

SUMMARY
We have previously proposed that selective inheritance, the limited transmission of damaging mtDNA mutations from mother to offspring, is based on replication competition in Drosophila melanogaster. This model, which stems from our observation that wild-type mitochondria propagate much more vigorously in the fly ovary than mitochondria carrying fitness-impairing mutations, implies that germ cells recognize the fitness of individual mitochondria, and selectively boost the propagation of healthy ones. Here, we demonstrate that the protein kinase PINK1 preferentially accumulates on mitochondria enriched for a deleterious mtDNA mutation. PINK1 phosphorylates Larp to inhibit protein synthesis on the mitochondrial outer membrane. Impaired local translation on defective mitochondria in turn limits the replication of their mtDNA, and hence the transmission of deleterious mutations to the offspring. Our work confirms that selective inheritance occurs at the organelle level during Drosophila oogenesis, and provides molecular entry points to test this model in other systems.
INTRODUCTION
The mitochondrial genome (mtDNA) follows unusual genetic rules that feature polyploidy, random segregation and maternal inheritance (Wallace, 2008). Due to the mitochondrial matrix’s highly-mutagenic environment and lack of efficient repair mechanisms, mtDNA is particularly prone to accumulating mutations (Pesole et al., 1999). Paradoxically, mtDNA mutations are exceedingly rare in populations (Vermulst et al., 2007), suggesting that deleterious mtDNA mutations are somehow eliminated in the germline. In mammals, mtDNA undergoes a genetic bottleneck during oogenesis, through which only a subpopulation of mtDNA is transferred from primordial germ cells to future oocytes (Chinnery et al., 2000). This bottleneck promotes the random segregation of mtDNA to individual oocytes and could lead to the elimination of oocytes inheriting a high proportion of mutant mtDNAs by simple Darwinian selection (Stewart and Larsson, 2014). However, the frequency of spontaneous mtDNA mutations, estimated at around 10−6 (Vermulst et al., 2007), is too low to elicit the kind of cell-level deficiencies on which Darwinian selection could act. Therefore, bottleneck inheritance and selection at the cell level may not be effective, or at least not sufficient, in preventing accumulation of mtDNA mutations over generations. Even when present at low level, mtDNA mutations are effectively prevented from passing through the female germline in humans and various animal models (Fan et al., 2008; Floros et al., 2018; Hill et al., 2014; Ma et al., 2014; Stewart et al., 2008). It is therefore likely that a potent selection system acts at the organelle level during oogenesis. However, the mechanism for this selection remains mostly mysterious.
Our previous work in Drosophila melanogaster (Dm) showed that mtDNA replication relies on mitochondrial respiratory activity during oogenesis (Hill et al., 2014). This observation led us to propose a replication-competition model whereby healthy organelles harboring wild-type genomes propagate more vigorously and outcompete defective ones afflicted by damaging mutations (Hill et al., 2014). While replication-competition provides a logically compelling mechanism for the elimination of unfit organelles, it does not explain how germ cells can discern the fitness of individual mitochondria and selectively stimulate the proliferation of healthy ones. Here we provide an answer to these remaining questions.
Oogenesis in Dm is marked by a prodigious increase in mitochondrial mass and mtDNA copy number (Hurd et al., 2016), much of which relies on protein translation at the mitochondrial outer membrane (Zhang et al., 2016). Mitochondria only encode a small subset of their own proteins, and this massive growth requires intense translation of mitochondrial proteins encoded in the nucleus. We have found that during Dm oogenesis, a large subset of nuclear-encoded mitochondrial proteins, including most mitochondrial ribosomal proteins, components of the electron transport chains (ETCs) and several factors involved in mtDNA replication and expression, are synthesized on the mitochondrial outer-membrane (Zhang et al., 2016). This local translation is thought to coordinate the activities of nuclear and mitochondrial genomes and facilitate the import of newly-synthesized mitochondrial proteins, which in turn increases the efficiency of mitochondrial biogenesis (Zhang et al., 2016). Furthermore, the mitochondrial outer-membrane is now recognized as a major platform for the regulation of various signaling pathways (Tait and Green, 2012). It also serves as the interface for communication between the mitochondrial compartment and the nuclear-cytoplasmic environment (Zhang and Xu, 2016). Thus, it seems quite plausible that oocytes would use components of the outer mitochondrial membrane, including those involved in local translation, to select against defective mitochondria.
Local translation at the mitochondrial outer membrane is partially mediated by the AKAP1 protein MDI, which recruits Larp, a translation stimulator, to mitochondria in Dm oocytes (Zhang et al., 2016). In mdi mutants, Larp diffuses to the cytoplasm, and both local translation and mtDNA replication are drastically reduced in the ovary (Zhang et al., 2016). As a result, mitochondria and mtDNA numbers are severely reduced in the eggs, which fail to progress through embryogenesis (Zhang et al., 2016). These observations demonstrate an essential role for local translation in mitochondrial biogenesis and mtDNA inheritance, and make MDI and Larp logical targets of our investigation into selective inheritance.
What remains mysterious is the mechanism that distinguishes between healthy and defective mitochondria during oogenesis. A well-known sensor of mitochondrial fitness in cultured cells is PTEN-induced kinase 1 (PINK1) (Pickrell and Youle, 2015). PINK1 is rapidly degraded in healthy mitochondria but selectively stabilized on the outer membrane of depolarized mitochondria to initiate mitophagy, the autophagic removal of mitochondria (Pickrell and Youle, 2015). While mitophagy can promote the reduction of mtDNA mutations in somatic tissues and cultured cells (Kandul et al., 2016; Suen et al., 2010), Parkin, the ubiquitin ligase downstream of PINK1 in the canonical mitophagy pathway, is not required for selective inheritance in the Dm female germline (Ma et al., 2014). However, PINK1 is versatile and involved in several processes unrelated to mitophagy, including promoting mitochondrial fission and arresting mitochondrial mobility (Ashrafi and Schwarz, 2013). Given PINK1’s many roles in mitochondrial biology and association with the mitochondrial outer membrane in somatic cells, we decided to test its potential role in selective mitochondrial inheritance during Dm oogenesis.
Here we report that PINK1 preferentially accumulates on mitochondria enriched in a deleterious mtDNA mutation, and is required for selective inheritance of mitochondria during Dm oogenesis. PINK1 phosphorylates Larp to inhibit local translation on defective mitochondria selectively. This regulation allows germ cells to specifically hinder the propagation of defective organelles afflicted by damaging mutations, thereby restricting their transmission to the next generation.
RESULTS
PINK1 Is Upregulated in Oocytes Carrying a mtDNA Mutation and Preferentially Localizes to Mitochondria Enriched for the Mutation.
PINK1 selectively accumulates on the surface of depolarized mitochondria in cultured cells treated with metabolic inhibitors that dissipate the mitochondrial membrane potential (Narendra et al., 2010). However, it is not clear whether PINK1 can recognize mitochondria with an energy defect stemming from a mtDNA mutation in vivo. To address this question, we took advantage of a temperature-sensitive lethal mutation, mt:CoIT300I (mt-ts), that maps to the cytochrome-C-oxidase-subunit-one (CoI) locus on the mtDNA (Hill et al., 2014). At a non-permissive temperature of 29 °C, the CoI subunit loses its heme A cofactor (Chen et al., 2015), which completely abolishes the activity of cytochrome C oxidase (Hill et al., 2014). To assess whether the mt-ts mutation affected mitochondrial polarization, we co-stained ovaries with TMRM and MitoTracker-green, fluorescent markers for mitochondrial membrane potential and mitochondrial mass, respectively (Lemasters and Ramshesh, 2007). The ratio of TMRM to MitoTracker fluorescence was much lower in mt-ts than in ovaries carrying wild type mtDNA (mt-wt) at 29 °C (Figure 1A and 1B), based on a ratiometric imaging assay described previously (Higuchi-Sanabria et al., 2016). However, mitochondrial morphology in mt-ts ovaries appeared normal compared to that in mt-wt (Figure S1). These observations demonstrate that mt-ts mitochondria are depolarized at restrictive temperature, and raise the possibility that they could be marked by PINK1 protein.
Figure 1. Homoplasmic mt-ts Ovaries Have Reduced Membrane Potential and Increased PINK1 Protein Level.

(A) Confocal images of egg chambers from mt-wt and homoplasmic mt-ts mutant flies grown at 29°C. Ovaries were stained alive with TMRM, a marker of mitochondrial membrane potential, and Mito-Tracker green (MTgreen), an indicator of mitochondrial mass. The ratio of TMRM to MTgreen are generated with Image J. Germ cells were outlined in dashed circles. Bar: 10 μm.
(B) Quantification of the ratios of TMRM to MTgreen in germ cells (dashed circles) of mt-wt (n = 24) and homoplasmic mt-ts mutant egg chambers (n = 24) in (A), showing a clear reduction in membrane potential in the mutant.
(C) Western blot analysis showing that PINK1 is upregulated, whereas MDI and Larp proteins are not altered, in mt-ts (ts) ovaries compared with mt-wt (wt) at 29°C. There is no dramatic difference in PINK1 levels between wt and ts ovaries at 18°C. VDAC and Actin are used as loading control for mitochondrial mass and total cellular proteins, respectively.
To follow PINK1 in ovaries, we inserted a gfp cDNA in-frame with the pink1 ORF at the endogenous locus, using CRISPR-mediated recombination. Homozygous pink1-gfp flies were viable and healthy, indicating that the fusion protein behaves as the native PINK1. In mt-wt ovaries, a relatively low-level of GFP signal was detected and co-localized with Tom20-mCherry (Zhang et al., 2016), a mitochondrial reporter (Figure 2A), confirming that PINK1-GFP localizes to mitochondria properly. Moreover, PINK1-GFP protein level was markedly increased in mt-ts ovaries compared to mt-wt ovaries at 29 °C, or to mt-wt and mt-ts ovaries at a permissive temperature of 18°C (Figure 1C), suggesting that the amount of PINK1-GFP protein is inversely correlated with mitochondrial fitness in vivo.
Figure 2. PINK1 Accumulates on Mitochondria Enriched for the mt-ts Genome in the Ovary of Heteroplasmic Flies.

(A) Confocal (left-two panels) and ratiometric images (right-two panels) of mid-stage egg chambers from mt-wt flies and heteroplasmic mt-ts (hetero.) flies expressing both PINK1-GFP and Tom20-mCherry at 29°C. Enlarged view of boxed areas shows PINK1-GFP colocalizing with Tom20-mCherry in mt-wt and heteroplasmic flies at 29°C. Ratiometric images (GFP/mCherry) show that PINK1-GFP level is elevated in heteroplasmic ovaries compared to mt-wt ovaries at 29°C. Enlarged view of boxed areas shows mitochondria (arrows) with much higher GFP/mCherry ratio than neighboring mitochondria in heteroplasmic flies at 29°C. Bars: 10 μm.
(B) Confocal imaging of two mitochondrial populations purified from pink1-GFP;; Tom20-mcherry (heteroplasmic) ovaries and sorted on the basis of GFP and mCherry intensity. Merged images are overlay of GFP and mCherry fluorescence with DIC channel. Note that GFP-high and GFP-low populations have similar level of Tom20-mCherry signal and are comparable in size. Bar: 1 μm.
(C) Mitochondria in two sorted populations have similar forward scatter (FSC-H) distribution, indicating they are comparable in size.
(D) Proportion of mt-ts genome in two sorted mitochondrial populations. High and low-GFP mitochondrial fractions from the same ovary are connected with a dotted line (n = 12). Note that the proportion of mt-ts genome is always higher in the high-GFP mitochondria fraction than the low-GFP fraction recovered from the same ovary.
In heteroplasmic ovaries containing both mt-wt and mt-ts (Hill et al., 2014), the ratio of PINK1-GFP to Tom20-mCherry fluorescence showed marked variation among different mitochondria at 29°C (Figure 2A). Some mitochondria had a much higher ratio than neighboring mitochondria in the same egg chamber or than mitochondria in wild-type ovaries. One may expect mitochondria in the heteroplasmic ovaries to have distinct fitness depending on their mtDNA composition. We therefore wondered whether PINK1-GFP had preferentially accumulated on mitochondria enriched in mt-ts mtDNA. To test this idea, we attempted to correlate PINK1-GFP protein level with mtDNA composition in heteroplasmic ovaries. Using fluorescence-based particle sorting, we recovered two mitochondrial populations with distinct PINK1-GFP fluorescence intensity but similar Tom20-mCherry intensity from extracts of heteroplasmic ovaries (Figure S2). Similar Tom20-mCherry intensity suggests similar mitochondria sizes. Indeed, forward scatter (FSC), which is proportional to the size of objects in FACS analysis, was similar in both populations (Figure 2C). Furthermore, mitochondria from these two populations indeed appeared comparable in sizes based on confocal imaging (Figure 2B). We are therefore confident that the difference in GFP signal between the two populations reflects different amounts of PINK1-GFP at the organelles’ surface, rather than differences in organelle sizes. Of primary significance, the proportion of mt-ts genomes was significantly higher in mitochondria with the higher GFP signal (81±8% vs 60±9%, Figure 2D), indicating that PINK1 preferentially accumulates on mitochondria enriched in mt-ts genomes.
mtDNA Selective Inheritance Is Impaired in pink1B9 Mutant Dm.
As the results above suggested that PINK1 marks defective mitochondria in vivo in oocytes, we next asked whether PINK1 was required for their counter-selection in the germline. To that end, we bred pink1B9, a mutation in the nuclear-encoded pink1 gene (Clark et al., 2006), into a heteroplasmic background combining both mt-ts and mt-wt genomes, and quantified heteroplasmy in the resulting mothers and their eggs. Under the restrictive temperature, eggs from mothers that did not carry the pink1 mutation had a smaller proportion of mt-ts genomes than their mothers did (wild type nuclear background, Figure 3A): on average, the proportion of mt-ts genomes decreased by 20% in the eggs, indicating a clear selection against this deleterious mtDNA mutation, as expected from previous results (Hill et al., 2014). By contrast, eggs from pink1 mutant mothers displayed a random mtDNA segregation pattern, with no observable overall decrease in the proportion of mt-ts genomes (Figure 3A). This result, consistent with a lack of counter-selection of the mutant genomes, demonstrates that PINK1 is indeed required for selective inheritance of mtDNA.
Figure 3. mtDNA Selective Inheritance is Impaired in pink1B9 and mdi1 Flies.

(A and B) Effect of pink1B9 (A) or mdi1 (B) mutations on mt-ts transmission from females to eggs. w1118 (wt, n = 14) and pink1B9 (n = 12) females (A), or w1118 (wt, n = 12), mdi1 (n = 11) females flies (B) with heteroplasmic mtDNA background consisting of mt-wt and mt-ts were cultured at 29 °C. The abundance of mt-ts in mothers and their eggs was quantified, and changes expressed as the ratio (in percentage) of abundance in the eggs to abundance in the mothers. Note that all eggs from wt flies have a reduced proportion of mt-ts genomes compared to their mothers, while in pink1B9 (A) or mdi1 flies (B), the pattern of transmission is random. Error bars represent standard deviation.
(C) Western blot showing that PINK1 levels are comparable between wt and mdi1 ovaries, whereas MDI level is sharply reduced in the mdi1 mutant. Actin is used as loading control.
(D) Representative images of egg chambers from mt-wt and heteroplasmic mt-ts flies expressing mito-GFP and mCherry-ATG8 (UASp-mitoGFP/UASp-mCherry-ATG8; nos-gal4) grown at 29 °C. mCherry-ATG8 puncta label autophagosomes (arrows). ATG8 puncta with more than 25% of their area overlapping with mitochondria are considered as mitophagosomes (arrowheads); arrows label other autophagosomes. Bars: 10 μm.
(E) Number of ATG8 puncta (total) and mitophagosomes (mito) in mt-wt and mt-ts heteroplamic (hetero.) ovaries. Puncta were quantified over 105-pixel egg-chamber areas in wild type (mt-wt, n = 8 ovaries) and heteroplasmic ovaries (hetero., n = 8 ovaries) and the number of total puncta (total) or mitophagic puncta (mito) plotted. Sizes of egg chambers range from 1.8 x105 to 3.1x105 pixels. Note that neither autophagy nor mitophagy is increased in the heteroplasmic background (p > 0.05).
(F) Area of total ATG8 puncta normalized to the area of whole egg chamber (total) and area of mitophagosomes normalized to the area of mitochondria (mito) in mt-wt (n = 8) and heteroplasmic ovaries (hetero., n = 8). Note that the density of total phagosomes and the density of mitophagosomes are similar in mt-wt and mt-ts heteroplasmic (hetero.) backgrounds, (p > 0.05), indicating that neither autophagy nor mitophagy are affected by the heteroplasmic mutation.
Autophagy does not Increase in Heteroplasmic mt-ts Ovaries
PINK1 is known to trigger autophagy of defective mitochondria, in a pathway that also includes Parkin (Pickrell and Youle, 2015). While the canonical mitophagy mediated by PINK1-Parkin is not required for selective inheritance (Ma et al., 2014), it is still possible that a Parkin-independent mitophagy pathway might be involved (Lim and Lim, 2017). To assess the level of mitophagy in heteroplasmic ovaries, we combined a heteroplasmic mt-ts background with a nuclear transgene expressing the mCherry reporter fused to ATG8 (Nakatogawa et al., 2007), a marker of autophagosomes. In both mt-wt and heteroplasmic ovaries, the mCherry-ATG8 fusion protein appeared as a diffuse, low-level signal throughout the cytoplasm, with a few bright puncta of various sizes corresponding to autophagosomes (Figure 3D). A small fraction of these puncta co-localized with mitochondria in both mt-wt and heteroplasmic mt-ts ovaries (Figure 3D), which may represent base-level mitochondrial turnover. Overall, the total amount of ATG8 puncta associated with mitochondria, as well as the mitochondrial area containing ATG8 puncta, were comparable in mt-wt and mt-ts heteroplasmic backgrounds (Figure 3E and 3F), indicating that mitophagy does not increase in heteroplasmic flies. We hence conclude that PINK1 restricts the transmission of mtDNA mutations through a process unrelated to mitophagy.
Targeting PINK1 to the Outer Membrane Mimics Loss-of-function Mutations in the MDI/Larp Pathway
If PINK1 does not mark defective oocyte mitochondria for elimination by mitophagy, it may regulate selective inheritance through other processes occurring at the mitochondrial surface. A logical candidate is local translation, the synthesis of nuclear-encoded mitochondrial proteins that drives mitochondrial biogenesis and mtDNA replication in ovaries (Zhang et al., 2016). However, the importance of local translation for selective inheritance has not yet been demonstrated.
Local translation on the mitochondrial outer membrane requires the nuclear-encoded AKAP protein MDI (Zhang et al., 2016). Mutating mdi impairs local translation and leads to severely reduced mtDNA replication in ovaries (Zhang et al., 2016). To test whether MDI was also important for selective inheritance of mtDNA, we bred a mdi mutation, located on the nuclear genome, into the heteroplasmic mt-ts background and examined mt-ts transmission from mothers to eggs. Selection against mt-ts mtDNA was completely abolished in the mdi mutant (Figure 3B and 3C), demonstrating an essential role for local translation in mtDNA selection.
We then asked whether PINK1 impinges on local translation at the outer membrane. To test this idea, we examined local translation in the presence of a Tom20-PINK1 fusion protein that is constitutively anchored to the outer mitochondrial membrane in ovaries. We assayed local translation by western blot after a brief chase with L-Azidohomoalanine (AHA), a methionine analog (Zhang et al., 2016). AHA incorporation associated with the mitochondrial fraction, which is predominantly carried out by cytosolic ribosomes (Zhang et al., 2016), was evidently reduced in Tom20-PINK1 ovaries compared to control ovaries (Figure 4A and 4B). These observations suggest that accumulation of PINK1 at the outer membrane inhibits local translation.
Figure 4. PINK1 Overexpression Inhibits Local Translation and Impairs mtDNA Replication in Ovaries.

(A) Western blot analyses of nascent protein synthesis labeled by AHA incorporation and detected by anti-biotin antibody in the mitochondrial fraction of control ovaries (ctrl.) and ovaries expressing Tom20-PINK1 (TP). Tom20 was used as a loading control.
(B) Quantification of nascent protein synthesis normalized to the Tom20 band in 3 independent experiments. Error bars represent standard deviation.
(C) Hatching rate and mtDNA content are greatly reduced in eggs laid by Tom20-PINK1-expressing (TP) flies compared to control eggs. N=3 X100 eggs/genotype for hatching rate. The relative mtDNA level was determined as the average of three biological repeats normalized to control. Error bars represent standard deviation.
(D) Confocal images of control and Tom20-PINK1-expressing (TP) ovaries that were labeled with EdU to monitor mtDNA replication. Bars: 10 μm.
(E) Area of EdU puncta (pixels) normalized to total pixels in eggs chambers of control (N = 10) and Tom20-PINK1 (N = 10) ovaries. Error bars represent standard deviation.
To further examine PINK1’s impact on the local translation of mitochondrial proteins, we analyzed the proteome of eggs laid by Tom20-PINK1 and wild-type flies. A total of 4220 proteins, including 645 mitochondrial proteins, were detected in both genotypes (Figure 5A and 5B). 136 mitochondrial proteins were reduced in Tom20-PINK1 eggs compared to wild type (Figure 5C). Mitochondrial ribosomal proteins (mRPs) and ETCs proteins were most significantly impacted (Figure 5A-5C). When we compared the targets of Tom20-PINK1 and of MDI-Larp (Zhang et al., 2016), we found a notable overlap. In fact, more than 70% of the mRPs and ETC proteins that were reduced in Tom20-PINK1 eggs were also reduced in the mdi mutant (Figure 5C). Additionally, mtDNA replication in Tom20-PINK1 ovaries was reduced (Figures 4D and 4E), as were the mtDNA content and hatching rate of the resulting eggs (Figure 4C). All these defects were also observed in mdi mutants (Zhang et al., 2016). This remarkable overlap between Tom20-PINK1 and mdi mutant phenotypes supports the idea that PINK1 inhibits MDI/Larp-mediated local translation and mtDNA replication.
Figure 5. PINK1 and MDI/Larp Share Mitochondrial Protein Targets.
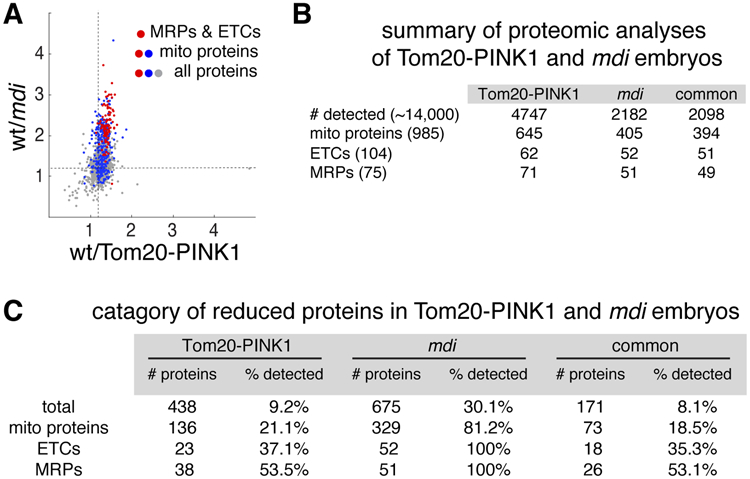
(A) Scatter plot of a total of 2098 proteins detected in both Tom20-PINK1 and mdi1 eggs (Zhang et al., 2016), showing the proteins’ abundance ratios in wt vs Tom20-PINK1 egg extracts (X-axis) and wt vs mdi1 egg extracts (Y-axis). Note that most proteins reduced in Tom20-PINK1 eggs are also reduced in mdi1 eggs, although the reduction is more pronounced in mdi1 eggs than in Tom20-PINK1 eggs. Dashed lines mark the 1.2 ratio. Mitochondrial proteins (blue), particularly mitochondrial ribosomal proteins and ETC subunits (red), are among the most severely reduced.
(B) Summary of proteomic analyses of Tom20-PINK1-expressing and mdi mutant eggs. The number of recovered proteins in each background and the number of proteins common to both (common) are shown. Total numbers of annotated proteins, mitochondrial proteins, mitochondrial ribosomal proteins and electron transport chain complex subunits in Drosophila melanogaster genome are shown in parentheses.
(C) Category of proteins reduced by more than 1.2-fold in Tom20-PINK1-expressing and mdi mutant eggs compared to wild type eggs. Absolute numbers of reduced proteins (# of proteins) and the ratio relative to the numbers of proteins detected in each category (% detected) are shown (P < 0.05 and FDR < 0.05). Note that most reduced proteins in Tom20-PINK-expressing and mdi mutant eggs are mitochondrial ribosomal proteins (MRPs) and subunits of electron transport chain complexes (ETCs).
Constitutively targeting PINK1 to the outer membrane could also deplete mitochondrial contents by triggering non-selective and excessive mitophagy (Narendra et al., 2010), which potentially could contribute to the phenotypes observed in Tom20-PINK1 ovaries. However, we did not observe increased mitophagy in Tom20-PINK1 ovaries compared to wild-type controls, based on mCherry-ATG8 imaging assay (Figure S3A and S3B). This result is also consistent with a previous study showing that anchoring PINK1 to the outer membrane is not sufficient to trigger mitophagy in Dm photoreceptor cells (Zhuang et al., 2016). We hence conclude that mitophagy does not play any role in the phenotype of the constitutively membrane-bound Tom20-PINK1 construct.
PINK1 Phosphorylates Larp on Defective Mitochondria
We noticed that PINK1 protein levels were comparable in wild-type and mdi mutant ovaries (Figure 3C), suggesting that PINK1 is not a downstream target of MDI. Instead we hypothesized that since PINK1 is a serine/threonine (S/T) kinase, it may act upstream of MDI, by directly phosphorylating MDI or Larp. To facilitate our biochemical analysis of Larp, we generated a Larp-gfp fusion gene by CRISPR-mediated recombination at the endogenous Larp locus. Using this transgene and a previously reported mdi-gfp line (Zhang et al., 2016), we immuno-purified MDI-GFP and Larp-GFP proteins from both mt-wt and mt-ts ovaries. Probing the purified MDI-GFP protein with an antibody against phosphorylated S/T revealed a very weak signal and no difference between mt-wt and mt-ts extracts at restrictive temperature (Figure S4B), suggesting that MDI phosphorylation status is not modified on defective mitochondria. Additionally, MDI protein still localizes to mitochondria (Figure S4A). By contrast, Larp was readily phosphorylated in mt-wt ovaries (Figure 6A), and the phospho-S/T signal was markedly increased in mt-ts ovaries at restrictive temperature (Figure 6A). Importantly, knockdown of pink1 in mt-ts flies greatly diminished Larp phosphorylation (Figure 6A). Additionally, a recombinant PINK1 protein, but not a kinase-dead PINK1 mutant, effectively phosphorylated Larp in an in-vitro assay (Figure 6B). These observations suggest that Larp protein is phosphorylated by PINK1 on defective mitochondria carrying the mt-ts genome.
Figure 6. Larp Is Phosphorylated and Inhibited by PINK1.

(A) Phosphorylation of a Larp-GFP protein immuno-purified from mt-wt and mt-ts ovaries with or without PINK1 knockdown (PINK1-KD), as revealed by a phosphorylated-serine/threonine (α–P-S/T) antibody. The same set of extracts was probed with anti-GFP (α–Larp) to ensure the same amount of loading. Quantification is shown on the lower panel (n = 3). Phosphorylation on Larp increases in mt-ts relative to mt-wt extracts. This increase is abolished in the PINK1 knock-down background (PINK1-KD).
(B) In vitro phosphorylation of Larp and a phosphorylation-resistant Larp mutant (Larp10A) in the presence of PINK1 or a kinase dead mutant (PINK1kd), as revealed by α–P-S/T and α–Larp antibody staining. Serine/threonine phosphorylation of Larp is abolished in the presence of the kinase-dead PINK1 or of the Larp10A mutation.
(C) mtDNA contents and hatching rates of eggs produced by mdi mutant flies expressing different Larp transgenes activated by a nanos-gal4 driver, relative to wild type. N = 3 X100 eggs/genotype for hatching rate. The relative mtDNA level was determined as the average of three biological repeats normalized to control. Error bars represent std. mtDNA levels and hatching rates, which are severely reduced in mdi mutant eggs, are partially restored by both Tom20-Larp (TL) and the phospho-resistant mutant, Tom20-LarpS1119A (TLS1119A), but not by the phospho-mimicking mutant, Tom20-LarpS1119D (TLS1119D).
To further test this hypothesis, we mapped the phosphorylation sites on Larp proteins that were purified from mt-wt, mt-ts and Tom20-PINK1 (in a mt-wt background) ovaries. A total of eight peptides derived from Larp showed a higher level of phosphorylation in mt-ts than mt-wt ovaries (fold change > 1.5, p < 0.001, Figure S6A and Table S3). Strikingly, seven of these eight peptides were also more phosphorylated in Tom20-PINK1 than in mt-wt ovaries (Figure S6A). To confirm the result of mass spectroscopy, we carried out in-vitro phosphorylation on a Larp recombinant protein with all 10 candidate residues mutated to Alanine. This Larp mutant (Larp10A) was completely insensitive to PINK1 kinase (Figure 6B), confirming that PINK1 indeed phosphorylates Larp on these residues. Taken together, these observations strongly suggest that PINK1 accumulating onto defective mitochondria leads to increased phosphorylation of Larp. Phosphorylation of Larp by PINK1 does not seem to affect Larp’s stability or localization, as Larp protein levels were comparable between mt-wt and mt-ts ovaries, and the protein remained closely associated to mitochondria in mt-ts ovaries (Figure S5). Therefore, phosphorylation by PINK1 must instead prevent Larp from acting as a translational activator.
Phosphorylation on Larp Inhibits Local Translation and is required for Selective Inheritance
A direct, genetic demonstration of Larp’s role in local translation at the outer membrane and in mtDNA inheritance is difficult because Larp mutants are either lethal or sterile, producing very few unfertilized eggs (Blagden et al., 2009). To overcome this problem, we took advantage of a Tom20-Larp fusion construct that constitutively localizes Larp on the outer membrane. Larp is normally recruited to the outer membrane by MDI, which acts as a scaffold for local translation (Zhang et al., 2016). But expression of Tom20-Larp can partially rescue mdi mutant phenotypes, including loss of local translation and reduced mtDNA replication (Zhang et al., 2016). We thus adopted this system to examine how phosphorylation on Larp impacts local translation on mitochondrial surface specifically.
We generated a series of Tom20-Larp transgenes carrying either phosphor-mimicking (Ser/Thr to Asp/Glu) or phosphor-resistant (Ser/Thr to Ala) mutations at all PINK1-phosphorylation sites on Larp (Figure S6A). When expressed in the mdi mutant background, all phosphor-mimicking mutants significantly restored both mtDNA level and hatching rate of mdi eggs, except for Tom20-LarpS1119D (Figures 6C and S6B). Thus, phosphorylation on S1119 by PINK1 appears to impair Larp’s activity.
We therefore predicted that the phosphor-resistant mutant at S1119, Tom20-LarpS1119A, would be insensitive to PINK1, and retain its ability to boost local translation even on defective mitochondria. To test this idea, we introduced Tom20-LarpS1119A into the mt-ts background and examined nascent protein synthesis by AHA incorporation. In homoplasmic mt-ts flies cultured at 29°C, AHA signal associated with mitochondria was lower than in mt-wt control flies (Figure 7A and 7B), consistent with the expectation that local translation is impaired in this background. However, expression of Tom20-LarpS1119A partially restored mitochondria-associated AHA signal in mt-ts flies (Figure 7A and 7B). We next checked the steady-state levels of Tamas, a target of the MDI-Larp complex (Zhang et al., 2016), by western blot. The amount of Tamas protein decreased in mt-ts ovaries (Figure 7C), but was partially restored by the expression of Tom20-LarpS1119A (Figure 7C). These rescue experiments support the hypothesis that phosphorylation of S1119 by PINK1 inhibits Larp’s ability to promote translation at the outer membrane.
Figure 7. The Phosphorylation of Larp by PINK1 Inhibits Larp-mediated Local Protein Synthesis and Limits the Transmission of Deleterious mtDNA Mutations.

(A) Nascent protein synthesis in the mitochondrial fraction of mt-wt ovaries, mt-ts ovaries and mt-ts ovaries expressing the phosphor-resistant Tom20-LarpS1119A protein under the control of a nanos-gal4 driver (TLS1119A). The nascent protein synthesis was labeled by AHA incorporation and detected by anti-biotin antibody. Tom20 was used as a loading control.
(B) Quantification of nascent protein synthesis shown in (A), normalized to Tom20, from 3 independent experiments. Nascent protein synthesis is reduced in mt-ts ovary, but partially restored by the expression of TLS1119A.
(C) Expression of Tamas, a target of the MDI-Larp complex, in mt-wt, mt-ts and mt-ts ovaries expressing TLS1119A, as revealed by western blot analysis of ovary extracts. Tamas expression is reduced in mt-ts ovaries, but restored by TLS1119A overexpression.
(D) Changes in mt-ts amounts from mothers to eggs in control (n = 12) and TLS1119A (n = 12) females harboring heteroplasmic mtDNA (mt-wt + mt-ts) cultured at 29 °C. The abundance of mt-ts in mothers and their eggs was quantified, and changes expressed as the ratio (in percentage) of abundance in the eggs to abundance in the mothers. Note that the proportion of mt-ts in is always smaller in the progeny than in the mothers in the control strain, while the expression of TLS1119A severely impedes the reduction of mt-ts in the progeny.
(E) Proposed model for PINK1’s role in regulating local translation and the selective inheritance that limits the transmission of damaging mutations.
Given that Tom20-LarpS1119A can promote local translation on defective mitochondria despite the presence of PINK1, one would expect it to interfere with selective inheritance. Indeed, in heteroplasmic flies expressing Tom20-LarpS1119A, the reduction of mt-ts load in progeny was clearly less pronounced than in control flies (Figure 7D), demonstrating a critical role of Larp phosphorylation on S1119 by PINK1 in selective inheritance.
DISCUSSION
We have previously proposed that replication-competition, the ability of healthy mitochondria carrying wild-type mtDNA to propagate much more vigorously than defective ones afflicted by damaging mtDNA mutations (Hill et al., 2014), could explain selective mtDNA inheritance during Dm oogenesis. However, this model left two key questions unanswered: how do germ cells recognize defective mitochondria, and how do they selectively prevent their propagation? In this study, we demonstrate that PINK1, a versatile player in mitochondrial quality control, is the likely sensor of defective mitochondria during Dm oogenesis. Moreover, we demonstrate that it promotes selective inheritance by specifically hindering the translation of mitochondrial proteins at the surface of defective mitochondria, eventually compromising the replication of their mtDNA.
While PINK1 is known to be stabilized on damaged mitochondria in cultured cells treated with metabolic poisons (Narendra et al., 2010), we demonstrate, for the first time to our knowledge, that PINK1 preferentially accumulates on depolarized mitochondria enriched for a deleterious mutation, mt-ts, in heteroplasmic ovaries. More importantly, selective inheritance, which would normally prevent the transmission of the mt-ts mutation, is completely abolished in heteroplasmic flies carrying a pink1 mutation. These observations establish PINK1 as a key marker of defective mitochondria and crucial player in selective inheritance during Dm oogenesis.
Despite PINK1’s celebrated role in mitophagy (Pickrell and Youle 2015), its action in selective inheritance appears unrelated to mitophagy. It is known that Parkin, the ubiquitin ligase downstream of PINK1 in the canonical mitophagy pathway, is not required for selective inheritance (Ma et al., 2014). We find that mitophagy, or autophagy in general, does not increase either in heteroplasmic ovaries or in ovaries overexpressing a form of PINK1 tethered to the mitochondrial outer membrane, Tom20-PINK1. These observations argue against the involvement of even a Parkin-independent form of mitophagy in selective inheritance. It is now recognized that mitophagy might act in a piecemeal fashion, to remove mitochondria debris containing damaged proteins (Hämäläinen et al., 2013, McLelland et al., 2014, Vincow et al., 2013, Yang and Yang, 2013) rather than to completely destroy whole organelles. Hence, mitophagy may not be effective to remove mutated mitochondrial genome. In Drosophila ovaries, increased autophagy in germ cells often leads to stress-induced apoptosis (Barth et al., 2011, Nezis et al., 2009). This may explain why mitophagy has not evolved as a suitable mechanism to limit the transmission of mtDNA mutations.
Rather than triggering mitophagy, we find that PINK1 phosphorylates Larp and inhibits the local translation of mitochondrial proteins at the mitochondrial outer membrane. Larp is recruited to the mitochondrial surface by the MDI protein to promote the local translation of many mitochondrial proteins, including most mitochondrial ribosomal proteins, most electron transport chains subunits and several factors required for mtDNA replication and expression (Zhang et al., 2016). Our evidence supporting PINK1’s effect on Larp and local translation is multifold. First, phosphorylation of Larp increases in mt-ts ovaries, but diminishes when PINK1 is reduced by knockdown. Second, seven out of the eight Larp sites whose phosphorylation increases in mt-ts ovaries also undergo potent phosphorylation increases in ovaries expressing Tom20-PINK1. Third, many translational targets of MDI-Larp are down-regulated in Tom20-PINK1-expressing eggs. Finally, functional genetic analyses on Larp’s phosphorylation sites provide direct evidence that PINK1 phosphorylates Larp and inhibits local translation. These data establish Larp as a key target of PINK1’s activity in the regulation of local translation.
Furthermore, our work demonstrates essential roles of local protein translation and mtDNA replication in selective inheritance. We show that selective inheritance is abolished in the mdi mutant flies, which have severely impaired mtDNA replication in the ovary specifically, but are otherwise completely healthy. This result proves an essential role for mtDNA replication in selective inheritance, supporting our previously proposed replication-competition model (Hill et al., 2014). Additionally, we find that expression of Tom20-PINK1 mimics many mdi mutant phenotypes, including impaired mtDNA replication, and reduced mtDNA content and egg hatching rate. These observations put PINK1 and the MDI/Larp complex on a shared pathway toward selective inheritance, which we summarize with a new model (Figure 7E). According to this model, in heteroplasmic germ cells, MDI and Larp located on healthy mitochondria promote the rapid synthesis and effective import of nuclear encoded mitochondrial proteins to support the prodigious biogenesis of these mitochondria and replication of their mtDNA during oogenesis. PINK1, stabilized on the surface of unhealthy mitochondria harboring deleterious mtDNA mutations, phosphorylates Larp and inhibits local translation, which consequently starves these mitochondria of the factors they need for their propagation and mtDNA replication. As a result, the proportion of mutant mtDNAs is reduced in mature oocytes. Our model explains the gradual decline of a deleterious mutation over generations and its eventual cleansing (Hill et al., 2014; Ma et al., 2014). In addition, by providing a mechanism for selection based on the fitness of individual mitochondria, it offers a convincing alternative to selection at the cell level for the elimination mtDNA mutations, even when present at low level.
Whether PINK1 controls selective inheritance through mechanisms other than local translation remains possible. Targeting a phosphor-resistant Larp mutant, Tom20-LarpS1119A, to the mitochondrial surface in the heteroplasmic mt-ts background, leads to weakened selective inheritance. This result confirms our model, as the mutation renders Larp impervious to phosphorylation by PINK1, and hence should interfere with selective interference by restoring translation on defective mitochondria. However, selective inheritance is only partially impaired in Tom20-LarpS1119A-expressing flies, whereas it is completely abolished in pink1 mutants. This discrepancy could be due to the partial restoration of local translation by Tom20-LarpS1119A on defective mitochondria. Thus, the healthy mitochondria would still propagate more vigorously than defective ones, although to a lesser extent. We noticed that even Tom20-Larp, which tethers the wild-type version of Larp to the outer membrane, can only partially rescue the phenotypes of mdi mutant flies. This observation implies that MDI could affect local translation via players other than Larp. These unidentified targets could themselves be targets of regulation by PINK1. In addition, PINK1 has been shown to regulate mitochondrial morphology (Tsai et al., 2018), mobility (Wang et al., 2011a), calcium homeostasis and protein import (Huang et al., 2017; Liu et al., 2018). Defects in any of these processes may, directly or indirectly, interfere with mtDNA replication and mitochondrial biogenesis, and thereby impair selective inheritance. Nonetheless, our work firmly establishes a critical role for PINK1-mediated phosphorylation on Larp in repressing local translation, ultimately allowing PINK1 to regulate the selective replication and inheritance of mtDNA.
Studies in human and mammalian models demonstrate a strong purifying selection that prevents the transmission of harmful mtDNA mutations (Fan et al., 2008; Floros et al., 2018; Stewart et al., 2008). Similar to our model, this selection has been shown to act in developing oocytes (Fan et al., 2008; Floros et al. 2018), although the underlying mechanism remains elusive. Interestingly, selective replication of a subpopulation of mitochondrial genome has been demonstrated in murine oocytes (Wai et al., 2008), although the physiological significance has not been explored. It is possible that selective inheritance mediated by replication-competition represents an evolutionally conserved mechanism safeguarding the transmission of mitochondrial genomes. The work we presented here should provide molecular entry points to test the relevance of the replication-competition model in organisms other than Drosophila melanogaster.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hong Xu (hong.xu@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fly genetics and husbandry
All flies were maintained on cornmeal medium at 25 °C or as indicated. w1118 was used as the wild-type nuclear genome control, and its mtDNA was used as wild-type mtDNA control and abbreviated as mt:wt. Flies carrying a temperature-sensitive lethal mutation, mt:CoIT300I(mt:ts), were combined with various nuclear genome backgrounds as described in the main text. pink1-gfp and larp-gfp fly lines were constructed by inserting GFP cDNA into endogenous loci by CRISPR/Cas9-mediated recombination (Gratz et al., 2013). mdi-gfp andTom20-mcherry flies were from previous study (Zhang et al., 2016). Embryo-hatching tests were carried out as previously described (Ren et al., 2017).
METHODS DETAILS
mtDNA selection in female germ line and quantification of heteroplasmy
mtDNA selection in the female germ line was performed as previously described with modifications (Ma et al., 2014). Briefly, the heteroplasmic flies with wt, mdi1, pink1B9 or UASp-Tom20-LarpS1119A ; nosgal4 nuclear backgrounds were transferred from 18 °C to 29 °C right after their eclosion. Individual heteroplasmic females were mated with 5 w1118 males, the vial was changed every other day. Eggs produced during the first 6 days at 29 °C were excluded from analyses. Eggs produced by each female on the 7th day were pooled and the heteroplasmy levels were compared between the mother and her eggs. Total DNA was extracted from flies, eggs or FACS-sorted mitochondria with QIAamp DNA Blood Mini Kit (Qiagen). Quantification of heteroplasmy was carried out as described previously (Hill et al., 2014). Briefly, a 4-kb fragment spanning the XhoI site on mtDNA was amplified by PCR. The PCR products were gel-purified using the GeneJET Gel Extraction Kit (Thermo Fisher Scientific), and digested with 20 units of XhoI (NEB) at 37 °C overnight. The digested DNA (500 ng) was analyzed on an Agilent 2100 Bioanalyzer using DNA 7500 kit (Agilent). The proportion of mt:CoIT300I DNA was calculated by normalizing the amount of undigested 4-kb band (mt:CoIT300I) to the sum of undigested 4-kb band (mt:CoIT300I) and two XhoI-digested bands of 1.6 kb and 2.4 kb (mt:wt).
CRISPR/Cas9-mediated recombination in flies
To insert GFP into the endogenous loci of pink1 and larp, chiRNA plasmids containing the targeting sequences GTCAATAATTATTGTACCGG (for pink1) and GATTGGATTGATATAGACTG (for larp) were injected into the embryos of PBac{y[+mDint2]=vas-Cas9}VK00027 flies for pink1, or M{vas-Cas9}ZH2A flies for larp along with the corresponding donor plasmids. Donor plasmids contained a fragment spanning 1kb upstream and 1kb downstream of the stop codon of each target gene inside a pOT2 vector. The GFP was inserted in front of the stop codons. G1 adults were screened for insertion events by PCR.
FACS mitochondria sorting
pink1-gfp;Tom20-mcherry flies carrying either mt:wt or heteroplasmic mtDNA were cultured at 29 °C for 2 days, individual ovaries were dissected and homogenized in 20 mM HEPES-KOH, pH 7.4, 0.25 M sucrose. The homogenates were centrifuged at 150 g for 10 mins at 4 °C to remove debris. Supernatants were then centrifuged at 9000 g for 15 min at 4 °C to separate mitochondrial pellets from soluble cytosolic fractions. The pellets (mitochondrial fraction) were resuspended in 20 mM HEPES-KOH, pH 7.4, 0.25 M sucrose, and sorted on Aria III based on their GFP and mCherry intensity. The isolated high-GFP and low-GFP mitochondrial fractions were visualized with confocal microscopy, and their DNA was extracted with QIAamp DNA Blood Mini Kit (Qiagen).
Live imaging
Live imaging of ovaries was performed as previously reported (Weil et al., 2012). To image mitochondrial membrane potential, ovaries dissected from mt:wt or mt-ts flies grown at 29 °C were stained with TMRM and MTgreen (both at 1:500) in PBS for 10 min then rinsed with PBS 3 times. Ovaries were transferred to a drop of PBS on a coverslip mounted to a custom-made metal frame, and then covered with a small piece of Saranwrap before imaging. To image autophagy, ovaries were dissected in PBS and immerged in halocarbon oil on a coverslip. Each ovary was oriented with forceps and an individual ovariole was dragged to the center of the oil drop using a dissection needle. The samples were visualized by Perkin Elmer Ultraview system and processed with Volocity software or by Instant Sim (iSIM) Super-Resolution Microscope and processed with ImageJ.
Mitochondrial area and ATG8 area were selected with ImageJ’s default method and max-Entropy “Color Threshold” method, respectively. Overlap between mitochondria and ATG8 areas was analyzed with the “Colocalization” method. Binary images from mitochondria in egg chambers were generated according to previous description (Course et al., 2017). To quantify the ratio between TMRM and MTgreen, the germ cell area in an egg chamber was outlined and isolated by “Clear Outside”. Mitochondrial area was selected from the MTgreen channel by default “Color Threshold”. The same area selection was made in the TMRM channel from the same image. “Image Calculator” was used to generate a grayscale ratiometric image between TMRM and MT green, followed by coloring with “Rainbow RGB”.
Molecular biology
Tom20-pink1 fusion gene was constructed by inserting fragment 451-2166 of the pink1 gene before the stop codon of Tom20. The fusion gene was then cloned into a pUASp-GFP expression vector using Drosophila Gateway Cloning system. PINK1 kinase-dead (KD) mutant plasmid was kindly provided by Dr. Jongkyeong Chung (Song et al., 2013). PINK1, PINK1 KD, Larp and Larp10A were cloned into pGEX 6p-1 plasmids by In-Fusion cloning (Takarabio). Tom20-Larp cDNAs carrying the phosphorylation-mimic and phosphorylation-resistant mutations were cloned between the Not1 and Xba1 sites of attB-pUASp expression vectors. All constructs were injected into the embryos of y1 w67 c23 ; P{CaryP}attP40 flies. Quantitative real-time PCR analysis of mtDNA level was performed as previously described (Zhang et al., 2015).
Immunohistochemistry
EdU incorporation was performed as previously described (Hill et al., 2014). Images were collected on a Perkin Elmer Ultraview system and processed with Volocity software. Antibodies used were rabbit α-mCherry (Abcam, ab157453, 1:1000) and Alexa Fluor 568 goat α-rabbit IgG (Invitrogen, 1:200).
Biochemistry
Mass spectrometry of embryo samples was performed as previously described (Zhang et al., 2016), Briefly, triplicates of wt or UASp-Tom20-pink1; nosgal4 eggs were collected and homogenized. Embryo lysates were sequentially reduced, alkylated, digested overnight with trypsin, and labeled with 6-plex Tandem Mass Tag (TMT) reagents (Thermo Fisher Scientific). Six labeled protein digests were pooled, and then separated into 12 fractions using high-pH reverse-phase liquid chromatography (Wang et al., 2011b).
To pull down GFP tagged proteins, ovaries (40 pairs for western, 500 pairs for mass spectrum) were lysed in 150 mM NaCl, 1% Triton X-100, 50 mM TrisHCl (pH 8.0) containing Phosphatase Inhibitor Cocktail (Sigma P5726, P0044) and incubated on ice for 30 min with occasional mixing. Lysates were centrifuged for 10 min at 10,000 g at 4°C. Supernatants were collected and incubated with 50 μl-GFP MicroBeads (Miltenyi Biotec) for 2h at 4 °C. The beads were washed four times with 150 mM NaCl, 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris–HCl (pH 8.0), and one time with 20 mM Tris-HCl (pH 7.5). The proteins were eluted with 50 mM Tris–HCl (pH 6.8), 50 mM DTT, 1% SDS, 1 mM EDTA, 0.005% bromophenol blue, 10% glycerol and subjected to SDS-PAGE. Coomassie-stained bands were excised from the gel, de-stained with acetonitrile, reduced with dithiothreitol, alkylated with iodoacetamide, and digested with trypsin overnight. All fractions of protein digests were analyzed using a nanoLCMS system equipped with an LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Peptide and protein IDs were assigned by searching LCMS raw data against Uniprot Drome database (http://www.uniprot.org) using Sequest HT algorithm on Proteome Discoverer 1.4 platform (Thermo Fisher Scientific). The results were compiled and quantitatively compared using Scaffold 4.0 software (Proteome Software, Inc, Portland OR). The relative protein abundance in corresponding bands from IP pull-down samples was quantified using spectral counting method: TMT-labelled samples were normalized using the total reported ion intensities of their corresponding channels, and then individual proteins were compared using the normalized reported ion intensities.
For the in vitro PINK1 kinase assay, proteins were expressed in BL21(DE3) E. coli competent cells (Thermo Fisher Scientific), and purified on Glutathione Sepharose 4B (GE Healthcare) according to the manufacturer’s protocol. The in vitro phosphorylation assay was performed as previously reported with modification (Wang et al., 2011a). Briefly, the kinases (PINK1 or PINK1 KD) and substrates (Larp or Larp 10A) were incubated with ATP in 1X kinase assay buffer at 30 °C for 15 minutes, and samples were boiled and centrifuged to collect the supernatants, which were further analyzed by immunoblotting.
Detecting nascent protein synthesis by western blot
AHA labeling of nascent protein synthesis in ovaries was carried out as described previously (Zhang et al., 2016). Briefly, ovaries (40 pairs) from 4- to 5-day-old female flies were dissected in methionine-free media (MFM) (Thermo Fisher Scientific) and washed three times with MFM. The ovaries were equilibrated in MFM for 45 min and then incubated in MFM containing 50 μM AHA for 4h. After labeling, ovaries were homogenized in 20 mM HEPES-KOH, pH 7.4, 0.25 M sucrose, and then centrifuged at 150 g for 10 mins at 4 °C to remove tissue debris. Supernatants were centrifuged at 9000 g for 15 min at 4 °C to separate mitochondrial pellets from soluble cytosolic fractions. The mitochondrial fraction was resuspended in 1% SDS in 50mM Tris-HCl (pH 8.0). AHA was labeled with biotin according to the manufacturer’s instructions (Thermo Fisher Scientific), and probed with α-biotin (#7075, CST).
QUANTIFICATION AND STATISTICAL ANALYSIS
All western blotting quantifications were conducted in ImageJ. Statistical analyses were performed using Prism (GraphPad Software). Error bars represent standard deviations in all the charts. P values were computed with Two‐tailed Student’s t test. The difference was considered statistically significant when P < 0.05. In the comparative proteomic analyses, Benjamini–Hochberg corrected P values were further calculated as the false discovery rates (FDR). For comparative proteomics in embryo, the difference was considered statistically significant when the fold change >1.2, P < 0.05 and FDR < 0.05.
DATA AND SOFTWARE AVAILABILITY
Source data files are deposited on Mendeley Data (https://data.mendeley.com/datasets/9vyphxf47r/draft?a=a3963d7d-d9fc-413c-8baf-6000d890b04e).
Supplementary Material
Table S3.List of All Phosphorylated Peptides on Larp Detected by Mass Spectrometry, Related to Figure 6.
Table S2.List of Proteins that Were Detected by Mass Spectrometry in Both Tom20-PINK1 Expressing and mdi Mutant Embryos, Related to Figure 5.
Table S1. List of All Proteins Detected by Mass Spectrometry in Tom20-PINK1 Expressing Embryos, Related to Figure 5.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GFP | Roche | 11814460001 |
| Rabbit polyclonal anti-VDAC | Cell Signaling Technology | 4866 |
| Rabbit polyclonal anti-Larp | Blagden et al., 2009 | https://doi.org/10.1016/j.ydbio.2009.07.016 |
| Rabbit polyclonal anti-MDI | Zhang et al., 2016 | https://doi.org/10.15252/embj.201592994 |
| Mouse monoclonal anti-actin, clone C4 | Millipore | MAB1501 |
| Goat anti-Biotin, HRP-linked | Cell Signaling Technology | 7075 |
| Rabbit polyclonal anti-Tom20 | Cell Signaling Technology | 13929 |
| Rabbit polyclonal anti-Phospho - (Ser/Thr) | Abcam | ab117253 |
| Rabbit polyclonal anti-Tamas | Zhang et al., 2015 | https://doi.org/10.15252/embr.201439636 |
| Rabbit polyclonal anti-mCherry | Biovision | 5993-100 |
| Chemicals and competent cells | ||
| TMRM | Thermo Fisher Scientific | I34361 |
| MitoTracker™ Green | Thermo Fisher Scientific | M7514 |
| Xho1 | NEB | R0146 |
| Click-IT™ AHA (L-Azidohomoalanine) | Thermo Fisher Scientific | C10102 |
| Biotin Alkyne | Thermo Fisher Scientific | B10185 |
| Glutathione Sepharose 4B | GE Life sciences | 17075601 |
| One Shot™ BL21(DE3) Chemically Competent E. coli | Thermo Fisher Scientific | C600003 |
| TMTsixplex™ Isobaric Label Reagent Set | Thermo Fisher Scientific | 90061 |
| Phosphatase Inhibitor Cocktail 2 | Sigma-Aldrich | P5726 |
| Phosphatase Inhibitor Cocktail 3 | Sigma-Aldrich | P0044 |
| ATP | Cell Signaling Technology | 9804 |
| Kinase Buffer (10X) | Cell Signaling Technology | 9802 |
| Grace's Insect Medium, Unsupplemented (methionine-free medium) | Thermo Fisher Scientific | 11595030 |
| Drosophila Gateway Vector Collection | DGRC | N/A |
| Critical Commercial Assays | ||
| Click-iT™ Plus EdU Alexa Fluor™ 555 Imaging Kit | Thermo Fisher Scientific | C10338 |
| Click-iT™ Protein Reaction Buffer Kit | Thermo Fisher Scientific | C10276 |
| GeneJET Gel Extraction Kit | Thermo Fisher Scientific | K0692 |
| QIAamp DNA Blood Mini Kit | Qiagen | 51106 |
| In-Fusion® HD EcoDry™ Cloning Plus | Takarabio | 638915 |
| DNA 7500 Kit | Agilent | 5067-1506 |
| μMACS GFP Starting Kit | Miltenyi Biotec | 130-091-288 |
| Experimental Models: D. melanogaster | ||
| w1118 (mt: wt) | Bloomington Drosophila Stock Center | N/A |
| w1118 (mt: ts) | Hill et al., 2014 | https://doi.org/10.1038/ng.2920 |
| pink1-gfp | This study | N/A |
| Tom20-mcherry | Zhang et al., 2016 | https://doi.org/10.15252/embj.201592994 |
| pink1-gfp;Tom20-mcherry | This study | N/A |
| pink1B9 | Clark et al., 2006 | https://doi.org/10.1038/nature04779 |
| mdi1 | Zhang et al., 2016 | https://doi.org/10.15252/embj.201592994 |
| UASp-Tom20-PINK1 | This study | N/A |
| larp-gfp | This study | N/A |
| mdi-gfp | Zhang et al., 2016 | https://doi.org/10.15252/embj.201592994 |
| UASp-Tom20-larp | This study | N/A |
| UASp-Tom20-larpS66D,T107E | This study | N/A |
| UASp-Tom20-larpS66A,T107A | This study | N/A |
| UASp-Tom20-larpS182D,S186D,S219D,S221D | This study | N/A |
| UASp-Tom20-larpS182A,S186A,S219A,S221A | This study | N/A |
| UASp-Tom20-larp T828E | This study | N/A |
| UASp-Tom20-larpT828A | This study | N/A |
| UASp-Tom20-larpS1119D | This study | N/A |
| UASp-Tom20-larpS1119A | This study | N/A |
| UASp-Tom20-larpS1510D,S1512D | This study | N/A |
| UASp-Tom20-larp S1510A,S1512A | This study | N/A |
| UASp-mCherry-ATG8 | Wang et al., 2016 | https://doi.org/10.1093/hmg/ddw067 |
| UASp-mito-GFP | Hill et al., 2014 | https://doi.org/10.1038/ng.2920 |
| Oligonucleotides | ||
|
pink1 chiRNA targeting sequence: 5’GTCAATAATTATTGTACCGG3’ |
This study | N/A |
|
pink1, genotyping Forward: 5’GATCACAGTCTTCGCGGCCT3’ |
This study | N/A |
|
pink1, genotyping Reverse 5’TTACTTGTACAGCTCGTCCATG3’ |
This study | N/A |
|
larp chiRNA targeting sequence: 5’GATTGGATTGATATAGACTG3’ |
This study | N/A |
|
larp, genotyping Forward: 5’GAAGTTCTGGGCCTTCCTGAAA3’ |
This study | N/A |
|
larp, genotyping Reverse: 5’TTACTTGTACAGCTCGTCCATG3’ |
This study | N/A |
|
mt:Co1, Xho1 site genotyping Forward: 5’TGGAGCTATTGGAGGACTAAATCA3’ |
Hill et al., 2014 | https://doi.org/10.1038/ng.2920 |
|
mt:Co1, Xho1 site genotyping Reverse: 5’GCTCCTGTTAATGGTCATGGACT3’ |
Hill et al., 2014 | https://doi.org/10.1038/ng.2920 |
| QPCR, his4, Forward: 5’TCCAAGGTATCACGAAGCC3’ |
Zhang et al., 2015 | https://doi.org/10.15252/embr.201439636 |
| QPCR, his4, Reverse: 5’AACCTTCAGAACGCCAC3’ |
Zhang et al., 2015 | https://doi.org/10.15252/embr.201439636 |
| QPCR, mt:Co1, Forward: 5’ATTGGAGTTAATTTAACATTTTTTCCTCA3’ |
Zhang et al., 2015 | https://doi.org/10.15252/embr.201439636 |
| QPCR, mt:Co1, Reverse: 5’AGTTGATACAATATTTCATGTTGTGTAAG3’ |
Zhang et al., 2015 | https://doi.org/10.15252/embr.201439636 |
| Recombinant DNA | ||
| PPWG-Tom20-PINK1 | This study | N/A |
| pGEX Larp | This study | N/A |
| pGEX PINK1 | This study | N/A |
| pGEX PINK1 KD | This study | N/A |
| pGEX Larp10A | This study | N/A |
| attB-UASp LarpS66D,T107E | This study | N/A |
| attB-UASp LarpS66A,T107A | This study | N/A |
| attB-UASp LarpS182D,S186D,S219D,S221D | This study | N/A |
| attB-UASp LarpS182A,S186A,S219A,S221A | This study | N/A |
| attB-UASp LarpT828E | This study | N/A |
| attB-UASp LarpT828A | This study | N/A |
| attB-UASp LarpS1119D | This study | N/A |
| attB-UASp LarpS1119A | This study | N/A |
| attB-UASp LarpS1510D,S1512D | This study | N/A |
| attB-UASp LarpS1510A,S1512A | This study | N/A |
| Software and Algorithms | ||
| GraphPad Prism7 | GraphPad Software, Inc. | http://www.graphpad.com/ |
| ImageJ2 | NIH | https://imagej.nih.gov/ij/ |
| Scaffold 4.0 | Proteome Software, Inc | http://www.proteomesoftware.com |
| Volocity | Quorum Technology | https://www.quorumtechnologies.com |
| Deposited Data | ||
| doi:10.17632/9vyphxf47r.1 | ||
ACKNOWLEDGEMENTS
We thank F. Chanut for comments and edits on the manuscript; N. Rusan, T. Finkel and R. Youle for comments on the work; the Bloomington Drosophila Stock Center and M. Guo for fly stocks; the Developmental Studies Hybridoma Bank and E. Matsuura for antibodies; J. Chung for the PINK1 kinase-dead mutant plasmid; NHLBI FACS core and Bestgene Inc. for technical assistance. Funding: This work is supported by NHLBI Intramural Research Program.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and three tables.
REFERENCES
- Abbaszadeh EK, and Gavis ER (2016). Fixed and live visualization of RNAs in Drosophila oocytes and embryos. Methods 98, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, and Schwarz TL (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth JM, Szabad J, Hafen E, and Koöhler K. 2011. Autophagy in Drosophila ovaries is induced by starvation and is required for oogenesis. Cell Death Differ. 18:915–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagden SP, Gatt MK, Archambault V, Lada K, Ichihara K, Lilley KS, Inoue YH, and Glover DM (2009). Drosophila Larp associates with poly(A)-binding protein and is required for male fertility and syncytial embryo development. Dev Biol 334, 186–197. [DOI] [PubMed] [Google Scholar]
- Chen Z, Qi Y, French S, Zhang G, Covian Garcia R, Balaban R, and Xu H (2015). Genetic mosaic analysis of a deleterious mitochondrial DNA mutation in Drosophila reveals novel aspects of mitochondrial regulation and function. Molecular biology of the cell 26, 674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, Thorburn DR, Samuels DC, White SL, Dahl H-HM, Turnbull DM, Lightowlers RN, and Howell N (2000). The inheritance of mitochondrial DNA heteroplasmy: random drift, selection or both? Trends in Genetics 16, 500–505. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, and Guo M (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166. [DOI] [PubMed] [Google Scholar]
- Course MM, Hsieh CH, Tsai PI, Codding-Bui JA, Shaltouki A, and Wang X (2017). Live Imaging Mitochondrial Transport in Neurons. Neuromethods 123, 49–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE, Vannan MA, Narula J, Macgregor GR, and Wallace DC (2008). A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 319, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floros VI, Pyle A, Dietmann S, Wei W, Tang WCW, Irie N, Payne B, Capalbo A, Noli L, Coxhead J, et al. (2018). Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nat Cell Biol 20, 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J and O’Connor-Giles KM (2013) Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 194: 1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hämäläinen RH, Manninen T, Koivumäki H, Kislin M, Otonkoski T, and Suomalainen A (2013). Tissue- and cell-type–specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. PNAS 110, E3622–E3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi-Sanabria R, Charalel JK, Viana MP, Garcia EJ, Sing CN, Koenigsberg A, Swayne TC, Vevea JD, Boldogh IR, Rafelski SM, et al. (2016). Mitochondrial anchorage and fusion contribute to mitochondrial inheritance and quality control in the budding yeast Saccharomyces cerevisiae. Mol Biol Cell 27, 776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JH, Chen Z, and Xu H (2014). Selective propagation of functional mitochondrial DNA during oogenesis restricts the transmission of a deleterious mitochondrial variant. Nature genetics 46, 389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E, Qu D, Huang T, Rizzi N, Boonying W, Krolak D, Ciana P, Woulfe J, Klein C, Slack RS, et al. (2017). PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat Commun 8, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd TR, Herrmann B, Sauerwald J, Sanny J, Grosch M, and Lehmann R (2016). Long Oskar Controls Mitochondrial Inheritance in Drosophila melanogaster. Dev. Cell 39, 560–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandul NP, Zhang T, Hay BA, and Guo M (2016). Selective removal of deletion-bearing mitochondrial DNA in heteroplasmic Drosophila. Nat Commun 7, 13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters J, Ramshesh V, 2007. Imaging of mitochondrial polarization and depolarization with cationic fluorophores. Meth. Cell Biol 80, 283–295. [DOI] [PubMed] [Google Scholar]
- Lim GG, and Lim K-L (2017). Parkin-independent mitophagy-FKBP8 takes the stage. EMBO Rep. 18, 864–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Duan X, Fang X, Shang W, and Tong C (2018). Mitochondrial protein import regulates cytosolic protein homeostasis and neuronal integrity. Autophagy 14, 1293–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Xu H, and O'Farrell PH (2014). Transmission of mitochondrial mutations and action of purifying selection in Drosophila melanogaster. Nature genetics 46, 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, and Fon EA (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO Journal 33, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen D-F, Gautier CA, Shen J, Cookson MR, and Youle RJ (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Ichimura Y, and Ohsumi Y (2007). Atg8, a Ubiquitin-like Protein Required for Autophagosome Formation, Mediates Membrane Tethering and Hemifusion. Cell 130, 165–178. [DOI] [PubMed] [Google Scholar]
- Nezis IP, Lamark T, Velentzas AD, Rusten TE, Bjørkøy G, Johansen T, et al. Cell death during Drosophila mela- nogaster early oogenesis is mediated through autophagy. Autophagy 2009; 5:298–302. [DOI] [PubMed] [Google Scholar]
- Pesole G, Gissi C, De Chirico A, and Saccone C (1999). Nucleotide substitution rate of mammalian mitochondrial genomes. J Mol Evol 48, 427–434. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, and Youle RJ (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Q, Zhang F, and Xu H (2017). Proliferation Cycle Causes Age Dependent Mitochondrial Deficiencies and Contributes to the Aging of Stem Cells. Genes (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Jang S, Park J, Bang S, Choi S, Kwon KY, Zhuang X, Kim E, and Chung J (2013). Characterization of PINK1 (PTEN-induced putative kinase 1) mutations associated with Parkinson disease in mammalian cells and Drosophila. J Biol Chem 288, 5660–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, and Larsson NG (2008). Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS biology 6, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JB, and Larsson NG (2014). Keeping mtDNA in shape between generations. PLoS genetics 10, e1004670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen DF, Narendra DP, Tanaka A, Manfredi G, and Youle RJ (2010). Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proceedings of the National Academy of Sciences of the United States of America 107, 11835–11840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SWG, and Green DR (2012). Mitochondria and cell signaling. J Cell Sci 125, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai P-I, Lin C-H, Hsieh C-H, Papakyrikos AM, Kim MJ, Napolioni V, Schoor C, Couthouis J, Wu R-M, Wszolek ZK, et al. (2018). PINK1 Phosphorylates MIC60/Mitofilin to Control Structural Plasticity of Mitochondrial Crista Junctions. Mol. Cell 69, 744–756.e6. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, and Loeb LA (2007). Mitochondrial point mutations do not limit the natural lifespan of mice. Nature genetics 39, 540–543. [DOI] [PubMed] [Google Scholar]
- Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, and Pallanck LJ (2013). The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proceedings of the National Academy of Sciences of the United States of America 110, 6400–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Teoli D, and Shoubridge EA (2008). The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nature genetics 40, 1484–1488. [DOI] [PubMed] [Google Scholar]
- Wallace DC (2008). Mitochondria as chi. Genetics 179, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, and Schwarz TL (2011a). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, Liu T, Shen Y, Monroe ME, Lopez-Ferrer D, Reno T, et al. (2011b). Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZH, Clark C, and Geisbrecht ER (2016). Drosophila clueless is involved in Parkin-dependent mitophagy by promoting VCP-mediated Marf degradation. Hum Mol Genet 25, 1946–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil TT, Parton RM, and Davis I (2012). Preparing individual Drosophila egg chambers for live imaging. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J-Y, and Yang WY (2013). Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nature Communications 4, 2428. [DOI] [PubMed] [Google Scholar]
- Zhang F, Qi Y, Zhou K, Zhang G, Linask K, and Xu H (2015). The cAMP phosphodiesterase Prune localizes to the mitochondrial matrix and promotes mtDNA replication by stabilizing TFAM. EMBO reports 16, 520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Zhang L, Qi Y, and Xu H (2016). Mitochondrial cAMP signaling. Cell. Mol. Life Sci 73, 4577–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen Y, Gucek M, and Xu H (2016). The mitochondrial outer membrane protein MDI promotes local protein synthesis and mtDNA replication. EMBO J 35, 1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Xu H Translational regulation of mitochondrial biogenesis. Biochem Soc Trans. 2016. December 15;44(6):1717–1724. [DOI] [PubMed] [Google Scholar]
- Zhuang N, Li L, Chen S, and Wang T (2016). PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death & Disease 7, e2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3.List of All Phosphorylated Peptides on Larp Detected by Mass Spectrometry, Related to Figure 6.
Table S2.List of Proteins that Were Detected by Mass Spectrometry in Both Tom20-PINK1 Expressing and mdi Mutant Embryos, Related to Figure 5.
Table S1. List of All Proteins Detected by Mass Spectrometry in Tom20-PINK1 Expressing Embryos, Related to Figure 5.
Data Availability Statement
Source data files are deposited on Mendeley Data (https://data.mendeley.com/datasets/9vyphxf47r/draft?a=a3963d7d-d9fc-413c-8baf-6000d890b04e).