

Abstract
Cell penetrating peptides (CPPs) are 9–35mer cationic and/or amphipathic peptides that are rapidly internalized across cell membranes. Importantly, they can be linked to a variety of cargo, including anti-cancer therapeutics, making CPPs an efficient, effective and non-toxic mechanism for drug delivery. In this review, we discuss a number of CPP conjugated therapies (CTTs) that are either patented are in the progress of patenting, and show strong promise for clinical efficacy. The CTTs discussed here target a number of different processes specific to cancer progression, including proliferation, survival and migration. In addition, many of these CTTs also increase sensitivity to current anti-cancer therapy modalities, including radiation and other DNA damaging chemotherapies, thereby decreasing the toxic dosage required for effective treatment. Mechanistically, these CTTs function in a dominant-negative manner by blocking tumor-specific protein-protein interactions with the CPP-conjugated peptide or protein. The treatment of both cell lines and mouse models demonstrates that this method of molecular targeting results in equal if not greater efficacy than current standards of care, including DNA damaging agents and topoisomerase inhibitors. For the treatment of invasive carcinoma, these CTTs have significant clinical potential to deliver highly targeted therapies without sacrificing the patient’s quality of life.
Keywords: Cell penetrating peptides, protein transduction domain, Tat, targeted therapies, patent
INTRODUCTION
Significant progress has been made in the identification of tumor specific cellular pathways which can be successfully blocked in vitro. Unfortunately, the safe, systemic delivery of these blocking agents to patients has been difficult and sometimes impossible due to difficulty with tissue penetration, toxicity and functional activity in vivo. The recent development of cell penetrating peptides (CPPs) has overcome these barriers, leading to the development of novel tumor-specific molecular therapeutics. Also known as protein transduction domains (PTD), CPPs consist of short amino acid sequences that are able to mediate the translocation of a conjugated cargo across the plasma membrane. Once inside the cell, CPP-associated cargoes are then able to interact with their intracellular targets, and block their tumorigenic properties. Importantly, the non-toxic mechanism of cell penetration allows for the safe and effective systemic delivery of cancer therapeutics.
Over the last decade, several laboratories have utilized naturally occurring and synthetic CPPs to deliver several types of therapeutic cargo, including proteins, antibodies, peptides, and nucleic acids directly into the cell’s cytoplasm [1–4]. CPPs are easily manipulated and optimized, and can be used to target cargo to a specific cell-type or tissue [5]. For example, one sub-group of CPPs can cross the blood-brain barrier, allowing for the development of therapies that target brain cancers which are normally difficult to treat with conventional chemotherapies [6, 7]. Importantly, CPP bound therapies offer a novel approach to treat cancer in a specific and non-toxic manner, as damage to the cell membrane, receptors or transporters is not required for entry into the cell. In this review we will discuss naturally occurring and optimized protein transduction domains and their utilization in the delivery of anti-cancer therapies into intracellular compartments of diseased cells. We have focused on studies with pending or recently awarded patents, whose functionality has been verified and published, and discuss the very promising future of these therapeutics for the treatment of cancer.
CELL PENETRATING PEPTIDES
CPPs were first discovered through the study of model organisms, such as Drosophila melanogaster (Antennapedia, Antp) and the Human Immunodeficiency Virus (HIV Tat) retrovirus, and there are now two fundamental types of CPPs that have been utilized for intracellular delivery of therapeutic molecules [6, 8]. The first and the most common are the cationic CPPs, which are short amino acid sequences that are mainly composed of arginine, lysine, and histidine. These amino acids are responsible for the cationic charge of the peptide that will mediate the interaction of the peptide with anionic motifs on the plasma membrane. Both naturally occurring and synthetic cationic CPPs have been described and are shown in Table 1. A second class of CPPs is the amphipathic peptides, which have lipophilic and hydrophilic tails that are responsible for mediating the peptide translocation across the plasma membrane (Table 1). Although these classes of CPPs are composed of different amino acids, and the mechanism of intracellular delivery is similar.
Table 1.
Commonly Utilized Cationic and Amphipathic Cell Penetrating Peptides. More specifically, this review will discuss recent patents that utilize CPPs 1, 2, 4 and 6. However, the remaining CPPs are also relevant to the field of peptide-based therapies.
| Name | Amino Acid Sequence | TypeofCPP | Ref | |
|---|---|---|---|---|
| 1 | HIV Tat (47–58) | YGRKKRRQRRR | Cationic | [6] |
| 1a | PTD-3 | YARKARRQARR | Cationic | [48] |
| 1b | PTD-4 | YARAAARQARA | Cationic | [48] |
| 1c | PTD-5 | YARAARRAARR | Cationic | [48] |
| 1d | PTD-6 | YARAARRAARA | Cationic | [48] |
| 1e | P3 | YGRKKRRQR | Cationic | [23] |
| 1f | Polyarginine | Rn | Cationic | [80] |
| 2 | Antennapedia (Antp) | RQIKIWFQNRRMKWKK | Cationic | [8] |
| 3 | VP-22 | DAATATRGRSAASRPTERPRAPARSASRPRRPVD | Cationic | [81] |
| 4 | PTD-SN1 | RKMLKSTRRQRR | Cationic | [13] |
| 5 | Bac | RRIRPRPPRLPRPRPRLPFPRPG | Cationic | [82] |
| 6 | DPT | VKKKKIKREIKI | Cationic | [83] |
| 7 | Transportan | GWTLNSAGYLLGKINLKALAALAKKIL | Amphipathic | [84] |
| 8 | Pep-1 | KETWWETWWTEWSQPKKKRKV | Amphipathic | [85] |
Mechanism of Cell Penetrating Peptides
The detailed mechanism of action by which CPPs transverse the plasma membrane and interact with cytosolic proteins was recently reviewed, and readers are encouraged to consult those publications for the details we will summarize here [9, 10]. Briefly, CPPs are composed of several cationic amino acids, such as lysine or arginine, and a group of these amino acids result in a highly positively charged peptide sequence. These positively charged sequences are able to interact with acidic motifs on the plasma membrane in a receptor-independent fashion. Acidic motifs can be several different substrates found on the plasma membrane, including proteoglycans and glycolipids [11, 12]. A recent patent has introduced the use of synthetic CPP, PTD-SN1, to improve the efficiency of this interaction and ultimately the peptide’s cytosolic translocation [13]. Following interaction with these substrates, the peptides undergo cell-type independent internalization [14]. The exact mechanism of this internalization process is yet to be elucidated but it is dependent on several peptide attributes, including type of CPP and the size and charge of the cargo. A recent study has indicated that Antp and Tat CPP internalization can be mediated by clathrin-dependent endocytosis, caveolin-dependent endocytosis, macropinocytosis, or direct intracellular translocation [11] Fig. (1). Another report used therapeutic cargo to alter the rate of endocytosis, which allowed the therapy to act in an extracellular fashion [15]. Following endosomal internalization, the CPP-tagged therapy (CTT) escapes lysosomal degradation by a process that is poorly understood. However, it is known that endosomal escape is the rate-limiting step in delivering therapeutic doses of CTTs to the cytosol [9]. Once the CTT is released into the cytosol, it is able to directly interact with its target. In vitro, a CPP (Tat) linked therapy has been demonstrated to enter the cell and start affecting their target within five minutes of the treatment [16]. In a mouse tumor model, a GFP-CTT (Tat) was observed to be localized at the tumor site within 30 minutes of the treatment, thus illustrating CPPs’ uptake speed and efficiency [3].
Fig. (1). Mechanism of Peptide Membrane Transduction.
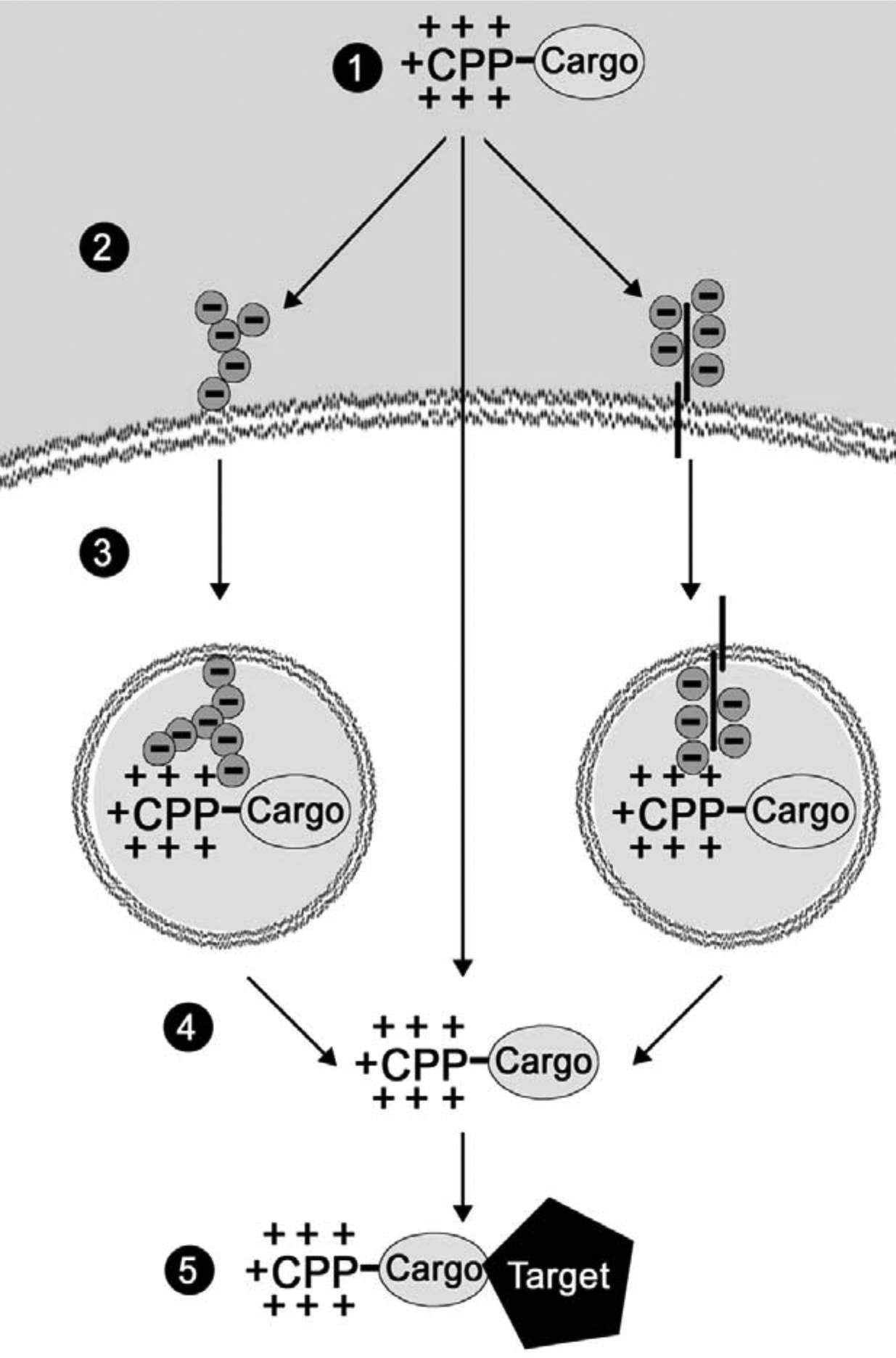
1) CPP-Cargo peptide is delivered to the site of disease. 2) The CPP’s highly cationic amino acids interact with proteoglycans (left) or glycolipids (right) on the cell surface. Note: There is an evidence that the CPP-cargo directly enters the cell (center arrow) 3) The peptide is endocytosed via clathrin or caveoloae or it is macropinocytosed. 4) Endosomal escape of peptide. 5) Interaction with target protein.
Non-Immunogenic
One of the benefits of using CPPs for therapeutic delivery is the lack of toxicity as compared to other cytoplasmic delivery devices, such as liposomes, etc. One potential drawback to using exogenous peptides, though, is the ability to induce a humoral immune response against the therapy, which can be highly detrimental for the subsequent treatments. A commonly used CPP derived from HIV’s Transcriptional Transactivator (Tat) was investigated for immunogenicity, and the portions of the Tat protein involved in eliciting a humoral immune response were determined. The authors discovered that the portion of Tat that conveys protein transduction, PTD, failed to induce an immune response [17]. In addition, our group has evaluated the induction of an immune response following repeated PTD-4 injection in immune intact mice, and observed no significant change in CD45-positive lymphocyte infiltration into tumors [3]. These studies indicate that the use of Tat’s PTD to deliver anti-cancer therapy does not induce a measurable immune response, although long-term toxicity studies in patients have not been performed yet. With respect to another PTD, Antp, several studies have determined that Antp efficiently elicits an immune response to the peptide cargo following processing by an antigen presenting cell [18, 19]. These examples demonstrate the possibility of modulating the immune response to the CTT based on the PTD used to deliver the therapeutic peptide.
ANTI-CANCER CELL PENETRATING PEPTIDES
Since 1988, when the first protein transduction domains were described, the use of CPPs to deliver anti-cancer therapies (CTTs) to the cytosol and nuclear compartments has become more common. Over the last decade numerous reports and patents have been published demonstrating the potential of CTTs for the treatment of leukemia and breast, lung, pancreatic, ovarian, colon cancers [1–3, 15, 20–23]. With respect to anti-cancer therapies, CTTs have been used to inhibit oncoprotein nuclear translocation, modulate oncoprotein signaling, and block metastases-promoting cell-ECM interactions Fig. (2) [2, 3, 15, 23]. The mechanism of inhibition is similar in most of the cases: a target interaction is determined and a portion or the whole interacting protein is attached to a CPP. The CTT is then transduced across the plasma membrane and delivered into the cytosol or nucleus to block the endogenous pro-cancerous interactions from occurring.
Fig. (2). Mechanism of CTT Anti-Cancer Therapy.
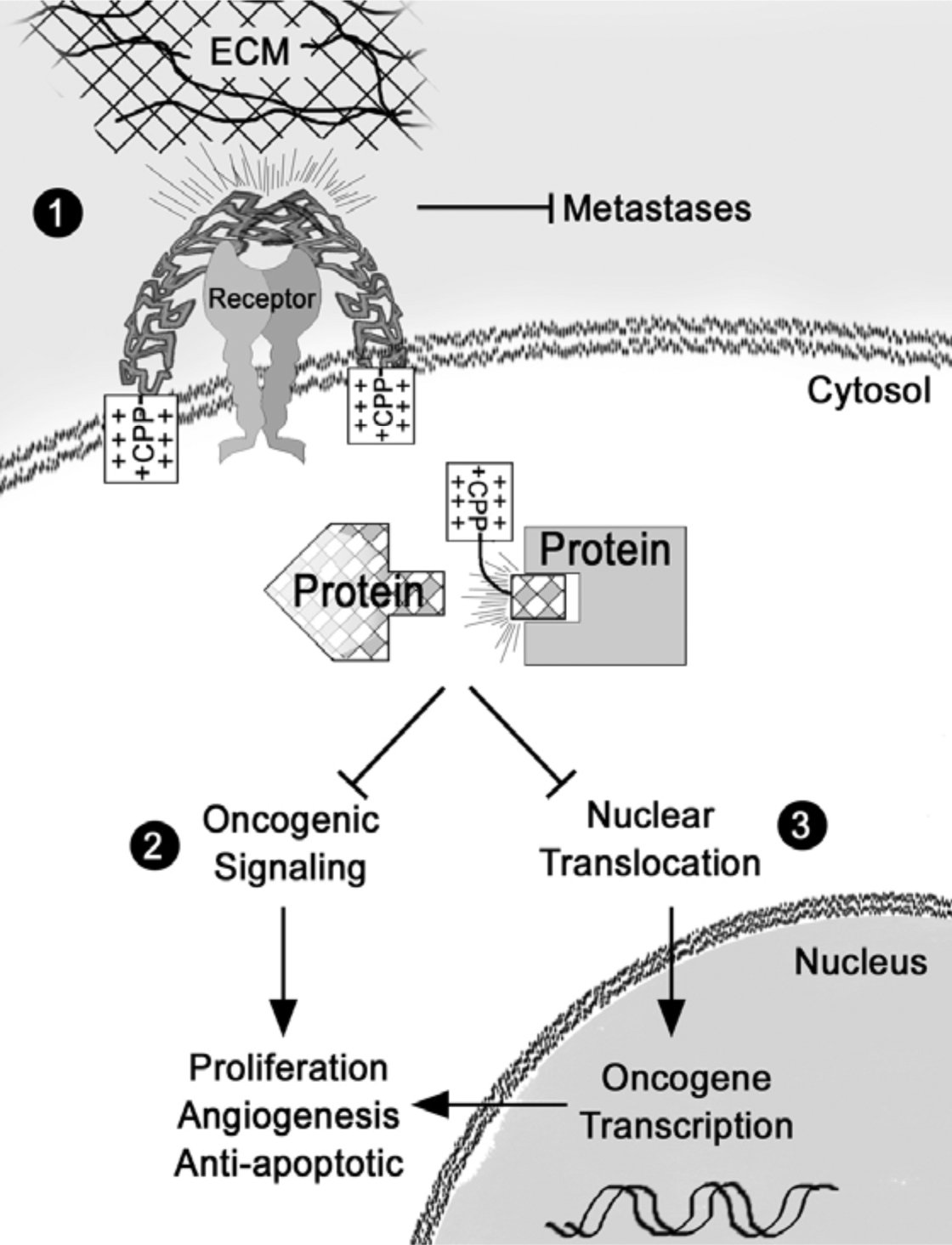
1) The CPP portion of the CTT attaches to the plasma membrane and directly inhibits ECM Receptor/ECM interaction. This inhibits the ability of metastatic cancer cells to establish secondary tumor sites. 2) CTTs block cancer dependent protein-protein interactions, resulting in the inhibition of proliferation, angiogenesis, and chemoresistance. 3) Additionally, inhibition of protein interactions can modulate nuclear translocation of oncogenic transcription factors.
Nuclear Translocation
Protein trafficking is a common cellular mechanism that regulates signal transduction and transcriptional activation. Following oncoprotein signaling, the translocation of activated proteins into the nucleus often results in the transcription of genes that are responsible for conveying many of the hallmark of cancers [24]. Increases in oncoprotein nuclear translocation are often observed in cancer cells and can further promote transformation and metastases.
Following DNA damage, there are a number of cell-cycle inhibitor proteins that become activated and are translocated to the nucleus. For example, in normal cells, DNA damage induces p21 nuclear translocation and cell cycle arrest, however, the extent of DNA damage determines, whether the cell undergoes caspase-dependent apoptosis or is able to repair its DNA [25, 26]. Current anti-cancer modalities, such as radiation and most chemotherapies cause irreparable DNA aberrations, which induces the cell to undergo apoptosis. However, these treatments can also result in the generation of a sub-population of cancer cells that aberrantly upregulate p21, which results in the inhibition of caspase 3-dependent apoptosis, thereby allowing the cells to survive the treatment [27, 28]. Blocking p21 nuclear translocation could inhibit the ability of p21 to convey chemoresistance, resulting in sensitizing cancer cells and allowing for a reduction in chemotherapy dosages. In an effort to sensitize cancer cells to the DNA damaging effects of radiation and chemotherapy, a recent study utilized a CPP conjugated to an anti-p21 antibody. The CPP-anti-p21 effectively blocked p21 translocation, allowing cells to progress through the cell cycle following chemotherapeutic exposure and replicate severe DNA adducts, inducing apoptosis [2]. These authors demonstrated that an IgG region could be conjugated to Tat’s PTD, (immunoconjugates; ICs) and the tat-anti-p21 IC was shown to inhibit the translocation of p21 into the nucleus [29]. Inhibition of nuclear trafficking of p21 resulted in the loss of the p21 dependent anti-apoptotic effect, and sensitized breast cancer cells to both -radiation and Camptothecin, a Topoisomerase inhibitor [2]. This study offers an example of how the tat-anti-p21 IC, in combination with chemotherapies, could result in lower and more effective dosages as well as less discomfort for the patient. The use of CPPs to deliver antibodies directly into the cytosol and nucleus of cancer cells has tremendous potential, allowing for the intracellular targeting of cancer-specific protein mutations.
Aberrant nuclear translocation of proteins is also observed, when cells are exposed to pro-inflammatory cytokines, an event frequently observed in epithelial cancers [Reviewed in [30]]. A protein commonly activated and translocated to the nucleus in the presence of these cytokines is the transcription factor, NF-κB /p65. NF-κB is normally found in the cytosol as a complex of several different proteins (p50, p65, and IκBα) and following an inflammatory signal, the inhibitor IκBα dissociates from the complex. Following dissociation, p65 is serine phosphorylated and translocated to the nucleus, where it activates transcription of several pro-cancerous genes such as, cyclin-D1, MMP-9 and COX-2 [31]. To inhibit the nuclear translocation of p65, a peptide was designed to block its serine phosphorylation sites. A recently approved patent utilized a p65 derived peptide conjugated with Antp (CPP) to inhibit p65 from activating transcription [32]. The peptide, PTD-p65–1, mimics the amino acid sequence of the p65 protein that spans two serine residues essential to nuclear translocation. In KBM-5 leukemia cells, treatment with PTD-p65–1 was shown to inhibit p65 phosphorylation, p65 nuclear translocation and p50-p65 DNA binding. These PTD-p65–1 treated leukemia cells also demonstrated an increase in the cytotoxic sensitivity following exposure to a chemotherapeutic agent, doxorubicin. Additionally, PTD-p65–1 was able to block the p65-dependent transcription of cyclin-D1, MMP-9, and COX-2 [22]. This indicates that peptides like the PTD-p65–1 can be utilized to sensitize chemotherapy resistant cancer cells to apoptosis or can be used to treat chronic inflammation.
Cancer cells often have deregulation of protein expression due to a variety of reasons, including gene amplification, epigenetic alterations, aberrant signaling or atypical protein translation. The altered protein expression of the eukaryotic initiation factor 4E (eIF4E) has been described to increase the translation of mRNAs with a long 5’ untranslated regions [33]. Increased eIF4E has been shown to be directly responsible for the altered protein expression of several oncogenes, such as cyclin D1, VEGF, and FGF2. Additionally, the overexpression of eIF4E has recently been described as an indicator of breast cancer recurrence [34].
eIF4E functions by entering the nucleus, interacting with mRNA, and assist in presenting the mRNA to the ribosome for translation. The nuclear translocation of eIF4E is vital for its function and blocking nuclear entry can promote cytoplasmic accumulation and apoptosis. A recent patent discusses the use of a CPP attached to a portion of the potato virus Y VPg protein to inhibit eIF4E nuclear translocation [35]. In Drosophila the authors have previously demonstrated that the VPg protein directly binds to eIF4e and inhibits cell proliferation. The conserved nature of imitation factors allowed a similar approach in mammalian cells. The authors demonstrated that treatment with CPP-VPg inhibited eIF4E nuclear translocation, loss of cell proliferation and cell death [35].
Nuclear translocation also plays an important role in signal transduction, transmitting signals from membrane-bound receptors to the nucleus. The erbB growth factor receptor family (erbB1/EGFR, erbB2/Her2, erbB3, and erbB4) has been implicated in the progression of several cancer types, including breast, lung, gastrointestinal stromal, and prostate [Reviewed in [36]]. For example, erbB2 is a common therapeutic target that is amplified and overexpressed in approximately 30% of all breast cancers [37]. One of the downstream signaling events following erbB2 activation is the tyrosine phosphorylation, dimerization, and nuclear trafficking of the signal transducers and activators of transcription 3 (STAT3) transcription factor. STAT3 is been found deregulated in most of the solid tumors (breast, ovarian, pancreatic, prostate, and melanoma) and hematological diseases (lymphoma and leukemia) [38–43]. Following STAT3 nuclear translocation, it activates the transcription of several oncogenes that play a role in conveying a chemo-resistant phenotype, such as VEGF, Bcl-2, CyclinD1, and MMP9 [44]. Therefore, nuclear translocation of STAT3 mediated by ErbB2 is an attractive therapeutic target.
In 2006, a report was published, which demonstrated that the CPP Tat could be manipulated to specifically target breast cancer cells that overexpress erbB2/Her2, by targeting cells with erbB2 binding peptide conjugated to Tat (P3-AHNP). By concentrating the P3-AHNP peptide to the cells that were erbB2-positive, the authors reported that cells overexpressing erbB2 preferentially absorbed the peptide [23]. P3-AHNP was then additionally conjugated to STAT3 binding peptide, STAT3BP, resulting in a CTT that could specifically localize to erbB2 overexpressing breast cancer cells and block STAT3 nuclear translocation, by blocking STAT3 dimerization [23]. Importantly, P3-AHNP-STAT3-BP inhibited STAT3-DNA interactions and its resulting oncogenic transcriptional activity. A significant decrease in proliferation and an increase in apoptosis was observed in P3-AHNP-STAT3BP treated breast cancer cell lines and xenograft tumors [23]. This is a novel example of how the protein expression profile of certain cancers can lead to the development of CTTs that specifically target certain cell-types.
Oncogenic Signaling Modulation
Oncogenic signaling, as a result of cancer-dependent protein-protein interactions can promote many aspects of cancer progression, including cell cycle progression, inhibition of apoptosis and insensitivity to anti-growth signals. In addition, changes in cellular polarity can result in novel protein-protein interactions that are present in cancer cells, but do not occur in polarized epithelium due to junctional separations. In some instances these cancer-dependent protein-protein interactions result in aberrant signaling and trafficking. These interactions and signaling events thus provide potential targets for a cancer-specific therapy.
Our laboratory has focused on targeting those interactions that occur in tumor cells, but not in normal polarized epithelium. Basolateral proteins, such as the Epidermal Growth Factor Receptor (erbB1/EGFR), and apical proteins, such as MUC1 are separated when epithelial cells maintain junctional formations. Loss of polarization in tumor cells, and in metastatic cancer specifically, results in novel protein-protein interactions that promote neoplasia. In the case of MUC1 and EGFR, interactions result in altered protein trafficking and oncogenic signal transduction events that promote breast cancer progression [45]. EGFR is a potent driver of tumor growth and when it interacts with MUC1, EGFR signaling is amplified [46]. Targeting the interaction domains between these proteins, we were able to demonstrate that a CTT, PMIP, blocks MUC1 and EGFR interactions resulting in significantly slower tumor growth [47]. PMIP was designed from a small portion of the MUC1 cytoplasmic tail and conjugated with the CPP, PTD4 [48]. In in vitro studies with breast cancer cell lines, PMIP’s inhibitory effect corresponded to a significant decrease in invasion and proliferation. In the xenograft mouse model, the PMIP treatment of the aggressive MDA-MB-231 breast cancer cell line resulted in slower growth and a reduction in distant metastases. Additionally, PMIP dramatically slowed down the tumor growth and in several instances caused regression of solids tumors in the genetically-driven mouse mammary tumor model (MMTV- py MT). Again, as others have observed, PMIP treated mice showed no weight loss or gross tissue damage, indicating a lack of toxic side-effects [3].
Immune response modulation against cancer has been the main focus of many cancer researchers because the concept that the body’s natural defense could target and eliminate tumor cells is an ideal therapeutic approach. Though there are many drawbacks to the immune modulation approach mainly autoimmunity. However, a recent patent has utilized a peptide to promote potential tumors antigens to the cell surface of dying tumor cells which elicits an anti-tumor response. The underlying concept of this patent is based on the finding that following anthracyclin (common type of chemotherapy, a DNA intercalating agent) the proteins calreticulin (CRT), KDEL receptor, and ERp57 are found at the plasma membrane. The membrane localization of these protein results in an immune response against these tumor antigens. However, this membrane localization is inefficient in dying cancer cells so a peptide was designed to promote membrane localization of these proteins. The authors found that inhibition of a protein phosphatase, GADD34, with a CTT (DPT-PP1 inhibitor) in colon and cervical cancer cells lines resulted in increased membrane localization of CRT, KDEL receptor, and ERp57. Additionally, this elicited a greater immune response as compared to peptide controls or no treatment [49]. This a novel use of a CTT to alter intracellular pathways that play an important role in extracellular processes, such as an immune response.
Most anti-cancer therapies used in the clinic take advantage of the replicative nature of cancer cells by disrupting DNA replication and synthesis. Radiotherapy (IR) is often used to cause double-strand breaks in proliferating tumor cells, thereby promoting apoptosis. However, following radiation exposure cell has an innate signaling mechanism to arrest the cell cycle to allow for DNA repair mechanism. For example, upon IR exposure the ataxia telangiectasia mutant (ATM) is activated through the interaction with another protein, Nijmegen breakage syndrome 1 (NSB-1), and this activation leads to downstream signaling events which result in cell cycle arrest [50]. The cell cycle arrest allows for DNA repair, which can promote the viability of the cell and inhibition of apoptosis. Although in normal cells this mechanism is important in preventing genomic instability however in cancer cells this pathway assists tumor cells in becoming radioinsensitive. A recent patent describes the use of a peptide based approach to inhibit the NSB-1/ATM interaction to sensitize tumor cells to IR [51]. The peptide designed utilized the polyarginine (R9) PTD attached to a NSB-1 mimetic peptide (wtNIP). A study found that the treatment of cervical and prostate cancer cells with wtNIP resulted in efficient cellular uptake and limited cytotoxicity. However, the authors found that the wtNIP treatment following radiation was able to sensitize these cells and increase cell death compared to peptide controls [52].
The drawback to the previous approach would be the non-specific nature of the CTT. In contrast, another patent has utilized a peptide-based therapy to specifically target the increased availability of single-stranded DNA (ssDNA) in cancer stem cells. In 2006, a report was published demonstrating the presence of “bell-shaped” nuclei in colorectal tumors and upon closer investigation the authors found this population of cells contained stem cell like properties [53]. The authors reported that “bell-shaped” nuclei undergo symmetrical and asymmetrical nuclear fission and this phenomenon results in increased availability of ssDNA. This led to the design of a peptide that could enter cells, bind ssDNA, and promote apoptosis of the cancer stem cells. This patent however, mentions the use of CPP to deliver the peptide but does not provide a specific CPP [54]. This a novel therapeutic approach which takes advantage of increased ssDNA to specifically target cancer stem cells for apoptosis.
Other laboratories have targeted protein-protein interactions that are important in anti-apoptotic signaling. An important step that leads to the inhibition of apoptosis in cancer cells is the increased signaling and stabilization of inhibitors of apoptosis proteins (IAP). One protein that has been found to stabilize IAPs is heat-shock protein 90 (Hsp90). Hsp90 is commonly deregulated in cancer and has been shown to assist the cell in adapting to environmental stresses [55]. Importantly, Hsp90 is able to directly interact with an IAP, survivin, and inhibit its proteosomal mediated degradation. Recently approved patents propose the use of CTTs that specifically targets the interaction between Hsp90 and survivin [56, 57]. The peptide, shepherdin, was designed to mimic a small domain of the survivin protein that interacts with the ATPase domain of Hsp90 and was then conjugated with the CPPs Antp or Tat. The authors demonstrated that in vitro treatment of several cancer cell lines (breast, prostate, colon, and cervical) with shepherdin caused caspase-dependent apoptosis and loss of membrane integrity [58]. In addition, shepherdin treated breast and prostate tumors grown superficially in immunocompromised mice showed slower tumor growth versus controls and no toxic side-effects were observed. Notably, this report revealed that shepherdin effects were cancer cell specific and did not have any effect on normal fibroblast [58]. Therefore, shepherdin demonstrates the inhibition of protein-protein interactions that results in the non-toxic targeting of tumors via the sensitization of cells to apoptotic signals.
The use of Hsp90 as a therapeutic peptide target has not been limited to IAPs. Another recent patent application describes the use of an Hsp90 peptide to promote the degradation of eNOS [59]. Activation of endothelial nitric-oxide synthase (eNOS) by Hsp90 results in the increased generation of Nitric Oxide (NO) which allows cancer cell to become insensitive to hypoxic and stress conditions. In this manner, activation of eNOS by Hsp90 is able to convey an anti-apoptotic response in several cancers, including oral squamous cell, colon, breast and prostate [60–63]. Therefore the interaction of Hsp90 and eNOS provides a potential anticancer target. An Hsp90 directed CTT was generated, and the treatment of endothelial cells resulted in a significant decrease in nitric-oxide production [64]. The loss of Nitric-Oxide production has been observed to slow the proliferation of a malignant melanoma cell line [65]. Thus, the targeting of Hsp90 may be useful in the treatment of multiple cancer types and further demonstrates that the blocking of cancer-specific protein-protein interactions may be a viable therapeutic approach.
Modulation of ECM Interactions
Aberrant interactions between tumor cells and the extracellular matrix is an important component of tumor progression. The ability of tumor cells to form productive interactions with components of the ECM is critical for movement from the primary organ to distant sites. For example, cells from the primary tumor of the breast disassociate from the tumor mass and utilize the surrounding ECM to migrate towards the blood and lymph vessels where they intravasate and are transported throughout the body, eventually forming secondary tumor sites in many tissues, including liver, bone, and brain [Reviewed in [66]]. This metastatic process is similar in the most solid tumors and demonstrates the critical role of the ECM interactions with the tumor in metastatic progression [Reviewed in [67]]. A recent study has utilized the Tat derived CPP linked with an elastin-like polypeptide (Tat-ELP) to inhibit ovarian cancer invasion, migration, and metastases [15]. Elastin is an extracellular matrix protein commonly found in connective tissue throughout the body that conveys elasticity to tissues, such as blood vessels, lungs, skin, and the bladder. This CTT was designed to block the interaction between elastin and circulating tumor cells, resulting in the loss of secondary tumor site establishment. The concept presented in this report is unique in the sense that the authors attached a CPP to a large elastin-like polypeptide (800aa), which resulted in Tat-ELP binding to the cell surface followed by a more gradual endocytosis [15]. In this way, Tat-ELP is attached to the cell membrane and is functioning to inhibit ECM interactions prior to internalization. The Treatment of ovarian cancer cells with Tat-ELP resulted in an inhibition of the circulating tumor cells from attaching and establishing secondary tumor sites [15]. The use of Tat to bind a therapy to the cell surface is a novel approach to inhibiting ECM Receptor/ECM interactions that could have potential in treating pre-metastatic and metastatic disease.
Novel Therapeutic Targets
The complexity of cancer biology and our understanding of mechanisms underlying cancer development are ever growing. As new areas of cancer research are being explored the discovery of new therapeutic targets has been uncovered. Recently, several patents applications have been submitted which plan on utilizing CPPs to deliver anti-cancer therapies directed against these novel targets. For example, the role of microRNA (miRNA) in cancer formation and progression is a relatively new field in cancer biology so a recent patent describes a miRNA processing protein as a potential therapeutic target [68]. Another patent plans to utilize CPPs to target a new area of cancer biology, epigenetics. Specifically, this patent describes the use of a CPP to deliver an antisense nucleic acid to block the translation of a splice variant of DNA methyltransferase 3B, which has been shown to promote hypermethylation and gene silencing in cancer [69–71].
CURRENT & FUTURE DEVELOPMENTS
In this review, we have examined the use of CTTs to target cancer in a manner that is non-toxic to normal cells. In vitro and in vivo CTTs have been shown to translocate across the plasma membrane, directly interact with their target in the cytosol or nucleus, and promote cancer cell death. Additionally, CTTs have also been used to induce sensitivities to chemotherapies and radiation. CTTs have shown to be effective in many cancer types, and while the shift from the laboratory to the clinic has been time consuming, recent progress should promote this translation.
Endosomal Escape
As more CTTs are being developed and tested in the clinic setting, there is a great need for a better understanding of how these peptides escape from the endosomal compartments, which limit their therapeutic potential. The ability to optimize endosomal escape would allow for higher therapeutic concentrations in the cytoplasm, resulting an increase in the CTT/target interaction. However, several studies examined in this reviewed show, a significant amount of CTT in the tumor several hours after injection. One possible explanation for this phenomenon is the increased bioavailability of the target found within tumor cells. The CTT fails to be degraded because it is interacting with its target [72].
CPP Stability and Modifications
One concern with using peptides as therapeutics is the potential effects of serum proteases on peptide degradation prior to cellular internalization. A potential modification to CTTs could involve incubation with SDS or polysialic acid, which has been shown to increase peptide stability and diffusion throughout the body [18]. Inhibiting serum protease activity in the blood would also assist in maintaining a higher concentration of therapeutic peptide. Although few studies have attempted this, Yi et al. discovered that the treatment of human serum samples with heparin or citrate could stabilize the serum peptides [73]. Another approach to stabilize peptides in the circulatory system is the manipulation of the amino-acid isomerization, which could have a dramatic effect on peptide bioavailability and potency. For example, the isomerization of an L-isoleucine to a D-isoleucine in an anti-angiogenic thrombospondin-1 peptide (Mal II D-Ile) resulted in better pharmocodynamic properties [74]. The proposed mechanism of this result was the limited protease activity towards the D-amino acid, thereby increasing the circulatory concentration of the peptide. Another biological characteristic of D-amino acids is that processing and presentation to the immune system of D-amino acid peptide is inefficient thereby limiting an immune response [75]. A recent patent describes the use of a CPP containing D-amino acid to deliver a targeted anti-cancer therapy to leukocytes and endothelial cells [76]. In addition to amino acid isomerization, cyclic peptides that contain a disulfide or amide bonds show an increased therapeutic advantage versus linear peptides [77]. These studies indicate that the amino acids of CTTs can be manipulated to optimize peptide bioavailability and efficacy.
Another potential drawback of CTTs is that most of the time the CTT is a low molecular weight peptide, which results in efficient renal clearance, and poor tumor penetration. To address this issue, one study utilized a hydrophilic polyethylene glycol coat to act as an efficient “macromolecular drug carrier” [78]. More recently the use of nanoparticles to delivery macromolecules drugs, such as, peptides and genes, offers a potential solution to the drawbacks of just treating with a CTT alone [79]. Theoretically these carriers would function by forming a “capsule” around the therapy, which would be able to deliver relatively larger doses of CTT through mucosal barriers to target tissues.
In conclusion, the use of CTTs to treat cancer is a relatively new field and there are still many areas that need to be explored. However CTTs show great promise in the field of anti-cancer therapy because of their many benefits including low toxicity, minimal modulation of immune system, tissue specificity, ability to target the most pro-cancerous interactions and regulation of oncogenic signal transduction. The therapeutic approaches/patents discussed in this review are only a snapshot of all new CTTs that have been reported, but it provides a glimpse of the potential these peptides have in the fight against cancer.
ACKNOWLEDGEMENTS
Thanks to Matt Hart, Jeanne Louderbough, Ina Menzl, Steven Su and Teresa White for critical reading of this review. The authors are supported by funding from the National Cancer Institute, National Institute of Health training grant, Department of Defense, and Arizona Biomedical Research Commission.
Footnotes
CONFLICT OF INTEREST
There are no conflicts of interest.
REFERENCES
- [1].Yu Z, Wu J, Wu S, Jia P, Tong Y, Wu X, et al. A recombinant cell-permeable p53 fusion protein is selectively stabilized under hypoxia and inhibits tumor cell growth. Cancer Lett 2009; 279: 101–7. [DOI] [PubMed] [Google Scholar]
- [2].Hu M, Wang J, Chen P, Reilly RM. HIV-1 Tat peptide immunoconjugates differentially sensitize breast cancer cells to selected antiproliferative agents that induce the cyclin-dependent kinase inhibitor p21WAF-1/CIP-1. Bioconjug Chem 2006; 17: 1280–7. [DOI] [PubMed] [Google Scholar]
- [3].Bitler BG, Menzl I, Huerta CL, Sands B, Knowlton W, Chang A, et al. Intracellular MUC1 peptides inhibit cancer progression. Clin Cancer Res 2009; 15: 100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chiu YL, Ali A, Chu CY, Cao H, Rana TM. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem Biol 2004; 11: 1165–75. [DOI] [PubMed] [Google Scholar]
- [5].Hong FD, Clayman GL. Isolation of a peptide for targeted drug delivery into human head and neck solid tumors. Cancer Res 2000; 60: 6551–6. [PubMed] [Google Scholar]
- [6].Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 1999; 285: 1569–72. [DOI] [PubMed] [Google Scholar]
- [7].Popiel HA, Nagai Y, Fujikake N, Toda T. Delivery of the aggregate inhibitor peptide QBP1 into the mouse brain using PTDs and its therapeutic effect on polyglutamine disease mice. Neurosci Lett 2009; 449: 87–92. [DOI] [PubMed] [Google Scholar]
- [8].Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem 1994; 269: 10444–50. [PubMed] [Google Scholar]
- [9].Edenhofer F Protein transduction revisited: Novel insights into the mechanism underlying intracellular delivery of proteins. Curr Pharm Des 2008; 14: 3628–36. [DOI] [PubMed] [Google Scholar]
- [10].Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br J Pharmacol 2009; 157: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R, Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007; 8: 848–66. [DOI] [PubMed] [Google Scholar]
- [12].Gump JM, Dowdy SF. TAT transduction: The molecular mechanism and therapeutic prospects. Trends Mol Med 2007; 13: 443–8. [DOI] [PubMed] [Google Scholar]
- [13].Karas M Intracellular delivery of small molecules, proteins, and nucleic acids. US20040209797 (2007).
- [14].Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, et al. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA 1994; 91: 664–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Massodi I, Bidwell GL 3rd, Davis A, Tausend A, Credit K, Flessner M, et al. Inhibition of ovarian cancer cell metastasis by a fusion polypeptide Tat-ELP. Clin Exp Metastasis 2009; 26: 251–60. [DOI] [PubMed] [Google Scholar]
- [16].Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med 2004; 10: 310–15. [DOI] [PubMed] [Google Scholar]
- [17].Kittiworakarn J, Lecoq A, Moine G, Thai R, Lajeunesse E, Drevet P, et al. HIV-1 Tat raises an adjuvant-free humoral immune response controlled by its core region and its ability to form cysteine-mediated oligomers. J Biol Chem 2006; 281: 3105–15. [DOI] [PubMed] [Google Scholar]
- [18].Schutze-Redelmeier MP, Gournier H, Garcia-Pons F, Moussa M, Joliot AH, Volovitch M, et al. Introduction of exogenous antigens into the MHC class I processing and presentation pathway by Drosophila antennapedia homeodomain primes cytotoxic T cells in vivo. J Immunol 1996; 157: 650–55. [PubMed] [Google Scholar]
- [19].Schutze-Redelmeier MP, Kong S, Bally MB, Dutz JP. Antennapedia transduction sequence promotes anti tumour immunity to epicutaneously administered CTL epitopes. Vaccine 2004; 22: 1985–91. [DOI] [PubMed] [Google Scholar]
- [20].Bidwell GL 3rd, Davis AN, Raucher D. Targeting a c-Myc inhibitory polypeptide to specific intracellular compartments using cell penetrating peptides. J Control Release 2009; 135: 2–10. [DOI] [PubMed] [Google Scholar]
- [21].Harada H, Hiraoka M, Kizaka-Kondoh S. Antitumor effect of TAT-oxygen-dependent degradation-caspase-3 fusion protein specifically stabilized and activated in hypoxic tumor cells. Cancer Res 2002; 62: 2013–8. [PubMed] [Google Scholar]
- [22].Takada Y, Singh S, Aggarwal BB. Identification of a p65 peptide that selectively inhibits NF-kappa B activation induced by various inflammatory stimuli and its role in down-regulation of NF-kappaB-mediated gene expression and up-regulation of apoptosis. J Biol Chem 2004; 279: 15096–104. [DOI] [PubMed] [Google Scholar]
- [23].Tan M, Lan KH, Yao J, Lu CH, Sun M, Neal CL, et al. Selective inhibition of ErbB2-overexpressing breast cancer in vivo by a novel TAT-based ErbB2-targeting signal transducers and activators of transcription 3-blocking peptide. Cancer Res 2006; 66: 3764–72. [DOI] [PubMed] [Google Scholar]
- [24].Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- [25].Huo JX, Metz SA, Li GD. p53-independent induction of p21(waf1/cip1) contributes to the activation of caspases in GTP-depletion-induced apoptosis of insulin-secreting cells. Cell Death Differ 2004; 11: 99–109. [DOI] [PubMed] [Google Scholar]
- [26].Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther 2002; 1: 639–49. [PubMed] [Google Scholar]
- [27].Waldman T, Zhang Y, Dillehay L, Yu J, Kinzler K, Vogelstein B, Williams J. Cell-cycle arrest versus cell death in cancer therapy. Nat Med 1997; 3: 1034–6. [DOI] [PubMed] [Google Scholar]
- [28].Liu ZM, Chen GG, Ng EK, Leung WK, Sung JJ, Chung SC. Upregulation of heme oxygenase-1 and p21 confers resistance to apoptosis in human gastric cancer cells. Oncogene 2004; 23: 503–13. [DOI] [PubMed] [Google Scholar]
- [29].Hu M, Chen P, Wang J, Chan C, Scollard DA, Reilly RM. Site-specific conjugation of HIV-1 tat peptides to IgG: A potential route to construct radioimmunoconjugates for targeting intracellular and nuclear epitopes in cancer. Eur J Nucl Med Mol Imaging 2006; 33: 301–10. [DOI] [PubMed] [Google Scholar]
- [30].Shan W, Yang G, Liu J. The inflammatory network: Bridging senescent stroma and epithelial tumorigenesis. Front Biosci 2009; 14: 4044–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shishodia S, Majumdar S, Banerjee S, Aggarwal BB. Ursolic acid inhibits nuclear factor-kappaB activation induced by carcinogenic agents through suppression of IkappaBalpha kinase and p65 phosphorylation: Correlation with down-regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1. Cancer Res 2003; 63: 4375–83. [PubMed] [Google Scholar]
- [32].Aggarwal BB, Singh S Selective inhibitors of nuclear factor-κB activation and uses thereof. US20050201976 (2008).
- [33].Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell G, Belsham GJ, et al. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5’ secondary structure. RNA 2001; 7: 382–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McClusky DR, Chu Q, Yu H, Debenedetti A, Johnson LW, Meschonat C, et al. A prospective trial on initiation factor 4E (eIF4E) overexpression and cancer recurrence in node-positive breast cancer. Ann Surg 2005; 242: 584–90; discussion 590–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chroboczek J, Zagorski W, Grezla R Cell-proliferation Inhibiting VPG Proteins, Fragments or Analogs Thereof and their applications. WO2008152527 (2008).
- [36].Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Exp Cell Res 2003; 284: 99–110. [DOI] [PubMed] [Google Scholar]
- [37].Kallioniemi OP, Holli K, Visakorpi T, Koivula T, Helin HH, Isola JJ. Association of c-erbB-2 protein over-expression with high rate of cell proliferation, increased risk of visceral metastasis and poor long-term survival in breast cancer. Int J Cancer 1991; 49: 650–55. [DOI] [PubMed] [Google Scholar]
- [38].Burke WM, Jin X, Lin HJ, Huang M, Liu R, Reynolds RK, et al. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene 2001; 20: 7925–34. [DOI] [PubMed] [Google Scholar]
- [39].Scholz A, Heinze S, Detjen KM, Peters M, Welzel M, Hauff P, et al. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 2003; 125: 891–905. [DOI] [PubMed] [Google Scholar]
- [40].Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, Bowman T, et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res 2002; 62: 6659–66. [PubMed] [Google Scholar]
- [41].Niu G, Bowman T, Huang M, Shivers S, Reintgen D, Daud A, et al. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene 2002; 21: 7001–10. [DOI] [PubMed] [Google Scholar]
- [42].Aoki Y, Feldman GM, Tosato G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003; 101: 1535–42. [DOI] [PubMed] [Google Scholar]
- [43].Nakajima K, Yamanaka Y, Nakae K, Kojima H, Ichiba M, Kiuchi N, et al. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J 1996; 15: 3651–8. [PMC free article] [PubMed] [Google Scholar]
- [44].Hsieh FC, Cheng G, Lin J. Evaluation of potential Stat3-regulated genes in human breast cancer. Biochem Biophys Res Commun 2005; 335: 292–9. [DOI] [PubMed] [Google Scholar]
- [45].Pochampalli MR, el Bejjani RM, Schroeder JA. MUC1 is a novel regulator of ErbB1 receptor trafficking. Oncogene 2007; 26: 1693–1701. [DOI] [PubMed] [Google Scholar]
- [46].Pochampalli MR, Bitler BG, Schroeder JA. Transforming growth factor alpha dependent cancer progression is modulated by Muc1. Cancer Res 2007; 67: 6591–8. [DOI] [PubMed] [Google Scholar]
- [47].Schroeder JA Therapeutic peptides for the treatment of metastatic cancer. US20060293234A1 (2006).
- [48].Ho A, Schwarze SR, Mermelstein SJ, Waksman G, Dowdy SF. Synthetic protein transduction domains: Enhanced transduction potential in vitro and in vivo. Cancer Res 2001; 61: 474–7. [PubMed] [Google Scholar]
- [49].Kroemer G, Zitvogel L Inhibitors of Protein Phosphatase 1, GADD34 and protein phosphatase 1/GADD34 complex, preparation and uses thereof. WO2008028965A2 (2006).
- [50].Cerosaletti K, Concannon P. Independent roles for nibrin and Mre11-Rad50 in the activation and function of Atm. J Biol Chem 2004; 279: 38813–9. [DOI] [PubMed] [Google Scholar]
- [51].Cariveau M,J, Xu B Targeting NBS1-ATM interaction to senistize cancer cells to radiotherapy and chemotherapy. WO2008054726A2 (2006).
- [52].Cariveau MJ, Tang X, Cui XL, Xu B. Characterization of an NBS1 C-terminal peptide that can inhibit ataxia telangiectasia mutated (ATM)-mediated DNA damage responses and enhance radiosensitivity. Mol Pharmacol 2007; 72: 320–6. [DOI] [PubMed] [Google Scholar]
- [53].Gostjeva EV, Zukerberg L, Chung D, Thilly WG. Bell-shaped nuclei dividing by symmetrical and asymmetrical nuclear fission have qualities of stem cells in human colonic embryogenesis and carcinogenesis. Cancer Genet Cytogenet 2006; 164: 16–24. [DOI] [PubMed] [Google Scholar]
- [54].Gostjeva EV, Thilly WG Methods and agents for inhibiting tumor growth. WO2008156629 (2007).
- [55].Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 2003; 3: 213–7. [DOI] [PubMed] [Google Scholar]
- [56].Altieri DC, Plescia J, Whitney S Compounds that inhibit Hsp90 protein-protein interactions with IAP proteins. US20060035837 (2008).
- [57].Altieri DC, Kang BH Mitochondria-targeted anti-tumor agents. WO2009036092 (2007).
- [58].Plescia J, Salz W, Xia F, Pennati M, Zaffaroni N, Daidone MG, Meli M, Dohi T, Fortugno P, Nefedova Y, Gabrilovich DI, Colombo G, Altieri DC. Rational design of shepherdin, a novel anticancer agent. Cancer Cell 2005; 7: 457–68. [DOI] [PubMed] [Google Scholar]
- [59].Pritchard K, Xu H, Shi Y HSP90 decoy peptides and uses thereof. US20070219129 (2006).
- [60].Shang ZJ, Li ZB, Li JR. In vitro effects of nitric oxide synthase inhibitor L-NAME on oral squamous cell carcinoma: A preliminary study. Int J Oral Maxillofac Surg 2006; 35: 539–43. [DOI] [PubMed] [Google Scholar]
- [61].Wang L, Shi GG, Yao JC, Gong W, Wei D, Wu TT, et al. Expression of endothelial nitric oxide synthase correlates with the angiogenic phenotype of and predicts poor prognosis in human gastric cancer. Gastric Cancer 2005; 8: 18–28. [DOI] [PubMed] [Google Scholar]
- [62].Vakkala M, Paakko P, Soini Y. eNOS expression is associated with the estrogen and progesterone receptor status in invasive breast carcinoma. Int J Oncol 2000; 17: 667–71. [DOI] [PubMed] [Google Scholar]
- [63].Tong X, Li H. eNOS protects prostate cancer cells from TRAIL-induced apoptosis. Cancer Lett 2004; 210: 63–71. [DOI] [PubMed] [Google Scholar]
- [64].Xu H, Shi Y, Wang J, Jones D, Weilrauch D, Ying R, et al. A heat shock protein 90 binding domain in endothelial nitric-oxide synthase influences enzyme function. J Biol Chem 2007; 282: 37567–74. [DOI] [PubMed] [Google Scholar]
- [65].Tao J, Tu YT, Li JW, Feng AP, Huang CZ, Wu Y, et al. Endogenous production of nitric oxide contributes to proliferation effect of vascular endothelial growth factor-induced malignant melanoma cell. Clin Exp Dermatol 2006; 31: 94–9. [DOI] [PubMed] [Google Scholar]
- [66].Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature 2001; 411: 375–9. [DOI] [PubMed] [Google Scholar]
- [67].O’Hayre M, Salanga CL, Handel TM, Allen SJ. Chemokines and cancer: migration, intracellular signalling and intercellular communication in the microenvironment. Biochem J 2008; 409: 635–49. [DOI] [PubMed] [Google Scholar]
- [68].Gregory RI, Daley GQ, Viswanathan SR Methods to regulate MIRNA Processing by targeting LIN-28. WO2009048932 (2007).
- [69].Godley LA, Ostler KR Inhibiting translation of aberrant DNMT3B transcripts in cancer cells using inhibitory nucleic acids. WO2008070858 (2008).
- [70].Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002; 416: 552–6. [DOI] [PubMed] [Google Scholar]
- [71].Ostler KR, Davis EM, Payne SL, Gosalia BB, Exposito-Cespedes J, Le Beau MM, et al. Cancer cells express aberrant DNMT3B transcripts encoding truncated proteins. Oncogene 2007; 26: 5553–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J Control Release 2000; 65: 271–84. [DOI] [PubMed] [Google Scholar]
- [73].Yi J, Liu Z, Craft D, O’Mullan P, Ju G, Gelfand CA. Intrinsic peptidase activity causes a Sequential Multi-Step Reaction (SMSR) in Digestion of human plasma peptides. J Proteome Res 2008; 7(12): 5112–8. [DOI] [PubMed] [Google Scholar]
- [74].Dawson DW, Volpert OV, Pearce SF, Schneider AJ, Silverstein RL, Henkin J, et al. Three distinct D-amino acid substitutions confer potent antiangiogenic activity on an inactive peptide derived from a thrombospondin-1 type 1 repeat. Mol Pharmacol 1999; 55: 332–8. [DOI] [PubMed] [Google Scholar]
- [75].Uebel S, Kraas W, Kienle S, Wiesmuller KH, Jung G, Tampe R. Recognition principle of the TAP transporter disclosed by combinatorial peptide libraries. Proc Natl Acad Sci USA 1997; 94: 8976–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Peer D, Shimaoka M Methods of delivery of agents to leukocytes and endothelial cells. WO2009026328 (2009).
- [77].Leduc AM, Trent JO, Wittliff JL, Bramlett KS, Briggs SL, Chirgadze NY, et al. Helix-stabilized cyclic peptides as selective inhibitors of steroid receptor-coactivator interactions. Proc Natl Acad Sci USA 2003; 100: 11273–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Prego C, Garcia M, Torres D, Alonso MJ. Transmucosal macromolecular drug delivery. J Control Release 2005; 101: 151–62. [DOI] [PubMed] [Google Scholar]
- [79].de la Fuente M, Csaba N, Garcia-Fuentes M, Alonso MJ. Nanoparticles as protein and gene carriers to mucosal surfaces. Nanomed 2008; 3: 845–57. [DOI] [PubMed] [Google Scholar]
- [80].Elferink JG. Changes of plasma membrane permeability in neutrophils treated with polycations. Inflammation 1991; 15: 103–15. [DOI] [PubMed] [Google Scholar]
- [81].Elliott G, O’Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 1997; 88: 223–33. [DOI] [PubMed] [Google Scholar]
- [82].Sadler K, Eom KD, Yang JL, Dimitrova Y, Tam JP. Translocating proline-rich peptides from the antimicrobial peptide bactenecin 7. Biochemistry 2002; 41: 14150–57. [DOI] [PubMed] [Google Scholar]
- [83].Guergnon J, Dessauge F, Dominguez V, Viallet J, Bonnefoy S, Yuste VJ, et al. Use of penetrating peptides interacting with PP1/PP2A proteins as a general approach for a drug phosphatase technology. Mol Pharmacol 2006; 69: 1115–24. [DOI] [PubMed] [Google Scholar]
- [84].Pooga M, Hallbrink M, Zorko M, Langel U. Cell penetration by transportan. FASEB J 1998; 12: 67–77. [DOI] [PubMed] [Google Scholar]
- [85].Morris MC, Depollier J, Mery J, Heitz F, Divita G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol 2001; 19: 1173–6. [DOI] [PubMed] [Google Scholar]