

Abstract
Oxidative stress and inflammation play key roles in development of pulmonary arterial hypertension. We previously reported that an endothelial cell-specific cyclophilin A overexpression mouse developed many characteristics of pulmonary arterial hypertension. In other models of cardiovascular disease, cyclophilin A stimulates smooth muscle proliferation and vascular inflammation, but mechanisms responsible for pulmonary arterial hypertension have not been defined. In particular, the contribution of endothelial-to-mesenchymal transition in cyclophilin A-mediated pulmonary arterial hypertension has not been studied.
We identified increased levels of cyclophilin A in endothelial and neointimal cells of pulmonary arteries in pulmonary arterial hypertension patients and animal pulmonary hypertension models. In the endothelial cell-specific cyclophilin A overexpression mouse that exhibited features characteristic of pulmonary arterial hypertension, lineage tracing showed high level expression of mesenchymal markers in pulmonary endothelial cells. A significant number of mesenchymal cells in media and perivascular regions of pulmonary arterioles and alveoli were derived from endothelial cells. Pulmonary endothelial cells isolated from these mice showed phenotypic changes characteristic of endothelial-to-mesenchymal transition in culture. Cultured pulmonary endothelial cells stimulated with extracellular cyclophilin A and acetylated cyclophilin A demonstrated functional changes associated with endothelial-to-mesenchymal transition such as increased cytokine release, migration, proliferation, and mitochondrial dysfunction. Acetylated cyclophilin A stimulated greater increases for most features of endothelial-to-mesenchymal transition. In conclusion, extracellular cyclophilin A (especially acetylated form) contributes to pulmonary arterial hypertension by mechanisms involving increased endothelial-to-mesenchymal transition, cytokine release, endothelial cell migration, proliferation, and mitochondrial dysfunction; strengthening the basis for studying cyclophilin A inhibition as a therapy for pulmonary arterial hypertension.
Keywords: cyclophilin A, pulmonary arterial hypertension, endothelial cell, endothelial-to-mesenchymal transition, inflammation
Graphical Abstract
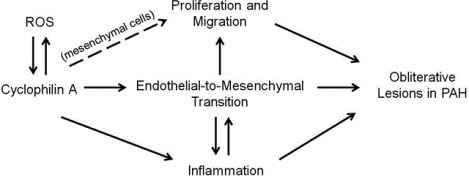
Introduction
Pulmonary arterial hypertension (PAH) is a progressive cardiovascular disease characterized by vascular remodeling including aberrant endothelial cell (EC) migration and proliferation, vascular smooth muscle cell (VSMC) proliferation and perivascular inflammation1–2. Cyclophilin A (CypA) is a ubiquitously expressed protein with peptidyl-prolyl cis-trans isomerase (PPIase) activity3. Our lab has identified CypA as a secreted oxidative stress induced factor that contributes to the generation of oxidative stress through autocrine and paracrine mechanisms4–5. CypA plays a key role in mediating vascular dysfunction and inflammation in several diseases including aortic aneurysm, atherosclerosis, intima formation and cardiac hypertrophy6–9. We have shown that acetylated CypA (AcK-CypA) is more potent than CypA for vascular cell activation10. Recently, we demonstrated that an EC-specific CypA overexpression mouse (ecCypA-Tg) developed many characteristics of PAH, including increased right ventricular systolic pressure, inflammatory cell infiltration and thickening of pulmonary arterioles11. We found that extracellular CypA promoted EC inflammation in vitro through activation of the NF-kB pathway, and identified a positive feedback loop between oxidative stress and CypA secretion that included stimulation of AcK-CypA production.
In PAH, it appears that muscularization and proliferation of cells to form an occlusive neointima represents an irreversible stage of PAH pathology12. There are several theories regarding the origin of the neointimal cells and the excessive VSMC. Sheikh et al. reported that the vast majority of distal arteriole VSMC in hypoxia-induced pulmonary hypertension (PH) derived from preexisting proximal VSMC13. They proposed that the VSMC dedifferentitate and migrate along the vessel and proliferate in the distal arterioles. Another theory implicates endothelial-to-mesenchymal transition (EndMT) as an important mechanism for muscularization and neointimal formation in pulmonary arterioles in both animal PH models and human PAH patients14–18. EndMT is a process in which EC lose key features such as tight junctions and highly regulated permeability while gaining features of mesenchymal cells, including increased contractile protein and secretion of extracellular matrix proteins. At the molecular level, these changes correlate with the increased expression of alpha smooth muscle actin (αSMA), smooth muscle protein 22 alpha (SM22α), vimentin and decreased expression of VE-Cadherin and platelet endothelial cell adhesion molecule 1 (PECAM-1)19. The process of EndMT involves interacting effects of extracellular matrix components, growth factors, inflammatory cytokines, and inflammatory cell adhesion molecules20. Two key pathologic features of EndMT are increased proliferation and migration due to a change in EC phenotype to a myofibroblast-like cell21–22.
In the present study we tested the hypothesis that extracellular CypA, especially AcK-CypA, contributes to PAH by mechanisms involving increased EndMT, cytokine release, EC migration, proliferation, and mitochondrial dysfunction.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. Further details can be found in the Supplemental Methods.
Generation of mice
To generate EC specific overexpression of CypA transgenic mice, the LacZflox-CypA transgenic mice and Cdh5-Cre mice (C57BL/6J background) were crossed. These EC specific CypA overexpression mice were then crossed with the B6.129(Cg)- Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J mice (The Jackson Laboratory, Stock No. 007676) to generate EC-specific reporter mice.
Cell isolation and culture
Mouse pulmonary microvascular endothelial cells (MPMECs) were isolated following the protocol from Dr. Simons Lab at Dartmouth Medical College and maintained in growth medium as we previously reported11. Human pulmonary microvascular endothelial cells (HPMECs) were purchased from ScienCell and maintained in the manufacturer’s recommended medium.
Results
Increased CypA expression in the endothelium of PAH patients and animal PH models.
To identify the cell-specific role of CypA in PAH, we stained for CypA using sections from human PAH patients and two different animal PH models: Sugen 5416/hypoxia mouse model and rat treated with monocrotaline after pneumonectomy (P-MCT). The information regarding human PAH patients are listed in Table S1. CypA expression was mildly enhanced in the medial layer, but greatly enhanced in the endothelium layer of pulmonary arterioles of PAH patients compared to controls (Fig.1A–B, indicated by thin black arrowheads). In addition, there was increased number of perivascular inflammatory cells with strong CypA expression in PAH patients compared to controls (Fig.1A–B, indicated by thick black arrows). CypA expression was also strongly enhanced in the endothelium and neointimal cells of obliterative pulmonary arterioles of different sizes in PAH patients (Figure.1B, indicated by thin black arrowheads). Similar to our observations in human PAH, there was increased CypA expression in the endothelium of thickened pulmonary arterioles of Sugen 5416/hypoxia mice and P-MCT rats compared to controls (Fig.1C–D, indicated by black arrowheads). Unlike human PAH, we did not see any increased CypA expression in the medium layer of pulmonary arterioles in Sugen 5416/hypoxia mice (Fig.1C). Interestingly, we observed a severity related increase of CypA expression in the medial layer of pulmonary arterioles in P-MCT rats. Specifically, compared to controls, there was strongly enhanced CypA expression in the medial layer of pulmonary arterioles that showed severe thickening; while no increase of CypA expression was observed in the medial layer of pulmonary arterioles with less severe thickening (Fig.1D). CypA expression was not only enhanced in the pulmonary arterioles, but also in the alveoli of PAH patients and animal PH models (Fig.S1A–C).
Figure 1. Increased CypA expression in the endothelium of PAH patients and animal PH models.
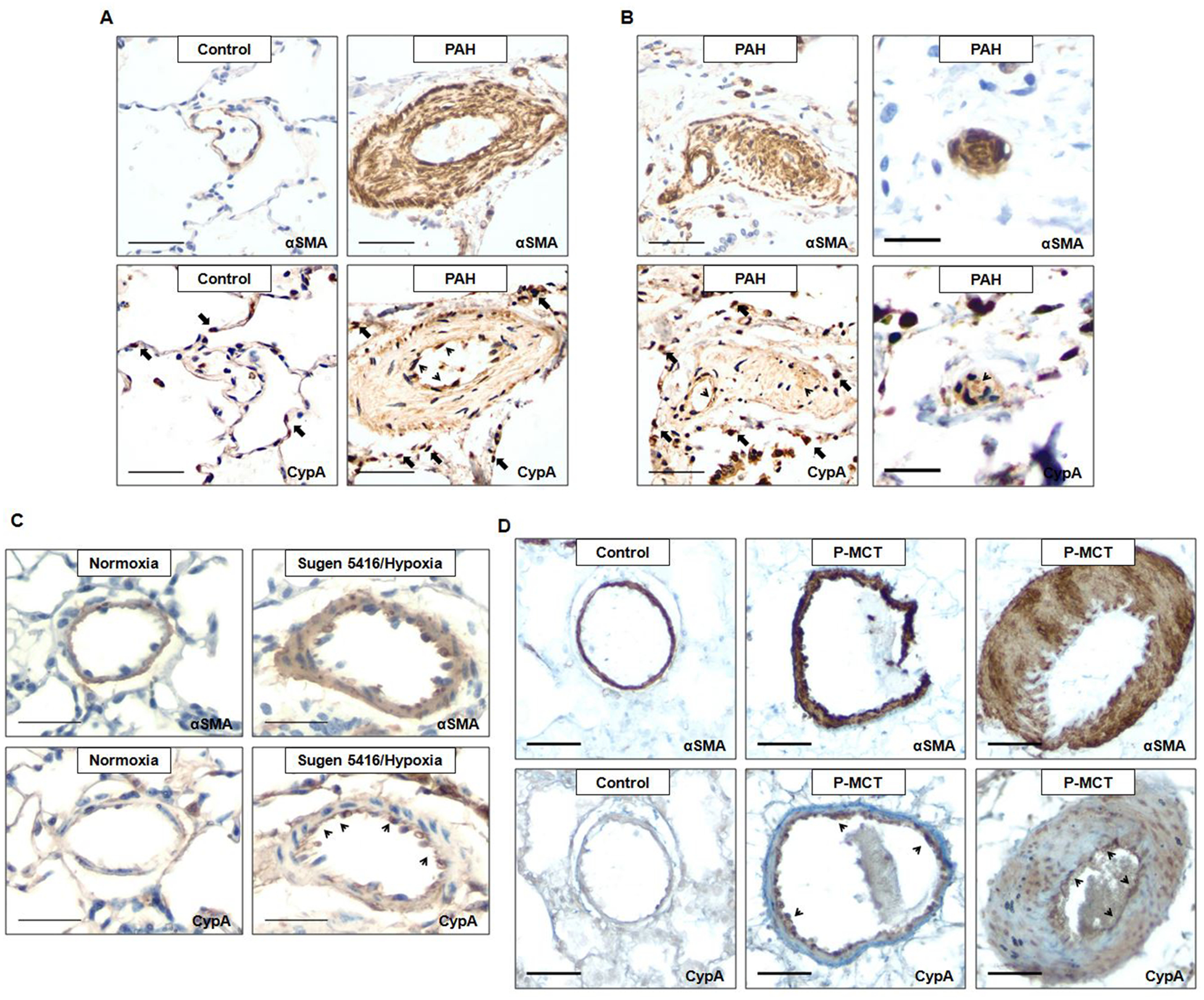
A, Representative lung IHC for αSMA and CypA in pulmonary arteries from controls (n=5) and PAH patients (n=3). Scale bar, 50μm. B, Representative lung IHC for αSMA and CypA in obliterative pulmonary arteries of different sizes from PAH patients. Scale bar, 50μm (left panel) and 20μm (right panel). C, Representative lung IHC for αSMA and CypA in pulmonary arteries from normoxia and Sugen 5416/hypoxia mice (n=4 each). Scale bar, 30μm. D, Representative lung IHC for αSMA and CypA in pulmonary arteries with different degrees of thickening from controls (n=3) and MCT plus pneumonectomy (P-MCT) rats (n=5). Scale bar, 50μm.
CypA overexpression increases EC-derived SM22α and vimentin positive cells in vivo, indicating EndMT.
To define the role of CypA in EndMT during the pathogenesis of PAH, we used dual fluorescence mTmG reporter mice to lineage trace the mesenchymal cells. Specifically, EC-specific CypA overexpression mice (ecCypA-Tg) were obtained by breeding FLAG-tagged CypA mice with Cdh5-Cre mice. These mice were then bred with ROSAmT/mG mice. In the resulting Cdh5-Cre/mTmG/CypA-Tg (ecCypA-Tg;mTmG) mice, cell membrane-localized GFP will be expressed in EC and all cells of endothelial origin, regardless of any further phenotypic changes. The features of PAH previously reported in ecCypA-Tg mice were still present. For example, Myosin-11 staining showed increased pulmonary arteriole thickening in ecCypA-Tg mice compared to control (Fig.S1F). In ecCypA-Tg;mTmG mice, immunofluorescence staining showed increased expression of the mesenchymal marker SM22α compared to compared to CypA negative or Cre negative littermate controls (referred to as WT;mTmG; Fig.2A). The greatest expression of SM22α was detected in cells of pulmonary arterioles (diameter <75 μm) at 6 months of age. These cells in the inner (endothelial) layer exhibited the greatest co-localization of GFP and SM22α (Fig.2A–B, indicated by white arrows); although larger vessels also had significant co-localization in the inner layer. We only observed GFP+;SM22α+ cells in the media and adventitia of pulmonary arteries with larger diameters (75–150μm) in ecCypA-Tg;mTmG mice (Fig.2B; Fig. S2A, indicated by white arrowheads), suggesting an EC origin of these vascular mesenchymal cells. In addition, there was a 1.9-fold increase in the total number of SM22α+ cells in the alveolar area of ecCypA-Tg;mTmG mice compared to controls (Fig.2C–E; Fig.S2B, indicated by white arrowheads). In control mice only 15% of SM22α+ cells expressed GFP, while in ecCypA-Tg;mTmG mice, 75% of SM22α+ cells expressed GFP (Fig.2C–F; Fig.S2B, indicated by orange arrowheads). These data demonstrate that the majority of SM22α+ cells in the alveolar area of the ecCypA-Tg;mTmG mice were derived from EC.
Figure 2. CypA overexpression increases EC-derived SM22α positive cells in vivo, indicating EndMT.
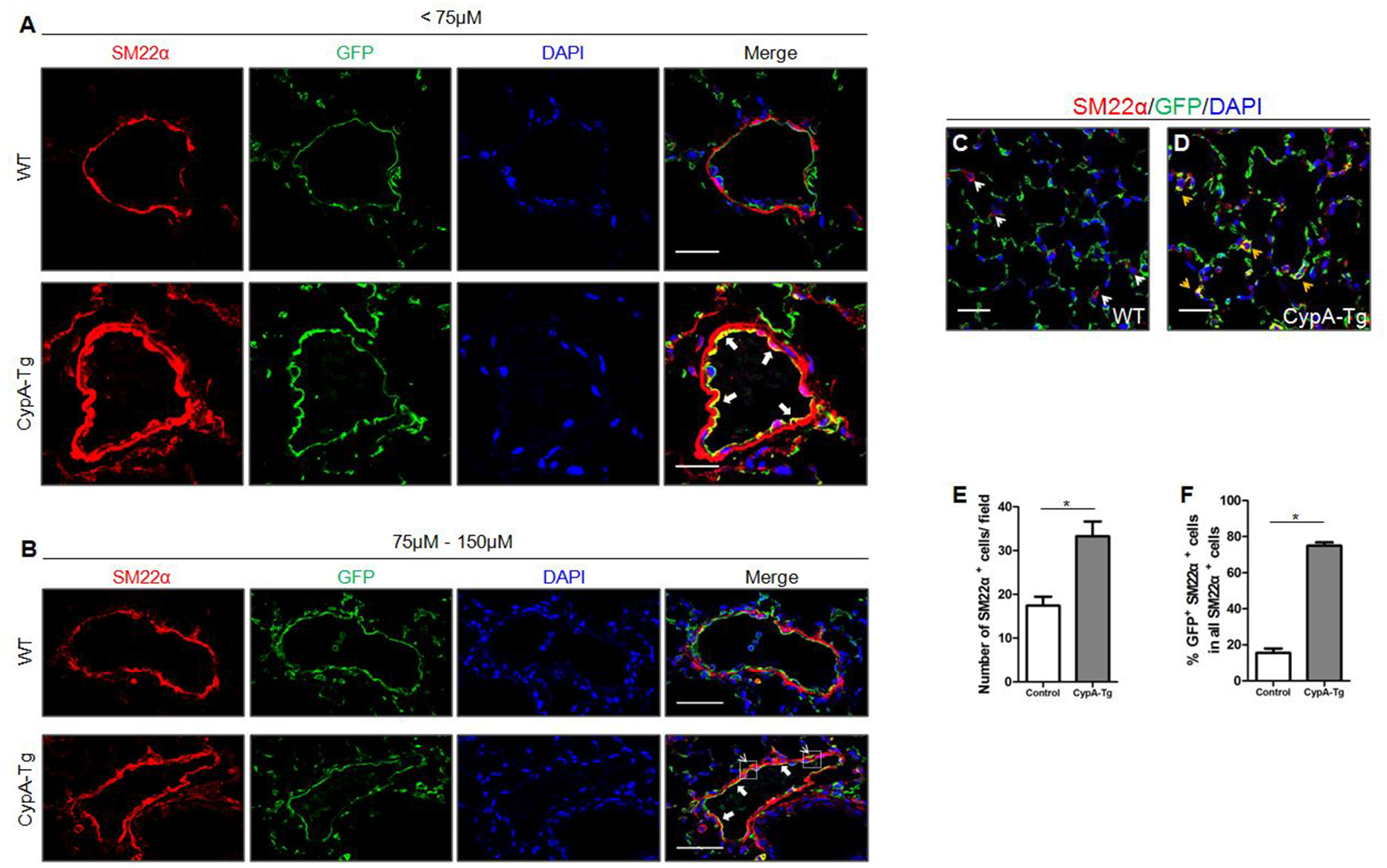
A-B, Representative immunofluoresence images for SM22α (red), GFP (green) and DAPI (blue) of different size pulmonary arteries in lung sections from WT;mTmG (n=5) and Cdh5-CypA;mTmG mice (n=8). Scale bar, 25μm(A) and 50μm(B). C-D, Representative immunofluoresence images for SM22α (red), GFP (green) and DAPI (blue) of alveoli in lung sections from WT;mTmG and Cdh5-CypA;mTmG mice. Scale bar, 25μm. E-F, Quantification of the total number of SM22α+ cells per field (E) and the percentage of SM22α+ cells that were GFP+ in all SM22α+ cells (F) as shown in C-D. Data are mean±SEM. *P<0.05.
We observed increased expression of vimentin, another marker for EndMT, in pulmonary arterioles (diameter <75μm) and co-localization with GFP in the inner (endothelial) layer of these vessels in ecCypA-Tg;mTmG mice at 6 months of age compared to littermate controls (Fig.S2C, indicated by white arrows). There were also GFP+; vimentin+ cells in the adventitia of pulmonary arteries (Fig.S2C, indicated by white arrowheads). Similar to SM22α+ cells, there was a 1.6-fold increase in the number of vimentin+ cells in the alveolar area of ecCypA-Tg;mTmG mice compared to littermate controls (Fig.S2D–E, indicated by white arrowheads). Surprisingly, in control mice, 70% of vimentin+ cells expressed GFP, while in ecCypA-Tg;mTmG mice, the proportion of these vimentin+ cells that expressed GFP was 81% (Fig.S2D–F, indicated by orange arrowheads). The difference between control and ecCypA-Tg;mTmG mice, however, was significant. No sex-based differences were observed. In summary, the data showed strong evidence of EndMT in pulmonary arterioles <75μm as measured by co-localization of SM22α and GFP. Arterioles of 75–150μm showed evidence of migration of EndMT cells into the media and adventitia. In addition to lung, we also examined EndMT in kidney, which is another organ that has many vessels of the same sizes as the lung. Interestingly, we observed a similar EndMT phenotype in the glomeruli of ecCypA-Tg;mTmG mice. Specifically, GFP+;SM22α+ and GFP+;vimentin+ cells were only present in the glomeruli of ecCypA-Tg;mTmG mice (Fig.S3A–B, indicated by white arrowheads).
CypA overexpression increases EC-derived αSMA positive cells in vivo, indicating EndMT.
To further confirm the EndMT phenotype in ecCypA-Tg mice, we stained for αSMA, which is known to be expressed in the neointimal cells in plexiform lesions of PAH. The ecCypA-Tg;mTmG mice demonstrated stronger αSMA staining in the pulmonary arterioles (diameter <75μm), indicating vessel thickening (Fig.3A). There was a 2.0-fold increase in the total number of αSMA+ cells in the alveolar area of ecCypA-Tg;mTmG mice compared to controls (Fig.3E). Unlike SM22α and vimentin, there were no GFP+; αSMA+ cells in the inner layer, media or adventitia of pulmonary arteries in either control or ecCypA-Tg;mTmG mice. However, we observed a significant number of GFP+; αSMA+ cells in the alveolar area of ecCypA-Tg;mTmG mice. Specifically, in control mice only 8% of αSMA+ cells expressed GFP, while in ecCypA-Tg;mTmG mice, 78% of αSMA+ cells expressed GFP (Fig.3B–F, indicated by orange arrowheads; Fig.S4A). No sex-based differences were present. In summary, the ecCypA-Tg mouse model of PAH showed that the vast majority of αSMA+ cells in the alveoli were derived from EC that had transitioned into a more mesenchymal cell type.
Figure 3. CypA overexpression increases EC-derived αSMA positive cells in vivo, indicating EndMT.
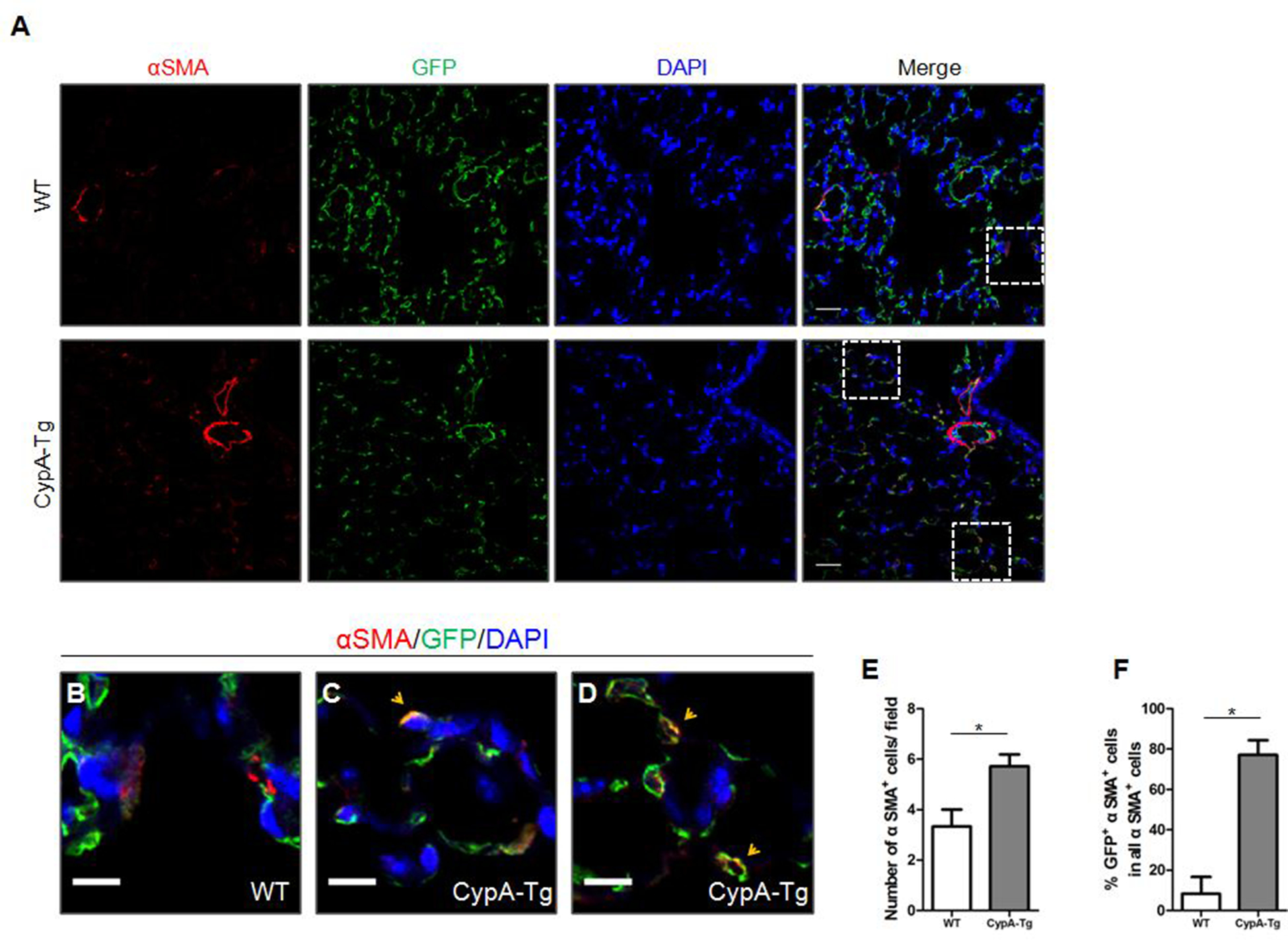
A, Representative immunofluoresence images for αSMA (red), GFP (green) and DAPI (blue) of pulmonary arteries and alveoli in lung sections from WT;mTmG (n=5) and Cdh5-CypA;mTmG mice (n=8). Scale bar, 25μm. B-D, Magnification of white boxes indicated in A. Scale bar, 10μm. E-F, Quantification of the total number of αSMA+ cells per field (E) and the percentage αSMA+ cells that were GFP+ in all αSMA+ cells (F) as shown in A. Data are mean±SEM. *P<0.05.
Pulmonary ECs from ecCypA-Tg mouse show an EndMT phenotype.
To investigate further the role of CypA in EndMT, we cultured mouse pulmonary microvascular EC (MPMEC) from WT and ecCypA-Tg mice for 3 passages. There was a significant decrease in PECAM-1 (2.1-fold), and a significant increase in the mesenchymal marker αSMA (2.1-fold) in MPMEC from ecCypA-Tg mice compared to those from WT mice (Fig.4A–B). There was a trend toward reduced VE-Cadherin expression (1.3-fold), although not significant. Expression of claudin-5, however, was similar (Fig.4B). Immunofluorescence staining showed localization of PECAM-1 at the cell membrane in WT MPMEC. In contrast, there was a more prominent cytoplasmic localization and reduced cell membrane association of PECAM-1 in ecCypA-Tg MPMEC (Fig.4C, indicated by white arrows). Immunofluorescence staining also revealed higher expression of αSMA in ecCypA-Tg MPMEC (Fig.4D). Since the overexpressed cyclophilin A is FLAG epitope-tagged, we stained for FLAG to explore further the connection between CypA overexpression and EC phenotypic change. Interestingly, EC with stronger staining of FLAG-tagged CypA had higher αSMA expression, and were more likely to have a spindle-shaped morphology (Fig.4D, indicated by white arrows), suggesting a direct effect of CypA overexpression on EC phenotypic change.
Figure 4. Pulmonary ECs from ecCypA-Tg mouse show an EndMT phenotype.
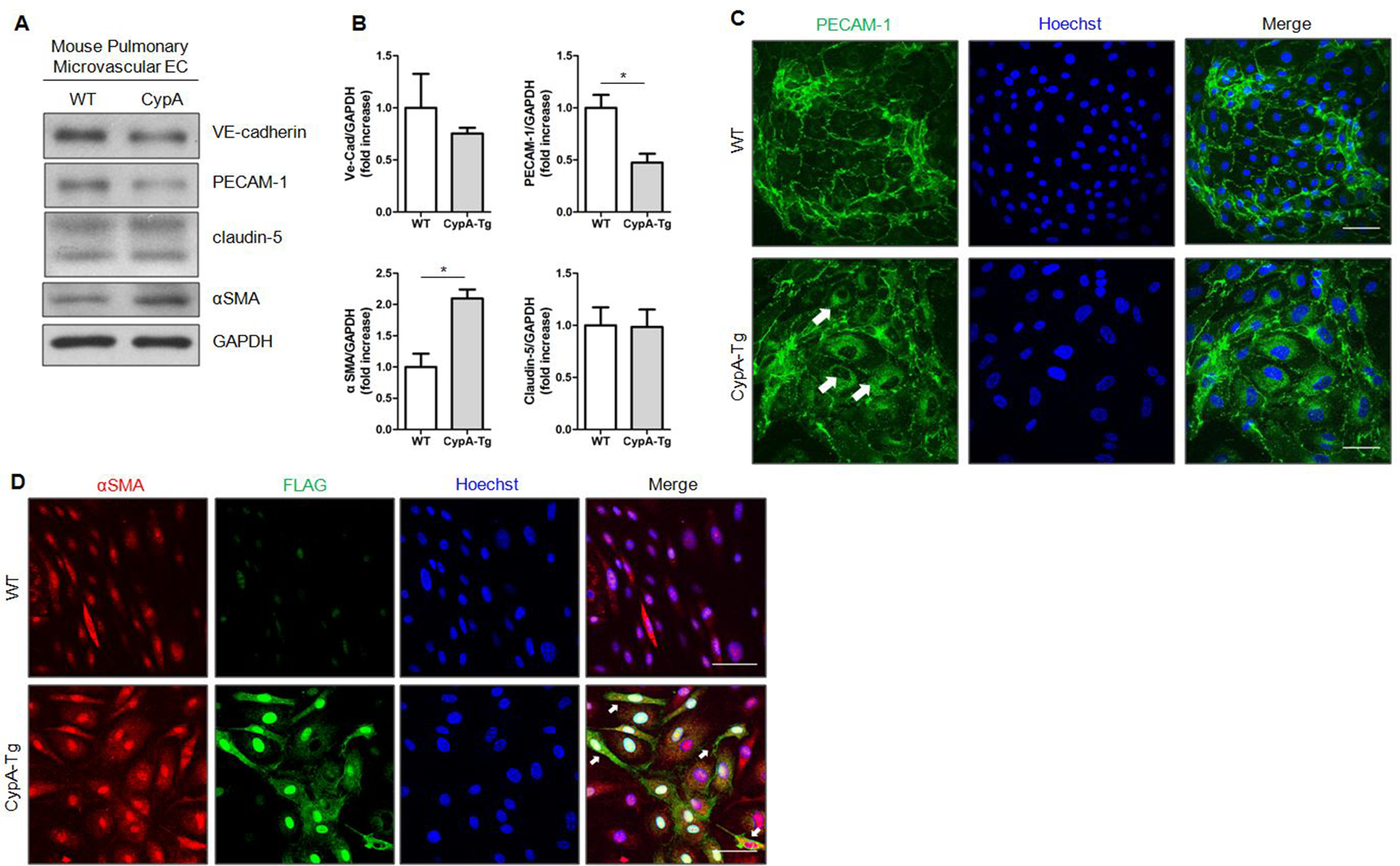
A, Expression of endothelial and mesenchymal markers in cultured MPMEC isolated from control and ecCypA-Tg mice. B, Quantification of protein expression shown in A. Data are mean±SEM. *P<0.05. C, Representative immunofluoresence images of cultured MPMEC isolated from control and ecCypA-Tg mice staining for PECAM-1(green) and Hoechst (blue). D, Representative immunofluoresence images of cultured MPMEC isolated from control and ecCypA-Tg mice staining for αSMA (red), FLAG (green) and Hoechst (blue). Scale bar, 50μm for all images.
CypA and Ack-CypA stimulate EndMT in pulmonary EC in vitro.
Our lab previously reported that secreted CypA caused EC dysfunction measured by apoptosis and expression of inflammatory markers11. To characterize the specific role of extracellular CypA in EndMT, we stimulated cultured mouse and human pulmonary EC with 50nmol/L CypA or AcK-CypA (synthesized as described in Detailed Methods) for 6 days, and analyzed the expression of EC markers (VE-Cadherin, PECAM-1 and claudin-5) and mesenchymal markers (phospho-vimentin and αSMA). We obtained concordant results between the two species; notably 1) the reduced expression of EC markers and increased expression of mesenchymal markers; 2) AcK-CypA was more potent than CypA. Specifically, PECAM-1 and claudin-5 expression were decreased by 1.4-fold and 1.2-fold in response to CypA, but 1.9-fold and 2.7-fold in response to AcK-CypA, respectively. Phospho-vimentin increased 1.1-fold in response to CypA, and 1.4-fold in response to AcK-CypA in MPMEC (Fig.5A–B). There was no change in phospho-vimentin in human pulmonary microvascular EC (HPMEC, data not shown). CypA had no detectable effect on αSMA expression in MPMEC, but increased 1.3-fold in HPMEC. AcK-CypA, however, significantly increased αSMA expression by 1.5-fold in both cell types. Neither CypA nor AcK-CypA had detectable effects on VE-Cadherin expression in MPMEC. AcK-CypA, however, caused a 1.2-fold decrease of VE-Cadherin expression in HPMEC (Fig.5A–B; Fig.S5A–E). To confirm the results, immunofluorescence staining was performed using MPMEC treated with 50nmol/L CypA or AcK-CypA for 6 days. Consistent with results from western blot, there was reduced PECAM-1 and claudin-5 at the cell membrane in a patchy distribution, as well as increased cytoplasmic αSMA in CypA and AcK-CypA treated cells. AcK-CypA had more potent effects than CypA on changes of these markers (Fig.5C–E). Reduced claudin-5 expression was also observed in HPMEC. PECAM-1 showed no effect of CypA while AcK-CypA caused translocation of PECAM-1 from membrane to the cytosol (Fig.S5F–G). For αSMA, we observed no effects of CypA or AcK-CypA on expression or location in HPMEC (data not shown). Significantly, treatment of MPMEC with CypA or AcK-CypA caused a dramatic change in MPMEC morphology from the endothelial cobblestone-like form to an elongated spindle-shaped form, which is characteristic of EndMT (Fig.5F).
Figure 5. CypA and AcK-CypA stimulate EndMT in pulmonary EC in vitro.
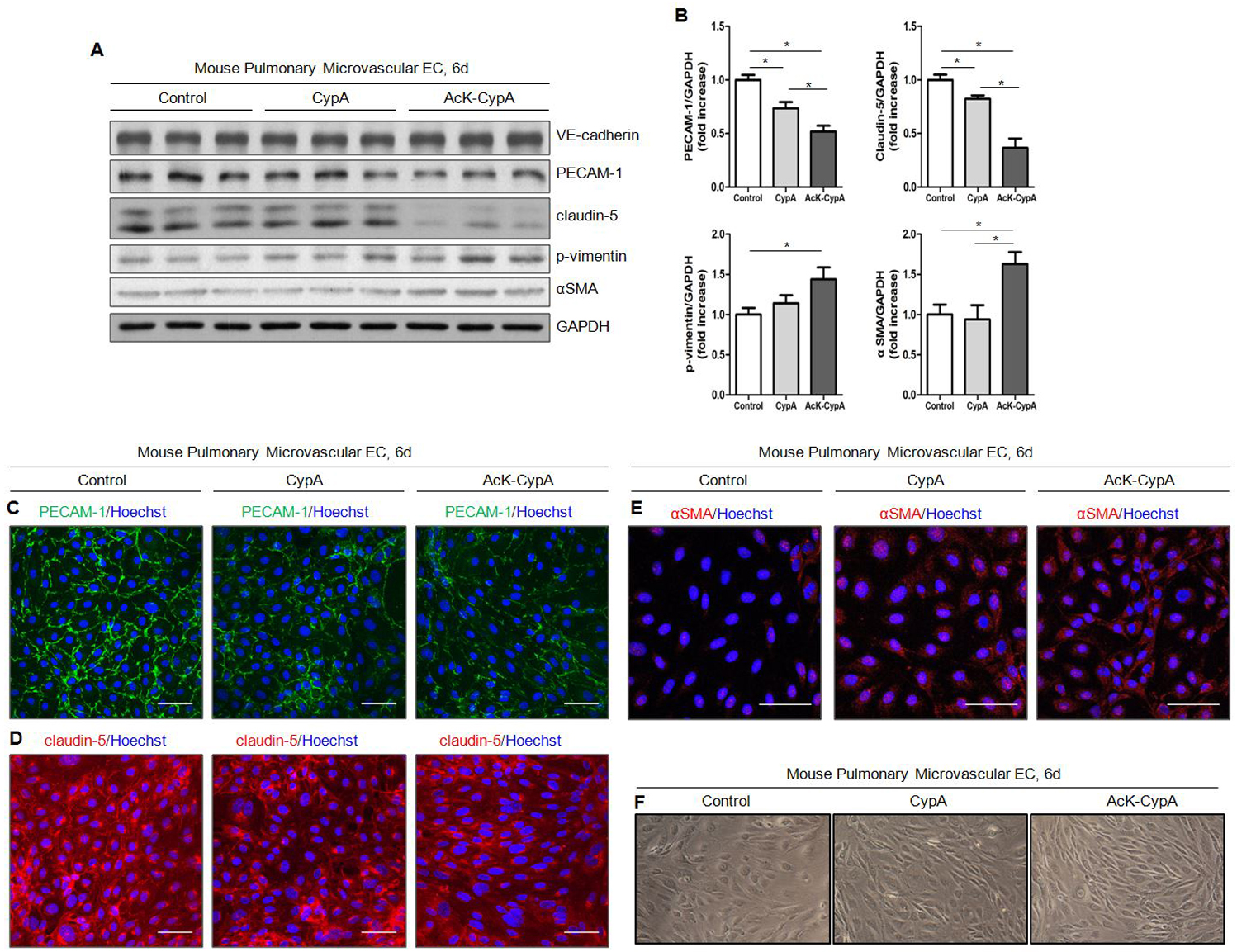
For all experiments CypA and AcK-CypA were used at 50nM. A, Expression of endothelial and mesenchymal markers in MPMEC treated with CypA or AcK-CypA. B, Quantification of protein expression shown in A. Data are mean±SEM. *P<0.05. C-E, Representative immunofluoresence images of MPMEC treated with CypA or AcK-CypA staining for PECAM-1(C), Claudin-5(D) and αSMA(E). Scale bar, 50μm. F, Representative light micrscopy images of MPMEC morphology after CypA or AcK-CypA treatment.
CypA and Ack-CypA stimulate Snail expression, but not Smad2/3 phosphorylation in pulmonary EC.
The transcription factors Snail, Slug and Twist have been implicated as critical mediators of EndMT23–24. To investigate the extracellular CypA and AcK-CypA dependent molecular pathway leading to EndMT, we measured the expression of these transcription factors in MPMEC treated with 50nmol/L CypA or AcK-CypA. Expression of Snail started to increase at 2 hours in MPMEC after AcK-CypA treatment, while CypA had no effect in the first 3 hours (Fig.6A). However, both CypA and AcK-CypA increased Snail expression after 24 hours (1.8-fold and 3.3-fold, respectively) and 6 days (1.6-fold and 4.0-fold, respectively). Consistent with previous observations, AcK-CypA induced more significant changes in expression compared to CypA (Fig.6B–E). We observed a similar increase of Snail in HPMEC after CypA and AcK-CypA treatment (Fig. S6A–B). There were no measurable changes in Slug and Twist expression in MPMEC after CypA or AcK-CypA treatment at any time point (Fig.6A–B and data not shown). In contrast, Slug was increased in HPMEC treated with CypA and AcK-CypA for 24 hours (Fig. S6B). The canonical transforming growth factor beta (TGF-β) pathway is well established for its role in Snail induction and resultant EndMT23. To examine the activation of TGF-β pathway, we measured phosphorylation of Smad2 (pSmad2) and Smad3 (pSmad3) at different time points. Surprisingly, there was no increase of either pSmad2 or pSmad3 at any time point after CypA and AcK-CypA treatment in either MPMEC or HPMEC (Fig.6A–E&; Fig.S6A–B). This suggested that CypA may be stimulating a non-canonical TGF-β pathway. It has been reported that mitogen-activated protein kinase kinase kinase 7 (TAK1) is activated by TGF-β and interleukin 1 beta in EC25. Interestingly, both CypA and AcK-CypA increased phospho-TAK1, with AcK-CypA promoting a more rapid phosphorylation (Fig.6F), supporting the involvement of a non-canonical TGF-β pathway in CypA-induced EndMT.
Fig 6. CypA and Ack-CypA stimulate Snail expression, but not Smad2/3 phosphorylation in pulmonary EC.
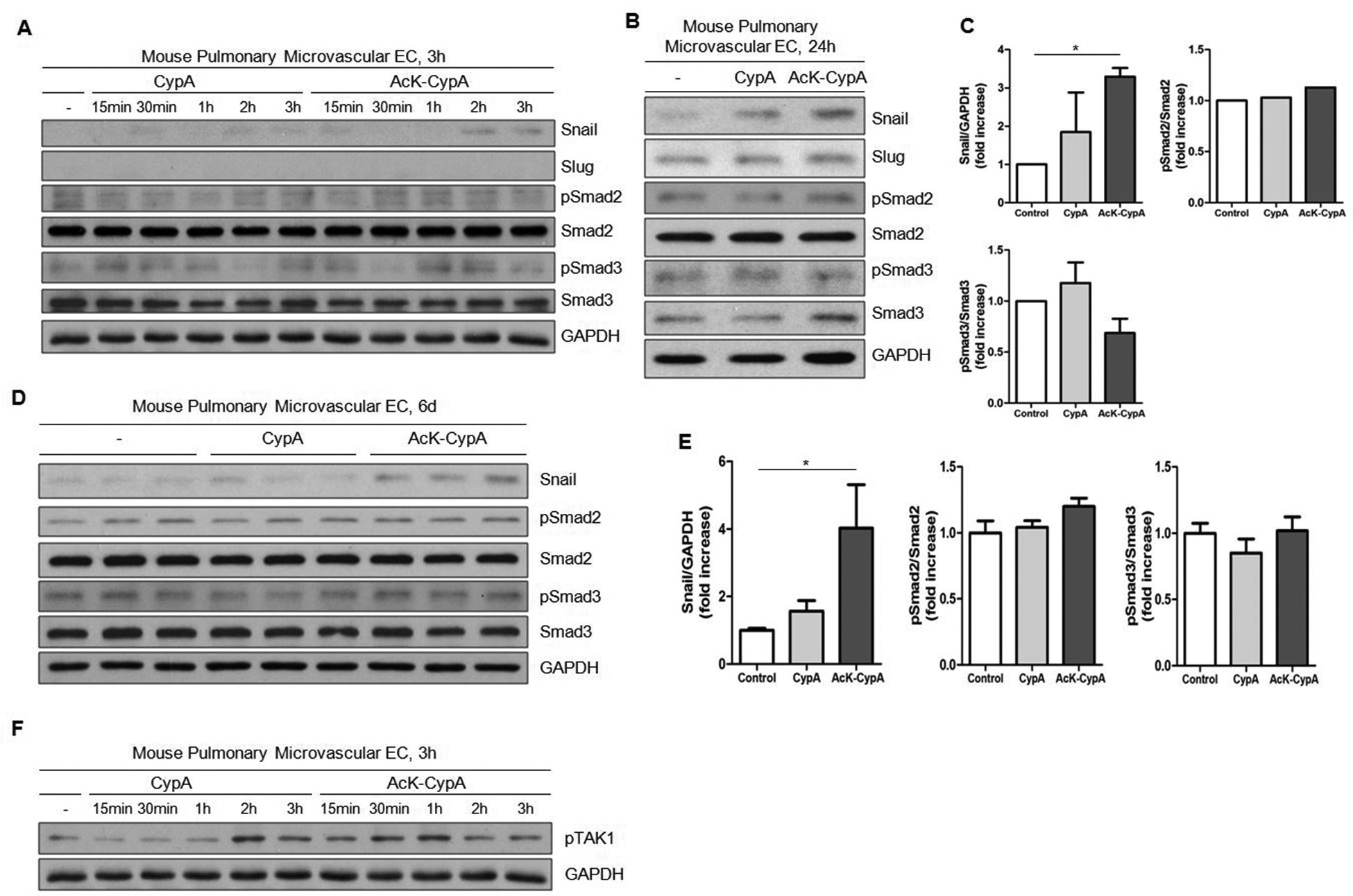
For all experiments CypA and AcK-CypA were used at 50nM. A, Time course of Snail expression and Smad2/3 activation in MPMEC over 3 hours. B-E, Snail expression and Smad2/3 activation in MPMEC treated with CypA or AcK-CypA for 24 hours (B) or 6 days (D). Quantification of protein expression is shown in C (24 hours) and E (6 days). Data are mean±SEM. *P<0.05. F, Time course of TAK1 phosphorylation in MPMEC over 3 hours.
CypA and Ack-CypA stimulate cytokine expression and secretion from pulmonary EC.
There is substantial evidence that multiple aspects of the inflammatory response, especially cytokines, contribute to PAH26–27. We previously reported that extracellular CypA and AcK-CypA induced EC inflammation11. Specifically, we showed that CypA stimulated expression of adhesion molecules (vascular cell adhesion molecule 1 (VCAM1) and intercellular adhesion molecule 1 (ICAM1)) through the NF-kB pathway. To characterize the secreted inflammatory mediators stimulated by extracellular CypA and AcK-CypA, MPMEC were treated with 50nmol/L CypA or AcK-CypA for 24 hours. Conditioned medium was collected and a cytokine array was performed to measure the levels of 40 inflammatory cytokines. We observed significantly elevated levels of 4 cytokines (granulocyte colony-stimulating factor (GCSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), chemokine (C-X-C motif) ligand 1 (CXCL1) and chemokine (C-X-C motif) ligand 5 (CXCL5)) in conditioned medium (Fig.S7A–B). Ack-CypA stimulated the levels of the identical set of chemokines GM-CSF, CXCL1 and CXCL5 but more significantly than CypA (Fig.S7C–G). To confirm these results, we also studied HPMEC which demonstrated significantly elevated levels of 7 cytokines (interleukin (IL)-6, monocyte chemoattractant protein 1 (MCP-1), macrophage colony stimulating factor (M-CSF), tumor necrosis factor beta (TNFβ), macrophage-derived chemokine (MDC), CCL5 and CXCL1) in conditioned medium after treatment with CypA or AcK-CypA (Fig.S8A–J). Among these cytokines, IL-6 and MCP-1 are frequently reported to be closely associated with PAH27–29. Immunohistochemistry showed a significant increase in both IL-6 and MCP-1 (indicated by black arrowheads) in the cells from lungs of the ecCypA-Tg mouse (Fig.S7H–K). ELISA using plasma also showed a significant increase of MCP-1 level in ecCypA-Tg mice compared to control (Fig.S7L). However, plasma IL-6 level showed no difference between control and ecCypA-Tg mice (Fig. S7M).
CypA and Ack-CypA stimulate pulmonary EC proliferation and migration in vitro.
Our data showed that EndMT is likely a pathogenic process for PAH mediated by CypA; its contribution to vessel muscularization likely involves both proliferation and migration15, 21, 30. To evaluate the effect of extracellular CypA and AcK-CypA on EC proliferation and migration we performed bromodeoxyuridine (BrdU) labeling and scratch wound assays in cultured MPMEC. MPMEC were seeded at a very low density, starved for 24 hours and then stimulated with 50nmol/L CypA or AcK-CypA for 2.5 hours in BrdU containing medium. Immunofluorescence staining showed a significantly higher percentage of BrdU labeled cells in response to CypA and AcK-CypA than control (1.5-fold and 1.8-fold increases respectively; Fig.S9A–B). To assay the combined effects of proliferation and migration, scratch wound assays were performed. 24-hour serum starved, confluent MPMEC were treated with 50nmol/L CypA or AcK-CypA for 72 hours in 0.1% serum. Under these conditions, CypA and AcK-CypA stimulated significantly more rapid wound closure compared to control (Fig. S10A&C). To assay migration only, anisomycin (10μM) was applied 2 hours prior to scratch to block cell proliferation. In the presence of anisomycin, 20% serum was required because the migration rate was very slow. While CypA and AcK-CypA both induced EC migration into the scratch area, AcK-CypA promoted significantly greater migration (Fig.S10B&D).
CypA and Ack-CypA stimulate mitochondria dysfunction in pulmonary EC.
In PAH, altered mitochondrial function has been frequently reported in both EC and VSMC31–32. Mitochondrial morphology is a commonly used indicator of mitochondria stress as mitochondria undergo fission in response to conditions of stress33. To study the effect of extracellular CypA on mitochondria function, MPMEC were treated with 50nmol/L CypA or AcK-CypA for 24 hours and then stained with MitoTracker™ Red CMXRos. Treatment caused a dramatic change in mitochondria morphology from the normal, branched, tubular form to a fragmented form (Fig.S11A). CypA and AcK-CypA treated MPMEC showed 5.7-fold and 6.0-fold increases in the percentage of cells with fragmented mitochondria (fragmentation ratio), respectively (Fig.S11B). To confirm these observations, we measured changes in mitochondrial membrane potential. When the mitochondrial membrane potential changes, the cationic dye JC-1 changes its peak excitation to emission ratios due to a switch from monomeric to aggregated form (485/520 for monomer and 570/620 for aggregated). Both CypA and AcK-CypA decreased the aggregated form and increased the monomeric form of JC-1 in MPMEC, consistent with mitochondria stress; confirming the Mitotracker data that demonstrated increased mitochondrial stress and fission stimulated by CypA and Ack-CypA (Fig.S11C–D). Results in HPMEC were similar (Fig.S11E–H).
Discussion
The 5 major findings of this study are 1) CypA expression is greatly enhanced in the endothelium and neointimal cells of pulmonary arteries of PAH patients and two animal PH models; 2) CypA is a novel mediator of EndMT in the pathogenesis of PAH as demonstrated by cell lineage tracing in mice that overexpress CypA in an EC-specific manner; 3) In vitro, both extracellular CypA and AcK-CypA stimulated EC to express an inflammatory EndMT phenotype including increased cytokine release, cell migration, proliferation and mitochondrial dysfunction, mechanisms known to be involved in the pathogenesis of PAH; 4) We provide evidence for a systemic effect of CypA mediated inflammation that includes increases in IL-6 and MCP-1; and 5) AcK-CypA was a more potent agonist than CypA for the phenotypic change. In summary, our data show that CypA can stimulate EC to develop many features of the muscularized cells found in PAH. While EndMT in PAH has been reported in other papers, this one is the first to report EndMT not caused by a BMPR2 mutation, hypoxia, or drugs (such as monocrotaline and Sugen 5416). This result more closely resembles the pathogenesis of idiopathic human PAH.
It was previously reported that CypA was increased in the plasma and vascular smooth muscle cells of pulmonary arteries in PAH patients34. Here we examined cell-specific expression of CypA in human PAH and two different animal PH models. In all three situations, we found there was significantly more CypA in EC than in VSMC, thus verified the increased presence of CypA in multiple species and multiple models of PAH. Importantly, the neointimal cells in human pulmonary arteries with obliterative lesions also showed strong expression of CypA. It is possible that these cells may have originated from EC via EndMT based on prior findings that they express endothelial markers (VE-Cadherin, PECAM-1) and mesenchymal marker αSMA16.
Substantial evidence suggests that EndMT contributes to a subset of neointimal cells in animal models of PH and human PAH14, 16–17. A number of inflammatory mediators including cytokines and transcription factors have been shown to play a role in EndMT20, 35–37. Considering the high level of CypA in EC and neointimal cells, as well as its ability to induce inflammation, we hypothesized that CypA could induce EndMT in pulmonary artery EC in PAH.
We previously demonstrated that an EC-specific CypA overexpression mouse developed many characteristics of PAH, including increased right ventricular systolic pressure and increased cellularity11. There were a large number of cells in the intima and alveoli that expressed inflammatory markers which were of unclear origin. To determine the lineage of these cells in our mouse model, we used the Cdh5-Cre/mTmG/CypA-Tg mouse in which all EC-derived cells express GFP, and then stained for markers of EndMT. 1) We found increased EndMT markers in these mice which differed in location in small(<75μm) versus medium(75–150μm) vessels. In the small vessels, co-expression of the mesenchymal markers SM22α and vimentin were only in the endothelial layer of pulmonary arterioles (Fig.2A; Fig.S2C). In the medium vessels, markers were present in both endothelial and smooth muscle layers (Fig.2B; Fig.S2A). 2) Of note, there was an increased number of SM22α and αSMA positive cells in the alveolar walls, a significant percentage of which had an EC origin. Together these data strongly suggest the presence of EndMT, and a critical role for CypA in driving this process in PAH in vivo. Future studies using single cell analysis will be performed to determine the phenotype of these alveolar EC-derived mesenchymal cells.
Interestingly, this in vivo EndMT phenotype was not restricted to the lung. There were increased GFP positive cells that were also positive for SM22α and vimentin in the glomeruli of ecCypA-Tg mice compared to control mice, suggesting a systemic effect of CypA and the associated inflammatory cytokines on EC to stimulate EndMT.
We studied the EndMT phenotype more closely using cultured MPMEC and HPMEC. Specifically, we took advantage of our ability to harvest pulmonary EC from WT and ecCypA-Tg mice. In MPMEC from ecCypA-Tg mice we observed decreased PECAM-1 and increased αSMA. The concurrent high level of αSMA in FLAG-CypA overexpressing EC indicated a correlation between CypA overexpression and appearance of mesenchymal markers in EC. Interestingly, we observed in the MPMEC from ecCypA-Tg mice that PECAM-1 translocated from membrane to cytoplasm, suggesting that a mechanism of EndMT is internalization of EC-specific plasma membrane proteins. Treatment with extracellular CypA and AcK-CypA in cultured MPMEC from WT mice also showed a reduction of EC specific proteins such as PECAM-1 and claudin-5, and an increase of mesenchymal proteins such as αSMA and phospho-vimentin. While extracellular CypA stimulated the EndMT phenotype, we cannot rule out a role for intracellular CypA, especially because in vivo the source of secreted CypA is intracellular CypA.
TGF-β signaling has a central role in promoting EndMT via both Smad-dependent and -independent signaling23, 25. The canonical pathway (Smad2/3-Snail/Slug) is the best characterized pathway causing EndMT in most types of EC24. Here we found that CypA increased Snail expression, but not phospho-Smad2/3 expression, which is not consistent with the canonical TGF-β pathway. In addition, we found that canonical Wnt/β-catenin signaling pathway, another pathway responsible for EndMT, was not affected by CypA or AcK-CypA treatment (data not shown). The transient increase of phospho-TAK1 suggests the participation of a non-canonical TGF-β pathway in this process; although the specific signaling pathway requires further study.
The loss of EC tight junctions due to decreased claudin-5 expression in EC undergoing EndMT appears to be an important change that enables EC proliferation and migration. These two processes are likely required for the formation of plexiform lesions in human PAH. We found that extracellular CypA stimulated proliferation and migration of EC. However, despite these changes ecCypA-Tg mice did not develop plexiform lesions, indicating that other processes are necessary.
Substantial data suggest that cytokine-driven inflammation is important to the development of PAH38. Specifically, in PAH patients, there are high circulating levels of cytokines such as IL-1β, IL-6 and TNFα39–40. Additionally, cytokines such as IL-1β and TNFα can act alone or synergistically with TGF-β to induce EndMT35. We found that extracellular CypA stimulated cultured EC secretion of several cytokines, including IL-6, MCP-1, GCSF, etc. The chemotactic effects of these cytokines combined with the EC expression of adhesion molecules likely accounts for the recruitment of CD45 positive inflammatory cells, as we previously reported11. Based upon clinical and experimental data, IL-6 is thought to be one of the most important cytokines in the pathogenesis of PAH28, 39, 41. Importantly, we observed higher IL-6 expression in lungs of ecCypA-Tg mice, and increased IL-6 secretion from HPMEC after extracellular CypA treatment. The discrepancy between increased IL-6 levels in lungs and similar IL-6 levels in plasma between control and ecCypA-Tg mice might reflect a localized biological function of this cytokine in PAH. It is also possible that IL-6 is rapidly degraded and/or taken up by circulating cells or endothelial cells. These results suggest that extracellular CypA is an important cause of PAH by its dual effects to stimulate EndMT and activate inflammatory cells.
We also found that exposure of MPMEC and HPMEC to CypA and AcK-CypA caused mitochondrial dysfunction as measured by Mitotracker staining and mitochondrial membrane potential assay. Mitochondria dysfunction creates oxidative stress, which has several effects that may promote PAH: 1) It stimulates an inflammatory genetic program mediated, in part, by activation of the NF-kB pathway. 2) Oxidative stress promotes EndMT because it stimulates the TGF-β pathway42–43. 3) We showed previously that oxidative stress increases AcK-CypA, which is a more potent mediator of the EndMT phenotype than CypA. We believe that AcK-CypA is more potent than CypA because of increased binding affinity to the putative CypA receptor.
Perspectives
Here we show that CypA is a novel mediator of EndMT and many features of the initial stage of PAH development. The mechanism appears to be primarily stimulation of a chronic inflammatory response that is mediated by EC expression of adhesion molecules and secretion of multiple cytokines, especially IL-6 and MCP-1. These results suggest that extracellular CypA (and likely other PAH mediators) causes PAH by its dual effects to stimulate EndMT and inflammation. Because CypA promotes multiple features of the PAH phenotype, and extracellular CypA can be selectively inhibited, our results strengthen the basis for inhibiting CypA activity (especially AcK-CypA) as a therapy for PAH.
Supplementary Material
Novelty and Significance.
What is new:
1) It describes a novel mechanism by which CypA induces PAH that involves EndMT and inflammation. 2) AcK-CypA was a more potent agonist for EndMT. 3) Since the primary stimulus for CypA secretion is oxidative stress, changes in redox state may be important in initiation of PAH.
What is relevant:
CypA causes vascular pathology by stimulating EndMT and inflammation. Because the CypA overexpression mouse is not hypertensive, CypA alone is not sufficient to cause systemic hypertension. However, with single cell RNA-seq, it should be possible to identify the specific receptors and signaling pathways used by CypA to cause pulmonary hypertension.
Summary:
the present study conclusively shows that EndMT is an important mechanism in development of PAH.
Acknowledgements
We are grateful to Deborah Haight and Amy Mohan for their expert technical assistance with tissue harvests. We are particularly grateful to Chen Yan, David Dean, Patricia Sime, Mary Wines-Samuelson, Slava Korshunov, Chia Hsu and members of the Aab Cardiovascular Research Institute for helpful suggestions.
Sources of Funding
This work was supported by NIH RO1 HL049192 to BCB.
Footnotes
Conflicts of Interest
None
Disclosures
None
References
- 1.Rabinovitch M Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S [DOI] [PubMed] [Google Scholar]
- 3.Marks AR. Cellular functions of immunophilins. Physiol Rev. 1996;76:631–649 [DOI] [PubMed] [Google Scholar]
- 4.Jin ZG, Lungu AO, Xie L, Wang M, Wong C, Berk BC. Cyclophilin a is a proinflammatory cytokine that activates endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:1186–1191 [DOI] [PubMed] [Google Scholar]
- 5.Jin ZG, Melaragno MG, Liao DF, Yan C, Haendeler J, Suh YA, Lambeth JD, Berk BC. Cyclophilin a is a secreted growth factor induced by oxidative stress. Circ Res. 2000;87:789–796 [DOI] [PubMed] [Google Scholar]
- 6.Satoh K, Nigro P, Matoba T, O’Dell MR, Cui Z, Shi X, Mohan A, Yan C, Abe J, Illig KA, Berk BC. Cyclophilin a enhances vascular oxidative stress and the development of angiotensin ii-induced aortic aneurysms. Nat Med. 2009;15:649–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nigro P, Satoh K, O’Dell MR, Soe NN, Cui Z, Mohan A, Abe J, Alexis JD, Sparks JD, Berk BC. Cyclophilin a is an inflammatory mediator that promotes atherosclerosis in apolipoprotein e-deficient mice. J Exp Med. 2011;208:53–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Satoh K, Nigro P, Zeidan A, Soe NN, Jaffre F, Oikawa M, O’Dell MR, Cui Z, Menon P, Lu Y, Mohan A, Yan C, Blaxall BC, Berk BC. Cyclophilin a promotes cardiac hypertrophy in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:1116–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xue C, Sowden MP, Berk BC. Extracellular and intracellular cyclophilin a, native and post-translationally modified, show diverse and specific pathological roles in diseases. Arterioscler Thromb Vasc Biol. 2018;38:986–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soe NN, Sowden M, Baskaran P, Kim Y, Nigro P, Smolock EM, Berk BC. Acetylation of cyclophilin a is required for its secretion and vascular cell activation. Cardiovasc Res. 2014;101:444–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xue C, Sowden M, Berk BC. Extracellular cyclophilin a, especially acetylated, causes pulmonary hypertension by stimulating endothelial apoptosis, redox stress, and inflammation. Arterioscler Thromb Vasc Biol. 2017;37:1138–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pietra GG, Edwards WD, Kay JM, Rich S, Kernis J, Schloo B, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Histopathology of primary pulmonary hypertension. A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the national heart, lung, and blood institute, primary pulmonary hypertension registry. Circulation. 1989;80:1198–1206 [DOI] [PubMed] [Google Scholar]
- 13.Sheikh AQ, Misra A, Rosas IO, Adams RH, Greif DM. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med. 2015;7:308ra159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiao L, Nishimura T, Shi L, Sessions D, Thrasher A, Trudell JR, Berry GJ, Pearl RG, Kao PN. Endothelial fate mapping in mice with pulmonary hypertension. Circulation. 2014;129:692–703 [DOI] [PubMed] [Google Scholar]
- 15.Suzuki T, Carrier EJ, Talati MH, Rathinasabapathy A, Chen X, Nishimura R, Tada Y, Tatsumi K, West J. Isolation and characterization of endothelial-to-mesenchymal transition cells in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2018;314:L118–L126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ, Dorfmuller P, Remy S, Lecerf F, Plante S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S, Perros F. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018 [DOI] [PubMed] [Google Scholar]
- 17.Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ, Holmes AM. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am J Pathol. 2015;185:1850–1858 [DOI] [PubMed] [Google Scholar]
- 18.Stenmark KR, Frid M, Perros F. Endothelial-to-mesenchymal transition: An evolving paradigm and a promising therapeutic target in pah. Circulation. 2016;133:1734–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: In vitro analysis. Circ Res. 2002;90:1189–1196 [DOI] [PubMed] [Google Scholar]
- 20.Cho JG, Lee A, Chang W, Lee MS, Kim J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol. 2018;9:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Welch-Reardon KM, Wu N, Hughes CC. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler Thromb Vasc Biol. 2015;35:303–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Souilhol C, Harmsen MC, Evans PC, Krenning G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc Res. 2018;114:565–577 [DOI] [PubMed] [Google Scholar]
- 23.Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for tgfbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci. 2008;121:3317–3324 [DOI] [PubMed] [Google Scholar]
- 24.Cooley BC, Nevado J, Mellad J, Yang D, St Hilaire C, Negro A, Fang F, Chen G, San H, Walts AD, Schwartzbeck RL, Taylor B, Lanzer JD, Wragg A, Elagha A, Beltran LE, Berry C, Feil R, Virmani R, Ladich E, Kovacic JC, Boehm M. Tgf-beta signaling mediates endothelial-to-mesenchymal transition (endmt) during vein graft remodeling. Sci Transl Med. 2014;6:227ra234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang YE. Non-smad pathways in tgf-beta signaling. Cell Res. 2009;19:128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price LC, Wort SJ, Perros F, Dorfmuller P, Huertas A, Montani D, Cohen-Kaminsky S, Humbert M. Inflammation in pulmonary arterial hypertension. Chest. 2012;141:210–221 [DOI] [PubMed] [Google Scholar]
- 27.Groth A, Vrugt B, Brock M, Speich R, Ulrich S, Huber LC. Inflammatory cytokines in pulmonary hypertension. Respir Res. 2014;15:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–244, 228p following 244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Itoh T, Nagaya N, Ishibashi-Ueda H, Kyotani S, Oya H, Sakamaki F, Kimura H, Nakanishi N. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology. 2006;11:158–163 [DOI] [PubMed] [Google Scholar]
- 30.Sakao S, Tatsumi K, Voelkel NF. Endothelial cells and pulmonary arterial hypertension: Apoptosis, proliferation, interaction and transdifferentiation. Respir Res. 2009;10:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan J, Dasgupta A, Huston J, Chen KH, Archer SL. Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med (Berl). 2015;93:229–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall JD, Bazan I, Zhang Y, Fares WH, Lee PJ. Mitochondrial dysfunction and pulmonary hypertension: Cause, effect, or both. Am J Physiol Lung Cell Mol Physiol. 2018;314:L782–L796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Picard M, Shirihai OS, Gentil BJ, Burelle Y. Mitochondrial morphology transitions and functions: Implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol. 2013;304:R393–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Satoh K, Satoh T, Kikuchi N, Omura J, Kurosawa R, Suzuki K, Sugimura K, Aoki T, Nochioka K, Tatebe S, Miyamichi-Yamamoto S, Miura M, Shimizu T, Ikeda S, Yaoita N, Fukumoto Y, Minami T, Miyata S, Nakamura K, Ito H, Kadomatsu K, Shimokawa H. Basigin mediates pulmonary hypertension by promoting inflammation and vascular smooth muscle cell proliferation. Circ Res. 2014;115:738–750 [DOI] [PubMed] [Google Scholar]
- 35.Perez L, Munoz-Durango N, Riedel CA, Echeverria C, Kalergis AM, Cabello-Verrugio C, Simon F. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017;33:41–54 [DOI] [PubMed] [Google Scholar]
- 36.Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L, Rabinovitch M. In pulmonary arterial hypertension, reduced bmpr2 promotes endothelial-to-mesenchymal transition via hmga1 and its target slug. Circulation. 2016;133:1783–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–1631 [DOI] [PubMed] [Google Scholar]
- 40.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, Walker C, Budd DC, Pepke-Zaba J, Morrell NW. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927 [DOI] [PubMed] [Google Scholar]
- 41.Heresi GA, Aytekin M, Hammel JP, Wang S, Chatterjee S, Dweik RA. Plasma interleukin-6 adds prognostic information in pulmonary arterial hypertension. Eur Respir J. 2014;43:912–914 [DOI] [PubMed] [Google Scholar]
- 42.Krstic J, Trivanovic D, Mojsilovic S, Santibanez JF. Transforming growth factor-beta and oxidative stress interplay: Implications in tumorigenesis and cancer progression. Oxid Med Cell Longev. 2015;2015:654594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Montorfano I, Becerra A, Cerro R, Echeverria C, Saez E, Morales MG, Fernandez R, Cabello-Verrugio C, Simon F. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a tgf-beta1 and tgf-beta2-dependent pathway. Lab Invest. 2014;94:1068–1082 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.