

Abstract
Although a previously developed bump-hole approach has proven powerful in generating specific inhibitors for mapping functions of protein kinases, its application is limited by the intolerance of the large-to-small mutation by certain kinases and the inability to control two kinases separately in the same cells. Herein, we describe the development of an alternative chemical-genetic approach to overcome these limitations. Our approach features the use of an engineered cysteine residue at a particular position as a reactive feature to sensitize a kinase of interest to selective covalent blockade by electrophilic inhibitors and is thus termed the Ele-Cys approach. We successfully applied the Ele-Cys approach to identify selective covalent inhibitors of a receptor tyrosine kinase EphB1 and solved cocrystal structures to determine the mode of covalent binding. Importantly, the Ele-Cys and bump-hole approaches afforded orthogonal inhibition of two distinct kinases in the cell, opening the door to their combined use in the study of multikinase signaling pathways.
Graphical Abstract
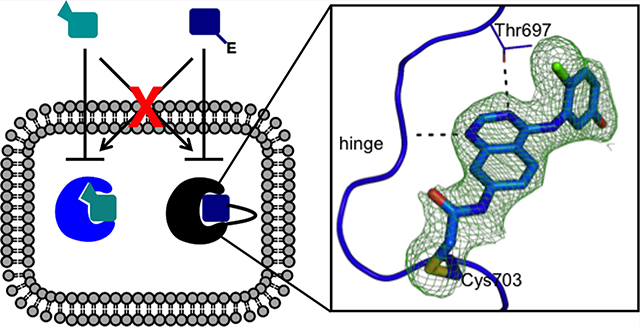
Protein kinases are essential regulators of protein functions and cell signaling by installing phosphate groups on serine, threonine, or tyrosine residues in substrate proteins.1,2 Because of the high conservation in the kinase superfamily, it is rather difficult to identify small molecules that can differentiate between closely related kinases and selectively inhibit individual ones (Figure 1A).3,4 A chemical-genetic strategy has previously been established to overcome this challenge. Shokat and colleagues demonstrated that a bulky “gatekeeper” residue in close contact with ATP within the kinase domain could be mutated to smaller residues (alanine or glycine) to create a “hole” where a “bumped” inhibitor can bind specifically (Figure 1B).5,6 This chemical-genetic approach is often referred to as the “bump-hole” approach with the engineered kinase termed analog-sensitive (AS) allele.5 The bump-hole approach has enabled selective chemical inhibition of numerous kinases from diverse organisms, allowing for determination of kinase functions and elucidation of a novel signaling mechanism.6,7 Despite the generality of the bump-hole approach, it does not work for all kinases. A portion of kinases either cannot tolerate the gatekeeper mutation or cannot be effectively inhibited by available bumped inhibitors.8,9 This has precluded application of the bump-hole approach to kinases of important functions such as MEK and RAF (C.Z. unpublished results). Moreover, different analog-sensitive kinases tend to be inhibited by the same bumped inhibitor because they harbor similar expanded active sites.9 For example, the bump-hole approach allowed for simultaneous inhibition of three Eph kinases but was unable to achieve separate inhibition of individual Eph kinases in the same cells.10 An approach that is complementary and ideally orthogonal to the bump-hole one is needed to facilitate a chemical-genetic analysis of additional kinases and to afford selective, separate inhibition of two kinases in the same cell, which is much desired for studying signaling pathways involving multiple kinases.
Figure 1.

Chemical-genetic strategies to distinguish among highly homologous protein kinases. (A) Conventional methods have difficulty generating specific inhibitors for individual members in the highly homologous protein kinase family. (B) The “bump-hole” approach accomplishes specificity based on steric complementarity between an engineered kinase and an inhibitor analog. (C) The novel “Ele-Cys” approach relies on a covalent interaction between the nucleophilic thiol group in a kinase and an electrophile in a small-molecule inhibitor to achieve specificity. (D) A partial sequence alignment of nine protein kinases within the subdomain V with the gatekeeper position and the gatekeeper+6 position highlighted. (E) Chemical structure of compound 1 and 3MB-PP1.
We sought to bridge the above gap by developing a chemical-genetic strategy that relies on a covalent reaction between an engineered cysteine (Cys) residue in the target protein and an electrophile in the inhibitor to achieve potent and specific binding. The irreversible nature of the reaction between reactive cysteine and electrophiles drives potency and target specificity of the binding.11,12 Because our approach features a covalent reaction between an electrophile in the small molecule and an engineered Cys in the target protein, we termed it the “Ele-Cys” approach (Figure 1C). A prior study showed that mutation of the gatekeeper residue to cysteine could sensitize kinases to electrophilic pyrazolopyrimidines, but the inhibitor potency was moderate, and the generality was not fully established.13 Other studies succeeded in conferring sensitivity to electrophilic inhibitors by introducing a cysteine residue near the end of the hinge region, but the inhibitors’ selectivity is limited by the fact that a significant number of human kinases including EGFR, HER2, and Btk contain a natural cysteine at this position.14,15 Here, by selecting a different position for introducing the cysteine mutation, we demonstrate that the Ele-Cys approach can yield kinase inhibitors of comparable potency and specificity to that of the bump-hole approach. Moreover, our study suggests that these two approaches can be combined to afford orthogonal inhibition of two kinases in the same cell.
To ensure maximal specificity of the Ele-Cys approach, we initiated a search for a position within the kinase active site where cysteine occurs extremely rarely among natural human kinases. We previously identified a position six residues after the gatekeeper position, where only a single human kinase EphB3 contains a natural cysteine residue (Figure 1D).16 Introduction of a cysteine residue at this position in a kinase of interest should allow for potent and specific inhibition by electrophilic inhibitors (Figure 1C). Location of the cysteine within a surface-exposed loop is expected to provide sufficient conformational flexibility for the cysteine thiol group to react with electrophiles in the inhibitors. Moreover, EphB3 expression was reported to be restricted to few tissues including select regions of the brain, which is echoed by our own observations that little EphB3 could be detected in a panel of cell lines of diverse tissue origins (Supporting Information Figure S1).17 This suggests that the off-target inhibition of EphB3 by electrophilic inhibitors will be minimal in most cell types.
We chose to apply the Ele-Cys approach to EphB1 for a proof of concept. To compare the performance of our Ele-Cys approach to that of the bump-hole approach, we created both the Cys mutant (G703C) and the analog-sensitive allele (T697G) of EphB1. Along with the wild-type protein, these mutants were expressed and purified from E. coli and evaluated for their enzymatic properties.18 The two mutants have kcat values 5.7- and 7.4-fold lower than that of the wild type (Table 1), indicating that the two mutations compromised the rate-limiting step of the EphB1-catalyzed phosphorylation reaction to similar extents. In contrast, the Km for ATP was not significantly affected by the G703C mutation but increased by ~3 fold by the T697G mutation. These results indicate that the G703C mutation caused less overall perturbation to the catalytic activity of EphB1 than the gatekeeper mutation T697G.
Table 1.
The Kinetic Parameters and Inhibitor IC50 Values for Three EphB1 Allelesaa
| EphB1 | ||||
|---|---|---|---|---|
| WT | G703C | T697G | ||
| κcat (min−1) | 20 ± 3 | 2.7 ± 0.6 | 3.5 ± 0.9 | |
| Km,ATP (μM) | 460 ± 57 | 490 ± 100 | 1770 ± 645 | |
| κcat/Km (min−1 μM−1) | 0.0444 ± 0.0088 | 0.0055 ± 0.0024 | 0.0020 ± 0.0005 | |
| in vitro | 1 (nM) | 220 ± 18 | 3.0 ± 1.2 | 15 ± 2 |
| 3MB (nM) | 580 ± 300 | 980 ± 190 | 5.0 ± 1.0 | |
| in cellulo | 1 (nM) | 2920 ± 860 | 27 ± 2 | 440 ± 220 |
| 3MB (nM) | 6100 ± 270 | 2600 ± 130 | 5.0 ± 2.0 | |
The top portion shows the steady-state kinetic parameters of the three EphB1 variants measured using radioactive disk kinase assay. The middle portion displays the IC50 values of 1 and 3MB-PP1 (3MB) against EphB1 in vitro. The bottom portion shows IC50 values of the same two compounds at inhibiting EphB1 autophosphorylation in cells. Errors shown are calculated as the standard deviation from three independent experiments.
We next measured the inhibition of the EphB1 alleles by electrophilic or bumped inhibitors. By screening a panel of electrophilic quinazolines previously developed for EphB3, we found that quinazoline 1 (Figure 1E) caused the most potent inhibition of EphB1G703C (Supporting Information Figure S2) with an IC50 value of 3 nM (Table 1).16 In comparison, 1 inhibited EphB1WT with an IC50 value of 220 nM, indicating that the Cys mutation enhanced this electrophilic inhibitor’s potency by nearly 100-fold. Unexpectedly, EphB1T697G was potently inhibited by 1. This might be due to a better fit of the 2-chloro-5-hydroxyaniline group in the expanded pocket in the analog-sensitive allele. A bumped inhibitor 3MB-PP1 (Figure 1E) selectively inhibited the analog-sensitive mutant out of the three EphB1 alleles.9 These data suggest that the Ele-Cys approach can generate inhibitors of similar potency and selectivity for the engineered kinase to that of the bump-hole approach. However, the substantial cross-inhibition of EphB1T697G by the electrophilic inhibitor 1 means that the two inhibitors are not sufficiently orthogonal to each other at inhibiting the two alleles of EphB1 in vitro.
To evaluate the specificity of the electrophilic inhibitor for the engineered EphB1, we profiled compound 1’s inhibition against a panel of 249 kinases. The profiling data revealed seven significant off-targets of the inhibitor (Supporting Information Table S1). Among the seven, only EphB3 was potently inhibited while the other six were only moderately inhibited by compound 1, providing useful information for guiding and evaluating future application of the Ele-Cys approach. We next examined the generality of the Ele-Cys approach. The homologous cysteine mutation was shown to confer another tyrosine kinase FGFR4 with high sensitivity to electrophilic inhibitors, although the effect on Abl is moderate (Supporting Information Table S2). Furthermore, a serine/threonine kinase RAF was found to be amenable to the Ele-Cys approach (manuscript in preparation). These results suggest that the Ele-Cys approach can be applied to divergent kinases beyond the Eph family.
To demonstrate that the electrophilic quinazoline compound 1 is capable of reacting covalently with the engineered Cys in the hinge region of the kinase, we determined the X-ray structure of wild-type and G703C EphB1 in complex with 1 (Supporting Information Table S3). The two alleles of the EphB1 kinase domain were overexpressed in E. coli with a noncleavable N-terminal hexahistidine tag and purified to homogeneity. Co-crystallization of the G703C mutant with 1 yielded a structure of the enzyme–inhibitor complex. At a resolution of 2.14 Å, the engineered Cys and the covalent adduct are defined unequivocally by electron density (Figure 2A). Solution of the EphB1WT–1 complex structure required soaking apoprotein crystals with the inhibitor to obtain full occupancy of the ligand, as the affinity for the wild-type enzyme is only moderate. The reactive warhead extends into the solvent and is poorly ordered in the complex with the wild-type enzyme (Figure 2B). The comparison of the two structures shows that the quinazoline core of the inhibitors overlays perfectly; likewise, there is no detectable difference in the conformation of the loop harboring the engineered C703 (Figure 2C). These results confirm that the nature and length of the chloroacetamide electrophile allows a sufficient degree of flexibility for reaction with the cysteine thiol group at the “gatekeeper+6” position of EphB1 without perturbing the kinase structure.
Figure 2.

Co-crystal structures verify the binding modes of the electrophilic quinazoline 1 to EphB1 variants. (A) Crystal structure of the EphB1G703C–1 complex. |2Fo–Fc| electron density map (contoured at 1 σ) shown as a green mesh supports the formation of a covalent bond between the inhibitor and the engineered cysteine (Cys703). (B) Crystal structure of the EphB1WT–1 complex. (C) Overlay of the two structures. The protein is shown as a cartoon representation (EphB1G703C in blue and EphB1WT in orange), while the ligand and C703 are shown as sticks.
Having characterized the catalytic activity and inhibitor sensitivity of the EphB1 alleles in vitro, we set out to determine the inhibition of the EphB1 alleles in cells. The full-length EphB1 variants were transfected into HEK293 cells, treated with the inhibitors at different concentrations, and measured for levels of autophosphorylation and total EphB1 using Western blotting. Both compound 1 and 3MB-PP1 inhibited EphB1WT autophosphorylation with micromolar IC50 values (Supporting Information Figure S3 and Table 1). In contrast, 1 and 3MB-PP1 inhibited the Cys mutant and the analog-sensitive allele of EphB1, respectively, with nanomolar IC50 values. 3MB-PP1 can differentiate between the two EphB1 mutants with a high selectivity factor (~500). Compound 1 exhibited a moderate selectivity (~16) for the Cys mutant over the gatekeeper mutant (Table 1). These results verify that the Ele-Cys and bump-hole approaches are both successful in sensitizing EphB1 to inhibition by appropriate inhibitors in cells. We next examined the cross-inhibition between the two chemical-genetic systems. When tested for inhibition of the three EphB1 alleles in cells, 3MB-PP1 at 100 nM only inhibited the analog-sensitive allele, while 1 at 300 nM selectively blocked the Cys mutant (Figure 3A). These results suggest that the cross-inhibition between the two systems is much lower in cells than in vitro, likely because of the enhanced efficacy of covalent inhibition in cells.19
Figure 3.

Orthogonal inhibition of two Eph kinases in cells afforded by the bump-hole and Ele-Cys approaches. (A) 1 at 300 nM only inhibited EphB1G703C, while 3MB-PP1 (3MB) at 100 nM only inhibited EphB1T697G when they were tested on three EphB1 alleles in cells. (B) 1 and 3MB afford orthogonal inhibition of EphB3WT and EphB1T697G in the same cell. The top band in both total Eph and phospho-Eph blots is BRIL-fused EphB3 and the bottom one is EphB1.
To test whether the two chemical-genetic approaches can afford orthogonal inhibition of two kinases in the same cell, we employed them to investigate the communications between two distinct Eph kinases upon activation.10 HEK293 cells were transfected to express both EphB3WT and EphB1T697G, which were predicted to be sensitive to inhibition by 1 and 3MB-PP1, respectively. EphB3WT was fused to a BRIL domain at the C-terminus to differentiate it from EphB1T697G in sizes in the Western blots. Upon stimulation by ephrin-B2, both Eph receptors exhibited enhanced autophosphorylation as expected (Figure 3B). The addition of 300 nM 1 abolished the autophosphorylation of EphB3WT with little effect on that of EphB1T697G. In contrast, 100 nM of 3MB-PP1 abolished the autophosphorylation of EphB1T697G but had little effect on that of EphB3WT. These results suggest that 1 and 3MB-PP1 cause orthogonal inhibition of two Eph kinases in the same cell. The selective abolishment of the autophosphorylation of just one Eph kinase by its cognate inhibitor indicates that there is little trans-phosphorylation occurring between EphB1 and EphB3. Combined with our previous finding that the autophosphorylation of EphB3 occurs in trans,16 the observation made using two orthogonal kinase inhibitors implies that EphB1 and EphB3 seem to prefer homodimerization with its own type to heterodimerization with each other. This is a striking result in view of the high homology between EphB1 and EphB3 (72% sequence identity).
In summary, we have succeeded in developing a reactivity-based chemical-genetic approach to generate selective covalent inhibitors of protein kinases. This approach appears to be fairly general as divergent kinases including EphB1, FGFR4, and RAF were shown to be amenable to it. This work is novel and significant for the following reasons. First, we chose to target a cysteine at a position of low natural occurrence in the human kinome, thus promising maximal inhibitor selectivity. Kinome profiling reveals that there are only a few moderate off-targets for compound 1 except for EphB3, whose expression is restricted to select human tissues. Second, we solved the crystal structures of one electrophilic inhibitor bound to the wild-type EphB1 and the cysteine mutant, allowing us to directly evaluate the impact of covalent interaction on inhibitor binding. None of the previous studies employing analogous strategies provided such a deep level of structural insight into the engineered covalent interactions.14,15 Finally, we demonstrate that our approach can afford orthogonal inhibition to the established bump-hole approach, opening the door to the combined usage of the two approaches in the functional studies of signaling pathways involving multiple protein kinases. We anticipate that the rapid advancement of genome-editing technologies such as CRISPR will dramatically accelerate the application of the Ele-Cys approach in functional studies of a large number of protein kinases.
METHODS
Recombinant Kinase Expression and Purification
EphB1 kinase was expressed and purified using a previously described procedure with minor modifications.18
EphB1 Kinase Assay
A phosphocellulose disk assay was used to determine the kinase activity of EphB1 by measuring the incorporation of γ−32P ATP into a peptide substrate, Btk-tide (NMPMYDY-LAVVKK).20 The kinase assays were conducted in 50 mM Tris (pH 8.0), 10 mM MgCl2, 1 mg mL−1 BSA, 1 nM EphB1 kinase, 100 μM Btk-tide, 5.5 nM [γ−32P]-ATP (PerkinElmer), and various concentrations of an inhibitor in a total volume of 30 μL for 30 min. Upon completion, 3 μL of the reaction was spotted onto a phosphocellulose paper disk and quenched with 10% acetic acid. This was followed by three washes with 0.5% phosphoric acid, one wash with acetone, and drying in the air. The radioactivity in paper disks was measured on a Beckman LC6500 scintillation counter. The data were analyzed using nonlinear regression (GraphPad Prism) to derive IC50 values.
Western Blotting
HEK293 cell lines transfected with EphB1, EphB3, and EphA4 variants were prepared using previously described procedures.16 Protein concentrations in cellular lysate were determined by a BCA assay (Pierce), normalized, and analyzed by Western blots for phospho-EphB (Ab61791, Abcam), EphB3 (Ab133742, Abcam), EphB1 (Ab129103, Abcam), β-actin (13E5, Cell-signaling), and EphA4 (D-4, Santa Cruz Biotech).
Protein Crystallography
EphB1 wild-type protein (11.0 mg mL−1) and G703C mutant (11.6 mg mL−1) in 50 mM MOPS at pH 7.5 and 50 mM NaCl was incubated with 1 mM compound 1 for 1 h at 4 °C, at a final DMSO concentration of 1%. Crystallization was carried out by sitting drop vapor diffusion against a reservoir of 18% PEG3350, 0.1–0.2 M ammonium sulfate, and 0.1 M PCTP (sodium propionate, sodium cacodylate trihydrate, Bis–Tris propane) buffer at pH 6–9. Crystals appeared within 1–5 days at 20 °C and were improved through microseeding. For wild-type EphB1, cocrystallization with 1 did not result in a complex. Therefore, crystals were soaked with 2 mM compound for 6 h. For data collection, crystals were briefly immersed in a cryoprotectant solution containing 15% glycerol in reservoir solution and flash-frozen in liquid nitrogen. X-ray diffraction experiments were carried out at Diamond Light Source beamlines I04 and I02. Data processing was performed with XDS.21 AIMLESS22 was used for scaling, and the structures were solved by molecular replacement with PHASER,23 using chain A of PDB entry 3ZFX (EphB1 apo structure) as the search model. Crystals belonged to space group P3121 and contained two molecules per asymmetric unit. Refinement was performed using BUSTER,24 with manual model building in Coot.25 Ligand topology information was generated with GRADE.26 Data collection and refinement statistics are shown in Table S3.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Greenberg (Harvard University) for providing the constructs of EphB1. This work was supported by National Science Foundation (CHE-1455306), American Cancer Society (IRG-58-007-51), and the University of Southern California.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.6b01083.
Details on experimental methods, additional figures, and analyses (PDF)
REFERENCES
- (1).Hunter T (1987) A thousand and one protein kinases. Cell 50, 823–829. [DOI] [PubMed] [Google Scholar]
- (2).Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S (2002) The Protein Kinase Complement of the Human Genome. Science 298, 1912. [DOI] [PubMed] [Google Scholar]
- (3).Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, and Zarrinkar PP (2008) A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol 26, 127–132. [DOI] [PubMed] [Google Scholar]
- (4).McLauchlan H, Elliott M, and Cohen P (2003) The specificities of protein kinase inhibitors: an update. Biochem. J. 371, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, and Shokat KM (2000) A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401. [DOI] [PubMed] [Google Scholar]
- (6).Bishop AC, Kung C. y., Shah K, Witucki L, Shokat KM, and Liu Y (1999) Generation of Monospecific Nanomolar Tyrosine Kinase Inhibitors via a Chemical Genetic Approach. J. Am. Chem. Soc. 121, 627–631. [Google Scholar]
- (7).Knight ZA, and Shokat KM (2007) Chemical Genetics: Where Genetics and Pharmacology Meet. Cell 128, 425–430. [DOI] [PubMed] [Google Scholar]
- (8).Zhang C, Kenski DM, Paulson JL, Bonshtien A, Sessa G, Cross JV, Templeton DJ, and Shokat KM (2005) A second-site suppressor strategy for chemical genetic analysis of diverse protein kinases. Nat. Methods 2, 435–441. [DOI] [PubMed] [Google Scholar]
- (9).Zhang C, Lopez MS, Dar AC, LaDow E, Finkbeiner S, Yun C-H, Eck MJ, and Shokat KM (2013) Structure-guided Inhibitor Design Expands the Scope of Analog-Sensitive Kinase Technology. ACS Chem. Biol 8, 1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Soskis MJ, Ho H-YH, Bloodgood BL, Robichaux MA, Malik AN, Ataman B, Rubin AA, Zieg J, Zhang C, Shokat KM, Sharma N, Cowan CW, and Greenberg ME (2012) A chemical genetic approach reveals distinct EphB signaling mechanisms during brain development. Nat. Neurosci. 15, 1645–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cohen MS, Zhang C, Shokat KM, and Taunton J (2005) Structural Bioinformatics-Based Design of Selective, Irreversible Kinase Inhibitors. Science 308, 1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhou W, Ercan D, Chen L, Yun C-H, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong K-K, Eck MJ, Gray NS, and Janne PA (2009) Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Garske AL, Peters U, Cortesi AT, Perez JL, and Shokat KM (2011) Chemical genetic strategy for targeting protein kinases based on covalent complementarity. Proc. Natl. Acad. Sci. U. S. A. 108, 15046–15052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Blair JA, Rauh D, Kung C, Yun C-H, Fan Q-W, Rode H, Zhang C, Eck MJ, Weiss WA, and Shokat KM (2007) Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat. Chem. Biol. 3, 229–238. [DOI] [PubMed] [Google Scholar]
- (15).Koch A, Rode HB, Richters A, Rauh D, and Hauf S (2012) A Chemical Genetic Approach for Covalent Inhibition of Analogue-Sensitive Aurora Kinase. ACS Chem. Biol. 7, 723–731. [DOI] [PubMed] [Google Scholar]
- (16).Kung A, Chen Y-C, Schimpl M, Ni F, Zhu J, Turner M, Molina H, Overman R, and Zhang C (2016) Development of Specific, Irreversible Inhibitors for a Receptor Tyrosine Kinase EphB3. J. Am. Chem. Soc. 138, 10554–10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pasquale EB (2008) Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 133, 38–52. [DOI] [PubMed] [Google Scholar]
- (18).Overman RC, Debreczeni JE, Truman CM, McAlister MS, and Attwood TK (2014) Completing the structural family portrait of the human EphB tyrosine kinase domains. Protein Sci. 23, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, and Moasser MM (2007) Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 445, 437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Witt JJ, and Roskoski R Jr (1975) Rapid protein kinase assay using phosphocellulose-paper absorption. Anal. Biochem. 66, 253–258. [DOI] [PubMed] [Google Scholar]
- (21).Kabsch W (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Evans PR, and Murshudov GN (2013) How good are my data and what is the resolution? Acta Crystallogr., Sect. D: Biol. Crystallogr. 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart O, Vonrhein C, and Womack T (2011) BUSTER version 2.11.6, Global Phasing Limited, Cambridge, United Kingdom. [Google Scholar]
- (25).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Smart OS, Womack TO, Sharff A, Flensburg C, Keller P, Paciorek W, Vonrhein C, and Bricogne G (2011) Grade, version 1.2.9, Global Phasing Ltd., Cambridge, United Kingdom. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.