

Abstract
Top-down proteomics by mass spectrometry (MS) involves the mass measurement of an intact protein followed by subsequent activation of the protein to generate product ions. Electron-based fragmentation methods like electron capture dissociation (ECD) and electron transfer dissociation (ETD) are widely used for these types of analysis. Recently, electron ionization dissociation (EID), which utilizes higher energy electrons (> 20 eV) has been suggested to be more efficient for top-down protein fragmentation compared to other electron-based dissociation methods. Here we demonstrate that the use of EID enhances protein fragmentation and subsequent detection of protein fragments. Protein product ions can form by either single cleavage events, resulting in terminal fragments containing the C-terminus or N-terminus of the protein, or by multiple cleavage events to give rise to internal fragments that include neither the C-terminus nor N-terminus of the protein. Conventionally, internal fragments have been disregarded as reliable assignments of these fragments were limited. Here, we demonstrate that internal fragments generated by EID can account for ~20–40% of the mass spectral signals detected by top-down EID-MS experiments. By including internal fragments, the extent of the protein sequence that can be explained from a single tandem mass spectrum increases from ~50% to ~99% for 29 kDa carbonic anhydrase II and 8.6 kDa ubiquitin. When searching for internal fragments during data analysis, previously unassigned peaks can be readily and accurately assigned to confirm a given protein sequence and to enhance the utility of top-down protein sequencing experiments.
Graphical Abstract
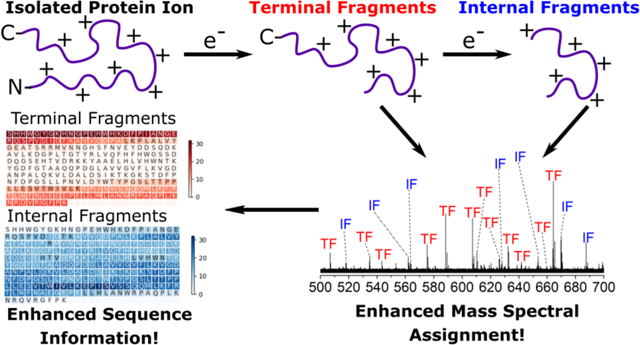
Electron ionization dissociation of proteins produces internal product ions that enhance sequence coverage for top-down mass spectrometry.
INTRODUCTION
Top-down mass spectrometry (MS) has emerged as a technique to characterize proteins and to elucidate unique proteoforms.1, 2 Typically, intact protein ions are generated using electrospray ionization (ESI), followed by dissociation of the intact protein ion within the mass spectrometer to generate product ions that can be used to return information about protein identification and primary structure, i.e., sequence. Electron-based dissociation techniques such as electron capture dissociation (ECD)3, 4 and electron transfer dissociation (ETD)5 employ low-energy electrons to generate protein fragment ions. ECD/ETD confers many advantages over other dissociation techniques (e.g., collision induced dissociation (CID),6 surface induced dissociation (SID),7 and ultraviolet photodissociation (UVPD)8), including but not limited to conserving post-translational modifications and non-selective fragmentation of the protein backbone.9, 10 Due to non-specific cleavage during electron-based dissociation, fragmentation by electron-based methods has the potential to generate more protein fragments that allow for richer sequence information.11, 12 Despite the many advantages and prevalent use of ECD/ETD (ExD),13 these fragmentation techniques can be limiting due to the reliance of generating protein ions in higher charge states.14 In addition, proteins have low electron capture efficiencies, thus potentially limiting ExD efficiency.3, 15
Electron ionization dissociation (EID) is a recently discovered alternative ExD fragmentation technique for peptide and protein characterization.16, 17, 18 EID utilizes high energy electrons (> 20 eV) to induce protein fragmentation along the backbone. In EID, the interaction of a multiply charged protein ion with a high energy electron results in the formation of the oxidized species. Subsequent rearrangement of the oxidized species and/or capture of a second electron promotes backbone fragmentation. Using EID, Zubarev and co-workers demonstrated that fragmentation efficiency for some proteins and peptides can be close to 100%.18 This data suggests that EID can result in more efficient fragmentation of polypeptides compared to ECD, which would be especially beneficial for the analysis of large proteins. Recently, Loo and co-workers demonstrated that by using EID for native top-down MS, extensive fragmentation of apo-human superoxide dismutase 1 homodimer complex (32 kDa) could be achieved, whereas ECD resulted only in charge reduced precursors and no protein fragmentation.17 In addition, EID can be used to probe the metal binding sites of proteins and protein complexes, which suggests that EID could be beneficial for investigating the binding of non-covalent ligands and labile PTMs.17 Despite the potential advantages conferred by EID compared to ECD/ETD, the use of EID for protein top-down MS has not been extensively explored.
Protein product ions can either be (i) a terminal fragment ion, where only a single cleavage event occurs to generate N-terminal-containing a, b, c fragments or C-terminal-containing x, y, z fragments,19 or (ii) an internal fragment ion, where two cleavage events occur generating ax, ay, az, bx, by, bz, cx, cy, cz fragment ions depending on the activation method occurring.20, 21, 22, 23 The number of theoretical internal products that can be generated is significantly greater than the number of possible terminal fragments that can be generated.20 Traditionally, internal fragments have been largely ignored due to the inability to reliably assign internal fragments.24 Due to this, a plethora of information that can be accessed has largely been ignored.
The analysis of internal fragments previously has been limited to peptides and small molecules.25, 26 Assignment of internal fragments for intact proteins has been relatively limiting owing to the complexity of the fragmentation spectra. Kelleher and co-workers showed that internal fragments from CID fragmentation of the common test protein, ubiquitin (8.6 kDa), can be assigned to result in significantly greater protein sequence coverage.20 Similarly, for other intact proteins, the inclusion of internal fragments that can be generated by CID could result in greater explanation of the protein sequence.27, 28 Our laboratory demonstrated that internal product ions can be generated from top-down MS of large, native protein complexes.29 These examples suggest that the inclusion of internal fragments in top-down protein sequencing experiments could significantly enhance the protein sequence coverage and the efficiency of top-down mass spectrometry experiments.
Here, we investigate the utility of EID and the inclusion of internal fragments for top-down protein sequencing experiments. By using EID for top-down MS of ubiquitin (Ubq) and 29 kDa carbonic anhydrase II (CAII), the number of product ions is significantly higher compared to ECD, and approximately 20–40% of the fragments in the mass spectra can be assigned to internal fragments. Inclusion of EID-generated internal fragment ions yields nearly complete sequence coverage for CAII.
EXPERIMENTAL
Materials.
Bovine carbonic anhydrase II and bovine ubiquitin were purchased from Sigma Aldrich (St. Louis, MO, USA) and used without further purification. LC/MS grade water and methanol were obtained from Fisher Chemical (Hampton, NH, USA). For electrospray ionization, aqueous solutions containing 10–20 μM protein, 49.5% water, 49.5% methanol, and 1% formic acid (v/v) were prepared.
Mass spectrometry.
All experiments were conducted on a 15-Tesla Bruker SolariX Fourier transform ion cyclotron resonance (FTICR)-MS equipped with an infinity ICR cell (Bruker Daltonics, Billerica, MA, USA). Protein solutions were loaded into metal-coated borosilicate capillaries (Au/Pd-coated, 1 μM inner diameter, Thermo Fisher Scientific, Waltham, MA, USA) and electrospray was initiated by applying a voltage between 0.9–1.4 kV on the ESI capillary. Charge states were isolated in the quadrupole, with an isolation window of 10 m/z, before EID/ECD fragmentation. For ECD fragmentation, the pulse length was set at 0.05s, with a lens voltage of 50 V, and an ECD bias voltage of 2 V. For EID fragmentation, the pulse length and lens voltage were kept constant and the bias voltage was altered between 20–30 V. For each spectrum, 200 scans were obtained.
Data analysis.
Peak assignments.
Deconvoluted mass lists were obtained from Bruker Data Analysis software, using the SNAP algorithm. Deconvoluted mass lists were uploaded into our in-house-developed python program that calculates all possible terminal and internal fragments written and compares them to the experimental deconvoluted masses. (A future report will describe the details of the program and the user interface.) The error for matching was set at 1 ppm error, and 42.0105603 for the mass of acetylation was added to all the N-terminal fragments for carbonic anhydrase II. Internal fragments searched and assigned were only for cz internal fragments.
Protein sequence confirmation.
Protein sequence elucidated were calculated by equation 1:
| [Eq. 1] |
where AAdet is the number of times an amino acid residue was detected. For internal fragments, amino acids were detected in at least 5 different fragments to ensure accurate detection similar to previous thresholds of Kelleher and coworkers.20 AAtot is the total number of amino acids in the protein. The sequence elucidated should give an indication of how much of the protein sequence can be defined by the fragments assigned.
Protein fragment coverage.
Protein fragment coverages were calculated by identifying the number of observed inter-residue sites divided by the total number of inter-residue cleavages on the protein backbone. For example, Ubq and CAII have 75 and 259 inter-residue cleavage sites, respectively.
RESULTS AND DISCUSSION
EID for protein tandem-MS experiments can result in greater fragmentation compared to ECD (Figure 1). EID (25 eV) of [CAII, 25H]25+ (i.e., [M+25H]25+) resulted in the formation of the [CAII, 25H]26+*· ion with measurable abundance (Figure 1b), which is in good agreement with Zubarev and coworkers’ observation that EID results in the formation of the oxidized species.30 Representative mass spectral signals for some product ions identifiable within the m/z 500–700 range are shown within the insets of Figure 1.
Figure 1.
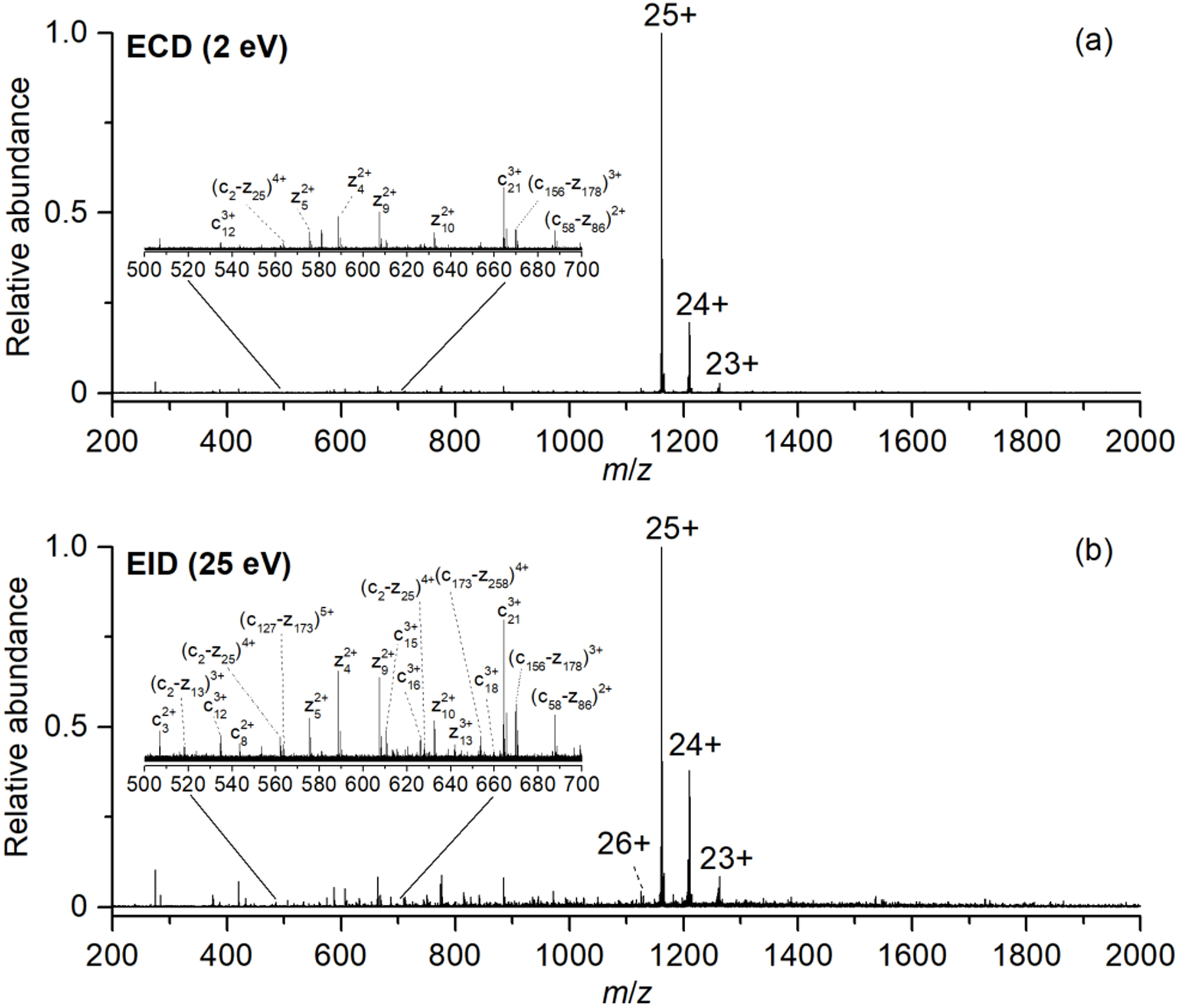
Representative top-down mass spectra of isolated [CAII, 25H]25+ formed from 10 μM carbonic anhydrase II in 50:50 water:methanol and 1% formic acid using (a) ECD (2eV), and (b) EID (25eV).
The product ions formed by EID have higher signal-to-noise ratios (S/N) compared to the fragments formed by ECD. For example, for the c123+ ion detected in both the ECD and EID spectra, the fragment within the EID spectrum has ~ 30% higher S/N compared to the fragment detected in the ECD spectrum. On average, there is ~ 30–40% increase in S/N of fragments formed by EID compared to ECD, suggesting that EID is a more efficient fragmentation method.
The number of fragment ions that are generated and detected by EID is also greater than that generated by ECD. For example, EID of [CAII, 25H]25+ resulted in the formation of 145 unique fragments, whereas ECD of [CAII, 25H]25+ resulted in the formation of 100 unique fragments (Figure 2a). Similarly, for [Ubq, 10H]10+, more fragments are formed by EID (159 unique fragments) compared to ECD (102 unique fragments) (Figure S1). Previous data from our lab demonstrated that EID generated more fragments than ECD for native SOD-1 protein.17
Figure 2.
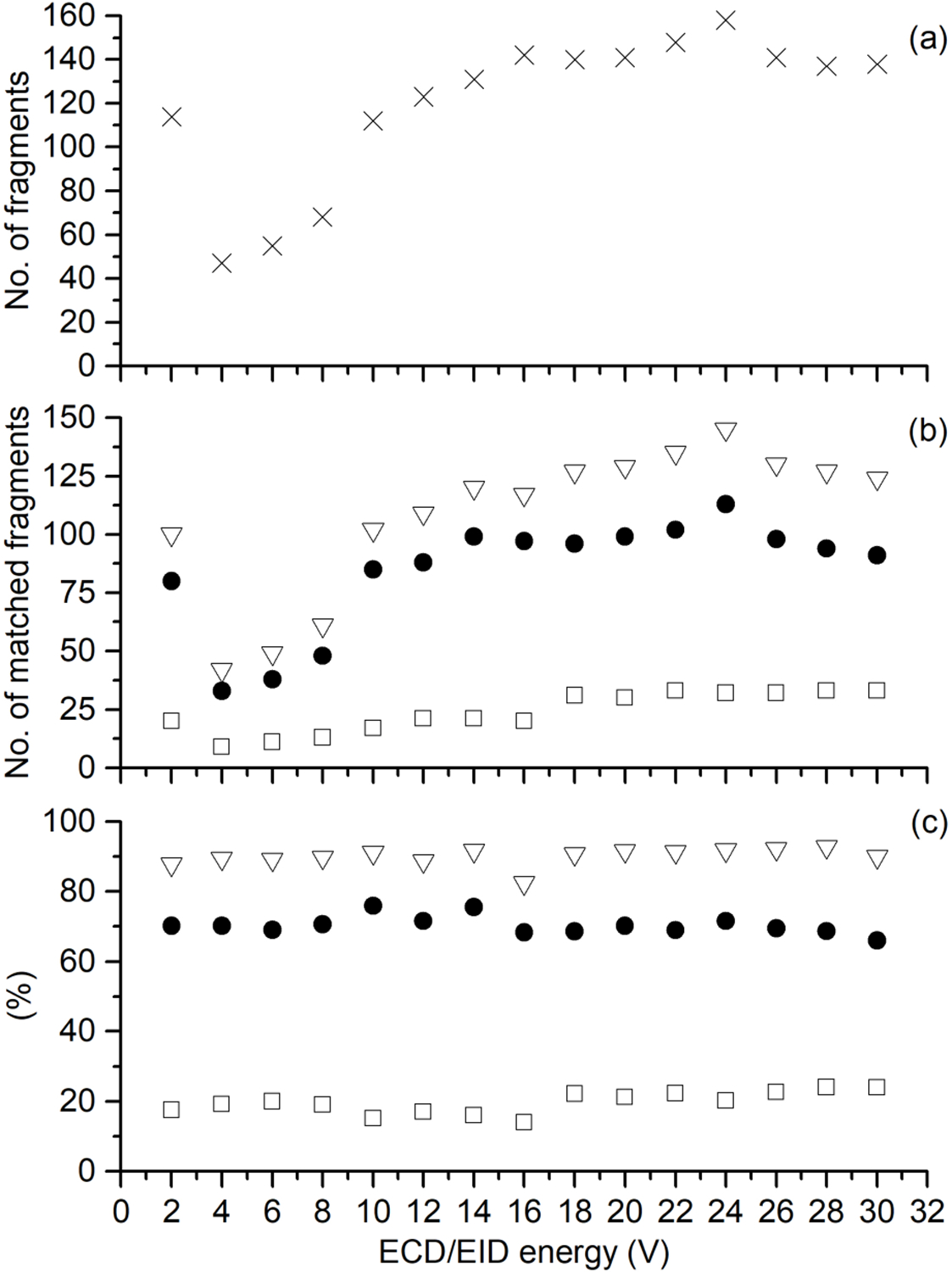
The effect of ECD/EID energy for [CAII, 25H]25+, where (a) is the number of fragments deconvoluted, and (b) is the total number of fragments identified (open triangles), total number of terminal fragments identified (closed circles), and the total number of internal fragments identified (open squares). The fraction of fragments matched is shown in (c), where the percentage of all fragments identified (open triangles), percentage of terminal fragments identified (closed circles), and the percentage of internal fragments identified (open squares) are shown.
Product ions generated for proteins can be classified as either terminal fragments (fragments containing the N-terminus or C-terminus), or internal fragments (fragments that contain neither the N-terminus nor C-terminus). The data shown here and in other reports suggest that internal fragments can account for many of the mass spectral signals within a mass spectrum regardless of the fragmentation method.31 Interestingly, the ion abundances of internal fragments are not significantly lower compared to low abundant terminal fragments formed. For example, the fragment assigned to the internal fragment (c58-z86)2+ is similar in ion abundance to low abundant c- and z-fragment ions detected within the spectrum. On average, the total intensity of terminal fragments is ~1.11 × 107 compared to ~6.74 × 106 for internal fragments (Table S1). This suggests that these fragments are identifiable and could be assigned. Therefore, the inclusion of these internal fragments could result in greater sequence information for the protein compared to terminal fragments alone.
Conventionally, internal fragments have been ignored, owing to the inability to reliably assign them due to instrument and computational limitations.24, 32 However, many ion signals within tandem mass spectra can potentially be explained by internal fragments (Figure S2b). Here, we attempt to assign previously unassigned mass spectral signals by calculating internal fragment masses using an in-house written program (see Materials and Methods); cz internal fragments can be uniquely assigned to the known protein sequence if MS2 mass calibration is achieved to ≤ 1 ppm error to limit false positives. For ECD of [CAII, 25H]25+, there were 100 unique fragments formed, of which 80 (80%) are assigned to terminal fragments and 20 (20%) are assigned to internal fragments. For EID, there were 145 unique fragments formed for [CAII, 25H]25+; 113 terminal (78%) and 32 internal fragments (22%). The number of internal fragment ions that are formed for CAII, and Ubq can account for 20–40 % of the ion signals within a mass spectrum (Figure 2; Figure S1).
The effect of electron energy on the ECD/EID efficiency of [CAII, 25H]25+ was investigated (Figure 2). At a conventional ECD energy (2 eV), 101 unique fragments were identified. However, at greater than 20 eV EID energies there are more fragments generated (> 125 unique fragments), with an energy range between 20–26 eV being optimal for generating the largest number of fragments (Figure 2a). Interestingly, as electron energy increases, the number of internal fragments formed also increases (Figure 2b); the number of internal fragments formed at 24 eV was 33 compared to only 20 at 2 eV. However, the percentage of fragments that are assigned as internal fragments do not increase significantly at 24 eV compared to 2 eV (18% to 24%, respectively). Similarly, for ubiquitin the percentage of fragments that can be explained by internal fragments formed at ECD energies is similar to the percentage of internal fragments formed at EID energies (Figure S1). The formation of internal fragments from EID of Ubq is in good agreement with previous data from Kelleher and co-workers who demonstrated that CID of Ubq results in the formation of internal fragments, with approximately 30% of the mass spectral signals attributed to internal fragments.20 As internal fragments are generated within conventional dissociation experiments (e.g., CID, SID, ECD/ETD), the assignment of internal fragments should be beneficial for characterizing the protein sequence as more of the mass spectral signals can be assigned to protein fragments.
The efficiency of ECD fragmentation has been shown to correlate with the charge state of the precursor ion.14, 33, 34, 35 Similarly, for EID the fragmentation efficiency increases with charge (Figure 3a). For CAII, as the precursor charge increases from 10+ to 38+, the number of fragments increases from 105 to 250, respectively. A similar trend is observed for Ubq; the number of fragments increases from 69 (7+ precursor ion charge) to 175 (13+) (Figure S1). EID’s greater fragmentation efficiency for higher charge states could be due to lowering the barriers for dissociation and larger reaction cross sections.
Figure 3.
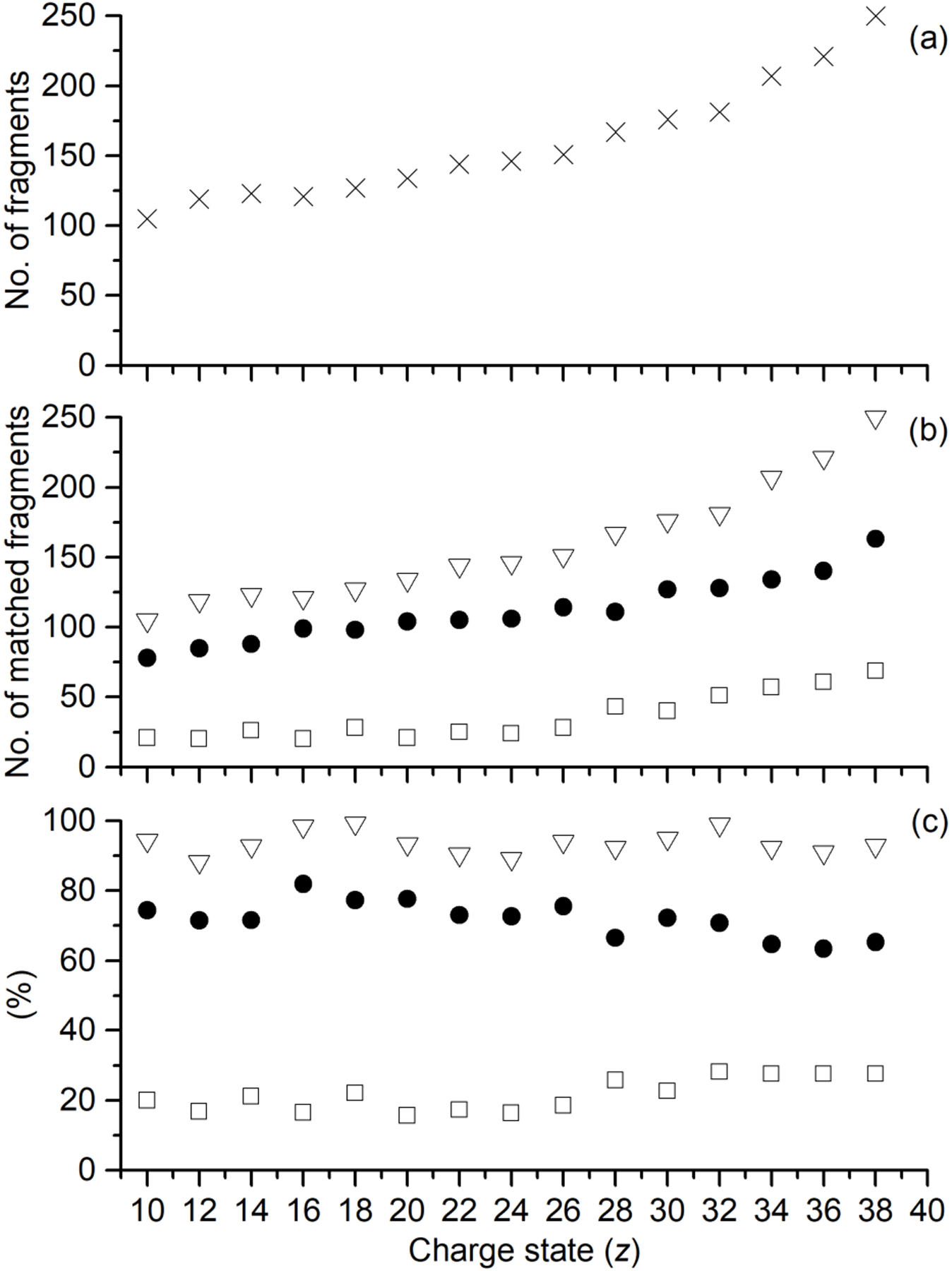
Performance of EID-MS for isolated [CAII, zH]z+ (z = 10+ to 38+), where (a) is the number of fragments deconvoluted, and (b) is the total number of fragments identified (open triangles), total number of terminal fragments identified (closed circles), and the total number of internal fragments identified (open squares). The percentage of the fragments matched is shown in (c), where the percentage of all fragments identified (open triangles), percentage of terminal fragments identified (closed circles), and the percentage of internal fragments identified (open squares) are depicted.
The theoretical total number of fragments, including internal fragments larger than 4 residues, scales with a second-order polynomial and with increasing number of amino acids (Figure S2). For example, the number of fragments that can be generated theoretically by fragmentation between each amino acid residue, based on cleavages of the Cα-carbonyl-C, N-Cα, and N-carbonyl-C bonds, ranges from 4 amino acid residues to one less than the entire protein length. For Ubq (76 amino acid residues), the total is 25,429 compared to 299,929 for CAII (259 amino acid residues), with the vast majority originating from internal fragments; for Ubq and CAII, the number of theoretical deisotoped internal fragment masses are 24,975 and 298,377, respectively. This is in close agreement with previous calculations by Kelleher and co-workers who demonstrated that the number of internal fragments that can theoretically be generated outnumber the number of terminal fragments generated.20 In addition, the formation of internal fragments should be especially beneficial for larger proteins because internal fragments are generally lighter than terminal fragments (Figure S2). For example, the average mass of a terminal fragment for CAII is 14.4 kDa, whereas the average mass of an internal fragment is 9.5 kDa (Figure S2b). This trend becomes significantly more important for larger proteins; the difference between the average mass of terminal fragments and internal fragments increases as protein size increases. By generating internal fragments that have smaller masses than terminal fragments, these internal fragments should fall within the mass range of most mass spectrometers, which should allow for greater confirmation of these amino acid sequence regions within the protein. Top-down MS analysis suffers from an upper mass limit;36, 37, 38 the inclusion of internal fragments could potentially help to overcome this limitation.
Internal fragment ion masses can be readily calculated and can be assigned to a target protein sequence if MS2 mass calibration is achieved to ≤ 1 ppm error to ensure precise assignments, and limit false positive assignments. As an example, to test the validity of these assignments, the theoretical fragment isotopic distribution was fitted over the corresponding fragment peak in the mass spectrum (Figure S3).39, 40 For both terminal and internal fragments, the theoretical isotopic distribution of the corresponding fragment are in good agreement with the peaks observed in the mass spectrum (Figure S3).
To estimate the false discovery rate of the internal fragment assignments, internal fragments masses of [CAII, 25H]25+ were shifted with a given ppm error from −500 to 500 to produce null data sets. These null data sets were then searched against the CAII sequence to estimate the amount of random matching to other internal fragments.20 The percentages of internal fragments that were assigned to a different internal fragment for the null data set are shown (Figure S4). When all possible internal fragments were considered (ax, ay, az, bx, by, bz, cx, cy, cz), ~20% of the internal fragment null set were assigned to a different internal fragment. However, considering that ExD fragmentation was utilized, internal fragments formed should be cz fragments that stem from multiple fragmentation events. When only cz internal fragments were considered for the null datasets, only 3.4% of the internal fragments were assigned to other cz internal fragments from the CAII sequence which indicates a low false discovery rate.
By including both internal fragments and terminal fragments, a larger fraction of the protein sequence can be explained (Figure 4). Heatmaps of the number of times a residue is represented by a product ion are plotted to give an indication of hotspots within the polypeptide sequence corresponding to where fragments are formed and detected. For ECD of CAII (Figure 4a), terminal fragments only account for a small fraction of the protein sequence (28%). Interestingly, the inclusion of internal fragments yields nearly complete sequence confirmation of carbonic anhydrase II (~90%). For EID, a larger percentage of the protein sequence can be explained by terminal fragments (47%), and similarly, the inclusion of internal fragments resulted in near complete protein sequence confirmation (~99%). The inclusion of internal fragments was also beneficial for the percentage of the inter-residue cleavages that were observed. For ECD, inclusion of the internal fragments showed that 200 of the 259 inter-residue cleavage sites were fragmented, and for EID, 234 of the 259 inter-residue cleavage sites were fragmented, indicating that a majority of the protein backbone was cleaved to form fragment ions (Figure S5). For Ubq, similar trends are observed with many internal fragments assigned to fragments that contain the center of the protein backbone (Figure S6). Further, for Ubq the majority of inter-residue cleavage sites were cleaved and assigned to protein fragments (Figure S7).
Figure 4.
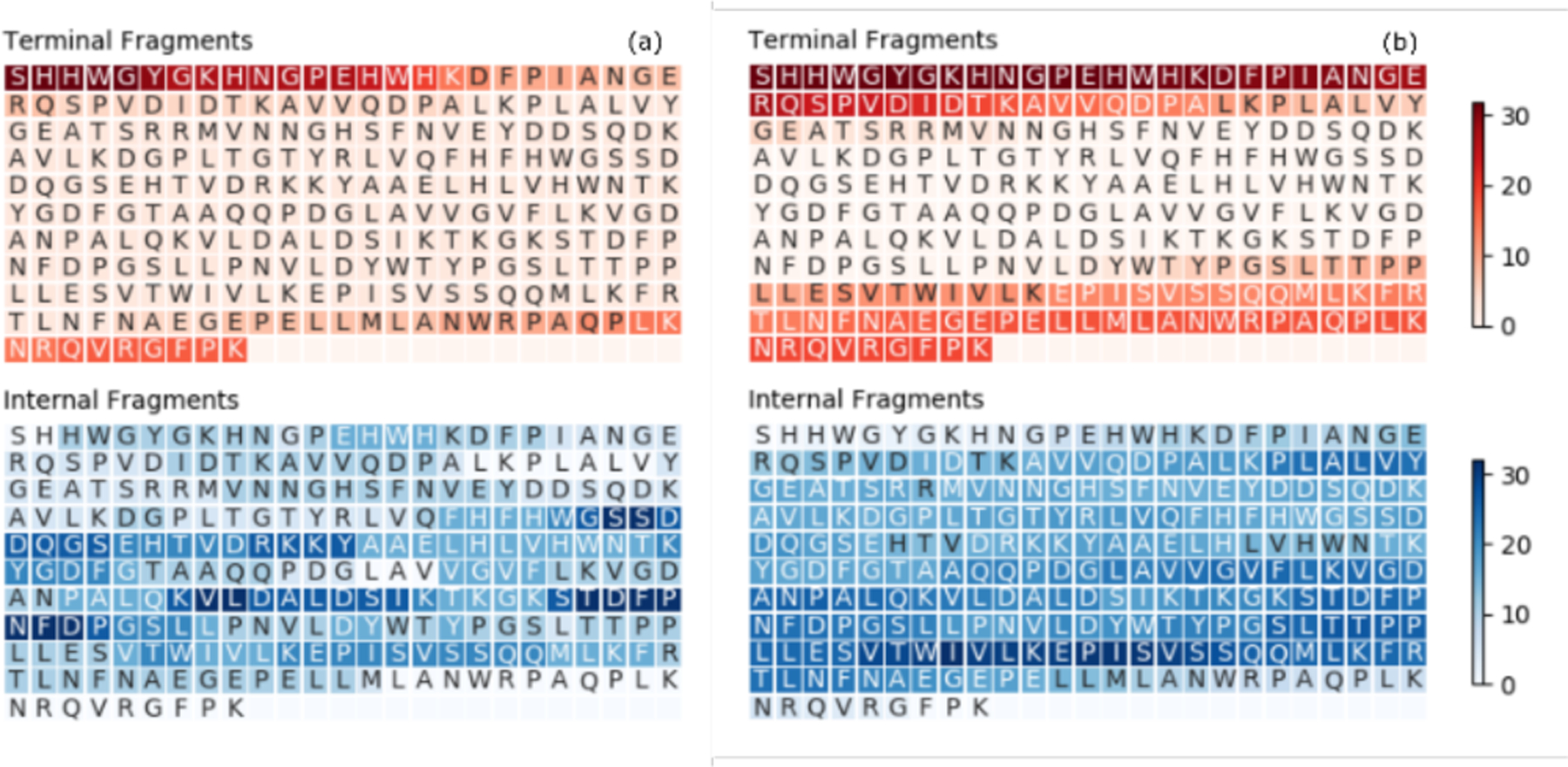
Heatmap depicting the number of times each residue is covered by a terminal fragment (top) or an internal fragment (bottom) for (a) ECD of [CAII, 25H]25+, and (b) EID of [CAII, 25H]25+. Darker colors indicate greater coverage.
CONCLUSIONS
EID for top-down MS can significantly enhance the efficiency of protein fragmentation. From this study, EID outperforms ECD with larger numbers of fragments generated, as well as higher fragment ion abundances. Most significantly, the use of internal fragment assignments resulted in the confirmation of a larger fraction of a given protein sequence. Because of some ambiguity in assigning internal fragments due to the large number of theoretical internal products, terminal fragments are ideal for protein identification and internal fragments are useful for sequence confirmation. Future work will define the types of internal fragments generated by ExD and other activation/dissociation methods and the limits of assigning internal fragments for larger proteins beyond 30 kDa. As the internal fragments formed contain amino acid sequences that are complementary to the terminal fragments, these internal fragments should be useful for localizing post-translational modifications41, 42 and protein-ligand binding sites43, 44, and for characterizing large protein complexes45 and membrane proteins46, 47 with native top-down MS. In general, incorporating the previously unassigned internal product ions generated by all activation/dissociation techniques, especially with EID, should greatly enhance the utility of top-down MS for protein sequence analysis to larger proteins.
Supplementary Material
ACKNOWLEDGMENTS
Support from the US National Institutes of Health (R01GM103479, S10RR028893), the US National Science Foundation (NSF) (CHE 1808492), and the US Department of Energy (DE-FC02–02ER63421) are gratefully acknowledged. C. L. acknowledges support from the Ruth L. Kirschstein National Research Service Award program (GM007185). We thank Drs. Ryan Julian (UC Riverside), Yury Tsybin (Spectroswiss), Mr. Benqian Wei (UCLA) and Mr. Zachary Hemminger (UCLA) for helpful discussions.
Footnotes
Supporting Information
The Supporting Information is included as a pdf file titled:
‘Supporting Information for Internal fragments generated by electron ionization dissociation enhance protein top-down mass spectrometry’
REFERENCES
- (1).Kelleher NL; Lin HY; Valaskovic GA; Aaserud DJ; Fridriksson EK; McLafferty FW, Top Down versus Bottom Up Protein Characterization by Tandem High-Resolution Mass Spectrometry. J. Am. Chem. Soc 1999, 121, 806–812. [Google Scholar]
- (2).Lermyte F; Tsybin YO; O’Connor PB; Loo JA, Top or Middle? Up or Down? Toward a Standard Lexicon for Protein Top-Down and Allied Mass Spectrometry Approaches. J. Am. Soc. Mass Spectrom 2019, 30, 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zubarev RA; Horn DM; Fridriksson EK; Kelleher NL; Kruger NA; Lewis MA; Carpenter BK; McLafferty FW, Electron Capture Dissociation for Structural Characterization of Multiply Charged Protein Cations. Anal Chem 2000, 72, 563–573. [DOI] [PubMed] [Google Scholar]
- (4).Zubarev RA; Kelleher NL; McLafferty FW, Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc 1998, 120, 3265–3266. [Google Scholar]
- (5).Good DM; Wirtala M; McAlister GC; Coon JJ, Performance Characteristics of Electron Transfer Dissociation Mass Spectrometry. Mol. Cell. Proteomics 2007, 6, 1942–1951. [DOI] [PubMed] [Google Scholar]
- (6).Katta V; Chowdhury SK; Chait BT, Use of a single-quadrupole mass spectrometer for collision-induced dissociation studies of multiply charged peptide ions produced by electrospray ionization. Anal Chem 1991, 63, 174–178. [DOI] [PubMed] [Google Scholar]
- (7).McCormack AL; Jones JL; Wysocki VH, Surface-Induced Dissociation of Multiply Protonated Peptides. J. Am. Soc. Mass Spectrom 1992, 3, 859–862. [DOI] [PubMed] [Google Scholar]
- (8).Brodbelt JS, Photodissociation mass spectrometry: new tools for characterization of biological molecules. Chem. Soc. Rev 2014, 43, 2757–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Brodbelt JS, Ion Activation Methods for Peptides and Proteins. Anal Chem 2016, 88, 30–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhurov KO; Fornelli L; Wodrich MD; Laskay UA; Tsybin YO, Principles of electron capture and transfer dissociation mass spectrometry applied to peptide and protein structure analysis. Chem. Soc. Rev 2013, 42, 5014–5030. [DOI] [PubMed] [Google Scholar]
- (11).Syrstad EA; Turecček F, Toward a general mechanism of electron capture dissociation. J. Am. Soc. Mass Spectrom 2005, 16, 208–224. [DOI] [PubMed] [Google Scholar]
- (12).Zubarev RA; Haselmann KF; Budnik B; Kjeldsen F; Jensen F, Towards an understanding of the mechanism of electron-capture dissociation: a historical perspective and modern ideas. Eur. J. Mass Spectrom 2002, 8, 337–350. [Google Scholar]
- (13).Lermyte F; Valkenborg D; Loo JA; Sobott F, Radical solutions: Principles and application of electron-based dissociation in mass spectrometry-based analysis of protein structure. Mass Spectrom. Rev 2018, 37, 750–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Iavarone AT; Paech K; Williams ER, Effects of Charge State and Cationizing Agent on the Electron Capture Dissociation of a Peptide. Anal Chem 2004, 76, 2231–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zubarev RA, Electron-capture dissociation tandem mass spectrometry. Curr. Opin. Biotechnol 2004, 15, 12–16. [DOI] [PubMed] [Google Scholar]
- (16).Kaczorowska MA; Cooper HJ, Electron induced dissociation (EID) tandem mass spectrometry of octaethylporphyrin and its iron(iii) complex. Chem. Commun 2011, 47, 418–420. [DOI] [PubMed] [Google Scholar]
- (17).Li H; Sheng Y; McGee W; Cammarata M; Holden D; Loo JA, Structural Characterization of Native Proteins and Protein Complexes by Electron Ionization Dissociation-Mass Spectrometry. Anal Chem 2017, 89, 2731–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zubarev RA; Yang H, Multiple Soft Ionization of Gas-Phase Proteins and Swift Backbone Dissociation in Collisions with ≤99 eV Electrons. Angew. Chem. Int. Ed 2010, 49, 1439–1441. [DOI] [PubMed] [Google Scholar]
- (19).Zubarev R, Protein primary structure using orthogonal fragmentation techniques in Fourier transform mass spectrometry. Exp. Rev. Proteomics 2006, 3, 251–261. [DOI] [PubMed] [Google Scholar]
- (20).Durbin KR; Skinner OS; Fellers RT; Kelleher NL, Analyzing internal fragmentation of electrosprayed ubiquitin ions during beam-type collisional dissociation. J. Am. Soc. Mass Spectrom 2015, 26, 782–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Holden DD; Brodbelt JS, Improving Performance Metrics of Ultraviolet Photodissociation Mass Spectrometry by Selective Precursor Ejection. Anal Chem 2017, 89, 837–846. [DOI] [PubMed] [Google Scholar]
- (22).Michalski A; Neuhauser N; Cox J; Mann M, A Systematic Investigation into the Nature of Tryptic HCD Spectra. J. Proteome Res 2012, 11, 5479–5491. [DOI] [PubMed] [Google Scholar]
- (23).Zinnel NF; Pai PJ; Russell DH, Ion mobility-mass spectrometry (IM-MS) for top-down proteomics: increased dynamic range affords increased sequence coverage. Anal Chem 2012, 84, 3390–3397. [DOI] [PubMed] [Google Scholar]
- (24).Xu C; Ma B, Complexity and scoring function of MS/MS peptide de novo sequencing. Computational systems bioinformatics. Computational Systems Bioinformatics Conference 2006, 361–369. [PubMed] [Google Scholar]
- (25).Ballard KD; Gaskell SJ, Sequential mass spectrometry applied to the study of the formation of “internal” fragment ions of protonated peptides. Int. J. Mass Spectrom. Ion Proc 1991, 111, 173–189. [Google Scholar]
- (26).Barran PE; Polfer NC; Campopiano DJ; Clarke DJ; Langridge-Smith PR; Langley RJ; Govan JR; Maxwell A; Dorin JR; Millar RP, Is it biologically relevant to measure the structures of small peptides in the gas-phase? Int. J. Mass Spectrom 2005, 240, 273–284. [Google Scholar]
- (27).Chen J; Shiyanov P; Green KB, Top-down mass spectrometry of intact phosphorylated β-casein: Correlation between the precursor charge state and internal fragments. J. Mass Spectrom 2019, 54, 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cobb JS; Easterling ML; Agar JN, Structural characterization of intact proteins is enhanced by prevalent fragmentation pathways rarely observed for peptides. J. Am. Soc. Mass Spectrom 2010, 21, 949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Li H; Nguyen HH; Ogorzalek Loo RR; Campuzano IDG; Loo JA, An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nature Chem. 2018, 10, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fung YME; Adams CM; Zubarev RA, Electron Ionization Dissociation of Singly and Multiply Charged Peptides. J. Am. Chem. Soc 2009, 131, 9977–9985. [DOI] [PubMed] [Google Scholar]
- (31).Lyon YA; Riggs D; Fornelli L; Compton PD; Julian RR, The Ups and Downs of Repeated Cleavage and Internal Fragment Production in Top-Down Proteomics. J. Am. Soc. Mass Spectrom 2018, 29, 150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zamdborg L; LeDuc RD; Glowacz KJ; Kim Y-B; Viswanathan V; Spaulding IT; Early BP; Bluhm EJ; Babai S; Kelleher NL, ProSight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucl. Acids Res 2007, 35, W701–W706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Iavarone AT; Jurchen JC; Williams ER, Effects of Solvent on the Maximum Charge State and Charge State Distribution of Protein Ions Produced by Electrospray Ionization. J. Am. Soc. Mass Spectrom 2000, 11, 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zenaidee MA; Donald WA, Electron capture dissociation of extremely supercharged protein ions formed by electrospray ionisation. Anal. Methods 2015, 7, 7132–7139. [Google Scholar]
- (35).Zenaidee MA; Donald WA, Extremely supercharged proteins in mass spectrometry: profiling the pH of electrospray generated droplets, narrowing charge state distributions, and increasing ion fragmentation. Analyst 2015, 140, 1894–1905. [DOI] [PubMed] [Google Scholar]
- (36).Catherman AD; Skinner OS; Kelleher NL, Top Down proteomics: facts and perspectives. Biochem Biophys Res Commun 2014, 445, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Reid GE; McLuckey SA, ‘Top down’ protein characterization via tandem mass spectrometry. J. Mass Spectrom 2002, 37, 663–675. [DOI] [PubMed] [Google Scholar]
- (38).Riley NMW, Michael S; Coon, Joshua J., Sequencing Larger Intact Proteins (30–70 kDa) with Activated Ion Electron Transfer Dissociation. J. Am Soc. Mass Spectrom 2018, 29, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kaur P; O’Connor PB, Algorithms for Automatic Interpretation of High Resolution Mass Spectra. J. Am. Soc. Mass Spectrom 2006, 17, 459–468. [DOI] [PubMed] [Google Scholar]
- (40).Beu SC; Senko MW; Quinn JP; Wampler FM; McLafferty FW, Fourier-transform electrospray instrumentation for tandem high-resolution mass spectrometry of large molecules. J. Am. Soc. Mass Spectrom 1993, 4, 557–565. [DOI] [PubMed] [Google Scholar]
- (41).Lin Z; Guo F; Gregorich ZR; Sun R; Zhang H; Hu Y; Shanmuganayagam D; Ge Y, Comprehensive Characterization of Swine Cardiac Troponin T Proteoforms by Top-Down Mass Spectrometry. J. Am Soc. Mass Spectrom 2018, 29, 1284–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wu Z; Jin Y; Chen B; Gugger MK; Wilkinson-Johnson CL; Tiambeng TN; Jin S; Ge Y, Comprehensive Characterization of the Recombinant Catalytic Subunit of cAMP-Dependent Protein Kinase by Top-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom 2019, 30, 2561–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Nshanian M; Lantz C; Wongkongkathep P; Schrader T; Klärner F-G; Blümke A; Despres C; Ehrmann M; Smet-Nocca C; Bitan G; Loo JA, Native Top-Down Mass Spectrometry and Ion Mobility Spectrometry of the Interaction of Tau Protein with a Molecular Tweezer Assembly Modulator. J. Am Soc. Mass Spectrom 2019, 30, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wongkongkathep P; Han JY; Choi TS; Yin S; Kim HI; Loo JA, Native Top-Down Mass Spectrometry and Ion Mobility MS for Characterizing the Cobalt and Manganese Metal Binding of α-Synuclein Protein. J. Am Soc. Mass Spectrom 2018, 29, 1870–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhou M; Yan J; Romano CA; Tebo BM; Wysocki VH; Pasa-Tolic L, Surface Induced Dissociation Coupled with High Resolution Mass Spectrometry Unveils Heterogeneity of a 211 kDa Multicopper Oxidase Protein Complex. J. Am Soc. Mass Spectrom 2018, 29, 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lippens JL; Nshanian M; Spahr C; Egea PF; Loo JA; Campuzano IDG, Fourier Transform-Ion Cyclotron Resonance Mass Spectrometry as a Platform for Characterizing Multimeric Membrane Protein Complexes. J. Am Soc. Mass Spectrom 2018, 29, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Susa AC; Lippens JL; Xia Z; Loo JA; Campuzano IDG; Williams ER, Submicrometer Emitter ESI Tips for Native Mass Spectrometry of Membrane Proteins in Ionic and Nonionic Detergents. J. Am Soc. Mass Spectrom 2018, 29, 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.