins MD. 1997. Staphylococcus lutrae sp. nov., a new coagulase-positive species isolated from otters. Int J Syst Bacteriol 47:724–726. doi: 10.1099/00207713-47-3-724. [DOI] [PubMed] [Google Scholar]
- 156.Król J, Wanecka A, Twardoń J, Mrowiec J, Dropińska A, Bania J, Podkowik M, Korzeniowska-Kowal A, Paściak M. 2016. Isolation of Staphylococcus microti from milk of dairy cows with mastitis. Vet Microbiol 182:163–169. doi: 10.1016/j.vetmic.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 157.Hajek V, Ludwig W, Schleifer KH, Springer N, Zitzelsberger W, Kroppenstedt RM, Kocur M. 1992. Staphylococcus muscae, a new species isolated from flies. Int J Syst Bacteriol 42:97–101. doi: 10.1099/00207713-42-1-97. [DOI] [PubMed] [Google Scholar]
- 158.Pantůček R, Sedláček I, Petráš P, Koukalová D, Švec P, Štětina V, Vancanneyt M, Chrastinová L, Vokurková J, Růžičková V, Doškař J, Swings J, Hájek V. 2005. Staphylococcus simiae sp. nov., isolated from South American squirrel monkeys. Int J Syst Evol Microbiol 55:1953–1958. doi: 10.1099/ijs.0.63590-0. [DOI] [PubMed] [Google Scholar]
- 159.Hauschild T, Stepanović S, Zakrzewska-Czerwińska J. 2010. Staphylococcus stepanovicii sp. nov., a novel novobiocin-resistant oxidase-positive staphylococcal species isolated from wild small mammals. Syst Appl Microbiol 33:183–187. doi: 10.1016/j.syapm.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 160.Chittpurna, Singh PK, Verma D, Pinnaka AK, Mayilraj S, Korpole S. 2011. Micrococcus lactis sp. nov., isolated from dairy industry waste. Int J Syst Evol Microbiol 61:2832–2836. doi: 10.1099/ijs.0.028043-0. [DOI] [PubMed] [Google Scholar]
- 161.Prakash O, Sharma A, Nimonkar Y, Shouche YS. 2015. Proposal for creation of a new genus Neomicrococcus gen. nov. to accommodate Zhihengliuella aestuarii Baik et al. 2011 and Micrococcus lactis Chittpurna et al. 2011 as Neomicrococcus aestuarii comb. nov. and Neomicrococcus lactis comb. nov. Int J Syst Evol Microbiol 65:3771–3776. doi: 10.1099/ijsem.0.000490. [DOI] [PubMed] [Google Scholar]
- 162.Ndiaye C, Bassene H, Cadoret F, Raoult D, Lagier JC, Sokhna C. 2018. “Citricoccus massiliensis” sp. nov., a new bacterial species isolated from human skin by culturomics. New Microbes New Infect 23:83–85. doi: 10.1016/j.nmni.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chng KR, Tay AS, Li C, Ng AH, Wang J, Suri BK, Matta SA, McGovern N, Janela B, Wong XF, Sio YY, Au BV, Wilm A, De Sessions PF, Lim TC, Tang MB, Ginhoux F, Connolly JE, Lane EB, Chew FT, Common JE, Nagarajan N. 2016. Whole metagenome profiling reveals skin microbiome-dependent susceptibility to atopic dermatitis flare. Nat Microbiol 1:16106. doi: 10.1038/nmicrobiol.2016.106. [DOI] [PubMed] [Google Scholar]
- 164.Pathom-Aree W, Nogi Y, Ward AC, Horikoshi K, Bull AT, Goodfellow M. 2006. Dermacoccus barathri sp. nov. and Dermacoccus profundi sp. nov., novel actinomycetes isolated from deep-sea mud of the Mariana Trench. Int J Syst Evol Microbiol 56:2303–2307. doi: 10.1099/ijs.0.64250-0. [DOI] [PubMed] [Google Scholar]
- 165.Pathom-Aree W, Nogi Y, Sutcliffe IC, Ward AC, Horikoshi K, Bull AT, Goodfellow M. 2006. Dermacoccus abyssi sp. nov., a piezotolerant actinomycete isolated from the Mariana Trench. Int J Syst Evol Microbiol 56:1233–1237. doi: 10.1099/ijs.0.64133-0. [DOI] [PubMed] [Google Scholar]
- 166.Takahashi N, Shinjoh M, Tomita H, Fujino A, Sugita K, Katohno Y, Kuroda T, Kikuchi K. 2015. Catheter-related blood stream infection caused by Dermacoccus barathri, representing the first case of Dermacoccus infection in humans. J Infect Chemother 21:613–616. doi: 10.1016/j.jiac.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 167.Ara I, Yamamura H, Tsetseg B, Daram D, Ando K. 2010. Luteipulveratus mongoliensis gen. nov., sp. nov., an actinobacterial taxon in the family Dermacoccaceae. Int J Syst Evol Microbiol 60:574–579. doi: 10.1099/ijs.0.009936-0. [DOI] [PubMed] [Google Scholar]
- 168.Kampfer P, Martin K, Schafer J, Schumann P. 2009. Kytococcus aerolatus sp. nov., isolated from indoor air in a room colonized with moulds. Syst Appl Microbiol 32:301–305. doi: 10.1016/j.syapm.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 169.Liu JC, Jenkins DR, Malnick H, Kovac J, Szostek J. 2012. Kytococcus schroeteri endocarditis successfully managed with daptomycin: a case report and review of the literature. J Med Microbiol 61:750–753. doi: 10.1099/jmm.0.035493-0. [DOI] [PubMed] [Google Scholar]
- 170.Chan JF, Wong SS, Leung SS, Fan RY, Ngan AH, To KK, Lau SK, Yuen KY, Woo PC. 2012. First report of chronic implant-related septic arthritis and osteomyelitis due to Kytococcus schroeteri and a review of human K. schroeteri infections. Infection 40:567–573. doi: 10.1007/s15010-012-0250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Roman-Ponce B, Wang D, Soledad Vasquez-Murrieta M, Feng Chen W, Estrada-de Los Santos P, Hua Sui X, Tao Wang E. 2016. Kocuria arsenatis sp. nov., an arsenic-resistant endophytic actinobacterium associated with Prosopis laegivata grown on high-arsenic-polluted mine tailing. Int J Syst Evol Microbiol 66:1027–1033. doi: 10.1099/ijsem.0.000830. [DOI] [PubMed] [Google Scholar]
- 172.Li WJ, Zhang YQ, Schumann P, Chen HH, Hozzein WN, Tian XP, Xu LH, Jiang CL. 2006. Kocuria aegyptia sp. nov., a novel actinobacterium isolated from a saline, alkaline desert soil in Egypt. Int J Syst Evol Microbiol 56:733–737. doi: 10.1099/ijs.0.63876-0. [DOI] [PubMed] [Google Scholar]
- 173.Park EJ, Kim MS, Roh SW, Jung MJ, Bae JW. 2010. Kocuria atrinae sp. nov., isolated from traditional Korean fermented seafood. Int J Syst Evol Microbiol 60:914–918. doi: 10.1099/ijs.0.014506-0. [DOI] [PubMed] [Google Scholar]
- 174.Tvrzova L, Schumann P, Sedlacek I, Pacova Z, Sproer C, Verbarg S, Kroppenstedt RM. 2005. Reclassification of strain CCM 132, previously classified as Kocuria varians, as Kocuria carniphila sp. nov. Int J Syst Evol Microbiol 55:139–142. doi: 10.1099/ijs.0.63304-0. [DOI] [PubMed] [Google Scholar]
- 175.Seo YB, Kim DE, Kim GD, Kim HW, Nam SW, Kim YT, Lee JH. 2009. Kocuria gwangalliensis sp. nov., an actinobacterium isolated from seawater. Int J Syst Evol Microbiol 59:2769–2772. doi: 10.1099/ijs.0.008482-0. [DOI] [PubMed] [Google Scholar]
- 176.Tang SK, Wang Y, Lou K, Mao PH, Xu LH, Jiang CL, Kim CJ, Li WJ. 2009. Kocuria halotolerans sp. nov., an actinobacterium isolated from a saline soil in China. Int J Syst Evol Microbiol 59:1316–1320. doi: 10.1099/ijs.0.006627-0. [DOI] [PubMed] [Google Scholar]
- 177.Mayilraj S, Kroppenstedt RM, Suresh K, Saini HS. 2006. Kocuria himachalensis sp. nov., an actinobacterium isolated from the Indian Himalayas. Int J Syst Evol Microbiol 56:1971–1975. doi: 10.1099/ijs.0.63915-0. [DOI] [PubMed] [Google Scholar]
- 178.Park EJ, Roh SW, Kim MS, Jung MJ, Shin KS, Bae JW. 2010. Kocuria koreensis sp. nov., isolated from fermented seafood. Int J Syst Evol Microbiol 60:140–143. doi: 10.1099/ijs.0.012310-0. [DOI] [PubMed] [Google Scholar]
- 179.Yun JH, Roh SW, Jung MJ, Kim MS, Park EJ, Shin KS, Nam YD, Bae JW. 2011. Kocuria salsicia sp. nov., isolated from salt-fermented seafood. Int J Syst Evol Microbiol 61:286–289. doi: 10.1099/ijs.0.021469-0. [DOI] [PubMed] [Google Scholar]
- 180.Cosseau C, Romano-Bertrand S, Duplan H, Lucas O, Ingrassia I, Pigasse C, Roques C, Jumas-Bilak E. 2016. Proteobacteria from the human skin microbiota: species-level diversity and hypotheses. One Health 2:33–41. doi: 10.1016/j.onehlt.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kandi V, Palange P, Vaish R, Bhatti AB, Kale V, Kandi MR, Bhoomagiri MR. 2016. Emerging bacterial infection: identification and clinical significance of Kocuria species. Cureus 8:e731. doi: 10.7759/cureus.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Xiong ZJ, Zhang JL, Zhang DF, Zhou ZL, Liu MJ, Zhu WY, Zhao LX, Xu LH, Li WJ. 2013. Rothia endophytica sp. nov., an actinobacterium isolated from Dysophylla stellata (Lour.) Benth. Int J Syst Evol Microbiol 63:3964–3969. doi: 10.1099/ijs.0.052522-0. [DOI] [PubMed] [Google Scholar]
- 183.Yassin AF, Hupfer H, Siering C, Klenk HP, Schumann P. 2011. Auritidibacter ignavus gen. nov., sp. nov., of the family Micrococcaceae isolated from an ear swab of a man with otitis externa, transfer of the members of the family Yaniellaceae Li et al. 2008 to the family Micrococcaceae and emended description of the suborder Micrococcineae. Int J Syst Evol Microbiol 61:223–230. doi: 10.1099/ijs.0.019786-0. [DOI] [PubMed] [Google Scholar]
- 184.Chen YG, Chen J, Chen QH, Tang SK, Zhang YQ, He JW, Li WJ, Liu YQ. 2010. Yaniella soli sp. nov., a new actinobacterium isolated from non-saline forest soil in China. Antonie Van Leeuwenhoek 98:395–401. doi: 10.1007/s10482-010-9453-1. [DOI] [PubMed] [Google Scholar]
- 185.Li WJ, Zhi XY, Euzeby JP. 2008. Proposal of Yaniellaceae fam. nov., Yaniella gen. nov. and Sinobaca gen. nov. as replacements for the illegitimate prokaryotic names Yaniaceae Li et al. 2005, Yania Li et al. 2004, emend Li et al. 2005, and Sinococcus Li et al. 2006, respectively. Int J Syst Evol Microbiol 58:525–527. doi: 10.1099/ijs.0.65792-0. [DOI] [PubMed] [Google Scholar]
- 186.Funke G, Hutson RA, Bernard KA, Pfyffer GE, Wauters G, Collins MD. 1996. Isolation of Arthrobacter spp. from clinical specimens and description of Arthrobacter cumminsii sp. nov. and Arthrobacter woluwensis sp. nov. J Clin Microbiol 34:2356–2363. doi: 10.1128/JCM.34.10.2356-2363.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ruckmani A, Kaur I, Schumann P, Klenk HP, Mayilraj S. 2011. Calidifontibacter indicus gen. nov., sp. nov., a member of the family Dermacoccaceae isolated from a hot spring, and emended description of the family Dermacoccaceae. Int J Syst Evol Microbiol 61:2419–2424. doi: 10.1099/ijs.0.025593-0. [DOI] [PubMed] [Google Scholar]
- 188.Wu C, Liang B, Gong Y, Zhang L, Zou Y, Ge J. 2014. Streptococcus acidominimus causing invasive disease in humans: a case series. J Med Case Rep 8:57. doi: 10.1186/1752-1947-8-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tammaro P, Richard N, Andre B, Andremont A, Mammeri H. 2017. First report of human infection due to Streptococcus devriesei. New Microbes New Infect 15:72–73. doi: 10.1016/j.nmni.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Glazunova OO, Raoult D, Roux V. 2006. Streptococcus massiliensis sp. nov., isolated from a patient blood culture. Int J Syst Evol Microbiol 56:1127–1131. doi: 10.1099/ijs.0.64009-0. [DOI] [PubMed] [Google Scholar]
- 191.Cattoir V, Kobal A, Legrand P. 2010. Aerococcus urinae and Aerococcus sanguinicola, two frequently misidentified uropathogens. Scand J Infect Dis 42:775–780. doi: 10.3109/00365548.2010.485576. [DOI] [PubMed] [Google Scholar]
- 192.Vela AI, García N, Latre MV, Casamayor A, Sánchez-Porro C, Briones V, Ventosa A, Domínguez L, Fernández-Garayzábal JF. 2007. Aerococcus suis sp. nov., isolated from clinical specimens from swine. Int J Syst Evol Microbiol 57:1291–1294. doi: 10.1099/ijs.0.64537-0. [DOI] [PubMed] [Google Scholar]
- 193.Lawson PA, Falsen E, Ohlen M, Collins MD. 2001. Aerococcus urinaehominis sp. nov., isolated from human urine. Int J Syst Evol Microbiol 51:683–686. doi: 10.1099/00207713-51-2-683. [DOI] [PubMed] [Google Scholar]
- 194.Cargill JS, Scott KS, Gascoyne-Binzi D, Sandoe JAT. 2012. Granulicatella infection: diagnosis and management. J Med Microbiol 61:755–761. doi: 10.1099/jmm.0.039693-0. [DOI] [PubMed] [Google Scholar]
- 195.Rahmati E, Martin V, Wong D, Sattler F, Petterson J, Ward P, Butler-Wu SM, She RC. 2017. Facklamia species as an underrecognized pathogen. Open Forum Infect Dis 4:ofw272. doi: 10.1093/ofid/ofw272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hoyles L, Foster G, Falsen E, Thomson LF, Collins MD. 2001. Facklamia miroungae sp. nov., from a juvenile southern elephant seal (Mirounga leonina). Int J Syst Evol Microbiol 51:1401–1403. doi: 10.1099/00207713-51-4-1401. [DOI] [PubMed] [Google Scholar]
- 197.Lécuyer H, Audibert J, Bobigny A, Eckert C, Jannière-Nartey C, Buu-Hoï A, Mainardi J-L, Podglajen I. 2007. Dolosigranulum pigrum causing nosocomial pneumonia and septicemia. J Clin Microbiol 45:3474–3475. doi: 10.1128/JCM.01373-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Aguirre M, Morrison D, Cookson BD, Gay FW, Collins MD. 1993. Phenotypic and phylogenetic characterization of some Gemella-like organisms from human infections: description of Dolosigranulum pigrum gen. nov., sp. nov. J Appl Bacteriol 75:608–612. doi: 10.1111/j.1365-2672.1993.tb01602.x. [DOI] [PubMed] [Google Scholar]
- 199.Collins MD, Rodriguez Jovita M, Hutson RA, Falsen E, Sjoden B, Facklam RR. 1999. Dolosicoccus paucivorans gen. nov., sp. nov., isolated from human blood. Int J Syst Bacteriol 49(Part 4):1439–1442. doi: 10.1099/00207713-49-4-1439. [DOI] [PubMed] [Google Scholar]
- 200.Collins MD, Lawson PA, Monasterio R, Falsen E, Sjoden B, Facklam RR. 1999. Ignavigranum ruoffiae sp. nov., isolated from human clinical specimens. Int J Syst Bacteriol 49(Part 1):97–101. doi: 10.1099/00207713-49-1-97. [DOI] [PubMed] [Google Scholar]
- 201.Collins MD, Falsen E, Lemozy J, Akervall E, Sjoden B, Lawson PA. 1997. Phenotypic and phylogenetic characterization of some Globicatella-like organisms from human sources: description of Facklamia hominis gen. nov., sp. nov. Int J Syst Bacteriol 47:880–882. doi: 10.1099/00207713-47-3-880. [DOI] [PubMed] [Google Scholar]
- 202.Caliendo AM, Jordan CD, Ruoff KL. 1995. Helcococcus, a new genus of catalase-negative, gram-positive cocci isolated from clinical specimens. J Clin Microbiol 33:1638. doi: 10.1128/JCM.33.6.1638-1638.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kutzer P, Schulze C, Engelhardt A, Wieler LH, Nordhoff M. 2008. Helcococcus ovis, an emerging pathogen in bovine valvular endocarditis. J Clin Microbiol 46:3291–3295. doi: 10.1128/JCM.00867-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Aguirre M, Collins MD. 1992. Phylogenetic analysis of Alloiococcus otitis gen. nov., sp. nov., an organism from human middle ear fluid. Int J Syst Bacteriol 42:79–83. doi: 10.1099/00207713-42-1-79. [DOI] [PubMed] [Google Scholar]
- 205.Ishikawa M, Tanasupawat S, Nakajima K, Kanamori H, Ishizaki S, Kodama K, Okamoto-Kainuma A, Koizumi Y, Yamamoto Y, Yamasato K. 2009. Alkalibacterium thalassium sp. nov., Alkalibacterium pelagium sp. nov., Alkalibacterium putridalgicola sp. nov. and Alkalibacterium kapii sp. nov., slightly halophilic and alkaliphilic marine lactic acid bacteria isolated from marine organisms and salted foods collected in Japan and Thailand. Int J Syst Evol Microbiol 59:1215–1226. doi: 10.1099/ijs.0.65602-0. [DOI] [PubMed] [Google Scholar]
- 206.Collins MD, Hutson RA, Falsen E, Sjoden B, Facklam RR. 1998. Description of Gemella sanguinis sp. nov., isolated from human clinical specimens. J Clin Microbiol 36:3090–3093. doi: 10.1128/JCM.36.10.3090-3093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Collins MD, Hutson RA, Falsen E, Sjoden B, Facklam RR. 1998. Gemella bergeriae sp. nov., isolated from human clinical specimens. J Clin Microbiol 36:1290–1293. doi: 10.1128/JCM.36.5.1290-1293.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Cai Y, Yang J, Pang H, Kitahara M. 2011. Lactococcus fujiensis sp. nov., a lactic acid bacterium isolated from vegetable matter. Int J Syst Evol Microbiol 61:1590–1594. doi: 10.1099/ijs.0.025130-0. [DOI] [PubMed] [Google Scholar]
- 209.Vesa T, Pochart P, Marteau P. 2000. Pharmacokinetics of Lactobacillus plantarum NCIMB 8826, Lactobacillus fermentum KLD, and Lactococcus lactis MG 1363 in the human gastrointestinal tract. Aliment Pharmacol Ther 14:823–828. doi: 10.1046/j.1365-2036.2000.00763.x. [DOI] [PubMed] [Google Scholar]
- 210.Milinovich GJ, Burrell PC, Pollitt CC, Bouvet A, Trott DJ. 2008. Streptococcus henryi sp. nov. and Streptococcus caballi sp. nov., isolated from the hindgut of horses with oligofructose-induced laminitis. Int J Syst Evol Microbiol 58:262–266. doi: 10.1099/ijs.0.65063-0. [DOI] [PubMed] [Google Scholar]
- 211.Clavel T, Charrier C, Haller D. 2013. Streptococcus danieliae sp. nov., a novel bacterium isolated from the caecum of a mouse. Arch Microbiol 195:43–49. doi: 10.1007/s00203-012-0846-6. [DOI] [PubMed] [Google Scholar]
- 212.Tappe D, Pukall R, Schumann P, Gronow S, Spiliotis M, Claus H, Brehm K, Vogel U. 2009. Streptococcus merionis sp. nov., isolated from Mongolian jirds (Meriones unguiculatus). Int J Syst Evol Microbiol 59:766–770. doi: 10.1099/ijs.0.65823-0. [DOI] [PubMed] [Google Scholar]
- 213.Vela AI, Sánchez V, Mentaberre G, Lavín S, Domínguez L, Fernández-Garayzábal JF. 2011. Streptococcus porcorum sp. nov., isolated from domestic and wild pigs. Int J Syst Evol Microbiol 61:1585–1589. doi: 10.1099/ijs.0.022657-0. [DOI] [PubMed] [Google Scholar]
- 214.Saito M, Shinozaki-Kuwahara N, Hirasawa M, Takada K. 2014. Streptococcus loxodontisalivarius sp. nov. and Streptococcus saliviloxodontae sp. nov., isolated from oral cavities of elephants. Int J Syst Evol Microbiol 64:3288–3292. doi: 10.1099/ijs.0.063263-0. [DOI] [PubMed] [Google Scholar]
- 215.Vela AI, Fernández E, Lawson PA, Latre MV, Falsen E, Domínguez L, Collins MD, Fernández-Garayzábal JF. 2002. Streptococcus entericus sp. nov., isolated from cattle intestine. Int J Syst Evol Microbiol 52:665–669. doi: 10.1099/00207713-52-2-665. [DOI] [PubMed] [Google Scholar]
- 216.Martin V, Manes-Lazaro R, Rodriguez JM, Maldonado-Barragan A. 2011. Streptococcus lactarius sp. nov., isolated from breast milk of healthy women. Int J Syst Evol Microbiol 61:1048–1052. doi: 10.1099/ijs.0.021642-0. [DOI] [PubMed] [Google Scholar]
- 217.Chen YS, Chang CH, Pan SF, Wang LT, Chang YC, Wu HC, Yanagida F. 2013. Lactococcus taiwanensis sp. nov., a lactic acid bacterium isolated from fresh cummingcordia. Int J Syst Evol Microbiol 63:2405–2409. doi: 10.1099/ijs.0.045757-0. [DOI] [PubMed] [Google Scholar]
- 218.Meucci A, Zago M, Rossetti L, Fornasari ME, Bonvini B, Tidona F, Povolo M, Contarini G, Carminati D, Giraffa G. 2015. Lactococcus hircilactis sp. nov. and Lactococcus laudensis sp. nov., isolated from milk. Int J Syst Evol Microbiol 65:2091–2096. doi: 10.1099/ijs.0.000225. [DOI] [PubMed] [Google Scholar]
- 219.Cho SL, Nam SW, Yoon JH, Lee JS, Sukhoom A, Kim W. 2008. Lactococcus chungangensis sp. nov., a lactic acid bacterium isolated from activated sludge foam. Int J Syst Evol Microbiol 58:1844–1849. doi: 10.1099/ijs.0.65527-0. [DOI] [PubMed] [Google Scholar]
- 220.Chen YS, Otoguro M, Lin YH, Pan SF, Ji SH, Yu CR, Liou MS, Chang YC, Wu HC, Yanagida F. 2014. Lactococcus formosensis sp. nov., a lactic acid bacterium isolated from yan-tsai-shin (fermented broccoli stems). Int J Syst Evol Microbiol 64:146–151. doi: 10.1099/ijs.0.052811-0. [DOI] [PubMed] [Google Scholar]
- 221.Kim J, Chun J, Han HU. 2000. Leuconostoc kimchii sp. nov., a new species from kimchi. Int J Syst Evol Microbiol 50(Part 5):1915–1919. doi: 10.1099/00207713-50-5-1915. [DOI] [PubMed] [Google Scholar]
- 222.Barreau C, Wagener G. 1990. Characterization of Leuconostoc lactis strains from human sources. J Clin Microbiol 28:1728–1733. doi: 10.1128/JCM.28.8.1728-1733.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lee SH, Park MS, Jung JY, Jeon CO. 2012. Leuconostoc miyukkimchii sp. nov., isolated from brown algae (Undaria pinnatifida) kimchi. Int J Syst Evol Microbiol 62:1098–1103. doi: 10.1099/ijs.0.032367-0. [DOI] [PubMed] [Google Scholar]
- 224.Ehrmann MA, Freiding S, Vogel RF. 2009. Leuconostoc palmae sp. nov., a novel lactic acid bacterium isolated from palm wine. Int J Syst Evol Microbiol 59:943–947. doi: 10.1099/ijs.0.005983-0. [DOI] [PubMed] [Google Scholar]
- 225.De Bruyne K, Franz CM, Vancanneyt M, Schillinger U, Mozzi F, de Valdez GF, De Vuyst L, Vandamme P. 2008. Pediococcus argentinicus sp. nov. from Argentinean fermented wheat flour and identification of Pediococcus species by pheS, rpoA and atpA sequence analysis. Int J Syst Evol Microbiol 58:2909–2916. doi: 10.1099/ijs.0.65833-0. [DOI] [PubMed] [Google Scholar]
- 226.Zhang B, Tong H, Dong X. 2005. Pediococcus cellicola sp. nov., a novel lactic acid coccus isolated from a distilled-spirit-fermenting cellar. Int J Syst Evol Microbiol 55:2167–2170. doi: 10.1099/ijs.0.63778-0. [DOI] [PubMed] [Google Scholar]
- 227.Tanasupawat S, Pakdeeto A, Thawai C, Yukphan P, Okada S. 2007. Identification of lactic acid bacteria from fermented tea leaves (miang) in Thailand and proposals of Lactobacillus thailandensis sp. nov., Lactobacillus camelliae sp. nov., and Pediococcus siamensis sp. nov. J Gen Appl Microbiol 53:7–15. doi: 10.2323/jgam.53.7. [DOI] [PubMed] [Google Scholar]
- 228.Wang L, Cui YS, Kwon CS, Lee ST, Lee JS, Im WT. 2011. Vagococcus acidifermentans sp. nov., isolated from an acidogenic fermentation bioreactor. Int J Syst Evol Microbiol 61:1123–1126. doi: 10.1099/ijs.0.022087-0. [DOI] [PubMed] [Google Scholar]
- 229.Lawson PA, Falsen E, Cotta MA, Whitehead TR. 2007. Vagococcus elongatus sp. nov., isolated from a swine-manure storage pit. Int J Syst Evol Microbiol 57:751–754. doi: 10.1099/ijs.0.64730-0. [DOI] [PubMed] [Google Scholar]
- 230.Killer J, Švec P, Sedláček I, Černohlávková J, Benada O, Hroncová Z, Havlík J, Vlková E, Rada V, Kopečný J, Kofroňová O. 2014. Vagococcus entomophilus sp. nov., from the digestive tract of a wasp (Vespula vulgaris). Int J Syst Evol Microbiol 64:731–737. doi: 10.1099/ijs.0.054940-0. [DOI] [PubMed] [Google Scholar]
- 231.Hoyles L, Lawson PA, Foster G, Falsen E, Ohlen M, Grainger JM, Collins MD. 2000. Vagococcus fessus sp. nov., isolated from a seal and a harbour porpoise. Int J Syst Evol Microbiol 50(Part 3):1151–1154. doi: 10.1099/00207713-50-3-1151. [DOI] [PubMed] [Google Scholar]
- 232.Jaffres E, Prevost H, Rossero A, Joffraud JJ, Dousset X. 2010. Vagococcus penaei sp. nov., isolated from spoilage microbiota of cooked shrimp (Penaeus vannamei). Int J Syst Evol Microbiol 60:2159–2164. doi: 10.1099/ijs.0.012872-0. [DOI] [PubMed] [Google Scholar]
- 233.Padonou SW, Schillinger U, Nielsen DS, Franz CM, Hansen M, Hounhouigan JD, Nago MC, Jakobsen M. 2010. Weissella beninensis sp. nov., a motile lactic acid bacterium from submerged cassava fermentations, and emended description of the genus Weissella. Int J Syst Evol Microbiol 60:2193–2198. doi: 10.1099/ijs.0.014332-0. [DOI] [PubMed] [Google Scholar]
- 234.Vela AI, Fernández A, Bernaldo de Quirós Y, Herráez P, Domínguez L, Fernández-Garayzábal JF. 2011. Weissella ceti sp. nov., isolated from beaked whales (Mesoplodon bidens). Int J Syst Evol Microbiol 61:2758–2762. doi: 10.1099/ijs.0.028522-0. [DOI] [PubMed] [Google Scholar]
- 235.Oh SJ, Shin NR, Hyun DW, Kim PS, Kim JY, Kim MS, Yun JH, Bae JW. 2013. Weissella diestrammenae sp. nov., isolated from the gut of a camel cricket (Diestrammena coreana). Int J Syst Evol Microbiol 63:2951–2956. doi: 10.1099/ijs.0.047548-0. [DOI] [PubMed] [Google Scholar]
- 236.Snauwaert I, Papalexandratou Z, De Vuyst L, Vandamme P. 2013. Characterization of strains of Weissella fabalis sp. nov. and Fructobacillus tropaeoli from spontaneous cocoa bean fermentations. Int J Syst Evol Microbiol 63:1709–1716. doi: 10.1099/ijs.0.040311-0. [DOI] [PubMed] [Google Scholar]
- 237.Fusco V, Quero GM, Cho GS, Kabisch J, Meske D, Neve H, Bockelmann W, Franz CM. 2015. The genus Weissella: taxonomy, ecology and biotechnological potential. Front Microbiol 6:155. doi: 10.3389/fmicb.2015.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Abriouel H, Lerma LL, Casado Muñoz MDC, Montoro BP, Kabisch J, Pichner R, Cho G-S, Neve H, Fusco V, Franz CMAP, Gálvez A, Benomar N. 2015. The controversial nature of the Weissella genus: technological and functional aspects versus whole genome analysis-based pathogenic potential for their application in food and health. Front Microbiol 6:1197. doi: 10.3389/fmicb.2015.01197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Patel R, Piper KE, Rouse MS, Steckelberg JM, Uhl JR, Kohner P, Hopkins MK, Cockerill FR, Kline BC. 1998. Determination of 16S rRNA sequences of enterococci and application to species identification of nonmotile Enterococcus gallinarum isolates. J Clin Microbiol 36:3399–3407. doi: 10.1128/JCM.36.11.3399-3407.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Carvalho M, Shewmaker PL, Steigerwalt AG, Morey RE, Sampson AJ, Joyce K, Barrett TJ, Teixeira LM, Facklam RR. 2006. Enterococcus caccae sp. nov., isolated from human stools. Int J Syst Evol Microbiol 56:1505–1508. doi: 10.1099/ijs.0.64103-0. [DOI] [PubMed] [Google Scholar]
- 241.Tyrrell GJ, Turnbull LAnn, Teixeira LM, Lefebvre J, Carvalho MDGS, Facklam RR, Lovgren M. 2002. Enterococcus gilvus sp. nov. and Enterococcus pallens sp. nov. isolated from human clinical specimens. J Clin Microbiol 40:1140–1145. doi: 10.1128/jcm.40.4.1140-1145.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Lebreton F, Manson AL, Saavedra JT, Straub TJ, Earl AM, Gilmore MS. 2017. Tracing the Enterococci from paleozoic origins to the hospital. Cell 169:849–861. doi: 10.1016/j.cell.2017.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Yassin AF, Hupfer H, Siering C, Schumann P. 2011. Comparative chemotaxonomic and phylogenetic studies on the genus Arcanobacterium Collins et al. 1982 emend. Lehnen et al. 2006: proposal for Trueperella gen. nov. and emended description of the genus Arcanobacterium. Int J Syst Evol Microbiol 61:1265–1274. doi: 10.1099/ijs.0.020032-0. [DOI] [PubMed] [Google Scholar]
- 244.Wauters G, Charlier J, Janssens M, Delmee M. 2000. Identification of Arthrobacter oxydans, Arthrobacter luteolus sp. nov., and Arthrobacter albus sp. nov., isolated from human clinical specimens. J Clin Microbiol 38:2412–2415. doi: 10.1128/JCM.38.6.2412-2415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Mages IS, Frodl R, Bernard KA, Funke G. 2008. Identities of Arthrobacter spp. and Arthrobacter-like bacteria encountered in human clinical specimens. J Clin Microbiol 46:2980–2986. doi: 10.1128/JCM.00658-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Chen YG, Tang SK, Zhang YQ, Li ZY, Yi LB, Wang YX, Li WJ, Cui XL. 2009. Arthrobacter halodurans sp. nov., a new halotolerant bacterium isolated from sea water. Antonie Van Leeuwenhoek 96:63–70. doi: 10.1007/s10482-009-9336-5. [DOI] [PubMed] [Google Scholar]
- 247.Heyrman J, Verbeeren J, Schumann P, Swings J, De Vos P. 2005. Six novel Arthrobacter species isolated from deteriorated mural paintings. Int J Syst Evol Microbiol 55:1457–1464. doi: 10.1099/ijs.0.63358-0. [DOI] [PubMed] [Google Scholar]
- 248.Ash C, Farrow JA, Dorsch M, Stackebrandt E, Collins MD. 1991. Comparative analysis of Bacillus anthracis, Bacillus cereus, and related species on the basis of reverse transcriptase sequencing of 16S rRNA. Int J Syst Bacteriol 41:343–346. doi: 10.1099/00207713-41-3-343. [DOI] [PubMed] [Google Scholar]
- 249.Xu D, Cote JC. 2003. Phylogenetic relationships between Bacillus species and related genera inferred from comparison of 3′ end 16S rDNA and 5′ end 16S-23S ITS nucleotide sequences. Int J Syst Evol Microbiol 53:695–704. doi: 10.1099/ijs.0.02346-0. [DOI] [PubMed] [Google Scholar]
- 250.Burgess SA, Flint SH, Lindsay D, Cox MP, Biggs PJ. 2017. Insights into the Geobacillus stearothermophilus species based on phylogenomic principles. BMC Microbiol 17:140. doi: 10.1186/s12866-017-1047-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Sáez-Nieto JA, Medina-Pascual MJ, Carrasco G, Garrido N, Fernandez-Torres MA, Villalón P, Valdezate S. 2017. Paenibacillus spp. isolated from human and environmental samples in Spain: detection of 11 new species. New Microbes New Infect 19:19–27. doi: 10.1016/j.nmni.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.von der Weid I, Duarte GF, van Elsas JD, Seldin L. 2002. Paenibacillus brasilensis sp. nov., a novel nitrogen-fixing species isolated from the maize rhizosphere in Brazil. Int J Syst Evol Microbiol 52:2147–2153. doi: 10.1099/00207713-52-6-2147. [DOI] [PubMed] [Google Scholar]
- 253.Tamai K, Akashi Y, Yoshimoto Y, Yaguchi Y, Takeuchi Y, Shiigai M, Igarashi J, Hirose Y, Suzuki H, Ohkusu K. 2018. First case of a bloodstream infection caused by the genus Brachybacterium. J Infect Chemother 24:998–1003. doi: 10.1016/j.jiac.2018.06.005. [DOI] [PubMed] [Google Scholar]
- 254.Schubert K, Ludwig W, Springer N, Kroppenstedt RM, Accolas JP, Fiedler F. 1996. Two coryneform bacteria isolated from the surface of French Gruyere and Beaufort cheeses are new species of the genus Brachybacterium: Brachybacterium alimentarium sp. nov. and Brachybacterium tyrofermentans sp. nov. Int J Syst Bacteriol 46:81–87. doi: 10.1099/00207713-46-1-81. [DOI] [PubMed] [Google Scholar]
- 255.Park SK, Kim MS, Jung MJ, Nam YD, Park EJ, Roh SW, Bae JW. 2011. Brachybacterium squillarum sp. nov., isolated from salt-fermented seafood. Int J Syst Evol Microbiol 61:1118–1122. doi: 10.1099/ijs.0.022517-0. [DOI] [PubMed] [Google Scholar]
- 256.Funke G, Carlotti A. 1994. Differentiation of Brevibacterium spp. encountered in clinical specimens. J Clin Microbiol 32:1729–1732. doi: 10.1128/JCM.32.7.1729-1732.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Roux V, Raoult D. 2009. Brevibacterium massiliense sp. nov., isolated from a human ankle discharge. Int J Syst Evol Microbiol 59:1960–1964. doi: 10.1099/ijs.0.007864-0. [DOI] [PubMed] [Google Scholar]
- 258.Wauters G, Haase G, Avesani V, Charlier J, Janssens M, Van Broeck J, Delmée M. 2004. Identification of a novel Brevibacterium species isolated from humans and description of Brevibacterium sanguinis sp. nov. J Clin Microbiol 42:2829–2832. doi: 10.1128/JCM.42.6.2829-2832.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Bernard K. 2012. The genus corynebacterium and other medically relevant coryneform-like bacteria. J Clin Microbiol 50:3152–3158. doi: 10.1128/JCM.00796-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Oliveira A, Oliveira LC, Aburjaile F, Benevides L, Tiwari S, Jamal SB, Silva A, Figueiredo HCP, Ghosh P, Portela RW, De Carvalho Azevedo VA, Wattam AR. 2017. Insight of genus Corynebacterium: ascertaining the role of pathogenic and non-pathogenic species. Front Microbiol 8:1937. doi: 10.3389/fmicb.2017.01937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Pascual C, Lawson PA, Farrow JA, Gimenez MN, Collins MD. 1995. Phylogenetic analysis of the genus Corynebacterium based on 16S rRNA gene sequences. Int J Syst Bacteriol 45:724–728. doi: 10.1099/00207713-45-4-724. [DOI] [PubMed] [Google Scholar]
- 262.Baek I, Kim M, Lee I, Na SI, Goodfellow M, Chun J. 2018. Phylogeny trumps chemotaxonomy: a case study involving Turicella otitidis. Front Microbiol 9:834. doi: 10.3389/fmicb.2018.00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.von Graevenitz A, Funke G. 2014. Turicella otitidis and Corynebacterium auris: 20 years on. Infection 42:1–4. doi: 10.1007/s15010-013-0488-x. [DOI] [PubMed] [Google Scholar]
- 264.Zhou Z, Yuan M, Tang R, Chen M, Lin M, Zhang W. 2012. Corynebacterium deserti sp. nov., isolated from desert sand. Int J Syst Evol Microbiol 62:791–794. doi: 10.1099/ijs.0.030429-0. [DOI] [PubMed] [Google Scholar]
- 265.Shin NR, Jung MJ, Kim MS, Roh SW, Nam YD, Bae JW. 2011. Corynebacterium nuruki sp. nov., isolated from an alcohol fermentation starter. Int J Syst Evol Microbiol 61:2430–2434. doi: 10.1099/ijs.0.027763-0. [DOI] [PubMed] [Google Scholar]
- 266.Goyache J, Ballesteros C, Vela AI, Collins MD, Briones V, Hutson RA, Potti J, García-Borboroglu P, Domínguez L, Fernández-Garayzábal JF. 2003. Corynebacterium sphenisci sp. nov., isolated from wild penguins. Int J Syst Evol Microbiol 53:1009–1012. doi: 10.1099/ijs.0.02502-0. [DOI] [PubMed] [Google Scholar]
- 267.Brown JM, Steigerwalt AG, Morey RE, Daneshvar MI, Romero LJ, McNeil MM. 2006. Characterization of clinical isolates previously identified as Oerskovia turbata: proposal of Cellulosimicrobium funkei sp nov and emended description of the genus Cellulosimicrobium. Int J Syst Evol Microbiol 56:801–804. doi: 10.1099/ijs.0.63882-0. [DOI] [PubMed] [Google Scholar]
- 268.Yoon JH, Kang SJ, Schumann P, Oh TK. 2007. Cellulosimicrobium terreum sp. nov., isolated from soil. Int J Syst Evol Microbiol 57:2493–2497. doi: 10.1099/ijs.0.64889-0. [DOI] [PubMed] [Google Scholar]
- 269.Kim H, Oh HW, Kim JA, Park DS, Park HM, Bae KS. 2014. Luteimicrobium xylanilyticum sp. nov., isolated from the gut of a long-horned beetle, Massicus raddei. Int J Syst Evol Microbiol 64:1401–1405. doi: 10.1099/ijs.0.048991-0. [DOI] [PubMed] [Google Scholar]
- 270.Kang MS, Im WT, Jung HM, Kim MK, Goodfellow M, Kim KK, Yang HC, An DS, Lee ST. 2007. Cellulomonas composti sp. nov., a cellulolytic bacterium isolated from cattle farm compost. Int J Syst Evol Microbiol 57:1256–1260. doi: 10.1099/ijs.0.63974-0. [DOI] [PubMed] [Google Scholar]
- 271.Funke G, Aravena-Roman M, Frodl R. 2005. First description of Curtobacterium spp. isolated from human clinical specimens. J Clin Microbiol 43:1032–1036. doi: 10.1128/JCM.43.3.1032-1036.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Funke G, Stubbs S, Pfyffer GE, Marchiani M, Collins MD. 1994. Characteristics of CDC group 3 and group 5 coryneform bacteria isolated from clinical specimens and assignment to the genus Dermabacter. J Clin Microbiol 32:1223–1228. doi: 10.1128/JCM.32.5.1223-1228.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Renvoise A, Aldrovandi N, Raoult D, Roux V. 2009. Helcobacillus massiliensis gen. nov., sp. nov., a novel representative of the family Dermabacteraceae isolated from a patient with a cutaneous discharge. Int J Syst Evol Microbiol 59:2346–2351. doi: 10.1099/ijs.0.003319-0. [DOI] [PubMed] [Google Scholar]
- 274.Martel A, Pasmans F, Hellebuyck T, Haesebrouck F, Vandamme P. 2008. Devriesea agamarum gen. nov., sp. nov., a novel actinobacterium associated with dermatitis and septicaemia in agamid lizards. Int J Syst Evol Microbiol 58:2206–2209. doi: 10.1099/ijs.0.65478-0. [DOI] [PubMed] [Google Scholar]
- 275.Zhang G, Zeng G, Cai X, Deng S, Luo H, Sun G. 2007. Brachybacterium zhongshanense sp. nov., a cellulose-decomposing bacterium from sediment along the Qijiang River, Zhongshan City, China. Int J Syst Evol Microbiol 57:2519–2524. doi: 10.1099/ijs.0.64968-0. [DOI] [PubMed] [Google Scholar]
- 276.Takahashi T, Fujisawa T, Tamura Y, Suzuki S, Muramatsu M, Sawada T, Benno Y, Mitsuoka T. 1992. DNA relatedness among Erysipelothrix rhusiopathiae strains representing all twenty-three serovars and Erysipelothrix tonsillarum. Int J Syst Bacteriol 42:469–473. doi: 10.1099/00207713-42-3-469. [DOI] [PubMed] [Google Scholar]
- 277.Verbarg S, Rheims H, Emus S, Fruhling A, Kroppenstedt RM, Stackebrandt E, Schumann P. 2004. Erysipelothrix inopinata sp. nov., isolated in the course of sterile filtration of vegetable peptone broth, and description of Erysipelotrichaceae fam. nov. Int J Syst Evol Microbiol 54:221–225. doi: 10.1099/ijs.0.02898-0. [DOI] [PubMed] [Google Scholar]
- 278.Chen X, Wang L, Zhou J, Wu H, Li D, Cui Y, Lu B. 2017. Exiguobacterium sp. A1b/GX59 isolated from a patient with community-acquired pneumonia and bacteremia: genomic characterization and literature review. BMC Infect Dis 17:508. doi: 10.1186/s12879-017-2616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Raichand R, Pareek S, Singh NK, Mayilraj S. 2012. Exiguobacterium aquaticum sp. nov., a member of the genus Exiguobacterium. Int J Syst Evol Microbiol 62:2150–2155. doi: 10.1099/ijs.0.035790-0. [DOI] [PubMed] [Google Scholar]
- 280.Weon HY, Kim BY, Schumann P, Kroppenstedt RM, Noh HJ, Park CW, Kwon SW. 2007. Knoellia aerolata sp. nov., isolated from an air sample in Korea. Int J Syst Evol Microbiol 57:2861–2864. doi: 10.1099/ijs.0.65293-0. [DOI] [PubMed] [Google Scholar]
- 281.Yu X, Du Y, Wang G. 2012. Knoellia flava sp. nov., isolated from pig manure. Int J Syst Evol Microbiol 62:384–389. doi: 10.1099/ijs.0.030932-0. [DOI] [PubMed] [Google Scholar]
- 282.Groth I, Schumann P, Schutze B, Augsten K, Stackebrandt E. 2002. Knoellia sinensis gen. nov., sp. nov. and Knoellia subterranea sp. nov., two novel actinobacteria isolated from a cave. Int J Syst Evol Microbiol 52:77–84. doi: 10.1099/00207713-52-1-77. [DOI] [PubMed] [Google Scholar]
- 283.Lim YK, Kweon OJ, Kim HR, Kim TH, Lee MK. 2017. First case of bacteremia caused by Janibacter hoylei. APMIS 125:665–668. doi: 10.1111/apm.12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Fernández-Natal MI, Sáez-Nieto JA, Medina-Pascual MJ, Valdezate-Ramos S, Guerra-Laso JM, Rodríguez-Pollán RH, Soriano F. 2015. First report of bacteremia by Janibacter terrae in humans. Infection 43:103–106. doi: 10.1007/s15010-014-0672-7. [DOI] [PubMed] [Google Scholar]
- 285.Loubinoux J, Rio B, Mihaila L, Fois E, Le Fleche A, Grimont PA, Marie JP, Bouvet A. 2005. Bacteremia caused by an undescribed species of Janibacter. J Clin Microbiol 43:3564–3566. doi: 10.1128/JCM.43.7.3564-3566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Li J, Long LJ, Yang LL, Xu Y, Wang FZ, Li QX, Zhang S, Li WJ. 2012. Janibacter alkaliphilus sp. nov., isolated from coral Anthogorgia sp. Antonie Van Leeuwenhoek 102:157–162. doi: 10.1007/s10482-012-9723-1. [DOI] [PubMed] [Google Scholar]
- 287.Hamada M, Shibata C, Tamura T, Yamamura H, Hayakawa M, Suzuki K. 2013. Janibacter cremeus sp. nov., an actinobacterium isolated from sea sediment. Int J Syst Evol Microbiol 63:3687–3690. doi: 10.1099/ijs.0.051532-0. [DOI] [PubMed] [Google Scholar]
- 288.Kageyama A, Takahashi Y, Yasumoto-Hirose M, Kasai H, Shizuri Y, Omura S. 2007. Janibacter corallicola sp. nov., isolated from coral in Palau. J Gen Appl Microbiol 53:185–189. doi: 10.2323/jgam.53.185. [DOI] [PubMed] [Google Scholar]
- 289.Porte L, Soto A, Andrighetti D, Dabanch J, Braun S, Saldivia A, Flores JC, Wozniak A, Garcia P, Weitzel T. 2012. Catheter-associated bloodstream infection caused by Leifsonia aquatica in a haemodialysis patient: a case report. J Med Microbiol 61:868–873. doi: 10.1099/jmm.0.037457-0. [DOI] [PubMed] [Google Scholar]
- 290.Han L, Lei JE, Wang X, Guo LT, Kang QY, He L, Xu JR. 2013. Septicemia caused by Leifsonia aquatica in a healthy patient after retinal reattachment surgery. J Clin Microbiol 51:3886–3888. doi: 10.1128/JCM.01339-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Jang YH, Kim SJ, Tamura T, Hamada M, Weon HY, Suzuki K, Kwon SW, Kim WG. 2013. Lysinimonas soli gen. nov., sp. nov., isolated from soil, and reclassification of Leifsonia kribbensis Dastager et al. 2009 as Lysinimonas kribbensis sp. nov., comb. nov. Int J Syst Evol Microbiol 63:1403–1410. doi: 10.1099/ijs.0.042945-0. [DOI] [PubMed] [Google Scholar]
- 292.Pindi PK, Kishore KH, Reddy GS, Shivaji S. 2009. Description of Leifsonia kafniensis sp. nov. and Leifsonia antarctica sp. nov. Int J Syst Evol Microbiol 59:1348–1352. doi: 10.1099/ijs.0.006643-0. [DOI] [PubMed] [Google Scholar]
- 293.Ganzert L, Bajerski F, Mangelsdorf K, Lipski A, Wagner D. 2011. Leifsonia psychrotolerans sp. nov., a psychrotolerant species of the family Microbacteriaceae from Livingston Island, Antarctica. Int J Syst Evol Microbiol 61:1938–1943. doi: 10.1099/ijs.0.021956-0. [DOI] [PubMed] [Google Scholar]
- 294.Soni DK, Dubey SK. 2014. Phylogenetic analysis of the Listeria monocytogenes based on sequencing of 16S rRNA and hlyA genes. Mol Biol Rep 41:8219–8229. doi: 10.1007/s11033-014-3724-2. [DOI] [PubMed] [Google Scholar]
- 295.Gneiding K, Frodl R, Funke G. 2008. Identities of Microbacterium spp. encountered in human clinical specimens. J Clin Microbiol 46:3646–3652. doi: 10.1128/JCM.01202-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Takeuchi M, Yokota A. 1994. Phylogenetic analysis of the genus Microbacterium based on 16S rRNA gene sequences. FEMS Microbiol Lett 124:11–16. doi: 10.1111/j.1574-6968.1994.tb07254.x. [DOI] [PubMed] [Google Scholar]
- 297.McNeil MM, Brown JM, Carvalho ME, Hollis DG, Morey RE, Reller LB. 2004. Molecular epidemiologic evaluation of endocarditis due to Oerskovia turbata and CDC group A-3 associated with contaminated homograft valves. J Clin Microbiol 42:2495–2500. doi: 10.1128/JCM.42.6.2495-2500.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Niamut SML, van der Vorm ER, van Luyn-Wiegers CGL, Gökemeijer JDM. 2003. Oerskovia xanthineolytica bacteremia in an immunocompromised patient without a foreign body. Eur J Clin Microbiol Infect Dis 22:274–275. doi: 10.1007/s10096-003-0900-z. [DOI] [PubMed] [Google Scholar]
- 299.Cruickshank JG, Gawler AH, Shaldon C. 1979. Oerskovia species: rare opportunistic pathogens. J Med Microbiol 12:513–515. doi: 10.1099/00222615-12-4-513. [DOI] [PubMed] [Google Scholar]
- 300.Stackebrandt E, Breymann S, Steiner U, Prauser H, Weiss N, Schumann P. 2002. Re-evaluation of the status of the genus Oerskovia, reclassification of Promicromonospora enterophila (Jager et al. 1983) as Oerskovia enterophila comb. nov. and description of Oerskovia jenensis sp. nov. and Oerskovia paurometabola sp. nov. Int J Syst Evol Microbiol 52:1105–1111. doi: 10.1099/00207713-52-4-1105. [DOI] [PubMed] [Google Scholar]
- 301.Hamada M, Tamura T, Shibata C, Yamamura H, Hayakawa M, Schumann P, Suzuki K. 2013. Paraoerskovia sediminicola sp. nov., an actinobacterium isolated from sea sediment, and emended description of the genus Paraoerskovia. Int J Syst Evol Microbiol 63:2637–2641. doi: 10.1099/ijs.0.043745-0. [DOI] [PubMed] [Google Scholar]
- 302.Khan ST, Harayama S, Tamura T, Ando K, Takagi M, Kazuo SY. 2009. Paraoerskovia marina gen. nov., sp. nov., an actinobacterium isolated from marine sediment. Int J Syst Evol Microbiol 59:2094–2098. doi: 10.1099/ijs.0.007666-0. [DOI] [PubMed] [Google Scholar]
- 303.Oyaert M, De Baere T, Breyne J, De Laere E, Marien S, Waets P, Laffut W. 2013. First case of Pseudoclavibacter bifida bacteremia in an immunocompromised host with chronic obstructive pulmonary disease (COPD). J Clin Microbiol 51:1973–1976. doi: 10.1128/JCM.00138-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Pailhories H, Lemarie C, Quinqueneau C, Eveillard M, Baufreton C, Rouleau F, Mahaza C, Joly-Guillou ML, Kempf M. 2014. First report of endocarditis caused by a Pseudoclavibacter species. J Clin Microbiol 52:3465–3467. doi: 10.1128/JCM.01388-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Lemaitre F, Stein A, Raoult D, Drancourt M. 2011. Pseudoclavibacter-like subcutaneous infection: a case report. J Med Case Rep 5:468. doi: 10.1186/1752-1947-5-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Srinivasan S, Kim HS, Kim MK, Lee M. 2012. Pseudoclavibacter caeni sp. nov., isolated from sludge of a sewage disposal plant. Int J Syst Evol Microbiol 62:786–790. doi: 10.1099/ijs.0.028951-0. [DOI] [PubMed] [Google Scholar]
- 307.Cho SL, Jung MY, Park MH, Chang YH, Yoon JH, Myung SC, Kim W. 2010. Pseudoclavibacter chungangensis sp. nov., isolated from activated sludge. Int J Syst Evol Microbiol 60:1672–1677. doi: 10.1099/ijs.0.015552-0. [DOI] [PubMed] [Google Scholar]
- 308.Kim MK, Jung HY. 2009. Pseudoclavibacter soli sp. nov., a beta-glucosidase-producing bacterium. Int J Syst Evol Microbiol 59:835–838. doi: 10.1099/ijs.0.65627-0. [DOI] [PubMed] [Google Scholar]
- 309.Mahobia N, Chaudhary P, Kamat Y. 2013. Rothia prosthetic knee joint infection: report and mini-review. New Microbes New Infect 1:2–5. doi: 10.1002/2052-2975.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Ramanan P, Barreto JN, Osmon DR, Tosh PK. 2014. Rothia bacteremia: a 10-year experience at Mayo Clinic, Rochester, Minnesota. J Clin Microbiol 52:3184–3189. doi: 10.1128/JCM.01270-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Adeolu M, Alnajar S, Naushad S, Gupta RS. 2016. Genome-based phylogeny and taxonomy of the “Enterobacteriales”: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int J Syst Evol Microbiol 66:5575–5599. doi: 10.1099/ijsem.0.001485. [DOI] [PubMed] [Google Scholar]
- 312.McAdam AJ. 2020. Enterobacteriaceae? Enterobacterales? What should we call enteric gram-negative bacilli? A micro-comic strip. J Clin Microbiol 58:e01888-19. doi: 10.1128/JCM.01888-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Alnajar S, Gupta RS. 2017. Phylogenomics and comparative genomic studies delineate six main clades within the family Enterobacteriaceae and support the reclassification of several polyphyletic members of the family. Infect Genet Evol 54:108–127. doi: 10.1016/j.meegid.2017.06.024. [DOI] [PubMed] [Google Scholar]
- 314.Hata H, Natori T, Mizuno T, Kanazawa I, Eldesouky I, Hayashi M, Miyata M, Fukunaga H, Ohji S, Hosoyama A, Aono E, Yamazoe A, Tsuchikane K, Fujita N, Ezaki T. 2016. Phylogenetics of family Enterobacteriaceae and proposal to reclassify Escherichia hermannii and Salmonella subterranea as Atlantibacter hermannii and Atlantibacter subterranea gen. nov., comb. nov. Microbiol Immunol 60:303–311. doi: 10.1111/1348-0421.12374. [DOI] [PubMed] [Google Scholar]
- 315.Khot PD, Fisher MA. 2013. Novel approach for differentiating Shigella species and Escherichia coli by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol 51:3711–3716. doi: 10.1128/JCM.01526-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Pupo GM, Lan R, Reeves PR. 2000. Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc Natl Acad Sci U S A 97:10567–10572. doi: 10.1073/pnas.180094797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Case RJ, Boucher Y, Dahllof I, Holmstrom C, Doolittle WF, Kjelleberg S. 2007. Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl Environ Microbiol 73:278–288. doi: 10.1128/AEM.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Kimura Z, Chung KM, Itoh H, Hiraishi A, Okabe S. 2014. Raoultella electrica sp. nov., isolated from anodic biofilms of a glucose-fed microbial fuel cell. Int J Syst Evol Microbiol 64:1384–1388. doi: 10.1099/ijs.0.058826-0. [DOI] [PubMed] [Google Scholar]
- 319.Joseph S, Cetinkaya E, Drahovska H, Levican A, Figueras MJ, Forsythe SJ. 2012. Cronobacter condimenti sp. nov., isolated from spiced meat, and Cronobacter universalis sp. nov., a species designation for Cronobacter sp. genomospecies 1, recovered from a leg infection, water and food ingredients. Int J Syst Evol Microbiol 62:1277–1283. doi: 10.1099/ijs.0.032292-0. [DOI] [PubMed] [Google Scholar]
- 320.Joseph S, Forsythe SJ. 2012. Insights into the emergent bacterial pathogen Cronobacter spp., generated by multilocus sequence typing and analysis. Front Microbiol 3:397. doi: 10.3389/fmicb.2012.00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Walterson AM, Stavrinides J. 2015. Pantoea: insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol Rev 39:968–984. doi: 10.1093/femsre/fuv027. [DOI] [PubMed] [Google Scholar]
- 322.Sato M, Miyazaki K. 2017. Phylogenetic Network analysis revealed the occurrence of horizontal gene transfer of 16S rRNA in the genus Enterobacter. Front Microbiol 8:2225. doi: 10.3389/fmicb.2017.02225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Giammanco GM, Grimont PA, Grimont F, Lefevre M, Giammanco G, Pignato S. 2011. Phylogenetic analysis of the genera Proteus, Morganella and Providencia by comparison of rpoB gene sequences of type and clinical strains suggests the reclassification of Proteus myxofaciens in a new genus, Cosenzaea gen. nov., as Cosenzaea myxofaciens comb. nov. Int J Syst Evol Microbiol 61:1638–1644. doi: 10.1099/ijs.0.021964-0. [DOI] [PubMed] [Google Scholar]
- 324.O'Hara CM, Brenner FW, Miller JM. 2000. Classification, identification, and clinical significance of Proteus, Providencia, and Morganella. Clin Microbiol Rev 13:534–546. doi: 10.1128/CMR.13.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.O'Hara CM, Brenner FW, Steigerwalt AG, Hill BC, Holmes B, Grimont PA, Hawkey PM, Penner JL, Miller JM, Brenner DJ. 2000. Classification of Proteus vulgaris biogroup 3 with recognition of Proteus hauseri sp. nov., nom. rev. and unnamed Proteus genomospecies 4, 5 and 6. Int J Syst Evol Microbiol 50(Part 5):1869–1875. doi: 10.1099/00207713-50-5-1869. [DOI] [PubMed] [Google Scholar]
- 326.Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB, Frankel G. 2014. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol 12:612–623. doi: 10.1038/nrmicro3315. [DOI] [PubMed] [Google Scholar]
- 327.Tindall BJ, Grimont PA, Garrity GM, Euzeby JP. 2005. Nomenclature and taxonomy of the genus Salmonella. Int J Syst Evol Microbiol 55:521–524. doi: 10.1099/ijs.0.63580-0. [DOI] [PubMed] [Google Scholar]
- 328.Lin CK, Hung CL, Chiang YC, Lin CM, Tsen HY. 2004. The sequence heterogenicities among 16S rRNA genes of Salmonella serovars and the effects on the specificity of the primers designed. Int J Food Microbiol 96:205–214. doi: 10.1016/j.ijfoodmicro.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 329.Sproer C, Mendrock U, Swiderski J, Lang E, Stackebrandt E. 1999. The phylogenetic position of Serratia, Buttiauxella and some other genera of the family Enterobacteriaceae. Int J Syst Bacteriol 49(Part 4):1433–1438. doi: 10.1099/00207713-49-4-1433. [DOI] [PubMed] [Google Scholar]
- 330.Geiger A, Fardeau ML, Falsen E, Ollivier B, Cuny G. 2010. Serratia glossinae sp. nov., isolated from the midgut of the tsetse fly Glossina palpalis gambiensis. Int J Syst Evol Microbiol 60:1261–1265. doi: 10.1099/ijs.0.013441-0. [DOI] [PubMed] [Google Scholar]
- 331.Huys G, Cnockaert M, Abbott SL, Janda JM, Vandamme P. 2010. Hafnia paralvei sp. nov., formerly known as Hafnia alvei hybridization group 2. Int J Syst Evol Microbiol 60:1725–1728. doi: 10.1099/ijs.0.018606-0. [DOI] [PubMed] [Google Scholar]
- 332.Janda JM, Abbott SL, Kroske-Bystrom S, Cheung WK, Powers C, Kokka RP, Tamura K. 1991. Pathogenic properties of Edwardsiella species. J Clin Microbiol 29:1997–2001. doi: 10.1128/JCM.29.9.1997-2001.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Galac MR, Lazzaro BP. 2012. Comparative genomics of bacteria in the genus Providencia isolated from wild Drosophila melanogaster. BMC Genomics 13:612. doi: 10.1186/1471-2164-13-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Khunthongpan S, Sumpavapol P, Tanasupawat S, Benjakul S, H-Kittikun A. 2013. Providencia thailandensis sp. nov., isolated from seafood processing wastewater. J Gen Appl Microbiol 59:185–190. doi: 10.2323/jgam.59.185. [DOI] [PubMed] [Google Scholar]
- 335.Reuter S, Connor TR, Barquist L, Walker D, Feltwell T, Harris SR, Fookes M, Hall ME, Petty NK, Fuchs TM, Corander J, Dufour M, Ringwood T, Savin C, Bouchier C, Martin L, Miettinen M, Shubin M, Riehm JM, Laukkanen-Ninios R, Sihvonen LM, Siitonen A, Skurnik M, Falcao JP, Fukushima H, Scholz HC, Prentice MB, Wren BW, Parkhill J, Carniel E, Achtman M, McNally A, Thomson NR. 2014. Parallel independent evolution of pathogenicity within the genus Yersinia. Proc Natl Acad Sci U S A 111:6768–6773. doi: 10.1073/pnas.1317161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Hurst MR, Becher SA, Young SD, Nelson TL, Glare TR. 2011. Yersinia entomophaga sp. nov., isolated from the New Zealand grass grub Costelytra zealandica. Int J Syst Evol Microbiol 61:844–849. doi: 10.1099/ijs.0.024406-0. [DOI] [PubMed] [Google Scholar]
- 337.Murros-Kontiainen A, Johansson P, Niskanen T, Fredriksson-Ahomaa M, Korkeala H, Björkroth J. 2011. Yersinia pekkanenii sp. nov. Int J Syst Evol Microbiol 61:2363–2367. doi: 10.1099/ijs.0.019984-0. [DOI] [PubMed] [Google Scholar]
- 338.Gomila M, Peña A, Mulet M, Lalucat J, García-Valdés E. 2015. Phylogenomics and systematics in Pseudomonas. Front Microbiol 6:214. doi: 10.3389/fmicb.2015.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Gellatly SL, Hancock RE. 2013. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis 67:159–173. doi: 10.1111/2049-632X.12033. [DOI] [PubMed] [Google Scholar]
- 340.Watanabe T, Yamazoe A, Hosoyama A, Fujihara H, Suenaga H, Hirose J, Futagami T, Goto M, Kimura N, Furukawa K. 2015. Draft genome sequence of Pseudomonas toyotomiensis KF710, a polychlorinated biphenyl-degrading bacterium isolated from biphenyl-contaminated soil. Genome Announc 3:e00223-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Tao Y, Zhou Y, He X, Hu X, Li D. 2014. Pseudomonas chengduensis sp. nov., isolated from landfill leachate. Int J Syst Evol Microbiol 64:95–100. doi: 10.1099/ijs.0.050294-0. [DOI] [PubMed] [Google Scholar]
- 342.Lafi FF, AlBladi ML, Salem NM, Al-Banna L, Alam I, Bajic VB, Hirt H, Saad MM. 2017. Draft genome sequence of the plant growth-promoting Pseudomonas punonensis strain D1-6 isolated from the desert plant Erodium hirtum in Jordan. Genome Announc 5:e01437-16. doi: 10.1128/genomeA.01437-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Uchino M, Kosako Y, Uchimura T, Komagata K. 2000. Emendation of Pseudomonas straminea Iizuka and Komagata 1963. Int J Syst Evol Microbiol 50(Part 4):1513–1519. doi: 10.1099/00207713-50-4-1513. [DOI] [PubMed] [Google Scholar]
- 344.Peix A, Berge O, Rivas R, Abril A, Velazquez E. 2005. Pseudomonas argentinensis sp. nov., a novel yellow pigment-producing bacterial species, isolated from rhizospheric soil in Cordoba, Argentina. Int J Syst Evol Microbiol 55:1107–1112. doi: 10.1099/ijs.0.63445-0. [DOI] [PubMed] [Google Scholar]
- 345.Zhang Y, Qiu S. 2016. Phylogenomic analysis of the genus Ralstonia based on 686 single-copy genes. Antonie Van Leeuwenhoek 109:71–82. doi: 10.1007/s10482-015-0610-4. [DOI] [PubMed] [Google Scholar]
- 346.Safni I, Cleenwerck I, De Vos P, Fegan M, Sly L, Kappler U. 2014. Polyphasic taxonomic revision of the Ralstonia solanacearum species complex: proposal to emend the descriptions of Ralstonia solanacearum and Ralstonia syzygii and reclassify current R. syzygii strains as Ralstonia syzygii subsp. syzygii subsp. nov., R. solanacearum phylotype IV strains as Ralstonia syzygii subsp. indonesiensis subsp. nov., banana blood disease bacterium strains as Ralstonia syzygii subsp. celebesensis subsp. nov. and R. solanacearum phylotype I and III strains as Ralstonia pseudosolanacearum sp. nov. Int J Syst Evol Microbiol 64:3087–3103. doi: 10.1099/ijs.0.066712-0. [DOI] [PubMed] [Google Scholar]
- 347.Sahin N, Tani A, Kotan R, Sedlacek I, Kimbara K, Tamer AU. 2011. Pandoraea oxalativorans sp. nov., Pandoraea faecigallinarum sp. nov. and Pandoraea vervacti sp. nov., isolated from oxalate-enriched culture. Int J Syst Evol Microbiol 61:2247–2253. doi: 10.1099/ijs.0.026138-0. [DOI] [PubMed] [Google Scholar]
- 348.Kokcha S, Bittar F, Reynaud-Gaubert M, Mely L, Gomez C, Gaubert JY, Thomas P, Rolain JM. 2013. Pandoraea pulmonicola chronic colonization in a cystic fibrosis patient, France. New Microbes New Infect 1:27–29. doi: 10.1002/2052-2975.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Yong D, Ee R, Lim YL, Yu CY, Ang GY, How KY, Tee KK, Yin WF, Chan KG. 2016. Complete genome sequence of Pandoraea thiooxydans DSM 25325(T), a thiosulfate-oxidizing bacterium. J Biotechnol 217:51–52. doi: 10.1016/j.jbiotec.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 350.Ee R, Yong D, Lim YL, Yin WF, Chan KG. 2015. Complete genome sequence of oxalate-degrading bacterium Pandoraea vervacti DSM 23571(T). J Biotechnol 204:5–6. doi: 10.1016/j.jbiotec.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 351.Ryan MP, Pembroke JT. 2018. Brevundimonas spp: emerging global opportunistic pathogens. Virulence 9:480–493. doi: 10.1080/21505594.2017.1419116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Estrela AB, Abraham WR. 2010. Brevundimonas vancanneytii sp. nov., isolated from blood of a patient with endocarditis. Int J Syst Evol Microbiol 60:2129–2134. doi: 10.1099/ijs.0.015651-0. [DOI] [PubMed] [Google Scholar]
- 353.Sly LI, Cox TL, Beckenham TB. 1999. The phylogenetic relationships of Caulobacter, Asticcacaulis and Brevundimonas species and their taxonomic implications. Int J Syst Bacteriol 49(Part 2):483–488. doi: 10.1099/00207713-49-2-483. [DOI] [PubMed] [Google Scholar]
- 354.Opota O, Ney B, Zanetti G, Jaton K, Greub G, Prod'hom G. 2014. Bacteremia caused by Comamonas kerstersii in a patient with diverticulosis. J Clin Microbiol 52:1009–1012. doi: 10.1128/JCM.02942-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Bayhan GI, Tanir G, Karaman I, Ozkan S. 2013. Comamonas testosteroni: an unusual bacteria associated with acute appendicitis. Balkan Med J 30:447–448. doi: 10.5152/balkanmedj.2013.9135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Tayeb LA, Lefevre M, Passet V, Diancourt L, Brisse S, Grimont PA. 2008. Comparative phylogenies of Burkholderia, Ralstonia, Comamonas, Brevundimonas and related organisms derived from rpoB, gyrB and rrs gene sequences. Res Microbiol 159:169–177. doi: 10.1016/j.resmic.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 357.Ma YF, Zhang Y, Zhang JY, Chen DW, Zhu Y, Zheng H, Wang SY, Jiang CY, Zhao GP, Liu SJ. 2009. The complete genome of Comamonas testosteroni reveals its genetic adaptations to changing environments. Appl Environ Microbiol 75:6812–6819. doi: 10.1128/AEM.00933-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Kobayashi T, Nakamura I, Fujita H, Tsukimori A, Sato A, Fukushima S, Ohkusu K, Matsumoto T. 2016. First case report of infection due to Cupriavidus gilardii in a patient without immunodeficiency: a case report. BMC Infect Dis 16:493. doi: 10.1186/s12879-016-1838-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Vandamme P, Coenye T. 2004. Taxonomy of the genus Cupriavidus: a tale of lost and found. Int J Syst Evol Microbiol 54:2285–2289. doi: 10.1099/ijs.0.63247-0. [DOI] [PubMed] [Google Scholar]
- 360.Kalka-Moll WM, LiPuma JJ, Accurso FJ, Plum G, van Koningsbruggen S, Vandamme P. 2009. Airway infection with a novel Cupriavidus species in persons with cystic fibrosis. J Clin Microbiol 47:3026–3028. doi: 10.1128/JCM.00846-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Bilgin H, Sarmis A, Tigen E, Soyletir G, Mulazimoglu L. 2015. Delftia acidovorans: a rare pathogen in immunocompetent and immunocompromised patients. Can J Infect Dis Med Microbiol 26:277–279. doi: 10.1155/2015/973284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Touchon M, Cury J, Yoon EJ, Krizova L, Cerqueira GC, Murphy C, Feldgarden M, Wortman J, Clermont D, Lambert T, Grillot-Courvalin C, Nemec A, Courvalin P, Rocha EP. 2014. The genomic diversification of the whole Acinetobacter genus: origins, mechanisms, and consequences. Genome Biol Evol 6:2866–2882. doi: 10.1093/gbe/evu225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Krizova L, McGinnis J, Maixnerova M, Nemec M, Poirel L, Mingle L, Sedo O, Wolfgang W, Nemec A. 2015. Acinetobacter variabilis sp. nov. (formerly DNA group 15 sensu Tjernberg & Ursing), isolated from humans and animals. Int J Syst Evol Microbiol 65:857–863. doi: 10.1099/ijs.0.000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Al-Anazi KA, Al-Jasser AM. 2014. Infections caused by Stenotrophomonas maltophilia in recipients of hematopoietic stem cell transplantation. Front Oncol 4:232. doi: 10.3389/fonc.2014.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Ryan RP, Monchy S, Cardinale M, Taghavi S, Crossman L, Avison MB, Berg G, van der Lelie D, Dow JM. 2009. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat Rev Microbiol 7:514–525. doi: 10.1038/nrmicro2163. [DOI] [PubMed] [Google Scholar]
- 366.Sawana A, Adeolu M, Gupta RS. 2014. Molecular signatures and phylogenomic analysis of the genus Burkholderia: proposal for division of this genus into the emended genus Burkholderia containing pathogenic organisms and a new genus Paraburkholderia gen. nov. harboring environmental species. Front Genet 5:429. doi: 10.3389/fgene.2014.00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Coenye T, Vandamme P. 2003. Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ Microbiol 5:719–729. doi: 10.1046/j.1462-2920.2003.00471.x. [DOI] [PubMed] [Google Scholar]
- 368.Peeters C, Zlosnik JE, Spilker T, Hird TJ, LiPuma JJ, Vandamme P. 2013. Burkholderia pseudomultivorans sp. nov., a novel Burkholderia cepacia complex species from human respiratory samples and the rhizosphere. Syst Appl Microbiol 36:483–489. doi: 10.1016/j.syapm.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 369.Dobritsa AP, Samadpour M. 2016. Transfer of eleven species of the genus Burkholderia to the genus Paraburkholderia and proposal of Caballeronia gen. nov. to accommodate twelve species of the genera Burkholderia and Paraburkholderia. Int J Syst Evol Microbiol 66:2836–2846. doi: 10.1099/ijsem.0.001065. [DOI] [PubMed] [Google Scholar]
- 370.Heylen K, Lebbe L, De Vos P. 2008. Acidovorax caeni sp. nov., a denitrifying species with genetically diverse isolates from activated sludge. Int J Syst Evol Microbiol 58:73–77. doi: 10.1099/ijs.0.65387-0. [DOI] [PubMed] [Google Scholar]
- 371.Malkan AD, Strollo W, Scholand SJ, Dudrick SJ. 2009. Implanted-port-catheter-related sepsis caused by Acidovorax avenae and methicillin-sensitive Staphylococcus aureus. J Clin Microbiol 47:3358–3361. doi: 10.1128/JCM.01093-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Orsborne C, Hardy A, Isalska B, Williams SG, Muldoon EG. 2014. Acidovorax oryzae catheter-associated bloodstream infection. J Clin Microbiol 52:4421–4424. doi: 10.1128/JCM.00657-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Shetty A, Barnes RA, Healy B, Groves P. 2005. A case of sepsis caused by Acidovorax. J Infect 51:e171–e172. doi: 10.1016/j.jinf.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 374.Li X, Hu Y, Gong J, Zhang L, Wang G. 2013. Comparative genome characterization of Achromobacter members reveals potential genetic determinants facilitating the adaptation to a pathogenic lifestyle. Appl Microbiol Biotechnol 97:6413–6425. doi: 10.1007/s00253-013-5018-3. [DOI] [PubMed] [Google Scholar]
- 375.Edwards BD, Greysson-Wong J, Somayaji R, Waddell B, Whelan FJ, Storey DG, Rabin HR, Surette MG, Parkins MD. 2017. Prevalence and outcomes of Achromobacter species infections in adults with cystic fibrosis: a North American cohort study. J Clin Microbiol 55:2074–2085. doi: 10.1128/JCM.02556-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Moremi N, Claus H, Hingi M, Vogel U, Mshana SE. 2017. Multidrug-resistant Achromobacter animicus causing wound infection in a street child in Mwanza, Tanzania. Diagn Microbiol Infect Dis 88:58–61. doi: 10.1016/j.diagmicrobio.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 377.Coenye T, Vanlaere E, Samyn E, Falsen E, Larsson P, Vandamme P. 2005. Advenella incenata gen. nov., sp. nov., a novel member of the Alcaligenaceae, isolated from various clinical samples. Int J Syst Evol Microbiol 55:251–256. doi: 10.1099/ijs.0.63267-0. [DOI] [PubMed] [Google Scholar]
- 378.Coenye T, Vancanneyt M, Cnockaert MC, Falsen E, Swings J, Vandamme P. 2003. Kerstersia gyiorum gen. nov., sp. nov., a novel Alcaligenes faecalis-like organism isolated from human clinical samples, and reclassification of Alcaligenes denitrificans Ruger and Tan 1983 as Achromobacter denitrificans comb. nov. Int J Syst Evol Microbiol 53:1825–1831. doi: 10.1099/ijs.0.02609-0. [DOI] [PubMed] [Google Scholar]
- 379.Matsuoka M, Park S, An SY, Miyahara M, Kim SW, Kamino K, Fushinobu S, Yokota A, Wakagi T, Shoun H. 2012. Advenella faeciporci sp. nov., a nitrite-denitrifying bacterium isolated from nitrifying-denitrifying activated sludge collected from a laboratory-scale bioreactor treating piggery wastewater. Int J Syst Evol Microbiol 62:2986–2990. doi: 10.1099/ijs.0.037440-0. [DOI] [PubMed] [Google Scholar]
- 380.Vandamme P, De Brandt E, Houf K, De Baere T. 2012. Kerstersia similis sp. nov., isolated from human clinical samples. Int J Syst Evol Microbiol 62:2156–2159. doi: 10.1099/ijs.0.037887-0. [DOI] [PubMed] [Google Scholar]
- 381.Lee YY, Lee JK, Park KH, Kim SY, Roh SW, Lee SB, Choi Y, Lee SJ. 2013. Paenalcaligenes hermetiae sp. nov., isolated from the larval gut of Hermetia illucens (Diptera: stratiomyidae), and emended description of the genus Paenalcaligenes. Int J Syst Evol Microbiol 63:4224–4229. doi: 10.1099/ijs.0.049098-0. [DOI] [PubMed] [Google Scholar]
- 382.Kampfer P, Falsen E, Langer S, Lodders N, Busse HJ. 2010. Paenalcaligenes hominis gen. nov., sp. nov., a new member of the family Alcaligenaceae. Int J Syst Evol Microbiol 60:1537–1542. doi: 10.1099/ijs.0.016576-0. [DOI] [PubMed] [Google Scholar]
- 383.Moon JY, Lim JM, Ahn JH, Weon HY, Kwon SW, Kim SJ. 2014. Paenalcaligenes suwonensis sp. nov., isolated from spent mushroom compost. Int J Syst Evol Microbiol 64:882–886. doi: 10.1099/ijs.0.058412-0. [DOI] [PubMed] [Google Scholar]
- 384.Tuuminen T, Heinasmaki T, Kerttula T. 2006. First report of bacteremia by Asaia bogorensis, in a patient with a history of intravenous-drug abuse. J Clin Microbiol 44:3048–3050. doi: 10.1128/JCM.00521-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Juretschko S, Beavers-May TK, Stovall SH. 2010. Nosocomial infection with Asaia lannensis in two paediatric patients with idiopathic dilated cardiomyopathy. J Med Microbiol 59:848–852. doi: 10.1099/jmm.0.019067-0. [DOI] [PubMed] [Google Scholar]
- 386.Suzuki R, Zhang Y, Iino T, Kosako Y, Komagata K, Uchimura T. 2010. Asaia astilbes sp. nov., Asaia platycodi sp. nov., and Asaia prunellae sp. nov., novel acetic acid bacteria isolated from flowers in Japan. J Gen Appl Microbiol 56:339–346. doi: 10.2323/jgam.56.339. [DOI] [PubMed] [Google Scholar]
- 387.Dourado MN, Andreote FD, Dini-Andreote F, Conti R, Araújo JM, Araújo WL. 2012. Analysis of 16S rRNA and mxaF genes revealing insights into Methylobacterium niche-specific plant association. Genet Mol Biol 35:142–148. doi: 10.1590/s1415-47572012005000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Read RC. 2014. Neisseria meningitidis; clones, carriage, and disease. Clin Microbiol Infect 20:391–395. doi: 10.1111/1469-0691.12647. [DOI] [PubMed] [Google Scholar]
- 389.Wolfgang WJ, Carpenter AN, Cole JA, Gronow S, Habura A, Jose S, Nazarian EJ, Kohlerschmidt DJ, Limberger R, Schoonmaker-Bopp D, Spröer C, Musser KA. 2011. Neisseria wadsworthii sp. nov. and Neisseria shayeganii sp. nov., isolated from clinical specimens. Int J Syst Evol Microbiol 61:91–98. doi: 10.1099/ijs.0.022426-0. [DOI] [PubMed] [Google Scholar]
- 390.Green O, Murray P, Gea-Banacloche JC. 2008. Sepsis caused by Elizabethkingia miricola successfully treated with tigecycline and levofloxacin. Diagn Microbiol Infect Dis 62:430–432. doi: 10.1016/j.diagmicrobio.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Mousavi SA, Österman J, Wahlberg N, Nesme X, Lavire C, Vial L, Paulin L, de Lajudie P, Lindström K. 2014. Phylogeny of the Rhizobium-Allorhizobium-Agrobacterium clade supports the delineation of Neorhizobium gen. nov. Syst Appl Microbiol 37:208–215. doi: 10.1016/j.syapm.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 392.Bjornstad ON, Harvill ET. 2005. Evolution and emergence of Bordetella in humans. Trends Microbiol 13:355–359. doi: 10.1016/j.tim.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 393.Wernli D, Emonet S, Schrenzel J, Harbarth S. 2011. Evaluation of eight cases of confirmed Bordetella bronchiseptica infection and colonization over a 15-year period. Clin Microbiol Infect 17:201–203. doi: 10.1111/j.1469-0691.2010.03258.x. [DOI] [PubMed] [Google Scholar]
- 394.Murphy TF, Parameswaran GI. 2009. Moraxella catarrhalis, a human respiratory tract pathogen. Clin Infect Dis 49:124–131. doi: 10.1086/599375. [DOI] [PubMed] [Google Scholar]
- 395.Vandamme P, Gillis M, Vancanneyt M, Hoste B, Kersters K, Falsen E. 1993. Moraxella lincolnii sp. nov., isolated from the human respiratory tract, and reevaluation of the taxonomic position of Moraxella osloensis. Int J Syst Bacteriol 43:474–481. doi: 10.1099/00207713-43-3-474. [DOI] [PubMed] [Google Scholar]
- 396.Pettersson B, Kodjo A, Ronaghi M, Uhlen M, Tonjum T. 1998. Phylogeny of the family Moraxellaceae by 16S rDNA sequence analysis, with special emphasis on differentiation of Moraxella species. Int J Syst Bacteriol 48(Part 1):75–89. doi: 10.1099/00207713-48-1-75. [DOI] [PubMed] [Google Scholar]
- 397.Bartosik D, Szymanik M, Baj J. 2003. Identification and distribution of insertion sequences of Paracoccus solventivorans. Appl Environ Microbiol 69:7002–7008. doi: 10.1128/aem.69.12.7002-7008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Bozal N, Montes MJ, Tudela E, Guinea J. 2003. Characterization of several Psychrobacter strains isolated from Antarctic environments and description of Psychrobacter luti sp. nov. and Psychrobacter fozii sp. nov. Int J Syst Evol Microbiol 53:1093–1100. doi: 10.1099/ijs.0.02457-0. [DOI] [PubMed] [Google Scholar]
- 399.Helsel LO, Hollis D, Steigerwalt AG, Morey RE, Jordan J, Aye T, Radosevic J, Jannat-Khah D, Thiry D, Lonsway DR, Patel JB, Daneshvar MI, Levett PN. 2007. Identification of “Haematobacter,” a new genus of aerobic Gram-negative rods isolated from clinical specimens, and reclassification of Rhodobacter massiliensis as “Haematobacter massiliensis comb. nov.” J Clin Microbiol 45:1238–1243. doi: 10.1128/JCM.01188-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Benedetti P, Rassu M, Pavan G, Sefton A, Pellizzer G. 2011. Septic shock, pneumonia, and soft tissue infection due to Myroides odoratimimus: report of a case and review of Myroides infections. Infection 39:161–165. doi: 10.1007/s15010-010-0077-1. [DOI] [PubMed] [Google Scholar]
- 401.Hu SH, Yuan SX, Qu H, Jiang T, Zhou YJ, Wang MX, Ming DS. 2016. Antibiotic resistance mechanisms of Myroides sp. J Zhejiang Univ Sci B 17:188–199. doi: 10.1631/jzus.B1500068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Coenye T, Goris J, Spilker T, Vandamme P, LiPuma JJ. 2002. Characterization of unusual bacteria isolated from respiratory secretions of cystic fibrosis patients and description of Inquilinus limosus gen. nov., sp. nov. J Clin Microbiol 40:2062–2069. doi: 10.1128/jcm.40.6.2062-2069.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Jung HM, Lee JS, Bae HM, Yi TH, Kim SY, Lee ST, Im WT. 2011. Inquilinus ginsengisoli sp. nov., isolated from soil of a ginseng field. Int J Syst Evol Microbiol 61:201–204. doi: 10.1099/ijs.0.018689-0. [DOI] [PubMed] [Google Scholar]
- 404.Khasawneh W, Yusef D. 2017. Ochrobactrum anthropi fulminant early-onset neonatal sepsis: a case report and review of literature. Pediatr Infect Dis J 36:1167–1168. doi: 10.1097/INF.0000000000001660. [DOI] [PubMed] [Google Scholar]
- 405.Trepa J, Mendes P, Goncalves R, Chaves C, Bras AM, Mesa A, Ramos I, Sa R, da Cunha JGS. 2018. Brucella vertebral osteomyelitis misidentified as an Ochrobactrum anthropi infection. IDCases 11:74–76. doi: 10.1016/j.idcr.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Tronel H, Plesiat P, Ageron E, Grimont PA. 2003. Bacteremia caused by a novel species of Sphingobacterium. Clin Microbiol Infect 9:1242–1244. doi: 10.1111/j.1469-0691.2003.00801.x. [DOI] [PubMed] [Google Scholar]
- 407.Schmidt VS, Wenning M, Scherer S. 2012. Sphingobacterium lactis sp. nov. and Sphingobacterium alimentarium sp. nov., isolated from raw milk and a dairy environment. Int J Syst Evol Microbiol 62:1506–1511. doi: 10.1099/ijs.0.036327-0. [DOI] [PubMed] [Google Scholar]
- 408.Holmes B, Segers P, Coenye T, Vancanneyt M, Vandamme P. 2006. Pannonibacter phragmitetus, described from a Hungarian soda lake in 2003, had been recognized several decades earlier from human blood cultures as Achromobacter groups B and E. Int J Syst Evol Microbiol 56:2945–2948. doi: 10.1099/ijs.0.64563-0. [DOI] [PubMed] [Google Scholar]
- 409.Bandyopadhyay S, Whitman WB, Das SK. 2017. Draft genome sequence of Pannonibacter indicus strain HT23(T) (DSM 23407(T)), a highly arsenate-tolerant bacterium isolated from a hot spring in India. Genome Announc 5:e00283-17. doi: 10.1128/genomeA.00283-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Faria JP, Edirisinghe JN, Davis JJ, Disz T, Hausmann A, Henry CS, Olson R, Overbeek RA, Pusch GD, Shukla M, Vonstein V, Wattam AR. 2015. Enabling comparative modeling of closely related genomes: example genus Brucella. Biotech 5:101–105. doi: 10.1007/s13205-014-0202-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Nymo IH, Tryland M, Godfroid J. 2011. A review of Brucella infection in marine mammals, with special emphasis on Brucella pinnipedialis in the hooded seal (Cystophora cristata). Vet Res 42:93. doi: 10.1186/1297-9716-42-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Scholz HC, Nockler K, Gollner C, Bahn P, Vergnaud G, Tomaso H, Al Dahouk S, Kampfer P, Cloeckaert A, Maquart M, Zygmunt MS, Whatmore AM, Pfeffer M, Huber B, Busse HJ, De BK. 2010. Brucella inopinata sp. nov., isolated from a breast implant infection. Int J Syst Evol Microbiol 60:801–808. doi: 10.1099/ijs.0.011148-0. [DOI] [PubMed] [Google Scholar]
- 413.Jiménez de Bagüés MP, Ouahrani-Bettache S, Quintana JF, Mitjana O, Hanna N, Bessoles S, Sanchez F, Scholz HC, Lafont V, Köhler S, Occhialini A. 2010. The new species Brucella microti replicates in macrophages and causes death in murine models of infection. J Infect Dis 202:3–10. doi: 10.1086/653084. [DOI] [PubMed] [Google Scholar]
- 414.Burstein D, Amaro F, Zusman T, Lifshitz Z, Cohen O, Gilbert JA, Pupko T, Shuman HA, Segal G. 2016. Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nat Genet 48:167–175. doi: 10.1038/ng.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Sánchez-Busó L, Olmos MP, Camaró ML, Adrián F, Calafat JM, González-Candelas F. 2015. Phylogenetic analysis of environmental Legionella pneumophila isolates from an endemic area (Alcoy, Spain). Infect Genet Evol 30:45–54. doi: 10.1016/j.meegid.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 416.Chambers ST, Murdoch D, Morris A, Holland D, Pappas P, Almela M, Fernandez-Hidalgo N, Almirante B, Bouza E, Forno D, del Rio A, Hannan MM, Harkness J, Kanafani ZA, Lalani T, Lang S, Raymond N, Read K, Vinogradova T, Woods CW, Wray D, Corey GR, Chu VH, International Collaboration on Endocarditis Prospective Cohort Study . 2013. HACEK infective endocarditis: characteristics and outcomes from a large, multi-national cohort. PLoS One 8:e63181. doi: 10.1371/journal.pone.0063181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Norskov-Lauritsen N. 2014. Classification, identification, and clinical significance of Haemophilus and Aggregatibacter species with host specificity for humans. Clin Microbiol Rev 27:214–240. doi: 10.1128/CMR.00103-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Fournier PE, Lelievre H, Eykyn SJ, Mainardi JL, Marrie TJ, Bruneel F, Roure C, Nash J, Clave D, James E, Benoit-Lemercier C, Deforges L, Tissot-Dupont H, Raoult D. 2001. Epidemiologic and clinical characteristics of Bartonella quintana and Bartonella henselae endocarditis: a study of 48 patients. Medicine 80:245–251. doi: 10.1097/00005792-200107000-00003. [DOI] [PubMed] [Google Scholar]
- 419.Dewhirst FE, Paster BJ, La Fontaine S, Rood JI. 1990. Transfer of Kingella indologenes (Snell and Lapage 1976) to the genus Suttonella gen. nov. as Suttonella indologenes comb. nov.; transfer of Bacteroides nodosus (Beveridge 1941) to the genus Dichelobacter gen. nov. as Dichelobacter nodosus comb. nov.; and assignment of the genera Cardiobacterium, Dichelobacter, and Suttonella to Cardiobacteriaceae fam. nov. in the gamma division of Proteobacteria on the basis of 16S rRNA sequence comparisons. Int J Syst Bacteriol 40:426–433. doi: 10.1099/00207713-40-4-426. [DOI] [PubMed] [Google Scholar]
- 420.Lawson PA, Malnick H, Collins MD, Shah JJ, Chattaway MA, Bendall R, Hartley JW. 2005. Description of Kingella potus sp. nov., an organism isolated from a wound caused by an animal bite. J Clin Microbiol 43:3526–3529. doi: 10.1128/JCM.43.7.3526-3529.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Houpikian P, Raoult D. 2001. Molecular phylogeny of the genus Bartonella: what is the current knowledge? FEMS Microbiol Lett 200:1–7. doi: 10.1111/j.1574-6968.2001.tb10684.x. [DOI] [PubMed] [Google Scholar]
- 422.Inoue K, Maruyama S, Kabeya H, Yamada N, Ohashi N, Sato Y, Yukawa M, Masuzawa T, Kawamori F, Kadosaka T, Takada N, Fujita H, Kawabata H. 2008. Prevalence and genetic diversity of Bartonella species isolated from wild rodents in Japan. Appl Environ Microbiol 74:5086–5092. doi: 10.1128/AEM.00071-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Rolain JM, Brouqui P, Koehler JE, Maguina C, Dolan MJ, Raoult D. 2004. Recommendations for treatment of human infections caused by Bartonella species. Antimicrob Agents Chemother 48:1921–1933. doi: 10.1128/AAC.48.6.1921-1933.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Naushad S, Adeolu M, Goel N, Khadka B, Al-Dahwi A, Gupta RS. 2015. Phylogenomic and molecular demarcation of the core members of the polyphyletic pasteurellaceae genera Actinobacillus, Haemophilus, and Pasteurella. Int J Genomics 2015:198560. doi: 10.1155/2015/198560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Avlami A, Papalambrou C, Tzivra M, Dounis E, Kordossis T. 1997. Bone marrow infection caused by Actinobacillus ureae in a rheumatoid arthritis patient. J Infect 35:298–299. doi: 10.1016/S0163-4453(97)93308-9. [DOI] [PubMed] [Google Scholar]
- 426.Ciantar M, Newman HN, Wilson M, Spratt DA. 2005. Molecular identification of Capnocytophaga spp. via 16S rRNA PCR-restriction fragment length polymorphism analysis. J Clin Microbiol 43:1894–1901. doi: 10.1128/JCM.43.4.1894-1901.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Gronow S, Munk C, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S, Cheng JF, Tapia R, Han C, Goodwin L, Pitluck S, Liolios K, Ivanova N, Mavromatis K, Mikhailova N, Pati A, Chen A, Palaniappan K, Land M, Hauser L, Chang YJ, Jeffries CD, Brambilla E, Rohde M, Goker M, Detter JC, Woyke T, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, Klenk HP. 2011. Complete genome sequence of Paludibacter propionicigenes type strain (WB4). Stand Genomic Sci 4:36–44. doi: 10.4056/sigs.1503846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Lawson PA, Carlson P, Wernersson S, Moore ER, Falsen E. 2010. Dysgonomonas hofstadii sp. nov., isolated from a human clinical source. Anaerobe 16:161–164. doi: 10.1016/j.anaerobe.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 429.Sjodin A, Svensson K, Ohrman C, Ahlinder J, Lindgren P, Duodu S, Johansson A, Colquhoun DJ, Larsson P, Forsman M. 2012. Genome characterisation of the genus Francisella reveals insight into similar evolutionary paths in pathogens of mammals and fish. BMC Genomics 13:268. doi: 10.1186/1471-2164-13-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Kuhnert P, Boerlin P, Emler S, Krawinkler M, Frey J. 2000. Phylogenetic analysis of Pasteurella multocida subspecies and molecular identification of feline P multocida subsp. septica by 16S rRNA gene sequencing. Int J Med Microbiol 290:599–604. doi: 10.1016/S1438-4221(00)80008-9. [DOI] [PubMed] [Google Scholar]
- 431.Davies RL. 2004. Genetic diversity among Pasteurella multocida strains of avian, bovine, ovine and porcine origin from England and Wales by comparative sequence analysis of the 16S rRNA gene. Microbiology 150:4199–4210. doi: 10.1099/mic.0.27409-0. [DOI] [PubMed] [Google Scholar]
- 432.Eisenberg T, Nicklas W, Mauder N, Rau J, Contzen M, Semmler T, Hofmann N, Aledelbi K, Ewers C. 2015. Phenotypic and genotypic characteristics of members of the genus Streptobacillus. PLoS One 10:e0134312. doi: 10.1371/journal.pone.0134312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Korczak BM, Stieber R, Emler S, Burnens AP, Frey J, Kuhnert P. 2006. Genetic relatedness within the genus Campylobacter inferred from rpoB sequences. Int J Syst Evol Microbiol 56:937–945. doi: 10.1099/ijs.0.64109-0. [DOI] [PubMed] [Google Scholar]
- 434.Debruyne L, On SL, De Brandt E, Vandamme P. 2009. Novel Campylobacter lari-like bacteria from humans and molluscs: description of Campylobacter peloridis sp. nov., Campylobacter lari subsp. concheus subsp. nov. and Campylobacter lari subsp. lari subsp. nov. Int J Syst Evol Microbiol 59:1126–1132. doi: 10.1099/ijs.0.000851-0. [DOI] [PubMed] [Google Scholar]
- 435.Rossi M, Debruyne L, Zanoni RG, Manfreda G, Revez J, Vandamme P. 2009. Campylobacter avium sp. nov., a hippurate-positive species isolated from poultry. Int J Syst Evol Microbiol 59:2364–2369. doi: 10.1099/ijs.0.007419-0. [DOI] [PubMed] [Google Scholar]
- 436.Kweon OJ, Lim YK, Yoo B, Kim HR, Kim TH, Lee MK. 2015. First case report of Campylobacter volucris bacteremia in an immunocompromised patient. J Clin Microbiol 53:1976–1978. doi: 10.1128/JCM.00442-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Hooi JKY, Lai WY, Ng WK, Suen MMY, Underwood FE, Tanyingoh D, Malfertheiner P, Graham DY, Wong VWS, Wu JCY, Chan FKL, Sung JJY, Kaplan GG, Ng SC. 2017. Global prevalence of Helicobacter pylori infection: systematic review and meta-analysis. Gastroenterology 153:420–429. doi: 10.1053/j.gastro.2017.04.022. [DOI] [PubMed] [Google Scholar]
- 438.Solnick JV, Schauer DB. 2001. Emergence of diverse Helicobacter species in the pathogenesis of gastric and enterohepatic diseases. Clin Microbiol Rev 14:59–97. doi: 10.1128/CMR.14.1.59-97.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Solnick JV, Vandamme P. 2001. Taxonomy of the Helicobacter genus In Mobley HLT, Mendz GL, Hazell SL (ed), Helicobacter pylori: physiology and genetics. ASM Press, Washington, DC. [PubMed] [Google Scholar]
- 440.Collado L, Figueras MJ. 2011. Taxonomy, epidemiology, and clinical relevance of the genus Arcobacter. Clin Microbiol Rev 24:174–192. doi: 10.1128/CMR.00034-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Voronina OL, Kunda MS, Aksenova EI, Ryzhova NN, Semenov AN, Petrov EM, Didenko LV, Lunin VG, Ananyina YV, Gintsburg AL. 2014. The characteristics of ubiquitous and unique Leptospira strains from the collection of Russian centre for leptospirosis. Biomed Res Int 2014:649034. doi: 10.1155/2014/649034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Slack AT, Khairani-Bejo S, Symonds ML, Dohnt MF, Galloway RL, Steigerwalt AG, Bahaman AR, Craig S, Harrower BJ, Smythe LD. 2009. Leptospira kmetyi sp. nov., isolated from an environmental source in Malaysia. Int J Syst Evol Microbiol 59:705–708. doi: 10.1099/ijs.0.002766-0. [DOI] [PubMed] [Google Scholar]
- 443.Smythe L, Adler B, Hartskeerl RA, Galloway RL, Turenne CY, Levett PN, International Committee on Systematics of Prokaryotes Subcommittee on the Taxonomy of Leptospiraceae . 2013. Classification of Leptospira genomospecies 1, 3, 4 and 5 as Leptospira alstonii sp. nov., Leptospira vanthielii sp. nov., Leptospira terpstrae sp. nov. and Leptospira yanagawae sp. nov., respectively. Int J Syst Evol Microbiol 63:1859–1862. doi: 10.1099/ijs.0.047324-0. [DOI] [PubMed] [Google Scholar]
- 444.Lotte R, Lotte L, Ruimy R. 2016. Actinotignum schaalii (formerly Actinobaculum schaalii): a newly recognized pathogen-review of the literature. Clin Microbiol Infect 22:28–36. doi: 10.1016/j.cmi.2015.10.038. [DOI] [PubMed] [Google Scholar]
- 445.Lawson PA, Falsen E, Akervall E, Vandamme P, Collins MD. 1997. Characterization of some Actinomyces-like isolates from human clinical specimens: reclassification of Actinomyces suis (Soltys and Spratling) as Actinobaculum suis comb. nov. and description of Actinobaculum schaalii sp. nov. Int J Syst Bacteriol 47:899–903. doi: 10.1099/00207713-47-3-899. [DOI] [PubMed] [Google Scholar]
- 446.Kononen E, Wade WG. 2015. Actinomyces and related organisms in human infections. Clin Microbiol Rev 28:419–442. doi: 10.1128/CMR.00100-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Yoon JH, Park YH. 2000. Phylogenetic analysis of the genus Thermoactinomyces based on 16S rDNA sequences. Int J Syst Evol Microbiol 50(Part 3):1081–1086. doi: 10.1099/00207713-50-3-1081. [DOI] [PubMed] [Google Scholar]
- 448.Oyaert M, Cools P, Breyne J, Heyvaert G, Vandewiele A, Vaneechoutte M, Vervaeke S, De Laere E. 2014. Sepsis with an atopobium-like species in a patient with Fournier's gangrene. J Clin Microbiol 52:364–366. doi: 10.1128/JCM.02310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Bilen M, Cadoret F, Dubourg G, Daoud Z, Fournier PE, Raoult D. 2017. “Olsenella congonensis” sp.nov., identified in human stool sample. New Microbes New Infect 19:132–133. doi: 10.1016/j.nmni.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Lugli GA, Mangifesta M, Duranti S, Anzalone R, Milani C, Mancabelli L, Alessandri G, Turroni F, Ossiprandi MC, van Sinderen D, Ventura M. 2018. Phylogenetic classification of six novel species belonging to the genus Bifidobacterium comprising Bifidobacterium anseris sp. nov., Bifidobacterium criceti sp. nov., Bifidobacterium imperatoris sp. nov., Bifidobacterium italicum sp. nov., Bifidobacterium margollesii sp. nov. and Bifidobacterium parmae sp. nov. Syst Appl Microbiol 41:173–183. doi: 10.1016/j.syapm.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 451.Milani C, Duranti S, Mangifesta M, Lugli GA, Turroni F, Mancabelli L, Viappiani A, Anzalone R, Alessandri G, Ossiprandi MC, van Sinderen D, Ventura M. 2018. Tracking the taxonomy of the genus Bifidobacterium based on a phylogenomic approach. Appl Environ Microbiol 84. doi: 10.1128/AEM.00706-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Durand GA, Pham T, Ndongo S, Traore SI, Dubourg G, Lagier JC, Michelle C, Armstrong N, Fournier PE, Raoult D, Million M. 2017. Blautia massiliensis sp. nov., isolated from a fresh human fecal sample and emended description of the genus Blautia. Anaerobe 43:47–55. doi: 10.1016/j.anaerobe.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 453.Traore SI, Azhar EI, Yasir M, Bibi F, Fournier PE, Jiman-Fatani AA, Delerce J, Cadoret F, Lagier JC, Raoult D. 2017. Description of “Blautia phocaeensis” sp. nov. and “Lachnoclostridium edouardi” sp. nov., isolated from healthy fresh stools of Saudi Arabia Bedouins by culturomics. New Microbes New Infect 19:129–131. doi: 10.1016/j.nmni.2017.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Lawson PA, Citron DM, Tyrrell KL, Finegold SM. 2016. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O'Toole 1935) Prevot 1938. Anaerobe 40:95–99. doi: 10.1016/j.anaerobe.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 455.Yutin N, Galperin MY. 2013. A genomic update on clostridial phylogeny: gram-negative spore formers and other misplaced clostridia. Environ Microbiol 15:2631–2641. doi: 10.1111/1462-2920.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Kageyama A, Benno Y, Nakase T. 1999. Phylogenetic evidence for the transfer of Eubacterium lentum to the genus Eggerthella as Eggerthella lenta gen. nov., comb. nov. Int J Syst Bacteriol 49(Part 4):1725–1732. doi: 10.1099/00207713-49-4-1725. [DOI] [PubMed] [Google Scholar]
- 457.Lee MR, Huang YT, Liao CH, Chuang TY, Wang WJ, Lee SW, Lee LN, Hsueh PR. 2012. Clinical and microbiological characteristics of bacteremia caused by Eggerthella, Paraeggerthella, and Eubacterium species at a university hospital in Taiwan from 2001 to 2010. J Clin Microbiol 50:2053–2055. doi: 10.1128/JCM.00548-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Douillard FP, Ribbera A, Jarvinen HM, Kant R, Pietila TE, Randazzo C, Paulin L, Laine PK, Caggia C, von Ossowski I, Reunanen J, Satokari R, Salminen S, Palva A, de Vos WM. 2013. Comparative genomic and functional analysis of Lactobacillus casei and Lactobacillus rhamnosus strains marketed as probiotics. Appl Environ Microbiol 79:1923–1933. doi: 10.1128/AEM.03467-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Yu J, Sun Z, Liu W, Bao Q, Zhang J, Zhang H. 2012. Phylogenetic study of Lactobacillus acidophilus gorup, L. casei group, and L. plantarum group based on partial hsp60, pheS and tuf gene sequences. Eur Food Res Technol 234:927–934. doi: 10.1007/s00217-012-1712-0. [DOI] [Google Scholar]
- 460.Sizova MV, Muller PA, Stancyk D, Panikov NS, Mandalakis M, Hazen A, Hohmann T, Doerfert SN, Fowle W, Earl AM, Nelson KE, Epstein SS. 2014. Oribacterium parvum sp. nov. and Oribacterium asaccharolyticum sp. nov., obligately anaerobic bacteria from the human oral cavity, and emended description of the genus Oribacterium. Int J Syst Evol Microbiol 64:2642–2649. doi: 10.1099/ijs.0.060988-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Veloo AC, Welling GW, Degener JE. 2011. Antimicrobial susceptibility of clinically relevant Gram-positive anaerobic cocci collected over a three-year period in the Netherlands. Antimicrob Agents Chemother 55:1199–1203. doi: 10.1128/AAC.01771-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Ulger-Toprak N, Lawson PA, Summanen P, O'Neal L, Finegold SM. 2012. Peptoniphilus duerdenii sp. nov. and Peptoniphilus koenoeneniae sp. nov., isolated from human clinical specimens. Int J Syst Evol Microbiol 62:2336–2341. doi: 10.1099/ijs.0.031997-0. [DOI] [PubMed] [Google Scholar]
- 463.Murphy EC, Frick IM. 2013. Gram-positive anaerobic cocci–commensals and opportunistic pathogens. FEMS Microbiol Rev 37:520–553. doi: 10.1111/1574-6976.12005. [DOI] [PubMed] [Google Scholar]
- 464.Scholz CF, Kilian M. 2016. The natural history of cutaneous propionibacteria, and reclassification of selected species within the genus Propionibacterium to the proposed novel genera Acidipropionibacterium gen. nov., Cutibacterium gen. nov. and Pseudopropionibacterium gen. nov. Int J Syst Evol Microbiol 66:4422–4432. doi: 10.1099/ijsem.0.001367. [DOI] [PubMed] [Google Scholar]
- 465.Piwowarek K, Lipińska E, Hać-Szymańczuk E, Kieliszek M, Ścibisz I. 2018. Propionibacterium spp.-source of propionic acid, vitamin B12, and other metabolites important for the industry. Appl Microbiol Biotechnol 102:515–538. doi: 10.1007/s00253-017-8616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Liu C, Finegold SM, Song Y, Lawson PA. 2008. Reclassification of Clostridium coccoides, Ruminococcus hansenii, Ruminococcus hydrogenotrophicus, Ruminococcus luti, Ruminococcus productus and Ruminococcus schinkii as Blautia coccoides gen. nov., comb. nov., Blautia hansenii comb. nov., Blautia hydrogenotrophica comb. nov., Blautia luti comb. nov., Blautia producta comb. nov., Blautia schinkii comb. nov. and description of Blautia wexlerae sp. nov., isolated from human faeces. Int J Syst Evol Microbiol 58:1896–1902. doi: 10.1099/ijs.0.65208-0. [DOI] [PubMed] [Google Scholar]
- 467.Nakazawa F, Hoshino E. 2004. DNA-DNA relatedness and phylogenetic positions of Slackia exigua, Slackia heliotrinireducens, Eggerthella lenta, and other related bacteria. Oral Microbiol Immunol 19:343–346. doi: 10.1111/j.1399-302x.2004.00162.x. [DOI] [PubMed] [Google Scholar]
- 468.Kim KS, Rowlinson MC, Bennion R, Liu C, Talan D, Summanen P, Finegold SM. 2010. Characterization of Slackia exigua isolated from human wound infections, including abscesses of intestinal origin. J Clin Microbiol 48:1070–1075. doi: 10.1128/JCM.01576-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Whitehead TR, Anoma C, McLaughlin RW. 2017. Rapid identification of Robinsoniella peoriensis using specific 16S rRNA gene PCR primers. Anaerobe 43:39–42. doi: 10.1016/j.anaerobe.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 470.Cotta MA, Whitehead TR, Falsen E, Moore E, Lawson PA. 2009. Robinsoniella peoriensis gen. nov., sp. nov., isolated from a swine-manure storage pit and a human clinical source. Int J Syst Evol Microbiol 59:150–155. doi: 10.1099/ijs.0.65676-0. [DOI] [PubMed] [Google Scholar]
- 471.Williams OM, Brazier J, Peraino V, Goldstein EJ. 2010. A review of three cases of Clostridium aldenense bacteremia. Anaerobe 16:475–477. doi: 10.1016/j.anaerobe.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 472.Park SK, Kim MS, Bae JW. 2013. Blautia faecis sp. nov., isolated from human faeces. Int J Syst Evol Microbiol 63:599–603. doi: 10.1099/ijs.0.036541-0. [DOI] [PubMed] [Google Scholar]
- 473.Zheng G, Summanen PH, Talan D, Bennion R, Rowlinson MC, Finegold SM. 2010. Phenotypic and molecular characterization of Solobacterium moorei isolates from patients with wound infection. J Clin Microbiol 48:873–876. doi: 10.1128/JCM.01381-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Bosshard PP, Zbinden R, Altwegg M. 2002. Turicibacter sanguinis gen. nov., sp. nov., a novel anaerobic, Gram-positive bacterium. Int J Syst Evol Microbiol 52:1263–1266. doi: 10.1099/00207713-52-4-1263. [DOI] [PubMed] [Google Scholar]
- 475.Karlsson FH, Ussery DW, Nielsen J, Nookaew I. 2011. A closer look at bacteroides: phylogenetic relationship and genomic implications of a life in the human gut. Microb Ecol 61:473–485. doi: 10.1007/s00248-010-9796-1. [DOI] [PubMed] [Google Scholar]
- 476.Vesth T, Ozen A, Andersen SC, Kaas RS, Lukjancenko O, Bohlin J, Nookaew I, Wassenaar TM, Ussery DW. 2013. Veillonella, Firmicutes: microbes disguised as Gram negatives. Stand Genomic Sci 9:431–448. doi: 10.4056/sigs.2981345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Mashima I, Theodorea CF, Thaweboon B, Thaweboon S, Nakazawa F. 2016. Identification of Veillonella species in the tongue biofilm by using a novel one-step polymerase chain reaction method. PLoS One 11:e0157516. doi: 10.1371/journal.pone.0157516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Shetty SA, Marathe NP, Lanjekar V, Ranade D, Shouche YS. 2013. Comparative genome analysis of Megasphaera sp. reveals niche specialization and its potential role in the human gut. PLoS One 8:e79353. doi: 10.1371/journal.pone.0079353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Devereux R, He SH, Doyle CL, Orkland S, Stahl DA, LeGall J, Whitman WB. 1990. Diversity and origin of Desulfovibrio species: phylogenetic definition of a family. J Bacteriol 172:3609–3619. doi: 10.1128/jb.172.7.3609-3619.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Baron EJ. 1997. Bilophila wadsworthia: a unique Gram-negative anaerobic rod. Anaerobe 3:83–86. doi: 10.1006/anae.1997.0075. [DOI] [PubMed] [Google Scholar]
- 481.Gupta RS, Sethi M. 2014. Phylogeny and molecular signatures for the phylum Fusobacteria and its distinct subclades. Anaerobe 28:182–198. doi: 10.1016/j.anaerobe.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 482.Eribe ERK, Olsen I. 2017. Leptotrichia species in human infections II. J Oral Microbiol 9:1368848. doi: 10.1080/20002297.2017.1368848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Both B, Buckel W, Kroppenstedt R, Stackebrandt E. 1992. Phylogenetic and chemotaxonomic characterization of Acidaminococcus fermentans. FEMS Microbiol Lett 76:7–11. doi: 10.1016/0378-1097(92)90355-r. [DOI] [PubMed] [Google Scholar]
- 484.Strompl C, Tindall BJ, Jarvis GN, Lunsdorf H, Moore ER, Hippe H. 1999. A re-evaluation of the taxonomy of the genus Anaerovibrio, with the reclassification of Anaerovibrio glycerini as Anaerosinus glycerini gen. nov., comb. nov., and Anaerovibrio burkinabensis as Anaeroarcus burkinensis [corrig.] gen. nov., comb. nov. Int J Syst Bacteriol 49(Part 4):1861–1872. doi: 10.1099/00207713-49-4-1861. [DOI] [PubMed] [Google Scholar]
- 485.Wu F, Guo X, Zhang J, Zhang M, Ou Z, Peng Y. 2017. Phascolarctobacterium faecium abundant colonization in human gastrointestinal tract. Exp Ther Med 14:3122–3126. doi: 10.3892/etm.2017.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Lagier JC, Armougom F, Mishra AK, Nguyen TT, Raoult D, Fournier PE. 2012. Non-contiguous finished genome sequence and description of Alistipes timonensis sp. nov. Stand Genomic Sci 6:315–324. doi: 10.4056/sigs.2685971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Mishra AK, Gimenez G, Lagier JC, Robert C, Raoult D, Fournier PE. 2012. Genome sequence and description of Alistipes senegalensis sp. nov. Stand Genomic Sci 6:1–16. doi: 10.4056/sigs.2625821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Hippe H, Hagelstein A, Kramer I, Swiderski J, Stackebrandt E. 1999. Phylogenetic analysis of Formivibrio citricus, Propionivibrio dicarboxylicus, Anaerobiospirillum thomasii, Succinimonas amylolytica and Succinivibrio dextrinosolvens and proposal of Succinivibrionaceae fam. nov. Int J Syst Bacteriol 49(Part 2):779–782. doi: 10.1099/00207713-49-2-779. [DOI] [PubMed] [Google Scholar]
- 489.Song Y, Liu C, Finegold SM. 2007. Peptoniphilus gorbachii sp. nov., Peptoniphilus olsenii sp. nov., and Anaerococcus murdochii sp. nov. isolated from clinical specimens of human origin. J Clin Microbiol 45:1746–1752. doi: 10.1128/JCM.00213-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Conrads G, Citron DM, Tyrrell KL, Horz HP, Goldstein EJ. 2005. 16S-23S rRNA gene internal transcribed spacer sequences for analysis of the phylogenetic relationships among species of the genus Porphyromonas. Int J Syst Evol Microbiol 55:607–613. doi: 10.1099/ijs.0.63234-0. [DOI] [PubMed] [Google Scholar]
- 491.Paster BJ, Dewhirst FE, Olsen I, Fraser GJ. 1994. Phylogeny of Bacteroides, Prevotella, and Porphyromonas spp. and related bacteria. J Bacteriol 176:725–732. doi: 10.1128/jb.176.3.725-732.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Srinivasan S, Morgan MT, Liu C, Matsen FA, Hoffman NG, Fiedler TL, Agnew KJ, Marrazzo JM, Fredricks DN. 2013. More than meets the eye: associations of vaginal bacteria with gram stain morphotypes using molecular phylogenetic analysis. PLoS One 8:e78633. doi: 10.1371/journal.pone.0078633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Hardham JM, King KW, Dreier K, Wong J, Strietzel C, Eversole RR, Sfintescu C, Evans RT. 2008. Transfer of Bacteroides splanchnicus to Odoribacter gen. nov. as Odoribacter splanchnicus comb. nov., and description of Odoribacter denticanis sp. nov., isolated from the crevicular spaces of canine periodontitis patients. Int J Syst Evol Microbiol 58:103–109. doi: 10.1099/ijs.0.63458-0. [DOI] [PubMed] [Google Scholar]
- 494.Sakamoto M, Takagaki A, Matsumoto K, Kato Y, Goto K, Benno Y. 2009. Butyricimonas synergistica gen. nov., sp. nov. and Butyricimonas virosa sp. nov., butyric acid-producing bacteria in the family 'Porphyromonadaceae' isolated from rat faeces. Int J Syst Evol Microbiol 59:1748–1753. doi: 10.1099/ijs.0.007674-0. [DOI] [PubMed] [Google Scholar]
- 495.Morotomi M, Nagai F, Watanabe Y. 2011. Parasutterella secunda sp. nov., isolated from human faeces and proposal of Sutterellaceae fam. nov. in the order Burkholderiales. Int J Syst Evol Microbiol 61:637–643. doi: 10.1099/ijs.0.023556-0. [DOI] [PubMed] [Google Scholar]
- 496.Mochon AB, Sussland D, Saubolle MA. 2016. Aerobic actinomycetes of clinical significance. Microbiol Spectr 4:DMIH2-0021-2015. doi: 10.1128/microbiolspec.DMIH2-0021-2015. [DOI] [PubMed] [Google Scholar]
- 497.Forbes BA. 2017. Mycobacterial taxonomy. J Clin Microbiol 55:380–383. doi: 10.1128/JCM.01287-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Parrish N. 2019. An update on mycobacterial taxonomy, 2016–2017. J Clin Microbiol 57:e01408-18. doi: 10.1128/JCM.01408-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Fatahi-Bafghi M. 2018. Nocardiosis from 1888 to 2017. Microb Pathog 114:369–384. doi: 10.1016/j.micpath.2017.11.012. [DOI] [PubMed] [Google Scholar]
- 500.McTaggart LR, Richardson SE, Witkowska M, Zhang SX. 2010. Phylogeny and identification of Nocardia species on the basis of multilocus sequence analysis. J Clin Microbiol 48:4525–4533. doi: 10.1128/JCM.00883-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.McTaggart LR, Doucet J, Witkowska M, Richardson SE. 2015. Antimicrobial susceptibility among clinical Nocardia species identified by multilocus sequence analysis. Antimicrob Agents Chemother 59:269–275. doi: 10.1128/AAC.02770-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Brown-Elliott BA, Brown JM, Conville PS, Wallace RJ Jr. 2006. Clinical and laboratory features of the Nocardia spp. based on current molecular taxonomy. Clin Microbiol Rev 19:259–282. doi: 10.1128/CMR.19.2.259-282.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Andalibi F, Fatahi-Bafghi M. 2017. Gordonia: isolation and identification in clinical samples and role in biotechnology. Folia Microbiol 62:245–252. doi: 10.1007/s12223-017-0491-1. [DOI] [PubMed] [Google Scholar]
- 504.Majidzadeh M, Fatahi-Bafghi M. 2018. Current taxonomy of Rhodococcus species and their role in infections. Eur J Clin Microbiol Infect Dis 37:2045–2062. doi: 10.1007/s10096-018-3364-x. [DOI] [PubMed] [Google Scholar]
- 505.Gupta RS, Lo B, Son J. 2018. Phylogenomics and comparative genomic studies robustly support division of the genus Mycobacterium into an emended genus Mycobacterium and four novel genera. Front Microbiol 9:67. doi: 10.3389/fmicb.2018.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Nouioui I, Carro L, García-López M, Meier-Kolthoff JP, Woyke T, Kyrpides NC, Pukall R, Klenk H-P, Goodfellow M, Göker M. 2018. Genome-based taxonomic classification of the phylum Actinobacteria. Front Microbiol 9:2007. doi: 10.3389/fmicb.2018.02007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Tortoli E. 2003. Impact of genotypic studies on mycobacterial taxonomy: the new mycobacteria of the 1990s. Clin Microbiol Rev 16:319–354. doi: 10.1128/cmr.16.2.319-354.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Brites D, Loiseau C, Menardo F, Borrell S, Boniotti MB, Warren R, Dippenaar A, Parsons SDC, Beisel C, Behr MA, Fyfe JA, Coscolla M, Gagneux S. 2018. A new phylogenetic framework for the animal-adapted Mycobacterium tuberculosis complex. Front Microbiol 9:2820. doi: 10.3389/fmicb.2018.02820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Orgeur M, Brosch R. 2018. Evolution of virulence in the Mycobacterium tuberculosis complex. Curr Opin Microbiol 41:68–75. doi: 10.1016/j.mib.2017.11.021. [DOI] [PubMed] [Google Scholar]
- 510.Sommerstein R, Kohler PP, Hasse B. 2017. Global Mycobacterium chimaera outbreak: challenges for the near future. J Hosp Infect 96:207–208. doi: 10.1016/j.jhin.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 511.van Ingen J, Kohl TA, Kranzer K, Hasse B, Keller PM, Katarzyna Szafrańska A, Hillemann D, Chand M, Schreiber PW, Sommerstein R, Berger C, Genoni M, Rüegg C, Troillet N, Widmer AF, Becker SL, Herrmann M, Eckmanns T, Haller S, Höller C, Debast SB, Wolfhagen MJ, Hopman J, Kluytmans J, Langelaar M, Notermans DW, Ten Oever J, van den Barselaar P, Vonk ABA, Vos MC, Ahmed N, Brown T, Crook D, Lamagni T, Phin N, Smith EG, Zambon M, Serr A, Götting T, Ebner W, Thürmer A, Utpatel C, Spröer C, Bunk B, Nübel U, Bloemberg GV, Böttger EC, Niemann S, Wagner D, Sax H. 2017. Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: a molecular epidemiological study. Lancet Infect Dis 17:1033–1041. doi: 10.1016/S1473-3099(17)30324-9. [DOI] [PubMed] [Google Scholar]
- 512.Tortoli E, Kohl TA, Brown-Elliott BA, Trovato A, Leao SC, Garcia MJ, Vasireddy S, Turenne CY, Griffith DE, Philley JV, Baldan R, Campana S, Cariani L, Colombo C, Taccetti G, Teri A, Niemann S, Wallace RJ Jr, Cirillo DM. 2016. Emended description of Mycobacterium abscessus, Mycobacterium abscessus subsp. abscessus and Mycobacterium abscessus subsp. bolletii and designation of Mycobacterium abscessus subsp. massiliense comb. nov. Int J Syst Evol Microbiol 66:4471–4479. doi: 10.1099/ijsem.0.001376. [DOI] [PubMed] [Google Scholar]
- 513.Nogueira CL, Whipps CM, Matsumoto CK, Chimara E, Droz S, Tortoli E, de Freitas D, Cnockaert M, Palomino JC, Martin A, Vandamme P, Leão SC. 2015. Mycobacterium saopaulense sp. nov., a rapidly growing mycobacterium closely related to members of the Mycobacterium chelonae–Mycobacterium abscessus group. Int J Syst Evol Microbiol 65:4403–4409. doi: 10.1099/ijsem.0.000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Tortoli E. 2003. Mycobacterium kansasii, species or complex? Biomolecular and epidemiological insights. Kekkaku 78:705–709. [PubMed] [Google Scholar]
- 515.Wallace RJ Jr, Steele LC, Sumter G, Smith JM. 1988. Antimicrobial susceptibility patterns of Nocardia asteroides. Antimicrob Agents Chemother 32:1776–1779. doi: 10.1128/aac.32.12.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Conville PS, Witebsky FG. 2007. Analysis of multiple differing copies of the 16S rRNA gene in five clinical isolates and three type strains of Nocardia species and implications for species assignment. J Clin Microbiol 45:1146–1151. doi: 10.1128/JCM.02482-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Safaei S, Fatahi-Bafghi M, Pouresmaeil O. 2018. Role of Tsukamurella species in human infections: first literature review. New Microbes New Infect 22:6–12. doi: 10.1016/j.nmni.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Cascio A, Mandraffino G, Cinquegrani M, Delfino D, Mandraffino R, Romeo O, Criseo G, Saitta A. 2011. Actinomadura pelletieri mycetoma–an atypical case with spine and abdominal wall involvement. J Med Microbiol 60:673–676. doi: 10.1099/jmm.0.027862-0. [DOI] [PubMed] [Google Scholar]
- 519.Vera-Cabrera L, Ortiz-Lopez R, Elizondo-Gonzalez R, Campos-Rivera MP, Gallardo-Rocha A, Molina-Torres CA, Ocampo-Candiani J. 2014. Draft genome sequence of Actinomadura madurae LIID-AJ290, isolated from a human mycetoma case. Genome Announc 2:e00201-14. doi: 10.1128/genomeA.00201-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Blaschke AJ, Bender J, Byington CL, Korgenski K, Daly J, Petti CA, Pavia AT, Ampofo K. 2007. Gordonia species: emerging pathogens in pediatric patients that are identified by 16S ribosomal RNA gene sequencing. Clin Infect Dis 45:483–486. doi: 10.1086/520018. [DOI] [PubMed] [Google Scholar]
- 521.Goodfellow M, Ferguson EV, Sanglier JJ. 1992. Numerical classification and identification of Streptomyces species–a review. Gene 115:225–233. doi: 10.1016/0378-1119(92)90563-5. [DOI] [PubMed] [Google Scholar]
- 522.Kapadia M, Rolston KV, Han XY. 2007. Invasive Streptomyces infections: six cases and literature review. Am J Clin Pathol 127:619–624. doi: 10.1309/QJEBXP0BCGR54L15. [DOI] [PubMed] [Google Scholar]
- 523.Schwartz MA, Tabet SR, Collier AC, Wallis CK, Carlson LC, Nguyen TT, Kattar MM, Coyle MB. 2002. Central venous catheter-related bacteremia due to Tsukamurella species in the immunocompromised host: a case series and review of the literature. Clin Infect Dis 35:e72. doi: 10.1086/342561. [DOI] [PubMed] [Google Scholar]
- 524.Miller RJ, Chow B, Pillai D, Church D. 2016. Development and evaluation of a novel fast broad-range 16S ribosomal DNA PCR and sequencing assay for diagnosis of bacterial infective endocarditis: multi-year experience in a large Canadian healthcare zone and a literature review. BMC Infect Dis 16:146. doi: 10.1186/s12879-016-1476-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Fenollar F, Roux V, Stein A, Drancourt M, Raoult D. 2006. Analysis of 525 samples to determine the usefulness of PCR amplification and sequencing of the 16S rRNA gene for diagnosis of bone and joint infections. J Clin Microbiol 44:1018–1028. doi: 10.1128/JCM.44.3.1018-1028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Verdier I, Gayet-Ageron A, Ploton C, Taylor P, Benito Y, Freydiere AM, Chotel F, Berard J, Vanhems P, Vandenesch F. 2005. Contribution of a broad range polymerase chain reaction to the diagnosis of osteoarticular infections caused by Kingella kingae: description of twenty-four recent pediatric diagnoses. Pediatr Infect Dis J 24:692–696. doi: 10.1097/01.inf.0000172153.10569.dc. [DOI] [PubMed] [Google Scholar]
- 527.Gomez E, Cazanave C, Cunningham SA, Greenwood-Quaintance KE, Steckelberg JM, Uhl JR, Hanssen AD, Karau MJ, Schmidt SM, Osmon DR, Berbari EF, Mandrekar J, Patel R. 2012. Prosthetic joint infection diagnosis using broad-range PCR of biofilms dislodged from knee and hip arthroplasty surfaces using sonication. J Clin Microbiol 50:3501–3508. doi: 10.1128/JCM.00834-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Meyer T, Franke G, Polywka SK, Lutgehetmann M, Gbadamosi J, Magnus T, Aepfelbacher M. 2014. Improved detection of bacterial central nervous system infections by use of a broad-range PCR assay. J Clin Microbiol 52:1751–1753. doi: 10.1128/JCM.00469-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Rampini SK, Bloemberg GV, Keller PM, Buchler AC, Dollenmaier G, Speck RF, Bottger EC. 2011. Broad-range 16S rRNA gene polymerase chain reaction for diagnosis of culture-negative bacterial infections. Clin Infect Dis 53:1245–1251. doi: 10.1093/cid/cir692. [DOI] [PubMed] [Google Scholar]
- 530.Relman DA, Falkow S. 1992. Identification of uncultured microorganisms: expanding the spectrum of characterized microbial pathogens. Infect Agents Dis 1:245–253. [PubMed] [Google Scholar]
- 531.Relman DA, Schmidt TM, MacDermott RP, Falkow S. 1992. Identification of the uncultured bacillus of Whipple's disease. N Engl J Med 327:293–301. doi: 10.1056/NEJM199207303270501. [DOI] [PubMed] [Google Scholar]
- 532.Lepidi H, Fenollar F, Dumler JS, Gauduchon V, Chalabreysse L, Bammert A, Bonzi MF, Thivolet-Bejui F, Vandenesch F, Raoult D. 2004. Cardiac valves in patients with Whipple endocarditis: microbiological, molecular, quantitative histologic, and immunohistochemical studies of 5 patients. J Infect Dis 190:935–945. doi: 10.1086/422845. [DOI] [PubMed] [Google Scholar]
- 533.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA. 2006. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A 103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Goldschmidt P, Rostane H, Sow M, Goepogui A, Batellier L, Chaumeil C. 2006. Detection by broad-range real-time PCR assay of Chlamydia species infecting human and animals. Br J Ophthalmol 90:1425–1429. doi: 10.1136/bjo.2006.096420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Millar BC, Xu J, Moore JE. 2002. Risk assessment models and contamination management: implications for broad-range ribosomal DNA PCR as a diagnostic tool in medical bacteriology. J Clin Microbiol 40:1575–1580. doi: 10.1128/jcm.40.5.1575-1580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Muhl H, Kochem AJ, Disque C, Sakka SG. 2010. Activity and DNA contamination of commercial polymerase chain reaction reagents for the universal 16S rDNA real-time polymerase chain reaction detection of bacterial pathogens in blood. Diagn Microbiol Infect Dis 66:41–49. doi: 10.1016/j.diagmicrobio.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 537.Klaschik S, Lehmann LE, Raadts A, Hoeft A, Stuber F. 2002. Comparison of different decontamination methods for reagents to detect low concentrations of bacterial 16S DNA by real-time-PCR. Mol Biotechnol 22:231–242. doi: 10.1385/MB:22:3:231. [DOI] [PubMed] [Google Scholar]
- 538.Kommedal O, Lekang K, Langeland N, Wiker HG. 2011. Characterization of polybacterial clinical samples using a set of group-specific broad-range primers targeting the 16S rRNA gene followed by DNA sequencing and RipSeq analysis. J Med Microbiol 60:927–936. doi: 10.1099/jmm.0.028373-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Chang SS, Hsu HL, Cheng JC, Tseng CP. 2011. An efficient strategy for broad-range detection of low abundance bacteria without DNA decontamination of PCR reagents. PLoS One 6:e20303. doi: 10.1371/journal.pone.0020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Kommedal O, Simmon K, Karaca D, Langeland N, Wiker HG. 2012. Dual priming oligonucleotides for broad-range amplification of the bacterial 16S rRNA gene directly from human clinical specimens. J Clin Microbiol 50:1289–1294. doi: 10.1128/JCM.06269-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Dixon P, Davies P, Hollingworth W, Stoddart M, MacGowan A. 2015. A systematic review of matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry compared to routine microbiological methods for the time taken to identify microbial organisms from positive blood cultures. Eur J Clin Microbiol Infect Dis 34:863–876. doi: 10.1007/s10096-015-2322-0. [DOI] [PubMed] [Google Scholar]
- 542.Graham R, Graham C, McMullan G. 2007. Microbial proteomics: a mass spectrometry primer for biologists. Microb Cell Fact 6:26. doi: 10.1186/1475-2859-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Mellmann A, Cloud J, Maier T, Keckevoet U, Ramminger I, Iwen P, Dunn J, Hall G, Wilson D, Lasala P, Kostrzewa M, Harmsen D. 2008. Evaluation of matrix-assisted laser desorption ionization-time-of-flight mass spectrometry in comparison to 16S rRNA gene sequencing for species identification of nonfermenting bacteria. J Clin Microbiol 46:1946–1954. doi: 10.1128/JCM.00157-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Nagy E, Becker S, Kostrzewa M, Barta N, Urban E. 2012. The value of MALDI-TOF MS for the identification of clinically relevant anaerobic bacteria in routine laboratories. J Med Microbiol 61:1393–1400. doi: 10.1099/jmm.0.043927-0. [DOI] [PubMed] [Google Scholar]
- 545.Bizzini A, Jaton K, Romo D, Bille J, Prod'hom G, Greub G. 2011. Matrix-assisted laser desorption ionization-time of flight mass spectrometry as an alternative to 16S rRNA gene sequencing for identification of difficult-to-identify bacterial strains. J Clin Microbiol 49:693–696. doi: 10.1128/JCM.01463-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Anhalt JP, Fenselau C. 1975. Identification of bacteria using mass spectrometry. Anal Chem 47:219–225. doi: 10.1021/ac60352a007. [DOI] [Google Scholar]
- 547.Clark AE, Kaleta EJ, Arora A, Wolk DM. 2013. Matrix-assisted laser desorption ionization-time of flight mass spectrometry: a fundamental shift in the routine practice of clinical microbiology. Clin Microbiol Rev 26:547–603. doi: 10.1128/CMR.00072-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Tran A, Alby K, Kerr A, Jones M, Gilligan PH. 2015. Cost savings realized by implementation of routine microbiological identification by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol 53:2473–2479. doi: 10.1128/JCM.00833-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Bizzini A, Durussel C, Bille J, Greub G, Prod'hom G. 2010. Performance of matrix-assisted laser desorption ionization-time of flight mass spectrometry for identification of bacterial strains routinely isolated in a clinical microbiology laboratory. J Clin Microbiol 48:1549–1554. doi: 10.1128/JCM.01794-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.van Belkum A, Chatellier S, Girard V, Pincus D, Deol P, Dunne WM Jr. 2015. Progress in proteomics for clinical microbiology: MALDI-TOF MS for microbial species identification and more. Expert Rev Proteomics 12:595–605. doi: 10.1586/14789450.2015.1091731. [DOI] [PubMed] [Google Scholar]
- 551.Marin M, Ruiz A, Iglesias C, Quiroga L, Cercenado E, Martin-Rabadan P, Bouza E, Rodriguez-Sanchez B. 2018. Identification of Nocardia species from clinical isolates using Maldi-Tof mass spectrometry. Clin Microbiol Infect 24:1342.e5–1342.e8. doi: 10.1016/j.cmi.2018.06.014. [DOI] [PubMed] [Google Scholar]
- 552.Mediavilla-Gradolph MC, De Toro-Peinado I, Bermúdez-Ruiz MP, García-Martínez MDLÁ, Ortega-Torres M, Montiel Quezel-Guerraz N, Palop-Borrás B. 2015. Use of MALDI-TOF MS for identification of nontuberculous Mycobacterium species isolated from clinical specimens. Biomed Res Int 2015:854078. doi: 10.1155/2015/854078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Tudo G, Monte MR, Vergara A, Lopez A, Hurtado JC, Ferrer-Navarro M, Vila J, Gonzalez-Martin J. 2015. Implementation of MALDI-TOF MS technology for the identification of clinical isolates of Mycobacterium spp. in mycobacterial diagnosis. Eur J Clin Microbiol Infect Dis 34:1527–1532. doi: 10.1007/s10096-015-2381-2. [DOI] [PubMed] [Google Scholar]
- 554.La Scola B, Fournier PE, Raoult D. 2011. Burden of emerging anaerobes in the MALDI-TOF and 16S rRNA gene sequencing era. Anaerobe 17:106–112. doi: 10.1016/j.anaerobe.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 555.Fedorko DP, Drake SK, Stock F, Murray PR. 2012. Identification of clinical isolates of anaerobic bacteria using matrix-assisted laser desorption ionization-time of flight mass spectrometry. Eur J Clin Microbiol Infect Dis 31:2257–2262. doi: 10.1007/s10096-012-1563-4. [DOI] [PubMed] [Google Scholar]
- 556.Jamal WY, Shahin M, Rotimi VO. 2013. Comparison of two matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry methods and API 20AN for identification of clinically relevant anaerobic bacteria. J Med Microbiol 62:540–544. doi: 10.1099/jmm.0.053256-0. [DOI] [PubMed] [Google Scholar]
- 557.Ndukum J, Atlas M, Datta S. 2011. pkDACLASS: open source software for analyzing MALDI-TOF data. Bioinformation 6:45–47. doi: 10.6026/97320630006045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Gibb S, Strimmer K. 2012. MALDIquant: a versatile R package for the analysis of mass spectrometry data. Bioinformatics 28:2270–2271. doi: 10.1093/bioinformatics/bts447. [DOI] [PubMed] [Google Scholar]
- 559.Böhme K, Fernández-No IC, Barros-Velázquez J, Gallardo JM, Cañas B, Calo-Mata P. 2012. SpectraBank: an open access tool for rapid microbial identification by MALDI-TOF MS fingerprinting. Electrophoresis 33:2138–2142. doi: 10.1002/elps.201200074. [DOI] [PubMed] [Google Scholar]
- 560.Jamal W, Albert MJ, Rotimi VO. 2014. Real-time comparative evaluation of bioMerieux VITEK MS versus Bruker Microflex MS, two matrix-assisted laser desorption-ionization time-of-flight mass spectrometry systems, for identification of clinically significant bacteria. BMC Microbiol 14:289. doi: 10.1186/s12866-014-0289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Jamal WY, Ahmad S, Khan ZU, Rotimi VO. 2014. Comparative evaluation of two matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) systems for the identification of clinically significant yeasts. Int J Infect Dis 26:167–170. doi: 10.1016/j.ijid.2014.05.031. [DOI] [PubMed] [Google Scholar]
- 562.Carbonnelle E, Beretti JL, Cottyn S, Quesne G, Berche P, Nassif X, Ferroni A. 2007. Rapid identification of staphylococci isolated in clinical microbiology laboratories by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol 45:2156–2161. doi: 10.1128/JCM.02405-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Seng P, Rolain JM, Fournier PE, La Scola B, Drancourt M, Raoult D. 2010. MALDI-TOF-mass spectrometry applications in clinical microbiology. Future Microbiol 5:1733–1754. doi: 10.2217/fmb.10.127. [DOI] [PubMed] [Google Scholar]
- 564.Fan WT, Qin TT, Bi RR, Kang HQ, Ma P, Gu B. 2017. Performance of the matrix-assisted laser desorption ionization time-of-flight mass spectrometry system for rapid identification of streptococci: a review. Eur J Clin Microbiol Infect Dis 36:1005–1012. doi: 10.1007/s10096-016-2879-2. [DOI] [PubMed] [Google Scholar]
- 565.Ruppitsch W, Stoger A, Indra A, Grif K, Schabereiter-Gurtner C, Hirschl A, Allerberger F. 2007. Suitability of partial 16S ribosomal RNA gene sequence analysis for the identification of dangerous bacterial pathogens. J Appl Microbiol 102:852–859. doi: 10.1111/j.1365-2672.2006.03107.x. [DOI] [PubMed] [Google Scholar]
- 566.Rotz LD, Khan AS, Lillibridge SR, Ostroff SM, Hughes JM. 2002. Public health assessment of potential biological terrorism agents. Emerg Infect Dis 8:225–230. doi: 10.3201/eid0802.010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Dunne WM Jr, Westblade LF, Ford B. 2012. Next-generation and whole-genome sequencing in the diagnostic clinical microbiology laboratory. Eur J Clin Microbiol Infect Dis 31:1719–1726. doi: 10.1007/s10096-012-1641-7. [DOI] [PubMed] [Google Scholar]
- 568.Deurenberg RH, Bathoorn E, Chlebowicz MA, Couto N, Ferdous M, Garcia-Cobos S, Kooistra-Smid AM, Raangs EC, Rosema S, Veloo AC, Zhou K, Friedrich AW, Rossen JW. 2017. Application of next generation sequencing in clinical microbiology and infection prevention. J Biotechnol 243:16–24. doi: 10.1016/j.jbiotec.2016.12.022. [DOI] [PubMed] [Google Scholar]
- 569.Didelot X, Bowden R, Wilson DJ, Peto TEA, Crook DW. 2012. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet 13:601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Dunne WM Jr, Jaillard M, Rochas O, Van Belkum A. 2017. Microbial genomics and antimicrobial susceptibility testing. Expert Rev Mol Diagn 17:257–269. doi: 10.1080/14737159.2017.1283220. [DOI] [PubMed] [Google Scholar]
- 571.Bertelli C, Greub G. 2013. Rapid bacterial genome sequencing: methods and applications in clinical microbiology. Clin Microbiol Infect 19:803–813. doi: 10.1111/1469-0691.12217. [DOI] [PubMed] [Google Scholar]
- 572.Buchan BW, Ledeboer NA. 2014. Emerging technologies for the clinical microbiology laboratory. Clin Microbiol Rev 27:783–822. doi: 10.1128/CMR.00003-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM. 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 574.Gargis AS, Kalman L, Lubin IM. 2016. Assuring the quality of next-generation sequencing in clinical microbiology and public health laboratories. J Clin Microbiol 54:2857–2865. doi: 10.1128/JCM.00949-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. 2012. Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012:251364. doi: 10.1155/2012/251364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ. 2012. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol 30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 577.Lefterova MI, Suarez CJ, Banaei N, Pinsky BA. 2015. Next-generation sequencing for infectious disease diagnosis and management: a report of the association for molecular pathology. J Mol Diagn 17:623–634. doi: 10.1016/j.jmoldx.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 578.Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y. 2012. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Loman NJ, Constantinidou C, Chan JZ, Halachev M, Sergeant M, Penn CW, Robinson ER, Pallen MJ. 2012. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol 10:599–606. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- 580.Fichot EB, Norman RS. 2013. Microbial phylogenetic profiling with the Pacific Biosciences sequencing platform. Microbiome 1:10. doi: 10.1186/2049-2618-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Davis BM, Chao MC, Waldor MK. 2013. Entering the era of bacterial epigenomics with single molecule real time DNA sequencing. Curr Opin Microbiol 16:192–198. doi: 10.1016/j.mib.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T, Di Ventra M, Garaj S, Hibbs A, Huang X, Jovanovich SB, Krstic PS, Lindsay S, Ling XS, Mastrangelo CH, Meller A, Oliver JS, Pershin YV, Ramsey JM, Riehn R, Soni GV, Tabard-Cossa V, Wanunu M, Wiggin M, Schloss JA. 2008. The potential and challenges of nanopore sequencing. Nat Biotechnol 26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Ashton PM, Nair S, Dallman T, Rubino S, Rabsch W, Mwaigwisya S, Wain J, O'Grady J. 2015. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat Biotechnol 33:296–300. doi: 10.1038/nbt.3103. [DOI] [PubMed] [Google Scholar]
- 584.Quick J, Ashton P, Calus S, Chatt C, Gossain S, Hawker J, Nair S, Neal K, Nye K, Peters T, De Pinna E, Robinson E, Struthers K, Webber M, Catto A, Dallman TJ, Hawkey P, Loman NJ. 2015. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol 16:114. doi: 10.1186/s13059-015-0677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Fricke WF, Rasko DA. 2014. Bacterial genome sequencing in the clinic: bioinformatic challenges and solutions. Nat Rev Genet 15:49–55. doi: 10.1038/nrg3624. [DOI] [PubMed] [Google Scholar]
- 586.Lynch T, Petkau A, Knox N, Graham M, Van Domselaar G. 2016. A primer on infectious disease bacterial genomics. Clin Microbiol Rev 29:881–913. doi: 10.1128/CMR.00001-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Rossen JWA, Friedrich AW, Moran-Gilad J, on behalf of the ESCMID Study Group for Genomic and Molecular Diagnostics (ESGMD) . 2018. Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin Microbiol Infect 24:355–360. doi: 10.1016/j.cmi.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 588.Sabat AJ, van Zanten E, Akkerboom V, Wisselink G, van Slochteren K, de Boer RF, Hendrix R, Friedrich AW, Rossen JWA, Kooistra-Smid A. 2017. Targeted next-generation sequencing of the 16S-23S rRNA region for culture-independent bacterial identification–increased discrimination of closely related species. Sci Rep 7:3434. doi: 10.1038/s41598-017-03458-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Angers A, Petrillo M, Patak A, Querci M, Van den Eede G. 2017. The role and implementation of next-generation sequencing technologies in the coordinated action plan against antimicrobial resistance. JRC conference and workshop reports. JRC Science Hub EU, Vill Borghi, Italy. [Google Scholar]
- 590.Clausen PT, Zankari E, Aarestrup FM, Lund O. 2016. Benchmarking of methods for identification of antimicrobial resistance genes in bacterial whole genome data. J Antimicrob Chemother 71:2484–2488. doi: 10.1093/jac/dkw184. [DOI] [PubMed] [Google Scholar]
- 591.McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, Bhullar K, Canova MJ, De Pascale G, Ejim L, Kalan L, King AM, Koteva K, Morar M, Mulvey MR, O'Brien JS, Pawlowski AC, Piddock LJ, Spanogiannopoulos P, Sutherland AD, Tang I, Taylor PL, Thaker M, Wang W, Yan M, Yu T, Wright GD. 2013. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Bradley P, Gordon NC, Walker TM, Dunn L, Heys S, Huang B, Earle S, Pankhurst LJ, Anson L, de Cesare M, Piazza P, Votintseva AA, Golubchik T, Wilson DJ, Wyllie DH, Diel R, Niemann S, Feuerriegel S, Kohl TA, Ismail N, Omar SV, Smith EG, Buck D, McVean G, Walker AS, Peto TE, Crook DW, Iqbal Z. 2015. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat Commun 6:10063. doi: 10.1038/ncomms10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Moran-Gilad J, Sintchenko V, Pedersen SK, Wolfgang WJ, Pettengill J, Strain E, Hendriksen RS, Global Microbial Identifier initiative's Working Group . 2015. Proficiency testing for bacterial whole genome sequencing: an end-user survey of current capabilities, requirements and priorities. BMC Infect Dis 15:174. doi: 10.1186/s12879-015-0902-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Bartels MD, Petersen A, Worning P, Nielsen JB, Larner-Svensson H, Johansen HK, Andersen LP, Jarlov JO, Boye K, Larsen AR, Westh H. 2014. Comparing whole-genome sequencing with Sanger sequencing for spa typing of methicillin-resistant Staphylococcus aureus. J Clin Microbiol 52:4305–4308. doi: 10.1128/JCM.01979-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Chan JZ, Pallen MJ, Oppenheim B, Constantinidou C. 2012. Genome sequencing in clinical microbiology. Nat Biotechnol 30:1068–1071. doi: 10.1038/nbt.2410. [DOI] [PubMed] [Google Scholar]
- 596.Vernet G, Saha S, Satzke C, Burgess DH, Alderson M, Maisonneuve JF, Beall BW, Steinhoff MC, Klugman KP. 2011. Laboratory-based diagnosis of pneumococcal pneumonia: state of the art and unmet needs. Clin Microbiol Infect 17(Suppl 3):1–13. doi: 10.1111/j.1469-0691.2011.03496.x. [DOI] [PubMed] [Google Scholar]
- 597.Tuite N, Reddington K, Barry T, Zumla A, Enne V. 2014. Rapid nucleic acid diagnostics for the detection of antimicrobial resistance in Gram-negative bacteria: is it time for a paradigm shift? J Antimicrob Chemother 69:1729–1733. doi: 10.1093/jac/dku083. [DOI] [PubMed] [Google Scholar]
- 598.Mellmann A, Harmsen D, Cummings CA, Zentz EB, Leopold SR, Rico A, Prior K, Szczepanowski R, Ji Y, Zhang W, McLaughlin SF, Henkhaus JK, Leopold B, Bielaszewska M, Prager R, Brzoska PM, Moore RL, Guenther S, Rothberg JM, Karch H. 2011. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One 6:e22751. doi: 10.1371/journal.pone.0022751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Gilmour MW, Graham M, Van Domselaar G, Tyler S, Kent H, Trout-Yakel KM, Larios O, Allen V, Lee B, Nadon C. 2010. High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Genomics 11:120. doi: 10.1186/1471-2164-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Koser CU, Fraser LJ, Ioannou A, Becq J, Ellington MJ, Holden MT, Reuter S, Torok ME, Bentley SD, Parkhill J, Gormley NA, Smith GP, Peacock SJ. 2014. Rapid single-colony whole-genome sequencing of bacterial pathogens. J Antimicrob Chemother 69:1275–1281. doi: 10.1093/jac/dkt494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Harris SR, Cartwright EJ, Torok ME, Holden MT, Brown NM, Ogilvy-Stuart AL, Ellington MJ, Quail MA, Bentley SD, Parkhill J, Peacock SJ. 2013. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis 13:130–136. doi: 10.1016/S1473-3099(12)70268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Naccache SN, Federman S, Veeraraghavan N, Zaharia M, Lee D, Samayoa E, Bouquet J, Greninger AL, Luk KC, Enge B, Wadford DA, Messenger SL, Genrich GL, Pellegrino K, Grard G, Leroy E, Schneider BS, Fair JN, Martinez MA, Isa P, Crump JA, DeRisi JL, Sittler T, Hackett J Jr, Miller S, Chiu CY. 2014. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res 24:1180–1192. doi: 10.1101/gr.171934.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Riesen A, Perreten V. 2010. Staphylococcus rostri sp. nov., a haemolytic bacterium isolated from the noses of healthy pigs. Int J Syst Evol Microbiol 60:2042–2047. doi: 10.1099/ijs.0.012443-0. [DOI] [PubMed] [Google Scholar]