

Harnessing the full therapeutic potential of the explosively growing universe of immuno‐oncology (IO) drug targets and diverse therapeutic modalities in a highly competitive clinical research landscape demands commitment to principled decisions through biologically sound quantitative translation. Model‐informed multidimensional optimization of dose, schedule, combination, and patient population remains an untapped opportunity. Herein, we offer perspectives on approaches to model‐informed decision making in early clinical development with a Bayesian mindset that exploits the totality of evidence.
Differentiating features of IO drug development include increased diversity of modalities, greater emphasis on combinations, including optimal sequencing, and patient selection strategies that require multidimensional characterization of the tumor microenvironment. Additionally, the complexity of human tumor immunology and questionable translatability of exposure‐response relationships for antitumor activity from preclinical in vivo models (e.g., syngeneic mice) to the clinical setting poses challenges for dose/schedule selection for early clinical development. This and other challenges are listed in Figure 1 .
Figure 1.

Key challenges in immuno‐oncology drug discovery, preclinical research, and clinical development. PD, pharmacodynamic; SCID, severe combined immunodeficiency.
Although there are many opportunities for quantitative disciplines to address the key translational challenges in IO drug discovery and development, in this Perspective, we highlight two major themes: fit‐for‐purpose mechanism‐informed modeling of the cancer‐immunity cycle (CIC), including the relevant mechanisms of action (MoAs) of the investigational treatment and potential combination partners, and novel phase I study designs that are best‐suited to the challenges of IO early development. Importantly, we emphasize the need to quantify both efficacy and safety to maximize the therapeutic index of the investigational treatment through optimal selection of dose, schedule, and combinations in context of the underlying mechanisms and patient population.
MECHANISM‐INFORMED MODELING OF THE CIC
The CIC is the process whereby dying tumor cells release tumor‐specific antigens (neoantigens), which are taken up by dendritic cells, causing them to mature and present the neoantigen to naïve T cells in the tumor draining lymph node, converting neoantigen‐specific T cells into cytotoxic T lymphocytes (CTLs). These CTLs traffic via peripheral blood into the tumor microenvironment, where they recognize and kill tumor cells displaying the neoantigen on their major histocompatibility complex class I molecules, causing further release of neoantigens, and the virtuous cycle repeats. 1 Development of a malignancy requires the CIC to be “broken” in at least one of these steps, representing potential opportunities for therapeutic intervention.
Fit‐for‐purpose mechanistic (also known as quantitative systems pharmacology (QSP)) modeling of the CIC accommodating the MoAs of the investigational treatment and its potential combination partners can address many of the key challenges outlined in Figure 1 . The first step in developing such a model is to visualize the underlying therapeutic hypothesis. A therapeutic hypothesis diagram should depict all relevant steps on the causal path from drug‐target engagement to desired antitumor activity, including mechanisms of resistance, accommodating modulation of biology at the biochemical, and cellular and organismal levels. Where feasible, we recommend approaching this diagram as a therapeutic index hypothesis, considering drivers of not only efficacy and resistance, but also safety (e.g., risk for cytokine release). This diagram (e.g., Figure 2a ) serves as a valuable communication tool between the biologist and modeler and is the basis for a fit‐for‐purpose model aimed at answering the key questions regarding dose, schedule, combinations, and/or patient population.
Figure 2.
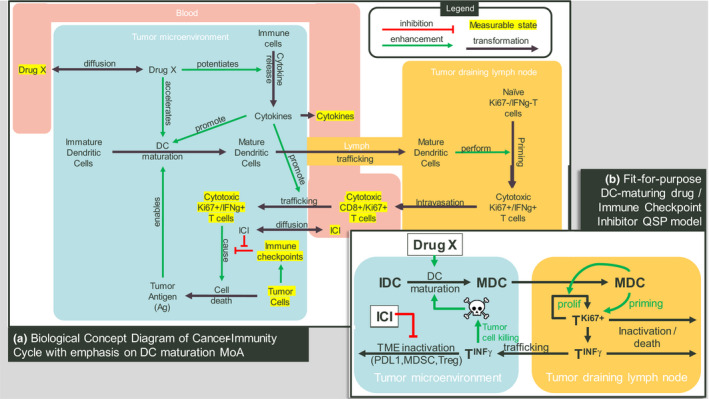
Modeling the cancer immunity cycle. (a) Biological concept diagram of cancer‐immunity cycle emphasizing a dendritic cell (DC) maturation enhancing mechanism of action. (b) Schematic of a fit‐for‐purpose mechanistic model for predicting antitumor effects of a DC activating agent either alone or in combination with an immune checkpoint inhibitor (ICI). In the tumor microenvironment (TME), immature DCs (IDCs) maturate into mature DCs (MDCs), requiring antigen released by dying tumor cells, and potentiated by the DC activating agent. MDCs traffic to the tumor draining lymph node (TDLN), where they prime T cells, causing T cell proliferation and interferon‐gamma expression. These activated T cells then traffic from TDLN to the TME and attack tumor cells, completing the tumor immune cycle. Additionally, inactivating factors, including immune checkpoints, may divert activated T cells in the TME to an inactive state, which, in some cases, can be overcome by an ICI. This fit‐for‐dose‐effect‐prediction quantitative systems pharmacology (QSP) model omits blood levels of cytokines and T cells, whereas a fit‐for‐clinical‐data‐interpretation QSP model would likely include these states in the blood compartment as represented in the original concept diagram a. MoA, mechanism of action.
That said, the resulting mathematical model need not incorporate every interaction in the therapeutic hypothesis diagram. Rather, we propose including only states and/or interactions that represent (i) direct targets of the study drug (in this case, dendritic cell maturation) and its potential combination partners (e.g., immune checkpoint inhibitors (ICIs)), (ii) in vivo measurable quantities relevant to the CIC, (iii) essential (possibly lumped) links to complete the CIC, (iv) known pathophysiology in the CIC that enable tumor growth in the indication of interest, and (v) tumor burden. Additionally, to emphasize therapeutic index, we propose the model include a normal tissue compartment if the drug target is not unique to the tumor microenvironment, or if CTLs activated by the drug recognize nontumor‐specific antigens.
At this point, the model must be calibrated to have parameter values that allow for uncontrolled tumor growth in the absence of treatment (or in the presence of an inadequate treatment). If molecular epidemiological data are available (rarely the case), it can be mapped to the functional mechanisms in the QSP model. Otherwise, one may use patient‐level tumor burden and pharmacodynamic data from patients progressing on the standard of care and use nonlinear mixed effect parameter estimation, restricting random effects to parameters that may vary among patients, to find parameter ranges corresponding to various disease states in the patient population. In this context, we note the immense reverse translational value of individual patient‐level data from prior clinical investigations that go beyond summary‐level associations between patient or tumor‐specific biology and clinical outcomes. Although the need for such patient‐level data preserving the connections across baseline, on‐treatment immunophenotype and clinical outcomes extend beyond IO, they are seldom available in IO due to the relative paucity of therapeutics with extensive clinical experience (e.g., ICIs 2 ).
Now we can apply the model (Figure 2b ) to address questions like determining minimally effective dose by asking what exposure time course of our agent is necessary to control tumor growth in ideal conditions, that is, in patients whose CICs are “broken” at a step amenable to the study drug’s MoA. Combination partners like ICIs can be evaluated by simulating what fraction of the virtual population of patients with CIC pathologies refractory to ICIs can be “rescued” by addition of study drug. Importantly, as tumor pharmacodynamics measurements (e.g., immunophenotype) in phase I IO studies are sparse and interrogate multidimensional end points in a limited number of patients with heterogeneous tumor immunology, biological variability and sparsity of tumor biopsies challenges interpretation. We posit that QSP models, by using known biology as a “filter” for data interpretation, are particularly well‐suited for using such data to assess proof‐of‐mechanism and to inform hypotheses for patient selection and combinations. Later in development, using the population distribution as a prior, the model parameter values and initial states could be recalibrated to a set of patient baseline (and early pharmacodynamic markers) and then simulated to predict whether the patient is likely to respond to the addition of the study drug.
NOVEL STUDY DESIGNS WELL‐SUITED TO INVESTIGATIONAL IO THERAPIES
The superior performance of adaptive dose‐finding in first‐in‐human oncology dose escalation studies relative to traditional rule‐based (e.g., “3 + 3”) designs is well‐established. Popular model‐assisted designs include the modified toxicity probability interval design and variations, Bayesian optimal interval design, and the Keyboard design, whereas commonly used model‐based designs include continual reassessment method and Bayesian logistic regression model with overdose control. 3 The unique challenges presented by IO drug development present many opportunities to further enhance these well‐developed dose finding designs. Selection of the starting dose as well as the Bayesian model prior can be informed by translational integration of the totality of data generated in the preclinical development phase. Inputs include exposure‐response relationships built from in silico (e.g., QSP), ex vivo (e.g., cytokine release assays), and preclinical (e.g., syngeneic mouse efficacy models and toxicology studies in cynomolgus monkeys) studies. Additionally, late onset of immune‐related adverse events beyond cycle 1 may be of concern. 4 Although the aforementioned dose‐finding designs can accommodate late onset toxicity by updating the model when it occurs, design improvements, including time‐to‐event continual reassessment method 5 and time‐to‐event Bayesian optimal interval design, 6 can take account of the late onset toxicity (e.g., as observed with ICIs) by assigning statistical weights to patients within a prespecified prolonged observation window. Such designs avoid enrollment pause and offer greater accuracy by formally integrating available information across dose and time.
Given that safety and efficacy typically exert opposite pressures on dose, a design that considers both safety and efficacy signals to identify the optimal biological dose may be more appropriate than maximum tolerated dose finding designs. Thall and Cook 7 proposed a model‐based dose‐finding design (EffTox) based on efficacy and safety data with a trade‐off to guide dose escalation decisions. Li et al. 8 proposed the toxicity and efficacy probability interval design for dose finding in adoptive cell therapy trial. Both designs demonstrate superior performance compared with safety driven designs and are applicable in seamless phase I/II settings for reduced development timelines.
Additional opportunities to inform definition of the recommended phase II dose range include timely incorporation of emerging pharmacokinetic and pharmacodynamic data for end points identified as central to the therapeutic hypothesis. Given the small sample sizes of individual dose escalation cohorts, inference based on exposure‐response relationships constructed using the totality of data across the dose/exposure range should be favored in lieu of cohort‐level inference based on summary statistics of pharmacodynamic effects. Of note, when formulated with an underlying Bayesian construct informed by priors from fit‐for‐purpose QSP models, the exposure‐response relationships for dose decision‐enabling pharmacodynamic biomarkers will mature as the trial progresses and more data are collected across a wide dose range. Taken together with dose/exposure‐response for safety, the recommended phase II dose range can be informed by calculations of the likelihood of achieving a desired extent and duration of pharmacodynamic modulation, as opposed to statistical significance of crossing a predefined threshold in a single expansion cohort at the single time point of measurement.
Unlike modalities in which the maximum tolerated dose paradigm may be more appropriate, IO therapies are particularly reliant on efficacy for dose determination and, therefore, more sensitive to heterogeneity in tumor types seen in first‐in‐human studies. A well‐calibrated QSP model could be used to define indications or tissue‐agnostic subpopulations most likely to respond to a given MoA. Otherwise, designs ignoring the heterogeneity of safety or efficacy profiles across different types of tumors could lead to misleading dose selection and become inefficient for hypothesis generation for target population selection. Li et al. 9 proposed a model‐based Bayesian Semi‐parametric Design for adaptive dose‐finding with multiple strata. Such a design allows data‐driven flexible information borrowing across multiple populations while allowing for heterogeneity. Supportive evidence to increase confidence in patient selection can be importantly gained through population exposure‐response modeling that incorporates patient/tumor‐specific features (e.g., cancer type, tumor mutational burden, and baseline immunophenotype) as covariates. Longitudinal models of tumor burden, including such covariate evaluations, when built on datasets from expansion cohorts or early “learning” phase II trials can particularly increase confidence in patient selection decisions ahead of entry into pivotal clinical development. 10 Although the analytical platforms for such analyses have been described and applied to larger scale datasets, routine application in early clinical development remains an untapped opportunity.
CONCLUDING REMARKS
The unique challenges of IO drug development offer many opportunities for model‐informed quantitative translation. Question‐driven, fit‐for‐purpose QSP models guided by the therapeutic hypothesis and appropriate study designs can help optimize dose, dosing schedule, combination, sequence, and patient population in real time, provided they are built proactively and updated frequently. Continuous learning of exposure‐response relationships for pharmacodynamics, safety, and efficacy as a function of the individual patient/tumor characteristics should be a key focus in early clinical development. Consistent application of these enablers will enhance probability of successful cancer immunotherapy development.
Funding
No funding was received for this work.
Conflict of Interest
D.B., R.L., and H.B. are full‐time employees of Takeda Pharmaceuticals. K.V. is a full‐time employee of EMD Serono. As an Associate Editor for Clinical Pharmacology & Therapeutics, K.V. was not involved in the review or decision process for this paper.
References
- 1. Chen, D.S. & Mellman, I. Oncology meets immunology: the cancer immunity cycle. Immunity 29, 1–10 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Kamphorst, A.O. et al Proliferation of PD‐1+ CD8 T cells in peripheral blood after PD‐1–targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA. 114, 4993–4998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou, H. , Murray, T.A. & Pan, H. Comparative review of novel model‐assisted designs for phase I clinical trials. Stat. Med. 37, 2208–2222 (2018). [DOI] [PubMed] [Google Scholar]
- 4. Puzanov, I. et al Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J. Immunother. Cancer 5, 95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheung, Y.K. & Chappell, R. Sequential designs for phase I clinical trials with late‐onset toxicities. Biometrics 56, 1177–1182 (2000). [DOI] [PubMed] [Google Scholar]
- 6. Yuan, Y. , Lin, R. , Li, D. , Nie, L. & Warren, K. Time‐to‐event Bayesian optimal interval design to accelerate phase I trials. Clin. Cancer Res. 24, 4921–4930 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thall, P.F. & Cook, J.D. Dose‐finding based on efficacy‐toxicity trade‐offs. Biometrics 60, 684–693 (2004). [DOI] [PubMed] [Google Scholar]
- 8. Li, D.H. , Whitmore, J.B. , Guo, W. & Ji, Y. Toxicity and efficacy probability interval design for phase I adoptive cell therapy dose‐finding clinical trials. Clin. Cancer Res. 23, 13–20 (2017). [DOI] [PubMed] [Google Scholar]
- 9. Li, M. , Liu, R. , Lin, J. , Bunn, V. & Zhao, H. Bayesian semi‐parametric design (BSD) for adaptive dose‐finding with multiple strata. J. Biopharm. Stat. 10.1080/10543406.2020.1730870 [DOI] [PubMed] [Google Scholar]
- 10. Williams, K. et al Precision medicine by modeling pharmacokinetic and biomarker drivers of tumor kinetics: assessing effects of alisertib exposure and target SNP status on antitumor activity. American Society for Clinical Pharmacology and Therapeutics Annual Meeting (2017). 10.1002/cpt.570 [DOI]