

Abstract
Metastasis contributes to the vast majority of cancer related mortality. Regulatory mechanisms of the multistep invasion-metastasis cascade are being unraveled. TP53 is the most frequently mutated gene across human cancers. Accumulating evidence has shown that mutations of TP53 not only lead to loss-of-function or dominant negative effect, but also promote a gain-of- function. Specifically, gain-of-function mutant p53 promotes cancer cell motility, invasion and metastasis. Here we summarize the mechanisms and functions of mutant p53 that foster metastasis in different types of cancers. We also discuss the prognostic value of mutant p53 and current status of therapeutic strategies targeting mutant p53. Future studies will shed light on discovering novel mechanisms of mutant p53 driven cancer metastasis, and developing innovative therapeutics to improve clinical outcome of patients harboring p53 mutations.
Keywords: mutant p53, metastasis, epithelial-to-mesenchymal transition (EMT), extracelular matrix (ECM), receptor tyrosine kinase (RTK), therapeutics
Overview of mutant p53 in metastasis
Despite significant progress in investigating the molecular mechanisms underlying cancer pathogenesis and advances in cancer therapeutics, patients diagnosed with metastatic disease are mostly detected at late stages. Indeed, metastatic disease contributes to over 90% of cancer-associated deaths [1]. Although a large body of studies has unveiled the mechanisms underlying primary tumor formation, understanding the how metastatic cancers emerge and to translate that knowledge into personalized therapeutics is critical, yet remains challenging. Metastasis involves a sequence of events, from cancer cell invasion at the primary tumor site to the outgrowth of metastatic colonies at distant organs [2]. To survive this multistep metastasis cascade, tumor cells reprogram gene expression and rewire metabolism, amongst other factors [3]. Regulation of intercellular and intracellular signaling is also required to mediate the interactions of tumor cells and extracellular matrix (ECM) (see Glossary), in order to adapt to the constantly changing tumor microenvironment (TME). To achieve these, genetic mutations and epigenetic alterations contribute significantly to metastasis [4].
The TP53 gene (equivalent to Trp53 gene in mice) is the most frequently mutated gene in human cancers [5] . TP53 encodes the p53 protein, which is among the most intensively investigated tumor suppressor gene for the last several decades. The majority of TP53 mutations are missense mutations. Mutations of TP53 are harbored in all coding exons, but predominantly cluster in exons 4-9 that represents the DNA binding domain. In almost all cancers, six “hotspot” residues are frequently mutated in the TP53 DNA binding domain, namely R175(4.8%), G245(3.12%), R248 (6.79%), R249 (2.59%), R273 (6.55%) and R282 (2.59%) (IARC TP53 Database, R20) [6]. Theses mutations may diminish the wild-type activity of p53, known as loss of function (LOF), and may act have dominant negative (DN) functions to antagonize the remaining wild-type p53 [6][7]. Furthermore, a growing body of evidence has revealed that many p53 mutants demonstrate gain of function (GOF) properties, whereby mutant p53 acquires “oncogenic” properties and results in a more aggressive tumor phenotype [6][7]. Li-Fraumeni(LFS) syndrome is a familial cancer syndrome caused by germline TP53 mutations and manifest by early-onset [8]. Furthermore, mice with germline Trp53 mutations have a different tumor spectrum compared to Trp53 null mice. Trp53R172H/− mice have increased incidence of carcinoma, whereas Trp53−/− mice frequently develop lymphomas and sarcomas [9].
Mutant p53 is a potent regulator of metastasis. Trp53R172H/+ mice harbor metastatic osteosarcomas and carcinomas, whereas tumors in Trp53+/− mice do not metastasize[10]. Indeed, mutant p53 promotes the metastasis cascade through multiple avenues (Figure 1) (Table 1). In this review, we focus on the mechanisms and functions of mutant p53 in promoting the canonical steps in the metastatic cascade. We also discuss the prognosis and therapeutic value of mutant p53 in cancers, specifically in metastasis (Figure 2).
Figure 1: Mechanisms of mutant p53 mediating metastasis.

Mutant p53 promotes cell motility, invasion and metastasis through multiple mechanisms, including regulating epithelial-to-mesenchymal transition (EMT) (labeled in yellow), regulating cell-extracellular matrix(ECM) interactions (labeled in purple), promoting receptor tyrosine kinase (RTK) signaling (labeled in green) and other recently revealed novel mechanisms (labeled in turquoise). Furthermore, interactions of mutant p53 with binding partners serve as an important mechanism to mediate these effects. For example, the binding of mutant p53 with p63, p73, Sp1, or ID4 transcriptionally activates downstream pathways to promote metastasis.
Table 1:
In vivo models of mutant p53 driven metastasis and related functional consequences.
| Mutant p53 | Model | Functional consequence(s) |
Metastatic site | Reference |
|---|---|---|---|---|
| Trp53R172H (Trp53R172H/+ vs Trp53+/−) | Li-Fraumeni | - | Liver, Lung and Brain | [10] |
| P53R28OK (ControlsiRNA vs p53siRNA) | Breast cancer cell line | Glycoprotein folding | Lung | [40] |
| Trp53R172H | KPC model | PDGFRβ signaling | Lung | [44] |
| Trp53R270H (KPC+sh.p53, +sh.Control, vs KPflC) | Pancreatic cancer cell line | |||
| P53R248W (p53R248W ectopically expressed vs p53−/−, p53+/+) | Colon cancer cell line | Exosome Macrophage reprogramming | Lung | [55] |
| R-72 p53R175H (R-72 p53R175H ectopically expressed vs p53−/−) | Lung cancer cell line | Mitochondrial function and oxidative phosphorylation | Lung and Bone | [66] |
Figure 2: Therapeutic strategies targeting mutant p53.
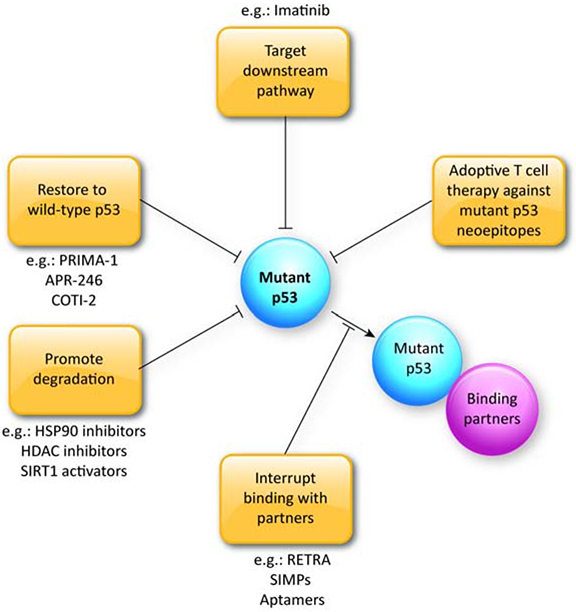
Strategies targeting mutant p53 include the following: (i) restoring mutant p53 to wild-type p53 structure and function; (ii) destabilizing mutant p53 to deplete the protein; (iii) inhibition of the interactions of mutant p53 with its binding partners to dampen transcriptional regulatory activity; and iv) targeting mutant p53 downstream pathways. Future strategy may involve adoptive T cell therapy against mutant p53 neoepitopes.
Epithelial-mesenchymal transition (EMT)
In epithelial carcinomas, Epithelial-mesenchymal transition (EMT) plays a crucial role in metastasis, and is involved in multiple steps of metastasis including malignant conversion, ECM degradation, primary tumor invasion, intravasation and extravasation [11]. These changes in cellular behavior are orchestrated by key EMT related transcription factors (TFs) such as basic helix-loop-helix (bHLH) TFs, zinc-finger E-box-binding (ZEB), and SNAIL [12][13].
Wild type p53 has been shown to negatively regulate the initiation and maintenance of EMT and to suppress metastasis [3]. By contrast, mutant p53 facilitates EMT and metastasis [14-23]. Several EMT TFs can be regulated by mutant p53 at transcriptional, post-transcriptional and translational levels. In endometrial cancer, Tp53R175H;R248Q;R273H GOF mutants induce ZEB1 expression through transcriptionally repressing miR-130b, a negative regulator of ZEB1. This leads to induction of EMT and increased endometrial cancer cell invasion [14]. Upregulation of ZEB1 is also observed in HCT116 colon carcinoma cell lines expressing a temperature-sensitive p53 (p53-A143) with mutant conformation [15]. In transformed human esophageal epithelial cells, Tp53R175H cooperates with epidermal growth factor receptor (EGFR) overexpression to induce increased expression of ZEB1 and ZEB2, and to promote EMT upon TGFβ stimulation [16]. Besides ZEB family of TFs, mutant p53 also affects SNAIL. One member of the SNAIL family of TFs, Slug, can be stabilized by Tp53R248W in non-small cell lung cancer (NSCLC). Whereas Slug is suppressed by wild type p53 through inducing MDM2-mediated degradation, Slug is maintained by mutant p53 through repression of MDM2 expression, leading to increased cancer cell invasiveness in NSCLC [17]. In addition, expression of p53 with mutant conformation also induces Slug and represses ECADHERIN expression in HCT116 colon carcinoma cell lines [15]. Furthermore, in immortalized human prostate epithelial cells, mutant p53 contributes to EMT and enhanced cell invasion[18]. Mechanistically, Tp53R175H mutant p53 specifically increases the expression of Twist1, a bHLH family TF, through attenuation of epigenetic repression [18].
In addition to regulating EMT TFs, the crosstalk between mutant p53 and TGFβ signaling is involved in regulating cell motility, invasion and metastasis [19-23]. However, mutant p53 cooperates or attenuates TGFβ signaling in different contexts [19-23]. In some cancers, mutant p53 is required for TGFβ-induced invasion and metastasis through diverse mechanisms [19][20]. In breast cancer cell lines, Tp53R248Q;R282W upregulates miR-155 and downregulates ZNF652, a zinc-finger transcriptional repressor of TGFB1, TGFB2, TGFBR2, SMAD2 and VIMENTIN. Activation of the mutant p53/miR-155 axis leads to increased invasion, migration and amoeboid transformation in vitro [20].
Mutant p53 (Tp53R175H) also affects TGFβ signaling through direct interactions with SMADs. SMAD2 functions as an essential platform to assemble mutant p53 and p63. Therefore, p63 is antagonized by the formation of this inhibitory complex through TGFβ signaling [19]. However, mutant p53 attenuates TGFβ pathway in other contexts. In human NSCLC H1299 cell line, repression of TGFBR2 expression by Tp53R175H is demonstrated. As a result, Tp53R175H attenuates the activity of TGFβ1 signaling pathway, including reduction in TGFβ1-induced phosphorylation of SMAD2/3 and inhibition of TGFβ1-mediated nuclear translocation of SMAD4 and its association with SMAD2. Functionally, Tp53R175H represses TGFβ1-mediated cell migration [21]. Tp53R175H also opposes TGFβ/SMAD3 pathway and protects the activity of REGy-proteasome pathway in H1299 cell line [22]. One possible explanation to partially reconcile these contradictory observations lies in the preferential binding of mutant p53 with SMAD3 than SMAD2 in the presence of ERK activation. Upon ERK activation, p53R175H preferentially binds to SMAD3, disrupts the SMAD3/SMAD4 complex, distorts the TGFβ pathway and unleashes SMAD2 specific signaling [23]. Despite functional redundancy, SMAD2 and SMAD3 play different roles in the TGFβ pathway [24]. For example, Slug expression is enhanced by SMAD2 but repressed by SMAD3 [23]. Therefore, mutant p53 may exerts parts of its effects through disrupting the balance of SMAD2 and SMAD3 transcriptional activity [23].
Cell motility and interaction with the ECM
Once EMT occurs, tumor cells form protrusive and invasive structures to navigate through the extracellular matrix (ECM), to break away from the invasive front and to migrate into the vasculature [25]. During extravasation, protrusive structures are also required to passage through endothelium into the ECM of the metastatic site [25]. The dynamic formation of protrusive structures such as lamellipodia and filopodia depends upon changes in the actin cytoskeleton. Therefore, actin cytoskeleton regulators are major controllers of cell motility, and play an important role in cancer cell invasion and metastasis [25]. The myosins family are actin-based motor proteins, and the founding member of class X myosins, Myo10, is well established for its induction of filopodia and contribution to cell migration [26]. In breast cancer cells, Myo10 is required for cell invasion, dissemination and metastasis through transporting integrins to filopodia tip. Furthermore, Myo10 expression is induced by mutant p53(Tp53R175H) in vitro and in vivo [27]. In patient samples, Myo10 expression also correlates with p53 mutation and poor prognosis, thereby suggesting that Myo10 is an important regulator in mutant p53-driven invasion [27]. In addition to myosins, small GTP-binding proteins RhoGTPases are also important regulators of cell motility. RhoA, a member of the Rho family, controls actomyosin based contractility and retraction [28][29]. In Trp53R172H driven invasive mouse pancreatic ductal adenocarcinoma (PDAC), RhoA activity is observed in both the leading edge and trailing edge of cells. However, this is not observed in p53 knockout PDAC cells [30]. These observations demonstrate the important role of mutant p53 in the spatial regulation of RhoA activity [30].
In addition to cancer cell motility, the interaction of cancer cells with ECM is indispensable for metastasis [31][32]. Integrins are the most predominant cell adhesion receptors for ECM components, and are involved in almost every step of cancer progression and metastasis [33][34]. In H1299 cells, fibronectin-binding α5β1 integrin is required for mutant p53-driven cell motility and invasion. The recycling rate of α5β1 integrin is increased by Tp53R175H;R273H through enhancing the interaction between α5β1 integrin and the Rab11 effector Rab-coupling protein (RCP or Rab11-FIP1), an important regulator of endocytic trafficking [35][36]. Taken together, this study demonstrates mutant p53 harnesses the endocytic recycling machinery to enhance integrin trafficking to the plasma membrane, and promotes tumor cell invasion and motility [35-37].
Glycosylation of cell surface proteins is another important contributor of cancer cell-ECM interactions and metastasis [38]. As the major cell adhesion glycoprotein, integrin’s glycosylation patterns affect cell-matrix adhesion and regulate breast cancer progression [39]. For prometastatic cell surface proteins such as integrins, mutant p53 not only enhances their expression level quantitatively, but also regulates their N-glycosylation state qualitatively. Tp53R175H;C176S;R248W;R273H;R280K induces the expression of ectonucleoside triphosphate dipsphohydrolase 5 (ENTPD5), an enzyme to promote folding of N-glycosylated membrane proteins in the endoplasmic reticulum (ER). As a result, Tp53R280K and ENTPD5 promote N-glycoprotein folding and maturation in ER to enhance invasion and lung colonization in breast cancer [40].
Receptor tyrosine kinase signaling
Receptor tyrosine kinases (RTKs) are a family of cell-surface receptors such as the epidermal growth factor receptor (EGFR), the platelet-derived growth factor receptor (PDGFR), and hepatocyte growth factor (HGF)/MET pathways. RTKs have been demonstrated as critical regulators in cancer cell proliferation, differentiation, and migration, and play an important role in metastasis [41][42]. Mutant p53 induces several RTK pathways to promote tumor invasion and metastasis. In epithelial malignancies, EGFR is a key regulator and therapeutic target in tumor growth, invasion and metastasis [43]. Tp53R175H;R273H promotes EGFR phosphorylation and AKT activation through enhancing EGFR trafficking to the plasma membrane. This contributes to increased invasive behavior in H1299 cells [35]. In addition to EGFR, Trp53R172H also promotes PDGFRβ signaling to enhance invasion and lung metastasis in PDAC. Furthermore, inhibition of PDGFRβ by imatinib decreases mutant p53 driven metastases, indicating PDGFRβ as a promising therapeutic target [44][45]. Another RTK pathway regulated by mutant p53 is the HGF/MET pathway. HGF is an important environmental inducer of tumor’s invasive growth, which binds to its receptor Met. HGF/MET signaling has been demonstrated to play a major role in promoting cell motility, proliferation, tumor formation, invasion and progression [46][47]. Suppression of HGF/MET and VEGF signaling decreases tumor invasion and metastasis in pancreatic neuroendocrine tumors [48]. In transformed primary esophageal epithelial cells, MET receptor is activated upon p53R175H expression. Inhibition of MET phosphorylation leads to decreased cell invasion, indicating that MET may be regulated by p53R175H to promote cell invasion [49]. Mechanistically, MET is an RCP-binding protein, and MET recycling is enhanced by p53R175H;R273H. Therefore, cells harboring mutant p53 are more sensitive to HGF, indicating a potential therapeutic strategy for mutant p53 driven invasion and metastasis [50].
New perspectives on mutant p53 and metastasis
Recently, emerging new mechanisms have been demonstrated in mutant p53 driven metastasis. While cell-to-cell interaction and cell-to-ECM interaction are important in invasion and metastasis, novel components of the tumor secretome such as exosomes also contribute to multiple steps of the metastasis cascade [51]. Exosomes are secreted nanovesicles, 40 to 100nm in diameter and round to cup-shaped in morphology. As end-products of the recycling pathway, they carry a variety of cargos including DNA, mRNA, miRNA, long non-coding RNA (lncRNA), proteins and lipids. Exosomes derived from cancer cells have been demonstrated to promote metastasis through mediating invasion into surrounding tissues, modulating immune responses, regulating angiogenesis and facilitating pre-metastatic niche formation [51–53]. In addition to the previous finding that that mutant p53 promotes cancer invasiveness through enhancing RCP-dependent integrin recycling [35], a recent study demonstrated that p53R175H;R273H expressing tumor cells release exosomes to upregulate RCP-dependent integrin recycling and cell migration in neighboring p53 null cells. Thus, this gain-of-function of mutant p53 may be potentially transferred inter-cellularly through exosomes [54]. Furthermore, the released exosomes also promote integrin trafficking in normal fibroblasts, leading to alterations in ECM structure and generation of a proinvasive microenvironment. In both subcutaneous tumor model and Pdx1-Cre, KrasG12D/+, Trp53R172H(or R270H)/+ (KPC) autochthonous PDAC model, even in the absence of lung metastatic cells, ECM organization in lung is altered by mutant p53 expressing primary tumors. These observations indicate a novel mechanism that mutant p53 expressing tumor cells generate pro-invasive niches through releasing exosomes [54]. In colon cancer, exosomes secreted by p53R248W expressing tumor cells are enriched with miR-1246. These exosomes can be taken up by neighboring tumor-associated macrophages (TAMs) and reprogram TAMs to a tumor supportive state [55].
Non-coding RNA regulatory networks also play an important role in regulating metastasis. These regulatory RNAs include small non-coding RNAs such as miRNA with transcript length less than 200bp, and lncRNAs with transcript length longer than 200bp [56]. MiRNAs and lncRNAs regulate gene expression in multiple ways including transcriptional regulation, post-transcriptional regulation and translational regulation [56][57]. In cancer cells, miRNA and lncRNA functions are significantly deregulated, and a large body of evidence has demonstrated their important role in metastasis [56][57]. P53R273H regulates miRNAs such as let-7i to affect a broad spectrum of gene expression [58]. Let-7i represses a network of proliferation and RNA post-transcriptional modification genes, and the inhibition of let-7i by mutant p53 leads to increased cell migration, invasion and metastasis [58]. LncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is known as an important regulator of lung cancer metastasis [59]. In breast cancer cells, p53R175H;R273H promotes the association of MALAT1 with chromatin and further modulates VEGFA isoforms expression. These observations demonstrate the regulation of lncRNA by mutant p53, and may indicate a potential mechanism of lncRNAs as mediator in mutant p53 driven metastasis [60].
Metabolism reprogramming is a hallmark of cancer [61]. To survive different stages of metastasis, tumor cells engage metabolic strategies to adapt to foreign metastatic microenvironments [62]. Mutant p53 rewires metabolism in tumor cells to meet the biosynthetic demands for tumor growth and progression, such as inducing the Warburg effect and regulating the mevalonate pathway [63-65]. Recently, one study demonstrated that a common TP53 polymorphism at codon 72 (R72) variant of p53R175H;R273H promotes migration and metastasis through regulating PGC-1a, a crucial regulator in mitochondrial biogenesis and oxidative phosphorylation. In multiple cancer cell lines, R72 variant of mutant p53 expressing cells demonstrate increased PGC-1a function, oxidative phosphorylation and enhanced metastatic capacity. Taken together, this study revealed a novel mechanism whereby mutant p53 reprograms metabolism pathway to enhance metastasis [66].
Binding partners of mutant p53 in metastasis
Mutant p53 exerts part of its functions through forming protein complexes with binding partners and affecting their transcriptional activity [65][67]. These binding partners include other members of the p53 family, such as p63 and p73, and TFs such as Sp1 and ETSs [67], Unlike p53, p63 somatic mutation is rarely detected in human cancers. In aggressive and metastatic tumors, loss of p63 expression occurs, indicating that p63 may function as a suppressor of tumor progression and metastasis [68]. P63 can be inhibited by mutant p53 through direct physical interaction [69][70]. In mutant p53 expressing H1299 cells, interaction of p63 and mutant p53 is observed [35]. Disruption of this interaction is associated with decreased invasion. Furthermore, depleting p63 increases α5β1-integrin and EGFR recycling, and promotes tumor cell invasion and random migration, suggesting that mutant p53 drives invasion and migration through inhibition of p63 [35]. Similarly, in p63 expressing cells, inhibition of p63 promotes mutant p53 driven HGF/MET dependent invasion [50]. In addition, mutant p53 binds to p63 using SMADs as a platform. The formation of this complex downregulates the expression of two endogenous p63 target genes, Sharp-1 and Cyclin G2. Low expression of these two genes in breast cancer patients is associated with decreased recurrence-free survival [19]. By contrast, the repression of another p63 target, miR-155, is alleviated by mutant p53, leading to upregulation of several key drivers in invasion and metastasis and increased migration, invasion and ameboid transformation [20]. Finally, the mutant p53-p63 complex represses the expression of let7i, which represses cell migration, invasion and metastasis through inhibiting a network of oncogenes [58]. Similar to p63, p73 also physically interacts with mutant p53 and its transcriptional activity is suppressed by this interaction [71][72]. Furthermore, inhibition of p73 by mutant p53 contributes to enhanced metastasis. In PDAC, mutant p53 sequesters p73 from forming the repressive p73/NF-Y complex, leading to increased transcription of PDGFRB and enhanced migration, invasion and lung metastasis [44].
Besides p63 and p73, mutant p53 interacts with other TFs to promote metastasis. Mutant p53 can form a complex with Sp1 [73]. In human PDAC cell lines, mutant p53 is recruited to ENTPD5 promoter by Sp1 and induces ENTPD5 expression to upregulate N-glycosylated membrane protein folding, eventually contributing to enhanced metastatic capacity [40]. In several breast cancer cell lines, inhibitor of DNA binding 4 (ID4), a transcriptional target of mutant p53, interacts with SKBR3 and together forms a complex with RNA splicing factor SRSF1. The formation of this complex promotes binding of SRSF1 to MALAT1 IncRNA, recruits MALAT1 on VEGFA pre-mRNA and affects VEGFA isoforms expression. Altogether, these events enhance tumor cell angiogenic potential, an important part of the metastasis cascade [60][74]. In H1299 and HaCaT cell lines, mutant p53 binds to and recruits p300, a coactivator, on REGy promoter to induce its expression, further affecting TGFβ signaling [22]. Finally, in an osteosarcoma model driven by mutant p53, depletion of Ets2, a TF previously shown to interact with mutant p53, abrogates the prometastatic phenotype [75][76]. This result indicates that Ets2 may be another TF partner of mutant p53 to drive metastasis [76].
Clinical implications of mutant p53’s functional roles in metastasis
Assessment of somatic p53 mutations has revealed the clinical value of p53 mutations in assessing the outcomes of cancers [77]. In a study analyzing matched primary tumors and lymph nodes, liver and pleural metastatic tumor samples, p53 mutations were found in either or both primary and metastatic tumors. These results suggest that in several cancers, some p53 mutations may occur prior to metastasis emergence [78]. One study performed whole-genome sequencing analyses of matched primary tumors, brain metastases and primary tumor derived xenografts from basal-like breast cancer patients. In primary and metastatic tumors, p53 mutation is detected at a relatively constant frequency, and is highly enriched in the xenografts [79]. Another retrospect study in primary early breast carcinomas revealed that p53 mutation is an independent predictor of distant metastasis and overall survival [80]. Furthermore, since p53 mutations often lead to stabilized protein, one study used immunohistochemistry (IHC) staining to evaluate p53 status in visceral metastasis breast cancer (VMBC). This study demonstrated that p53 expression is correlated with time from primary breast cancer diagnosis to death, but not for time from metastasis to death [81]. Taken together, these and other studies show that in breast cancer, p53 mutations are associated with tumor aggressiveness [82].
Dual mutations in KRAS and TP53 are an independent predictor of decreased overall survival in metastatic colorectal cancer [83]. In prostate cancer, despite the classic role of prostate-specific antigen (PSA) as a biomarker, screening for p53 mutations in 90 tumor samples from radical prostatectomy demonstrated that mutations in exon 7 and 8 contribute to tumor recurrence more significantly than the pre-therapeutic PSA level and tumor stage [84]. Furthermore, detection of mutant p53 at DNA and/or protein levels also suggests its role as a biomarker for metastatic diseases in endometrial endometrioid carcinoma and epithelial ovarian cancer [85][86].
Therapeutic strategies to target mutant p53 in metastasis
Several therapeutic strategies have been developed to potentially target mutant p53. These efforts include developing agents to restore wild-type p53 structure and activity, to promote mutant p53 degradation, to disrupt interactions of mutant p53 and binding partners, and to exploit synthetic lethality in mutant p53 tumors [87-89]. To re-establish wild-type p53 function, the small molecule p53 reactivation and induction of massive apoptosis (PRIMA-1) has been identified from a screening of 2,000 compounds in suppressing mutant p53-dependent cell growth [90][91]. PRIMA-1 and its methylated analogue APR-246 not only restore p53 mutants to a wild-type protein structure, but also revoke the wild-type transcriptional activity such as inducing Bax and PUMA expressions [92][93]. Furthermore, in preclinical studies, APR246 demonstrated promising tumor suppressive effects in breast cancer, small cell lung cancer, and multiple myeloma mouse models [93-95]. A phase I/IIa clinical trial of APR-246 was conducted in 22 patients with hematologic malignancies and prostate cancer, where p53-dependent effects were induced in tumor cells [96]. Currently, more clinical trials of APR-246 are in progress (ClinVar: NCT03931291i, NCT03745716ii). In addition, the combination of PRIMA-1 with Nutlin-3, an MDM2 inhibitor, revealed synergistic effects in a pancreatic cancer cell line [97]. Given the availability of MDM2 inhibitors in clinical trials (e.g.: ClinVar: NCT03041688iii, NCT03107780iv, NCT03217266v, NCT03031730vi), this combinatorial treatment may enhance the efficiency of targeting mutant p53 [98][99]. Zn2+-chelating compounds represent another category of agents to restore wild-type p53 function. Zn2+ is a critical factor for folding of wild-type p53. Some Zn2+ metallochaperones, such as ZMC1, promote intracellular Zn2+ transport and optimize intracellular Zn2+ concentrations to enhance Zn2+ binding to mutant p53 especially p53R175H. Therefore, p53 mutants that are deficient in Zn2+ binding are reactivated [99]. Recently, a novel Zn2+ chelator COTI-2 has been developed to restore wild-type p53 activities in ovarian cancer, breast cancer and head and neck squamous cell carcinoma (HNSCC) cell lines [100-102]. A phase I study of COTI-2 is active ( NCT02433626vii).
Strategies to destabilize mutant p53 include HSP90 inhibitors, HDAC inhibitors and SIRT1 activators [87][103]. These agents synergize the mutant p53 degradation effects with others and reduce mutant p53 enrichment and activity at both transcriptional and post-translational levels. Several pre-clinical and clinical studies are being performed to evaluate their tumor suppressive effects [87].
A large body of evidence has demonstrated the interactions of mutant p53 with p63, p73 and other TFs. These interactions serve as important mechanisms for the oncogenic functions of mutant p53, including promoting cancer metastasis [65][67]. Therefore, disrupting these interactions, such as inhibiting the formations of mutant p53/p73 and mutant p53/p63 complexes could be a promising strategy [88]. For example, small molecules such as RETRA, short interfering peptides SIMPs and peptide aptamers have demonstrated the capacity to block mutant p53/p73 interaction and to induce promising tumor suppressive effects [104-106]. However, translating the application of small molecules and short peptides to clinic may be challenging. More studies are needed to ensure the efficient delivery to the tumor site and minimize toxicity in normal cells [88].
Finally, other emerging therapies have shown promising results. Targeting downstream pathways driven by mutant p53 contributes to tumor suppressive effects such as inhibiting metastasis. For example, mutant p53 upregulates PDGFRβ to promote lung metastasis in a PDAC mouse model. Pharmacological inhibition of PDGFRβ by imatinib reduces the occurrence of lung metastasis in a PDAC model harboring mutant p53 [44]. In addition, p53 mutations as tumor associated oncogenic events may influence the immune landscape [107][108]. Results from a TP53-specific screening assay in common epithelial tumors demonstrated that autologous mutant p53 neoantigens can be recognized by tumor-infiltrating lymphocytes (TILs), indicating that TP53 mutations are immunogenic [107]. In ovarian cancer metastases, infiltration of TP53 mutation-reactive T cells was identified [108]. Together, these results indicate that adoptive T cell therapy against neoantigens found in TP53 mutations may become a potential therapeutic approach to target mutant p53 mediated metastasis [108][107].
Concluding remarks
Despite a recent study questioning the gain-of-function properties of mutant p53[109], a large body of evidence has demonstrated the role of mutant p53 in promoting cancer metastasis. To achieve this, mutant p53 harnesses multiple mechanisms (Figure 1). To reduce the integrity of cell-cell junctions and disrupt epithelial polarity, mutant p53 induces the EMT program through upregulating certain TFs and interacting with the TGFβ pathway. To promote cell dissemination from the primary tumor site, mutant p53 enhances cell motility through regulation of cell-ECM interactions and cytoskeleton machinery. Enhanced cell invasion, motility, and metastatic capacity is also contributed through activated RTK pathways, which in turn are regulated by mutant p53. Mechanistically, mutant p53 interacts with several binding partners such as p63 and p73.
Recent studies have revealed novel mechanisms of mutant p53 in promoting metastasis. To survive the multi-step metastatic cascade, metastatic cells reprogram gene expression rewire metabolism to adapt to foreign microenvironments. Non-coding RNAs, such as miRNAs and lncRNAs can be regulated by mutant p53 to affect a broad spectrum of gene expression. Mutant p53 also affects mitochondrial function to control the Warburg effect in cancer cells. In addition, mutant p53 promotes exosome production, indicating that mutant p53 may actively regulate the tumor microenvironment in pre-metastatic and metastatic niches. Furthermore, recent studies have shown that mutant p53 plays an important role in epigenetic regulation [110-112]. Given the emerging findings that epigenetic reprogramming contributes to metastasis [113][114], an interesting topic for future study is to investigate whether the regulation of epigenome by mutant p53 enhances metastasis. Furthermore, many interesting aspects of mutant p53 driven metastasis remain to be elucidated (See “Outstanding Questions” section). For example, how does mutant p53 affect individual steps of metastasis such as cancer cell intravasation, circulation, extravasation? What is the role of mutant p53 in regulating metastatic cell dormancy? Does immune regulation involve in mutant p53 driven metastasis? Besides hematogenous metastasis, how does mutant p53 affect lymphatic dissemination?
Outstanding questions:
How does mutant p53 affect intravasation, circulating tumor cell survival and extravasation in the metastatic cascade?
How does mutant p53 regulate the immune system in tumor metastasis?
What is the possible relationship of the epigenome and mutant p53 in tumor metastasis?
What is the role of mutant p53 in regulating dormancy of metastatic cells?
How does mutant p53 affect chemoresistance of metastatic cells?
What is the efficacy of therapeutics in targeting mutant p53 in tumor metastasis?
How does mutant p53 regulate lymph node metastasis?
Finally, therapeutic strategies have been developed to target mutant p53 from different aspects. These include strategies to restore wild-type p53 functions, to destabilize mutant p53, to interfere binding partner interactions, and to exploit synthetic lethality though combining other pathway inhibitors (Figure 2). Future studies may provide insights into therapeutic approaches to target mutant p53 in primary tumors versus metastatic tumors, and demonstrate the role of mutant p53 in metastatic cell chemoresistance.
Highlights:
TP53 is the most frequently mutated gene in human cancers. Mutant p53 can exert oncogenic effects and enhance metastasis in diverse cancers.
Mutant p53 promotes cell motility and metastasis through epithelial-to-mesenchymal transition (EMT), receptor tyrosine kinase (RTK) signaling, extracellular matrix (ECM) interactions, mitochondrial metabolism, non-coding RNAs and exosome secretion, and other biological processes.
Mechanistically, mutant p53 interacts with several transcriptional factors (TFs) to influence gene expression in the promotion of metastasis.
Mutant p53 is an important prognosis factor in metastasis and clinical outcomes. Therapeutics targeting mutant p53 are under investigation.
Acknowledgements
We apologize to the authors whose work we could not cite due to space limitations. This work was funded by NIH grant P01-CA098101 (AKR) and the American Cancer Society Research Professorship (AKR).
Glossary
- ECM
A non-cellular component that is composed of extracellular macromolecules, providing physical, biochemical and biomechanical support for surrounding cells.
- EMT
A cellular process that cell-cell adhesion and cell polarity are disrupted in epithelial cells, and mesenchymal transcriptional programs are activated. It is an important process in development, wound healing, fibrosis and cancer progression.
- Exosome
Nano-sized extracellular vesicles that are derived from endosomal system and released into extracellular environment. Secreted exosomes carry various cargo such as RNAs and cytokines.
- Metastasis
A multi-step process whereby cancer cells spread from primary tumor and colonize in distant organs by way of blood or lymph system.
- Mutant p53 “Gain-of-function”
The function of mutant p53 to act as an oncogenic protein and promote cancer malignancy such as tumor growth, invasion and metastasis, beyond the function of sequestering wild-type p53 from DNA binding.
- Non-coding RNA
RNA transcripts that do not encode protein products. This group of RNA includes miRNA, siRNA, piRNA and lncRNA.
- RTK
A large family of receptors for cytokines and growth factors, playing important roles in cellular communication and signal transduction.
- Transcription factors
Proteins that activate gene transcription. They recognize and bind to promoter or enhancer regions of DNA, directly interact with RNA polymerase, and regulate mRNA production.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Resources
i https://clinicaltrials.gov/ct2/show/NCT03931291
ii https://clinicaltrials.gov/ct2/show/NCT03745716
iii https://clinicaltrials.gov/ct2/show/NCT03041688
iv https://clinicaltrials.gov/ct2/show/NCT03107780
v https://clinicaltrials.gov/ct2/show/NCT03217266
References
- 1.Gupta GP and Massagué J (2006) Review Cancer Metastasis: Building a Framework. DOI: 10.1016/j.cell.2006.11.001 [DOI] [PubMed]
- 2.Lambert AW et al. (2016) Review Emerging Biological Principles of Metastasis. Cell 168, 670–691. DOI: 10.1016/j.cell.2016.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Powell E et al. (2014) Contribution of p53 to metastasis. Cancer Discov. 4, 405–414. DOI: 10.1158/2159-8290.CD-13-0136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alderton GK (2017) Tumour evolution: Epigenetic and genetic heterogeneity in metastasis. Nat. Rev. Cancer 17, 141 DOI: 10.1038/nrc.2017.11 [DOI] [PubMed] [Google Scholar]
- 5.Kandoth C et al. (2013) Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. DOI: 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivlin N et al. (2011) Mutations in the p53 Tumor Suppressor Gene : Important Milestones at the Various Steps of Tumorigenesis. 2, 466–474. DOI: 10.1177/1947601911408889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller PAJ and Vousden KH (2014) Perspective Mutant p53 in Cancer : New Functions and Therapeutic Opportunities. Cancer Cell 25, 304–317. DOI: 10.1016/j,ccr.2014.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malkin D (2011) Li-Fraumeni Syndrome. DOI: 10.1177/1947601911413466 [DOI] [PMC free article] [PubMed]
- 9.Olive KP et al. (2004) Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860. DOI: 10.1016/j.cell.2004.11.004 [DOI] [PubMed] [Google Scholar]
- 10.Lang GA et al. (2004) Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119, 861–872. DOI: 10.1016/j.cell.2004.11.006 [DOI] [PubMed] [Google Scholar]
- 11.Tsai JH and Yang J (2013) Epithelial – mesenchymal plasticity in carcinoma metastasis. DOI: 10.1101/gad.225334.113.2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamouille S et al. (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. DOI: 10.1038/nrm3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dongre A and Weinberg RA (2019) New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol 20, DOI: 10.1038/S41580-018-0080-4 [DOI] [PubMed] [Google Scholar]
- 14.Dong P et al. (2013) Mutant p53 gain-of-function induces epithelial – mesenchymal transition through modulation of the miR-130b – ZEB1 axis. DOI: 10.1038/onc.2012.334 [DOI] [PMC free article] [PubMed]
- 15.Roger L et al. (2010) Gain of oncogenic function of p53 mutants regulates E-cadherin expression uncoupled from cell invasion in colon cancer cells. DOI: 10.1242/jcs.061002 [DOI] [PubMed]
- 16.Ohashi S et al. (2010) Epidermal Growth Factor Receptor and Mutant p53 Expand an Esophageal Cellular Subpopulation Capable of Epithelial-to-Mesenchymal Transition through ZEB Transcription Factors. 70, 4174–4185. DOI: 10.1158/0008-5472.CAN-09-4614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang S et al. (2009) p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. 11, DOI: 10.1038/ncb1875 [DOI] [PubMed] [Google Scholar]
- 18.Kogan-Sakin I et al. (2010) Mutant p53 R175H upregulates Twist1 expression and promotes epithelial – mesenchymal transition in immortalized prostate cells. Cell Death Differ. 18, 271–281. DOI: 10.1038/cdd.2010.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adorno M et al. (2009) A Mutant-p53/Smad Complex Opposes p63 to Empower TGFbeta -Induced Metastasis. Cell 137, 87–98. DOI: 10.1016/j.cell.2009.01.039 [DOI] [PubMed] [Google Scholar]
- 20.Neilsen PM et al. (2012) Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene 32, 2992–3000. DOI: 10.1038/onc.2012.305 [DOI] [PubMed] [Google Scholar]
- 21.Kalo E et al. (2007) Mutant p53 Attenuates the SMAD-Dependent Transforming Growth Factor beta 1 ( TGF- beta 1 ) Signaling Pathway by Repressing the Expression of TGF- beta Receptor Type II . Mol Cell Biol, 27, 8228–8242. DOI: 10.1128/MCB.00374-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ali A et al. (2013) Differential regulation of the REGγ-proteasome pathway by p53/TGF-β signalling and mutant p53 in cancer cells. DOI: 10.1038/ncomms3667 [DOI] [PMC free article] [PubMed]
- 23.Ji L et al. (2015) Mutant p53 Promotes Tumor Cell Malignancy by Both Positive and Negative Regulation of the Transforming Growth Factor β ( TGF- β ) Pathway J Biol Chem. 290, 11729–11740. DOI: 10.1074/jbc.M115.639351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng XM et al. (2010) Smad2 Protects against TGF-β / Smad3-Mediated Renal Fibrosis. DOI: 10.1681/ASN.2009121244 [DOI] [PMC free article] [PubMed]
- 25.Machesky LM (2008) Lamellipodia and filopodia in metastasis and invasion. 582, 2102–2111. DOI: 10.1016/j.febslet.2008.03.039 [DOI] [PubMed] [Google Scholar]
- 26.Kerber ML and Cheney RE (2011) Myosin-X : a MyTH-FERM myosin at the tips of filopodia. DOI: 10.1242/jcs.023549 [DOI] [PMC free article] [PubMed]
- 27.Arjonen A et al. (2014) Mutant p53 – associated myosin-X upregulation promotes breast cancer invasion and metastasis. J Clin Invest. 124,1069–82. DOI: 10.1172/JCI67280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ridley AJ et al. (1992) The Small GTP-Binding Protein rat Regulates Growth Factor-Induced Membrane. Cell, 70, 401–410. DOI: 10.1016/0092-8674(92)90164-8 [DOI] [PubMed] [Google Scholar]
- 29.Ridley AJ and Hall A (1992) The Small GTP-Binding Protein rho Regulates the Assembly of Focal Adhesions and Actin Stress Fibers in Response to Growth Factors. Cell, 70, 389–399. DOI: 10.1016/0092-8674(92)90163-7 [DOI] [PubMed] [Google Scholar]
- 30.Timpson P et al. (2011) Spatial Regulation of RhoA Activity during Pancreatic Cancer Cell Invasion Driven by Mutant p53. DOI: 10.1158/0008-5472.CAN-10-2267 [DOI] [PMC free article] [PubMed]
- 31.Eble JA and Niland S (2019) The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 36, 171–198. DOI: 10.1007/s10585-019-09966-1 [DOI] [PubMed] [Google Scholar]
- 32.Lu P et al. (2012) The extracellular matrix : A dynamic niche in cancer progression. J Cell Biol. 196, 395–406. DOI: 10.1083/jcb.201102147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamidi H and Ivaska J Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer DOI: 10.1038/s41568-018-0038-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ganguly KK et al. (2013) Integrins and metastasis. Cell Adh Migr 7, 251–61. DOI: 10.4161/cam.23840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller PAJ et al. (2009) Mutant p53 Drives Invasion by Promoting Integrin Recycling. Cell 139, 1327–1341. DOI: 10.1016/j.cell.2009.11.026 [DOI] [PubMed] [Google Scholar]
- 36.Caswell P and Norman J (2008) Endocytic transport of integrins during cell migration and invasion. DOI: 10.1016/j.tcb.2008.03.004 [DOI] [PubMed]
- 37.Selivanova G and Ivaska J (2009) Previews Integrins and Mutant p53 on the Road to Metastasis. Cell 63, 1220–1222. DOI: 10.1016/j.cell.2009.12.016 [DOI] [PubMed] [Google Scholar]
- 38.Kolbl AC et al. (2015) The role of glycosylation in breast cancer metastasis and cancer. Front Oncol. 5:219 DOI: 10.3389/fonc.2015.00219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh C et al. (2018) Biochemical and Biophysical Research Communications Integrin expression and glycosylation patterns regulate cell-matrix adhesion and alter with breast cancer progression. Biochem. Biophys. Res. Commun. 499, 374–380. DOI: 10.1016/j.bbrc.2018.03.169 [DOI] [PubMed] [Google Scholar]
- 40.Vogiatzi F et al. (2016) Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum. Proc Natl Acad Sci USA 53,E8433–E8442.DOI: 10.1073/pnas.1612711114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muller-Tidow C et al. (2005) Identification of Metastasis-Associated Receptor Tyrosine Kinases in Non – Small Cell Lung Cancer. DOI: 10.1158/0008-5472.CAN-04-3388 [DOI] [PubMed]
- 42.Lemmon MA and Schlessinger J (2010) Review Cell Signaling by Receptor Tyrosine Kinases. DOI: 10.1016/j.cell.2010.06.011 [DOI] [PMC free article] [PubMed]
- 43.Sasaki T et al. (2013) The Role of Epidermal Growth Factor Receptor in Cancer Metastasis and Microenvironment. Biomed Res Int. 2013: 546318 DOI: 10.1155/2013/546318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weissmueller S et al. (2013) Mutant p53 Drives Pancreatic Cancer Metastasis through Cell-Autonomous PDGF Receptor β Signaling. Cell 157, 382–394. DOI: 10.1016/j.cell.2014.01.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai Y (2010) Platelet-derived growth factor receptor tyrosine kinase inhibitors : a review of the recent patent literature. Expert Opin Ther Pat. 20(7):885–97. DOI: 10.1517/13543776.2010.493559 [DOI] [PubMed] [Google Scholar]
- 46.Trusolino L et al. (2010) MET signalling : principles and functions in development , organ regeneration and cancer. Nat. Rev Mol Cell Biol 11, 834–848. DOI: 10.1038/nrm3012 [DOI] [PubMed] [Google Scholar]
- 47.Benvenuti S and Comoglio PM (2007) The MET Receptor Tyrosine Kinase in Invasion and Metastasis. J Cell Physiol. 213(2): 316–25. DOI: 10.1002/JCP [DOI] [PubMed] [Google Scholar]
- 48.Sennino B et al. (2012) Suppression of Tumor Invasion and Metastasis by Concurrent Inhibition of c-Met and VEGF Signaling in Pancreatic Neuroendocrine Tumors. Cancer Discov. 2(3): 270–87. DOI: 10.1158/2159-8290.CD-11-0240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grugan KD et al. (2013) A common p53 mutation (R175H) activates c-Met receptor tyrosine kinase to enhance tumor cell invasion. Cancer Biol Ther. 14(9) 835–9. DOI: 10.4161/cbt.25406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muller PAJ et al. (2013) Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene 32, 1252–1265. DOI: 10.1038/onc.2012.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lobb RJ et al. (2017) Exosomes : Key mediators of metastasis and pre-metastatic niche formation. Semin Cell Dev Biol. 67, 3–10. DOI: 10.1016/j.semcdb.2017.01.004 [DOI] [PubMed] [Google Scholar]
- 52.Ulrich Weidle H. et al. (2017) The Multiple Roles of Exosomes in Metastasis. Cancer Genomics Proteomics. 14(1): 1–16. DOI: 10.21873/cgp.20015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steinbichler TB et al. (2017) The role of exosomes in cancer metastasis. Semin Cancer Biol. 44, 170–181. DOI: 10.1016/j.semcancer.2017.02.006 [DOI] [PubMed] [Google Scholar]
- 54.Novo D et al. (2018) Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat Commun. 9(1): 5069 DOI: 10.1038/s41467-018-07339-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cooks T et al. (2018) Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat Commun. 9(1):771 DOI: 10.1038/s41467-018-03224-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang Q et al. (2018) Long non-coding RNAs in metastasis. Cancer Metastasis Rev. 37(1):75–81. DOI: 10.1007/s10555-017-9713-x [DOI] [PubMed] [Google Scholar]
- 57.Kim J et al. (2018) MicroRNAs and metastasis: small RNAs play big roles. Cancer Metastasis Rev. 37, 5–15. DOI: 10.1007/s10555-017-9712-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Subramanian M et al. (2015) A mutant p53 / let-7i-axis-regulated gene network drives cell migration, invasion and metastasis. Oncogene. 34(9):1094–104 DOI: 10.1038/onc.2014.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gutschner T et al. (2013) The Noncoding RNA MALAT1 Is a Critical Regulator of the Metastasis Phenotype of Lung Cancer Cells. Cancer Res. 73, 1180–1190. DOI: 10.1158/0008-5472.CAN-12-2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pruszko M et al. The mutant p53-ID4 complex controls VEGFA isoforms by recruiting lncRNA MALAT1. EMBO Rep. 18(8): 1331–1351. DOI:15252/embr.201643370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heiden Vander MG and Deberardinis RJ (2017) Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. DOI: 10.1016/j.cell.2016.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lehuede C et al. (2016) Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res. 76 (18): 5201–8. DOI: 10.1158/0008-5472.CAN-16-0266 [DOI] [PubMed] [Google Scholar]
- 63.Mantovani F, et al. (2019) Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 26 (2): 199–212. DOI: 10.1038/s41418-018-0246-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang C et al. (2013) Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 4: 2935 DOI: 10.1038/ncomms3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Freed-pastor WA and Prives C (2012) Mutant p53 : one name , many proteins. Genes Dev. 26(12): 1268–86. DOI: 10.1101/gad.190678.112.1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Basu S et al. (2018) Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1 α. Genes Dev. 32(3-4): 230–243. DOI: 10.1101/gad.309062.117.mouse [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim MP and Lozano G (2018) Mutant p53 partners in crime. Cell Death Differ. 25, 161–168. DOI: 10.1038/cdd.2017.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Melino G (2011) p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 18, 1487–1499. DOI: 10.1038/cdd.2011.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gaiddon C et al. (2001) A Subset of Tumor-Derived Mutant Forms of p53 Down-Regulate p63 and p73 through a Direct Interaction with the p53 Core Domain. 21, 1874–1887. DOI: 10.1128/MCB.21.5.1874-1887.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Strano S et al. (2002) Physical Interaction with Human Tumor-derived p53 Mutants Inhibits p63 Activities. J Biol Chem. 277, 18817–18826. DOI: 10.1074/jbc.M201405200 [DOI] [PubMed] [Google Scholar]
- 71.Como CJDI et al. (1999) p73 Function Is Inhibited by Tumor-Derived p53 Mutants in Mammalian Cells. Mol Cell Biol 19, 1438–1449. DOI: 10.1128/mcb.19.2.1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Strano S et al. (2000) Physical and Functional Interaction between p53 Mutants and Different Isoforms of p73. J Biol Chem 275, 29503–29512. DOI: 10.1074/jbc.M003360200 [DOI] [PubMed] [Google Scholar]
- 73.Chicas A et al. (2000) Mutant p53 Forms a Complex with Sp1 on HIV-LTR DNA. Biochem Biophys Res Commun 390, 383–390. DOI: 10.1006/bbrc.2000.3965 [DOI] [PubMed] [Google Scholar]
- 74.Fontemaggi G et al. (2009) The execution of the transcriptional axis mutant p53 , E2F1 and ID4 promotes tumor neo-angiogenesis. Nat. Struct. Mol. Biol 16, 1086–1093. DOI: 10.1038/nsmb.1669 [DOI] [PubMed] [Google Scholar]
- 75.Do PM et al. (2012) Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 26(8):830–45 DOI: 10.1101/gad.181685.111.830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pourebrahim R et al. (2017) Integrative genome analysis of somatic p53 mutant osteosarcomas identifies Ets2-dependent regulation of small nucleolar RNAs by mutant p53 protein. Genes Dev. 31, 1847–1857. DOI: 10.1101/gad.304972.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robles AI and Harris CC (2010) Clinical Outcomes and Correlates of TP53 Mutations and Cancer. Cold Spring Harb Perspect Biol 2(3):a001016 DOI: 10.1101/cshperspect.a001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peller S et al. (1995) p53 Mutations in Matched Primary and Metastatic Human Tumors. Mol Carcinog. 13(3):166–72. [DOI] [PubMed] [Google Scholar]
- 79.Ding L et al. (2010) Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005. DOI: 10.1038/nature08989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alsner J et al. (2000) Heterogeneity in the Clinical Phenotype of TP53 Mutations in Breast Cancer Patients. Clin Cancer Res 6(10): 3923–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang P et al. (2013) The impact of p53 in predicting clinical outcome of breast cancer patients with visceral metastasis. Sci Rep. 3:2246 DOI: 10.1038/srep02246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huszno J and Grzybowska EWA (2018) TP53 mutations and SNPs as prognostic and predictive factors in patients with breast cancer. Oncol Lett. 16(1):34–40. DOI: 10.3892/ol.2018.8627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chun YS et al. (2019) Deleterious Effect of RAS and Evolutionary High-risk TP53 Double Mutation in Colorectal Liver Metastases. Ann Surg. 269, 917–923. DOI: 10.1097/SLA.0000000000002450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ecke TH et al. (2010) TP53 Gene Mutations in Prostate Cancer Progression. Anticancer Res. 30(5):1579–1586. [PubMed] [Google Scholar]
- 85.Ono M et al. (2017) Dual expression of immunoreactive estrogen receptor β and p53 is a potential predictor of regional lymph node metastasis and postoperative recurrence in endometrial endometrioid carcinoma. Plos One. 12(11):e0188641 DOI: 10.1371/journal.pone.0188641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang Y et al. (2016) TP53 mutations in epithelial ovarian cancer. Transl Cancer Res. 5(6): 650–663. DOI: 10.21037/tcr.2016.08.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kastenhuber ER and Lowe SW (2017) Putting p53 in Context. Cell 170(6), 1062–1078. DOI: 10.1016/j.cell.2017.08.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blandino G and Agostino Di S (2018) New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J Exp Clin Cacner Res. 37(1):30 DOI: 10.1186/s13046-018-0705-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou X et al. (2019) Mutant p53 in cancer therapy — the barrier or the path. J Mol Cell Biol. 11(4), 293–305. DOI: 10.1093/jmcb/mjy072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bykov VJ, et al. (2002) Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med 8(3):282–8. DOI: 10.1038/nm0302-282 [DOI] [PubMed] [Google Scholar]
- 91.Bykov VJ, et al. (2018) Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs : a statistical analysis of information in the National Cancer Institute database. Carcinogenesis. 23(12), 2011–2018. DOI: 10.1093/carcin/23.12.2011 [DOI] [PubMed] [Google Scholar]
- 92.Wang T et al. (2007) PRIMA-1 induces apoptosis by inhibiting JNK signaling but promoting the activation of Bax. Biochem Biophys Res Commun. 352(1), 203–212. DOI: 10.1016/j.bbrc.2006.11.006 [DOI] [PubMed] [Google Scholar]
- 93.Zandi R et al. (2011) Cancer Therapy : Preclinical PRIMA-1 Met / APR-246 Induces Apoptosis and Tumor Growth Delay in Small Cell Lung Cancer Expressing Mutant p53. Clin Cancer Res. 17(9) 2830–2842. DOI: 10.1158/1078-0432.CCR-10-3168 [DOI] [PubMed] [Google Scholar]
- 94.Liang Y et al. (2009) PRIMA-1 inhibits growth of breast cancer cells by re-activating mutant p53 protein. Int J Oncol. 35(5): 1015–23. DOI: 10.3892/ijo [DOI] [PubMed] [Google Scholar]
- 95.Saha MN et al. (2013) PRIMA-1 Met / APR-246 Displays High Antitumor Activity in Multiple Myeloma By Induction of p73 and Noxa. Mol Cancer Ther 12(11):2331–41. DOI: 10.1158/1535-7163.MCT-12-1166 [DOI] [PubMed] [Google Scholar]
- 96.Bykov VJN et al. (2019) Targeting p53 in Vivo : A First-in-Human Study With p53-Targeting Compound APR-246 in Refractory Hematologic Malignancies and Prostate Cancer. J Clin Oncol. 30(29): 3633–3639. DOI: 10.1200/JCO.2011.40.7783 [DOI] [PubMed] [Google Scholar]
- 97.Izetti P et al. (2014) PRIMA-1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Invest. New Drugs 32, 783–794. DOI: 10.1007/s10637-014-0090-9. [DOI] [PubMed] [Google Scholar]
- 98.Hoe KK et al. (2014) Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 13, 217–236. DOI: 10.1038/nrd4236 [DOI] [PubMed] [Google Scholar]
- 99.Bykov VJN et al. (2018) Targeting mutant p53 for efficient cancer therapy. Nat. Rev Cancer. 18(2): 89–102. DOI: 10.1038/nrc.2017.109 [DOI] [PubMed] [Google Scholar]
- 100.Salim KY et al. COTI-2 , a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget. 7(27):41363–41379. DOI: 10.18632/oncotarget.9133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vareki SM et al. (2018) Novel anti-cancer drug COTI-2 synergizes with therapeutic agents and does not induce resistance or exhibit cross-resistance in human cancer cell lines. PLoS One. 13(1): e019766 DOI: 10.1371/journal.pone.0191766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lindemann A, et al. (2019) COTI-2, A novel thiosemicarbazone derivative, exhibits antitumor activity in HNSCC through p53-dependent and -independent mechanisms. Clin Cancer Res. 25(18):5650–5662 DOI: 10.1158/1078-0432.CCR-19-0096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Parrales A and Iwakuma T (2015) Targeting Oncogenic Mutant p53 for Cancer Therapy. Front Oncol. 5:288 DOI: 10.3389/fonc.2015.00288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kravchenko JE et al. (2008) Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent. Proc Natl Acad Sci USA. 105(17):6302–7. DOI: 10.1073/pnas.0802091105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Agostino S Di et al. (2008) The disruption of the protein complex mutant p53/p73 increases selectively the response of tumor cells to anticancer drugs. Cell Cycle. 7(21):3440–7. DOI: 10.4161/cc.7.21.6995 [DOI] [PubMed] [Google Scholar]
- 106.Guida E et al. (2008) Peptide Aptamers Targeting Mutant p53 Induce Apoptosis in Tumor Cells. Cancer Res. 68(16):6550–8. DOI: 10.1158/0008-5472.CAN-08-0137 [DOI] [PubMed] [Google Scholar]
- 107.Malekzadeh P et al. (2019) Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Invest 129, 1109–1114. DOI: 10.1172/JCI123791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Deniger DC et al. (2018) T-cell responses to TP53 “Hotspot” Mutations and unique neoantigens expressed by human ovarian cancers. Clin. Cancer Res. 24, 5562–5573. DOI: 10.1158/1078-0432.CCR-18-0573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Giacomelli AO et al. (2018) Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet 50, 1381–1387. DOI: 10.1038/s41588-018-0204-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhu J, et al. (2015) Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 525 (7568):206–11. DOI: 10.1038/nature15251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pfister NT et al. (2015) Mutant p53 cooperates with the SWI / SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 29(12):1298–315. DOI: 10.1101/gad.263202.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rahnamoun H et al. (2018) Mutant p53 regulates enhancer-associated H3K4 monomethylation through interactions with the methyltransferase MLL4. J Biol Chem. 293(34):13234–13246. DOI: 10.1074/jbc.RA118.003387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mcdonald OG et al. (2017) Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet 49(3): 367–376. DOI: 10.1038/ng.3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chatterjee A et al. (2018) Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 51: 149–159. DOI: 10.1016/j.semcancer.2017.08.004 [DOI] [PubMed] [Google Scholar]