

Abstract
Protein digestion is a key challenge in mass spectrometry (MS)-based structural proteomics. Although using hydrogen deuterium exchange kinetics with mass spectrometry (HDX-MS) to interrogate high order structure of proteins is now established, it can be challenging for ß-barrel proteins, which are important in cellular transport. These proteins contain a continuous chain of H-bonds that impart stability, causing difficulty in digestion for bottom-up measurements. To overcome this impediment, we tested organic solvents as denaturants during on-line pepsin digestion of soluble ß-barrel proteins. We selected green fluorescent protein (GFP), siderocalin (Scn), and retinol-binding protein 4 (RBP4) as model proteins and screened six different polar-aprotic and polar-protic solvent combinations to disrupt the H-bonds and hydrophobic interactions holding together the ß-sheets. Use of organic solvents improves digestion, generating more peptides from the rigid ß-barrel regions, without compromising the ability to predict the retinol binding site on RBP4 when adopting this proteolysis in hydrogen/deuterium exchange.
Graphical Abstract

Structure and conformational dynamics determine protein function.1 While X-ray crystallography and NMR routinely provide high-resolution structural information, not all protein structural questions are addressable by these methods1,2 X-ray crystallography, for example, provides information on static, crystallized proteins but provides very limited information on dynamics. Alternative approaches, including MS-based structural proteomics, have become indispensable for these problems. Unlike traditional MS-based proteomics that detect changes in protein expression and quantify post-translational modifications,3 MS-based structural proteomics use chemistry to characterize higher order protein structure, interactions, and dynamics.4 The constituent methodologies, including hydrogen deuterium exchange (HDX),5 footprinting,6 cross-linking,7 affinity labeling,8 and limited proteolysis,9 now have wide application.
Although MS-based proteomics and structural proteomics have improved over the past decade, many challenges remain. Prominent among these is high coverage protein digestion. Many sets of peptide are needed to achieve good detection limits, accurate quantification, adequate spatial resolution, and isoform differentiation.10 Although high sequence coverage is helpful for protein identification and quantification in traditional MS-based proteomics, it is essential for structural proteomics.
To ensure high coverage, investigators have used several enzymes (e.g., trypsin, chymotrypsin, LysC, LysN, AspN, GluC, ArgC, WaLP, and MaLP) individually or in combination to give overlapping patterns. Another approach is optimized activation methods (e.g., CID, ETD, and HCD).3,10,11 Various surfactants, solvents, and chaotropes are available to denature membrane proteins,12 and pressure-enhanced online digestion can improve digestion efficiency.13
The digestion problem becomes more serious for the HDX-MS platform. One way to improve spatial resolution is to generate overlapping peptides in the digestion so that the HDX of overlapped segments can pinpoint on amino acid residues. This approach can be challenging because on-line digestion of proteins must be fast under exchange-quenched conditions (low T and pH of 2.5). The requirements of short digestion time and acidic proteases require robust methods for digestion.5,14,15 Several approaches for improving the digestion efficiency in HDX include using acidic proteases with different specificities to generate more and overlapping peptides,16–19 using TCEP or electrochemical reduction to break disulfide bonds,20 using detergents21 or incorporating high-pressure on-line digestion to unfold the protein.22
When analyzing proteolytically resistant proteins, denaturation is a primary concern. Denaturation methods include physical treatment (e.g., heat, pressure, freezing, irradiation) and chemical treatment (e.g., low pH, organic solvents, detergents, and organic solutes (i.e., urea and guanidine chloride)).23 High-pressure on-line digestion systems require special equipment, heating the protein accelerates the HD back exchange, and using detergents may contaminate the mass spectrometer.
Here, we evaluated the use of MS-friendly organic solvents to improve on-line digestion. Organic solvents are considered “turbodigestion” reagents because they efficiently break both H-bonds and hydrophobic interactions within the polypeptide chain and between the chain and the surrounding medium.24,25 Although use of aqueous organic solvents improves protein digestion in tandem mass spectrometry (MS/MS)-based peptide sequencing and reduces back exchange in HDX experiments,26 there have been no systematic studies, to our knowledge, in which organic solvents were applied to improve digestion on the HDX platform.24 Here, we fill this gap with a systematic study of the utility of organic solvents for on-line digestion in HDX-MS.
We encountered the digestion problem when applying HDX in studies of ligand-induced conformational changes of ß-barrel proteins.27 These proteins are responsible for solubilization, flux, binding, and translocation of ions and small molecules. One of the most common alignments is an “up-and-down” motif formed by antiparallel ß-strands that are H-bonded to neighboring strands.28 The vast majority of ß-barrel proteins are endowed with stable, rigid structures, rendering them resistant to proteolysis.
To overcome this problem, we evaluated the ability of several organic solvents to unfold three unique water-soluble ß-barrel proteins on the HDX platform and facilitate on-line digestion. Two of the proteins, siderocalin (Scn) and retinol-binding protein 4 (RBP4), are from the lipocalin family; the third is green fluorescent protein (GFP). Because HDX is often used to identify ligand binding sites, we assessed the binding site between RBP4 and retinol in the presence and absence of organic solvent to ensure that this approach did not compromise labeling.
EXPERIMENTAL SECTION
Quench solutions.
Normal quench buffer was 4 M guanidine chloride, 50 mM TCEP in PBS buffer at pH 2.5. Quench solutions containing organic solvents were prepared by mixing 250 μL buffer (8 M guanidine chloride, 100 mM TCEP in PBS buffer) with 250 μL organic solvent. The pH was adjusted to 2.5.
HD Exchange (HDX) experiment.
For mapping, 80 pmol of protein (GFP, Scn, or RBP4, all in 1 μL), was diluted in PBS buffer (24 μL) at 25 °C for 1 min (GFP) or 5 min (Scn and RBP4) then quenched by adding 25 μL quench solution. After 3 min denaturation in quench buffer at 37 °C (Scn and RBP4) or 1 min denaturation in quench buffer at 25 °C (GFP), the sample solution was diluted to 500 μL with 1% TFA in water (pH 2.5) and then injected into a custom-fabricated, online pepsin column. The peptides from digestion were trapped on a C8 column and separated with a C18 analytical column.
To initiate exchange, 80 pmol of protein (in 1 μL) was diluted with deuterated PBS buffer (24 μL) at 25 °C. After several exchange times, from 10 s to 4 h, the exchange was stopped by adding 25 μL quench solution at 37 °C for 3 min (for SCN and RBP4) or 25 °C for 1 min (for GFP). The quenched samples were then diluted to 500 μL by using 1% TFA in water (pH 2.5). The HDX data were collected on an LTQ FT mass spectrometer (Thermo, Waltham, MA).
While HDX quenching is normally conducted at low temperature to minimize back-exchange, highly stable proteins (like the β-barrels studied here) do not efficiently denature and digest under these conditions. We therefore used 25 °C (GFP) and 37 °C (Scn and RBP4) to improve digestion at the expense of increased back-exchange. Although the higher T digestion (37 °C) will cause ~2.3-fold increase of back exchange compared to room temperature,29 that back exchange contributes little to the deuterium uptake differences between apo- and holo- protein.26
The elution gradient for the peptic peptides started with 95% solvent A and decreased to 50% solvent A in 5.5 min, followed by a linear gradient to 0% in 0.5 min. Solvent B was then maintained at 100% for 1.5 min before column re-equilibration. [Solvent A: water with 0.1% formic acid, solvent B: 80% acetonitrile, 20% water, 0.1% formic acid. The flow rate was 100 μL/min].
RESULTS AND DISCUSSION
Green fluorescent protein (GFP).
We first selected GFP (~27 kDa), consisting of eleven β-strands with a pleated-sheet arrangement, to test whether organic solvents can facilitate denaturation and improve digestion efficiency compatible with the needs of fast digestion for HDX. We selected polar aprotic (dimethylformamide (DMF), and acetonitrile (MeCN)), and polar protic solvents (methanol (MeOH), trifluoroethanol (TFE), isopropanol (i-PrOH), and 1,1,1,3,3,3-hexafluoro-2-propanol (H2P)). We prepared the quench buffers to contain 4 M guanidine, 50% organic solvent, and pH 2.5. Because a pepsin column cannot tolerate organic solvents at > 5%, we diluted the sample with 1% TFA (pH 2.5) so that the organic solvent was 2.5% just before injecting the sample.
We evaluated sequence coverage, the number of MS/MS scans, and unique peptides (the number of peptide sequences unique to a protein group) for each solvent system. Normal quench (i.e., 4 M guanidine) (Figure 1, Figure S1a) showed 92% coverage of GFP. The mean GFP coverage when an additional organic solvent was included was 97%, indicating improvement over normal quench.
Figure 1.
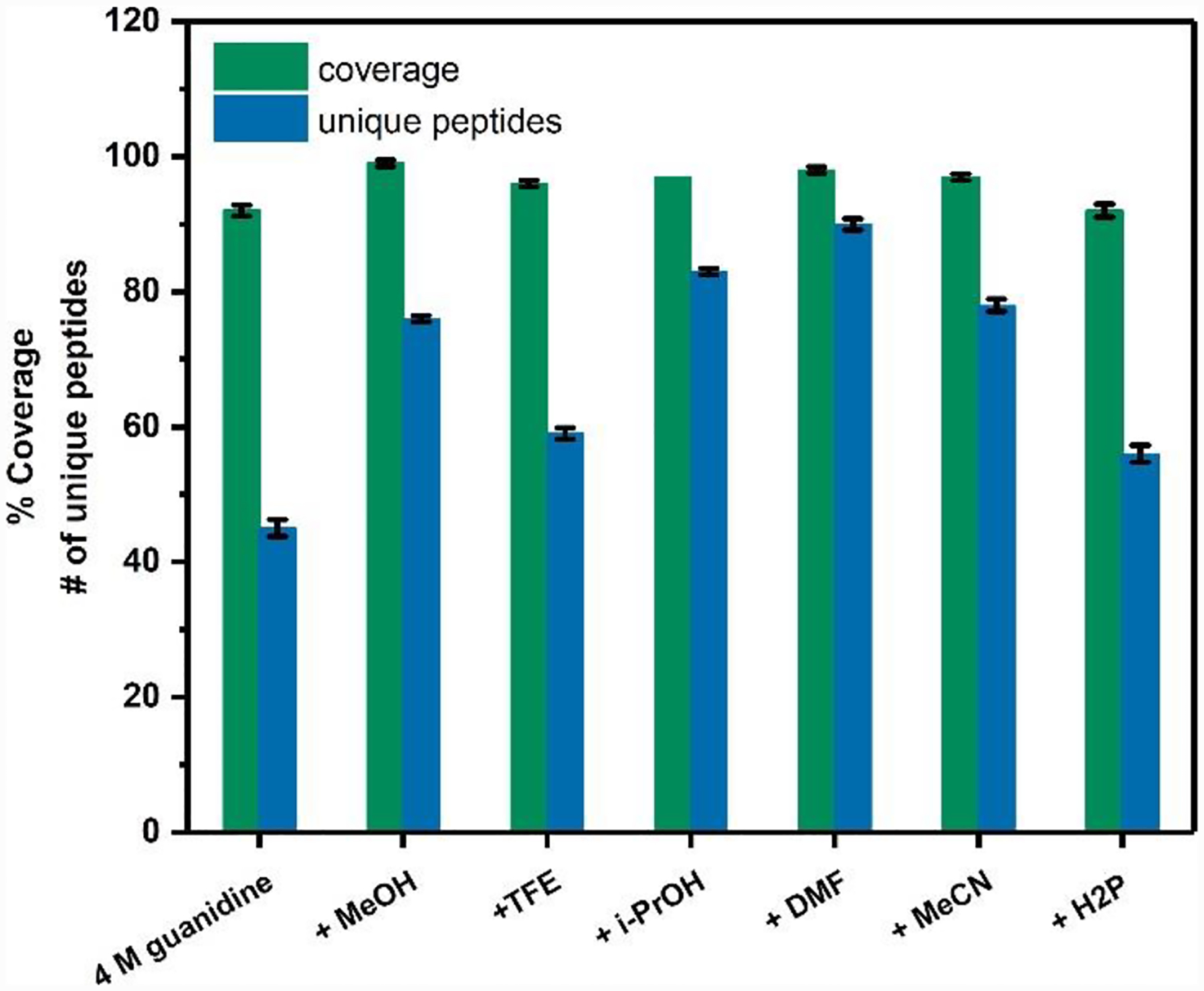
Scan statistics for GFP under quench conditions with various organic solvents. The % coverage and number of unique peptides are annotated on the y axis.
Digestion with organic solvents not only increases coverage, but also the mean number of unique peptides and MS/MS scans (by ~63% and 110%, respectively, in GFP). Among the organic solvents, DMF gave the most improved GFP digestion, with two times the number of unique peptides, and nearly three times the spectral number compared with the normal quench (Figure 1, Figure S1b).
We analyzed peptide length and segment length to evaluate pepsin digestion performance under various organic solvent conditions. An appropriate peptide length for MS sequencing is 7–35 amino acids.30 The average peptic peptide length is similar for normal quench compared to that with organic solvents (~15 for normal quench vs. ~ 17 for addition of organic solvent) (Figure S2). Segment length is another measure of digestion effectiveness (Figure S3). Average segment length (ASL) (Figure 2a and equ 1) is an approximate measure of the amino acid level resolution that can be deduced from overlapped peptides22. Taking the region 130–151 as an example (Figure 2b), we find two peptides with an ASL of 7.3 in the absence of DMF. Digestion in DMF generates significantly more overlapping peptides, and the ASL decreases to 4.4. The presence of organic solvents increases pepsin digestion efficiency, providing improved spatial resolution, sometimes even to the amino-acid level.
Figure 2.
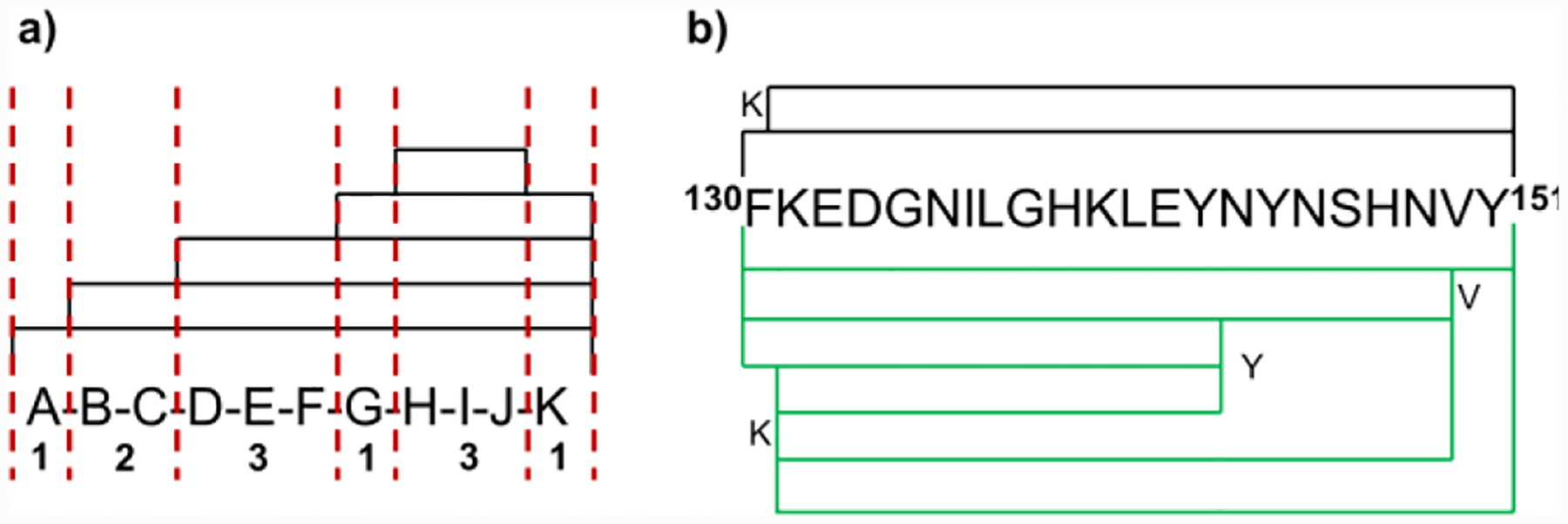
(a) An example of segment-length determination. The observed peptides are covered by black lines; the red dashed lines illustrate the ability to pinpoint residues or small sequences (segments) based on peptide overlap. Six segments are present in this example; their lengths are indicated. (b) Digestion coverage of representative peptic peptide 130–151 from GFP in the presence (green lines) and absence (black lines) of DMF.
| equ 1 |
We then analyzed the degree of pepsin specificity in each condition. As shown in Figure S4, the most frequent cleavage events occur at the C-terminus of Phe, Leu, Tyr, and Trp, in accord with specificity rules.13 Our analysis shows that the use of organic solvents changes the digestion pattern presumably by facilitating protein unfolding and exposure of cleavage sites without changing the specificity of pepsin.
Finally, we mapped the observed pepsin cleavage sites onto 3D structures of GFP, Scn, and RBP4. Many of the sites that are cleaved more frequently in the presence of organic solvent are located on the β-barrels (Figure 3). This suggests that organic solvents facilitate unfolding of the β-barrel, presumably by breaking H-bonds and disrupting hydrophobic interactions between the β-sheets, thus increasing exposure of these sites to pepsin.
Figure 3.
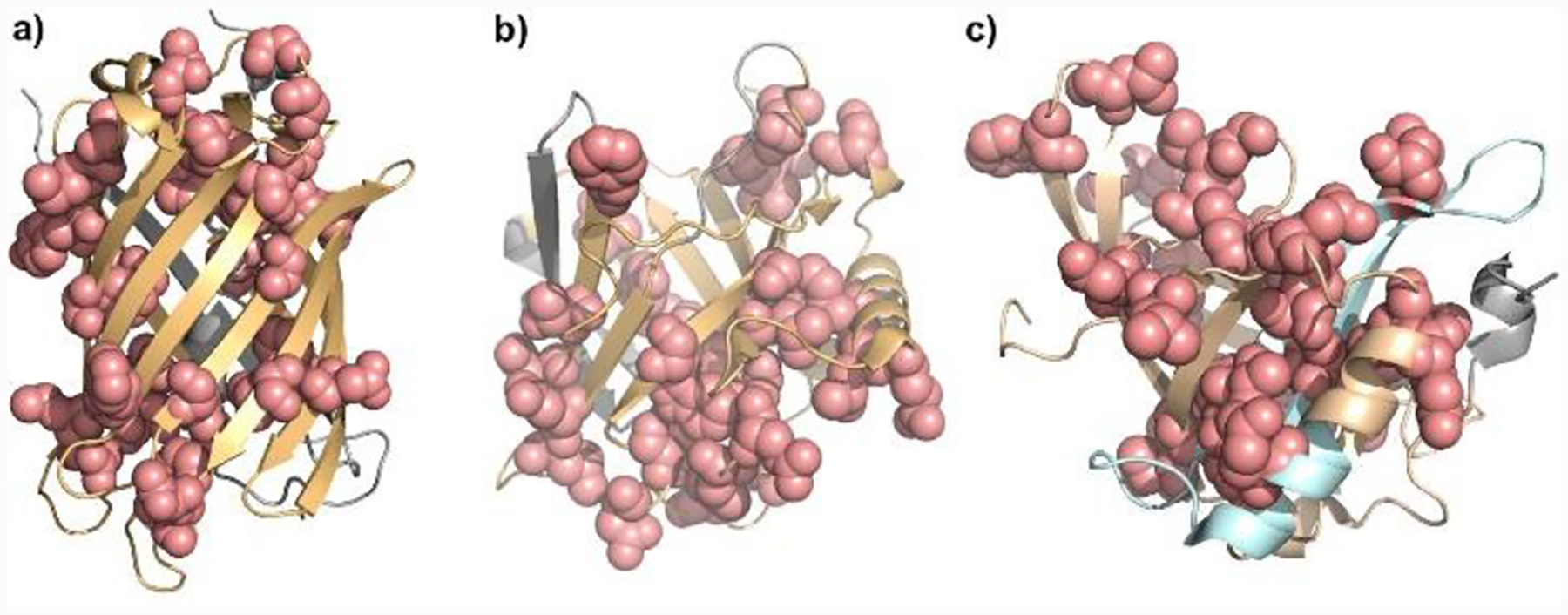
3D structure of a) GFP (PDB: 1GFL), b) Scn (PDB: 4K19), c) RBP4 (PDB: 5NU7). Sites that are cleaved more frequently in the presence of organic solvent (light pink) are located on beta-barrels peptides (light orange). Sites that are cleaved only in the presence of organic solvent are shown in light blue.
Siderocalin (Scn).
Given the digestion efficiency increase for GFP in organic solvents, we tested a more challenging β-barrel protein, siderocalin (Scn, ~25 kDa), from the lipocalin family. It consists of an eight-stranded, continuously H-bonded, antiparallel ß-barrel that encapsulates a ligand-binding region. The binding of siderophores or of iron and catecholate cofactors to this site is key to the antibiotic activity of Scn.31–34
The differences in conformation between ferric-siderophore bound- and unbound-Scn are revealed by HDX-MS. Previous literature reported that Scn maintains its secondary structure at a pH range of 2–7, consistent with the high stability of this and other ß-barrel proteins.31 Although the coverage is an acceptable 92% under normal quench (Figure S5), a large amount of the Scn remains undigested (Figure S6a and b; the broad chromatographic peak at 5.28 min shows undigested protein, a possible source of bias in HDX.27)
Scn digestion improved dramatically (Figure S5) in each organic solvent tested except H2P and i-PrOH. When Scn was quenched in 4 M guanidine in MeCN, we saw higher signal peaks for peptic peptides than when quenched in 4 M guanidine alone. More importantly, the chromatogram peak at ~5.3 min, representing the undigested protein, decreased significantly (Figure S6c). Compared with normal quench, which gave 44 unique peptides and 50 spectra (Figure S5, Figure S7a), the addition of acetonitrile gave 65 unique peptides, 104 MS/MS scans, and 98% coverage (Figure S5, Figure S7b).
Although the average Scn peptide length, at ~14 amino-acids, does not increase with organic solvents, there is ~18% increase in the number of segments, leading to ~18% decrease of the segment length (38 segments under conditions without organic solvent vs. 45 segments for conditions with MeCN) (Figure S7). These short and overlapped peptides are invaluable for improving spatial resolution.
We then analyzed the degree of specificity of pepsin on Scn under various conditions (Figure S8). The digestion pattern is similar to that of GFP. Mapping the sites that are cleaved more frequently in the presence of organic solvent shows that most are from six of the eight rigid β-strands and one is from the α-helix region (Figure 3b).
Retinol-binding Protein 4 (RBP4).
To test whether the use of organic solvents interferes with locating a ligand binding site, we performed HDX on bound vs. unbound RBP4 (21 kDa MW) with nonaqueous solvents in the quench buffer.
Retinol-binding protein 1 (RBP1) (15 kDa) and RBP4 belong to a protein family that binds hydrophobic ligands including retinol (vitamin A alcohol). RBP1 consists of an “up-and-down” β-strand motif giving a continuous, nearly cylindrical barrel. Several groups35,36 used HDX/ESI MS to reveal the overall deuterium incorporation of RBP1 at the intact protein level. Careri and coworkers37 measured HDX by MALDI and ESI at the peptide level to provide localized exchange information, but they saw only 13 and 17 peptic peptides with ~78% coverage for bound and unbound, respectively.
To test the ability of organic solvents to improve digestion and binding-site identification, we first identified which solvent provided the best coverage of RBP4. The data show that 4 M guanidine with 50% methanol affords the best results with 97% coverage, 47 unique peptides, and 77 spectra (Figure S9, Figure S10). We found that: (1) the peptide length remains at ~13 residues under all solvent conditions (Figure S2); (2) the segment length of the protein with methanol decreases by 30% compared to no organic solvent; (3) the addition of methanol does not change the digestion specificity (Figure S11).
Consistent with the results for GFP and Scn digestion, the sites with increased cleavage in the presence of organic solvent are on the β-barrels of RBP4. Furthermore, peptides 116–134 and 138–152, which cannot be found in aqueous quench, are produced using non-aqueous solvents (Figure 3, Figure S10). The improvement of digestion efficiency increases the spatial resolution, allowing more specific identification of protein regions whose HDX rates change from retinol-free to retinol-bound states.
Next, we measured the differential HDX kinetics of retinol-free and retinol-bound RBP4 at 37 °C from 10 s to 4 h. The HDX data are consistent with earlier NMR and X-ray crystallography results;38 that is, Leu35, Phe36, Ala43, Met88, Tyr90, Gln98, His104, and Phe135 interact with the retinol (V61 was also identified as interacting with retinol by NMR and X-ray crystallography, but not by HDX). There are six regions that show HDX differences (peptides 24–36, 40–44, 72–77, 77–96, 97–108, 126–136, Figure 4). To ensure that the deuterium-uptake differences between bound and unbound in these regions are significant, we compared the cumulative changes with 3 times the propagation error (Figure S12). If the changes exceed 3 times the error, HDX at that region is significantly different between bound and unbound.
Figure 4.
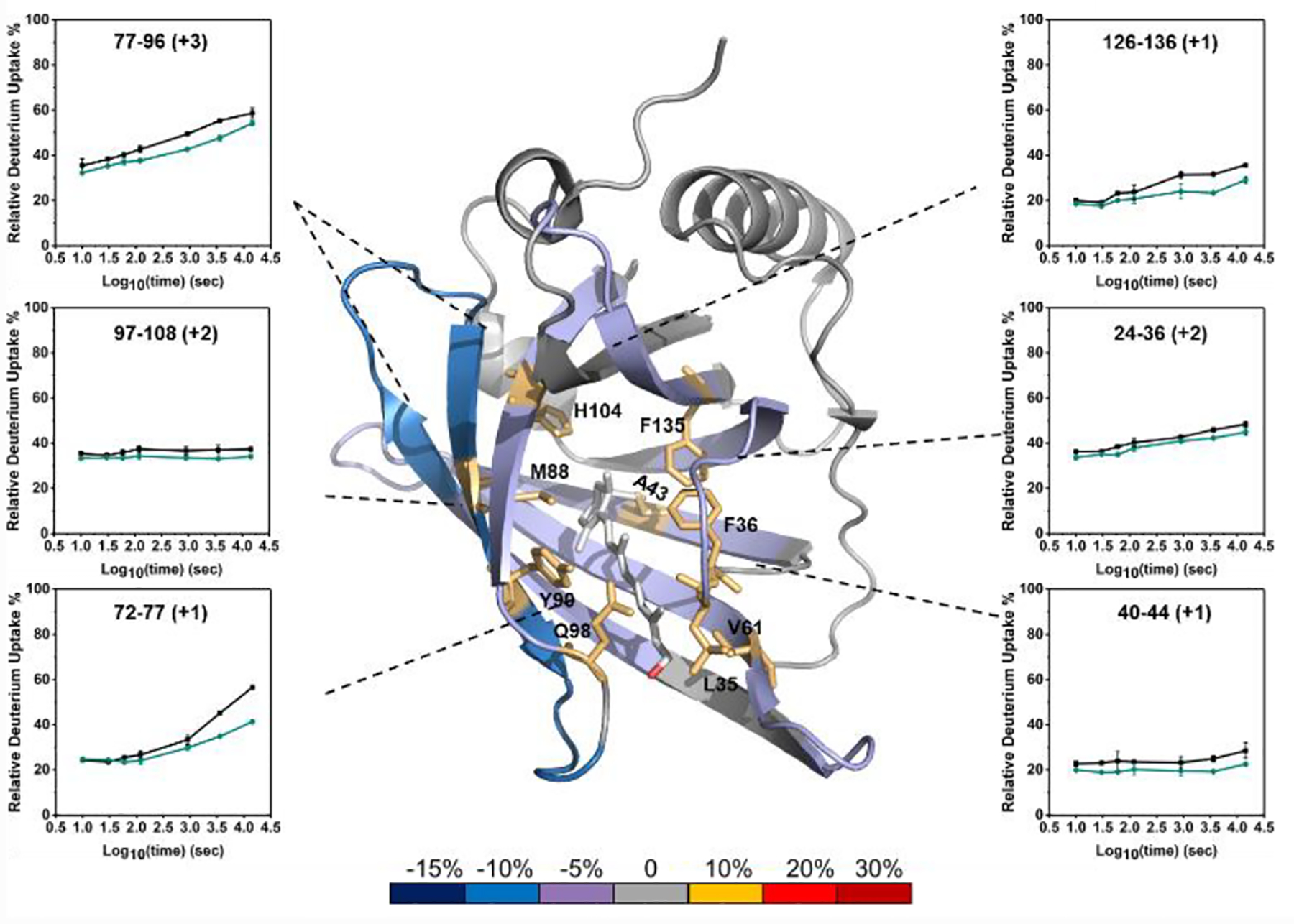
Average percentage differences in deuterium uptake with MeOH addition between retinol-free and retinol-bound RBP4 mapped onto the X-ray crystal structure (PDB 5NU7) of retinol-bound RBP4. Insets: retinol-free (black) and retinol-bound RBP4 (dark-green).
We detected eight of the nine known binding residues as well as two additional regions that likely undergo remote conformational changes upon binding. Peptides 24–36, 40–44, 77–96, 97–108, 126–136 contain the eight identified critical binding residues. The respective kinetic curves and accompanying statistical analysis demonstrate that these regions are more protected in the bound state. Peptide 72–77 shows distinctive HDX differences between the two states, but this region does not contain a known binding residue. We conclude the change in this region is due to a remote conformational change upon binding. The kinetic curves of peptide 24–36 (containing binding residues Leu35 and Phe36) do not show convincing HDX differences, but the cumulative changes are more than 3 times the propagation error. Thus, this region gains measurable protection upon ligand binding. We did not detect any measurable HDX differences for region 47–67 containing binding residue V61. That residue is part of an 11 amino-acid long peptide, and the changes at V61 may be small and/or diluted by no changes elsewhere in that region. Alternatively, it may be that the ligand binding interaction does not change the conformation of the adjoining peptide backbone. Overall, the results show that MeOH improves mapping without interfering with the ability of HDX to report most of the binding regions between retinol and RBP4.
Compared to previous work with older technology36 that covered 75% of the protein sequence, we obtained nearly full coverage of RBP4, and revealed two additional regions that show significant deuterium uptake difference (peptides 40–44, 126–136). Further, the additional MS/MS increased the confidence of peptide identification, and the ~ 4 times greater number of unique overlapping peptides gave shorter segment-length and improved spatial resolution (e.g., our identification of peptide 72–77 is more precise than the previously reported 70–84).
In conclusion, the use of organic solvents to facilitate digestion of highly stable β-barrel proteins provides increased protein coverage (~95%), twice the number of unique peptides, and three times the number of MS/MS spectra, mostly from the rigid β-barrels. These improvements allow a less ambiguous determination of regions where HDX differs between ligand-free and ligand-bound states. The results are consistent with the organic solvent weakening H-bonds and disrupting hydrophobic interactions within the protein. Unfortunately, the optimum solvent is protein-dependent, as demonstrated by DMF, acetonitrile, and methanol being the best of the tested denaturation solvents for GFP, Scn, and RBP4, respectively. The successful footprinting of RBP4 suggests that this approach does not compromise the ability to detect conformational changes. The next tests of the improved platform are challenging β-barrel membrane proteins and other proteins resistant to proteolysis on the HDX platform.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by the National Institutes of Health, Grants No P41GM103422 and R24GM136766 to M.L.G., 1R01GM131008 to M.L.G. and Weikai Li, and RO1DK099534 and RO1DK111930 to J.P.H. We also thank Dr Ming Cheng and Dr Nicole Wagner for discussion.
Footnotes
Supporting Information
The Supporting Information of experiment detail, scan statistics of each protein digested with various organic solvents, HDX mapping of each protein, chromatograms and mass spectra of the Scn peptides generated when quenched with and without organic solvent is available free of charge on the ACS Publications website.
REFERENCES
- [1].Engen J Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal. Chem 2009, 81, 7870–7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Percya A; Reyb M; Burnsb K; Schriemer D Probing protein interactions with hydrogen/deuterium exchange and mass spectrometry—A review. Anal. Chim. Acta 2012, 721, 6, 7–21. [DOI] [PubMed] [Google Scholar]
- [3].Giansanti P; Tsiatsiani L; Low T; Heck A Six alternative proteases for mass spectrometry–based proteomics beyond trypsin. Nat. Protoc 2016, 11, 993–1006. [DOI] [PubMed] [Google Scholar]
- [4].Serpa J; Parker C; Petrotchenko E; Han J; Pan J; Borchersa C Mass spectrometry-based structural proteomics. Eur. J. Mass Spectrom 2012, 18, 251–267. [DOI] [PubMed] [Google Scholar]
- [5].Chalmers M; Busby S; Pascal B; West G; Griffin P Differential hydrogen/deuterium exchange mass spectrometry analysis of protein–ligand interactions. Expert Rev. Proteomics 2011, 8, 43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mendoza V; Vachet R Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom. Rev 2009, 28, 785–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sinz A Cross-linking/mass spectrometry for studying protein structures and protein-protein interactions: Where are we now and where should we go from here? Angew. Chem. Int. Ed 2018, 57, 6390–6396. [DOI] [PubMed] [Google Scholar]
- [8].Jessani N; Cravatt B The development and application of methods for activity-based protein profiling. Curr. Opin. Chem. Biol 2004, 8, 54–59. [DOI] [PubMed] [Google Scholar]
- [9].Schopper S; Kahraman A; Leuenberger P; Feng Y; Piazza I; Muller O; Boersema P; Picotti P Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nat. Protoc 2017, 12, 2391–2410. [DOI] [PubMed] [Google Scholar]
- [10].Guo X; Trudgian D; Lemoff A; Yadavalli S; Mirzaei H Confetti: A multiprotease map of the HeLa proteome for comprehensive proteomics. Mol. Cell Proteomics 2014, 13,1573–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Meyer J; Kim S; Maltby D; Ghassemian M; Bandeira N; Komives E Expanding proteome coverage with orthogonal-specificity α-lytic proteases. Mol. Cell Proteomics 2014, 13, 823–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Waas M; Bhattacharya S; Chuppa S; Wu X; Jensen D; Omasits U; Wollscheid B; Volkman B; Noon K; Gundry R Combine and conquer: surfactants, solvents, and chaotropes for robust mass spectrometry based analyses of membrane proteins. Anal. Chem 2014, 86, 1551–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lόpez-Ferrer D; Petritis K; Robinson E; Hixson K; Tian Z; Lee J; Lee S; Tolić N; Weitz K; Belov M; Smith R; Paša-Tolić L Pressurized pepsin digestion in proteomics. Mol. Cell Proteomics 2011, 10, M110.001479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Busenlehner L; Armstrong R Insights into enzyme structure and dynamics elucidated by amide H/D exchange mass spectrometry. Arch. Biochem. Biophys 2005, 433, 34–46. [DOI] [PubMed] [Google Scholar]
- [15].Woods V; Hamuro Y High resolution, high-throughput amide deuterium exchange-mass spectrometry (DXMS) determination of protein binding site structure and dynamics: utility in pharmaceutical design. J. Cell Biochem. Suppl 2001, 37, 89–98. [DOI] [PubMed] [Google Scholar]
- [16].Cravello L; Lascoux D; Forest E Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Commun. Mass Spectrom 2003, 17, 2387–2393. [DOI] [PubMed] [Google Scholar]
- [17].Mazon H; Marcillat O; Forest E; Vial C Local dynamics measured by hydrogen/deuterium exchange and mass spectrometry of creatine kinase digested by two proteases. Biochimie. 2005, 87, 1101–1110. [DOI] [PubMed] [Google Scholar]
- [18].Zhang H; Kazazic S; Schaub T; Tipton J; Emmett M; Marshall A Enhanced digestion efficiency, peptide ionization efficiency, and sequence resolution for protein hydrogen/deuterium exchange monitored by fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem 2008, 80, 9034–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tsiatsiani L; Akeroyd M; Olsthoorn M; Heck A Aspergillus niger prolyl endoprotease for hydrogen−deuterium exchange mass spectrometry and protein structural studies. Anal. Chem 2017, 89, 7966–7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Trabjerg E; Jakobsen R; Mysling S; Christensen; S., Jørgensen, T.; Rand, K. Conformational analysis of large and highly disulfide-stabilized proteins by integrating online electrochemical reduction into an optimized H/D exchange mass spectrometry workflow. Anal. Chem 2015, 87, 8880–8888. [DOI] [PubMed] [Google Scholar]
- [21].Zhang X; Chien E; Chalmers M; Pascal B; Gatchalian J; Stevens R; Griffin P Dynamics of the ß (2)-adrenergic G-protein coupled receptor revealed by hydrogendeuterium exchange. Anal. Chem 2010, 82, 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jones L; Zhang H; Vidavsky I; Gross M Online, high-pressure digestion system for protein characterization by hydrogen/deuterium exchange and mass spectrometry. Anal. Chem 2010, 82, 1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Neurath H; Greenstein J; Putnam F; Erickson J The Chemistry of Protein Denaturation. Chem. Rev 1944, 34, 2157–265. [Google Scholar]
- [24].Slysz G; Schriemer D On-column digestion of proteins in aqueous-organic solvents. Rapid Commun. Mass Spectrom 2003,17, 1044–1050. [DOI] [PubMed] [Google Scholar]
- [25].Kauzmann W Some factors in the interpretation of protein denaturation. 1959, 14, 1–63. [DOI] [PubMed] [Google Scholar]
- [26].Valeja S; Emmett M; Marshall A Polar aprotic modifiers for chromatographic separation and back-exchange reduction for protein hydrogen/deuterium exchange monitored by fourier transform ion cyclotron resonance mass spectrometry. J. AM. Soc. Mass Spectrom 2012, 23, 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Guo C; Steinberg L; Cheng M; Song J, Henderson J Site-specific siderocalin binding to ferric and ferric-free enterobactin as revealed by mass spectrometry. ACS Chem. Biol 2020, 15, 1154–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].LaLonde J; Bernlohr D; Banaszak L The up-and-down beta-barrel proteins. FASEB J. 1994, 8, 1240–1247. [DOI] [PubMed] [Google Scholar]
- [29].Lim X; Chandramohan A; Lim X; Bag N; Sharma K; Wirawan M; Wohland T; Lok S; Anand G Conformational changes in intact dengue virus reveal serotype-specific expansion. Nat. Commun 2017, 8, 14339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Swaney D; Wenger C; Coon J Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J. Proteome Res 2010, 9, 1323–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Abergel R; Clifton M; Pizarro J; Warner J; Shuh D; Strong R; Raymond K The siderocalin/enterobactin interaction: A link between mammalian immunity and bacterial iron transport. J Am Chem Soc 2008, 130, 11524–11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Raymond K; Dertz E; Kim S Enterobactin: An archetype for microbial iron transport. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 3584–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shields-Cutler R; Crowley J; Hung C; Stapleton A; Aldrich C; Marschall J; Henderson J Human urinary composition controls antibacterial activity of siderocalin. J. Biol. Chem 2015, 290, 15949–15960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Clifton M; Rupert P; Hoette T; Raymondc K; Abergel R; Strong R Parsing the functional specificity of Siderocalin/Lipocalin 2/NGAL for siderophores and related small-molecule ligands. J. Struct. Biol, 2019, X, 100008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xiao H; Kaltashov I Indirect assessment of small hydrophobic ligand binding to a model protein using a combination of ESI MS and HDX/ESI MS. J. Am. Soc. Mass Spectrom 2003, 14, 506–515. [DOI] [PubMed] [Google Scholar]
- [36].Torta F; Elviri L; Careri M; Mangia A; Cavazzini D; Rossi G Mass spectrometry and hydrogen/deuterium exchange measurements of alcohol‐induced structural changes in cellular retinol‐binding protein type I. Rapid Commun. Mass Spectrom 2008, 22, 330–336. [DOI] [PubMed] [Google Scholar]
- [37].Careri M; Elviri L; Mangia A; Zagnoni I; Torta F; Cavazzini D; Rossi G Mass spectrometry techniques for detection of ligand-dependent changes in the conformational flexibility of cellular retinol-binding protein type I localized by hydrogen/deuterium exchange. Rapid Commun. Mass Spectrom 2006, 20, 1973–1980. [DOI] [PubMed] [Google Scholar]
- [38].Perducaa M; Nicolisb S; Mannuccic B; Gallianod M; Monaco H Human plasma retinol-binding protein (RBP4) is also a fatty acid-binding protein. Biochimica et Biophysica Acta, Molecular and Cell Biology of Lipids, 2018, 1863, 458–466. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.