

Abstract
The formation of disulfide bonds in proteins is an essential process in both prokaryotes and eukaryotes. In Gram-negative bacteria including E. coli, the proteins DsbA and DsbB mediate the formation of disulfide bonds in the periplasm. DsbA acts as the periplasmic oxidant of periplasmic substrate proteins. DsbA is reoxidized by transfer of reducing equivalents to the 4 TM helix membrane protein DsbB which transfers reducing equivalents to ubiquione or menaquinone. Multiple structural studies of DsbB have provided detailed structural information on intermediates in the process of DsbB catalyzed oxidation of DsbA. These structures and the insights gained are described. In proteins with more than one pair of Cys residues, there is the potential for formation of non-native disulfide bonds, making it necessary for the cell to have a mechanism for the isomerization of such non-native disulfide bonds. In E. coli, this is mediated by the proteins DsbC and DsbD. DsbC reduces mis-formed disulfide bonds. The 8 TM helix protein DsbD reduces DsbC and is itself reduced by cytoplasmic thioredoxin. DsbD also contributes reducing equivalents for the reduction of cytochrome c to facilitate heme attachment. The DsbD functional homolog CcdA is a 6 TM helix membrane protein that provides reducing equivalents for the reduction of cytochrome c. A recent structure determination of CcdA has provided critical insights into how reducing equivalents are transferred across the membrane that likely also provides understanding how this is achieved by DsbD as well. This structure and the insights gained are described.
Keywords: DsbB, CcdA, DsbD, DsbA, DsbC, DsbG
Graphical Abstract
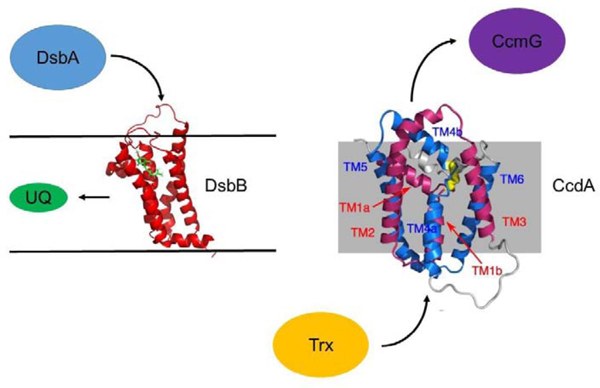
Membrane proteins involved in disulfide bond formation
Protein disulfide formation in the periplasm of E.coli (DsbA and DsbB).
The classical studies of Anfinsen and co-workers [1] on the oxidative refolding of ribonuclease were originally taken to indicate that there was no necessity for an enzymatic activity for disulfide bond formation. However, the extremely slow rate of oxidative refolding in vitro strongly suggested a need for such an enzymatic activity. This led to the discovery in eukaryotic cells of protein disulfide isomerase (PDI) which enables correct protein disulfide formation by catalyzing thiol-disulfide exchange reactions [2, 3]. In prokaryotes, some 30 years after Anfinsen’s initial studies, a mutation in the DsbA gene was identified which leads to a defect in disulfide formation for secreted proteins such as beta-lactamase and alkaline phosphatase [4]. Subsequent screening efforts based either on defects in folding of bacterial cell envelope proteins or altered sensitivity to DTT resulted in the identification of all the members of the Dsb family, namely DsbA, DsbB, DsbC, and DsbD [5, 6].
In Gram-negative bacteria including E. coli, the proteins DsbA and DsbB work in concert to mediate the formation of disulfide bonds (see Figure 1). DsbA is a soluble protein that functions as the primary oxidant for proteins in the periplasm of E. coli whereas DsbB is an integral membrane protein that reoxidizes DsbA and is itself re-oxidized by various quinones. As mentioned above, DsbA mutants result in a lack of disulfide bond formation resulting in rapid degradation of proteins such as alkaline phosphatase and OmpA. DsbB mutants are not motile due to improper assembly of the flagellar motor [7] and are hypersensitive to DTT, benzylpenicillin, and metals [8, 9]. Interestingly, mutations of DsbA in pathogenic bacteria lead to avirulence [10], since many virulence components contain disulfide bonds, suggesting this pathway may be a useful target for development of novel antibiotics. DsbA is 21 kDa protein possessing a thioredoxin fold [11] and CPHC redox-active active site reminiscent of the active sites seen in thioredoxins and glutaredoxins. DsbA, with a redox potential of −120 mV, is one of the most highly oxidizing thiol-disulfide oxidoreductases identified. Structures of both the oxidized and reduced forms of the protein have been determined [12, 13]. As has also been seen for the thioredoxins and glutaredoxins, DsbA has one active-site thiol with a dramatically depressed pKa of ca. 3.5, a result of stabilization by a helix dipole and hydrogen bonding.
Figure 1. The disulfide bond formation system in E. coli.
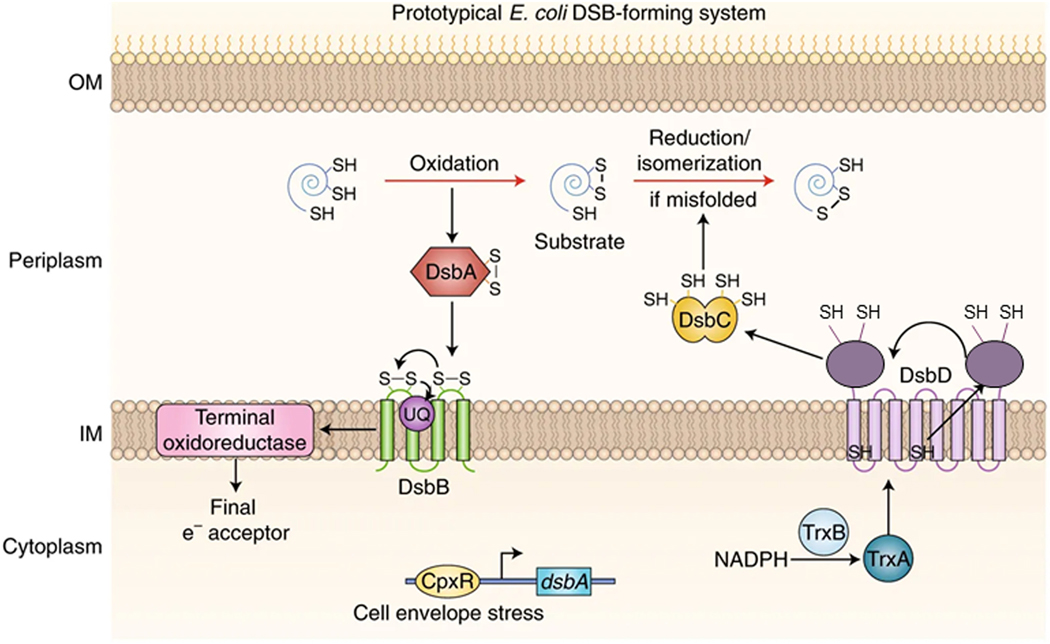
Figure from reference 37.
Mutations of DsbB cause DsbA to accumulate in the reduced form and show the same phenotypes as mutants of DsbA [7, 8, 14]. DsbB takes advantage of the oxidizing power of the electron transport chain to re-oxidize DsbA [15, 16]. Under aerobic conditions, DsbB transfers electrons to ubiquinone [16, 17] and under anaerobic conditions to menaquinone [17]. Bardwell and co-workers demonstrated binding of ubiquinone to DsbB indicating there is a binding site on DsbB [17].
DsbB homologs in other prokaryotes.
DsbB homologs are pervasive throughout prokaryotes. A recent search of the NCBI non-redundant database yielded 112 homologs [18]. Based on predicted topology and functional characteristics, these have been grouped into 3 classes. Group I, the largest group, is characterized by 4 TM helices, one pair of Cys residues at the N-terminal end of TM2, and an additional pair of Cys residues in a periplasmic loop connecting TM3 and TM4. E. coli DsbB falls into this class. Group II is predicted to have 5 TM helices, has the N-terminal Cys pair at the end of TM2, but differs from Group I in having its second pair of Cys residues on a C-terminal loop. Among the members of Group II is the DsbB homolog from Francisella tularensis, a potent human pathogen. Group III resembles Group II but lacks the second pair of Cys residues seen I both Groups I and II. Examples of group III are the DsbB homologs of Helicobacter pylori and Bordetella pertussis. There is currently no structural data available for any member of the Group II or III DsbB homologs, leading to an incomplete picture of the mechanistic differences between these three classes. .
Interestingly, in a subset of prokaryotes DsbA re-oxidation is not mediated by DsbB but by the membrane protein VKOR, a homolog of the human vitamin K epoxide reductase (VKOR) [19–22]. DsbB and VKOR do not show homology in their primary sequences, but they are structurally quite similar and both utilize a quinone cofactor to mediate disulfide bond formation. Furthermore, the mechanism of interaction of DsbA with VKOR proceeds via the same steps as seen for DsbA and DsbB [22]. VKOR is the subject of a separate chapter in this issue on Intramembrane Enzymes.
DsbB functional homologs in eukaryotes.
As described above, protein disulfide formation in gram-negative bacteria takes place in the periplasm and is mediated by the integral membrane protein DsbB and the soluble protein DsbA. In contrast, disulfide bond formation in eukaryotes takes place in the ER. The soluble protein PDI is the analog of DsbA in the ER, where it is the direct catalyst for disulfide bond formation. PDI is subsequently re-oxidized by members of either the Ero1 or SOX/ALR families of proteins. Ero1 is a flavoprotein which is bound to the lumenal surface of the ER membrane and uses oxygen as the terminal electron acceptor. Members of the SOX/ALR family are also flavoproteins which use oxygen as the terminal electron acceptor, but unlike Ero1 they are found in several cellular compartments [23, 24]. This clear distinction in the enzymes used for disulfide formation in prokaryotes and eukaryotes along with the demonstrated need for this pathway in pathogenesis makes DsbB an interesting potential target for the development of novel antibiotics.
The structures of a functional domain of Ero1 and of Erv2, a SOX/ALR family member, have been solved by X-ray crystallography [25, 26]. While showing little to no sequence homology, the two enzymes clearly show a striking structural similarity [18]. Four striking parallels were noted between the two enzymes: 1) both contain 4 α-helices arranged in an antiparallel 4-helix bundle to create the active site, 2) a conserved Cys-x-x-Cys motif at one end of one of the helices with one Cys in close proximity to the FAD cofactor, 3) conserved aromatic amino acids which play a role in FAD binding, and 4) a second essential pair of cysteines located in a relatively unstructured portion of the protein which are critical for enzyme function. Indeed, in the crystal structures this loop in Erv2 and Ero1 has been seen in multiple conformations, likely indicative of increased mobility for this region of the protein. A comparison of DsbB to Erv2 and Ero1 suggested these conserved features may also be present in DsbB [18], a prediction which our structural data and that of others subsequently confirmed (described below): 1) There are 4 transmembrane helices which create the active site, 2) The Cys41-Cys44 pair in E. coli DsbB has been shown to be in close proximity to the bound quinone, 3) the Cys-104-Cys130 pair in E. coli DsbB functions as the site for disulfide exchange with DsbA, and 4) significant mobility for the loop containing Cys-104-Cys130, similar to what has been reported for Ero1 and Erv2.
Role of protein disulfide formation in bacterial virulence.
Bacterial resistance to current classes of antibiotics is rapidly reaching a crisis point. Indeed, this has already become a primary cause of death in intensive care units of hospitals [27, 28]. As a result of actual killing of the bacteria, current classes of antibiotics create an evolutionary pressure for bacteria to evolve to escape their effects [29, 30]. An alternative approach has been proposed which targets bacterial virulence [31–34]. In this approach, the goal is to inhibit specific bacterial functions that promote infection and are essential to persistence such as binding, invasion, subversion of host defenses, and chemical signaling. Such an approach has the advantage that inhibition of these functions will not kill the bacteria and therefore should result in a reduced selection pressure for drug-resistant mutations. This approach would also avoid the destruction of the normal host bacteria that is associated with current antibiotics.
Disulfide bond formation is critical for the functioning of many proteins that mediate virulence functions, therefore the Dsb proteins have been suggested to be appropriate targets for the development of antivirulence agents [35–37]. Uropathogenic E. coli (UPEC) require pili (fimbriae), hair-like extensions from the cell surface, to adhere to eukaryotic cells and thereby mediate urinary tract infection. Lack of DsbA function results in degradation of the chaperone PapD and the PapG subunit of the pilus, resulting in disruption of pilus formation in UPEC [38]. The DsbA homolog in P. aeruginosa is essential for the activity of elastase, a secreted metalloprotease that enhances virulence in the lungs of patients with cystic fibrosis [39]. DsbA homologs are also required for production of cholera toxin in V. cholerae, pertussis toxin of B. pertussis, and enterotoxin of enterotoxigenic E. coli [10, 40–42]. Numerous studies have shown that bacterial mutants defective in the DsbA-DsbB oxidation system have reduced virulence in infection models [10, 40, 43–51]. These results clearly support efforts to develop small molecule inhibitors of DsbB, as well as its functional homologs, as a novel approach to inhibit bacterial virulence.
Structural insights into the mechanism of DsbB mediated oxidation of DsbA
E. coli DsbB is a 21 kDa integral membrane protein with 4 membrane spanning helices and the N- and C-termini facing the cytoplasm. There are two periplasmic loops containing redox-active cysteine pairs, C41/C44 and C104/C130, that have been directly implicated in the mechanism of DsbA oxidation [52, 53]. Through studies by Kadokura and Beckwith [54], a coordinated pathway for the flow of electrons through these thiols was proposed (see Figure 2). Such a pathway was also suggested by the lab of Koreaki Ito and co-workers based on studies in their lab [55]. Specifically, DsbA reacts with C104 in the second of the 2 periplasmic loops. The resulting free thiol at C130 then reacts with C41 in the first periplasmic loop to form an inter-loop disulfide and a free thiol at C44. The free thiol at position 44 is well-positioned to interact with the bound quinone and initiate the re-oxidation of DsbB and release of oxidized DsbA. It is not entirely clear if this final step is a concerted process, however there is good evidence from mutagenesis for the proximity of C44 to the quinone. Specifically, mutation of R48 to C or H results in a protein that retains the ability to oxidize DsbA under aerobic conditions but is dysfunctional under anaerobic conditions [56]. This defect has been traced to a pronounced increase in the Km for menaquinone which is employed under anaerobic growth conditions, strongly suggesting the quinone binding site is in spatial proximity. In addition, insertion of additional alanine residues between C44 and R48 leads to a complete loss of quinone-based oxidation of DsbB [57].
Figure 2. Pathway for DsbB mediated oxidation of DsbA and structures of intermediates on the pathway.
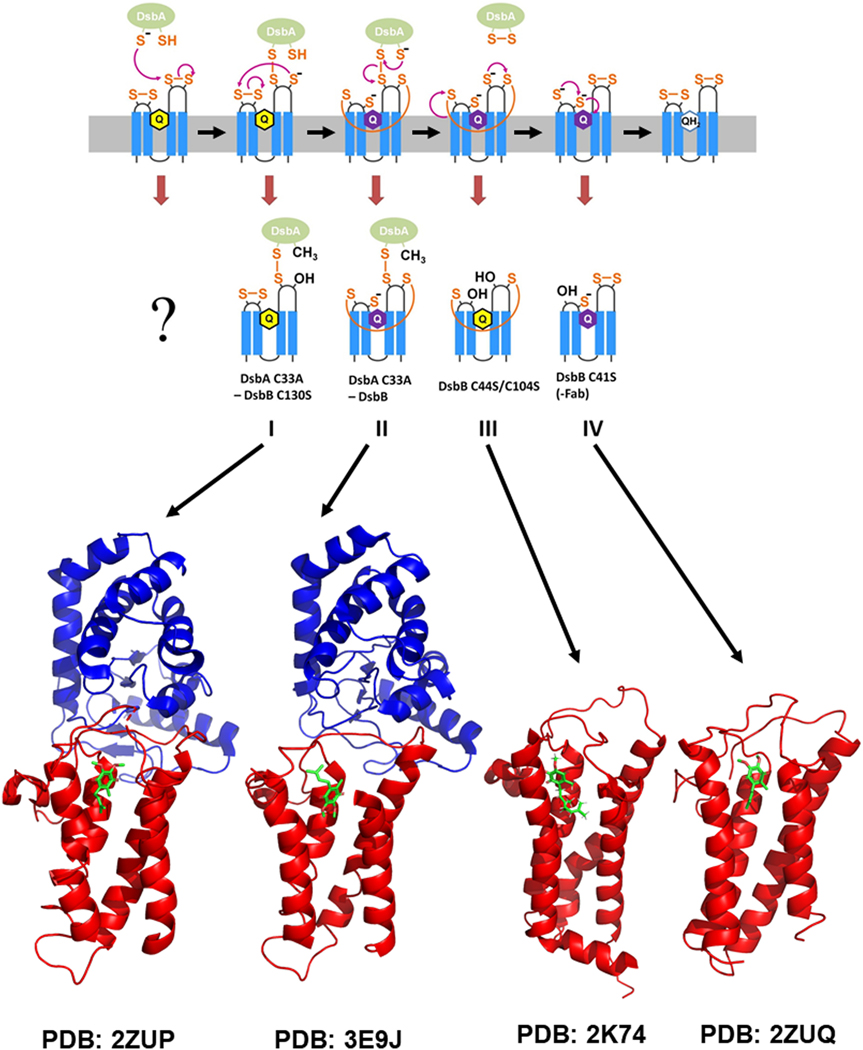
Top: Schematic illustrating the pathway and DsbB intermediates for transfer of reducing equivalents by DsbB from DsbA to cellular quinones. Bottom: 3D structures of DsbB intermediates along the pathway that have been determined by NMR and X-ray crystallography.
To date, the structure of DsbB has been determined in four different forms, using NMR for one and X-ray crystallography for 3 others (Figure 2). These structures provide a nearly complete view of DsbB at different stages of its catalytic mechanism. Structure I, a 3.7 Å resolution crystal structure of a DsbA C33A-DsbB C130S disulfide bonded complex [58], represents the intermediate in which reduced DsbA has reacted with DsbB to generate an intermolecular disulfide bond but disulfides inside DsbB are not rearranged. The structure of DsbB was shown to consist of 4 transmembrane helices arranged in a bundle with two pairs of essential Cys residues (Cys41-Cys44, Cys104-Cys130) in its two periplasmic loops. This structure provided important details of the interaction between DsbA and DsbB and an approximate location of the ubiquinone (UQ) cofactor. Residues Pro100-Phe106 of DsbB, which includes Cys104 which is disulfide bonded to DsbA is found bound in a groove on the surface of DsbA where substrates bind. In the absence of DsbA, DsbB harbors Cys41-Cys44 and Cys104-Cys130 disulfide bonds [53, 59, 60]. While there was a lack of electron density for the loop region around Cys130, SeMet labeling made it possible to show that the distance between Cys104 and Cys130 in the complex was ~8 Å and that it was located in proximity to the Cys41-Cys44 disulfide pair. Such a structural rearrangement will reduce the back reaction of Cys130 reacting with Cys104 to unproductively release reduced DsbA. In addition, the placement of Cys130 in proximity to the Cys41-Cys44 pair will enhance the rate of attack of Cys130 on Cys41 to generate a charge transfer complex with the bound quinone that will result in quinone reduction and re-oxidation of DsbB. The authors suggested that this structural rearrangement is what allows the protein to perform a series of thiol-disulfide reactions that are uphill thermodynamically based on measurement of the individual redox potentials [60, 61]. Based on observation of an area of electron density consistent with the quinone ring, a binding site for the quinone was suggested at the N-terminal end of TM2.
Structure II is a 3.7 Å resolution crystal structure of the DsbA C33A-DsbB complex [62] which contains the intermolecular disulfide bond between DsbA and DsbB, the intramolecular inter-loop disulfide bond in DsbB, and the DsbB Cys44-UQ charge transfer complex. The authors showed based on UV spectroscopy (absorbance max at 580 nm) that that the charge transfer complex was indeed present in this complex which proved to be quite stable, likely due to stabilization of the charge on Cys44 by the quinone and Arg48. A difference Fourier map comparing this structure to the crystal structure described above showed a significant difference for Cys44, with Cys44 oriented toward the quinone in this structure but not in the structure described above. The authors go on to suggest a mechanism for nucleophilic attack of Cys44 on the quinone ring followed by Cys41 attack on Cys44 to achieve reduction of the quinone. However, details of the inter-loop disulfide bond and the Cys44-UQ interaction are lacking due to low resolution and a lack of electron density in the relevant portions of the structure.
Structure III is the NMR solution structure of the DsbB C44S/C104S double cysteine mutation solved by our lab [63], corresponding to an intermediate in which oxidized DsbA has departed but the inter-loop disulfide bond between Cys41 and Cys130 remains. This structure was solved using a combination of sparse NOE data, dihedral angle restraints from chemical shifts, paramagnetic relaxation enhancement (PRE) restraints, and residual dipolar coupling (RDC) restraints. The overall fold of the left-handed 4 helix bundle was similar to that seen in the crystal structures described above. Unlike the structures described above, detailed structural information was obtained for the loop containing Cys104 and Cys130 which showed the N-terminal portion of this loop is quite mobile whereas the C-terminal portion of the loop is well-structured. EPR spectroscopy was used to measure the depth of insertion in the membrane of nitroxide labels at multiple sites on DsbB in order to dock the structure of DsbB into a bilayer, showing the burial of the transmembrane segments in the bilayer and the periplasmic loop containing Cys104, site of reaction with DsbA, located in the aqueous phase where it can interact with DsbA. Chemical shift perturbation and saturation transfer difference NMR methods indicate that amino acids 97–110 and 126–131 of DsbB, in the periplasmic loop containing Cys104 and Cys130, are involved in binding to DsbA. Consistent with this, the structure described above identified Pro100-Phe106 as DsbB residues in contact with DsbA. Our data suggest that the contacts made with DsbA aid in placing Cys130 in proximity to Cys41, thereby enhancing the rate of formation of the inter-loop disulfide bond. Interestingly, the Cβ carbon chemical shift of Cys41 was found to be unusually downfield (50.7 ppm compared to average value of 41.6 ppm), indicative of an environment around the Cys sulfur that is strongly electron withdrawing, resulting in this being the favored leaving group in a thiol-disulfide exchange reaction and likely helping to direct the proper directionality for transfer of reducing equivalents. Indeed, using the crystal structure described above in combination with our structure of the inter-loop disulfide for of DsbB, we created a model of a DsbA-DsbB complex with the inter-loop disulfide present which showed all 6 functionally relevant Cys residues aligned in a straight line making a concerted reaction series feasible, consistent with previous stopped flow kinetics studies [64]. Using NOE and PRE information to a bound molecule of UQ2 (ubiquinone with 2 isoprenyl units), we defined the binding site and orientation of the bound quinone with the benzoquinone ring located at the C-terminal end of TM1 and the N-terminal end of TM2 with the isoprenyl chains extending down a groove between TM1 and TM4. This mapping of the quinone binding site was consistent with previous mutagenesis studies.
Structure IV is a 3.4 Å resolution crystal structure of the DsbB Cys41Ser mutant stabilized by an Fab [65], which provides a view of the DsbB intermediate just prior to full re-oxidation of the protein. Additional residues in the Cys104 and Cys130 containing periplasmic loop two were observed in this crystal structure relative to the crystal structure of the DsbA-DsbB complex described above. The structure confirmed two key elements of the NMR structure described above, namely the location of the amphipathic helix in periplasmic loop two and the second interaction site on DsbB near Cys130 for binding of DsbA. This structure confirmed a significant conformational change for the periplasmic loop that separates Cys104 and Cys130 from one another. Not unexpectedly, comparison of this structure to the inter-loop disulfide NMR structure showed a significant conformational change for the Cys130 region of periplasmic loop two. Mutagenesis studies of the amphipathic helix in periplasmic loop two demonstrated its importance to DsbB function leading the authors to suggest that the mobility of this periplasmic loop is modulated by this amphipathic helix to restrain it to a level that is productive for catalysis.
While these structures have provided many novel insights into the mechanism of DsbB, all the structures lack significant sidechain information, limiting our ability to assess the details of, for example, the arrangement of the inter-loop disulfide bond or specific sidechain interactions with the bound quinone. In the case of the X-ray structures, this results from the very limited resolution of the structures and in the case of the NMR structure it is the result of inadequate experimental data to define the sidechain conformations, even though the backbone was determined to high resolution.
Membrane proteins involved in disulfide bond reduction
DsbC and DsbD catalyze disulfide isomerase activity in the periplasm of E. coli
As described above, DsbA catalyzes the formation of protein disulfide bonds in the periplasm of Gram-negative bacteria such as E. coli. DsbA is re-oxidized by the membrane protein DsbB. This DsbA catalyzed disulfide bond formation can result in the formation of non-native disulfide bonds in substrate proteins containing more than one pair of Cys residues, making it necessary for the cell to also have a protein disulfide isomerase which can facilitate the formation of native disulfide bonds (Figure 1). In. E. coli, the protein carrying out this function is DsbC [66]. The oxidized DsbC that results from its reduction of non-native disulfide bonds is reduced by the membrane protein DsbD. The folding of a number of E. coli proteins, including the periplasmic RNase I (4 disulfide bonds) and MepA (3 disulfide bonds), has been shown to be dependent on the presence of DsbC [67].
DsbC function
Biochemical studies indicate that DsbC acts as a protein disulfide reductase, resulting in reduction of nonnative disulfide bonds [68, 69]. Reduction of non-native disulfide bonds by DsbC makes it possible for DsbA to subsequently catalyze the formation of native disulfide pairings. DsbC has also been shown to have chaperone activity [70]. A homolog of DsbC, DsbG, is structurally homologous to DsbC [71] and also is a substrate of DsbD. DsbG functions to protect Cys thiols against oxidative damage [72, 73]. DsbC is a homodimer with an N-terminal dimerization domain and a C-terminal catalytic domain harboring a thioredoxin-like CXXC motif [74, 75]. With both the DsbA/DsbB and DsbC/DsbD redox systems operating in the periplasm, there is a need for these two systems to be decoupled from one another to avoid endless cycles of futile redox cycling. The dimeric structure of DsbC serves to sterically block the ability of its redox active Cys residues to interact with DsbB [76]. Similarly, the helical domain of DsbA was suggested to sterically block the interaction of DsbA with DsbD [77]. Thus, the two pathways are well insulated from one another, allowing both activities to function in the periplasm.
DsbD function
DsbD was originally identified as a protein required for the maturation of cytochrome c in the periplasm of Gram-negative bacteria [78]. This occurs via DsbD reduction of CcmG which reduces any oxidized Cys residues in cytochrome c which is essential for heme binding [79, 80]. DsbD was also shown to be essential for maintaining DsbC, as well as its homolog DsbG, in the reduced state [69, 72, 81]. DsbD is comprised of 3 domains (Figure 1): an N-terminal periplasmic domain with two essential Cys residues, a transmembrane domain with 8 predicted TM helices and two essential Cys residues, and a C-terminal domain with two essential Cys residues. Structures of the N- and C-terminal domains have been determined which showed the C-terminal domain to be similar to thioredoxin [82, 83] and the N-terminal domain to be an immunoglobulin fold [77, 84] that has not been observed for other proteins catalyzing Cys redox reactions, likely due to the need for this protein to interact in a specific manner with substrate proteins such as DsbC and DsbG. DsbD is reduced by intracellular thioredoxin. The pathway for electron transfer through the three DsbD domains has been elucidated – thioredoxin reduces the Cys pair in the transmembrane domain, followed by reduction of the Cys pair in the C-terminal periplasmic domain, reduction of the Cys pair in the N-terminal periplasmic domain, and finally reduction of DsbC via transfer of reducing equivalents from the N-terminal DsbD periplasmic domain [69, 85, 86]. As the redox potential for each pair of Cys residues in this cascade is progressively higher than the previous pair, this cascade is thermodynamically downhill [87, 88]. The most intriguing aspect of this mechanism is how the electron transfer occurs across the transmembrane domain of DsbD. There is currently no structural data for the transmembrane domain of DsbD. Beckwith and co-workers showed that a disulfide bond is formed in the transmembrane domain in cells lacking thioredoxin but this is found in the reduced state when thioredoxin is present [89]. They suggested [90], and Collet and co-workers provided experimental evidence [91], that the transmembrane domain may undergo a significant conformational change between the reduced and oxidized states, an effect we have supporting evidence for via structural studies of the functional homolog CcdA (see below).
ScsB function
The suppressor of copper sensitivity (Scs) proteins are homologs of Dsb proteins which contribute to the virulence property of resistance to copper stress [92–94]. In relevant bacteria, four Scs proteins are coded for, all of which have catalytic motifs with two Cys residues. The soluble periplasmic ScsC protein has been most extensively characterized [92, 94, 95]. The oligomerization state of ScsC seems to vary among different bacteria, with monomeric, dimeric, and trimeric forms observed. ScsB is a putative redox partner of ScsC which strongly resembles E. coli DsbD. Very similar to DsbD, ScsB was predicted to have an N-terminal periplasmic domain, a transmembrane domain comprised of eight transmembrane helices, and a C-terminal thioredoxin-like domain. In addition, both DsbD and ScsB have two catalytic Cys residues in each of the three domains. As described above, DsbD uses a cascade of transfers of reducing equivalents from cytoplasmic thioredoxin to the membrane domain, from the membrane domain to the thioredoxin-like domain, and from the thioredoxin-like domain to the N-terminal domain to mediate reduction of periplasmic DsbC. Based on the strong similarity of the architecture of ScsB to DsbD, it is reasonable to expect that a similar mechanism applies for ScsB. Indeed, C. crescentus ScsB has been shown to be essential for maintaining ScsC in the reduced state necessary for activity [92, 95]. The N-terminal domains of DsbD and ScsB differ significantly, with the domain from ScsB significantly larger than the corresponding domain from DsbD, likely to mediate specific interactions with substrate proteins. Recent structural studies of the N-terminal domain from P. mirabilis ScsB have shown it to harbor two Ig folds, unlike the one Ig fold found for DsbD [96]. As for DsbD, there is no structural data currently for the transmembrane domain of ScsB.
CcdA function
Most classes of bacteria express cytochrome c’s but lack DsbC/DsbD, requiring another protein to provide the reducing equivalents necessary for cytochrome c reduction. In Rhodobacter capsulatas, this function is provided by the membrane protein CcdA [97], a functional homolog of the DsbD membrane domain but with only 6 transmembrane helices unlike the 8 predicted for DsbD. Indeed, mutants of Rhodobacter capsulatas lacking CcdA are unable to make cytochrome c [97]. Mutants of Bacillis subtilis lacking CcdA are defective in cytochrome c production as well [98]. As with DsbD, biochemical studies are consistent with transfer of reducing equivalents from intracellular thioredoxin to the membrane embedded CcdA which then reduces periplasmic substrates. Interestingly, while Rhodobacter CcdA can complement a DsbD mutation in E. coli to restore cytochrome c reduction, it cannot restore DsbC function, indicating it can reduce CcmG but not DsbC [99].
Structural insights into the mechanism of CcdA mediated disulfide exchange across the membrane
▪. Ccda fold comprised of two 3 TM helix repeats
CcdA, like the DsbD membrane domain, is unique among membrane electron transporters in that it uses a pair of Cys residues to move electrons across the membrane without the aid of any cofactor [88]. As such, structural insights into how this is achieved are critical. We recently determined the solution structure of oxidized CcdA from Thermus thermophilus using limited NOE constraints, dihedral angle restraints from backbone chemical shifts, paramagnetic relaxation effects, and residual dipolar couplings [100]. The reduced form of the protein gave very poor quality NMR spectra, perhaps indicative of conformational heterogeneity. Similar to DsbD where sequence similarity was observed for TM helices 1–3 and 4–6 [101] of the 8 TM helices, TM helices 1–3 and 4–6 in CcdA also show sequence similarity. Our structure showed that these two 3 TM repeats adopt similar structures and are arranged in an antiparallel arrangement relative to one another in the structure (Figure 3). TM1 and TM4 are central transport helices and are connected by the active site disulfide bond. The overall architecture can be described as two kinked transport helices inserted into an O-scaffold channel with flexible loops connecting them. In this structure Cys127 is more exposed to the periplasm than Cys20, suggesting that it is Cys127 on TM4 that interacts with periplasmic substrates. This structure is consistent with the inverted symmetry solvent-accessibility profile seen in the functional homolog E. coli DsbD [101, 102]. The similar sequence pattern in DsbD along with the the solvent-accessibility data suggests the same fold could be found in DsbD, although how the additional 2 TM helices in DsbD would participate in this structure remains an open question. The structure of CcdA is open to the periplasmic side but blocked from the cytoplasmic side as a result of the different orientations of TM1 and TM4 resulting in TM1b and TM4a blocking access (Figure 3). This conformation shows how periplasmic substrates can gain access to the active site but does not explain how cytoplasmic thioredoxin can gain access.
Figure 3. Structure and mechanism of CcdA.
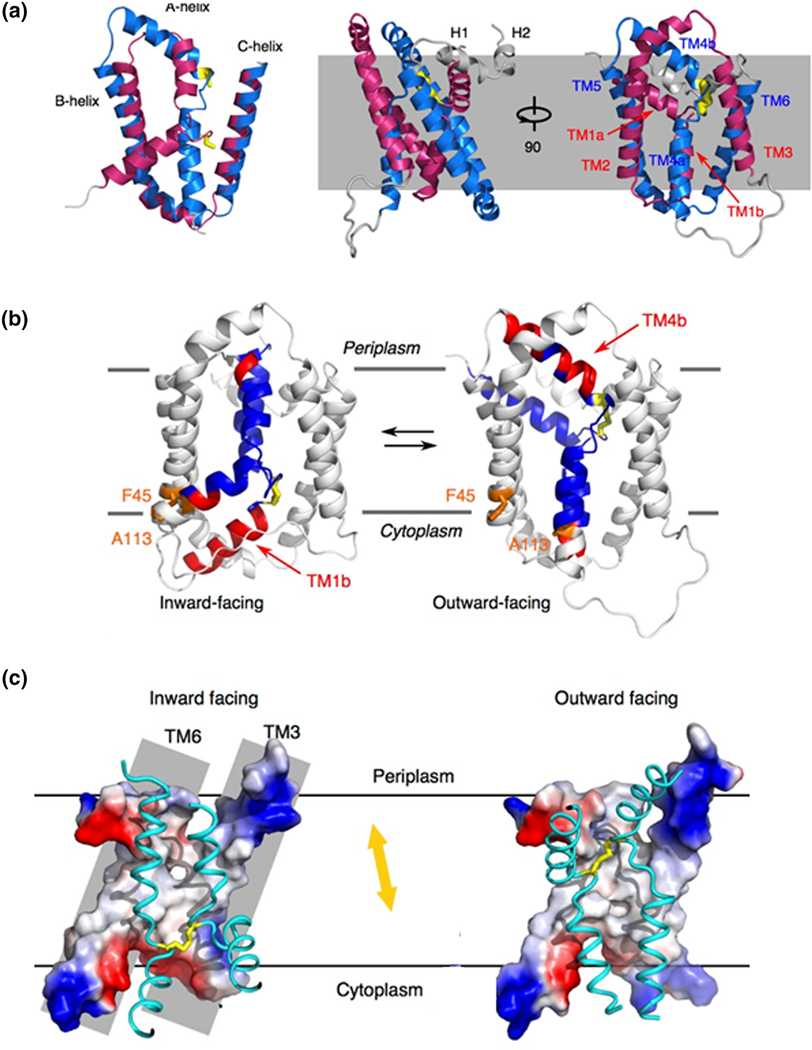
A. Overlay of the 3 TM helix repeats that comprise the structure of Thermus thermophilus CcdA. Structure of CcdA in two separate orientiations. B. Ribbon representations of the structure of the outward facing conformation of CcdA and the model of the inward facing conformation. The positions of Phe45 and Ala113, used for the cross-linking experiments, are indicated. C. Surface representations of the structure of the outward facing conformation of CcdA and the model of the inward facing conformation of CcdA, illustrating the conformational change that moves the active site Cys residues up and down within the protein.
Structural insights into the mechanism of CcdA mediated disulfide exchange across the membrane
▪. Model of cytoplasm facing conformation suggests an elevator mechanism for transport of reducing equivalents
In order for cytoplasmic thioredoxin to gain access to the active site, CcdA must undergo a conformational change to a conformation in which the disulfide is exposed to the cytoplasm. The imperfect structural symmetry of the two repeat elements we observe in the structure strongly suggests conformational flexibility. Based on this, we used an established repeat-swap homology modeling approach [103] to swap the conformations of the two repeat elements and show that this predicts a conformation that is now open to the cytoplasm (Figure 3). In this modeled conformation Cys20 of TM1 is more exposed to the cytoplasm than Cys127 indicating that Cys20 interacts with cytoplasmic thioredoxin, consistent with previous biochemical studies of DsbD [86]. Additionally, TM1a and TM4b now seal the disulfide bond off from the periplasmic side, providing a detailed picture of how the necessary alternating access is achieved. To confirm that this cytoplasm facing conformation is accessed by the protein, we carried out a Cys cross-linking experiment [104]. Two additional Cys residues were introduced, one into one of the transport helices and one in the scaffold, at positions that are distant in the periplasm facing conformation but predicted by the model of the cytoplasm facing conformation to be close in space. Treatment with one equivalent of HgCl2 protected these two new Cys residues from alkylation, indicative of formation of a cross-link between the two, which is only possible if they sample a conformation where the two Cys residues are close in space. In addition, the 15N-1H HSQC spectrum of the Hg treated mutant showed substantial changes relative to the untreated protein, indicative of a significant conformational change.
The outward-facing structure and inward-facing model suggest that CcdA uses an elevator-type transport mechanism [105]. Interestingly, CcdA is the smallest of the identified elevator transporters [105–107]. The O scaffold is immobile in the membrane, while TM1 and TM4 use a rotation movement to transfer the disulfide bond ~12 Å along the direction of the membrane norm (Figure 3). This rotation movement minimizes the hydrophobic mismatch as the result of TM1 and TM4 moving in and out of the membrane and maximizes the vertical translation of the disulfide bond. The disulfide bond runs against the inside surface of TM3 and TM6, which is tightly packed and hydrophobic in the middle to prevent solvent leakage while loosely packed and hydrophilic at both ends to encourage water penetration, which is important for the thiol/ disulfide-exchange reactions between CcdA and its substrates.
CcdA structure resembles other transporters
CcdA belongs to the LysE family superfamily of transporters which includes 6 families of divalent ion transporters, 3 families of amino acid transporters, 1 family of peptidoglycolipid transporter, and the CcdA family. The internal 3 TM repeats observed in CcdA are also observed in some members of the LysE superfamily [108]. In addition, conserved and functionally important motifs have been identified in TM1 and TM4 in other family members as well [108, 109]. Based on this CcdA may serve as a model to understand other members of the superfamily.
The use of 3 TM repeats as a building block is not unique to CcdA but is observed in a number of other transporters. semiSWEET and Pnu vitamin transporters also utilize 3 TM repeats, but they are arranged in a parallel manner [106, 110]. The major facilitator superfamily (MFS) is a large group of transporters and facilitators whose architectures are built up from four left-handed 3 TM repeats [111, 112], thus sharing the use of a left-handed 3 TM building block found in CcdA. In addition, within the N- and C-terminal halves of MFS family proteins, the two 3 TM units are antiparallel to one another, as seen in CcdA. It is quite intriguing that a wide range of transporters have evolved to utilize this same basic building block to mediate such a wide variety of functions across the membrane.
Perspectives
The wealth of structural information on both DsbB and CcdA has provided important insights into function. Future efforts are likely to focus both on the holes in our structural knowledge as well as efforts to harness the structure/function studies to help advance efforts to target these proteins for inhibitor development. E. coli DsbB is a member of the Group I DsbB family. We currently lack structural information on members of Group II and III DsbB proteins. In combination with functional studies on the identity of the proteins whose activity is regulated by these DsbB proteins, it is likely that opportunities to target essential functions of various pathogenic bacteria can be identified. In the context of the functional homologs DsbD and CcdA, we still lack structural information on the membrane domain of DsbD. As this is an 8 TM helix structure, it differs from that of CcdA and only a structure can define how the additional 2 TM helices contribute to the structure and the degree to which the structure of CcdA explains the mechanism of DsbD.
As described above for DsbB, and to a lesser extent for CcdA, these proteins contribute to essential functions of bacteria that mediate virulence. As the eukaryotic functional homologs are quite different, they represent intriguing targets for antibacterial drug development. There have already been some initial efforts to identify small molecules which inhibit DsbB function [113, 114]. Indeed, Beckwith and co-workers demonstrated that loss of DsbB in P. aeruginosa, a bacteria whose infections are particularly resistant to antibiotics and have a high mortality rate, resulted in a substantial reduction in virulence using two mouse models including a mouse model of P. aeruginosa induced pneumonia [114]. Based on this result they screened a large compound library for inhibitors of P. aeruginosa DsbBb1 and M. tuberculosis VKOR. As the authors point out, DsbA is a member of the thioredoxin family with many homologs in eukaryotes whereas DsbB does not have eukaryotic homologs, making it a more attractive target for drug development. The screen identified molecules which inhibit P. aeruginosa DsbB and at least one of which has a specificity of action that warrants further development. These results are highly encouraging, but further development as well as testing using in vivo models of bacterial infection will be required to evaluate the utility of this approach.
Highlights.
Protein disulfide bond formation is catalyzed by DsbA and the membrane protein DsbB.
Multiple structural studies of DsbB provided key insights into DsbB function.
Protein disulfide reduction is essential for isomerization and cytochrome c activity.
Membrane protein DsbD provides reducing equivalents to periplasmic substrates.
Structure of DsbD homolog CcdA provides key insights into CcdA, DsbD function.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Anfinsen CB, haber E, Sela M, White FH. Proc Natl Acad Sci USA. 1961;47:1309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Goldberger RF, Epstein CJ, Anfinsen CB. Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. The Journal of biological chemistry. 1963;238:628–35. [PubMed] [Google Scholar]
- [3].Venetianer P, Straub FB. The enzymic reactivation of reduced ribonuclease. Biochimica et biophysica acta. 1963;67:166–8. [DOI] [PubMed] [Google Scholar]
- [4].Bardwell JC, McGovern K, Beckwith J. Identification of a protein required for disulfide bond formation in vivo. Cell. 1991;67:581–9. [DOI] [PubMed] [Google Scholar]
- [5].Raina S, Missiakas D. Making and breaking disulfide bonds. Annu Rev Microbiol. 1997;51:179–202. [DOI] [PubMed] [Google Scholar]
- [6].Rietsch A, Beckwith J. The genetics of disulfide bond metabolism. Annu Rev Genet. 1998;32:163–84. [DOI] [PubMed] [Google Scholar]
- [7].Dailey FE, Berg HC. Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli. Proc Natl Acad Sci U S A. 1993;90:1043–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Missiakas D, Georgopoulos C, Raina S. Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc Natl Acad Sci U S A. 1993;90:7084–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stafford SJ, Humphreys DP, Lund PA. Mutations in dsbA and dsbB, but not dsbC, lead to an enhanced sensitivity of Escherichia coli to Hg2+ and Cd2+. FEMS Microbiol Lett. 1999;174:179–84. [DOI] [PubMed] [Google Scholar]
- [10].Peek JA, Taylor RK. Characterization of a periplasmic thiol:disulfide interchange protein required for the functional maturation of secreted virulence factors of Vibrio cholerae. Proc Natl Acad Sci U S A. 1992;89:6210–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Martin JL. Thioredoxin--a fold for all reasons. Structure. 1995;3:245–50. [DOI] [PubMed] [Google Scholar]
- [12].Schirra HJ, Renner C, Czisch M, Huber-Wunderlich M, Holak TA, Glockshuber R. Structure of reduced DsbA from Escherichia coli in solution. Biochemistry. 1998;37:6263–76. [DOI] [PubMed] [Google Scholar]
- [13].Guddat LW, Bardwell JC, Martin JL. Crystal structures of reduced and oxidized DsbA: investigation of domain motion and thiolate stabilization. Structure. 1998;6:757–67. [DOI] [PubMed] [Google Scholar]
- [14].Bardwell JC, Lee JO, Jander G, Martin N, Belin D, Beckwith J. A pathway for disulfide bond formation in vivo. Proc Natl Acad Sci U S A. 1993;90:1038–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kobayashi T, Kishigami S, Sone M, Inokuchi H, Mogi T, Ito K. Respiratory chain is required to maintain oxidized states of the DsbA-DsbB disulfide bond formation system in aerobically growing Escherichia coli cells. Proc Natl Acad Sci U S A. 1997;94:11857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bader M, Muse W, Ballou DP, Gassner C, Bardwell JC. Oxidative protein folding is driven by the electron transport system. Cell. 1999;98:217–27. [DOI] [PubMed] [Google Scholar]
- [17].Bader MW, Xie T, Yu CA, Bardwell JC. Disulfide bonds are generated by quinone reduction. J Biol Chem. 2000;275:26082–8. [DOI] [PubMed] [Google Scholar]
- [18].Sevier CS, Kadokura H, Tam VC, Beckwith J, Fass D, Kaiser CA. The prokaryotic enzyme DsbB may share key structural features with eukaryotic disulfide bond forming oxidoreductases. Protein Sci. 2005;14:1630–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dutton RJ, Boyd D, Berkmen M, Beckwith J. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature. 2010;463:507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Singh AK, Bhattacharyya-Pakrasi M, Pakrasi HB. Identification of an atypical membrane protein involved in the formation of protein disulfide bonds in oxygenic photosynthetic organisms. The Journal of biological chemistry. 2008;283:15762–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang X, Dutton RJ, Beckwith J, Boyd D. Membrane topology and mutational analysis of Mycobacterium tuberculosis VKOR, a protein involved in disulfide bond formation and a homologue of human vitamin K epoxide reductase. Antioxidants & redox signaling. 2011;14:1413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sevier CS, Cuozzo JW, Vala A, Aslund F, Kaiser CA. A flavoprotein oxidase defines a new endoplasmic reticulum pathway for biosynthetic disulphide bond formation. Nat Cell Biol. 2001;3:874–82. [DOI] [PubMed] [Google Scholar]
- [24].Thorpe C, Hoober KL, Raje S, Glynn NM, Burnside J, Turi GK, et al. Sulfhydryl oxidases: emerging catalysts of protein disulfide bond formation in eukaryotes. Arch Biochem Biophys. 2002;405:1–12. [DOI] [PubMed] [Google Scholar]
- [25].Gross E, Sevier CS, Vala A, Kaiser CA, Fass D. A new FAD-binding fold and intersubunit disulfide shuttle in the thiol oxidase Erv2p. Nat Struct Biol. 2002;9:61–7. [DOI] [PubMed] [Google Scholar]
- [26].Gross E, Kastner DB, Kaiser CA, Fass D. Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell. 2004;117:601–10. [DOI] [PubMed] [Google Scholar]
- [27].Alekshun MN, Levy SB. Molecular mechanisms of antibacterial multidrug resistance. Cell. 2007;128:1037–50. [DOI] [PubMed] [Google Scholar]
- [28].Livermore DM. Minimising antibiotic resistance. Lancet Infect Dis. 2005;5:450–9. [DOI] [PubMed] [Google Scholar]
- [29].Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5:175–86. [DOI] [PubMed] [Google Scholar]
- [30].Mwangi MM, Wu SW, Zhou Y, Sieradzki K, de Lencastre H, Richardson P, et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci U S A. 2007;104:9451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. The biology and future prospects of antivirulence therapies. Nat Rev Microbiol. 2008;6:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Finlay BB, Falkow S. Common themes in microbial pathogenicity revisited. Microbiol Mol Biol Rev. 1997;61:136–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee YM, Almqvist F, Hultgren SJ. Targeting virulence for antimicrobial chemotherapy. Curr Opin Pharmacol. 2003;3:513–9. [DOI] [PubMed] [Google Scholar]
- [34].Marra A. Can virulence factors be viable antibacterial targets? Expert Rev Anti Infect Ther. 2004;2:61–72. [DOI] [PubMed] [Google Scholar]
- [35].Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. DSB proteins and bacterial pathogenicity. Nat Rev Microbiol. 2009;7:215–25. [DOI] [PubMed] [Google Scholar]
- [36].Lasica AM, Jagusztyn-Krynicka EK. The role of Dsb proteins of Gram-negative bacteria in the process of pathogenesis. FEMS Microbiol Rev. 2007;31:626–36. [DOI] [PubMed] [Google Scholar]
- [37].Landeta C, Boyd D, Beckwith J. Disulfide bond formation in prokaryotes. Nat Microbiol. 2018;3:270–80. [DOI] [PubMed] [Google Scholar]
- [38].Jacob-Dubuisson F, Pinkner J, Xu Z, Striker R, Padmanhaban A, Hultgren SJ. PapD chaperone function in pilus biogenesis depends on oxidant and chaperone-like activities of DsbA. Proc Natl Acad Sci U S A. 1994;91:11552–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Braun P, Ockhuijsen C, Eppens E, Koster M, Bitter W, Tommassen J. Maturation of Pseudomonas aeruginosa elastase. Formation of the disulfide bonds. J Biol Chem. 2001;276:26030–5. [DOI] [PubMed] [Google Scholar]
- [40].Yu J. Inactivation of DsbA, but not DsbC and DsbD, affects the intracellular survival and virulence of Shigella flexneri. Infect Immun. 1998;66:3909–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wulfing C, Rappuoli R. Efficient production of heat-labile enterotoxin mutant proteins by overexpression of dsbA in a degP-deficient Escherichia coli strain. Arch Microbiol. 1997;167:280–3. [DOI] [PubMed] [Google Scholar]
- [42].Yu J, Webb H, Hirst TR. A homologue of the Escherichia coli DsbA protein involved in disulphide bond formation is required for enterotoxin biogenesis in Vibrio cholerae. Mol Microbiol. 1992;6:1949–58. [DOI] [PubMed] [Google Scholar]
- [43].Bringer MA, Rolhion N, Glasser AL, Darfeuille-Michaud A. The oxidoreductase DsbA plays a key role in the ability of the Crohn’s disease-associated adherent-invasive Escherichia coli strain LF82 to resist macrophage killing. J Bacteriol. 2007;189:4860–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Burall LS, Harro JM, Li X, Lockatell CV, Himpsl SD, Hebel JR, et al. Proteus mirabilis genes that contribute to pathogenesis of urinary tract infection: identification of 25 signature-tagged mutants attenuated at least 100-fold. Infect Immun. 2004;72:2922–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Coulthurst SJ, Lilley KS, Hedley PE, Liu H, Toth IK, Salmond GP. DsbA plays a critical and multifaceted role in the production of secreted virulence factors by the phytopathogen Erwinia carotovora subsp. atroseptica. J Biol Chem. 2008;283:23739–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ha UH, Wang Y, Jin S. DsbA of Pseudomonas aeruginosa is essential for multiple virulence factors. Infect Immun. 2003;71:1590–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lee SH, Butler SM, Camilli A. Selection for in vivo regulators of bacterial virulence. Proc Natl Acad Sci U S A. 2001;98:6889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lin D, Rao CV, Slauch JM. The Salmonella SPI1 type three secretion system responds to periplasmic disulfide bond status via the flagellar apparatus and the RcsCDB system. J Bacteriol. 2008;190:87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Meima R, Eschevins C, Fillinger S, Bolhuis A, Hamoen LW, Dorenbos R, et al. The bdbDC operon of Bacillus subtilis encodes thiol-disulfide oxidoreductases required for competence development. J Biol Chem. 2002;277:6994–7001. [DOI] [PubMed] [Google Scholar]
- [50].Qin A, Scott DW, Mann BJ. Francisella tularensis subsp. tularensis Schu S4 disulfide bond formation protein B, but not an RND-type efflux pump, is required for virulence. Infect Immun. 2008;76:3086–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tomb JF. A periplasmic protein disulfide oxidoreductase is required for transformation of Haemophilus influenzae Rd. Proc Natl Acad Sci U S A. 1992;89:10252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jander G, Martin NL, Beckwith J. Two cysteines in each periplasmic domain of the membrane protein DsbB are required for its function in protein disulfide bond formation. Embo J. 1994;13:5121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kobayashi T, Ito K. Respiratory chain strongly oxidizes the CXXC motif of DsbB in the Escherichia coli disulfide bond formation pathway. The EMBO journal. 1999;18:1192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kadokura H, Beckwith J. Four cysteines of the membrane protein DsbB act in concert to oxidize its substrate DsbA. Embo J. 2002;21:2354–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Inaba K, Takahashi YH, Fujieda N, Kano K, Miyoshi H, Ito K. DsbB elicits a red-shift of bound ubiquinone during the catalysis of DsbA oxidation. J Biol Chem. 2004;279:6761–8. [DOI] [PubMed] [Google Scholar]
- [56].Kadokura H, Bader M, Tian H, Bardwell JC, Beckwith J. Roles of a conserved arginine residue of DsbB in linking protein disulfide-bond-formation pathway to the respiratory chain of Escherichia coli. Proc Natl Acad Sci U S A. 2000;97:10884–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kobayashi T, Takahashi Y, Ito K. Identification of a segment of DsbB essential for its respiration-coupled oxidation. Mol Microbiol. 2001;39:158–65. [DOI] [PubMed] [Google Scholar]
- [58].Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, et al. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell. 2006;127:789–801. [DOI] [PubMed] [Google Scholar]
- [59].Kishigami S, Ito K. Roles of cysteine residues of DsbB in its activity to reoxidize DsbA, the protein disulphide bond catalyst of Escherichia coli. Genes Cells. 1996;1:201–8. [DOI] [PubMed] [Google Scholar]
- [60].Inaba K, Takahashi YH, Ito K. Reactivities of quinone-free DsbB from Escherichia coli. The Journal of biological chemistry. 2005;280:33035–44. [DOI] [PubMed] [Google Scholar]
- [61].Inaba K, Ito K. Paradoxical redox properties of DsbB and DsbA in the protein disulfide-introducing reaction cascade. The EMBO journal. 2002;21:2646–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Malojcic G, Owen RL, Grimshaw JP, Glockshuber R. Preparation and structure of the charge-transfer intermediate of the transmembrane redox catalyst DsbB. FEBS letters. 2008;582:3301–7. [DOI] [PubMed] [Google Scholar]
- [63].Zhou Y, Cierpicki T, Jimenez RH, Lukasik SM, Ellena JF, Cafiso DS, et al. NMR solution structure of the integral membrane enzyme DsbB: functional insights into DsbB-catalyzed disulfide bond formation. Molecular cell. 2008;31:896–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tapley TL, Eichner T, Gleiter S, Ballou DP, Bardwell JC. Kinetic characterization of the disulfide bond-forming enzyme DsbB. The Journal of biological chemistry. 2007;282:10263–71. [DOI] [PubMed] [Google Scholar]
- [65].Inaba K, Murakami S, Nakagawa A, Iida H, Kinjo M, Ito K, et al. Dynamic nature of disulphide bond formation catalysts revealed by crystal structures of DsbB. The EMBO journal. 2009;28:779–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hatahet F, Boyd D, Beckwith J. Disulfide bond formation in prokaryotes: history, diversity and design. Biochimica et biophysica acta. 2014;1844:1402–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hiniker A, Bardwell JC. In vivo substrate specificity of periplasmic disulfide oxidoreductases. The Journal of biological chemistry. 2004;279:12967–73. [DOI] [PubMed] [Google Scholar]
- [68].Rietsch A, Belin D, Martin N, Beckwith J. An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:13048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Rietsch A, Bessette P, Georgiou G, Beckwith J. Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. Journal of bacteriology. 1997;179:6602–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chen J, Song JL, Zhang S, Wang Y, Cui DF, Wang CC. Chaperone activity of DsbC. The Journal of biological chemistry. 1999;274:19601–5. [DOI] [PubMed] [Google Scholar]
- [71].Heras B, Edeling MA, Schirra HJ, Raina S, Martin JL. Crystal structures of the DsbG disulfide isomerase reveal an unstable disulfide. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8876–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bessette PH, Cotto JJ, Gilbert HF, Georgiou G. In vivo and in vitro function of the Escherichia coli periplasmic cysteine oxidoreductase DsbG. The Journal of biological chemistry. 1999;274:7784–92. [DOI] [PubMed] [Google Scholar]
- [73].Depuydt M, Leonard SE, Vertommen D, Denoncin K, Morsomme P, Wahni K, et al. A periplasmic reducing system protects single cysteine residues from oxidation. Science. 2009;326:1109–11. [DOI] [PubMed] [Google Scholar]
- [74].McCarthy AA, Haebel PW, Torronen A, Rybin V, Baker EN, Metcalf P. Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli. Nature structural biology. 2000;7:196–9. [DOI] [PubMed] [Google Scholar]
- [75].Zapun A, Missiakas D, Raina S, Creighton TE. Structural and functional characterization of DsbC, a protein involved in disulfide bond formation in Escherichia coli. Biochemistry. 1995;34:5075–89. [DOI] [PubMed] [Google Scholar]
- [76].Segatori L, Murphy L, Arredondo S, Kadokura H, Gilbert H, Beckwith J, et al. Conserved role of the linker alpha-helix of the bacterial disulfide isomerase DsbC in the avoidance of misoxidation by DsbB. The Journal of biological chemistry. 2006;281:4911–9. [DOI] [PubMed] [Google Scholar]
- [77].Haebel PW, Goldstone D, Katzen F, Beckwith J, Metcalf P. The disulfide bond isomerase DsbC is activated by an immunoglobulin-fold thiol oxidoreductase: crystal structure of the DsbC-DsbDalpha complex. The EMBO journal. 2002;21:4774–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Crooke H, Cole J. The biogenesis of c-type cytochromes in Escherichia coli requires a membrane-bound protein, DipZ, with a protein disulphide isomerase-like domain. Mol Microbiol. 1995;15:1139–50. [DOI] [PubMed] [Google Scholar]
- [79].Beckett CS, Loughman JA, Karberg KA, Donato GM, Goldman WE, Kranz RG. Four genes are required for the system II cytochrome c biogenesis pathway in Bordetella pertussis, a unique bacterial model. Mol Microbiol. 2000;38:465–81. [DOI] [PubMed] [Google Scholar]
- [80].Deshmukh M, Brasseur G, Daldal F. Novel Rhodobacter capsulatus genes required for the biogenesis of various c-type cytochromes. Mol Microbiol. 2000;35:123–38. [DOI] [PubMed] [Google Scholar]
- [81].Andersen CL, Matthey-Dupraz A, Missiakas D, Raina S. A new Escherichia coli gene, dsbG, encodes a periplasmic protein involved in disulphide bond formation, required for recycling DsbA/DsbB and DsbC redox proteins. Mol Microbiol. 1997;26:121–32. [DOI] [PubMed] [Google Scholar]
- [82].Kim JH, Kim SJ, Jeong DG, Son JH, Ryu SE. Crystal structure of DsbDgamma reveals the mechanism of redox potential shift and substrate specificity(1). FEBS letters. 2003;543:164–9. [DOI] [PubMed] [Google Scholar]
- [83].Rozhkova A, Stirnimann CU, Frei P, Grauschopf U, Brunisholz R, Grutter MG, et al. Structural basis and kinetics of inter- and intramolecular disulfide exchange in the redox catalyst DsbD. The EMBO journal. 2004;23:1709–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Goulding CW, Sawaya MR, Parseghian A, Lim V, Eisenberg D, Missiakas D. Thiol-disulfide exchange in an immunoglobulin-like fold: structure of the N-terminal domain of DsbD. Biochemistry. 2002;41:6920–7. [DOI] [PubMed] [Google Scholar]
- [85].Stewart EJ, Katzen F, Beckwith J. Six conserved cysteines of the membrane protein DsbD are required for the transfer of electrons from the cytoplasm to the periplasm of Escherichia coli. The EMBO journal. 1999;18:5963–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Katzen F, Beckwith J. Transmembrane electron transfer by the membrane protein DsbD occurs via a disulfide bond cascade. Cell. 2000;103:769–79. [DOI] [PubMed] [Google Scholar]
- [87].Collet JF, Riemer J, Bader MW, Bardwell JC. Reconstitution of a disulfide isomerization system. The Journal of biological chemistry. 2002;277:26886–92. [DOI] [PubMed] [Google Scholar]
- [88].Rozhkova A, Glockshuber R. Thermodynamic aspects of DsbD-mediated electron transport. Journal of molecular biology. 2008;380:783–8. [DOI] [PubMed] [Google Scholar]
- [89].Katzen F, Beckwith J. Role and location of the unusual redox-active cysteines in the hydrophobic domain of the transmembrane electron transporter DsbD. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10471–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Porat A, Cho SH, Beckwith J. The unusual transmembrane electron transporter DsbD and its homologues: a bacterial family of disulfide reductases. Res Microbiol. 2004;155:617–22. [DOI] [PubMed] [Google Scholar]
- [91].Hiniker A, Vertommen D, Bardwell JC, Collet JF. Evidence for conformational changes within DsbD: possible role for membrane-embedded proline residues. Journal of bacteriology. 2006;188:7317–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Cho SH, Parsonage D, Thurston C, Dutton RJ, Poole LB, Collet JF, et al. A new family of membrane electron transporters and its substrates, including a new cell envelope peroxiredoxin, reveal a broadened reductive capacity of the oxidative bacterial cell envelope. mBio. 2012;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Gupta SD, Wu HC, Rick PD. A Salmonella typhimurium genetic locus which confers copper tolerance on copper-sensitive mutants of Escherichia coli. Journal of bacteriology. 1997;179:4977–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Shepherd M, Heras B, Achard ME, King GJ, Argente MP, Kurth F, et al. Structural and functional characterization of ScsC, a periplasmic thioredoxin-like protein from Salmonella enterica serovar Typhimurium. Antioxidants & redox signaling. 2013;19:1494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Furlong EJ, Lo AW, Kurth F, Premkumar L, Totsika M, Achard MES, et al. A shape-shifting redox foldase contributes to Proteus mirabilis copper resistance. Nature communications. 2017;8:16065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Furlong EJ, Choudhury HG, Kurth F, Duff AP, Whitten AE, Martin JL. Disulfide isomerase activity of the dynamic, trimeric Proteus mirabilis ScsC protein is primed by the tandem immunoglobulin-fold domain of ScsB. The Journal of biological chemistry. 2018;293:5793–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Deshmukh M, Brasseur G, Daldal F. Novel Rhodobacter capsulatus genes required for the biogenesis of various c-type cytochromes. Molecular Microbiology. 2000;35:123–38. [DOI] [PubMed] [Google Scholar]
- [98].Schiott T, Hederstedt L. Efficient spore synthesis in Bacillus subtilis depends on the CcdA protein. Journal of bacteriology. 2000;182:2845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Katzen F, Deshmukh M, Daldal F, Beckwith J. Evolutionary domain fusion expanded the substrate specificity of the transmembrane electron transporter DsbD. The EMBO journal. 2002;21:3960–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zhou Y, Bushweller JH. Solution structure and elevator mechanism of the membrane electron transporter CcdA. Nature structural & molecular biology. 2018;25:163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Cho SH, Porat A, Ye J, Beckwith J. Redox-active cysteines of a membrane electron transporter DsbD show dual compartment accessibility. The EMBO journal. 2007;26:3509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Cho SH, Beckwith J. Two snapshots of electron transport across the membrane: insights into the structure and function of DsbD. The Journal of biological chemistry. 2009;284:11416–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Vergara-Jaque A, Fenollar-Ferrer C, Kaufmann D, Forrest LR. Repeat-swap homology modeling of secondary active transporters: updated protocol and prediction of elevator-type mechanisms. Front Pharmacol. 2015;6:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Mulligan C, Fenollar-Ferrer C, Fitzgerald GA, Vergara-Jaque A, Kaufmann D, Li Y, et al. The bacterial dicarboxylate transporter VcINDY uses a two-domain elevator-type mechanism. Nature structural & molecular biology. 2016;23:256–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Drew D, Boudker O. Shared Molecular Mechanisms of Membrane Transporters. Annual review of biochemistry. 2016;85:543–72. [DOI] [PubMed] [Google Scholar]
- [106].Xu Y, Tao Y, Cheung LS, Fan C, Chen LQ, Xu S, et al. Structures of bacterial homologues of SWEET transporters in two distinct conformations. Nature. 2014;515:448–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lee Y, Nishizawa T, Yamashita K, Ishitani R, Nureki O. Structural basis for the facilitative diffusion mechanism by SemiSWEET transporter. Nature communications. 2015;6:6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Tsu BV, Saier MH Jr. The LysE Superfamily of Transport Proteins Involved in Cell Physiology and Pathogenesis. PloS one. 2015;10:e0137184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Colinet AS, Thines L, Deschamps A, Flemal G, Demaegd D, Morsomme P. Acidic and uncharged polar residues in the consensus motifs of the yeast Ca(2+) transporter Gdt1p are required for calcium transport. Cell Microbiol. 2017;19. [DOI] [PubMed] [Google Scholar]
- [110].Jaehme M, Guskov A, Slotboom DJ. Crystal structure of the vitamin B3 transporter PnuC, a full-length SWEET homolog. Nature structural & molecular biology. 2014;21:1013–5. [DOI] [PubMed] [Google Scholar]
- [111].Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301:610–5. [DOI] [PubMed] [Google Scholar]
- [112].Yan N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends in biochemical sciences. 2013;38:151–9. [DOI] [PubMed] [Google Scholar]
- [113].Fruh V, Zhou Y, Chen D, Loch C, Ab E, Grinkova YN, et al. Application of fragment-based drug discovery to membrane proteins: identification of ligands of the integral membrane enzyme DsbB. Chemistry & biology. 2010;17:881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Landeta C, McPartland L, Tran NQ, Meehan BM, Zhang Y, Tanweer Z, et al. Inhibition of Pseudomonas aeruginosa and Mycobacterium tuberculosis disulfide bond forming enzymes. Mol Microbiol. 2019;111:918–37. [DOI] [PMC free article] [PubMed] [Google Scholar]