

Abstract
Signaling proteins exemplified by calmodulin usually bind cooperatively to multiple ligands. Intermediate states and allosteric behavior are difficult to characterize. Here we extend a recently reported mass spectrometry (MS)-based method named LITPOMS (ligand titration, fast photochemical oxidation of proteins and mass spectrometry) that characterizes complex binding systems typically found as signaling proteins. As reported previously, calmodulin’s response to binding four Ca2+ can be determined by LITPOMS to reveal binding sites, binding order, and most importantly composite binding behavior. Modeling this behavior provides site-specific binding affinities. In this article, we dissect the composite, peptide-level conformational changes at several regions either by digestion with a different protease or by tandem MS of LITPOMS behavior at the amino-acid residue level. Such dissection greatly elevates spatial resolution and increases the confidence of binding order assignment. These complementary views of complex protein conformational change recapitulate the cumulative understanding via a single approach, providing new insights on poorly understood yet important allostery and underpin an approach applicable for exploring other signaling systems.
Graphical Abstract
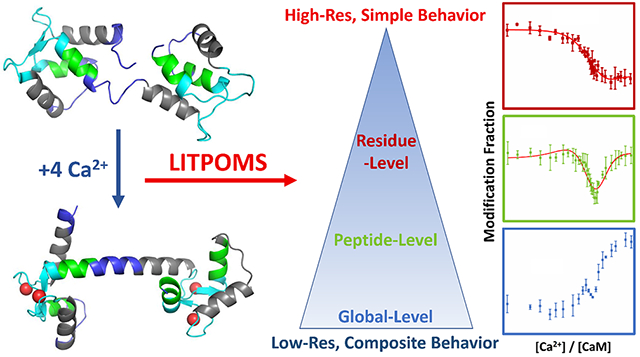
Cell signaling adapts to changing environments, requiring messengers that change their concentrations in response to local stimulation. Ca2+ signaling system includes hundreds of cellular proteins.1 Their Kd’s with Ca2+ range from nanomolar to millimolar while binding to multiple Ca2+ ions in a cooperative fashion, as exemplified by neuronal calcium sensor family,2 synaptotagmin,3 and calmodulin1–2, 4. The binding, chelation with negatively charged residues, alters the protein conformation and facilitates signal transduction by opening new binding regions.
The canonical Ca2+ chelating motif is an EF-hand, composed of a “helix-loop-helix” domain.5–6 During binding, Ca2+ binds with the negatively charged residues in the calcium loop, further triggering a structural change whereby the two helices alter their interhelical angles. As a result, the EF-hand binding transforms the protein from a closed to an open conformation during which the negatively charged chelating residues become less solvent-accessible and the two helices become more. Among all EF-hand-containing Ca2+ signaling proteins, calmodulin (CaM) is the archetype owing to its ubiquitous presence in eukaryotic systems and its role as a signaling protein.7–8 Within its 148-amino-acid sequence reside four canonical EF-hands, two each in the N-terminal and C-terminal lobes.9 Macroscopically, CaM binds with four Ca2+ ions with μM affinity, changing its high order structure.1, 10 Upon binding, CaM opens to a ‘dumbbell-shaped’ conformation, exposing hydrophobic residues that interact with targets to cause signaling and regulation.6, 11
Understanding these structural transformations is vital and has required decades of effort and the use of several approaches. High resolution structures for the calcium-free and calcium-bound states of CaM, as resolved by NMR12 and X-ray crystallography13, respectively, underpin additional research on binding, dynamics, and allostery 10, 14. Park et al.15 used microfluidics to track the kinetics of CaM conformational change with calcium at low resolution. Isothermal titration calorimetry affords binding thermodynamics.16–17 Protein engineering of the calcium loops leads to their intrinsic binding affinities.18 Although the complex binding scheme of CaM and Ca2+ have been revealed by many approaches, important features including allostery and binding orders remain unclear.
Although lower in resolution, MS uses smaller sample amounts, allowing studies with the native protein and eliminating the need for isotopic enrichment, mutation, or addition of a reporter.19–21 To follow the conformational changes of proteins upon metal binding, investigators have developed and applied synchrotron-based hydroxyl radical labeling,22–23 hydrogen/deuterium exchange (HDX)-based titration including SUPREX (stability of unpurified proteins from rates of H/D exchange)21 and PLIMSTEX (protein ligand interactions by MS, titration and H/D exchange)19, footprinting of specific residues by slow labeling reagents such as glycine ethyl ester (GEE)24 and benzhydrazide (BHD)25 that targets solvent-accessible carboxyl groups on aspartic (D) and glutamic acids (E). They all contribute to the understanding of complex metal binding proteins.
Moreover, recent advances of MS fragmentation methods such as electron transfer dissociation26–27 and electron capture dissociation28 allow following HDX at amino acid residue resolution. Fast photochemical oxidation of protein (FPOP) was also extensively demonstrated to extend the spatial resolution to single residue level.29–31 These advances can be extended to study the composite behavior of metal binding proteins. Recently, we communicated a new MS-based approach, “ligand titration, fast photochemical oxidation of proteins and mass spectrometry (LITPOMS)” that resolves complex binding dynamics by means of a ligand titration.32–33 The titration is monitored by FPOP that labels irreversibly the solvent-accessible surface at sub-millisecond times,34–35 enabling a “snapshot” of the system state. LITPOMS, first applied to a 1:1 binding system, revealed binding sites, site-specific binding affinities and even critical binding residues.32
Because LITPOMS greatly elevates spatial resolution compared to optical approaches, we applied it to CaM-Ca2+ binding and found that it not only recapitulates the cumulative understanding via a single approach but also reveals the composite binding dynamics as a function of [Ca2+] and the accompanying allosteric changes. Encouraged by these preliminary findings,33 we now report, in this full article, an expansion of that study whereby we increase the spatial resolution, even to the amino-acid level, using different protease digestion and MS/MS, describe additional data processing, and provide new insights on the results. The outcomes indicate that LITPOMS can characterize complex binding dynamics via a single approach, providing insights that are not easily accessible from any single or simple combination of conventional methods.
EXPERIMENTAL
2.1. Materials
Calcium-free bovine calmodulin was purchased from Ocean Biologics (Seattle, WA, USA). Catalase (from bovine liver) was from Sigma-Aldrich (St. Louis, MO, USA). Concentrations for each were determined by UV absorption with a Thermo Scientific NanoDrop OneC (Thermo Scientific, Waltham, MA, USA). Trizma base, urea, potassium chloride (anhydrous, ≥ 99.0%), calcium chloride (≥ 99.0%), L-methionine (≥ 99.5%), L-histidine (≥ 99.0%), hydrogen peroxide solution (containing inhibitor, 30% wt. in H2O), hydrochloric acid (36.5% - 38%) were acquired from Sigma-Aldrich (St. Louis, MO, USA). Formic acid (LCMS Grade) was from CovaChem (Loves Park, IL, USA). Trypsin/Lys-C and chymotrypsin digestion kits were purchased from Promega (Madison, WI, USA). All these chemicals were used without further purification. Tris buffer solution (10 mM) was made by dissolving Trizma base in water. Hydrochloric acid solution was added to obtain a desired pH of 7.4 (measured with an Orion Star A211 pH meter, Thermo Scientific, Waltham, MA, USA). Potassium chloride was added to the tris buffer to give a concentration of 100 mM to ensure physiologically relevant ionic strength of the buffer solution.
2.2. LITPOMS
To start, a solution of calmodulin was diluted with Tris buffer. Calcium chloride solution was added stepwise in a titration. The mixtures were kept at room temperature for 45 min to ensure that the CaM and Ca2+ had equilibrated. The other components were added prior to the laser irradiation that formed the OH radicals.
To extract binding affinities, experiments were performed at protein concentration of 1 μM. Aliquots of 100 μL with a final concentration of [CaM] = 1 μM, [L-histidine] = 0.1 mM, [H2O2] = 3 mM, [Ca2+] = 0 – 60 μM (titrant) were prepared for laser irradiation. A mixture of 10 μL 7 mM L-methionine (in Tris buffer) and 0.3 μL 500 nM catalase (in Tris Buffer) was used as a quench for each sample aliquot. All samples were prepared in duplicate to assess precision, followed by FPOP labeling, where the sample was introduced through a capillary tubing (I.D. of 150 μm) by a syringe pump (Pump 11 Elite, Harvard Apparatus, Holliston, MA). The beam from a KrF excimer laser (EX50/250, GAM Laser, Orlando, FL) with wavelength of 248 nm and frequency of 7.2 Hz was introduced through a transparent window in the capillary tubing. The average laser energy was tuned to ~ 25 mJ/pulse as measured with a pyroelectric energy sensor (PE25-SH-V2, Ophir Optronics Solutions, North Logan, UT) The flow rate was 22-25 μL/min so that the laser spot width and the operating frequency ensured a 25% exclusion volume. The irradiated sample was collected at the end of capillary tubing in the quenching solution to ensure that there is no further oxidative labeling of the protein by residual radicals or left-over H2O2.
2.3. Protein Digestion
To measure the footprinting extent at the peptide level, samples were digested by using two different proteases, trypsin/Lys-C mixture and chymotrypsin.
For trypsin/Lys-C digestion, a total of 60 μL of each labeled protein solution was added to an Eppendorf tube. The solvent was evaporated with a SpeedVac (Thermo Scientific, Waltham, MA, USA), leaving a protein pellet at the bottom of the Eppendorf. Urea (10 μL of 8 M) was added to the top of the protein pellet to dissolve and denature the protein. The mixture was kept at room temperature for 30 min to ensure that the protein had been fully denatured. Tris buffer (90 μL of 10 mM) was then added to dilute the urea. Finally, 0.35 μL of 0.2 μg/μL trypsin/Lys-C (in H2O) was added to the solution, which was then kept at 37 °C overnight so that the protein can been fully digested. Similar procedures were adopted for chymotrypsin digestion, where a total of 30 μL labeled protein solution was used. After evaporating the solvent, 5 μL of 8 M urea was added to denature the protein, followed by addition of 44.6 μL of 10 mM Tris buffer and 0.35 μL of 0.1 μg/μL chymotrypsin solution (in H2O) for overnight incubation. Under both digestion conditions, formic acid was added to give 1% (by volume) to quench the proteolysis.
2.4. Data Acquisition and Analysis
A global-level (intact protein) response was determined with a Quadrupole Time-of-Flight instrument (Bruker MaXis, Billerica, MA) in the ESI mode coupled to an HPLC system (FLX. 10371, Leap Technologies, Morrisville, NC). A C-8 column (Agilent, Santa Clara, CA) was used for sample loading and separation.
Peptide-level responses were determined with a Thermo Scientific Q Executive Plus Orbitrap mass spectrometer coupled with a Dionex UltiMate 3000 RSLCnano UPLC system (Waltham, MA). A custom-packed C-18 column was for peptide separation. Fragmentation was in the HCD cell.
MS/MS data were identified by Byonic (Protein Metrics, San Carlos, CA) at peptide and amino-acid residue levels, and the data were processed with Byologic (Protein Metrics). Modification fractions at peptide level was calculated by integrating the areas of extracted ion chromatograms (example shown in Figure S1 in Supporting Information) for each individual species as:
Similarly, residue-level modification fractions were calculated by using the peak area of a specific modified species versus the overall peak area as:
To extract the site-specific binding affinities, all peptide level responses for eight tryptic peptides were co-fitted by using a previously developed algorithm19–20 implemented by Mathcad 14.0 (Parametric Technology Corporation, Needham, MA). To be specific, EF-1 and EF-2 were grouped into the N-terminal whereas EF-3 and EF-4 in the C-terminal lobes. A fitting model was constructed,10 where positive cooperativity in each lobe and independent binding in the two lobes were assumed. Each binding state (Apo-CaM, CaM-1-Ca2+, CaM-2-Ca2+…) had a distinct modification fraction. Under these assumptions, a high order partition polynomial was constructed as a function of calcium concentration. A search utilized the experimental data to determine an answer for the polynomial, after which binding affinities and peptide-specific fitting curves were obtained. Only eight tryptic peptides (1-13, 14-30, 31-37, 38-74, 76-90, 91-106, 107-126 and 127-148) were brought into the search owing to the limitation of the current searching algorithm. Red lines in other peptide (90-99, 125-138 and 139-145) and residue level (F16, F19, K21, M51, M71, M72/R74, F92, Y99, M109, M124, Y138, M144 and M145) LITPOMS responses were reconstructed from search results and agreed with the experimental data.
RESULTS
3.1. Global Level LITPOMS Response
Upon binding of CaM and Ca2+, the interhelical angles alter to facilitate a ‘closed’ to ‘open’ transition for each EF-hand. CaM responds by assuming a ‘dumbbell’ conformation from a protected form.6, 11 This structural transition leads to an increase of the overall solvent accessible surface area (SASA), which is clearly captured as an increase in modification fraction as a function of calcium concentration at the global or whole protein level (Figure S2 in SI). We observed a decrease in modification fraction at [Ca2+]/[CaM] between 2 to 5, suggesting one or more steps of Ca2+ binding induced protection, thus motivating digestion and analysis at the peptide and amino-acid levels. Formula for global level modification fraction calculation and examples of global level MS of CaM are provided in Supporting Information (Figure S2).
3.2. Four Classes of LITPOMS Behaviors on Trypsin Digested Calmodulin
As reported previously, we can identify four distinct classes of conformational change among eight tryptic peptides.33 (A sequence coverage map, Figure 1, shows 99% tryptic sequence coverage was achieved by using MS/MS incorporating a precursor-ion inclusion list.) The irreversible labeling of FPOP 35–37 allowed extension of LITPOMS to the amino-acid level for some residues and improved the spatial resolution. Here, we present a new, highly resolved view of the conformational changes induced by Ca2+ binding.
Figure 1.

Sequence coverage map of the CaM protein sequence. Black lines under the sequence indicate tryptic peptides whereas red lines designate chymotryptic peptides. The four EF-hands are super-scored by red (EF-1), magenta (EF-2), olive (EF-3) and navy (EF-4). Residues shaded in green, cyan and dark yellow represent the E helix, calcium loop and F helix of each EF-hand, respectively. Residues boxed in red are resolved by MS/MS analysis.
A protein region exhibiting Class I behavior undergoes a loss of protection upon binding (with Ca2+). Peptide 76-90, which covers the central linker together with part of helix-E of EF-3 (Figure S3a in SI), opens as calmodulin transitions from a compact conformation to a ‘dumbbell-shape’ upon Ca2+binding. LITPOMS captures this change and that for region 107-126 (Figure S3b in SI), which covers the linker between EF-3 and EF-4. To increase further the spatial resolution, we analyzed the amino-acid residue data (results in Figure 2a and 2b) to show that residues M109, in the F-helix of EF-3, and M124, in the E-helix of EF-4, become more exposed upon binding with Ca2+.
Figure 2.

Residue-level LITPOMS responses for residue (a) M109 and (b) M124. Data points represent averages of two runs, and error bars are standard deviations. Red solid lines are re-constructed based on fitting results of eight tryptic peptides by an algorithm reported previously (See Experimental).
Class II behavior epitomizes locations where the modification fraction remains relatively constant as Ca2+ interacts with CaM. Peptide 1-13 (Figure S3c in SI), part of the N-terminus of CaM, and peptide 31-37 (Figure S3d in SI), covering the F-helix of EF-1, represent two regions not involved in binding or remote conformational changes. Because the only FPOP-modified residue in peptide 31-37 is M36, the behavior (Figure S3d in SI) represents the M36 residue level LITPOMS response.
Class III behavior represents classic binding (peptide 14-30, Figure 3a). This region remains relatively unprotected at early stage of the titration (until [Ca2+]:[CaM] ≅ 2). As the titration proceeds, peptides representing binding regions becomes protected owing to Ca2+ binding, consistent with a decrease in modification fraction at [Ca2+]:[CaM] of 2–10. At the later titration stages, region 14-30 stays protected because it is saturated with Ca2+.
Figure 3.
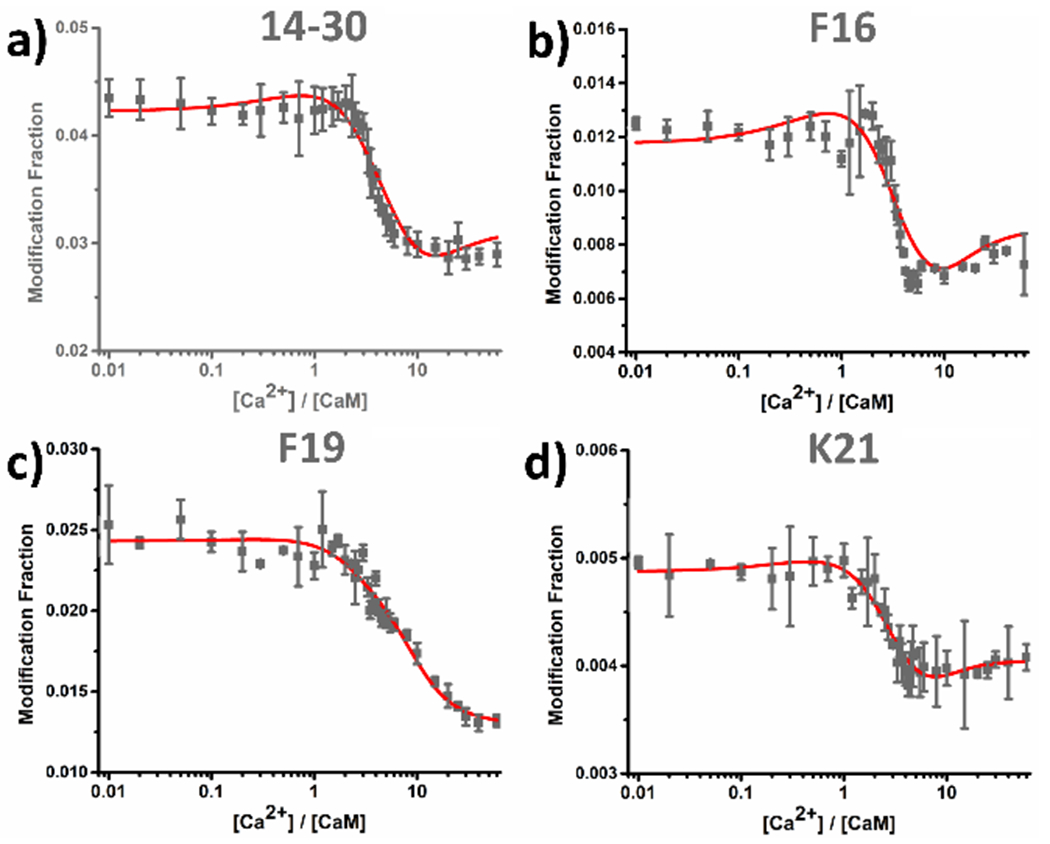
(a) Class III peptide-level LITPOMS responses as illustrated by peptide 14-30. Data adopted from ref. 33. Copyright 2019 American Chemical Society. (b) Residue-level LITPOMS response for residue F16. (c) Residue-level LITPOMS response for residue F19. (d) Residue-level LITPOMS response for residue K21. Red solid line in (a) is from the fitting (see Experimental) and the results from fitting were adopted to reconstruct the red solid lines in (b), (c) and (d). Data points are averages of two runs; error bars are standard deviations (see SI for a discussion of error).
Magnifying the view to the residue level, we resolved three amino acid residues: F16 (Figure 3b) and F19 (Figure 3c) reside on the E-helix of EF-1 whereas K21 (Figure 3d) occupies the calcium loop of EF-1. LITPOMS curves for these residues are similar to those for peptide 14-30, indicating that the Ca2+ loop together with part of the E-helix are involved in either the Ca2+ binding of EF-1 or conformational changes induced by binding. Although the resolved residues are not negatively charged or directly involved in binding, they reside in proximity with the actual binding residues, thus reporting binding-induced conformational changes. Such detailed spatial resolution is difficult to achieve, usually only by an X-ray crystal determination.38 NMR can utilize changes of chemical shifts upon titrating with a ligand,39 but NMR is time and labor consuming and challenging to resolve high-resolution structure at each titration point. LITPOMS fills the gap by resolving complex binding of signaling proteins with amino-acid residue resolution.
Regions represented by peptides 38-74, 91-106 and 127-148 that cover the remaining three EF-hands behave differently than peptide 14-30 and are termed Class IV. The curves reveal composite behavior including a combination of binding and remote conformational change induced by binding, some of which is allostery.15, 40 Region 127-148, covering the calcium loop and F-helix of EF-4 (Figure 1, S4b), shows a gain in protection at the initial stage of the LITPOMS curve, followed by a loss of protection upon adding more Ca2+. The initial gain-of-protection originates from Ca2+ binding by EF-4, during which the loop in EF-4 chelates Ca2+, thus becoming more protected. Upon reaching saturation with calcium at [Ca2+]:[CaM] of ca. 1.5, the region, represented by peptide 127-148, becomes less protected. This loss in protection can be reasonably explained by a binding-induced conformational change of F-helix in the EF-4. Once the calcium loop becomes protected, the F-helix re-orientates to a more exposed configuration, preparing CaM to take a subsequent Ca2+. Such cooperativity and allosteric behavior will be discussed in greater detail in later sections.
Among all regions of CaM, three peptides representing 38-74, 91-106 and 127-148, show composite behavior.33 Composite LITPOMS curves are highly informative, revealing CaM binding dynamics as a function of [Ca2+] at peptide-level resolution and providing new information. Composite-behavior information can be enhanced with additional protein digestion. To dissect the composite behavior, we chose chymotrypsin to give different cleavage sites from those of trypsin and yield peptides 125-138 and 139-145 (Figure 4c and 4d, respectively), which cover the majority of calcium loop of EF-4 and the F-helix of EF-4 (see Figure 1), respectively. Indeed, region 125-138 exhibits classical binding behavior, where the modification fraction starts to decrease in a similar manner as tryptic peptide 127-148. At later stages of the titration where [Ca2+]:[CaM] > 1.5, the modification fraction of chymotryptic peptide 125-138 stays relatively constant until the very end of the titration, unlike the increasing modification for peptide 127-148. Peptide 125-138 must contain the critical binding region of EF-4, consistent with canonical calcium-loop binding behavior. On the other hand, the modification fraction of chymotryptic peptide 139-145 remains stable until [Ca2+]:[CaM] is ~ 1.5, after which a clear loss of protection occurs (Figure 4d). Because region 139-145 includes the F-helix of EF-4, the loss of protection is expected.
Figure 4.
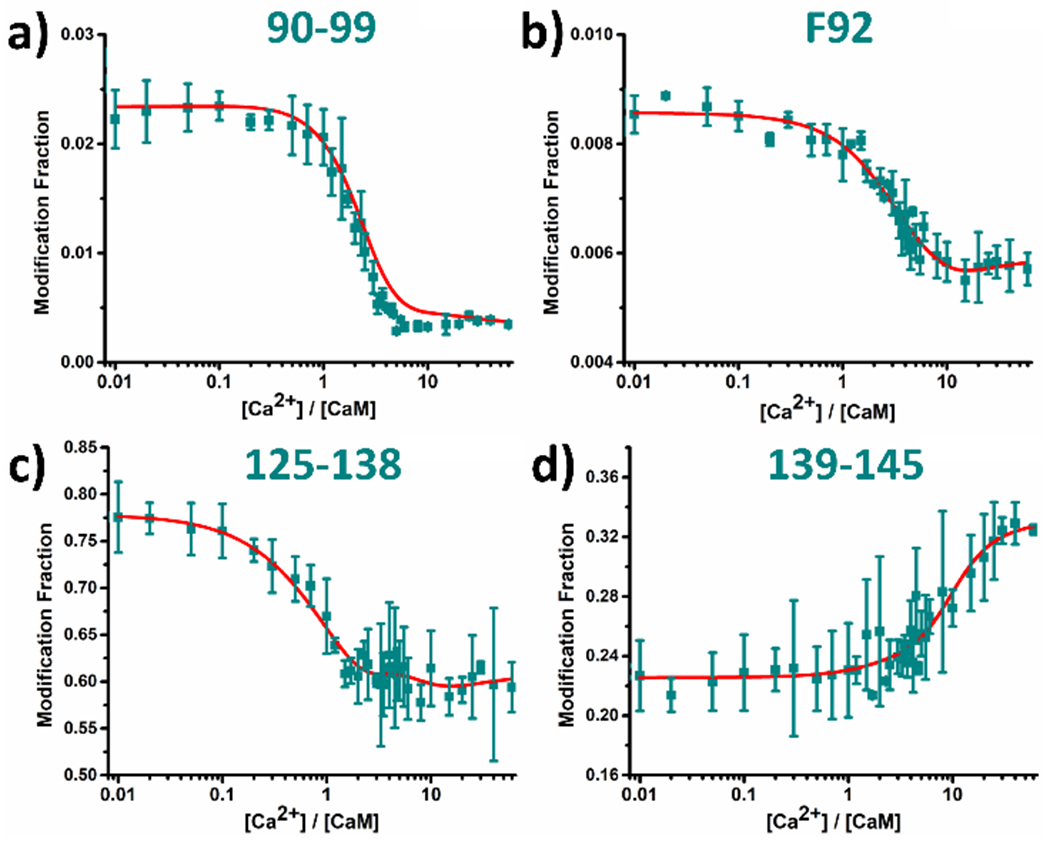
LITPOMS responses for (a) chymotryptic peptide 90-99, (b) residue F92, (c) chymotryptic peptide 125-138 and (d) chymotryptic peptide 139-145. Data points represent average of two runs, and error bars are standard deviations. Red solid lines are re-constructed based on fitting results of eight tryptic peptides by an algorithm reported previously (See Experimental). The relatively large standard deviations in (c) and (d) are discussed in SI.
Like peptide 127-148, tryptic peptide 91-106 falls into Class IV (Figure S4a). Its modification fraction stays relatively constant until [Ca2+]:[CaM] is ~ 0.7, after which significant binding-induced protection occurs. An increase of modification happens at later stages of the titration owing to a binding-induced conformational change that facilitates binding of other EF-hands. Residue Y99 on the calcium loop of EF-3 shows a composite LITPOMS curve (Figure S4c in SI). The chymotryptic peptide 90-99 better illustrates the actual binding event (Figure 4a). A decrease in modification occurs between [Ca2+]:[CaM] of 0.7 and 5.0, resembling the binding of the calcium loop in EF-3. Once again, the use of a combination of different proteases dissects the composite LITPOMS curve and elevates the spatial resolution. Two residues for peptide 91-106 (F92 in Figure 4b, Y99 in Figure S4c) and three residues in peptide 127-148 (Y138, M144 and M145 in Figure S5d–f) demonstrate the improved spatial resolution.
Although also assigned to Class IV, tryptic peptide 38-74 behaves differently than the other two. It loses protection at the initial stage of titration, seen by an increase followed by a decrease in modification at [Ca2+]:[CaM] of 1.7, suggesting binding. When [Ca2+] is > 5 equivalents of CaM, peptide 38-74 becomes less protected. Given that the peptide covers the central linker between EF-1 and EF-2 and the entire EF-2 (Figure 1), its complex behavior is expected. Unfortunately, we found no chymotryptic peptide in this region. The LITPOMS responses for selected residues, however, do capture some of the features (Figure 5b–d). Residues M71 (Figure 5c) and M72/R74 (Figure 5d) lose protection simultaneously at [Ca2+]:[CaM] of ~ 1.5. These residues sit in the F-helix of EF-2, altering its conformation from ‘closed’ to ‘open’ and facilitate the structural transformation of CaM upon Ca2+ binding. Residue M51, however, loses protection initially followed by a significant gain and remains protected till the end of titration (Figure 5b). The Loss of protection is consistent with a binding-induced conformational change whereas the gain corresponds to the actual binding of Ca2+ at EF-2. M51, in proximity to the calcium loop of EF-2, shows protection consistent with that of the loop (see SI for discussion of Met footprinting).
Figure 5.

Class IV peptide-level LITPOMS responses as illustrated for peptide 38-74 (a). Three residues were successfully resolved and corresponding residue-level LITPOMS responses were plotted as (b) residue F16, (c) residue F19 and (d) K21. The red solid lines in (a) are from fitting (see Experimental), and the fitting results were used to reconstruct data in (b), (c) and (d). Data are averages of two runs, and error bars are standard deviations. Data in (a) are adopted from ref 33. Copyright 2019 American Chemical Society.
DISCUSSION
4.1. Determining the CaM Allostery
To access the allosteric behavior of CaM upon Ca2+binding, we take a combined view of the Ca2+-dependent behavior of peptides covering four EF-hands. Upon interacting with Ca2+, EF-4 first responds by taking-up a Ca2+ as seen by an increase in protection for tryptic peptide 127-148 (Figure S4b) and chymotryptic peptide 125-138 (Figure 4c). The protection then decreases at [Ca2+]:[CaM] of ~ 1.5, when the two flexible linkers represented by peptides 76-90 (Figure S3a) and 107-126 (Figure S3b) become more exposed. Moreover, modification of region 38-74 keeps increasing until [Ca2+]:[CaM] ~ 1.7 (Figure 5a), where EF-4 finishes binding Ca2+ (Figure 4c and Figure S4b) and binding at EF-3 is still underway (Figure 4a and S4a). Importantly, loss of protection for peptides 38-74, 76-90 and 107-126 occurs during the Ca2+ binding of EF-4 and EF-3. Peptide 38-74 covers EF-2 (Figure 1), and its behavior reveals the CaM allostery upon Ca binding. The LITPOMS results suggest that Ca2+ binding at EF-3 and EF-4 triggers a conformational change for EF-2 (and other parts of the protein), preparing it for the subsequent Ca2+ binding.
As the titration proceeds to [Ca2+]:[CaM] ~ 5.0, the majority of CaM has opened as captured by LITPOMS for peptides 38-74, 76-90, 91-106, 107-126, and 127-148. Meanwhile, EF-1 (covered by tryptic peptide 14-30) is starting to bind Ca2+ (Figure 3a). The curves also reveal the cooperative nature of four EF-hands in CaM binding to Ca2+. At later titration stages when [Ca2+]:[CaM] is > 15, all LITPOMS responses become nearly constant, indicating saturation with Ca2+. Tracking LITPOMS responses as a function of [Ca2+] informs us on the Ca2+ binding dynamics at the peptide and even at amino-acid levels (M51 in Figure 5b), which cannot be achieved with low-to-medium resolution approaches (e.g., proteolytic footprinting 40 and fluorescence15).
4.2. Defining Calcium Binding Order
There has been a long-standing debate on the order of Ca2+ binding to CaM. Its four EF-hands exist in two lobes, EF-1 and EF-2 in the N-terminal lobe and EF-3 and EF-4 in the C-terminal lobe. Early tyrosine fluorescence and terbium luminescent studies suggested a binding first at the N-terminal lobe then at the C-terminal lobe.41 Later, a binding assay with a chromophoric chelator gives the four binding affinities; the six-fold higher calcium affinity at the C-terminal than at the N-terminal lobe suggests the C-terminal lobe binds first.10 Subsequently, the results of mutational studies and tryptophan fluorescence allowed the binding order to be adjusted as EF-3 > EF-4 > EF-1 > EF-2.42 Using the same four-site mutants, isothermal titration calorimetry confirmed that binding occurs first at the C-terminal lobe.17 So far, this is the accepted binding order, although other mutations may cause adjustments. Low resolution fluorescence, however, limits the confidence of these conclusions. Ye et al.18 grafted four calcium loops individually onto a scaffold protein and concluded that the intrinsic binding affinity of four loops rank as EF-1 > EF-3 > EF-2 > EF-4. It is, however, risky to assign calcium binding orders by comparing their intrinsic binding affinities because the four EF-hands often work cooperatively in Ca2+ binding. Thus, the calcium binding order remains unsettled.
LITPOMS offers a novel approach to address this important question. Ca2+ binding increases protection (decreases reactivity) at a binding site owing to spatial hinderance. Thus, comparing the onset point of the LITPOMS decay as a function of [Ca2+] affords the binding order. Although previously we suggested a binding order of EF-4 > EF-3 > EF-2 > EF-1 by ranking four tryptic peptides,33 the order for EF-3 and EF-2 is competitive and thus ambiguous owing to entanglement of LITPOMS decay curves for regions 91-106 and 38-74. The entanglement is due to broad coverages of these peptides, where part of a region opens and part tightens. The order was eventually assigned by the early onset of EF-3.
Elevated spatial resolution for each EF-hand can give more accuracy. We chose peptides 125-138, 90-99 and 14-30 that cover primarily calcium loops of EF-4, EF-3 and EF-1, respectively (no peptides or resolved residues for calcium loop of EF-2 were captured). Fortunately, residue M51 is close to the calcium loop of EF-2 and exhibits binding-induced protection, and we chose it for comparison.
To illustrate better calcium binding orders for each EF-hand, plots of the decay region for four candidates were normalized to 1 and plotted together (Figure 6). Clearly, the first binding occurs at EF-4 (peptide 125-138) whereas EF-1 binds last (peptide 14-30). Curves for EF-3 and EF-2 are now well separated at the early stage of the titration, clearly indicating that EF-3 binds prior to EF-2. With improvement of LITPOMS, the overall binding order can be confidently assigned as EF-4 > EF-3 > EF-2 > EF-1.
Figure 6.

Normalized (to 1) peptide-level LITPOMS responses for selected peptides and residue showing increases in protection (decreases in modification) for peptides 125-138, peptide 90-99, residue M51 and peptide 14-30 representing EF hands 4 – 1 of CaM, respectively. The insert shows expanded decreases in modification for peptide 90-99, residue M51 and peptide 14-30. Data are averages of two independent runs, and error bars are standard deviations and are normalized accordingly. The relatively large standard deviations for EF-3 (90-99) are discussed in SI.
4.3. Measuring Site-specific Binding Affinity
In addition to assessing binding orders, another LITPOMS advance is the determination of site-specific binding affinities. This is particularly important for signaling proteins whose binding stoichiometry is usually greater than 1, and the affinities for different binding sites may differ significantly owing to allostery.1 Current approaches are mainly based on optical methods, requiring specific fluorophores for signal enhancement but giving limited spatial resolution.10 LITPOMS, however, delivers site-specific binding affinities with mid-to-high structural resolution. We previously communicated that affinities for EF-2 and 3 agree with literature values within a factor of 1.6, and EF-1 and 4 are within a factor of 20.10, 33 Binding within each lobe (N-terminal lobe of EF-1 and EF-2, C-terminal lobe of EF-3 and EF-4) is positively cooperative, consistent with previous conclusions.
In this work, the binding affinities obtained from fitting the eight original tryptic peptides were adopted to reconstruct simulation curves for the newly obtained LITPOMS curves of chymotryptic peptides (Figure 4a, 4c and 4d) and residues (Figures 2a–b, 3b–d, 4b, 5b–d, S4c–f). The current fitting algorithm cannot fit all available LITPOMS responses; thus, the newly obtained LITPOMS responses were not part of the fitting. The reconstructed simulations for these newly obtained LITPOMS responses agree well with the data, indicating confidence in the binding affinities and the LITPOMS approach. It is possible that the values determined here are more accurate than those in the literature.
4.4. Dissecting the Composite Behavior
In general, spatial resolution can be elevated by tracking smaller peptides, obtainable by either a combination of multiple enzymes to cleave differently the protein or by fragmentation in MS2 accompanied by improved LC separation.
Class IV tryptic peptides 38-74, 91-106 and 127-148 behave compositely because the peptides are long and represent regions undergoing multiple conformational changes (e.g., peptide 127-148 covers the calcium loop and F-helix of EF-4). These two structural motifs behave differently in the Ca2+ titration, resulting in a composite curve. By digesting with a different protease, however, the two motifs can be cleaved into two peptides that cover each region and reveal single behavior.
Furthermore, given the irreversible labeling by FPOP, the resolution can be enhanced by tracking at the amino-acid residue level.35–37 The response of peptide 38-74 can be dissected by three residue-level LITPOMS curves, each showing simpler behavior. Residue M51, however, is either involved with or near an allosteric site, as seen from its “up and down” modification behavior. The promise of increased spatial resolution motivates the development of new FPOP reagents that provide complementary labeling to OH radicals,43–45 even of residues directly involved in binding.
4.5. Probing Local Conformational Changes in Calcium Loop
The elevated spatial resolution allows differentiating direct binding sites from those not directly involved in binding, even in a calcium loop. Residue Y138 sits in the calcium loop of EF-4. Binding of Ca2+ tightens the loop by chelating with negatively charged residues and protecting them. Tyrosine, however, is not involved in Ca2+ chelation and becomes less protected upon binding owing to a structural transition of the calcium loop whereby the SASA of the unbound at 23 Å2 becomes 50 Å2 in the bound form. Residue-level LITPOMS successfully captures these transitions, as demonstrated in Figure S4d.
4.6. Advancing LITPOMS over Other Approaches
Another hydroxyl radical-based protein footprinting approach utilizing synchrotron radiation was previously applied to a Ca2+ binding protein. With an X-ray exposure time of 80 ms, Chance and coworkers followed structural transitions of gelsolin upon Ca2+ titration and successfully identified a three-state activation process.22–23 Moreover, three classes of residues/peptides were identified, which become more and less protected while titrating with Ca2+. Although conceptually similar, the synchrotron footprinting does not reveal composite behavior or the relative timing of change from one region to another. This may be because the timescale for footprinting by LITPOMS is shorter by two orders of magnitude.
Quantitative measurements of protein ligand interactions by HDX were pioneered in the SUPREX21 and PLIMSTEX19 methods, both of which were brought to regional-level spatial resolution by protease digestion.46 HDX detects changes of SASA by probing the changes in hydrogen bonding. Protein conformational changes, however, do not necessarily associate with re-organization of its hydrogen bonding pattern. In an HDX study that compares Ca2+-free and Ca2+-bound CaM, kinetic curves successfully reveal the binding regions of Ca2+ in CaM, whose deuterium uptake decreases upon Ca2+ binding. HDX fails to probe regions that open-up, whose SASA increases upon Ca2+ binding.47 Ca2+-binding tightens the calcium loop in an EF-hand, which strengthens the hydrogen bonds in the backbone. Structural opening of E and F helices, however, is due to changes in their interhelical angles that is not easily captured by HDX. FPOP is more sensitive to subtle changes in the SASA of amino acid side chains, making it suitable to probe such structural transitions.
In addition to HDX and free radicals, slower reacting reagents such as GEE24 and BHD25 also shed light on this field. Negative-charged side chains of D and E in the calcium loop of EF-hands are the actual binding motifs that chelate with the positive-charged Ca2+. GEE48 and BHD25 footprinting of CaM reveal decreases in modification extents for D and E that located in the calcium loop upon Ca2+ binding, but these changes have not yet been followed in a titration format and composite behavior not seen.
A binding site that has a fast off-rate can show noticeable convergence in footprinting if the labeling time is slow. The result is a reduction in the difference in labeling between bound and unbound states. Calcium off-rate constants for interacting with CaM range from 10 to 4000 s−1,15, 49–50 making the off-rate timescale for Ca2+ binding fast, in the range of 200 μs to 100 ms. For HDX, which labels the protein over exchange times of minutes to hours, problems in convergence occur.19 Similar slow timescales apply to GEE labeling, where the reaction times for protein and reagent can be up to an hour.24 Other spectroscopic methods such as fluorescence and circular dichroism, even though their detection timescales are often shorter than the ligand off-rates, cannot afford the detailed structural information afforded by LITPOMS.10 LITPOMS, for the first time, combines high resolution with an ultra-short timescale for footprinting. The labeling time of hydroxyl radical-based FPOP is in the sub-millisecond range, which is comparable or even shorter than the calcium off-rates, 34–35 allowing structural details to be revealed at high spatial resolution without diminishing the footprint between bound and unbound states.
CONCLUSION
LITPOMS can provide unprecedented structural details in a single approach to determine Ca2+ binding sites, Ca2+ binding orders, site-specific binding affinities, allostery, and cooperativity. That LITPOMS can accomplish this is seen by its ability to reveal the composite behavior of CaM upon binding with Ca2+ and to uncover allostery and cooperativity accompanying binding. Moreover, the composite behavior can be further dissected by using a different protease digestion or a residue-level MS/MS analysis.
High-resolution approaches such as NMR and X-ray crystallography can resolve protein structures in solution and solid state, respectively. Most of these resolved structures, however, are either in their ligand-unbound or ligand-bound states, and the requirement to look at many intermediate states places high demands on time and sample quantity. For signaling proteins, whose binding usually involves multiple ligands in a cooperative fashion,1–2 understanding the intermediate states between unbound and bound is difficult38 and perhaps impossible for large proteins that don’t crystallize. Understanding binding intermediates with mid-to-high resolution makes it possible to explore complex allostery during binding, which is challenging for optical approaches but feasible for LITPOMS, as demonstrated in the current study. Moreover, by tailoring labeling conditions51 or utilizing new labeling reagents43–45, it should be possible to monitor selectively or nearly all amino-acid residues, enabling the direct and complete spatial tracking of ligand binding.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH NIGMS Grant 2P41GM103422 and 1S10OD016298-01A1 (to MLG.). Authors are grateful to Ms. Mengru Mira Zhang and Dr. Jagat Adhikari for helpful discussions and Protein Metrics for software.
Footnotes
The authors declare no competing financial interests.
Supporting Information
Detailed example of modification fraction calculation, discussion of error measurements, global-level LITPOMS response and sample spectra, the remaining peptide- & residue-level LITPOMS curves and discussion of Met footprinting are provided in Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- 1.Clapham DE, Calcium Signaling. Cell 2007, 131 (6), 1047–1058. [DOI] [PubMed] [Google Scholar]
- 2.Burgoyne RD, Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nature Reviews Neuroscience 2007, 8, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geppert M; Goda Y; Hammer RE; Li C; Rosahl TW; Stevens CF; Südhof TC, Synaptotagmin I: A major Ca2+ sensor for transmitter release at a central synapse. Cell 1994, 79 (4), 717–727. [DOI] [PubMed] [Google Scholar]
- 4.Hoeflich KP; Ikura M, Calmodulin in Action: Diversity in Target Recognition and Activation Mechanisms. Cell 2002, 108 (6), 739–742. [DOI] [PubMed] [Google Scholar]
- 5.S Nakayama a.; Kretsinger RH, Evolution of the EF-Hand Family of Proteins. Annual Review of Biophysics and Biomolecular Structure 1994, 23 (1), 473–507. [DOI] [PubMed] [Google Scholar]
- 6.Lewit-Bentley A; Réty S, EF-hand calcium-binding proteins. Current Opinion in Structural Biology 2000, 10 (6), 637–643. [DOI] [PubMed] [Google Scholar]
- 7.Cheung W, Calmodulin plays a pivotal role in cellular regulation. Science 1980, 207 (4426), 19–27. [DOI] [PubMed] [Google Scholar]
- 8.Means AR; Dedman JR, Calmodulin—an intracellular calcium receptor. Nature 1980, 285, 73. [DOI] [PubMed] [Google Scholar]
- 9.Watterson DM; Sharief F; Vanaman TC, The complete amino acid sequence of the Ca2+-dependent modulator protein (calmodulin) of bovine brain. Journal of Biological Chemistry 1980, 255 (3), 962–975. [PubMed] [Google Scholar]
- 10.Linse S; Helmersson A; Forsén S, Calcium binding to calmodulin and its globular domains. Journal of Biological Chemistry 1991, 266 (13), 8050–8054. [PubMed] [Google Scholar]
- 11.Zhang M; Tanaka T; Ikura M, Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nature Structural Biology 1995, 2, 758. [DOI] [PubMed] [Google Scholar]
- 12.Kuboniwa H; Tjandra N; Grzesiek S; Ren H; Klee CB; Bax A, Solution structure of calcium-free calmodulin. Nature Structural Biology 1995, 2, 768. [DOI] [PubMed] [Google Scholar]
- 13.Rajagopal Chattopadhyayal WEM, Anthony R Means, Florante A. Quiocho, Calmodulin Structure Refined at l.7 A Resolution. J. Mol. Biol 1992, 228, 1177–1192. [DOI] [PubMed] [Google Scholar]
- 14.Miyawaki A; Llopis J; Heim R; McCaffery JM; Adams JA; Ikura M; Tsien RY, Fluorescent indicators for Ca2+based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882. [DOI] [PubMed] [Google Scholar]
- 15.Park HY; Kim SA; Korlach J; Rhoades E; Kwok LW; Zipfel WR; Waxham MN; Webb WW; Pollack L, Conformational changes of calmodulin upon Ca2+ binding studied with a microfluidic mixer. Proceedings of the National Academy of Sciences 2008, 105 (2), 542–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henzl MT; Larson JD; Agah S, Estimation of parvalbumin Ca2+- and Mg2+-binding constants by global least-squares analysis of isothermal titration calorimetry data. Analytical Biochemistry 2003, 319 (2), 216–233. [DOI] [PubMed] [Google Scholar]
- 17.Gilli R; Lafitte D; Lopez C; Kilhoffer MC; Makarov A; Briand C; Haiech J, Thermodynamic Analysis of Calcium and Magnesium Binding to Calmodulin. Biochemistry 1998, 37 (16), 5450–5456. [DOI] [PubMed] [Google Scholar]
- 18.Ye Y; Lee H-W; Yang W; Shealy S; Yang JJ, Probing Site-Specific Calmodulin Calcium and Lanthanide Affinity by Grafting. Journal of the American Chemical Society 2005, 127 (11), 3743–3750. [DOI] [PubMed] [Google Scholar]
- 19.Zhu MM; Rempel DL; Du Z; Gross ML, Quantification of Protein–Ligand Interactions by Mass Spectrometry, Titration, and H/D Exchange: PLIMSTEX. Journal of the American Chemical Society 2003, 125 (18), 5252–5253. [DOI] [PubMed] [Google Scholar]
- 20.Zhu MM; Rempel DL; Gross ML, Modeling data from titration, amide H/D exchange, and mass spectrometry to obtain protein-ligand binding constants. J Am Soc Mass Spectrom 2004, 15 (3), 388–97. [DOI] [PubMed] [Google Scholar]
- 21.Powell KD; Ghaemmaghami S; Wang MZ; Ma L; Oas TG; Fitzgerald MC, A General Mass Spectrometry-Based Assay for the Quantitation of Protein–Ligand Binding Interactions in Solution. Journal of the American Chemical Society 2002, 124 (35), 10256–10257. [DOI] [PubMed] [Google Scholar]
- 22.Kiselar JG; Janmey PA; Almo SC; Chance MR, Visualizing the Ca2+-dependent activation of gelsolin by using synchrotron footprinting. Proceedings of the National Academy of Sciences 2003, 100 (7), 3942–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiselar JG; Janmey PA; Almo SC; Chance MR, Structural Analysis of Gelsolin Using Synchrotron Protein Footprinting. Molecular & Cellular Proteomics 2003, 2 (10), 1120–1132. [DOI] [PubMed] [Google Scholar]
- 24.Wen J; Zhang H; Gross ML; Blankenship RE, Membrane orientation of the FMO antenna protein from Chlorobaculum tepidum as determined by mass spectrometry-based footprinting. Proceedings of the National Academy of Sciences 2009, 106 (15), 6134–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo C; Cheng M; Gross ML, Protein-Metal-Ion Interactions Studied by Mass Spectrometry-Based Footprinting with Isotope-Encoded Benzhydrazide. Analytical Chemistry 2019, 91 (2), 1416–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zehl M; Rand KD; Jensen ON; Jørgensen TJD, Electron Transfer Dissociation Facilitates the Measurement of Deuterium Incorporation into Selectively Labeled Peptides with Single Residue Resolution. Journal of the American Chemical Society 2008, 130 (51), 17453–17459. [DOI] [PubMed] [Google Scholar]
- 27.Huang RY-C; Krystek SR Jr; Felix N; Graziano RF; Srinivasan M; Pashine A; Chen G In Hydrogen/deuterium exchange mass spectrometry and computational modeling reveal a discontinuous epitope of an antibody/TL1A Interaction, MAbs, Taylor & Francis: 2018; pp 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rand KD; Adams CM; Zubarev RA; Jørgensen TJD, Electron Capture Dissociation Proceeds with a Low Degree of Intramolecular Migration of Peptide Amide Hydrogens. Journal of the American Chemical Society 2008, 130 (4), 1341–1349. [DOI] [PubMed] [Google Scholar]
- 29.Li KS; Rempel DL; Gross ML, Conformational-Sensitive Fast Photochemical Oxidation of Proteins and Mass Spectrometry Characterize Amyloid Beta 1–42 Aggregation. Journal of the American Chemical Society 2016, 138 (37), 12090–12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson DT; Di Stefano LH; Jones LM, Fast photochemical oxidation of proteins (FPOP): A powerful mass spectrometry–based structural proteomics tool. Journal of Biological Chemistry 2019, 294 (32), 11969–11979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X; Grant OC; Ito K; Wallace A; Wang S; Zhao P; Wells L; Lu S; Woods RJ; Sharp JS, Structural Analysis of the Glycosylated Intact HIV-1 gp120–b12 Antibody Complex Using Hydroxyl Radical Protein Footprinting. Biochemistry 2017, 56 (7), 957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu XR; Zhang MM; Rempel DL; Gross ML, Protein-Ligand Interaction by Ligand Titration, Fast Photochemical Oxidation of Proteins and Mass Spectrometry: LITPOMS. Journal of The American Society for Mass Spectrometry 2019, 30 (2), 213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu XR; Zhang MM; Rempel DL; Gross ML, A Single Approach Reveals the Composite Conformational Changes, Order of Binding, and Affinities for Calcium Binding to Calmodulin. Analytical Chemistry 2019, 91 (9), 5508–5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gau BC; Sharp JS; Rempel DL; Gross ML, Fast Photochemical Oxidation of Protein Footprints Faster than Protein Unfolding. Analytical Chemistry 2009, 81 (16), 6563–6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hambly DM; Gross ML, Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom 2005, 16 (12), 2057–63. [DOI] [PubMed] [Google Scholar]
- 36.Li KS; Shi L; Gross ML, Mass Spectrometry-Based Fast Photochemical Oxidation of Proteins (FPOP) for Higher Order Structure Characterization. Accounts of Chemical Research 2018, 51 (3), 736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang B; Cheng M; Rempel D; Gross ML, Implementing fast photochemical oxidation of proteins (FPOP) as a footprinting approach to solve diverse problems in structural biology. Methods 2018, 144, 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slade DJ; Fang P; Dreyton CJ; Zhang Y; Fuhrmann J; Rempel D; Bax BD; Coonrod SA; Lewis HD; Guo M; Gross ML; Thompson PR, Protein Arginine Deiminase 2 Binds Calcium in an Ordered Fashion: Implications for Inhibitor Design. ACS Chemical Biology 2015, 10 (4), 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fielding L, NMR methods for the determination of protein–ligand dissociation constants. Progress in Nuclear Magnetic Resonance Spectroscopy 2007, 51 (4), 219–242. [Google Scholar]
- 40.Sorensen BR; Shea MA, Interactions between Domains of Apo Calmodulin Alter Calcium Binding and Stability. Biochemistry 1998, 37 (12), 4244–4253. [DOI] [PubMed] [Google Scholar]
- 41.Kilhoffer M-C; Demaille JG; Gerard D, Terbium as luminescent probe of calmodulin calcium-binding sites. FEBS Letters 1980, 116 (2), 269–272. [DOI] [PubMed] [Google Scholar]
- 42.Kilhoffer MC; Kubina M; Travers F; Haiech J, Use of engineered proteins with internal tryptophan reporter groups and perturbation techniques to probe the mechanism of ligand-protein interactions: investigation of the mechanism of calcium binding to calmodulin. Biochemistry 1992, 31 (34), 8098–8106. [DOI] [PubMed] [Google Scholar]
- 43.Cheng M; Zhang B; Cui W; Gross ML, Laser-Initiated Radical Trifluoromethylation of Peptides and Proteins: Application to Mass-Spectrometry-Based Protein Footprinting. Angewandte Chemie International Edition 2017, 56 (45), 14007–14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang MM; Rempel DL; Gross ML, A Fast Photochemical Oxidation of Proteins (FPOP) platform for free-radical reactions: the carbonate radical anion with peptides and proteins. Free Radical Biology and Medicine 2019, 131, 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang B; Rempel DL; Gross ML, Protein Footprinting by Carbenes on a Fast Photochemical Oxidation of Proteins (FPOP) Platform. Journal of The American Society for Mass Spectrometry 2016, 27 (3), 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang H; Rempel DL; Giblin D; Frieden C; Gross ML, Peptide-Level Interactions between Proteins and Small-Molecule Drug Candidates by Two Hydrogen–Deuterium Exchange MS-Based Methods: The Example of Apolipoprotein E3. Analytical Chemistry 2017, 89 (20), 10687–10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sperry JB; Huang RYC; Zhu MM; Rempel DL; Gross ML, Hydrophobic peptides affect binding of calmodulin and Ca2+ as explored by H/D amide exchange and mass spectrometry. International Journal of Mass Spectrometry 2011, 302 (1), 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H; Wen J; Huang RYC; Blankenship RE; Gross ML, Mass spectrometry-based carboxyl footprinting of proteins: Method evaluation. International Journal of Mass Spectrometry 2012, 312, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.ANDERSSON T; DRAKENBERG T; FORSÉN S; THULIN E, Characterization of the Ca2+ Binding Sites of Calmodulin from Bovine Testis Using 43Ca and 113Cd NMR. European Journal of Biochemistry 1982, 126 (3), 501–505. [DOI] [PubMed] [Google Scholar]
- 50.Bayley P; Ahlstrőm P; Martin SR; Forsen S, The kinetics of calcium binding to calmodulin: Quin 2 and ANS stopped-flow fluorescence studies. Biochemical and Biophysical Research Communications 1984, 120 (1), 185–191. [DOI] [PubMed] [Google Scholar]
- 51.Liu XR; Zhang MM; Zhang B; Rempel DL; Gross ML, Hydroxyl-Radical Reaction Pathways for the Fast Photochemical Oxidation of Proteins Platform As Revealed by 18O Isotopic Labeling. Analytical Chemistry 2019, 91 (14), 9238–9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.