

Abstract
Chemical approaches, in combination with mass-spectrometry analysis, have emerged as powerful tools to enable a broad range of biological applications. This approach is called structural proteomics. The key to success is a biocompatible reagent that modifies a protein without affecting its high-order structure. Fluorine, well-known to exert profound effects on the physical and chemical properties of reagents, should have an impact on structural proteomics. Here, we describe several fluorine-containing reagents that can be applied in structural proteomics. We organize their applications around four MS-based techniques: a) affinity labeling b) activity-based protein profiling (ABPP) c) protein footprinting, and d) protein cross-linking. Our aim is an overview of the research, development, and application of fluorine-containing reagents in protein structural studies.
Keywords: mass spectrometry, affinity-based protein profiling, affinity labeling, protein crosslinking, protein footprinting
Graphical Abstract
Combined with mass spectrometry, chemical labelling has become a powerful approach in structural proteomics. Fluorine substitution enables development of a wide range of biocompatible reagents for interrogating high-order structure and functions of proteins. This Mini review focuses on the application of fluorine-containing reagents that are playing a role at the forefront of structural proteomics.
1. Introduction
The term proteomics, proposed by Marc Wilkins in 1996, denotes the “PROTein complement of a genOME.[1] Two transforming technologies are crucial to the advancement of proteomics: (1) strategies for peptide sequencing that use mass spectrometry (MS) coupled with “soft” electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI), and (2) improvement, miniaturization and automation of liquid chromatography (LC). Together, these technologies allow the rapid identification of peptides and enable not only proteomics, aimed at primary structure but also structural proteomics for higher order structure.
Chemical labeling (e.g. footprinting) of proteins is the foundation of structural proteomics permitting the investigation of biological phenomena both in vitro and even in vivo. With MS analysis, investigators can validate therapeutic targets, interrogate cellular process, probe solution structures and protein–ligand complexes, modulate their biophysical properties, understand disordered proteins, uncover hidden conformations, locate binding sites, and determine affinity. The ultimate design of these approaches must eschew perturbing protein function while working in solution or a complex cellular milieu where other components can interfere with the desired chemical reaction. These challenges have captured the imagination of chemists.
Fluorinated molecules, which occupy a niche in the specialty-material, pharmaceutical, and agrochemical areas, may also play a role in structural proteomics. Their properties depend on the strong electronegativity, high hydrophobicity, good thermal and chemical stability, and strength of the X─F bond [2] These properties enable fine-tuning molecular properties to meet challenges in structure determination.
In this review, we describe several important fluorinated species and their application in structural proteomics. Some reagents are photoactivable (e.g., diazirines, azides, trifluoromethyl radical) and electrophilic (e.g., sulfur (VI) fluoride, fluorophosphonate, fluoromethylketone). To emphasize opportunities for chemical design, we organize this review according to four applications using MS-based proteomic approaches: (1) affinity labeling, (2) activity-based protein profiling (ABPP), (3) protein footprinting, and (4) protein cross-linking. In lieu of an encyclopedic review, we chose papers that illustrate key developments and applications.
2. Applications of Affinity Labelling
The term “affinity labeling” was first coined in 1962 by Singer in his pioneering work on proteins, including affinity-labeling of antibodies.[3] The concept is to place a chemically reactive “handle” on a protein ligand and initiate covalent modification of a target protein upon specific-ligand recognition. Basically, ligand recognition affords a noncovalent ligand complex in which the probe concentration is high, enhancing covalent bond formation between the protein and the probe.
2.1. Fluorinated diazirine reagents
2.1.1. 3-Trifluoromethyl-3-phenyldiazirine Derivatives
Diazirine is the most commonly used photo-activatable group for photo-affinity labeling. The group activates upon irradiation with ~360 nm UV light to react by expelling N2. The resulting singlet carbene has a ~ ns half-life and rapidly inserts into O-H, N-H or C-H bonds of the target. Brunner and coworkers [4] developed the first fluorinated diazirines, the 3-trifluoromethyl-3-phenyldiazirines (TPDs), for photolabeling. The effectiveness of diazirines in affinity-labelling was recently reviewed.[5] As compared to other diazirines, TPDs offer better chemical and thermal stability (Fig.1a). The adjacent aryl and trifluoromethyl groups (−CF3) stabilize the diazo intermediate formed by UV-light irradiation.[6] The corresponding carbenes do not undergo a Wolff rearrangement, avoiding unintended labeling.[7] Further, as the maximal absorption is ~ 360 nm, photodamage to protein is minimal.
Figure 1.
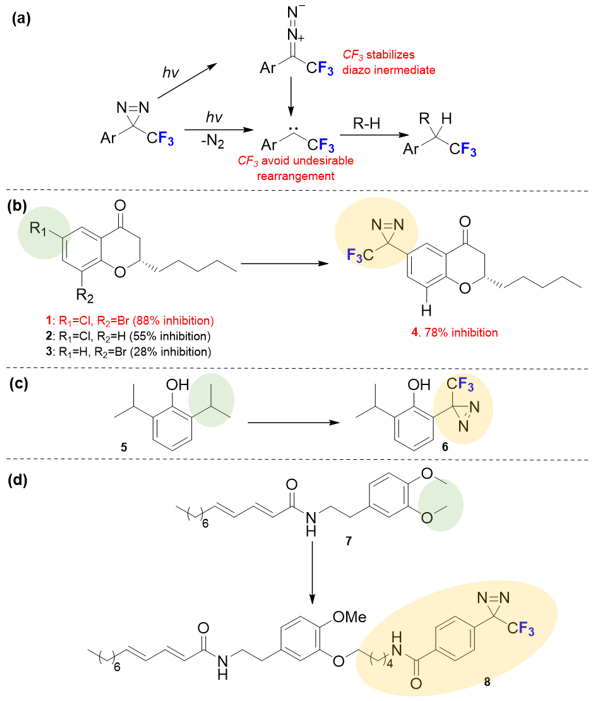
(a) Photochemical pathways for TPDs. The adjacent aryl and trifluoromethyl groups (−CF3) stabilize the diazo intermediate and avoid side rearrangement reaction. (b-d) Three strategies involving diazirines structural optimization for TPDs based probes.
One key for developing a potent TPD-based probe is to accommodate the reactive moiety or “warhead” in a pharmacophore. As TPDs have steric hinderance, their installation needs to be carefully considered. One way is to attach the CF3-diazirine group directly to an aromatic ring. Luthman and coworkers [8] developed TPD-based probes containing a chromon-4-one scaffold (Fig.1b). They found that the CF3-diazirine motif directly replaces chloride at R1, affording a more potent inhibitor (4 vs. 2). Further, even drug efficacy can profit from the CF3-diazirine substitution, as seen for compound 3 that has a deceased inhibitory effect when R1 = H.
Another way is to modulate the side chain involves the CF3-diazirine group. In 2013, Franks, Evers and coworkers [9] developed a new propofol analog by using CF3-diazirine as the “warhead”. Although the direct attachment of CF3-diazirine at the para or meta positions gives a compound that fails to mimic the parent compound, replacing one isopropyl side chain at the ortho position allowed efficient photolabeling while retaining drug efficacy (Fig.1c). Instead of directly modulating the pharmacophore, a linker can be used when a modified drug is challenged to undergo direct attachment of the CF3-diazirine to the pharmacophore (Fig.1d). Chicca and co-workers [10] synthesized probe 8 by replacing a methyl ether with an amide-derived TPD, showing that use of a linker can maintain efficacy. The design of TPD-based probes needs to be evaluated on a case-by-case basis. TPDs with aromatic rings may be analogs for pharmacophores that also contain aromatic rings to which a CF3-diazirine group can be attached. Even when the latter group is directly incorporated, structure-reactivity relationships (SAR) should be carefully considered.
2.1.2. Fluorinated Aliphatic Diazirines
Fluorine-containing compounds are often used as inhaled anesthetics. Fluorination increases anesthetic potency and preserves pharmacological properties. Fluorine-substituted compounds are often nonflammable, lipid-soluble, and resistant to metabolism, facilitating the induction of anesthesia, and permitting precise control of anesthetic concentrations.[11] To locate the protein binding sites for alkyl anesthetics, several investigators took advantage of fluorine properties and placed the F’s adjacent to a terminal diazirine group to enhance labeling efficiencies, as reviewed previously.[12]
An undesirable reaction for carbene is an intramolecular C-H insertion to generate alkenes (Fig.2a, pathway B). This undesired reaction is avoided if the two adjacent hydrogens are replaced by fluorine atoms or aromatic rings (Fig.2a, pathway A).[13]
Figure 2.

(a) The undesired intramolecular rearrangement of diazirines to an alkene (Pathway B) upon activation can be avoided when two adjacent hydrogens are replaced by fluorine (Pathway A). (b) Examples of anaesthetic photoaffinity probes containing terminal diazirine.
Eckenhoff and coworkers [14] developed an isoflurane analog, azi-isoflurane (Fig.2b), for photoaffinity labeling. This isoflurane analog shows even higher potency in tadpoles than the parent isoflurane, suggesting that drug efficacy can be maintained in photoaffinity labeling, and the location of binding is relevant for the parent compound. Azithane and azisevoflurane are additional examples of photolabels (Fig.2b).[15]
2.2. Fluorinated Azide Reagents
Azides are the most common precursors to deliver nitrenes in photolabeling studies. Aryl azides predominate in photolabeling compared to alkyl azides, which have poor stability. One of the drawbacks of aryl azides, however, is the incipient singlet nitrene undergoes rearrangement to a ketenimine that reacts only with nucleophiles and not with ubiquitous C-H bonds (Fig. 3).[16] This undesired rearrangement reduces labeling efficiency.
Figure 3.

The photochemical reaction of fluorinated phenyl azide. The substitution of fluorine at the aromatic ring can retard the undesired side rearrangement reaction to form a ketenimine.
Fluorinated aryl azides overcome this limitation because electron-withdrawing fluorine substituents favor bond insertion for an aryl nitrene and impair ring expansion.[17] Keana and coworkers[18] confirmed that the C-H or N-H insertions (Fig.3a) are the main pathways for fluorinated aryl azides and that ring expansion (Fig.3b) is retarded. Platz and coworkers [19] reported the C─H insertion of fluorinated aryl nitrenes is almost quantitative, and fluorine substituents when located at the two ortho positions relative to the azide group are critical to retarding ring expansion and stabilizing the singlet nitrene. Based on these advantageous photochemical properties, Peng and coworkers [20] developed an array of fluorinated phenyl azides as probes to examine lipid-protein interactions. The analysis was by 18F-NMR, but the approach may made more general and sensitive when using MS for analysis.
2.3. Sulfur (VI) Fluoride Exchange (SuFEx) Reaction
Although sulfonyl fluoride (SF) (Fig.4) can be traced back 100 years in Germany for applications in dyes,[21] its application has been revitalized as sulfur (VI) fluoride exchange (SuFEx) reagents by Sharpless and coworkers.[22] These exchange reactions were reviewed[23] and may be emerging as the next click chemistry in chemical biology. SuFEx chemistry includes several important reagents, SF, sulfonimidoyl fluorides, fluorosulfates, and sulfamoyl fluoride, which show specific reactivity to several amino acids (e.g., Tyr, Ser, Thr, His, Lys and Cys). SF has at least four features:[22] 1) resistance to reduction, (2) thermodynamic stability, (3) nearly exclusive reactivity at sulfur, and (4) strong H bonding to F. Sulfonimidoyl fluorides, an imino sibling of SF, possess similar click properties but with an additional “handle” at nitrogen for tuning its chemical properties. Compared to SF, fluorosulfates and the analogous sulfamoyl fluorides are even less reactive.
Figure 4.

(a) Mechanism of SuFEx reaction. The SuFEx reagents undergo a click-chemistry type reaction via nucleophilic substitution (b) Sulfur (VI) fluoride reagents.
The most intriguing property for SuFEx is its proximity-driven reactivity. That is, SuFEx reagents are reluctant to react with a protein unless a binding pocket provides proper solvation to activate the fluoride group.[24] Using computational modeling, Kelly and coworkers [25] found that Lys15 and Glu54 form a salt bridge in the transthyretin (TTR) pocket and provide a hydrogen bond that activates SF via a selective SN2 reaction of Lys15 (Fig.5). The pocket also catalyzes a side reaction, SF hydrolysis, if a nearby reactive residue is absent.
Figure 5.

Potential fate of SF probe in the TTR binding pocket. TTR and 1,3,4-oxadiazoles initially form noncovalent complexes. The functional groups on the sides chains that line the binding pocket can catalyze either hydrolysis (path a) or covalent adduction (path b) by stabilization of the fluoride leaving group. The figure is adapted from reference [25] with permission.
SF reacting with Lys forms a stable adduct. With serine, however, nature’s design of the protein catalytic site is so intricate that it also permits a cascade reaction. Liskamp and coworkers [26] incubated peptidic sulfonyl fluoride (PSF) with crystalline immunoproteasome (iCP) and found an unexpected mass shift (loss of a water) by MS analysis, rather than a covalent adduct of PSF. A cascade mechanism generates the crosslinking at the active site (Fig.6). Noteworthy is that this unexpected modification (a net loss of water) can be even observed in the X-ray crystal structure, revealing an intramolecular crosslink in the proteasomal subunit β5.
Figure 6.
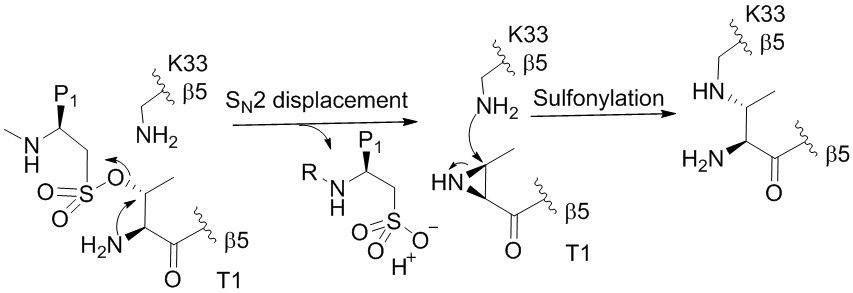
Mechanism of the PSF in the proteasomal active site of iCP to yield an aziridine. Therione (T1) was sulfonylated by PSF, followed by an intramolecular substitution by amine group of T1 to yield an aziridine. Aziridine attack on proximate Lys give a cross-linking product with a net loss of water.
3. Applications in Activity-based Protein Profiling (ABPP)
Activity-based protein profiling (ABPP), pioneered by Cravatt [27], is a chemical proteomic strategy for broadly profiling enzyme activities in native biological systems. Although ABPP is conceptually like traditional affinity labeling (in vitro), its aim is to characterize enzyme function on a proteome scale. The design has a probe molecule that contains a core unit, a linker, and a reporter tag (Fig.7a). The core unit includes a substrate (pharmacophore) that binds at enzymatic sites and a “warhead” that reacts with a characteristic amino-acid residue at catalytic site. Sometimes, a linker is included to modulate the space between pharmacophore and reporter tag. The goal is to capture a family of enzymes at the proteome level by chemically targeting and tagging them and finally enriching them for MS analysis.
Figure 7.

(a) A cartoon structure of an ABPP probe containing a core unit (triangle), a linker, and a reporter tag (oval). The core unit contains a pharmacophore and a “warhead” that enable covalent attachment of the probe to the complex. Linker is used to modulate the space between core unit and reporter tag, which facilitates downstream protein enrichment and analysis. (b) Common ABPP probe warheads containing fluorine.
There are three types of electrophilic and fluorine-containing ABPP reagents, based on their strategies to forge covalent bonds with enzymes: (1) fluorophosphonates (FP) (2) fluoromethylketone (FMK), and (3) SuFEx reagents (Fig.7b).
3.1. Fluorophosphonate in Serine Hydrolase Profiling
FPs are a class of electrophilic phosphonates that target serine hydrolases (SH) (see review).[28] FP use can be traced back to the early 1950s when diisopropyl fluorophosphate 9 (Fig.8) was first used for inhibition of SH.[29] Fluorophosphonate probes (e.g., 11) were introduced by Cravatt [30] who laid the foundation for further development. Later, that group [31] developed FP-peg-biotin 12 that generates similar “maximal coverage” as probes 11 for SH activity profiles, when incubated with both soluble and membrane proteomes.
Figure 8.

Structure of selected FP probes. FP derivatives are relatively stable but can undergo hydrolysis when subjected to moisture or to a basic solution, producing hydrofluoric acid.
Two factors make FPs excellent ABBP probes for SH. (1) FPs show broad reactivity for SH but little or no cross-reactivity with other enzymes (i.e., cysteine proteases are not labeled by a FP probe).[27] (2) In terms of PF chemical reactivity, they are stable for hours in buffer from pH 5 to 9, although in strongly basic conditions like 1% (w/v) NaOD, they are hydrolyzed. One PF 10 is even stable in the presence of 1 mM of dithiothreitol (DTT).[32]
3.2. Fluoromethylketones in Cysteine Hydrolase Profiling
Although FPs show reactivity to SH, cysteine proteases (CP) are not reactive to FP. Using FMK for CP profiling (Fig.9), Taunton[33] developed an adenine 13 analog containing FMK, 14, that specifically modifies human kinases at the conserved cysteine. Compared to chloromethylketone (CMK), FMK has greater chemical stability in biological milieu. To improve FMK cell-membrane permeability, that group [34] developed FMK-BODIPY probe 15 containing a membrane-permeable fluorescent tag.
Figure 9.

Structure of selected FMK probes starting with adenine, 13. FMK is incorporated at adenine 2-position to make 14. A compact gatekeeper (e.g., threonine) in a kinase allows compounds with bulky aromatics at the 3-position (14) to enter the kinase hydrophobic pocket. Cell-membrane permeability is improved after installing a BODIPY group to give 15.
3.3. SuFEx Reaction in ABPP
Among sulfur (VI) fluorides (Fig.4b), SF and aryl fluorosulfates (AF) have distinguished themselves in ABPP. SF was developed to probe lysine, tyrosine, or serine. Taunton and coworkers [35] optimized probe 16 (Fig.10) to modify lysine and enable broad-spectrum kinase profiling in live cells. Jones and coworkers developed probe 17 that, for the first time, targets tyrosine as exemplified by labeling the enzyme DcpS.[36]
Figure 10.

Structures of selected SuFEx reagents for ABPP. 16 enables kinase profiling by reacting with lysine residues.17 labels tyrosine residue in the enzyme DcPS. 18 is the first reagent used to label serine proteases. 19 reacts with serine, generating a product that loses water.
For serine labeling, Fahrney and Gold [37] are the first to develop probe 18 (PMSF) to label serine residues at the active site of serine proteases. The PMSF, however, was unstable in aqueous solutions. Grimster and coworkers [38] evaluated the reactivity of aryl SF by using protected amino acids, and they demonstrated no reaction other than SF hydrolysis for the reaction with N-acetylated serine. N-Acetylcysteine (NAC) is the most reactive to SF, but the product is susceptible to hydrolysis. The reactivities of SF toward N-acetyl tyrosine and N-α-acetyl lysine are lower than that of NAC, but the latter generate stable products in both cases.
AF probes are less reactive than SF counterparts and preferentially modify lysine in protein pockets.[39] Recently, Jones[40] found that probe 19 reacts with tyrosine and serine, to generate products that lose water. Dehydration occurs by beta-elimination following serine modification. The AF is so stable that fluorosulfate-L-tyrosine can be genetically encoded into proteins as a latent electrophile for protein cross-linking, demonstrating its intrinsically low chemical reactivity and metabolic stability.[41]
The promise of ABPP is that it can capture the temporal variability of the proteome, but it remains to be seen whether the method is sufficiently robust for the complexity of whole proteomes.
4. Applications in Protein Footprinting
“Footprinting” characterizes protein structure by using chemical reactions to determine solvent accessibility of protein backbone or side chains.[42] Footprinting encompasses different labeling strategies depending on chemistry used. Hydroxyl radical footprinting (HRF) is the most common of the approaches that irreversibly modify proteins,[43] and it is used commonly on synchrotron or photolysis-based systems (e.g., fast photochemical oxidation of proteins or FPOP is the most common photolysis platform). Other chemical footprinting reagents range modestly reactive (e.g., carbenes, iodide, and carbonate radicals) that can be generated on the FPOP platform to highly specific amino-acid labeling reagents (e.g., diethylpyrocarbonate, glycine ethyl ester, benzyl hydrazide) that require no special platform. Instead, the latter are generated slowly by chemical reactions in solution.[44] Interest in new reagent development is growing as investigators search for broad coverage. No single reagent will be equally reactive with all amino-acid residues, and as a result, modifications concentrate on reactive residues. Reagents with complementary reactivity are needed for high coverage.
4.1. Radical Trifluoromethylation
Although the trifluoromethyl group (−CF3) is a versatile motif that has widespread applications in pharmaceuticals, agrochemicals, and specialty materials, its installation into macromolecules (i.e., proteins and peptides) remains rare. Its interest is growing owing in part to the work of Baran45] who discovered that sodium triflinate (Langlois’ reagent) modifies caffeine when spiked into a cell lysate. In another example, beta-lactamase in Tris buffer is labeled with •CF3 and retains its functional activity. [45]
Recently, Gross and coworkers [46] capitalized on trifluoromethyl radical (•CF3) as a footprinting reagent generated by pulsed laser photolysis on the FPOP platform. The highly reactive •CF3 inserts into various X-H bonds (X is C, O, and N) of protein amino-acid residues, resulting in a mass shift of +67.987 Da that can be tracked with high certainty by MS.
When applied to membrane proteins, the observed modifications are localized on extra-membrane regions, demonstrating its aqueous accessibility and compatibility with detergent milieu. Subsequently, Davis[47a] and Krska[47b] incubated the Langlois reagent with t-butyl hydroperoxide (TBHP) to form •CF3 for peptide or protein engineering. To establish specific trifluoromethylation, the •CF3 reactivity was tuned by adjusting the concentration of the Langlois’ reagent to constrain modification to a narrow range of amino acids.
Protein trifluoromethylation has several advantages and disadvantages for footprinting and manipulation. (1) Despite wide accessibility of trifluoromethylation reagents, most are susceptible to hydrolysis or air oxidation that restrict applications in proteomics. Langlois’ reagent, however, is stable and water-soluble. Radical generation and reaction can be performed in open-air. Further, under certain conditions, addition of the reagent is biocompatible with protein function as illustrated by modification of beta-lactamase that maintains enzymatic function after labeling,[45] suggesting that Langlois’ reagent is suitable for structural proteomics and protein engineering.
(2) •CF3 is reactive. For example, styrene reacts 440 times faster with •CF3 than with the methyl radical; several investigators demonstrated that perfluoroalkyl radicals not only undergo addition reactions with alkenes and aromatic rings but also abstract hydrogen from alkanes.[48] Factors governing the high reactivity are the pyramidalization of •CF3 and the strong σ-inductive effect of fluorine.[49] Even with quenchers like DMSO or detergents, which often are in protein solutions, •CF3 maintains its reactivity with proteins and small organic molecules.[46,50]
(3) The Langlois’ reagent, however, cannot be directly photolyzed to give the radical. Given that formation of •CF3 relies on primary oxyl radicals, formed by photolysis of peroxide or incubation with TBHP, the reaction can be muted if reductants (e.g., TECP or DTT) that quench the primary radical precursor are present. Because the •CF3 reaction is driven by •OH, this chemistry can be applied to any platform provided •OH or equivalent radicals can be generated by pulsed radiation; an example is protein footprinting via synchrotron radiation.[51]
(4) More specific protein trifluoromethylation via Langlois’ reagent can be achieved by restricting reagent quantities,[47] or using a specific CF3 modification on cysteine,[52] whereby 19F was installed by applying a halogen exchange 18F-fluorination with 18F-fluoride followed by oxidative cyclization to form the 19F-Umemoto reagent. This new 18F-reagent allows for the direct chemoselective 18F-labeling of unmodified peptides at Cys.
4.2. 3-Trifluoromethyl-3-phenyldiazirine Derivatives
Carbenes, generated from the photolysis of diazirine precursors, are broad-based and alternatives for protein footprinting. Diazomethane (CH2N2), the smallest carbene precursor, was first used for protein footprinting by Richards.[44a] Although the reagent gives extensive labeling, it is difficult to handle especially quantitatively. Recently, photoleucine (PL), [44b] a mimic of leucine, and other diazirines were introduced by several groups as more tractable footprinting reagents that can be generated by photolysis and used in static or flow (FPOP) systems[44c].
The use of fluorous chemistry has advantages in carbene footprinting. Oldham [53a] reported TPD-based diazirine with an ionic carboxyl group at para- position of TPD. The design of TPDs as footprinters is based on two principles: (1) the aromatic ring interacts with hydrophobic patches on a protein, and the carboxyl group associates with polar amino-acid sidechains. Both interactions increase the local concentration of the footprinter on the surface of a protein, thereby improving photolabeling efficiency. (2) The −CF3 group reduces rearrangement side reactions and stabilizes the diazo intermediate. Oldham[53a] compared TPDs to PL for several different proteins and showed that TPDs exhibit higher reaction efficiency than PL. Given the amphipathic nature of the TPDs, Oldham, Moses and coworkers [53b] successfully demonstrated that this probe can label the hydrophobic transmembrane domains of a membrane protein.
In summary, several features can be attributed to carbene protein footprinting: (1) carbenes insert into various X-H bonds (i.e., C-H, N-H, O-H, S-H), allowing the reagent to probe many exposed sites of the protein. (2) The lifetime of carbenes in aqueous solution is nanoseconds owing to its rapid reaction with water, pushing carbene labeling to be faster than protein unfolding. (3) On the other hand, the reactivity of carbene reagents is protein-dependent. Further, steric hindrance of the carbene precursor needs to be carefully considered; for example, a region accessible to small solvent molecules may not be accessible to the reagent.
5. Applications in Protein Cross-linking
In contrast to a footprinter that contains one reactive group, cross-linking reagents comprise two or more functional groups that spatially tether amino acids residues through the formation of covalent bonds. The goal of cross-linking is to determine the topology of protein complexes and identify adjoining regions. The length of the cross-linker is a spatial constraint for the distance between two reactive sites, and some cross-linking studies use a variety of linkers. The development of cross-linking reagents follows two main trends: (1) improving chemical reactivities to capture more amino acids sites, and (2) increasing the identification confidence and sensitivity to detect low-abundance cross-linked products. In this regard, strategies like cleavable crosslinkers, affinity tags for crosslink enrichment, and heterobifunctional cross-linkers with photoactivable headgroups, are valuable. The field of cross-linking was expertly reviewed by Sinz [54].
Fluorine-containing cross-linkers can play a role as shown by Radford, Wilson and coworkers [55] who developed a novel “tag and transfer” approach that incorporates a methanethiosulfonate (MTS) group and a photoactivable diazirine group (one of them a TPD) into a heterobifunctional photocrosslinker. The MTS group first reacts with a single Cys introduced into the “bait” protein, fixing the cross-linker with the “bait” protein. Subsequent incubation allows the target protein to form a complex, followed by photochemical crosslinking between these two proteins. The “bait” protein is removed by reduction of a disulfide bond with SDS-PAGE (Fig.12). Because the location of the crosslink in the bait protein is known, MS analysis can be focused on the target upon removal of the “bait” peptides, thereby reducing the search space for crosslinked products. This approach also demonstrates that absolute crosslinking efficiencies are higher for the MTS-TFMD tagged peptides (26–53%) relative to MTS-diazirine (9-40%). This is expected because TPD-based diazirine usually shows high labeling efficiency.
Figure 12.

(a) Structures of MTS-diazirine and MTS-TFMD. (b) Crosslinking workflow schematic (c) Image of the UV LED lamp and custom-built acrylic chip comprising a 33 mL sample well.
Another example, reported by Wang[56] who utilized proximity-driven properties of SuFEx (see section 2.3) to develop a heterobifunctional cross-linker that contains a reactive succinimide ester and a less reactive sulfonyl fluoride. The succinimide ester reacts with Lys that “plants” the reagent on the protein surface. The sulfur (VI) fluoride is then “cast” to explore nearby surfaces by nucleophilic-enhanced SuFEx substitution. This “plant-and-cast” strategy (fishing analogy) enables a cross-linker to capture unactivated Ser, Thr, His, and Tyr by using SF as the “hook”.
6. Summary and Prospects
Fluorine chemistry is revitalized! Advances enhance our understanding of how “F” profoundly affects molecular structure, reactivity, and function. This revitalization bears fruit in materials, polymers, agrochemicals, and pharmaceuticals and undoubtedly will extend beyond these fields. Here, we describe how fluorine-containing reagents can influence structural proteomics, a new frontier for fluorine chemistry. We identify four contributing factors:
(1) The X-F bond is strong, enabling reagents that contain X-F to retain appropriate aqueous stability commensurate with biochemical experiments. Protein modification must occur under physiologically relevant conditions (usually <37 ⁰C, pH 6–8, in aqueous media with mM buffer and other salts). Successful uses of sulfur (VI) fluorides, fluorophosphonate, and fluoromethylketone are convincing that fluorine-containing reagents are compatible with aqueous environments and are more robust than their halide counterparts.
(2) The presence of fluorine can improve labeling efficiency. Introduction of TPDs and fluorinated azides avoids side rearrangements because strong C-F bonds are reluctant to undergo rearrangement. The •CF3 is more reactive than its methyl radical analog owing to pyramidalization of •CF3 and the strong σ-inductive effect.
(3) Fluorine substitution can retain drug efficacy (e.g., inhaled anesthetics), suggesting a partnership between fluorine therapeutics and structural proteomics. In fact, drugs with one or more fluorine have become commonplace. [57]
(4) The use of fluorine has some issues. The synthesis of fluorine-containing reagents is usually more difficult than their non-fluorine siblings. Increasing degree of fluorination in a reagent normally reduces reagent solubility in aqueous milieu.
A goal in structural proteomic is the determination of endogenous protein structure in live cells.[58] The efficient isolation of the targeted peptide subset from the crude sample is crucial to success in reaching that goal. Peters and coworkers [59] introduced the concept of “fluorous proteomics” to extract fluorinated analytes from biological origins. A promising future direction is to combine fluorous affinity chromatography to enrich targeted peptide subsets from complicated living organisms. The target peptides tagged with perfluorinated labels can be enriched by fluorinated solid phase extraction (FSPE).[60] Further, as fluorinated reagents have improved cell permeability and metabolic/proteolytic stability, they can better serve as reagents to probe protein structure in vivo. Efforts in these directions may evoke a paradigm for fluorine chemistry in structural proteomics.
Figure 11.

Mechanism of radical trifluoromethylation. •OH is formed by laser photolysis of HO-OH (pathway a), followed by •OH oxidation of CF3SO2− to form the reactive intermediate CF3SO2•, which generates •CF3 via entropy-driven loss of SO2. The electron-deficient •CF3 readily adds to proteins (pathway b). In alternatively minor pathway c, HO• directly abstracts H• from the protein to produce a protein radical that either couples with •CF3 to provide a CF3-modified product or reacts with HO• to generate oxygen-containing products.
Acknowledgements
The preparation of this article and some of the reported research was supported by the National Institutes of Health, Grant P41GM103422. We also thank Xuecheng Liao, from Guian elementary school, Guizhou Normal University, for assistance on the GA.
Biography

Ming Cheng obtained his B.E. in pharmaceutical engineering in 2009 from Northwest University for Nationality working with Prof. Liang Xian. From 2009 to 2014, he was a research assistant in Youhong Hu research group at Shanghai Institute of Materia Medica, where he developed synthetic methodology for drug discovery. From 2014 he began his PhD studies with Michael L Gross at Washington University in St. Louis (WUSTL). His research interest at WUSTL centers on mass spectrometry based structural proteomics.

Chunyang Guo obtained her B.S. in applied chemistry from Jinan University (Guangzhou, China). Subsequently, she went to Nankai University (Tianjin, China) to pursue her master’s degree in material science. Currently, she is a PhD candidate in Michael L Gross’s lab at Washington University in St. Louis (WUSTL). Her research focuses on employing footprinting coupled with mass spectrometry-based tools to characterize protein structure and to study protein-ligand interactions.

Michael L. Gross has been an independent mass spectrometrist for 50 y, contributing approximately 12 articles and book chapters per year for that time. His interests began with ion chemistry, instrumentation, and environmental chemistry, and they evolved to structural proteomics, specifically HDX, protein footprinting, and native MS. He is a winner of the ACS Midwest, Field and Franklin, and Analytical Awards, the JJ Thomson medal, and the Commonwealth of Massachusetts Pioneer Award and is a fellow of AAAS.
References
- [1].Wilkins MR, Sanchez JC, Gooley AA, Appel RD, Smith IH, Hochstrasser DF, Williams KL, Biotechnol Genet Eng Rev.1996, 13, 19. [DOI] [PubMed] [Google Scholar]
- [2].Hagan DO, Chem. Soc. Rev, 2008, 37, 308.18197347 [Google Scholar]
- [3].Wofsy L, Metzger H, Singer SJ, Biochemistry 1962, 1, 1031. [DOI] [PubMed] [Google Scholar]
- [4].Brunner J, Serin SH, Richards FM, J Biol Chem.1980, 255, 3313. [PubMed] [Google Scholar]
- [5].Halloran MW, Lumb JP, Chem. Eur. J 2019, 25, 4885. [DOI] [PubMed] [Google Scholar]
- [6].Blencowe A, Hayes W, Soft Matter, 2005, 1, 178. [DOI] [PubMed] [Google Scholar]
- [7].Chowdhry V, Vaughan R, Westheimer FH, Proc. Nati. Acad. Sci. USA, 1976,73, 1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Seifert T, Malo M, Lengqvist J, Sihlbom C, Jarho EM, Luthman K, J. Med. Chem 2016, 59, 10794. [DOI] [PubMed] [Google Scholar]
- [9].Yip GM, Chen ZW, Edge CJ, Smith EH, Dickinson R, Hohenester E, Townsend RR, Fuchs K, Sieghart W, Evers AS, Franks NP, Nat. Chem. Biol 2013, 9, 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chicca A, Nicolussi S, Bartholomaus R, Blunder M, Rey AA, Petrucci V, Reynoso-Moreno ID, Viveros-Paredes JM, Gens MD, Lutz B, Schioth HB, Soeberdt M, Abels C, Charles RP, Altmann KH, Gertsch J, Proc. Natl. Acad. Sci. USA, 2017, 114, E5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Herrera LD, Ostroff RD, and Rogers SA, CNS Drug Rev. 2001, 7, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Woll KA, Dailey WP, Eckenhoff RG, Methods Enzymol. 2018, 602,231. [DOI] [PubMed] [Google Scholar]
- [13].Brahms DLS, Dailey WP, Chem. Rev 1996, 96, 1585. [DOI] [PubMed] [Google Scholar]
- [14].Eckenhoff RG, Xi J, Shimaoka M, Bhattacharji A, Covarrubias M, and Dailey WP, ACS Chem. Neurosci 2010, 1, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Woll KA, Peng W, Liang Q, Zhi L, Jacobs JA, Maciunas L, Bhanu N, Garcia BA, Covarrubias M, Loll PJ, Dailey WP, Eckenhoff RG, ACS Chem. Biol 2017, 12, 1353. [DOI] [PubMed] [Google Scholar]; b) Eckenhoff R, Knoll FJ, Greenblatt EP, Dailey WP, J. Med. Chem 2002, 45, 1879. [DOI] [PubMed] [Google Scholar]
- [16].Schrock AK, Schuster GB, J. Am. Chem. Soc 1984, 106, 5228. [Google Scholar]
- [17].Poe R, Schnapp K, Young MJT, Grayzar J, Platz MS, J. Am. Chem. Soc 1992, 114, 5054. [Google Scholar]
- [18].Keana JFW, Cai SX, J. Org. Chem 1990, 55, 3640. [Google Scholar]
- [19].Leyva E, Young MJT, Platz MS, J. Am. Chem. Soc 1986, 108, 8307. [DOI] [PubMed] [Google Scholar]
- [20].a) Xia Y, Qu F, Maggiani A, Sengupta K, Liu C, Peng L, Org. Lett, 2011. 13, 4248. [DOI] [PubMed] [Google Scholar]; b) Xia Y, Sengupta K, Maggiani A, Qu F, Peng L, Org. Biomol. Chem, 2013, 11, 5000. [DOI] [PubMed] [Google Scholar]
- [21].Steinkopf W, J. Prakt. Chem 1927, 117, 1–82. [Google Scholar]
- [22].Dong J, Krasnova L, Finn MG, Sharpless KB, Angew. Chem. Int. Ed 2014, 53, 9430; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 9584. [Google Scholar]
- [23].a) Narayanan A, Jones LH, Chem. Sci, 2015, 6, 2650. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jones LH, Angew. Chem. Int. Ed 2018, 57, 9220; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 9362. [Google Scholar]; c) Gago PM, Olsen CA, Angew. Chem. Int. Ed 2019, 58, 957; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 969. [Google Scholar]; d) Gehringer M, Laufer SA, 2018, J. Med. Chem DOI: 10.1021/acs.jmedchem.8b01153. [DOI] [Google Scholar]
- [24].Baker RB, Acc. Chem. Res 1969, 2, 129. [Google Scholar]
- [25].Grimster NP, Connelly S, Baranczak A, Dong J, Krasnova LB, Sharpless KB, Powers ET, Wilson IA, Kelly JW, J. Am. Chem. Soc 2013, 135, 5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dubiella C, Cui H, Gersch M, Brouwer AJ, Sieber SA, Krgger A, Liskamp RMJ, Groll M, Angew. Chem. Int. Ed 2014, 53, 11969; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 12163. [Google Scholar]
- [27].Cravatt BF, Wright AT, Kozarich JW, Annu. Rev. Biochem 2008. 77, 383. [DOI] [PubMed] [Google Scholar]
- [28].Chen B, Ge SS, Zhao YC, Chen C, Yang S, RSC Adv, 2016, 6, 113327. [Google Scholar]
- [29].Jansen EF, Curl AL, Balls AKJ, J. Biol. Chem 1951, 190, 557. [PubMed] [Google Scholar]
- [30].Liu Y, Patricelli MP, Cravatt BF, Proc. Natl. Acad. Sci. USA 1999, 96, 14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kidd D, Liu Y, Cravatt BF, Biochemistry 2001, 40, 4005. [DOI] [PubMed] [Google Scholar]
- [32].Wagner S, Accorsi M, Rademann J, Chem. Eur. J 2017, 23, 15387. [DOI] [PubMed] [Google Scholar]
- [33].Cohen MS, Zhang C, Shokat KM, Taunton J, Science 2005, 308, 1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].S Cohen M, Hadjivassiliou H, Taunton J, Nat. Chem. Biol 2007, 3, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhao Q, Ouyang X, Wan X, Gajiwala KS, Kath JC, Jones LH, Burlingame AL, Taunton J, J. Am. Chem. Soc 2017, 139, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hett EC, Xu H, Geoghegan KF, Gopalsamy A, Kyne RE, Menard CA, Narayanan A, Parikh MD, Liu S, Roberts L, Robinson RP, Tones MA, Jones LH, ACS Chem. Biol 2015, 10, 1094. [DOI] [PubMed] [Google Scholar]
- [37].Fahrney DE, Gold AM, J. Am. Chem. Soc, 1963, 85, 997. [Google Scholar]
- [38].Mukherjee H, Debreczeni J, Breed J, Tentarelli S, Aquila B, Dowling JE, Org. Biomol. Chem, 2017, 15, 9685. [DOI] [PubMed] [Google Scholar]
- [39].Chen W, Dong J, Plate L, Mortenson DE, Brighty GJ, Li S, Liu Y, Galmozzi A, Lee PS, Hulce JJ, Cravatt BF, Saez E, Powers ET, Wilson IA, Sharpless KB, Kelly JW, J. Am. Chem. Soc 2016, 138, 7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fadeyi OO, Hoth LR, Choi C, Feng X, Gopalsamy A, Hett EC, Kyne RE, Robinson RP, Jones LH, ACS Chem. Biol 2017,12, 2015. [DOI] [PubMed] [Google Scholar]
- [41].Wang N, Yang B, Fu C, Zhu H, Zheng F, Kobayashi T, Liu J, Li S, Ma C, Wang PG, Wang Q, Wang L, J. Am. Chem. Soc 2018, 140, 4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang L, Chance MR, Mol. Cell. Proteomics 2017, 16, 706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].a) Maleknia SD, Brenowitz M, Chance MR, Anal. Chem 1999, 71, 3965; [DOI] [PubMed] [Google Scholar]; b) Hambly DM, Gross ML, J. Am. Soc. Mass. Spectrom 2005, 16, 2057; [DOI] [PubMed] [Google Scholar]; c) Watson C, Janik I, Zhuang T, Charvátová O, Woods RJ, Sharp JS, Anal. Chem 2009, 81, 2496; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Minkoff BB, Blatz JM, Choudhury FA, Benjamin D, Shohet JL, Sussman MR, Sci. Rep 2017, 7, 12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].a) Richards FM, Lamed R, Wynn R, Patel D, Olack G, Protein Sci. 2000. 9, 2506. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang B, Rempel DL, Gross ML, J. Am. Soc. Mass Spectrom 2016, 27, 552; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jumper CC, Schriemer DC, Anal. Chem 2011, 83, 2913; [DOI] [PubMed] [Google Scholar]; d) Chen J, Cui W, Giblin D, Gross ML, J. Am. Soc. Mass Spectrom, 2012, 23, 1306; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang MM, Rempel DL, Gross ML, Free Radic Biol Med, 2019, 131, 126; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zhang H, Wen J, Huang R, Blankenship RE, Gross ML, Int J Mass Spectrom. 2012, 312 78; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Guo C, Cheng M, Gross ML, Anal. Chem 2019, 91, 1416; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Limpikirati P, Hale JE, Hazelbaker M, Huang Y, Jia Z, Yazdani M, Graban EM, Vaughan RC, Vachet RW, mAbs, 2019, 11, 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fujiwara Y, Dixon JA, QHara FO, Funder ED, Dixon DD, Rodriguez RA, Baxter RD, Herle B, Sach N, Collins MR, Ishihara Y, Baran PS, Nature 2012, 492, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cheng M, Zhang B, Cui W, Gross ML, Angew. Chem. Int. Ed 2017, 56, 14007; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 14195. [Google Scholar]
- [47].a) Imiołek M, Karunanithy G, Ng WL, Baldwin AJ, Gouverneur V, Davis BG, J. Am. Chem. Soc 2018, 140, 1568; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ichiishi N, Caldwell JP, Lin M, Zhong W, Zhu X, Streckfuss E, Kim HY, Parish CA, Krska SW, Chem. Sci, 2018, 9, 4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].a) Dolbier WR, Chem. Rev 1996, 96, 1557; [DOI] [PubMed] [Google Scholar]; b) Liguori L, Bjørsvik HR, Bravo A, Fontana F, Minisci F, Chem. Commun 1997, 1501; [Google Scholar]; c) Shtarev AB, Tian F, Dolbier WR, Smart BE, J. Am. Chem. Soc 1999, 121, 7335. [Google Scholar]
- [49].Studer A, Angew. Chem. Int. Ed 2012, 51, 8950. [DOI] [PubMed] [Google Scholar]; Angew. Chem 2012, 124, 9082. [Google Scholar]
- [50].Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS, Proc. Natl. Acad. Sci. USA 2011, 108, 14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Unpublished results: ·CF3 can be generated by radiolysis of water in synchrotron.
- [52].Verhoog S, Kee CW, Wang Y, Khotavivattana T, Wilson TC, Kersemans V, Smart S, Tredwell M, Davis BG, Gouverneur V, J. Am. Chem. Soc 2018, 140, 1572. [DOI] [PubMed] [Google Scholar]
- [53].a) Manzi L, Barrow AS, Scott D, Layfield R, Wright TG, Moses JE, Oldham NJ, Nat. Commun 2016, 7, 13288; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Manzi L, Barrow AS, Hopper JTS, Kaminska R, Kleanthous C, Robinson CV, Moses John E., Oldham NJ, Angew. Chem. Int. Ed 2017, 56, 14873. [DOI] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 15069. [Google Scholar]
- [54].Sinz A, Angew. Chem. Int. Ed 2018, 57, 6390; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 6498. [Google Scholar]
- [55].Horne JE, Walko M, Calabrese AN, Levenstein MA, Brockwell DJ, Kapur N, Wilson AJ, Radford SE; Angew. Chem. Int. Ed 2018, 57, 16688, [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16930. [Google Scholar]
- [56].Yang B, Wu H, Schnier PD, Liu Y, Liu J, Wang N, DeGradoa WF, Wang L, Proc. Natl. Acad. Sci. USA, 2018, 115, 11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Aceña JL, Soloshonok VA, Izawa K, Liu H, Chem. Rev 2016, 116, 422. [DOI] [PubMed] [Google Scholar]
- [58].Espino JA, Mali VS, Jones LM, Anal. Chem 2015, 87, 7971. [DOI] [PubMed] [Google Scholar]
- [59].Brittain SM, Ficarro SB, Brock A, Peters EC, Nat Biotechnol. 2005, 3, 463. [DOI] [PubMed] [Google Scholar]
- [60].Song Z, Huang W, Zhang Q, Chem. Commun, 2012, 48, 3339. [DOI] [PubMed] [Google Scholar]