

Abstract
Although postmarketing studies conducted in population‐based databases often contain information on patients in the order of millions, they can still be underpowered if outcomes or exposure of interest is rare, or the interest is in subgroup effects. Combining several databases might provide the statistical power needed. A multi‐database study (MDS) uses at least two healthcare databases, which are not linked with each other at an individual person level, with analyses carried out in parallel across each database applying a common study protocol. Although many MDSs have been performed in Europe in the past 10 years, there is a lack of clarity on the peculiarities and implications of the existing strategies to conduct them. In this review, we identify four strategies to execute MDSs, classified according to specific choices in the execution: (A) local analyses , where data are extracted and analyzed locally, with programs developed by each site; (B) sharing of raw data, where raw data are locally extracted and transferred without analysis to a central partner, where all the data are pooled and analyzed; (C) use of a common data model with study‐specific data, where study‐specific data are locally extracted, loaded into a common data model, and processed locally with centrally developed programs; and (D) use of general common data model, where all local data are extracted and loaded into a common data model, prior to and independent of any study protocol, and protocols are incorporated in centrally developed programs that run locally. We illustrate differences between strategies and analyze potential implications.
It is widely accepted that information on medicines’ safety, which emerges from premarketing clinical trials, is incomplete and needs to be supplemented by studies with larger and more heterogeneous populations over longer observation periods. 1 , 2 Indeed, several agents, such as rofecoxib, troglitazone, and valdecoxib, were withdrawn from the market because of adverse drug reactions that were not observed or poorly described in premarketing clinical trials. 3 The role of postmarketing safety studies becomes even more important for an increasing number of medicines, such as orphan drugs that are marketed through accelerated approval procedures before a sufficient body of efficacy and safety evidence is available. 4 , 5
In Europe, the pharmacovigilance directive 2010/84/EU came into force in July 2012 accompanied by the Commission Implementing Regulation (European Union) No. 520/2012, with the aim of increasing the quality of postmarketing data on medicines’ safety and promoting the rapid and thorough evaluation of safety issues throughout the product life‐cycle. Among other requirements, these pharmacovigilance regulations made it mandatory for European marketing authorization holders to adopt a risk‐management plan for all new marketing authorization and to conduct postauthorization safety or postauthorization effectiveness safety studies if requested by competent authorities. Drug regulatory authorities may be central, such as the European Medicines Agency (EMA), or they may belong the member states of the European Union. For centrally approved products, the Pharmacovigilance Risk Assessment Committee is mandated to be involved in the assessment of the protocol and results of the studies, to ensure they contribute meaningfully to regulatory decision making. 6
Even prior to the new pharmacovigilance legislation, the increasing number of postauthorization studies around the world, including in Europe, called for a better monitoring of the quality of such studies. In this context, in 2007, EMA launched the European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP), which is composed of public and private research institutions with an interest in pharmacoepidemiology and pharmacovigilance. 7 ENCePP’s mission is the promotion of high‐quality, multicenter postmarketing medicines benefit‐risk research as well as the support of transparency and scientific independence in research. As such, ENCePP developed the ENCePP e‐Register (renamed later EU PAS Register), a public online repository of medicines‐related studies, where both imposed and nonimposed postauthorization studies can be registered. A review of studies registered in the EU PAS Register between 2012 and 2015 showed that one‐third of all studies were conducted using secondary data. 8 Another review of the EU PAS Register reported that studies investigating drug safety and effectiveness used more commonly primary data, whereas drug utilization studies were more likely to be conducted using secondary data. 9 In this study, primary data capture referred to the collection of data specifically for a study, secondary data was the use of data already collected for another purpose.
Although postmarketing studies conducted in population‐based databases often contain information on patients numbering in the order of millions, they can still be underpowered if outcomes or exposure of interest are rare, or when the interest is in subgroup effects. In such cases, combining several databases serves to try and provide the statistical power needed. Moreover, combining different databases is often necessary when evidence is needed from different countries, both to study specific patterns of utilization and to produce evidence with more robust external validity.
We define a multi‐database study (MDS) as a study using at least two healthcare databases, which are not linked with each other at an individual person level, either because they insist on different populations, or because, even if populations overlap, local regulations forbid record linkage. Analyses are carried out in parallel across each database applying a common study protocol. The core difference compared to a “traditional” meta‐analysis of observational studies is that an MDS attempts to reduce the possible sources of heterogeneity across participating databases by applying a common study protocol and study design, including common exposure, outcome, and covariate definitions, as opposed to summarizing the results of different studies a posteriori. Indeed, a disadvantage of traditional meta‐analyses is that the studies being pooled usually have a high methodological variability. An MDS has the additional advantage that the whole study team can profit from the expertise of all the sites to develop the study protocol: this may be particularly relevant when the study question refers to a special population (e.g., pediatric) and the international availability of centers having the relevant methodological and clinical expertise is scarce. Platt et al. 10 describe some features of specific distributed networks that improve replicability, increase transparency, and reduce bias. With similar aims, Schneeweiss et al. explore the implications of adopting different flavors of common data models. 11
Although many MDSs have been performed in Europe, there is a lack of clarity and consistency on terminology describing the main features of the existing approaches to execute them. Hence, the aim of this commentary is to define and compare the strategies that can be adopted to execute MDSs and analyze their potential implications from a European perspective.
Definitions of strategies to execute an MDS
The ENCePP Methodological Guide 12 describes four strategies to execute an MDS. The strategies are classified according to specific choices in the steps needed to execute a study: protocol development and agreement, approval by the relevant governance bodies; database transformation into the analytical dataset; and analysis of the analytical dataset. To appreciate the differences between strategies, it must be noted that a database in itself does not contain all study variables per each subject, such as presence or absence of a health condition of interest, but rather raw data, that is, a longitudinal sequence of observations generated for purposes different from the research question at hand. Study variables are obtained by manipulating raw data, 13 for instance, by identifying patterns of diagnostic codes associated with a condition of interest for the study, 14 , 15 by detecting associated treatments, 16 or by running natural language processing or machine‐learning algorithms on free‐text data items. 17 Creating an analytical dataset from a database for a specific study requires, therefore, first creating study variables from raw data, and second, using the study variables to apply the study design to: select the study population, associate an observation period to each study subject, possibly with censoring criteria, an exposure status, confounding variable(s), and outcome(s) occurrence. The creation of the analytic dataset may occur after extracting a subset of the data from the original database, which is commonly conditional on permission for data access, based on general and possibly local regulations. As an optional step, some strategies entail converting some or all of the data into a common data model (CDM). For the purpose of this document, “Common Data Model” means a common structure and format for data: if two datasets have the same CDM, a single program can run on both. Sometimes the CDM imposes a common terminology as well, and other noteworthy differences between different CDMs can be highlighted. 11 This level of detail goes beyond the scope of this paper: for our purposes, a CDM allows the same program to run on all the datasets formatted accordingly. It must be noted that if two datasets have the same structure and format, but different terminology, the same program can run on both, provided that the terminology is recorded. For instance, the program may specify conditional clauses such as: “if terminology = ‘X,’ then select ‘A’; or else if terminology = ‘Y,’ then select ‘B’”. Moreover, note that a CDM is a conversion of the database, therefore, it contains raw data (possibly selected and/or recoded with some clinical insight), not study variables: see in the next section (paragraph discussing “time to create the study variable”) a discussion on different options to create study variables based on a CDM.
The four strategies to execute an MDS are classified by the ENCePP Methodological Guide as local analyses (hereafter referred to as strategy A), sharing of raw data (hereafter referred to as strategy B), use of CDM with study‐specific data (hereafter referred to as strategy C), and use of general CDM (hereafter referred to as strategy D). The main characteristics of the strategies are depicted in Figure 1 and described in detail as follows.
Figure 1.
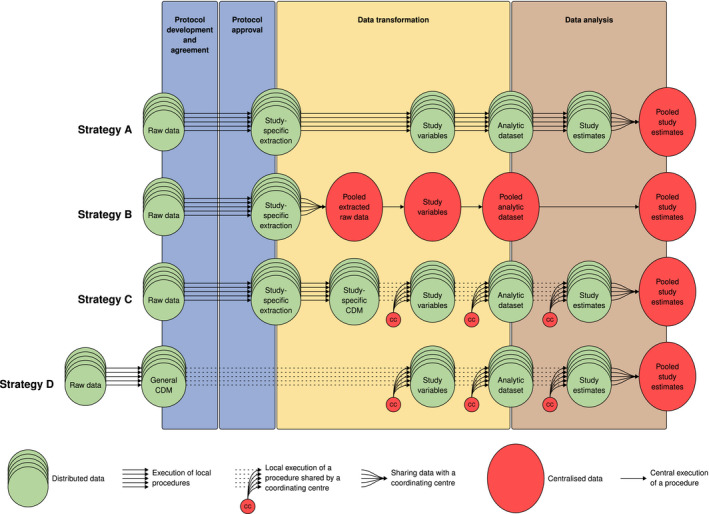
Graphical representation of the four strategies. For simplicity, we did not graphically represent the possibility that in strategies A, C, and D analytic datasets or an aggregated version thereof may be shared. It is intended that the data transformation of raw data into the general CDM in strategy D happens independently of a specific study. CDM, common data model.
Local analyses (strategy A)
In this approach, a protocol is mutually agreed by study partners and data intended to be used for the study are extracted and analyzed locally, with site‐specific programs that are developed individually by each site. No conversion of the source data to a CDM is required. The output of these individual programs may then be transferred to a specific central partner. The output to be shared may be data at a patient level (analytic dataset) or at an aggregate level, or at the level of study estimates, depending on the governance of the network. The PROTECT project 10 , 18 as well as the CNODES project use this approach, although the latter is experimenting with a CDM. 19
Sharing of raw data (strategy B)
In this approach, a mutually agreed protocol is agreed by study partners and raw data intended to be used for the study are locally extracted and transferred without analysis to a specific central partner where the individual site data are pooled and analyzed with programs that are specific for the protocol. Again, no conversion of the data source to a CDM is required. Examples for this approach are collaborative studies based on the registries in the Nordic countries, or on the Italian regional databases, 20 , 21 , 22 or when studies are based on data purchased from more than one vendor (e.g., IQVIA, IBM, or pharmaceutical manufacturers).
CDM with study‐specific data (strategy C)
In this approach, a mutually agreed protocol is agreed by study partners and data intended to be used for the study are locally extracted and loaded into a CDM; data in the CDM are then processed locally in all the sites with one common program, which first creates the study variables from the CDM, then creates the analytical dataset, and possibly proceeds with analysis. 13 The output of the common program is transferred to a specific partner. The output to be shared may be data at a person level (analytic dataset) or at an aggregate level, or at the level of study estimates, depending on the governance of the network. Many examples of studies executed with this approach exist, 23 such as those performed in the following projects: EU‐ADR, 24 SOS, 25 , 26 ARITMO, 27 VAESCO, 28 SOMNIA, 29 EMIF, 16 ADVANCE, 30 and others. 31
General CDM (strategy D)
In this approach, all the local data are extracted and loaded into a CDM, prior to and independent of any study protocol. When a study is required, a protocol is agreed by study partners, and data in the CDM are processed locally in all the sites using one centrally developed common program, which first creates the study variables from the CDM, then creates the analytical dataset, and possibly proceeds with analysis. 13 The output of the common programs is shared, similarly as in the strategy C. To our knowledge, so far, there are no examples of multinational European studies carried out based on this strategy. Examples of initiatives that apply this strategy are the Vaccine Safety Datalink (launched in 1990), 32 the Sentinel initiative (launched in 2008 as MiniSentinel), 33 and PCORnet (launched in 2014) all from the United States, 34 as well as the Observational Health Data Sciences and Informatics community (launched in 2012), which is a global initiative. 35
The technical differences among the strategies are summarized in Table 1 .
Table 1.
Comparison between the four strategies to execute a multi‐database study
| Strategy | First step of the strategy | Data extraction | Conversion to CDM | Data transformation and analysis | Level of data sharing | Examples |
|---|---|---|---|---|---|---|
| A: Local analysis | An agreed study protocol | Each site extracts a dataset specific for the study | Not done | Programmed locally by each site, not necessarily shared by design | Anonymized analytic dataset or aggregated data or final results | CNODES, PROTECT, EU PE, & PV Research network |
| B: Sharing of raw data | As for A | As for A | Not done | Programmed by one site, not necessarily shared by design | Raw extracted data | Collaborative studies on similar countries, or when data are licensed from more than one vendor |
| C: CDM with study‐specific data | As for A | As for A | Once procedures for conversion to a CDM have been programmed, they can be re‐used for subsequent studies to load extracted data into the same CDM | Programmed by one site, existing standard programs can be re‐used, shared with sites | As for A | EU‐ADR, SOS, ARITMO, VAESCO, SOMNIA, EMIF, ADVANCE |
| D: General CDM | Periodically, local data are extracted and loaded to the CDM. An agreed protocol is then the starting point of each individual study. | Each site extracts and converts the entire database periodically; the program that executes the extraction from CDM for each individual study is then programmed centrally | Once procedures for conversion to a CDM have been programmed, they can be re‐used to refresh the data in the CDM | As for C | As for A | Sentinel, OHDSI, no multinational European study completed as yet |
ADVANCE, advancing collaborative vaccine benefits and safety research in Europe; ARITMO, arrhythmogenic potential of drugs; CDM, common data model; CNODES, Canadian Network for Observational Drug Effect Studies; EMIF, European Medical Information Framework; EU PE & PV (European Pharmacoepidemiology & Pharmacovigilance) research network; EU‐ADR, exploring and understanding adverse drug reactions by integrate mining of clinical records and biomedical knowledge; OHDSI, Observational Health Data Sciences and Informatics; PROTECT, Pharmacoepidemiological Research on Outcomes of Therapeutics by a European Consortium; SOMNIA, Influenza Immunization Assessment; SOS, safety of non‐steroidal anti‐inflammatory drugs; VAESCO, Vaccine Adverse Event Surveillance and Communication.
The four strategies are further depicted in Figure 1 .
Potential impact of the four strategies on relevant dimensions
In this section, we describe the potential impact of each of the four strategies on the requirements for an infrastructure to inform regulatory decision making in Europe. We used as a reference the description from a recent keynote speech: “timely access to data that is high quality, is actionable and relevant for benefit‐risk assessment, which supports multiple use cases, is representative of the whole of Europe, of sufficient quality and generated through a transparent methodology and is delivered through a platform with robust data governance.” 36 A comparative description of the potential impact of each strategy on the fundamental ENCePP principles of scientific independence and transparency is summarized in Table 2 .
Table 2.
Potential impact of the four strategies on relevant dimensions
| Strategy | Timeliness | Compliance with data regulations | Data processing quality control | Scientific independence and transparency |
|---|---|---|---|---|
| A: Local analysis |
Time to protocol agreement: full involvement of the local expertise is supported by design. Time to approval of protocol: depends on each site. Time for local extraction: according to local resources. Time to create study variables and analytic dataset: according to local resources. Time to develop analytic procedures: done in all sites. |
Each site has full control on who uses the data and for which purpose |
Data extraction quality: does not allow common data quality checks. Data transformation quality: difficult to check. |
Transparency: is not necessarily ensured by design. Scientific independence: control by local researchers is complete. |
| B: Sharing of raw data |
Time to protocol agreement: full involvement of the local expertise is not ensured by this design, which may speed up the process. Time to approval of protocol: as for A. Time for local extraction: as for A. Time to create study variables and analytic dataset: lengthy when one site needs to harmonize all the different data. Time to develop analytic procedures: depends on whether programs are available or easily adapted, or study‐specific programs must be developed. |
Local sites retain minimum control of data re‐use after data sharing: a specific agreement is needed between data controller and data processors |
Data extraction quality: as for A. Data transformation quality: depends on the quality of a single site, full involvement of the local expertise is not ensured by design. |
Transparency: as for A. Scientific independence: control of local researchers may be minimal, researchers from the coordinating center have control. |
| C: CDM with study specific data |
Time to protocol agreement: as for B. Time to approval of protocol: as for A. Time for local extraction: as for A. Time to load the data into the CDM: depends on whether there is previous experience with the same CDM. Time to create study variables and analytic dataset: depends on whether standard tools are used (fast) or a specific refined strategy is developed (longer). Time to develop analytic procedures: as for B. |
Local sites can require that the type of data to be shared (pseudoanonymized analytic dataset or aggregated data, or final results) complies with their local obligations/laws |
Data extraction quality: checks may apply for each database. Data conversion quality: quality framework can be put in place to ensure accurate data conversion into the CDM. Data transformation quality: can be checked by all the partners. |
Transparency: among partners is partly ensured by design, although not necessarily clear to the external audience. Scientific independence: control of local researchers is high. |
| D: General CDM |
Time to protocol agreement: as for B. Time to approval of protocol: as for A. Time for local extraction: centralized as it builds on the CDM. Time to load the data into the CDM: happens periodically, prior to the study. Time to create study variables and analytic dataset: as for C. Time to develop analytic procedures: as for B. |
As for C, except for some countries where strategy D cannot be applied because data cannot be accessed independently of a protocol |
Data conversion quality: as for C. Data extraction quality: checks may apply centrally. Data transformation quality: as for C. |
Transparency: as for C. Scientific independence: control of local researchers is high, but there is a dependence on a source of funding to extract, transform, and load periodically data into a CDM. |
Timeliness is split between the execution time of the three steps: finalization of the protocol, data transformation, and development of analytic procedures (see Figure 1 ). The time needed for the finalization of the protocol is further split in time to development and time to approval: whereas the latter is not dependent on the strategy, the former may in principle be longer in strategy A, as full involvement of all the partners is supported by design. Time for transformation (local extraction, creation of study variables, and of analytic dataset) is difficult to quantify in strategies A and B, as it depends, for the former, on the local resources and, for the latter, on how complex harmonization of the data transferred to the unique central site is. As for strategies C and D, time for local extraction depends on local resources in strategy C, whereas in strategy D it is optimized, as it builds on the previous extraction to the CDM; and data transformation entails the additional step of transforming and loading the data to the CDM, which in strategy D is carried out prior to any study and, hence, does not increase the study duration, whereas in strategy C it is carried out anew as for each study and the delay is shortened if code from previous studies is reusable. In both strategies C and D, time to create the study variables depends on strategy‐independent choices. Indeed, variables creation may be may be “quick and dirty” if there is urgency to produce preliminary results, because in both strategies the common program running on the CDM may be developed in such a way to create the study variables irrespective of the specific characteristic of the study and of the local data characteristics; or it may take longer when variables creation is study‐specific and database‐tailored, possibly taking into account previous validation studies. Time to develop analytic procedures is not efficient in strategy A, as all the sites are developing their own program. In the other three strategies, where analytic procedure are programmed centrally, time depends on whether programs are available or easily adapted, or study‐specific programs must be developed: many studies conducted in Europe with the strategy C, for instance, have progressively adapted a Java‐based software called Jerboa 27 in order to streamline the process.
Compliance with data privacy regulations is different for all strategies. In strategy A, each site retains total control, so there is no additional impact of the strategy used on data protection obligations, in particular the amount of data that is shared can be tuned to the local data privacy regulations. In strategy B, the impact could be significant, as raw patient‐level data is shared, as the local sites retain minimum control on possible re‐use of shared data. In this case, an agreement between the local data controller and the central data processor is required. In general, raw data contain hundreds of records per person, and even if personal direct identifiers, such as birth dates, are stripped off, potential chances for re‐identification may be high, and it is difficult to argue that sharing this amount of data is necessary to specifically address the research question. On the contrary, an analytic dataset (normally one record per study subject) is tailored to the research question and the chances of re‐identification are often negligible. Based on this, in strategies A, C, and D, the amount of data that is shared can be tuned to the local data privacy regulations; it may range from sharing of the analytic dataset, down to sharing of only study estimates, and hybrid approaches can be used. Finally, in countries where large datasets are tightly controlled by law and processors from different institutions are only allowed to access them based on a study protocol, strategy D, which requires that “all the local data are extracted and loaded into a CDM, prior to and independent of any study protocol,” is only viable when the transformation into the general CDM is executed by the original data owner. This may hamper record linkage between datasets stored with different owners (e.g., different governmental agencies in the same country). Strategies A, B, and C would be easier because data request is based on the specific study by design. Examples of such datasets are registries from Denmark, Finland, Sweden, Norway, and Iceland, 37 , 38 , 39 , 40 , 41 the French national insurance data, 42 or the data from German Social Health Insurances. 43
Quality control of data extraction and transformation is possible in all strategies, but is easier in strategies C and D as working on a CDM allows common standard procedures to be run to check data extraction and transformation quality, and easiest if the CDM adopted by the MDS has its own tradition and a comprehensive program of data quality checks is maintained. Strategies A and B do not natively support data extraction quality checks with a common program, due to the absence of a CDM. In particular, in strategy B, the coordinating center has access to all the extractions and may perform centralized quality checks, whereas in strategy A, even if programs are shared, quality of data transformation and/or analysis is difficult to check, because all the data transformation programs are different.
In strategies A and B, transparency of the data transformation process is not supported by design, because data transformation programs are not necessarily shared across sites. However, in strategy A, blinding of study results between network sites may minimize the chance that post hoc changes to analyses bias study results. 10 Moreover, statistical analysis plans, which are documents having a higher level of specificity than a protocol, may be shared and agreed upon to increase transparency. This can help reducing the risk of small variations, which may have a large impact. 44 In strategies C and D, transparency between sites is supported by design because of the shared and distributed data transformation processes. As for scientific independence of sites, control by local partners is complete in strategy A, high in strategies C and D, and low in strategy B. In strategy D, there is by design a dependence on a source of funding independent of studies, to enable the regular extraction of the source data to the general CDM. Because public funding available in Europe is, as for now, less substantial than, for instance, in the United States, 45 this may warrant scrutiny.
We did not include in this analysis a comparison of costs associated with strategies. Study cost indeed will depend on many factors. A full analysis would need to split costs into study‐specific elements and those associated with any permanent infrastructure required for the strategy. Moreover, for strategies with more infrastructure, the average cost would depend on the study throughput. Such a full analysis is beyond the scope of this paper.
Final thoughts and future perspectives
When organizing an MDS, a strategy should be chosen a priori, considering the study‐specific characteristics and requirements. Based on the seminal classification of strategies illustrated in this commentary, the Working Group 3 of ENCePP has designed a comprehensive research plan to systematically assess which strategies for the conduct of MDS have been usually used in Europe and what was the impact from regulatory perspective based on revision of the EU PAS Register, 46 relevant scientific literature, and publicly available information from European regulatory agencies.
Our final aim is to have a transparent assessment of what is the most suitable approach in Europe to conduct multiple database studies to address different research questions of regulatory interest, an activity which is vital for the benefit of patients in Europe.
Funding
This study was funded by the authors’ institutions.
Conflict of Interests
R.G.’s institution participates in multi‐database studies, which have adopted each of the strategies described in this paper. Some studies are funded by the Innovative Medicines Initiative, Novartis, Eli Lilly, and Daiichi Sankyo, and all are compliant with the ENCePP Code of Conduct for transparency and scientific independence. M.C.J.S. is coordinating and has coordinated/led several EU and global publicly funded projects developing and using common data models to assess medication safety in multiple data sources, these include IMI‐Conception, IMI‐ADVANCE, FP7‐GRIP, FP7‐SOS, FP7‐ARITMO, FP7‐SAFEGUARD, ECDC‐VAESCO, CDC‐SOMNIA, FP‐7 EU‐ADR, and FP‐6 TEDDY. She is/was also principal investigator in several multi‐database postauthorization safety studies required or commissioned by the EMA, sponsored by Novartis, Servier, and GSK, and all are compliant with the ENCePP Code of Conduct for transparency and scientific independence. G.R. works as scientific consultant for Agenzia regionale di sanità della Toscana, which participates in different multi‐database studies. Some studies are funded by the Innovative Medicines Initiative, Novartis, Eli Lilly, and Daiichi Sankyo, and all are compliant with the ENCePP Code of Conduct for transparency and scientific independence. T.S. is working at an independent, nonprofit research institute, the Leibniz Institute for Prevention Research and Epidemiology—BIPS. Unrelated to this paper, BIPS occasionally conducts studies financed by the pharmaceutical industry. Almost exclusively, these are post‐authorization safety studies requested by health authorities. All studies are compliant with the ENCePP Code of Conduct for transparency and scientific independence. G.T. coordinates an academic team at University of Messina that received in the last 2 years unconditional grants for the conduct of observational studies from Daiichi Sankyo, AstraZeneca, PTC Therapeutics, and Amgen. All other authors declared no competing interests for this work.
Disclaimer
The views expressed in this paper are the personal views of the author(s) and may not be understood or quoted as being made on behalf of or reflecting the position of the regulatory agency/agencies or organizations with which the author(s) is/are employed/affiliated.
Acknowledgments
The authors thank Olaf Klungel and the Working Group Health Data of TEDDY, the European Network of Excellence for Paediatric Clinical Research, for useful comments. The manuscript has been reviewed and endorsed by the ENCePP Steering Group.
References
- 1. Skovlund, E. , Leufkens, H.G.M. & Smyth, J.F. The use of real‐world data in cancer drug development. Eur. J. Cancer 101, 69–76 (2018). [DOI] [PubMed] [Google Scholar]
- 2. Jarow, J.P. , LaVange, L. & Woodcock, J. Multidimensional evidence generation and FDA regulatory decision making. Defining and using "real‐world" data. JAMA 318, 703–704 (2017). [DOI] [PubMed] [Google Scholar]
- 3. Sharrar, R.G. & Dieck, G. Monitoring product safety in the postmarketing environment. Ther. Adv. Drug Saf. 4, 211–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pitts, P.J. , Louet, H.L. , Moride, Y. & Conti, R.M. 21st century pharmacovigilance: efforts, roles, and responsibilities. Lancet Oncol. 17, e486–e492 (2016). [DOI] [PubMed] [Google Scholar]
- 5. Klungel, O.H. et al. Multi‐centre, multi‐database studies with common protocols: lessons learnt from the IMI PROTECT project. Pharmacoepidemiol. Drug Saf. 25 (Suppl. 1), 156–165 (2016). [DOI] [PubMed] [Google Scholar]
- 6. Arlett, P. et al. Proactively managing the risk of marketed drugs: experience with the EMA Pharmacovigilance Risk Assessment Committee. Nat. Rev. Drug Discovery 13, 395–397 (2014). [DOI] [PubMed] [Google Scholar]
- 7. Kurz, X. & Perez‐Gutthann, S. ENCePP Steering Group. Strengthening standards, transparency, and collaboration to support medicine evaluation: ten years of the European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP). Pharmacoepidemiol. Drug Saf. 27, 245–252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Engel, P. , Almas, M.F. , De Bruin, M.L. , Starzyk, K. , Blackburn, S. & Dreyer, N.A. Lessons learned on the design and the conduct of post‐authorization safety studies: review of 3 years of PRAC oversight. Br. J. Clin. Pharmacol. 83, 884–893 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carroll, R. , Ramagopalan, S.V. , Cid‐Ruzafa, J. , Lambrelli, D. & McDonald, L. An analysis of characteristics of post‐authorisation studies registered on the ENCePP EU PAS Register. Version 2. F1000Res. 6, 1447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Platt, R.W. , Platt, R. , Brown, J.S. , Henry, D.A. , Klungel, O.H. & Suissa, S. How pharmacoepidemiology networks can manage distributed analyses to improve replicability and transparency and minimize bias. Pharmacoepidemiol Drug Saf. 29, 3–7 (2020). [DOI] [PubMed] [Google Scholar]
- 11. Schneeweiss, S. , Brown, J. , Bate, A. , Trifirò, G. & Bartels, D. Choosing among common data models for real world data analyses fit for making decisions about the effectiveness of medical products. Clin. Pharmacol. Ther. 107, 827–833 (2020). [DOI] [PubMed] [Google Scholar]
- 12. The European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP) . Guide on methodological standards in pharmacoepidemiology (Revision 7) <http://www.encepp.eu/standards_and_guidances/methodologicalGuide.shtml> (July 2018). Accessed February 2020.
- 13. Gini, R. et al. Data Extraction And Management In Networks Of Observational Health Care Databases For Scientific Research: A Comparison Among EU‐ADR, OMOP, Mini‐Sentinel And MATRICE Strategies. EGEMS (Wash DC) 4, 1189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hripcsak, G. , Levine, M.E. , Shang, N. & Ryan, P.B. Effect of vocabulary mapping for conditions on phenotype cohorts. J. Am. Med. Inform. Assoc. 25, 1618–1625 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gini, R. et al. Quantifying outcome misclassification in multi‐database studies: the case study of pertussis in the ADVANCE project. Vaccine. 10.1016/j.vaccine.2019.07.045. [e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 16. Roberto, G. et al. Identifying cases of type 2 diabetes in heterogeneous data sources: strategy from the EMIF Project. PLoS One 11, e0160648 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schuemie, M.J. , Sen, E. , 't Jong, G.W. , Soest, E.M. , Sturkenboom, M.C. & Kors, J.A. Automating classification of free‐text electronic health records for epidemiological studies. Pharmacoepidemiol. Drug Safety 21, 651–658 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Reynolds, R.F. et al. The IMI PROTECT project: purpose, organizational structure, and procedures. Pharmacoepidemiol Drug Saf. 25 (suppl. 1), 5–10 (2016). [DOI] [PubMed] [Google Scholar]
- 19. Sketris, I.S. , Carter, N. , Traynor, R.L. , Watts, D. & Kelly, K. Building a framework for the evaluation of knowledge translation for the Canadian Network for Observational Drug Effect Studies. Pharmacoepidemiol. Drug Saf. 29 (S1), 8–25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Furu, K. et al. Selective serotonin reuptake inhibitors and venlafaxine in early pregnancy and risk of birth defects: population based cohort study and sibling design. BMJ 350, h1798 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ingrasciotta, Y. et al. How much are biosimilars used in clinical practice? A retrospective Italian population‐based study of erythropoiesis‐stimulating agents in the years 2009–2013. BioDrugs 29, 275–284 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sultana, J. et al. All‐cause mortality and antipsychotic use among elderly persons with high baseline cardiovascular and cerebrovascular risk: a multi‐center retrospective cohort study in Italy. Expert Opin. Drug Metab. Toxicol. 15, 179–188 (2019). [DOI] [PubMed] [Google Scholar]
- 23. Trifirò, G. et al. Combining multiple healthcare databases for postmarketing drug and vaccine safety surveillance: why and how? J. Intern. Med. 275, 551–561 (2014). [DOI] [PubMed] [Google Scholar]
- 24. Coloma, P.M. et al. Combining electronic healthcare databases in Europe to allow for large‐scale drug safety monitoring: the EU‐ADR Project. Pharmacoepidemiol. Drug Saf. 20, 1–11 (2011). [DOI] [PubMed] [Google Scholar]
- 25. Valkhoff, V.E. et al. Safety of non‐steroidal anti‐inflammatory drugs (SOS) project. Population‐based analysis of non‐steroidal anti‐inflammatory drug use among children in four European countries in the SOS project: what size of data platforms and which study designs do we need to assess safety issues? BMC Pediatr. 13, 192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arfe, A. et al. Non‐steroidal anti‐inflammatory drugs and risk of heart failure in four European countries: nested case‐control study. BMJ 354, i4857 (2016). [DOI] [PubMed] [Google Scholar]
- 27. Trifirò, G. et al. Use of azithromycin and risk of ventricular arrhythmia. CMAJ 189, E560–E568 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dieleman, J. , Romio, S. , Johansen, K. , Weibel, D. , Bonhoeffer, J. & Sturkenboom, M. Guillain‐Barre syndrome and adjuvanted pandemic influenza A (H1N1) 2009 vaccine: multinational case‐control study in Europe. BMJ 343, d3908 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dodd, C.N. et al. Incidence rates of narcolepsy diagnoses in Taiwan, Canada, and Europe: the use of statistical simulation to evaluate methods for the rapid assessment of potential safety issues on a population level in the SOMNIA study. PLoS One 13, e0204799 ( 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Becker, B.F.H. et al. CodeMapper: semiautomatic coding of case definitions. A contribution from the ADVANCE project. Pharmacoepidemiol. Drug Saf. 26, 998–1005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Coloma, P.M. et al. Risk of cardiac valvulopathy with use of bisphosphonates: a population‐based, multi‐country case‐control study. Osteoporos Int. 27, 1857–1867 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Geier, D.A. , Kern, J.K. & Geier, M.R. Increased risk for an atypical autism diagnosis following Thimerosal‐containing vaccine exposure in the United States: a prospective longitudinal case‐control study in the VaccineSafety Datalink. J. Trace Elem. Med. Biol. 42, 18–24 (2017). [DOI] [PubMed] [Google Scholar]
- 33. Ball, R. , Robb, M. , Anderson, S.A. & Dal Pan, G. The FDA's sentinel initiative–a comprehensive approach to medical product surveillance. Clin. Pharmacol. Ther. 99, 265–268 (2016). [DOI] [PubMed] [Google Scholar]
- 34. Wiese, A.D. et al. Performance of a computable phenotype for identification of patients with diabetes within PCORnet: The Patient‐Centered Clinical Research Network. Pharmacoepidemiol Drug Saf. 28, 632–639 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hripcsak, G. et al Observational Health Data Sciences and Informatics (OHDSI): opportunities for observational researchers. Stud. Health Technol. Inform. 216, 574–578 (2015). [PMC free article] [PubMed] [Google Scholar]
- 36. Cave, A. Keynote session: opportunities for ‘Big Data’ in medicines development and regulatory science. 34th International Conference on Pharmacoepidemiology & Therapeutic Risk Management. Prague <https://webed.pharmacoepi.org/products/34th‐icpe‐keynote‐session‐opportunities‐for‐big‐data‐in‐medicines‐development‐and‐regulatory‐science> (2018).
- 37. Wettermark, B. et al. The Nordic prescription databases as a resource for pharmacoepidemiological research—a literature review. Pharmacoepidemiol. Drug Saf. 22, 691–699 (2013). [DOI] [PubMed] [Google Scholar]
- 38. Schmidt, M. et al. The Danish health care system and epidemiological research: from health care contacts to database records. Clin. Epidemiol. 11, 563–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kilpeläinen, K. et al. Finnish experiences of health monitoring: local, regional, and national data sources for policy evaluation. Global Health Action 9, 28824 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. How to use register‐based research <https://www.registerforskning.se/en/>. Accessed February 2020.
- 41. Norwegian Center for Research Data . Individual level data <https://nsd.no/nsd/english/index.html>. Accessed February 2020.
- 42. Bezin, J. et al. The national healthcare system claims databases in France, SNIIRAM and EGB: powerful tools for pharmacoepidemiology. Pharmacoepidemiol. Drug Saf. 26, 954–962 (2017). [DOI] [PubMed] [Google Scholar]
- 43. Bundesrepublik Deutschland . Zehntes Buch Sozialgesetzbuch ‐ Sozialverwaltungsverfahren und Sozialdatenschutz <https://dejure.org/gesetze/SGB_X>.
- 44. Izem, R. , Huang, T.‐Y. , Hou, L. , Pestine, E. , Nguyen, M. & Maro, J.C. Quantifying how small variations in design elements affect risk in an incident cohort study in claims. Pharmacoepidemiol. Drug Saf. 29, 84–93 (2020). [DOI] [PubMed] [Google Scholar]
- 45. Gini, R. et al. The ENCePP Code of Conduct: a best practise for scientific independence and transparency in noninterventional postauthorisation studies. Pharmacoepidemiol. Drug Saf. 28, 422–433 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. EU PAS Register <http://www.encepp.eu/encepp/studiesDatabase.jsp>. Accessed February 2020.