

Abstract
Hepatocellular carcinoma (HCC) is a devastating and prevalent cancer with limited treatment options. Technological advances have enabled genetic screens to be employed in HCC model systems to characterize genes regulating tumor initiation and growth. Relative to traditional methods for studying cancer biology, such as candidate gene approaches or expression analysis, genetic screens have several advantages: they are unbiased, with no a priori selection, can directly annotate gene function, and can uncover gene-gene interactions. In HCC, three main types of screens have been conducted and are reviewed here: (1) Transposon-based mutagenesis screens, (2) knock-down screens using RNA interference (RNAi) or the CRISPR/Cas9 system, and (3) overexpression screens using CRISPR activation (CRISPRa) or cDNAs. These methods will be valuable in future genetic screens to delineate the mechanisms underlying drug resistance and to identify new treatments for HCC.
Keywords: Hepatocellular Carcinoma, genetic screening, Sleeping Beauty Transposon, CRISPR, hydrodynamic injection
HCC is a unique cancer that needs unique approaches to treatment
Hepatocellular carcinoma (HCC) is the third most common cause of cancer-related mortality worldwide and is growing in incidence1,2. HCC is unique among cancers: a recent analysis of 17 human cancer types revealed substantial overlap in the gene expression patterns of all the cancers with the single exception of HCC3. This is consistent with the observation that several treatments, though effective against other cancers, have largely failed to improve outcomes for patients with HCC4. Therefore, the discovery of new treatments will depend on the development of models that accurately recapitulate HCC.
Most cases of HCC arise in cirrhotic livers; hence it is not surprising that several major underlying risk factors are characterized by chronic liver injury. Worldwide, the most common etiologies are chronic hepatitis B or C infections. Though the prevalence of these infections is decreasing in many countries due to the availability of an effective vaccine and treatment, the incidence of Hepatitis C-associated HCC in the United States is still not falling due to a large number of ‘baby boomers’ who have been chronically infected for decades. Non-alcoholic fatty liver disease is also rising in incidence in Western countries and is likely to soon overtake Hepatitis C as the major cause of HCC in the United States5–7. Additional etiologies of HCC include biliary tract diseases and exposure to toxins such as alcohol.
The advent of high throughput sequencing (HTS) technology has accelerated the identification of mutations and gene expression changes in cancers. The genomes of thousands of patients’ cancers have been sequenced to date, including hundreds of HCCs (a large collection of data is available publicly from the Cancer Genome Atlas at http://cancergenome.nih.gov/). These data have helped define the diverse genetic landscape of mutations and expression changes that occur in HCCs8–11. Nevertheless, additional complementary data are necessary to provide evidence of causation, i.e. that genetic changes induce tumor formation rather than occurring secondarily during the process of cancer development12.
HCC is challenging to treat due to several factors including its genetic diversity, the underlying liver dysfunction in most patients, the difficulty in detecting early stage disease, and the lack of effective treatments. Only 30% of patients with HCC are eligible for potentially curative interventions like liver transplantation at the time of diagnosis13. In general, classical chemo- and radiotherapies have either low efficacy or undesirable toxicity4,13. Only a single drug is approved in the United States to treat HCC as a first-line therapy: sorafenib, a multi-specific kinase inhibitor14,15. Sorafenib treatment has a marginal median survival benefit of just 2–3 months, and there are no good predictors of response16. In patients whose disease progresses during sorafenib treatment, second-line options were recently approved. One is another multikinase inhibitor, regorafenib, which improves survival over placebo by just under three months17. The other second-line option is Nivolumab, an immune-therapeutic with remarkable efficacy in a fraction of patients18–20. However, the search must continue for more broadly-effective therapies.
The goal of this review is to provide an overview of how innovations in gene delivery and in genetic screening technology in mice can significantly expand our knowledge of the genes driving or modulating the development of HCC, which can be applied to discover new therapeutic targets. The main methods and most notable studies are summarized in Table 1.
Table 1.
Three genomic screening methods discussed in this review and notable examples in hepatocellular carcinoma.
| Molecular screening tool | Method for identifying genes of interest | Notable studies | |
|---|---|---|---|
| Mouse model | Reference | ||
| Transposon insertional mutagenesis | High throughput sequencing to identify insertions at genomic loci above rate expected by chance | Rosa26-lsl-SB11;T2/OncTg/+;Alb-CreTg/+;p53- lsl-R270H | 56 |
| Rosa26-lsl-SB11;T2/Onc2Tg/+;Alb-CreTg/+;HBsAgTg/+ | 60 | ||
| RosaSBaseLsL/+;T2/Onc3Tg/+;Alb-CreTg/+ treated with CCl4 | 63 | ||
| shRNA | Relative abundance of shRNA transgenes in tumor lesions | Mosaic liver populated with Trp53−/− immortalized embryonic liver progenitor cells infected with shRNA-retroviral library | 72 |
| Arf−/− mice underwent HTVI with SB13 transposase and NrasG12V/shRNA transposon library plasmids, treated with sorafenib or vehicle | 76 | ||
| CRISPR/Cas9 | Relative abundance of gRNAs transgenes in tumor lesions | Trp53−/−;Myc;Cas9 liver progenitor cells infected with mGeCKOa lentiviral library subcutaneously transplanted into nude mice | 95 |
| HTVI of PB transposon library encoding GeCKOv2 gRNAs and plasmids encoding Cdkn2a-gRNA and NrasG12V | 101 | ||
| Fah−/−;dCas9+ mice underwent HTVI with AAV-Cre and plasmids encoding scFv-VP64, an Fah expression cassette, and gRNAs targeting promoters of genes of interest | 45 | ||
Genetic screens are a powerful method for annotating gene function
A genetic screen is an assay in which the genetic codes are mutated or gene expression is systematically perturbed in order to identify which changes result in a phenotype of interest21. Whole organism genetic screening has been employed frequently in the nematode Caenorhabditis elegans, in the fruit fly Drosophila melanogaster, in the zebrafish Danio rerio, and in mice (reviewed in 22). Internet resources for many model organisms, including wormbase.org, flybase.org, and ZFIN.org, catalog detailed information linking specific genetic changes to precise phenotypic manifestations. Screens have used a variety of methods to manipulate gene expression, including chemical mutagenesis, transposon insertional mutagenesis, RNA interference (RNAi), complementary DNA (cDNA) expression, and the prokaryotic clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated protein 9 (Cas9) system. More recent efforts have combined genetic screens with chemical library screens to explore therapeutic potential or identify mechanisms responsible for drug efficacy22. While useful in identifying phenotypes in a whole organism, these types of studies do not easily translate to complex, tissue-specific human diseases such as HCC.
Genetic screens in cancer cells can provide mechanistic evidence linking genetic changes to the important phenotypes of tumor initiation, morphology, growth, or drug sensitivity or resistance. Specific functional insights, as compared to correlative genetic associations, better facilitate therapeutic development and selection.
To efficiently identify cancer-driving genes, transposon mutagenesis, cDNA, RNAi, and CRISPR screens all heavily rely on high-throughput sequencing (HTS). For transposon mutagenesis, the molecular readout is the flanking genomic sequence, which precisely identifies the insertion site of the transposon. cDNA screens may use barcoding of plasmids to enable precise quantification of the proportion of plasmids before and after the screen23. RNAi and CRISPR screens depend on identifying and quantifying the RNA coding sequences.
Data generated by genetic screens in mice can be integrated with the large collection of available human cancer databases to provide insights into the multiple alterations found in HCC. Such studies can significantly contribute to the identification of potential new drug targets and to the ability to predict which signals should be targeted in patients.
Mouse models of HCC
While most strains of mice do not spontaneously develop HCC, liver cancer in mice can be induced through chemical stimulus, transplantation of tumor cells, or genetic alteration. Administration of the chemical diethylnitrosamine (DEN) causes DNA damage, which leads to the formation of tumors by proliferating hepatocytes harboring mutations. DEN does not induce tumorigenesis in adult mice unless it is combined with an additional toxin, such as carbon tetrachloride (CCl4), a hepatotoxin that triggers inflammation and fibrosis. Chemical stimulus is also frequently used in combination with genetically engineered mouse models (GEMMs) of HCC.
A number of GEMMs have been generated to study tumorigenesis in the liver. Due to the numerous etiologies contributing to hepatocellular carcinogenesis, no single GEMM is capable of completely recapitulating the disease. However, by reproducing genetic alterations that are common in HCC (Figure 1A), such as by inactivating tumor suppressors, including transformation related protein 53 (Trp53) and adenomatous polyposis coli (Apc), or ectopically expressing oncogenes, including Myc and catenin beta 1 (Ctnnb1), in hepatocytes, transgenic mice develop tumors that mimic features of the disease (reviewed in 24–27).
Figure 1: Mouse models to examine specific genes involved in the initiation and progression of hepatocellular carcinoma.

(A) Examples of traditional genetically engineered mouse models (GEMMs). Transgenic and knock-in mice express oncogenes in the liver, often using liver-specific promoters (Top). Conditional expression systems such as the tTA/TRE system, in which administration of doxycycline turns off expression (middle). Loss-of-function models target tumor suppressor genes for mutations such as deletion of an exon (bottom). (B) The hydrodynamic tail vein injection (HTVI) technique leads to expression of oncogene cDNA (top) or knock-down of tumor suppressors using shRNA (bottom) in hepatocytes. The transposon systems enable integration and stable expression of the oncogenic sequences in a subset of the hepatocytes.
The use of GEMMs for combinatorial studies has led to the discovery of important genetic synergisms, such as concomitant MYC and transforming growth factor α (TGFα) overexpression, which accelerates liver carcinogenesis28. A GEMM with temporally controlled oncogene expression was used to demonstrate that activated MYC drives the development of HCC and that MYC-driven tumors involute when MYC expression is removed29. Several mouse models carrying alterations of receptor tyrosine kinase (RTK) signaling components recapitulate alterations frequently observed in HCC patient subgroups, including transcriptional, epigenetic, and signaling modifications. Recently, a unique genetic system found that mice with slightly enhanced wild-type MET expression (Alb-R26Met mice) spontaneously develop HCC, illustrating a striking vulnerability of the liver to subtly increased RTK levels30.
The downside of the GEMM approach is that models can take months to develop and often require complicated breeding schemes in order to examine gene-gene interactions in cancer development. Furthermore, the expression of oncogenes may not be restricted to the adult stage of development, as the promoters – most commonly alpha-fetoprotein (AFP) or albumin (Alb) – may be active during early liver development, unless expression is controlled with inducible systems such as tTA/TRE or Cre-LoxP (Figure 1A). Thus, while transgenic mouse models are useful in studying liver cancer, several technical challenges must be taken into consideration prior to implementation.
A powerful method for transgene delivery to the mouse liver is the hydrodynamic tail vein injection (HTVI) technique (Figure 1B)31–34. HTVI introduces DNA sequences into the nuclei of up to 40% of hepatocytes upon the injection of a large volume of the DNA in saline or lactated ringer’s solution (a 1:10 ratio of volume to mouse mass) into the tail vein of mice. The solution is delivered rapidly, in fewer than 10 seconds, which is believed to cause a transient right-sided venous congestion that engorges and floods the liver with fluid and mechanically disrupts cell membranes35–39. The technique is highly specific to hepatocytes, with little expression in other cell types of the liver or in other organs. When the HTVI technique is used to inject plasmids encoding transposon systems such as Sleeping Beauty (SB)37,40,41, piggyBac (PB)42, or Tol243, the transposon sequences and the “cargo” – i.e. the genes of interest – are integrated into the hepatocyte genome, which enables stable expression for the life of the cell. This achieves stable integration in 0.1% to 2% of all hepatocytes with SB37,44. In an adult mouse there are roughly 108 hepatocytes; therefore, HTVI of transposons could in theory examine more than 106 independent clonal events in a single mouse45.
In previous studies of liver cancer, HTVI was used to transfect hepatocytes in vivo in order to assess the effects of specific changes in gene expression on tumorigenesis. For example, in one study, wild type mice underwent HTVI with an SB-overexpression plasmid encoding activated thymoma viral proto-oncogene 1 (Akt), which was stably integrated into the hepatocyte genomic DNA46. The upregulation of AKT induced increased lipogenesis and hepatocarcinogenesis. A subsequent study in which an SB-overexpression plasmid encoding the intracellular domain of the NOTCH1 receptor was coinjected with the AKT overexpression plasmid showed that intrahepatic cholangiocarcinomas arise from mature hepatocytes that have undergone malignant transformation and not from biliary epithelial or liver progenitor cells47. These studies discovered mechanisms of HCC tumorigenesis and pointed toward new target genes for the development of new therapies. Thus, HTVI is a powerful alternative to generating genetically modified animals in order to study the role of hepatocytes in liver cancer.
In combination with HTVI, a useful tool for performing high throughput genetic screens in the mouse liver is the murine genetic model of hereditary tyrosinemia type 1 (HT-1)48. Humans with HT-1 have mutations in the gene encoding fumarylacetoacetate hydrolase (FAH), an enzyme required for proper tyrosine metabolism. Lack of FAH activity results in severe liver injury from the accumulation of the toxic metabolite fumarylacetoacetate (FAA) in hepatocytes. The drug nitisinone (chemical name 2-(2-nitro-4-(trifluoromethyl)benzoyl)cyclohexane-1,3-dione, or NTBC) restores healthy liver function in HT-1 patients by inhibiting an upstream enzyme in tyrosine catabolism and preventing the formation of FAA49.
In the Fah−/− mouse, liver toxicity can be alleviated by the stable genomic integration of a transgene encoding the functional FAH enzyme32,44. Transposons expressing Fah are delivered by HTVI to Fah−/− mice that have been continuously administered nitisinone, which is then withdrawn to induce liver injury and create a selective environment for any Fah-corrected hepatocytes to repopulate the liver. Although the initial integration of the Fah transgene by SB transposase is estimated to occur in approximately 0.1–2% of hepatocytes, these FAH-positive cells are able to expand and restore liver function without tumor formation44. The rescue of Fah-null hepatocytes by functional FAH expression has been leveraged to perform genetic screens by linking a functional copy of Fah to libraries of cDNAs, short hairpin RNAs (shRNAs), or guide RNAs (gRNAs) in order to study their effects on liver repopulation and cancer41,50,51.
Transposon-based mutagenesis
Several forward genetic screens aimed at identifying HCC drivers have been performed via the mobilization of mutagenic transposons by the SB transposase (Figure 2). SB excises transposon DNA that is flanked by inverted repeat/direct repeat sequences and reintegrates the transposon at random TA target dinucleotides52. The system is well-suited for insertional mutagenesis screens in mice using engineered transposon DNA cassettes, as the transposase is highly active in mammalian cells.
Figure 2: Transposon-based mutagenesis systems.
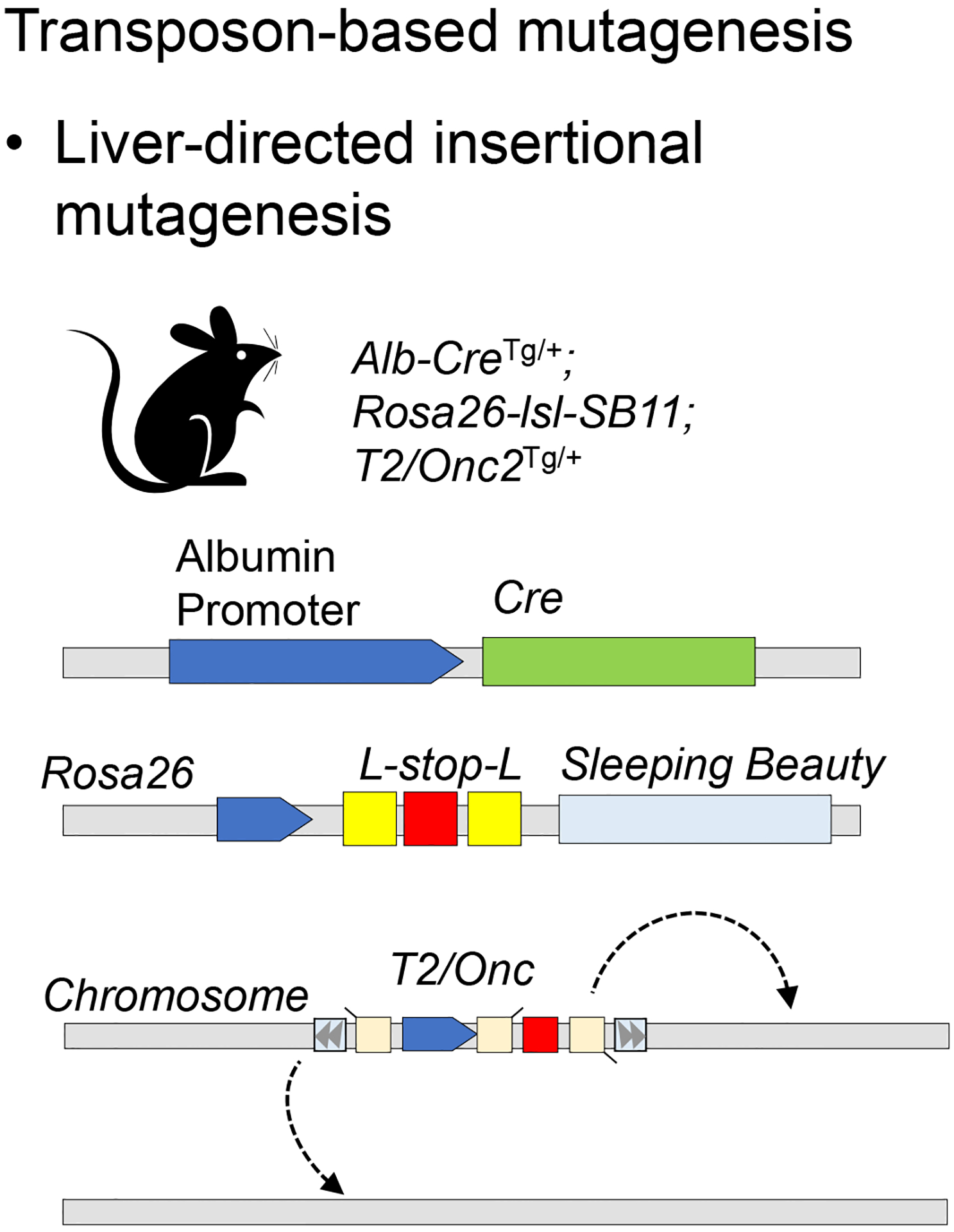
The transposase enzyme mobilizes an engineered mutagenic transposon (e.g. T2/onc), which can disrupt tumor suppressor gene expression or activate oncogene expression, according to the position and the orientation of its insertion. Liver-specific expression of the transposase is regulated by albumin promoter-driven Cre recombinase.
Three different iterations of the SB mutagenic transposon have been engineered, T2/Onc, T2/Onc2, and T2/Onc3, all of which are capable of inducing both gain- and loss-of-function mutations depending on the orientation of their insertion relative to coding sequence. These transposons all comprise the following elements: (a) a strong promoter and enhancer followed by a splice donor site, (b) a splice acceptor site joined to a polyadenylation signal positioned upstream of the promoter/enhancer in the sense orientation, and (c) a second splice acceptor site joined to a polyadenylation signal, which is positioned downstream of the promoter/enhancer in the antisense orientation (Figure 2). Integration of a transposon cassette in the sense orientation can drive overexpression of downstream coding sequence, while its insertion in either orientation downstream of coding sequence truncates the transcript. By altering the gene sequence, the transposon mutagenesis system can uncover genomic mutations that lead to oncogenic transformation, a unique advantage over RNAi or cDNA screens, which instead aim to modulate expression levels of endogenous genes.
Thousands of disruptive transposition events can occur in a tissue expressing the SB transposase as a consequence of multiple transposon mobilizations from the parent transposon concatemer53–55. Sites where transposons integrate more often than would be expected by chance in independent tumors are termed common insertion sites (CISs). Potential drivers of tumorigenesis are identified based on increased frequency of CISs in tumors, which is indicative of selection for a tumorigenic event.
The first version of the SB mutagenesis screen used the SB/T2Onc transposon in a p19Arf-deficient (Arf−/−) mouse strain that ubiquitously expresses the SB transposase (SB10). The majority of mice formed tumors, mostly sarcomas, by 6 months of age. The most common CIS identified was in the ninth intron of Braf, an alteration predicted to produce a constitutively activated form of this known oncogene. This result demonstrated that SB insertional mutagenesis can identify disease-causing mutations that synergize with common mutations in cancer models53.
Subsequent refinements made to the SB-T2/Onc system have improved its ability to promote tumorigenesis. The second-generation mutagenic transposon (T2/Onc2) included a larger splice acceptor and optimized SB transposase binding sites, along with an improved SB allele (SB11). T2/Onc2;SB11 mice showed insertional mutations at the embryonic stage, exhibited high prenatal lethality, and spontaneously developed up to three separate, metachronous tumors by 17 weeks of age, the most common of which were T-cell lymphomas54. However, when investigating epithelial-derived tumors, this high frequency of lymphomas is undesirable. To increase the frequency of carcinomas, the chicken β-actin (CAG) promoter, which is highly active in epithelial cells, was introduced in the T2/Onc3 system55. This change increased the frequency of tissue-specific carcinomas, including HCC. More recently, the Rosa26-lsl-SB11 mouse was developed to enable spatial and temporal activation of SB11 allele using Cre recombinase enzyme56.
To identify mutations that synergize with loss of function of tumor protein 53 (TP53; ortholog of mouse Trp53), which is often mutated in human HCCs, insertions of T2/Onc were screened in mice expressing a dominant negative Trp53 specifically in the liver56. Similar to what was seen in the Arf−/− genetic background, mice expressing mutant Trp53 together with the transposon/transposase displayed a significantly increased tumor burden compared to mice expressing mutant Trp53 alone. Importantly, as is observed in human HCC, there was a strong sex bias toward male mice, which showed greater tumor incidence and decreased latency. Epidermal growth factor receptor (Egfr) had the highest number of insertions, most of which truncated the transcript and resulted in increased kinase activity of the translated protein56. HCC tumors in males displayed higher rates of chromosome 7 polysomy (the locus encoding EGFR) and higher EGFR mRNA levels compared to females, suggesting elevated susceptibility to EGFR-driven HCC in male patients57. As both p53 and EGFR are linked to human HCC, these results are noteworthy and suggest that simultaneous dysregulation of these genes can promote HCC in a sex-specific manner.
Transposon insertional mutagenesis has also been used to examine epithelial-mesenchymal transition (EMT), which is thought to play a prominent role in HCC metastasis and resistance to cancer drugs like sorafenib58. Kodama et al. generated immortalized hepatoblast cell lines from fetal liver cells harvested from mice with liver-specific transposase activation. When injected into the flanks of immunodeficient mice, these cells produced tumors that coexpressed epithelial and mesenchymal markers, including Epcam and vimentin, respectively, despite their epithelial origin. The authors examined CIS-associated genes from the tumors using KEGG pathway analysis and identified the involvement of pathways already known to be involved in EMT, supporting the accuracy of their model. Unexpectedly, however, the most highly enriched pathway was ubiquitin-mediated proteolysis, which was not previously associated with EMT. The authors went on to confirm that this pathway is involved in EMT using in vitro assays, and showed that inactivation of the HUWE1, KDM6A, and PTPN12 tumor suppressors led to the EMT phenotype of sorafenib resistance. Thus, the use of an insertional mutagenesis screen led to the identification of candidate genes driving EMT in human HCC58.
Although clinically relevant discoveries have been made through SB transposon insertional mutagenesis, the system has several drawbacks. One disadvantage is the long latency to tumor development, which can take well over 100 days53,57,59. Additionally, the system requires that engineered mice express the mutagenic transposon, the transposase, and a liver-specific Cre recombinase for the system to be functional. Performing these screens on a cancer-predisposed genetic background adds another layer of complexity. Furthermore, in order to obtain sufficient statistical power, large cohorts of mice must be used. For example, a screen by Collier et al. utilized 64 T2/Onc;SB10;Arf−/− animals53. Given the complicated mating schemes required for such experiments, obtaining sufficient animals of the correct genotype requires significant effort and time.
Another issue to consider while implementing insertional mutagenesis screens is the “local hopping” phenomenon in which transposition is more likely to occur near the locus of the parent concatemer (the series of multiple plasmids inserted into a genomic locus), than would be expected by chance. Insertions that occur on the same chromosome as the parent transposon concatemer must be excluded in downstream analysis of CIS loci to reduce false-positive CISs, except when supported by another transposon line in which the concatemer is located on a different chromosome. The exclusion of a chromosome from analysis imposes limits on the ability to fully exploit the findings of a screen. For example, in a study using SB and T2/Onc2 to identify drivers of liver tumorigenesis in the context of HBV surface antigen (HBsAg) expression, CISs identified in chromosome 1 were filtered from the results to account for local hopping60. Thus, a large number of genes on chromosome 1, a gene-rich chromosome harboring the orthologs of numerous known human cancer genes, could not be assessed. Therefore, a follow-up screen was performed in HBsAg mice in which the transposons were mobilized from a transposon concatemer located on chromosome 961. As an alternative to SB, the PB transposon system has less local hopping and may be an improved insertional mutagenesis transposon system, but it has not yet been characterized in HCC models62.
Collectively, these studies using SB to study HCC have shown that the identification of candidate cancer genes is dependent on the components and genetic background used in the screen. A comprehensive comparison of the genes identified by several SB HCC screens found dissimilarities in sample number, genetic background, SB transposon version, sequencing, and bioinformatics methodologies63. Despite these limitations, eight candidate genes were identified as significantly enriched for transposon insertions in at least five of the eight SB-induced HCC models examined63. Human homologs of three of the genes, staphylococcal nuclease and tudor domain containing 1 (Snd1), StAR-related lipid transfer (START) domain containing 13 (Stard13; also known as deleted in liver cancer 2), and thyroid hormone receptor interactor 12 (Trip12), have already been implicated in HCC64–66. Two other candidate genes, dihydropyrimidine dehydrogenase (Dpyd) and glycogen synthase kinase 3 beta (Gsk3b) may mediate tumor chemoresistance67,68. While the relevance of these eight genes to HCC is not yet entirely clear, the regularity with which they are associated with CISs, as well as previous studies linking many to cancer, strongly supports their involvement in the disease. Future studies will be needed to identify cooperative interactions, genetic context dependence, and strengths of these candidate genes as drivers of HCC. Mapping of cooperative interactions has been described in mouse models of lung adenocarcinoma69, in which a variety of tumor-predisposing GEMMs were used for CRISPR-based tumor suppressor inactivation screens in the lung to interrogate gene-gene interactions.
RNAi screening
Rather than disrupting gene expression at the DNA level, RNA interference (RNAi) knocks down expression of specific genes by targeting degradation of their transcripts (Figures 1B and 3A). In an RNAi screen, mRNAs are targeted for degradation by RISC by base pairing with the processed products of shRNAs expressed from a vector library or small interfering RNAs (siRNAs) transfected as a pool into cells70. This method has been used in numerous large genetic screens in cancer cell lines to identify tumor suppressor genes and genes that confer resistance to drug treatment (reviewed in 71).
Figure 3: Genetic screens performed by hydrodynamic tail vein injection (HTVI) of plasmid pools.
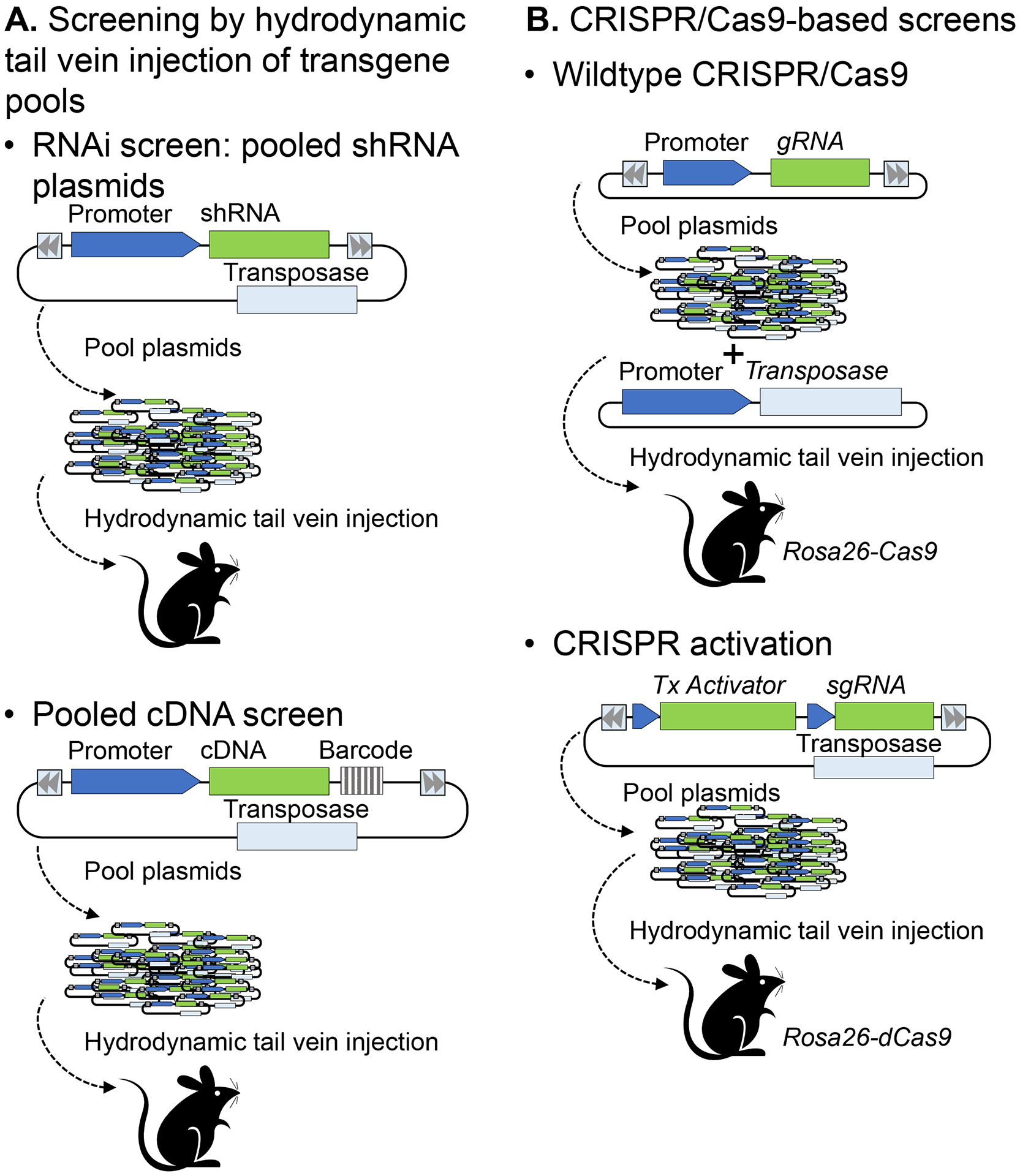
(A) RNAi screens by HDI of a pool of plasmids, each carrying a unique shRNA sequence (top) or a cDNA expression vector (bottom). The shRNA or cDNA linked to the resultant tumors is determined by sequencing the shRNA coding sequence or the unique barcode linked to each cDNA. (B) CRISPR/Cas9 screening systems use either a wild type Cas9 enzyme to cleave target gene DNA (for loss-of-function studies, top), or a dead Cas9 (dCas9) that binds to transcriptional activators (to enhance target gene expression, bottom).
RNAi screens have been performed in vivo to study liver cancer as well. Zender et al. tested the hypothesis that tumor suppressor genes are likely to reside in chromosomal regions that are deleted in HCC72. They first analyzed DNA copy number variations in 100 human HCCs that resulted from different etiologies, then identified the mouse orthologs of the genes of interest in deleted regions. Candidate genes were targeted using mir30-based shRNA sequences. Pools of Retroviruses expressing shRNAs were used to infect Trp53−/− embryonic hepatocytes overexpressing MYC, which were then injected intrasplenically into mice to generate a “mosaic” liver. The tumor lesions resulting in these animals were collected in order to identify the shRNAs through sequencing. The most highly enriched shRNA targeted was exportin 4 (Xpo4), which follow-up studies in human HCC cell lines and tissues have confirmed functions as a tumor suppressor73,74. Xpo4 encodes exportin 4, which is required for the nuclear export of the signal transduction molecule SMAD3, a mediator of TGF-β signaling75. Thus, this report demonstrated the utility of RNAi screens in identifying bona fide tumor suppressor genes.
A separate investigation used the same shRNA library to identify gene targets that could increase the regenerative capacity of hepatocytes in the Fah−/− mouse model50. The shRNAs were subcloned into Fah-SB transposon plasmids. To further model chronic liver damage beyond the induction of tyrosinemia, following repopulation, the Fah−/− mice were treated for six weeks with CCl4. Hairpins targeting mitogen-activated protein kinase kinase 4 (Mkk4; also called Map2k4) became enriched several thousand-fold during both repopulation and CCl4 treatment. Furthermore, the hepatocytes of mice that were injected with an Mkk4-shRNA transposon and then underwent partial hepatectomy (PH) re-entered the cell cycle more quickly than those that were injected with a noncoding shRNA transposon. Hepatocytes that expressed Mkk4-shRNA were also more resistant to induced apoptosis, had an increased proliferation rate, and an increased survival rate in vitro, after which they could repopulate the mouse liver. Notably, Mkk4-shRNA-expressing mice did not develop Mkk4-shRNA-expressing tumors, even after a year50, illustrating how, in some genetic contexts, knockdown of a tumor suppressor can improve liver regeneration without causing cancer.
In contrast to these studies examining genes that are deleted in HCC, Rudalska et al. targeted genes that undergo amplifications76. In an in vivo RNAi screen, HCC-susceptible Arf−/− mice underwent HTVI with a plasmid expressing SB transposase and a transposon vector expressing oncogenic neuroblastoma ras oncogene (NrasG12V) plus a library of shRNAs against the genes of interest in order to uncover a possible mechanism leading to sorafenib resistance. Half of the mice receiving shRNA libraries were treated with sorafenib, while the other half were treated with vehicle. Notably, two mitogen-activated protein kinase 14 (Mapk14) shRNAs were depleted over 100-fold in sorafenib-treated livers, suggesting that Mapk14 expression promotes tumor growth and sorafenib resistance. Encouragingly, Arf−/− mice with tumor growth triggered by NrasG12V that were treated with a combination of sorafenib and the MAPK14 inhibitor BIRB796 had a lower tumor burden and longer survival than mice treated by sorafenib alone. Together, this suggests that sorafenib does not completely inhibit kinase signaling in rodent models and that the addition of a second kinase inhibitor is a potential strategy to treat HCC76.
RNAi screening has led to a better understanding of HCC mechanisms and possible treatments, but like insertional mutagenesis, it has limitations that must be considered. In particular, RNAi activation in vivo has the potential to overwhelm the endogenous RNAi machinery, leading to interference with normal microRNA processing that may be critical for hepatocyte function, causing severe hepatotoxicity77–79. Furthermore, there may be off-target effects from binding of shRNAs to similar target sequences. Therefore, all of the most promising hits from these screens must undergo extensive validation, such as with additional specific shRNAs80. A potential advantage of RNAi is that it could potentially translate to a direct therapeutic in the form of siRNAs that are targeted to genes in the liver (reviewed in 81).
CRISPR-based screening
Due to its ease of use, specificity, and scalability, the CRISPR/Cas9 system is now widely used for gene editing and to modulate endogenous gene expression in cancer studies, including studies on tumor initiation and progression in mice (Figure 3B).
The CRISPR/Cas9 system was adapted from bacteria for genome and epigenome editing in eukaryotes (reviewed in depth in 82 and 83) and in genome-wide screens, particularly in cancer (reviewed in 84 and 85). Briefly, wild type Cas9 is an RNA-guided DNA endonuclease that forms a complex with a gRNA molecule, which then targets the complex to a complementary DNA sequence where it cleaves both strands of DNA. This system been adapted to target the induction of double strand breaks (DSBs) in order to precisely edit the genome by homology-directed repair, or to mutate the genome by insertion or deletions of local nucleotides (indels) introduced by non-homologous end joining82. A catalytically dead Cas9 (dCas9), which cannot cleave DNA, can be used to target genomic loci for activation (known as CRISPR activation, or CRISPRa) or for inhibition (known as CRISPR inhibition or CRISPRi).
CRISPR screens performed in vitro are capable of identifying mediators of cell viability and potential drug targets in HCC. For example, a recent screen used the human genome-scale CRISPR knockout library version 2 (GeCKO v2), which comprises gRNAs targeting 19,050 genes, to perform a loss-of-function screen in human HCC-derived Huh7 cells being treated with sorafenib86. Among cells that were resistant to sorafenib treatment, gRNAs targeting shugoshin 1 (SGOL1) were the most enriched, suggesting that expression of this gene is necessary for sorafenib to induce cell death. Intriguingly, the authors found that high SGOL1 expression in HCCs predicts worse survival. Future studies will need to examine whether patients with high SGOL1 are more likely to respond to sorafenib. Another recent screen used the HCC cell lines Hep3B and Huh7 and transduced them with a lentiviral gRNA library targeting the full complement of human kinases87. gRNAs targeting cyclin-dependent kinase 7 (CDK7) were significantly depleted in both cell lines, suggesting that this gene is essential for HCC cell survival. Consistent with these results, CDK7 is upregulated in human HCC, and the expression correlates with worse survival. Importantly, HCC cell lines and mouse HCC xenografts were sensitive to a CDK7 inhibitor as long as MYC was upregulated in these cells, which is consistent with previous studies of CDK7 inhibitors and MYC-driven cancer88–90. Thus, these studies highlight the power of in vitro screens to identify promising therapeutic targets, but the results must be validated by comparison to human data, such as datasets from TCGA, in order to determine their clinical relevance.
A number of very large genetic screens using CRISPR have been performed in ex vivo transduced cancer cell lines, derived from leukemia, melanoma, intestine and lung, which could be xenografted into recipient mice to examine genes associated with phenotypes such as metastasis91–94. In these cases, the cell lines undergo a drug selection step to ensure that the gRNAs are linked to the resultant tumors, which can be easily sequenced and used as a surrogate for the genetic changes that become enriched in resultant tumors. The disadvantage of this approach is that these screens cannot examine tumor initiation, as the cells have already been transformed.
An ex vivo knockout screen was performed by Song et al. using cancer predisposed Trp53−/− mouse embryonic liver progenitor cells expressing oncogenic MYC and Cas995. The cells were stably transduced with the mGeCKOa lentiviral library expressing gRNAs targeting 20,611 mouse genes and transplanted subcutaneously into nude mice. The relative enrichment of gRNAs in the resultant tumors was measured by high throughput sequencing of pre- and post-implantation cells, revealing significant enrichment of all three gRNAs for only one target, neurofibromin 1 (Nf1). Mutations in NF1 cause neurofibromatosis and have been reported in cholangiocarcinoma. To validate the function of Nf1 as a tumor suppressor in hepatocytes, the authors examined human HCC data and identified point mutations in NF1. Accelerated tumorigenesis in mice injected with plasmids encoding Myc, Cas9, and Nf1-gRNA in multiple tumor-predisposed genetic backgrounds further validated the tumor suppressor function of Nf1. Notably, HCC patients with low NF1 mRNA levels also had shorter survival times than those with high NF1 mRNA levels. Despite the clinical relevance of this discovery, tumor initiation may be influenced by the liver microenvironment. Thus, in vivo screens with the same lentiviral library may result in enrichment of gRNAs against a different set of targets95.
In vivo modeling of oncogenesis using CRISPR systems has developed rapidly over the past few years. In a few of the first studies, mice were shown to develop lung cancer after intra-tracheal injection of viruses delivering CRISPR/Cas9 components that targeted a variety of tumor suppressor genes, caused chromosomal translocation, or activated Kirsten rat sarcoma viral oncogene homolog (Kras) via homology directed repair to a mutant form96–98.
In the liver, HTVI of CRISPR plasmids targeting mutations in Trp53 and phosphate and tensin homolog (Pten) led to the development of liver tumors by 3 months99. The liver tumors had bile duct features that were similar to tumors that develop in mouse models of ablation of these two genes. CRISPR/Cas9 was also used to generate activated β-catenin, one of the most common features of HCC, by HTVI of plasmids providing Cas9, a gRNA targeting the β-catenin gene, Ctnnb1, and a repair cassette to insert four activating point mutations into the gene via homology-directed repair. After injection, nuclear β-catenin, a marker of activation, was detected in a number of hepatocytes, demonstrating the ability of CRISPR systems to model HCC99.
The first true in vivo multiplexed CRISPR/Cas9 screen was performed by HTVI of plasmids into cancer predisposed KrasG12D mice100. The plasmids consisted of SB transposase and transposons containing a Cas9 expression cassette plus gRNAs targeting up to 18 different putative liver cancer genes. The mice developed multiple tumors by 20–30 weeks of age. Similarly, wild type mice injected with the Cas9-gRNA library and treated with CCl4 developed multiple tumors. Interestingly, the transposon sequences were found to be integrated in the genomic DNA of only 5% of tumors, indicating that transient gRNA expression could lead to tumor formation. Therefore, rather than determining the enrichment of the gRNA sequences in each tumor, the target sites were instead examined for indels. Numerous indels were found in each tumor, with significant enrichment of mutations at Pten and tet methylcytosine dioxygenase 2 (Tet2) loci, which was correlated with a cholangiocarcinoma-type tumor phenotype. Sequencing also showed that Cas9-induced large intrachromosomal deletions between gRNA target sites. Thus, this was the first demonstration that CRISPR/Cas9 could be used for a multiplexed screen in vivo to induce specific genetic mutations, and to correlate the genetic changes with the tumor phenotype. Importantly, however, the study highlighted the challenges to scaling up, as even transient gRNA expression could lead to dramatic changes to the genome. Use of larger gRNA libraries with wild type Cas9 would potentially require whole exome sequencing of tumors to characterize all of the target site changes, which is costly and labor-intensive.
A large, fully in vivo loss-of-function CRISPR screen in HCC was performed by Xu et al. more recently101. They built a PB transposon library using the GeCKOv2 genome-scale mouse CRISPR/Cas9 knockout library containing over 130,000 gRNAs targeting all mouse protein coding genes and miRNAs, which was delivered by HTVI to recipient mice together with vectors expressing cyclin dependent kinase inhibitor 2A (Cdkn2a)-gRNA and NrasG12V to accelerate tumorigenesis. This resulted in the development of tumors in 9 out of 27 mice at 45 weeks. The gRNAs linked to tumors were identified by sequencing, revealing that 271 gRNAs were present in tumor genomic DNA, including 26 gRNAs targeting 21 known tumor suppressor genes. In support of the efficacy of this technique, two gRNAs targeted the well characterized tumor suppressor Trp53. Unexpectedly, Cdkn2b, which was not previously recognized as having a role in liver cancer, was targeted by three gRNAs. Mutations in the target region of Cdkn2b, as well as the development of tumors in mice injected with Cdkn2b-gRNA in the absence of a sensitizing background, confirmed that Cdkn2b functions as a tumor suppressor gene in the mouse liver. The identification of a new tumor suppressor gene supports the potential for CRISPR/Cas9 to be used for large-scale screens in vivo to identify drivers of liver cancer102. What was not clear in this manuscript was whether, similar to what was seen in the first in vivo multiplexed CRISPR/Cas9 screen103, substantial genetic changes from gRNA-mediated chromosomal cleavages were missed because of transient gRNA expression102.
With a similar aim of targeting multiple tumor suppressors throughout the liver to study HCC, Wang et al. developed a CRISPR system that used pools of adeno-associated viruses (AAVs)104. AAV virus is highly effective at infecting hepatocytes, approaching expression in 100% hepatocytes for certain serotypes105,106. As it only rarely integrates sequences into the genome, expression with AAV is transient. In the Wang study, each AAV was designed to express Cre, a gRNA targeting Trp53, and a gRNA targeting one of 49 putative tumor suppressor genes or 7 housekeeping control genes, with 4–5 gRNAs for each target104. The authors injected the AAV library into lox-stop-lox-Cas9 mice, which developed multiple large liver tumor nodules within four months. Because gRNA sequences likely had dissipated, the target genomic sites were examined to discover which mutations were linked to the tumors. The authors used an approach called molecular inversion probe (MIP) capture sequencing107 to assess for mutations in the ±70–base pair regions surrounding the predicted cut site for each of the gRNAs in their library. Analysis of co-occurring mutations revealed that beta-2 microglobulin (B2m) and KAT8 regulatory NSL complex subunit 1 (Kansl1) mutations were often found together. Notably, individually targeting B2m or Kansl1 using AAV-CRISPR did not induce tumorigenesis, while targeting both together accelerated tumorigenesis, validating their approach for the identification of gene-gene interactions that drive HCC. The drawbacks of this approach are that many mutations – such as those secondary to Trp53 knockout or due to large chromosomal deletions or off target effects – may be missed by this technique. Indeed, the causative mutations were undetectable by MIP capture sequencing in a number of the tumors analyzed in this study. Furthermore, scale up to include hundreds or thousands of genes may not be feasible with this methodology.
Instead of inducing permanent alterations in the genome with CRISPR/Cas9 to induce loss of function, or overexpressing cDNAs to assess gain of function, expression of specific genes can either be repressed (CRISPRi) or activated (CRISPRa) through systems employing dCas982,84 (Figure 3B). CRISPRi, which comprises dCas9 and gRNAs complementary to the target gene promoters, represses transcription either by steric inhibition of the transcriptional machinery by dCas9 alone or through fusion of dCas9 to the Kruppel-associated box (KRAB) transcriptional repressor82. Conversely, in the CRISPRa system, a transcriptional activator (TA) is either tethered to the dCas9 or recruited to minimal hairpin aptamers appended to the to the stem loop regions of the gRNA. CRISPRa induces the expression of the endogenous gene, which circumvents limitations of cDNA overexpression screens such as difficulty in building comprehensive libraries due to the variability in length of the cDNA sequences, and in capturing all of the gene isoforms82,108–110.
The “first-generation” CRISPRa system consists of the TA VP64, derived from four tandem copies of the Herpes Simplex Viral Protein 16 (VP16), fused directly to the C terminus of dCas9. Subsequent refined versions are referred to as “second-generation” activators111. Two significant “second-generation” activators are the SunTag system and synergistic activation mediator (SAM). The SunTag version is composed of dCas9 fused to a multimer of short peptide epitopes that binds multiple copies of its cognate single-chain variable fragment (scFv) domain, which is fused to VP64112. SAM harnesses the synergistic effects of multiple activation domains: VP64 is tethered to dCas9, while NF-kB trans-activating subunit p65 and human heat shock factor 1 (HSF1) are bound to MS2, a bacteriophage coat protein that selectively binds the hairpin aptamers that are appended to the gRNA scaffold109. Both SAM and SunTag produce robust transcriptional activation from a single gRNA, making them practical for multiplexed, genome-wide gain-of-function screens111.
In order to streamline the CRISPRa system for use in in vivo screens targeted to specific tissues, including hepatocytes, we derived mice encoding a nuclease-deficient dCas9 allele fused to the ‘SunTag’ domain at the Rosa26 locus, termed dCas9+, with an upstream floxed stop cassette45. We then crossed them to Fah−/− mice and activated dCas9 expression specifically in hepatocytes by injecting AAV-Cre. Using SB transposon vectors, we generated a library of plasmids containing the TA scFv-VP64, an Fah expression cassette, and gRNAs targeting the promoters of Myc, tumor necrosis factor superfamily member 1a (Tnfrsf1a), solute carrier family 7 member 11 (Slc7a11), and Trp53, which we intravenously injected into the Fah−/−;dCas9+ mice. Nodules of MYC-expressing, FAH-positive hepatocytes were found in Fah−/−;dCas9+ livers, indicating a robust and specific activation of expression of the endogenous gene. Myc gRNA sequences were highly significantly enriched in tumors, while Trp53 and Tnfrsf1a gRNAs were significantly depleted. Thus, the in vivo dCas9 system activated the Myc locus in hepatocytes and accelerated repopulation by these cells. The potent activation of MYC expression by CRISPRa strongly supports the potential for this system to be expanded to perform large scale screens of sets of genes belonging to a specific pathway simultaneously in vivo45. Furthermore, the dCas9+ mouse can be crossed to GEMMs of HCC, such as the HBsAg transgenic mouse113, to identify context-specific drivers of tumorigenesis associated with the multitude of HCC risk factors, as well as potential drug targets and mediators of resistance to chemotherapeutics.
General considerations with genetic screens
The following are important considerations for genetic screening in general:
Genetic screens must take into account statistical power and the rate of false discovery. In the case of transposon insertional mutagenesis, mutation events in a particular gene must be observed in independently a number of times114. For cDNA, CRISPR, and RNAi experiments, the results also need to be observed independently in multiple replicates. There is the additional consideration of the “coverage” of the sequencing. In producing a screening library, there is a natural distribution of the components of the screen, and the lowly represented components may be lost by chance115. The standard for CRISPR screening on a genome scale, which often includes 100,000 plasmids or more84,116, is to screen and sequence to a depth of greater than 100-fold of the median amount of the plasmid117,118.
Off-target or non-specific effects also need to be considered. In the case of transposons, “local hopping” is overcome by excluding genes located on the same chromosome as the donor transposon from the analyses, or by using independent transgenic mice with transposon donors on different chromosomes. In the case of RNAi and CRISPR screening, multiple shRNA or gRNA sequences are typically designed to target the same gene, which helps ensure that the observed effect is specific80,118.
Finally, positive hits from a screen should be further validated in isolation, ideally with multiple modalities such as gene deletion, temporal or conditional control, or drug targeting117.
Future directions
Techniques for performing high-throughput genetic screens of cancer candidate genes have evolved dramatically over the past decade, with new technology enabling an exponential growth in the number and types of genes that can be screened. Liver cancer screens will continue to evolve as we develop new strategies to more accurately model HCC, classify its subtypes, and determine the responses to specific drug treatments. Transposon mutagenesis studies on different cancer-predisposed backgrounds have demonstrated the power of a predisposing mutation to influence which specific mutations become enriched in tumors. SB transposon and RNAi screens have provided clues to the mechanisms of sorafenib resistance, and CRISPR screens will likely be powerful tools in future studies to examine drivers and tumor suppressors in the setting of different types of liver injury, as well as drug resistance and sensitivity patterns. Re-framing the concept of HCC as a group of diseases, based on discoveries from these previous studies, will increase the power of screens to identify the clusters of genes cooperatively driving HCC in subsets of patients, and the alterations in genes leading to sensitivity or resistance to specific treatments, enabling a personalized approach to HCC treatment with pharmaceuticals.
Finally, the technologies that have been applied so far – HTVI of plasmids, transposable elements, RNAi, and various types of CRISPR – are likely to evolve and improve in the decades to come. For example, future screens may examine cooperation between technologies to discover how combinations of gene activators and gene inhibitors interact. They may also include screens of libraries of expressed peptides or RNA molecules, which could accelerate the development of drugs that significantly improve the survival of patients with HCC.
Main Concepts and Learning Points.
Genetic screens in animal models shed light on functional alterations in HCC
Advances in sequencing technology allow for increasingly complex screens
Liver genetic screens are revealing the heterogeneity of genetic alterations in HCC patients
Future work may apply genetic screening to discover new treatments for HCC
Financial Support
This work was supported by the National Institutes of Health (K08-DK106478 to K.J.W.) and by the NIH Physical Sciences in Oncology Network center PSOC@Penn (U54-CA193417).
List of Abbreviations:
- AAV-Cre
serotype 8 adeno-associated virus with thyroxin-binding globulin promoter driving Cre recombinase enzyme expression
- AFP
alpha-fetoprotein
- AKT
thymoma viral proto-oncogene 1
- ARF
alternative reading frame protein; human: p14ARF, mouse: p19ARF
- Apc
adenomatous polyposis coli
- B2m
beta-2 microglobulin
- Cas9
CRISPR associated protein 9
- CDK7
cyclin-dependent kinase 7
- Cdkn2a
cyclin dependent kinase inhibitor 2A
- cDNA
complementary DNA
- CIS
common insertion site
- CRISPR
clustered regularly interspaced short palindromic repeats
- CRISPRa
CRISPR activation
- CRISPRi
CRISPR inhibition
- dCas9
catalytically dead Cas9
- Dpyd
dihydropyrimidine dehydrogenase
- EGFR
epidermal growth factor receptor
- FAH
fumarylacetoacetate hydrolase
- GeCKO
genome-scale CRISPR knockout
- GEMM
genetically engineered mouse model
- Gsk3b
glycogen synthase kinase 3 beta
- gRNA
guide RNA
- HBsAg
HBV surface antigen
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HTVI
hydrodynamic tail vein injection
- HSF1
heat shock factor 1
- HTS
high-throughput sequencing
- Kansl1
KAT8 regulatory NSL complex subunit 1
- KRAB
Kruppel-associated box
- Kras
Kirsten rat sarcoma viral oncogene homolog
- Mapk14
mitogen-activated protein kinase 14
- Mkk4
mitogen-activated protein kinase kinase 4; also called Map2k4
- mRNA
messenger RNA
- Nf1
neurofibromin 1
- Nras
neuroblastoma ras oncogene
- PB
piggyBac
- PH
partial hepatectomy
- Pten
phosphate and tensin homolog
- RISC
RNA-induced silencing complex
- RNAi
RNA interference
- SAM
synergistic activation mediator
- SB
Sleeping Beauty
- scFv
single-chain variable fragment
- shRNA
short hairpin RNA
- Slc7a11
solute carrier family 7 member 11
- Snd1
staphylococcal nuclease and tudor domain containing 1
- Stard13
StAR-related lipid transfer (START) domain containing 13; also known as deleted in liver cancer 2
- TA
transcriptional activator
- TBG
thyroxin-binding globulin
- Tet2
tet methylcytosine dioxygenase 2
- TGFα
transforming growth factor α
- Tnfrsf1a
tumor necrosis factor superfamily member 1a
- TP53
tumor protein 53; ortholog of Trp53
- Trp53
transformation related protein 53
- Trip12
thyroid hormone receptor interactor 12
- tTA/TRE
tetracycline transactivator/tetracycline response element
- VP16
Herpes Simplex Viral Protein 16
- Xpo4
exportin 4
References
- 1.European Association for the study of the liver. EASL–EORTC Clinical Practice Guidelines: Management of hepatocellular carcinoma. J Hepatol [Internet]. 2012;56(4):908–943. Available from: http://www.journal-of-hepatology.eu/article/S0168-8278(11)00873-7/pdf [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global Cancer Statistics, 2002. CA Cancer J Clin [Internet]. 2005;55(2):74–108. Available from: http://doi.wiley.com/10.3322/canjclin.55.2.74 [DOI] [PubMed] [Google Scholar]
- 3.Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, Von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnström H, Glimelius B, Sjöblom T, Edqvist PH, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F. A pathology atlas of the human cancer transcriptome. Science (80-) 2017;357(6352). [DOI] [PubMed] [Google Scholar]
- 4.Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: Reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res. 2014;20(8):2072–2079. [DOI] [PubMed] [Google Scholar]
- 5.Wangensteen L, Wangensteen KJ, Evans SG, Everts LE, Trooskin SB. Hepatitis C: How to fine-tune your approach. J Fam Pr. 2015; [PubMed] [Google Scholar]
- 6.Goldberg D, Ditah IC, Saeian K, Lalehzari M, Aronsohn A, Gorospe EC, Charlton M. Changes in the Prevalence of Hepatitis C Virus Infection, Nonalcoholic Steatohepatitis, and Alcoholic Liver Disease Among Patients With Cirrhosis or Liver Failure on the Waitlist for Liver Transplantation. Gastroenterology. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Thiel DH, Ramadori G. Non-viral causes of hepatocellular carcinoma. Journal of Gastrointestinal Cancer. 2011. p. 191–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015;149(5):1226–1239. [DOI] [PubMed] [Google Scholar]
- 9.Ally A, Balasundaram M, Carlsen R, Chuah E, Clarke A, Dhalla N, Holt RA, Jones SJM, Lee D, Ma Y, Marra MA, Mayo M, Moore RA, Mungall AJ, Schein JE, Sipahimalani P, Tam A, Thiessen N, Cheung D, Wong T, Brooks D, Robertson AG, Bowlby R, Mungall K, Sadeghi S, Xi L, Covington K, Shinbrot E, Wheeler DA, Gibbs RA, Donehower LA, Wang L, Bowen J, Gastier-Foster JM, Gerken M, Helsel C, Leraas KM, Lichtenberg TM, Ramirez NC, Wise L, Zmuda E, Gabriel SB, Meyerson M, Cibulskis C, Murray BA, Shih J, Beroukhim R, Cherniack AD, Schumacher SE, Saksena G, Pedamallu CS, Chin L, Getz G, Noble M, Zhang H, Heiman D, Cho J, Gehlenborg N, Saksena G, Voet D, Lin P, Frazer S, Defreitas T, Meier S, Lawrence M, Kim J, Creighton CJ, Muzny D, Doddapaneni HV, Hu J, Wang M, Morton D, Korchina V, Han Y, Dinh H, Lewis L, Bellair M, Liu X, Santibanez J, Glenn R, Lee S, Hale W, Parker JS, Wilkerson MD, Hayes DN, Reynolds SM, Shmulevich I, Zhang W, Liu Y, Iype L, Makhlouf H, Torbenson MS, Kakar S, Yeh MM, Jain D, Kleiner DE, Jain D, Dhanasekaran R, El-Serag HB, Yim SY, Weinstein JN, Mishra L, Zhang J, Akbani R, Ling S, Ju Z, Su X, Hegde AM, Mills GB, Lu Y, Chen J, Lee JS, Sohn BH, Shim JJ, Tong P, Aburatani H, Yamamoto S, Tatsuno K, Li W, Xia Z, Stransky N, Seiser E, Innocenti F, Gao J, Kundra R, Zhang H, Heins Z, Ochoa A, Sander C, Ladanyi M, Shen R, Arora A, Sanchez-Vega F, Schultz N, Kasaian K, Radenbaugh A, Bissig KD, Moore DD, Totoki Y, Nakamura H, Shibata T, Yau C, Graim K, Stuart J, Haussler D, Slagle BL, Ojesina AI, Katsonis P, Koire A, Lichtarge O, Hsu TK, Ferguson ML, Demchok JA, Felau I, Sheth M, Tarnuzzer R, Wang Z, Yang L, Zenklusen JC, Zhang J, Hutter CM, Sofia HJ, Verhaak RGW, Zheng S, Lang F, Chudamani S, Liu J, Lolla L, Wu Y, Naresh R, Pihl T, Sun C, Wan Y, Benz C, Perou AH, Thorne LB, Boice L, Huang M, Rathmell WK, Noushmehr H, Saggioro FP, Tirapelli DP da C, Junior CGC, Mente ED, Silva O de C, Trevisan FA, Kang KJ, Ahn KS, Giama NH, Moser CD, Giordano TJ, Vinco M, Welling TH, Crain D, Curley E, Gardner J, Mallery D, Morris S, Paulauskis J, Penny R, Shelton C, Shelton T, Kelley R, Park JW, Chandan VS, Roberts LR, Bathe OF, Hagedorn CH, Auman JT, O’Brien DR, Kocher JPA, Jones CD, Mieczkowski PA, Perou CM, Skelly T, Tan D, Veluvolu U, Balu S, Bodenheimer T, Hoyle AP, Jefferys SR, Meng S, Mose LE, Shi Y, Simons JV., Soloway MG, Roach J, Hoadley KA, Baylin SB, Shen H, Hinoue T, Bootwalla MS, Van Den Berg DJ, Weisenberger DJ, Lai PH, Holbrook A, Berrios M, Laird PW. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169(7):1327–1341.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calderaro J, Couchy G, Imbeaud S, Amaddeo G, Letouzé E, Blanc JF, Laurent C, Hajji Y, Azoulay D, Bioulac-Sage P, Nault JC, Zucman-Rossi J. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J Hepatol. 2017;67(4):727–738. [DOI] [PubMed] [Google Scholar]
- 11.Hoshida Y, Nijman SMB, Kobayashi M, Chan JA, Brunet J-P, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, Watanabe G, Gabriel S, Friedman SL, Kumada H, Llovet JM, Golub TR. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res [Internet]. 2009;69(18):7385–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19723656%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC3549578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nature Reviews Clinical Oncology. 2015. p. 408–424. [DOI] [PubMed] [Google Scholar]
- 14.Llovet JM, Ducreux M, Lencioni R, Di Bisceglie AM, Galle PR, Dufour JF, Greten TF, Raymond E, Roskams T, De Baere T, Mazzaferro V, Bernardi M, Bruix J, Colombo M, Zhu A. EASL-EORTC Clinical Practice Guidelines: Management of hepatocellular carcinoma. Eur J Cancer. 2012;48(5):599–641. [DOI] [PubMed] [Google Scholar]
- 15.Xie B, Wang DH, Spechler SJ. Sorafenib for treatment of hepatocellular carcinoma: A systematic review. Dig Dis Sci. 2012;57(5):1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med [Internet] 2008;359(4):378–390. Available from: http://www.nejm.org/doi/pdf/10.1056/NEJMoa0708857 [DOI] [PubMed] [Google Scholar]
- 17.Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, Gerolami R, Masi G, Ross PJ, Song T, Bronowicki JP, Ollivier-Hourmand I, Kudo M, Cheng AL, Llovet JM, Finn RS, LeBerre MA, Baumhauer A, Meinhardt G, Han G. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66. [DOI] [PubMed] [Google Scholar]
- 18.Sprinzl MF, Galle PR. Current progress in immunotherapy of hepatocellular carcinoma. J Hepatol. 2017;66(3):482–484. [DOI] [PubMed] [Google Scholar]
- 19.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20(19):5064–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling TH, Meyer T, Kang YK, Yeo W, Chopra A, Anderson J, dela Cruz C, Lang L, Neely J, Tang H, Dastani HB, Melero I. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet [Internet]. Elsevier Ltd; 2017;389(10088):2492–2502. Available from: 10.1016/S0140-6736(17)31046-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White R, Rose K, Zon L. Zebrafish cancer: The state of the art and the path forward. Nature Reviews Cancer. 2013. p. 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giacomotto J, Ségalat L. High-throughput screening and small animal models, where are we? British Journal of Pharmacology. 2010. p. 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wangensteen KJ, Zhang S, Greenbaum LE, Kaestner KH. A genetic screen reveals Foxa3 and TNFR1 as key regulators of liver repopulation. Genes Dev. 2015;29(9):904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santos NP, Colaço AA, Oliveira PA. Animal models as a tool in hepatocellular carcinoma research: A Review. Tumor Biol. 2017;39(3). [DOI] [PubMed] [Google Scholar]
- 25.He L, Tian D-A, Li P-Y, He X-X. Mouse models of liver cancer: Progress and recommendations. Oncotarget [Internet]. 2015;6(27):23306–23322. Available from: http://www.oncotarget.com/fulltext/4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newell P, Villanueva A, Friedman SL, Koike K, Llovet JM. Experimental models of hepatocellular carcinoma. Journal of Hepatology. 2008. p. 858–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caviglia JM, Schwabe RF. Mouse Models of Cancer. 2015;1267 Available from: http://link.springer.com/10.1007/978-1-4939-2297-0 [DOI] [PubMed]
- 28.Murakami H, Sanderson ND, Nagy P, Marino PA, Merlino G, Thorgeirsson SS. Transgenic Mouse Model for Synergistic Effects of Nuclear Oncogenes and Growth Factors in Tumorigenesis - Interaction of c- myc and Transforming Growth Factor-alpha in Hepatic Oncogenesis. Cancer Res. 1993;53:1719–1723. [PubMed] [Google Scholar]
- 29.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson Å, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, Yang Q, Bishop JM, Contag CH, Felsher DW. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431(7012):1112–1117. [DOI] [PubMed] [Google Scholar]
- 30.Fan Y, Arechederra M, Richelme S, Daian F, Novello C, Calderaro J, Di Tommaso L, Morcrette G, Rebouissou S, Donadon M, Morenghi E, Zucman-Rossi J, Roncalli M, Dono R, Maina F. A phosphokinome-based screen uncovers new drug synergies for cancer driven by liver-specific gain of nononcogenic receptor tyrosine kinases. Hepatology. 2017;66(5):1644–1661. [DOI] [PubMed] [Google Scholar]
- 31.Liu F, Song YK, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6(7):1258–1266. [DOI] [PubMed] [Google Scholar]
- 32.Zhang G, Budker V, Wolff JA. High Levels of Foreign Gene Expression in Hepatocytes after Tail Vein Injections of Naked Plasmid DNA. Hum Gene Ther [Internet]. 1999;10(10):1735–1737. Available from: http://www.liebertonline.com/doi/abs/10.1089/10430349950017734 [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. American Journal of Pathology. 2014. p. 912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ju HL, Han KH, Lee JD, Ro SW. Transgenic mouse models generated by hydrodynamic transfection for genetic studies of liver cancer and preclinical testing of anti-cancer therapy. Int J Cancer. 2016;138(7):1601–1608. [DOI] [PubMed] [Google Scholar]
- 35.Andrianaivo F, Lecocq M, Wattiaux-De Coninck S, Wattiaux R, Jadot M. Hydrodynamics-based transfection of the liver: Entrance into hepatocytes of DNA that causes expression takes place very early after injection. J Gene Med. 2004;6(8):877–883. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi N, Nishikawa M, Hirata K, Takakura Y. Hydrodynamics-based procedure involves transient hyperpermeability in the hepatic cellular membrane: Implication of a nonspecific process in efficient intracellular gene delivery. J Gene Med. 2004;6(5):584–592. [DOI] [PubMed] [Google Scholar]
- 37.Yant SR, Meuse L, Chiu W, Ivics Z, Izsvak Z, Kay MA. Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat Genet. 2000;25(1):35–41. [DOI] [PubMed] [Google Scholar]
- 38.Zhang G, Gao X, Song YK, Vollmer R, Stolz DB, Gasiorowski JZ, Dean DA, Liu D. Hydroporation as the mechanism of hydrodynamic delivery. Gene Ther. 2004;11(8):675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilber A, Frandsen JL, Wangensteen KJ, Ekker SC, Wang X, McIvor RS. Dynamic gene expression after systemic delivery of plasmid DNA as determined by in vivo bioluminescence imaging. Hum Gene Ther. 2005;16(11):1325–1332. [DOI] [PubMed] [Google Scholar]
- 40.Mikkelsen JG, Yant SR, Meuse L, Huang Z, Xu H, Kay MA. Helper-independent Sleeping Beauty transposon-transposase vectors for efficient nonviral gene delivery and persistent gene expression in vivo. Mol Ther. 2003;8(4):654–665. [DOI] [PubMed] [Google Scholar]
- 41.Wangensteen KJ, Wilber A, Keng VW, He Z, Matise I, Wangensteen L, Carson CM, Chen Y, Steer CJ, McIvor RS, Largaespada DA, Wang X, Ekker SC. A facile method for somatic, lifelong manipulation of multiple genes in the mouse liver. Hepatology. 2008;47(5):1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saridey SK, Liu L, Doherty JE, Kaja A, Galvan DL, Fletcher BS, Wilson MH. PiggyBac transposon-based inducible gene expression in vivo after somatic cell gene transfer. Mol Ther. 2009;17(12):2115–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balciunas D, Wangensteen KJ, Wilber A, Bell J, Geurts A, Sivasubbu S, Wang X, Hackett PB, Largaespada DA, McIvor RS, Ekker SC. Harnessing a high cargo-capacity transposon for genetic applications in vertebrates. PLoS Genet. 2006;2(11):1715–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Montini E, Held PK, Noll M, Morcinek N, Al-Dhalimy M, Finegold M, Yant SR, Kay MA, Grompe M. In vivo correction of murine tyrosinemia type I by DNA-mediated transposition. Mol Ther [Internet]. American Society for Gene Therapy; 2002;6(6):759–769. Available from: 10.1006/mthe.2002.0812 [DOI] [PubMed] [Google Scholar]
- 45.Wangensteen KJ, Wang YJ, Dou Z, Wang AW, Mosleh-Shirazi E, Horlbeck MA, Gilbert LA, Weissman JS, Berger SL, Kaestner KH. Combinatorial genetics in liver repopulation and carcinogenesis with a novel in vivo CRISPR activation platform. Hepatology. 2017. November; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, Destefanis G, Delogu S, Zimmermann A, Ericsson J, Brozzetti S, Staniscia T, Chen X, Dombrowski F, Evert M. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011;140(3):1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, Gores GJ, Dombrowski F, Evert M, Chen X, Willenbring H. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122(8):2911–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grompe M, Lindstedt S, Al-Dhalimy M, Kennaway NG, Papaconstantinou J, Torres-Ramos CA, Ou CN, Finegold M. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1995;10(4):453–460. [DOI] [PubMed] [Google Scholar]
- 49.Grompe M The Pathophysiology and Treatment of Hereditary Tyrosinemia Type 1. Semin Liver Dis [Internet]. 2001;21(04):563–572. Available from: http://www.thieme-connect.de/DOI/DOI?10.1055/s-2001-19035 [DOI] [PubMed] [Google Scholar]
- 50.Wuestefeld T, Pesic M, Rudalska R, Dauch D, Longerich T, Kang TW, Yevsa T, Heinzmann F, Hoenicke L, Hohmeyer A, Potapova A, Rittelmeier I, Jarek M, Geffers R, Scharfe M, Klawonn F, Schirmacher P, Malek NP, Ott M, Nordheim A, Vogel A, Manns MP, Zender L. A direct in vivo RNAi screen identifies MKK4 as a key regulator of liver regeneration. Cell. 2013;153(2):389–401. [DOI] [PubMed] [Google Scholar]
- 51.Wang AW, Wangensteen KJ, Wang YJ, Zahm AM, Moss NG, Erez N, Kaestner KH. TRAP-seq identifies cystine/glutamate antiporter as a driver of recovery from liver injury. J Clin Invest [Internet] 2018;128(6):1–49. Available from: 10.1172/JCI95120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ivics Z, Hackett PB, Plasterk RH, Izsvák Z. Molecular reconstruction of sleeping beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91(4):501–510. [DOI] [PubMed] [Google Scholar]
- 53.Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436(7048):272–276. [DOI] [PubMed] [Google Scholar]
- 54.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436(7048):221–226. [DOI] [PubMed] [Google Scholar]
- 55.Dupuy AJ, Rogers LM, Kim J, Nannapaneni K, Starr TK, Liu P, Largaespada DA, Scheetz TE, Jenkins NA, Copeland NG. A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res. 2009;69(20):8150–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keng VW, Villanueva A, Chiang DY, Dupuy AJ, Ryan BJ, Matise I, Silverstein KAT, Sarver A, Starr TK, Akagi K, Tessarollo L, Collier LS, Powers S, Lowe SW, Jenkins NA, Copeland NG, Llovet JM, Largaespada DA. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat Biotechnol. 2009;27(3):264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keng VW, Sia D, Sarver AL, Tschida BR, Fan D, Alsinet C, Solé M, Lee WL, Kuka TP, Moriarity BS, Villanueva A, Dupuy AJ, Riordan JD, Bell JB, Kevin KA, Llovet JM, Largaespada DA. Sex bias occurrence of hepatocellular carcinoma in Poly7 molecular subclass is associated with EGFR. Hepatology. 2013;57(1):120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kodama T, Newberg JY, Kodama M, Rangel R, Yoshihara K, Tien JC, Parsons PH, Wu H, Finegold MJ, Copeland NG, Jenkins NA. Transposon mutagenesis identifies genes and cellular processes driving epithelial-mesenchymal transition in hepatocellular carcinoma. Proc Natl Acad Sci [Internet]. 2016;113(24):E3384–E3393. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1606876113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bard-Chapeau EA, Nguyen A-T, Rust AG, Sayadi A, Lee P, Chua BQ, New L-S, de Jong J, Ward JM, Chin CKY, Chew V, Toh HC, Abastado J-P, Benoukraf T, Soong R, Bard FA, Dupuy AJ, Johnson RL, Radda GK, Chan ECY, Wessels LFA, Adams DJ, Jenkins NA, Copeland NG. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. Nat Genet [Internet]. Nature Publishing Group; 2013;46(1):24–32. Available from: http://www.nature.com/doifinder/10.1038/ng.2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bard-Chapeau EA, Nguyen A-T, Rust AG, Sayadi A, Lee P, Chua BQ, New L-S, de Jong J, Ward JM, Chin CKY, Chew V, Toh HC, Abastado J-P, Benoukraf T, Soong R, Bard FA, Dupuy AJ, Johnson RL, Radda GK, Chan ECY, Wessels LFA, Adams DJ, Jenkins NA, Copeland NG. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. Nat Genet. Nature Publishing Group; 2013;46(1):24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kodama T, Bard-Chapeau EA, Newberg JY, Kodama M, Rangel R, Yoshihara K, Ward JM, Jenkins NA, Copeland NG. Two-Step Forward Genetic Screen in Mice Identifies Ral GTPase-Activating Proteins as Suppressors of Hepatocellular Carcinoma. Gastroenterology. 2016;151(2):324–337.e12. [DOI] [PubMed] [Google Scholar]
- 62.Rad R, Rad L, Wang W, Cadinanos J, Vassiliou G, Rice S, Campos LS, Yusa K, Banerjee R, Li MA, De La Rosa J, Strong A, Lu D, Ellis P, Conte N, Yang FT, Liu P, Bradley A. PiggyBac transposon mutagenesis: A tool for cancer gene discovery in mice. Science (80-) 2010;330(6007):1104–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Riordan JD, Feddersen CR, Tschida BR, Beckmann PJ, Keng VW, Linden MA, Amin K, Stipp CS, Largaespada DA, Dupuy AJ. Chronic liver injury alters driver mutation profiles in hepatocellular carcinoma in mice. Hepatology. 2018;67(3):924–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yoo BK, Santhekadur PK, Gredler R, Chen D, Emdad L, Bhutia S, Pannell L, Fisher PB, Sarkar D. Increased RNA-induced silencing complex (RISC) activity contributes to hepatocellular carcinoma. Hepatology. 2011;53(5):1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cai J Bin, Shi GM, Dong ZR, Ke AW, Ma HH, Gao Q, Shen ZZ, Huang XY, Chen H, Yu DD, Liu LX, Zhang PF, Zhang C, Hu MY, Yang LX, Shi YH, Wang XY, Ding Z Bin, Qiu SJ, Sun HC, Zhou J, Shi YG, Fan J. Ubiquitin-specific protease 7 accelerates p14ARF degradation by deubiquitinating thyroid hormone receptor-interacting protein 12 and promotes hepatocellular carcinoma progression. Hepatology. 2015;61(5):1603–1614. [DOI] [PubMed] [Google Scholar]
- 66.Ching YP, Wong CM, Chan SF, Leung THY, Ng DCH, Jin DY, Ng IOL. Deleted in liver cancer (DLC) 2 encodes a RhoGAP protein with growth suppressor function and is underexpressed in hepatocellular carcinoma. J Biol Chem. 2003;278(12):10824–10830. [DOI] [PubMed] [Google Scholar]
- 67.Grassilli E, Narloch R, Federzoni E, Ianzano L, Pisano F, Giovannoni R, Romano G, Masiero L, Leone BE, Bonin S, Donada M, Stanta G, Helin K, Lavitrano M. Inhibition of GSK3B bypass drug resistance of p53-null colon carcinomas by enabling necroptosis in response to chemotherapy. Clin Cancer Res. 2013;19(14):3820–3831. [DOI] [PubMed] [Google Scholar]
- 68.Wei X, McLeod HL, McMurrough J, Gonzalez FJ, Fernandez-Salguero P. Molecular basis of the human dihydropyrimidine dehydrogenase deficiency and 5-fluorouracil toxicity. J Clin Invest [Internet]. 1996;98(3):610–5. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=507468&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rogers ZN, McFarland CD, Winters IP, Seoane JA, Brady JJ, Yoon S, Curtis C, Petrov DA, Winslow MM. Mapping the in vivo fitness landscape of lung adenocarcinoma tumor suppression in mice. Nat Genet. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rao DD, Vorhies JS, Senzer N, Nemunaitis J. siRNA vs. shRNA: Similarities and differences. Advanced Drug Delivery Reviews. 2009. p. 746–759. [DOI] [PubMed] [Google Scholar]
- 71.Diehl P, Tedesco D, Chenchik A. Use of RNAi screens to uncover resistance mechanisms in cancer cells and identify synthetic lethal interactions. Drug Discov Today Technol. 2014;11(1):11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zender L, Xue W, Zuber J, Semighini CP, Krasnitz A, Ma B, Zender P, Kubicka S, Luk JM, Schirmacher P, Richard McCombie W, Wigler M, Hicks J, Hannon GJ, Powers S, Lowe SW. An Oncogenomics-Based In Vivo RNAi Screen Identifies Tumor Suppressors in Liver Cancer. Cell. Elsevier Inc; 2008;135(5):852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liang X-T, Pan K, Chen M-S, Li J-J, Wang H, Zhao J-J, Sun J-C, Chen Y-B, Ma H-Q, Wang Q-J, Xia J-C. Decreased expression of XPO4 is associated with poor prognosis in hepatocellular carcinoma. J Gastroenterol Hepatol. 2011;26(3):544–549. [DOI] [PubMed] [Google Scholar]
- 74.Zhang H, Wei S, Ning S, Jie Y, Ru Y, Gu Y. Evaluation of TGFβ, XPO4, elF5A2 and ANGPTL4 as biomarkers in HCC. Exp Ther Med. 2013;5(1):119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kurisaki A, Kurisaki K, Kowanetz M, Sugino H, Yoneda Y, Heldin C-H, Moustakas A. The Mechanism of Nuclear Export of Smad3 Involves Exportin 4 and Ran. Mol Cell Biol [Internet]. 2006;26(4):1318–1332. Available from: http://mcb.asm.org/cgi/doi/10.1128/MCB.26.4.1318-1332.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rudalska R, Dauch D, Longerich T, McJunkin K, Wuestefeld T, Kang TW, Hohmeyer A, Pesic M, Leibold J, Von Thun A, Schirmacher P, Zuber J, Weiss KH, Powers S, Malek NP, Eilers M, Sipos B, Lowe SW, Geffers R, Laufer S, Zender L. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat Med. 2014;20(10):1138–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441(7092):537–541. [DOI] [PubMed] [Google Scholar]
- 78.Grimm D, Wang L, Lee JS, Schürmann N, Gu S, Börner K, Storm TA, Kay MA. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J Clin Invest. 2010;120(9):3106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Valdmanis PN, Gu S, Chu K, Jin L, Zhang F, Munding EM, Zhang Y, Huang Y, Kutay H, Ghoshal K, Lisowski L, Kay MA. RNA interference-induced hepatotoxicity results from loss of the first synthesized isoform of microRNA-122 in mice. Nat Med. 2016;22(5):557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Munoz DM, Cassiani PJ, Li L, Billy E, Korn JM, Jones MD, Golji J, Ruddy DA, Yu K, McAllister G, Deweck A, Abramowski D, Wan J, Shirley MD, Neshat SY, Rakiec D, De Beaumont R, Weber O, Kauffmann A, Robert McDonald E, Keen N, Hofmann F, Sellers WR, Schmelzle T, Stegmeier F, Schlabach MR. CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discov. 2016;6(8):900–913. [DOI] [PubMed] [Google Scholar]
- 81.Wittrup A, Lieberman J. Knocking down disease: A progress report on siRNA therapeutics. Nature Reviews Genetics. 2015. p. 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Komor AC, Badran AH, Liu DR, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen Z-Y, Liu DR, Al E CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell [Internet]. 2017;168(1–2):20–36. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27866654%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC5235943%5Cnhttp://linkinghub.elsevier.com/retrieve/pii/S0092867416314659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nature Reviews Genetics. 2015. p. 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moses C, Garcia-Bloj B, Harvey AR, Blancafort P. Hallmarks of cancer: The CRISPR generation. European Journal of Cancer. 2018. p. 10–18. [DOI] [PubMed] [Google Scholar]
- 86.Sun W, He B, Yang B, Hu W, Cheng S, Xiao H, Yang Z, Wen X, Zhou L, Xie H, Shen X, Wu J, Zheng S. Genome-wide CRISPR screen reveals SGOL1 as a druggable target of sorafenib-treated hepatocellular carcinoma. Lab Investig [Internet]. Springer US; 2018;1–11. Available from: 10.1038/s41374-018-0027-6 [DOI] [PubMed] [Google Scholar]
- 87.Wang C, Jin H, Gao D, Wang L, Evers B, Xue Z, Jin G, Lieftink C, Beijersbergen RL, Qin W, Bernards R. A CRISPR screen identifies CDK7 as a therapeutic target in hepatocellular carcinoma. Cell Res. 2018;(January):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, Jenkins CE, Hannett NM, McMillin D, Sanda T, Sim T, Kim ND, Look T, Mitsiades CS, Weng AP, Brown JR, Benes CH, Marto JA, Young RA, Gray NS. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511(7511):616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, Perez-Atayde A, Wong KK, Yuan GC, Gray NS, Young RA, George RE. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159(5):1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, Zhang J, Shimamura T, Capelletti M, Reibel JB, Cavanaugh JD, Gao P, Liu Y, Michaelsen SR, Poulsen HS, Aref AR, Barbie DA, Bradner JE, George RE, Gray NS, Young RA, Wong K-K. Targeting Transcriptional Addictions in Small Cell Lung Cancer with a Covalent CDK7 Inhibitor. Cancer Cell [Internet] 2014;26(6):909–922. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25490451%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4261156%0Ahttp://linkinghub.elsevier.com/retrieve/pii/S1535610814004231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Drost J, Van Jaarsveld RH, Ponsioen B, Zimberlin C, Van Boxtel R, Buijs A, Sachs N, Overmeer RM, Offerhaus GJ, Begthel H, Korving J, Van De Wetering M, Schwank G, Logtenberg M, Cuppen E, Snippert HJ, Medema JP, Kops GJPL, Clevers H Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521(7550):43–47. [DOI] [PubMed] [Google Scholar]
- 92.Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, Scott DA, Song J, Pan JQ, Weissleder R, Lee H, Zhang F, Sharp PA. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160(6):1246–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science [Internet]. 2014;343(6166):80–4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24336569%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC3972032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Braun CJ, Bruno PM, Horlbeck MA, Gilbert LA, Weissman JS, Hemann MT. Versatile in vivo regulation of tumor phenotypes by dCas9-mediated transcriptional perturbation. Proc Natl Acad Sci. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Song CQ, Li Y, Mou H, Moore J, Park A, Pomyen Y, Hough S, Kennedy Z, Fischer A, Yin H, Anderson DG, Conte D, Zender L, Wang XW, Thorgeirsson S, Weng Z, Xue W. Genome-Wide CRISPR Screen Identifies Regulators of Mitogen-Activated Protein Kinase as Suppressors of Liver Tumors in Mice. Gastroenterology [Internet]. Elsevier, Inc; 2017;152(5):1161–1173.e1. Available from: 10.1053/j.gastro.2016.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng G, Sharp PA, Zhang F. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159(2):440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, Ogrodowski P, Crippa A, Rekhtman N, Stanchina E De, Lowe SW, Ventura A In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516(7531):423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sanchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, Joshi NS, Subbaraj L, Bronson RT, Xue W, Jacks T. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature. 2014;516(7531):428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R, Crowley DG, Zhang F, Anderson DG, Sharp PA, Jacks T. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. 2014;514(7522):380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weber J, Öllinger R, Friedrich M, Ehmer U, Barenboim M, Steiger K, Heid I, Mueller S, Maresch R, Engleitner T, Gross N, Geumann U, Fu B, Segler A, Yuan D, Lange S, Strong A, de la Rosa J, Esposito I, Liu P, Cadiñanos J, Vassiliou GS, Schmid RM, Schneider G, Unger K, Yang F, Braren R, Heikenwälder M, Varela I, Saur D, Bradley A, Rad R. CRISPR/Cas9 somatic multiplex-mutagenesis for high-throughput functional cancer genomics in mice. Proc Natl Acad Sci [Internet]. 2015;112(45):13982–13987. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1512392112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu C, Qi X, Du X, Zou H, Gao F, Feng T, Lu H, Li S, An X, Zhang L, Wu Y, Liu Y, Li N, Capecchi MR, Wu S. piggyBac mediates efficient in vivo CRISPR library screening for tumorigenesis in mice. Proc Natl Acad Sci [Internet]. 2017;114(4):722–727. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1615735114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu C, Qi X, Du X, Zou H, Gao F, Feng T, Lu H, Li S, An X, Zhang L, Wu Y, Liu Y, Li N, Capecchi MR, Wu S. piggyBac mediates efficient in vivo CRISPR library screening for tumorigenesis in mice. Proc Natl Acad Sci. 2017;114(4):722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weber J, Öllinger R, Friedrich M, Ehmer U, Barenboim M, Steiger K, Heid I, Mueller S, Maresch R, Engleitner T, Gross N, Geumann U, Fu B, Segler A, Yuan D, Lange S, Strong A, de la Rosa J, Esposito I, Liu P, Cadiñanos J, Vassiliou GS, Schmid RM, Schneider G, Unger K, Yang F, Braren R, Heikenwälder M, Varela I, Saur D, Bradley A, Rad R. CRISPR/Cas9 somatic multiplex-mutagenesis for high-throughput functional cancer genomics in mice. Proc Natl Acad Sci. 2015;112(45):13982–13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang G, Chow RD, Ye L, Guzman CD, Dai X, Dong MB, Zhang F, Sharp PA, Platt RJ, Chen S. Mapping a functional cancer genome atlas of tumor suppressors in mouse liver using AAV-CRISPR-mediated direct in vivo screening. Sci Adv. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, Thung SN, Wells RG, Greenbaum LE, Stanger BZ. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013;27(7):719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shin S, Wangensteen KJ, Teta-Bissett M, Wang YJ, Mosleh-Shirazi E, Buza EL, Greenbaum LE, Kaestner KH. Genetic lineage tracing analysis of the cell of origin of hepatotoxin-induced liver tumors in mice. Hepatology. 2016;64(4):1163–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hiatt JB, Pritchard CC, Salipante SJ, O’Roak BJ, Shendure J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liao H-K, Hatanaka F, Araoka T, Reddy P, Wu M-Z, Sui Y, Yamauchi T, Sakurai M, O’Keefe DD, Núñez-Delicado E, Guillen P, Campistol JM, Wu C-J, Lu L-F, Esteban CR, Izpisua Belmonte JC. In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans -epigenetic Modulation. Cell [Internet]. 2017;1495–1507. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867417312473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517(7536):583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell [Internet]. Elsevier Inc.; 2014;159(3):647–661. Available from: 10.1016/j.cell.2014.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chavez A, Tuttle M, Pruitt BW, Ewen-Campen B, Chari R, Ter-Ovanesyan D, Haque SJ, Cecchi RJ, Kowal EJK, Buchthal J, Housden BE, Perrimon N, Collins JJ, Church G. Comparison of Cas9 activators in multiple species. Nat Methods. 2016;13(7):563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell [Internet]. Elsevier Inc.; 2014;159(3):635–646. Available from: 10.1016/j.cell.2014.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, Pinkert CA, Brinster RL, Palmiter RD. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell. 1989;59(6):1145–1156. [DOI] [PubMed] [Google Scholar]
- 114.Riordan JD, Drury LJ, Smith RP, Brett BT, Rogers LM, Scheetz TE, Dupuy AJ. Sequencing methods and datasets to improve functional interpretation of sleeping beauty mutagenesis screens. BMC Genomics. 2014;15(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: Key considerations in genomic analyses. Nature Reviews Genetics. 2014. p. 121–132. [DOI] [PubMed] [Google Scholar]
- 116.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods [Internet]. Nature Publishing Group; 2014;11(8):783–784. Available from: 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Miles LA, Garippa RJ, Poirier JT. Design, execution, and analysis of pooled in vitro CRISPR/Cas9 screens. FEBS J. 2016;283:3170–3180. [DOI] [PubMed] [Google Scholar]
- 118.Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509(7501):487–491. [DOI] [PubMed] [Google Scholar]