

Abstract
This thorough QT (TQT) study evaluated the effect of zanubrutinib on electrocardiogram (ECG) parameters by using concentration‐QTc (C‐QTc) analysis as the primary analysis for this study. Part A of the study determined the safety and tolerability of a single supratherapeutic dose of zanubrutinib (480 mg) in healthy volunteers. Part B was a randomized, blinded, placebo‐controlled and positive‐controlled, four‐way crossover, TQT study of single therapeutic (160 mg) and supratherapeutic (480 mg) doses of zanubrutinib, placebo, and open‐label moxifloxacin 400 mg. Thirty‐two participants received at least 1 dose of zanubrutinib, and 26 participants completed all 4 periods. Zanubrutinib did not have any effect on heart rate or cardiac conduction (pulse rate, QRS interval, or T‐wave morphology) and was generally well‐tolerated. Using C‐QTc analysis, the predicted placebo‐corrected change‐from‐baseline QT interval using Fridericia’s formula (ΔΔQTcF) was −3.4 msec (90% confidence interval: −4.9 to −1.9 msec) at peak concentrations of the 480 mg dose. A QT effect (ΔΔQTcF) exceeding 10 msec could be excluded within the observed concentration range at 160 and 480 mg doses. Assay sensitivity was established by moxifloxacin with 90% lower bound exceeding 5 msec. Implementing a C‐QTc analysis prospectively in this TQT study resulted in a substantially smaller sample size to maintain a similar study power as shown in the traditional time‐point analysis. A single 160‐mg or 480‐mg zanubrutinib dose did not prolong the QTc interval or have any other clinically relevant effects on ECG parameters.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ The effects of zanubrutinib on QT/QTc interval are unknown and there are limited published examples using concentration‐QTc (C‐QTc) analysis as the primary analysis in a thorough QT (TQT) study.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The effect of zanubrutinib on the QT/QTc interval was investigated in a TQT study in healthy subjects in which C‐QTc analysis was used as the primary analysis.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Zanubrutinib does not increase QT/QTc interval up to doses of 480 mg and this study increased understanding of the cardiovascular safety profile of zanubrutinib, a BTK inhibitor that has been granted a breakthrough therapy designation and received accelerated approval by US Food and Drug Administration (FDA) for the treatment of patients with relapsed or refractory mantle cell lymphoma.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Using a concentration‐QTc analysis as the primary analysis in the dedicated TQT study can substantially reduce the sample size compared with the typical by time‐point analysis for the TQT study.
Bruton’s tyrosine kinase (BTK), a member of the Tec kinase family, is a critical component of the B‐cell receptor signaling cascade. Activation of BTK in B cells initiates a series of signaling events that includes recruitment of BTK to the plasma membrane, autophosphorylation at Tyr223, activation of phospholipase Cγ2, subsequent activation of nuclear factor kappa‐light‐chain‐enhancer of activated B cells, and expression of genes involved in proliferation and survival. 1 , 2 , 3 Inhibition of BTK has emerged as a promising strategy for targeting B‐cell malignancies, including mantle cell lymphoma (MCL), chronic lymphocytic leukemia, small lymphocytic lymphoma, follicular lymphoma, Waldenström’s macroglobulinemia, and diffuse large B‐cell lymphoma. 4 Ibrutinib, the first‐in‐class US Food and Drug Administration (FDA)‐approved BTK inhibitor, has demonstrated promising antitumor activities in several B‐cell malignancies.
Zanubrutinib (BGB‐3111) is a potent, specific, and irreversible BTK inhibitor designed to maximize BTK occupancy and minimize off‐target inhibition. Zanubrutinib has been granted a breakthrough therapy designation and recently received accelerated approval by the FDA for the treatment of adult patients with MCL who have previously received at least one prior therapy. 5 Like ibrutinib, zanubrutinib forms an irreversible covalent bond at Cys481 within the adenosine triphosphate–binding pocket of BTK. However, zanubrutinib is more selective than ibrutinib against off‐target kinases, including epidermal growth factor receptor, Janus tyrosine kinase 3, human epidermal growth factor receptor 2, Tec protein–tyrosine kinase, and inducible tyrosine kinase. 6 The increased selectivity of zanubrutinib for BTK may result in a lower incidence and severity of off‐target toxicities linked to inhibition of the aforementioned kinases. Emerging clinical data have shown that zanubrutinib as a single agent and in combination with other therapies resulted in high rates of objective response in patients with B‐cell malignancies, including Waldenström’s macroglobulinemia, MCL, and chronic lymphocytic leukemia, 7 , 8 , 9 consistent with its near‐complete BTK occupancy and target engagement in target tissues (median occupancy of 100% in lymph nodes).
Following oral administration, zanubrutinib is eliminated with a mean terminal elimination half‐life (t ½) of ~ 2–4 hours. Median time to peak zanubrutinib plasma concentration (Tmax) is 2 hours. There is a dose‐proportional increase in maximum concentration (Cmax) and area under the plasma concentration–time curve (AUC) from time 0 extrapolated to infinity (AUC0–∞) at doses from 40 mg to 320 mg. 9 Following multiple‐dose administrations of zanubrutinib at doses from 40 to 320 mg, limited systemic accumulation is observed, which is consistent with the observed t ½. Zanubrutinib is primarily metabolized by cytochrome P450 3A (CYP3A) enzymes. There are no major active metabolites in circulation.
The preclinical cardiovascular safety profile of zanubrutinib has been assessed in in vitro and in vivo studies. In the in vitro human ether‐à‐go‐go related gene (hERG) ion channel assay, an inhibitory effect on hERG channels was noted with half‐maximal inhibitory concentration of 9.11 μM. This represents an ~ 217‐fold exposure margin based on the clinical steady‐state unbound Cmax of 0.042 μM at the recommended phase III dose of 160 mg twice daily. Based on the nonclinical studies, the potential risk of QT interval prolongation in humans by zanubrutinib is considered low.
This thorough QT (TQT) study was conducted in healthy volunteers to support overall assessment of risk of QT prolongation for zanubrutinib based on International Conference on Harmonisation (ICH) E14 guidelines. 10 , 11 Early knowledge of QTc risk for zanubrutinib is critical in supporting design of late‐phase studies in patients with B‐cell malignancies, including providing guidance on the eligibility criteria and on the use of concomitant medications that may prolong the QT/QTc interval and whether intensive late‐stage monitoring is needed. Unlike traditional TQT studies, 12 concentration‐QTc (C‐QTc) analysis 13 was specified prospectively as the primary analysis for this TQT study based on recommendations from the International Consortium for Innovation and Quality in Pharmaceutical Development‐Cardiac Safety Research Consortium (IQ‐CSRC) study, 14 , 15 so a substantially smaller sample size could be used to achieve similar power. 16 As described in a white paper on C‐QTc analysis, 13 this type of analysis is appropriate for evaluation of the QTc effects of new drugs, unless indirect effects (e.g., autonomic effects or QTc prolongation) caused by so‐called trafficking is expected.
Zanubrutinib is administered orally at a dose of 160 mg twice daily in ongoing clinical studies in patients. A single dose of 480 mg was selected as the supratherapeutic dose in this TQT study, based on a 2.6‐fold increase in zanubrutinib Cmax when co‐administered with a strong CYP3A inhibitor, itraconazole from a clinical drug‐drug interaction study. 17 With 320 mg having been the maximum zanubrutinib dose tested in humans prior to this study and clinical safety data unavailable for the supratherapeutic dose, an initial assessment of the safety and tolerability of zanubrutinib 480 mg was conducted in healthy volunteers (part A of the study) prior to the crossover TQT study in part B.
METHODS
This study (NCT03432884) was conducted in accordance with the ethical principles communicated in the Declaration of Helsinki, ICH Good Clinical Practice guidelines, and applicable regulatory requirements and in compliance with the protocol. All participants provided written informed consent as defined by the protocol prior to all screening procedures. The design of part B of this study followed the general design principles outlined in the ICH E14 guidance for the clinical evaluation of the QT/QTc interval with nonanti‐arrhythmic drugs. 11 , 18
Study participants
Participants were healthy men and women between the ages of 18 and 55 years, inclusive, with a body mass index of ≥ 18 to ≤ 33 kg/m2, who provided written informed consent. Women were required to be of nonchildbearing potential (either surgically sterile or postmenopausal). Participants had to be in stable health without any evidence of cardiovascular disease, other significant medical conditions, or clinically significant laboratory test abnormalities. Key exclusion criteria included subjects who used prescription medications, including nonsteroidal anti‐inflammatory drugs or sucralfate and medications known to prolong the QT/QTc interval or herbal preparations within 14 days or 5 half‐lives (whichever was longer) before study drug dosing, or use of an over‐the‐counter medication, vitamins, or supplements (including omega‐3 fish oils) within 7 days before study drug dosing.
Study design and treatments
Part A was a double‐blind, placebo‐controlled study to evaluate the safety, tolerability, and pharmacokinetics (PK) of a supratherapeutic dose (480 mg) of zanubrutinib. Eight healthy participants were randomized 3:1 to receive a single oral dose of 480 mg of zanubrutinib or placebo under fasted conditions. An initial cohort of four participants was dosed, and the next cohort of four participants was dosed after a safety review of the initial cohort. The safety and tolerability of zanubrutinib 480 mg was considered acceptable if there were no serious adverse events (SAEs) or grade 3 or higher adverse events (AEs) related to zanubrutinib administration. If this criterion was met, this dose was to be used as the supratherapeutic dose in part B of the study. The primary end point was the incidence of treatment‐emergent adverse events (TEAEs) reported following administration of zanubrutinib compared with placebo.
Part B was a placebo‐controlled and positive‐controlled, four‐way crossover TQT study. The primary objective was to evaluate the effects of single doses of zanubrutinib on heart rate (HR)‐corrected QT interval using Fridericia’s formula (QTcF) compared with placebo. The secondary objectives were to assess the effects of single doses of zanubrutinib 160 and 480 mg on echocardiogram (ECG) parameters (HR, pulse rate (PR), and QRS intervals), PK, and the safety and tolerability of zanubrutinib. Participants were randomly assigned to 1 of 12 sequences to receive, under fasted conditions, the following four different treatments orally: a single dose of 160 mg zanubrutinib (two 80‐mg capsules plus four placebo capsules); six 80‐mg zanubrutinib capsules totaling a single cumulative dose of 480 mg zanubrutinib; placebo to match zanubrutinib (six placebo capsules); and one 400‐mg moxifloxacin tablet (the positive control). Zanubrutinib and placebo were administered in a double‐blind manner, and moxifloxacin was administered in an open‐label manner. Treatments were separated by a washout period of 7 ± 2 days.
The primary ECG end point was placebo‐corrected change‐from‐baseline QT interval using Fridericia’s formula (ΔΔQTcF), and secondary ECG end points included change‐from‐baseline HR, QTcF, PR, and QRS intervals (ΔHR, ΔPR, and ΔQRS), placebo‐corrected intervals (ΔΔHR, ΔQTcF, ΔΔPR, and ΔΔQRS), the frequency of treatment‐emergent T‐wave morphology abnormalities, and the presence of U‐waves. Participants rested in the supine position for at least 10 minutes prior to the administration of ECGs.
In both parts of the study, standard assessments (e.g., clinical laboratory tests, vital signs, safety ECGs, and physical examinations) were performed and demographics, medical history, and details on concomitant medication use were obtained. AEs and SAEs were assessed and graded based upon the NCI Common Terminology Criteria for Adverse Events (CTCAE) version 4.03.
Sample size calculation
A total of 32 subjects were enrolled to ensure that at least 24 subjects completed all four treatment periods. The sample size for part B of the study (n = 24) was selected based on experience from the IQ‐CSRC study. 10 Simulations were used to evaluate the power of small studies with C‐QTc analysis, 15 as well as 25 recent TQT studies performed by the central ECG laboratory (data on file, eResearchTechnology) using C‐QTc analysis. A sample size of 24 provided > 90% power to exclude the possibility that zanubrutinib causes a > 10‐msec QTc effect at clinically relevant plasma levels, as shown by the upper bound of the two‐sided 90% confidence interval (CI) of the model‐predicted QTc effect (ΔΔQTcF) at the observed mean Cmax of zanubrutinib. The sample size was also approximated using a simple paired t‐test for equivalence.
Electrocardiogram and QTc analyses
In part B, on day 1 in each period, a continuous ECG recording was performed for 25 hours, starting 1 hour prior to dosing. All ECG data were collected using a Global Instrumentation (Manlius, NY) M12R ECG continuous 12‐lead digital recorder. ECG intervals were measured blindly by iCardiac Technologies (Rochester, NY) using a high‐precision QT technique, 19 and the ECG database was locked before any statistical analysis was undertaken. The 12‐lead ECGs were extracted in up to 10 replicates by the central ECG laboratory at the following time points: −45, −30, and −15 minutes prior to dosing, and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 12, and 24 hours after dosing. ECGs were extracted from the continuous recording during a window of 5 minutes preceding the nominal time point, immediately preceding blood sampling for PK determination.
Plasma concentrations and PK analyses
Blood samples for PK analysis of moxifloxacin and zanubrutinib in plasma were collected immediately before dosing on day 1 of each period and at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 12, and 24 hours after dosing. The determination of plasma concentrations of zanubrutinib and moxifloxacin were performed by XenoBiotic Laboratories (Plainsboro, NJ), using validated liquid chromatography tandem mass spectrometry methods. The method utilizes a reverse‐phase high performance liquid chromatography column to elute zanubrutinib with the IS (BGB‐4257) and an liquid chromatography tandem mass spectrometry instrument (AB Sciex API‐4000) with positive electrospray ionization multiple reaction monitoring mode to quantify zanubrutinib. The lower limit of quantification was 1.00 ng/mL and 20 ng/mL for the plasma zanubrutinib and moxifloxacin concentration, respectively. The precision and accuracy of the analytical methods for zanubrutinib and moxifloxacin were within the acceptance criteria.
PK analyses were performed based on actual time of sample collection, using noncompartmental methods with Phoenix WinNonlin, version 6.3.1 (Certara USA, Princeton, NJ). All plasma concentrations below the lower limit of quantification were treated as missing, except those measured prior to the first quantifiable concentration on the day of dosing or after the last quantifiable concentration, which was treated as “0.” PK parameters included Cmax, Tmax, AUC0–t, AUC0–inf, and t ½. AUC was estimated using the linear trapezoidal method (linear up log down). If the adjusted R‐squared value (Rsq_adjusted) was < 0.8, no value of AUC0–inf or t ½ was reported.
Analyses for 12‐lead ECG data and C‐QTc analysis
All statistical analyses of ECG data, including the C‐QTc analyses, were performed using SAS version 9.3 (SAS Institute, Cary, NC). Baseline was the average of the derived ECG intervals from the three time points prior to dosing on day 1 for the respective period. Categorical data were summarized by participant and time point.
The C‐QTc analysis was based on time‐matched ∆∆QTcF. The relationship between plasma concentrations of zanubrutinib and ∆∆QTcF was investigated using a linear mixed‐effects modeling approach, with separate analyses for zanubrutinib and moxifloxacin. The following three linear models were considered: a linear model with an intercept (model 1); a linear model with mean intercept fixed to 0 (with variability; model 2); and a linear model with no intercept (model 3). Time‐matched concentrations of zanubrutinib were included in the model as a covariate, with centered baseline QTcF as an additional covariate and participant as a random effect for both intercept and slope, when applicable. The model that fit the data best was used for predicting population average ∆∆QTcF and its corresponding 90% two‐sided CI at the observed geometric mean Cmax at each dose of zanubrutinib.
The plot of the observed median‐quantile zanubrutinib concentrations and the associated mean ΔΔQTcF (90% CI), together with the mean (90% CI) predicted ΔΔQTcF as described by Tornøe et al., 20 was used to evaluate the adequacy of the model fit to the assumption of linearity and the impact on quantifying the C‐QTc relationship. The delay between peak plasma levels and any potential QTc effect, also known as hysteresis, was assessed based on joint graphical displays of the least‐squares mean ΔΔQTcF for each postdose time point and the mean concentrations of zanubrutinib at the same time points. The scatter plots of standardized residuals vs. concentration and centered baseline QTcF by locally estimated scatter plot smoothing fitting, as described by Cleveland, 21 were also produced with optimal smoothing parameters selected by the Akaike information criterion. 22 Zanubrutinib was to be deemed to have no clinically relevant QT effect if the upper bound of the two‐sided 90% CI of the predicted effect at the observed geometric mean Cmax concentrations was below 10 msec.
Categorical outlier analysis
The number (percentage) of participants and time points with increases in absolute QTcF interval values > 450 msec and ≤ 480 msec, > 480 msec and ≤ 500 msec, and > 500 msec, and changes from predose baseline of > 30 and ≤ 60 msec, and > 60 msec were determined. Additionally, an increase in PR interval from predose baseline > 25% to > 200 msec, an increase in QRS interval from predose baseline > 25% to > 120 msec, a decrease in HR from predose baseline > 25% to < 50 bpm, and an increase in HR from predose baseline > 25% to > 100 bpm were determined.
RESULTS
Participants
In part A, six healthy participants received zanubrutinib 480 mg and two received placebo. All eight participants were male, completed study treatment, and were included in the safety population. Overall mean age was 36.4 years and racial composition was 75.0% black/African American and 25.0% white. Two participants reported AEs: one, who received zanubrutinib 480 mg, reported diarrhea, proteinuria, thrombocytopenia, and viral infection, each of mild (grade 1) or moderate severity (grade 2); and the other, who received placebo, reported mild increase in blood creatinine. The investigator assessed all AEs as related to study drug. There was no SAEs or grade 3 or higher AEs related to zanubrutinib administration. Thus, the safety and tolerability of the 480‐mg dose were considered acceptable, and 480 mg was used as the supratherapeutic dose in part B.
In part B, 32 healthy participants (31 men and 1 woman) were enrolled and received at least one dose of study treatment and 26 participants completed all treatment periods. Six participants in part B discontinued prematurely from the study (i.e., three withdrew consent, and three were withdrawn due to noncompliance). Overall mean age was 42.9 years, and racial composition was 59.4% black/African American and 40.6% white.
Pharmacokinetics
A summary of zanubrutinib PK parameters for part A and part B is presented in Table 1 , and the concentration profiles of zanubrutinib from part B are presented in Figure 1 . Zanubrutinib was rapidly absorbed, with a median Tmax of 1.5–2.8 hours postdose. There was a 1.9‐fold increase in Cmax and a 2.5‐fold increase in AUC0–∞ following a 3‐fold increase in dose of zanubrutinib (from 160 mg to 480 mg in part B). The mean t ½ seemed to be higher with the 480‐mg dose (8.1–11 hours) than with the 160‐mg dose (5.3 hours; Table 1 ).
Table 1.
Summary of zanubrutinib PK parameters in healthy TQT study participants
| Parameter | Zanubrutinib dose | ||
|---|---|---|---|
| Part A | Part B | ||
| 480 mg (n = 6) | 160 mg (n = 28) | 480 mg (n = 30) | |
| Tmax, median (min, max), hour | 2.8 (1.0, 3.5) | 1.5 (1.0, 6.0) | 2.0 (0.5, 6.0) |
| Cmax, mean (CV%), ng/mL | 353 (33.5) | 216 (24.2) | 406 (30.7) |
| AUC0–t, mean (CV%), hour × ng/mL | 2,570 (36.2) | 1,160 (25.1) | 2,770 (28.9) |
| AUC0–∞, mean (CV%), hour × ng/mL | 2,670 (39.1) | 1,230 (23.5) | 3,060 (25.9) |
| t ½, mean (CV%), hour | 11 (50) | 5.3 (47) | 8.1 (68) |
| CL/F, mean (CV%), L/hour | 180 (39.2) | 126 (29.0) | 140 (38.8) |
| Vz/F, mean (CV%), L | 2,840 (41.0) | 966 (36.7) | 1,630 (55.8) |
AUC0– t, area under the concentration‐time curve from time zero to the last quantifiable concentration; AUC0–∞, area under the concentration‐time curve from time zero extrapolated to infinity; CL/F, apparent systemic clearance; Cmax, maximum observed concentration; CV, coefficient of variation; PK, pharmacokinetic; t ½, apparent terminal elimination half‐life; Tmax, time to maximum observed concentration; TQT, thorough QT; Vz/F, apparent volume of distribution during the terminal elimination phase.
Figure 1.
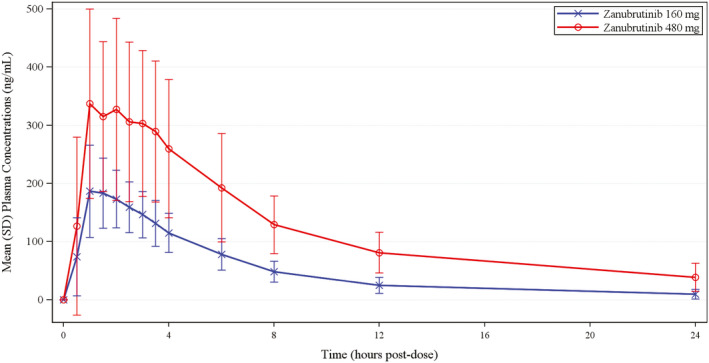
Mean (SD) zanubrutinib plasma concentrations (ng/mL) vs. time. Blue line, zanubrutinib 160 mg (n = 28); red line, zanubrutinib 480 mg (n = 30).
Moxifloxacin was absorbed rapidly, with a median Tmax of 1 hour postdose. Geometric mean (coefficient of variation) Cmax was 1,960 (24.1) ng/mL, and geometric mean (coefficient of variation) AUC0–t was 20,920 (20.0) hour × ng/mL. The mean t ½ of moxifloxacin was 12 hours.
ECG results
HR changes on zanubrutinib and moxifloxacin closely followed the diurnal pattern seen with placebo, with very small differences of mean change from baseline (ΔHR; Figure 2a ). Mean placebo‐corrected change from baseline (ΔΔHR) for zanubrutinib was smaller than ± 5 bpm at all postdose time points, with values ranging from − 0.4 to − 3.6 bpm for zanubrutinib 160 mg and from 0.7 bpm to − 2.9 bpm for zanubrutinib 480 mg. There was no participant with HR outlier values. At most postdose time points, patients on zanubrutinib or placebo exhibited mean decreases in ΔQTcF interval, with the decreases somewhat more pronounced in the patients who were on zanubrutinib. In contrast, moxifloxacin resulted in a mean increase in ΔQTcF interval at most postdose time points (Figure 2b ).
Figure 2.
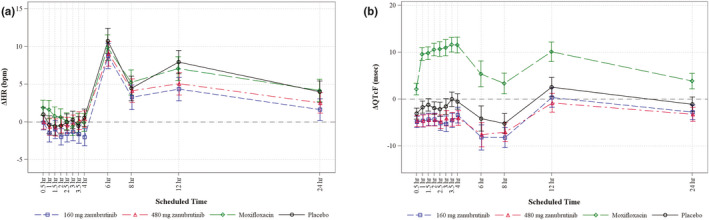
Effects of zanubrutinib on ECG parameters. (a) Change‐from‐baseline heart rate (ΔHR) across time points and (b) mean change from baseline in QTcF interval over time using Fridericia’s formula (ΔQTcF). Study participants received zanubrutinib 160 mg (open squares), zanubrutinib 480 mg (open triangles), moxifloxacin (open diamonds), or placebo (open circles).
A small shortening in ΔΔQTcF was seen at both zanubrutinib doses, with mean values ranging between − 1.6 and − 4.5 msec across all postdose time points without clear correlation to dosage or time of dosing. The upper bound of the 90% CI of ΔΔQTcF did not exceed 1.2 msec at any postdose time point. No participants had outlier QTcF values (i.e., > 450 and ≤ 480 msec, > 480 and ≤ 500 msec, or > 500 msec).
C‐QTc analysis
The relationship between individual observed zanubrutinib concentrations and ΔΔQTcF is shown in Figure 3a . The mean zanubrutinib concentration at the 480 mg dose was 406 ng/mL (Table 1 ), whereas the concentration range observed in this TQT study was as high as ≈ 800 ng/mL; this concentration range was included in the C‐QTc modeling (Figure 3a ). Given the absence of a QT effect (ΔΔQTcF), hysteresis was not accounted for in the modeling. The linear model with an intercept had the smallest Akaike information criterion value of the three models tested, and this model was, therefore, chosen as the final model and used for further analysis. The goodness‐of‐fit plot (Figure 3b ) showed that the model‐predicted ΔΔQTcF values were close to the observed values across deciles of zanubrutinib plasma concentrations. Based on this, the proposed model was deemed to provide an acceptable representation of the relationship between ΔΔQTcF and zanubrutinib concentrations. The estimated population slope of the C‐QTc relationship was − 0.001 msec (90% CI, − 0.0058 to – 0.0035 msec) per ng/mL with an intercept of − 2.9 msec (90% CI, − 1.2 to –4.7 msec). The slope was not statistically significant from 0. The predicted ΔΔQTcFs at the geometric mean peak zanubrutinib concentrations after single oral doses of zanubrutinib 160 mg and 480 mg are − 3.16 msec and − 3.38 msec, respectively (Table 2 ).
Figure 3.
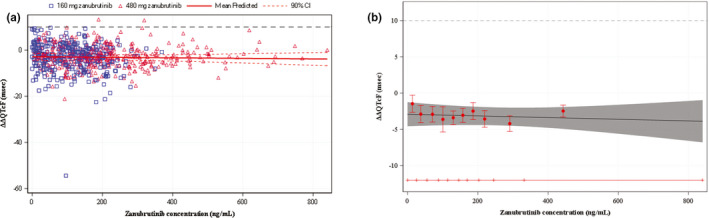
Zanubrutinib C‐QTc analysis. (a) Scatter plot of observed zanubrutinib plasma concentrations and mean change from baseline in QTcF interval over time using Fridericia’s formula (ΔΔQTcF) and (b) mean (90% confidence interval (CI)) model‐predicted and observed ΔΔQTcF across deciles of zanubrutinib plasma concentrations. (a) The solid red line with dashed red curves above and below denotes the model‐predicted mean (90% CI) ΔΔQTcF. The blue squares and red triangles denote the pairs of observed zanubrutinib plasma concentrations and ΔΔQTcF by participants for the 160‐mg and 480‐mg doses of zanubrutinib, respectively. (b) The red dots with vertical bars denote the observed mean (90% CI) ΔΔQTcF at the median zanubrutinib plasma concentration within each decile. The solid black line within the gray shaded area denotes the model‐predicted mean (90% CI) ΔΔQTcF. The horizontal red line with notches shows the range of concentrations divided into deciles for zanubrutinib. The area between each decile represents the point at which 10% of the data are present; the first notch to second notch denotes the first 10% of the data, the second notch to third notch denotes the second 10%, and so on.
Table 2.
Predicted ΔΔQTcF interval at geometric mean peak zanubrutinib concentration (PK/QTc population)
| Treatment | n | Geometric mean Cmax (90% CI), ng/mL | Predicted (90% CI) ΔΔQTcF, msec |
|---|---|---|---|
| Zanubrutinib 160 mg | 27 | 215.4 (198.96 to 233.29) | −3.16 (−4.51 to −1.81) |
| Zanubrutinib 480 mg | 28 | 401.1 (363.37 to 442.82) | −3.38 (−4.86 to −1.89) |
ΔΔQTcF, placebo‐corrected change‐from‐baseline QT interval corrected for heart rate with Fridericia’s formula; CI, confidence interval, Cmax, maximum observed concentration; PK, pharmacokinetic, QTc, QT interval corrected for heart rate.
QTc assay sensitivity: Moxifloxacin
Moxifloxacin treatment resulted in the expected level of QTc prolongation with mean ΔΔQTcF above 10 msec between 1 and 4 hours postdose (Figure 2b ) and a peak effect of 12.9 msec (90% CI, 10.71–15.02 msec) observed at 2.5 hours postdose. Using the same C‐QTc model as for the primary analysis, the estimated population slope of the moxifloxacin C‐QTc relationship was 0.005 msec per ng/mL, with a large intercept of 3.1 msec. The predicted ΔΔQTcF at the geometric mean peak moxifloxacin concentration was 13.2 msec (95% CI, 10.1–14.3 msec) at the mean peak moxifloxacin concentration of 1,952 ng/mL (90% CI, 1,800–2,116 msec). The lower bound of the 90% CI of the predicted effect at the observed Cmax (12.1 msec) is clearly above 5 msec, thereby demonstrating assay sensitivity.
Other ECG parameters
Zanubrutinib did not have an effect on PR or QRS intervals. Mean ΔΔPR was < 5 msec across all time points after dosing with zanubrutinib. Mean ΔΔQRS was within ± 1.0 msec at all postdose time points regardless of zanubrutinib dose. No participants exhibited a > 25% increase in PR from baseline to a PR > 200 msec or a > 25% increase in QRS from baseline to a QRS > 120 msec. There were a few observations of flat T‐waves (i.e., two participants each in the moxifloxacin and placebo groups) and biphasic T‐waves (i.e., one participant each in the zanubrutinib and placebo groups).
Categorical outlier analysis
No participant had outlier QTcF values (i.e., > 450 and ≤ 480 msec, > 480 and ≤ 500 msec, or> 500 msec when not present at baseline) in any treatment group. No participant had outlier ΔQTcF values (i.e., > 30 and ≤ 60 msec or > 60 msec) in the zanubrutinib or placebo groups. No participant had outlier PR or QRS values.
Safety
In part A, data for all eight enrolled participants were included in the safety analysis. Two participants reported five AEs, including diarrhea, proteinuria, thrombocytopenia, and viral infection, in 1 participant receiving zanubrutinib 480 mg and mild blood creatinine increased in 1 subject receiving placebo. All AEs were mild (grade 1) or moderate severity (grade 2), assessed as related to study drug by the investigator.
In part B, data for all 32 enrolled participants were included in the safety analysis. All TEAEs occurred in < 10% of participants. The overall percentage of TEAEs for all of the four treatment periods was 22.2%, 10.0%, 13.3%, and 7.1% for moxifloxacin 400 mg, zanubrutinib 160 mg, zanubrutinib 480 mg, and placebo, respectively. Twelve participants distributed across all 4 treatments reported 24 AEs, including abdominal pain, diarrhea, dry mouth, dyspepsia, hyperglycemia, hypophosphatemia, polydipsia, back pain, musculoskeletal pain, myalgia, headache, presyncope, dysuria, hematuria, proteinuria, neutropenia, fatigue, urinary tract infection, increase in alanine aminotransferase, increase in aspartate aminotransferase, and dermatitis. All AEs were mild (grade 1) in intensity except for the urinary tract infection, which was assessed as moderate (grade 2) in intensity. All AEs were assessed as related to study drug by the investigator, with the exception of dermatitis and presyncope, which the investigator deemed unrelated to study drug.
DISCUSSION
This study demonstrated that single oral doses of zanubrutinib at an expected therapeutic dose of 160 mg and a supratherapeutic dose of 480 mg did not have a clinically relevant effect on ECG parameters, including QTc intervals and other parameters. The result of this study is consistent with a low risk of QTc prolongation with zanubrutinib assessed from the preclinical data and accumulated safety data in patients with B‐cell malignancy. 5 It is also consistent with available clinical profiles of other BTK inhibitors, in which both ibrutinib 12 and acalabrutinib 23 have been shown not to prolong the QTc interval at therapeutic and supratherapeutic concentrations.
The sample size for part B of the study (n = 24) was selected based on the experience from the IQ‐CSRC study; 14 Simulations were used to demonstrate that a sample of 24 participants would have enough study power to exclude a small QTc effect (10 msec) by using C‐QTc analysis. 15 With the traditional “by time point” analysis, described in the ICH E14 document, 18 the effect of the drug is tested at each postdose time point, using the Intersection Union Test (IUT). 24 Because the objective is to exclude a 10 msec effect at each postdose time point, repeated statistical tests were performed on data from each time point separately. Therefore, substantially less data are used in several separate tests with IUT as compared with C‐QTc analysis, which analyzes QTc values and concentration data from all subjects and postdose time points in one statistical test. In addition to the underlying effect of the drug and the variability of the data, the power of a study to exclude an effect is largely determined by the sample size. For the reasons described above, the C‐QTc analysis has much smaller variability of the estimated QTc effect, which results in more narrow width of the 90% CI, as compared with the by time point analysis with IUT. Because the objective of the study is to exclude a small effect (10 msec), the sample size can, therefore, be substantially reduced with C‐QTc analysis. TQT studies that required ~ 48 evaluable subjects with the by time point analysis 24 can thereby be performed with 20 to 24 subjects, 16 provided data are analyzed with C‐QTc. It was possible to reduce the sample size of this TQT study by approximately half, thereby making the ECG evaluation more resource efficient.
The limitations of the supratherapeutic dose (480 mg) in this TQT study are acknowledged. It has been shown that oral clearance of CYP3A substrates displays considerable interindividual variability due to environmental, dietary, pathologic, and genetic modulation of enzyme activity at both hepatic and intestinal sites. 25 Zanubrutinib is a sensitive CYP3A substrate, and moderate to high PK variability (between subject and between study) has been observed in clinical studies. The mean Cmax of zanubrutinib 160 mg in patients with B‐cell malignancy was 346 ng/mL 9 compared with 216 and 406 ng/mL at 160 and 480 mg, respectively, in this study. As such, the highest clinically relevant plasma concentrations observed in this TQT study may not substantially exceed drug concentrations seen in patients and the potential increase by intrinsic or extrinsic factors, such as drug‐drug interactions. In addition, following the completion of this TQT study, zanubrutinib has received accelerated approval by the FDA for zanubrutinib 320 mg once daily in addition to 160 mg twice daily dose for patients with R/R MCL. 5 With mean Cmax at 320 mg ~ 2 times higher than that of 160 mg, the 480 mg dose cannot be considered a supratherapeutic dose for 320‐mg once‐daily dose.
Despite challenges around selecting the appropriate supratherapeutic dose, the use of C‐QTc analysis in the study rather than the traditional time point analysis enabled the assessment of QTc changes over a wide range of concentrations. Additionally, the totality of evidence for assessment of the risk of QT prolongation based on review of nonclinical data, categorical analyses of outliers in this TQT study, and AEs in targeted patient population 5 signals low risk for potential proarrhythmic effects of zanubrutinib. Of note, the C‐QTc analysis has been shown to be useful in predicting effects in specific populations or conditions (e.g., drug interactions) not directly evaluated in the TQT study. 26 Although the mean Cmax at 480 mg was 406 ng/mL, a wide range of exposure up to ~ 800 ng/mL was observed in this TQT study. Based on the C‐QTc analysis for zanubrutinib, an effect on the QTc interval exceeding 10 msec can be excluded within this observed plasma concentration range. In addition, given that the slope of C‐QTc relationship is not significant from 0 with trend for a negative slope (the estimated mean slope was − 0.001 msec (90% CI, −0.0058 to 0.0035 msec) per ng/mL), it seems very unlikely that zanubrutinib will cause concerning QTc prolongation at clinically relevant plasma concentrations and even in patients on a strong CYP3A inhibitor where peak concentrations around 1,000 mg/mL could be expected.
A shortening of QTc with a mean of 3 msec relative to predose and the placebo group was observed in participants receiving zanubrutinib treatment at both the 160‐mg and 480‐mg doses, even though a small shortening of QTc was also observed in the placebo group. Although the clinical implication of a small decrease in QTc (i.e., < 20 msec) is either null or poorly understood, 27 it is recognized that if a drug causes a truly large QTc shortening (e.g., by an average of 50–60 msec), its safety may need to be seriously considered, as cardiac electrophysiology may be dramatically affected. 27 A shortening QTc has been observed for another BTK inhibitor, ibrutinib. 12 It has been shown that ibrutinib can cause a concentration‐dependent mild shortening of QTc and mild PR prolongation, although these effects have not been considered clinically meaningful. In this study, there was not a clear concentration‐dependent effect of zanubrutinib on QTc shortening. QTc shortening has been seen frequently in placebo controls in TQT studies, most likely because of autonomic conditioning due to the stress of the restricted environment of clinical units. 27
In conclusion, in this TQT study, single doses of zanubrutinib 160 and 480 mg did not prolong the QTc interval to any clinically relevant extent. There was no effect on HR or cardiac conduction (PR, QRS intervals, or T‐wave morphology), and no participant had outlier values (QTcF > 450 msec or ΔQTcF > 30 msec) for absolute QTcF or ΔQTcF with zanubrutinib treatment. The plasma C‐QTc analysis showed no relationship between zanubrutinib exposure and ΔΔQTcF. The predicted mean ΔΔQTcF at mean zanubrutinib C max was < 10 msec for both 160 and 480 mg doses. It can be concluded that zanubrutinib at the therapeutic dose of 160 mg twice daily or 320 mg once daily is unlikely to have a clinically relevant effect on cardiac repolarization in patients.
Funding
This study was supported by BeiGene, Inc.
Conflicts of Interest
B.D. owns stock and is eligible for stock options in E.R.T., Rochester, NY. S.M., Z.T., W.N., M.T., L.L., Y.O., and S.S. own stock and are eligible for stock options in BeiGene, Inc., San Mateo, CA. All other authors declared no competing interests for this work.
Author Contributions
S.M., B.D., Z.T., W.N., M.T., H.X., M.W., L.L., S.S., and Y.O. wrote the manuscript. S.M., B.D., Z.T., H.X., S.S., and Y.O designed the research. S.M., B.D., Z.T., W.N., M.T., H.X., M.W., L.L., S.S., and Y.O performed the research. B.D., Z.T., and H.X analyzed the data. B.D., H.X and Z.T. contributed new reagents/analytical tools.
Compliance with Ethical Standards
Ethical approval: All the procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This paper does not contain any studies with animals performed by any of the authors.
Informed consent: Informed consent was obtained from all individual participants included in the study.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Acknowledgments
The authors thank the study participants who volunteered for this study. Copyediting support, funded by BeiGene, Inc., was provided by Erin Spohr and Joshua Safran of Ashfield Healthcare Communications (Lyndhurst, NJ).
Data Availability Statement
Upon request, and subject to certain criteria, conditions, and exceptions, BeiGene will provide access to individual de‐identified participant data from BeiGene‐sponsored global interventional clinical studies conducted for medicines (i) for indications that have been approved or (ii) in programs that have been terminated. BeiGene will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data requests may be submitted to medicalinformation@beigene.com.
References
- 1. Humphries, L.A. et al Tec kinases mediate sustained calcium influx via site‐specific tyrosine phosphorylation of the phospholipase Cgamma Src homology 2‐Src homology 3 linker. J. Biol. Chem. 279, 37651–37661 (2004). [DOI] [PubMed] [Google Scholar]
- 2. Advani, R.H. et al Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J. Clin. Oncol. 31, 88–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petro, J.B. & Khan, W.N. Phospholipase C‐gamma 2 couples Bruton’s tyrosine kinase to the NF‐kappa B signaling pathway in B lymphocytes. J. Biol. Chem. 276, 1715–1719 (2001). [DOI] [PubMed] [Google Scholar]
- 4. Rickert, R.C. New insights into pre‐BCR and BCR signalling with relevance to B cell malignancies. Nat. Rev. Immunol. 13, 578–591 (2013). [DOI] [PubMed] [Google Scholar]
- 5. BRUKINSA™ (zanubrutinib) [package insert] (BeiGene USA, Inc, San Mateo, CA, 2019). [Google Scholar]
- 6. Guo, Y. et al Discovery of zanubrutinib (BGB‐3111), a novel, potent, and selective covalent inhibitor of Bruton’s tyrosine kinase. J. Med. Chem. 62, 7923–7940 (2019). [DOI] [PubMed] [Google Scholar]
- 7. Dimopoulos, M.A. et al Major responses in MYD88 wildtype (MYD88WT) Waldenstrom macroglobulinemia (WM) patients treated with Bruton tyrosine kinase inhibitor zanubrutinib (BGB‐3111). Poster presented at: 24th Annual Congress of the European Hematology Association (EHA). June 13–16, 2019; Amsterdam, Netherlands. [Google Scholar]
- 8. Song, Y. et al Zanubrutinib in patients with relapsed or refractory mantle cell lymphoma: a single‐arm, multicenter, pivotal phase 2 study. Presented at: 15th International Conference on Malignant Lymphoma. June 18–22, 2019; Lugano, Switzerland. [Google Scholar]
- 9. Tam, C.S. et al Phase 1 study of selective BTK inhibitor zanubrutinib in B‐cell malignancies and safety and efficacy evaluation in CLL. Blood 134, 851–859 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Darpo, B. , Garnett, C. , Keirns, J. & Stockbridge, N. Implications of the IQ‐CSRC prospective study: time to revise ICH E14. Drug Saf. 38, 773–780 (2015). [DOI] [PubMed] [Google Scholar]
- 11. ICH E14 Implementation Working Group ICH E14 guideline: the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs. Questions and answers (R3). <https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Q_As_R3__Step4.pdf> (2015). Accessed June 7, 2019.
- 12. de Jong, J. et al Ibrutinib does not prolong the corrected QT interval in healthy subjects: results from a thorough QT study. Cancer Chemother. Pharmacol. 80, 1227–1237 (2017). [DOI] [PubMed] [Google Scholar]
- 13. Garnett, C. et al Scientific white paper on concentration‐QTc modeling. J. Pharmacokinet. Pharmacodyn. 45, 383–397 (2018). [DOI] [PubMed] [Google Scholar]
- 14. Darpo, B. et al Results from the IQ‐CSRC prospective study support replacement of the thorough QT study by QT assessment in the early clinical phase. Clin. Pharmacol. Ther. 97, 326–335 (2015). [DOI] [PubMed] [Google Scholar]
- 15. Ferber, G. , Zhou, M. & Darpo, B. Detection of QTc effects in small studies—implications for replacing the thorough QT study. Ann. Noninvasive Electrocardiol. 20, 368–377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang, D.P. et al Assay sensitivity in “hybrid thorough QT/QTc (TQT)” study. J. Biopharm. Stat. 29, 378–384 (2019). [DOI] [PubMed] [Google Scholar]
- 17. Mu, S. et al Effect of rifampin and itraconazole on the pharmacokinetics of zanubrutinib (a Bruton's tyrosine kinase inhibitor) in Asian and non‐Asian healthy subjects. Cancer Chemother. Pharmacol. 85, 391–399 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. U.S. Department of Health and Human Services . Guidance for industry: E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs. <https://www.fda.gov/media/71372/download> (2005). Accessed June 7, 2019.
- 19. Darpo, B. et al Improving the precision of QT measurements. Cardiol. J. 18, 401–410 (2011). [PubMed] [Google Scholar]
- 20. Tornøe, C.W. , Garnett, C.E. , Wang, Y. , Florian, J. , Li, M. & Gobburu, J.V. Creation of a knowledge management system for QT analyses. J. Clin. Pharmacol. 51, 1035–1042 (2011). [DOI] [PubMed] [Google Scholar]
- 21. Cleveland, W.S. Robust locally weighted regression and smoothing scatter plots. J. Am. Stat. Assoc. 74, 829–836 (1979). [Google Scholar]
- 22. Hurvich, C.M. , Simonoff, J.S. & Tsai, C.L. Smoothing parameter selection in nonparametric regression using an improved Akaike information criterion. J. R. Stat. Soc. Series B. Stat. Methodol. 60, 271–293 (1998). [Google Scholar]
- 23. CALQUENCE® (acalabrutinib) [package insert] (AstraZeneca Pharmaceuticals LP, Wilmington, DE, 2017). [Google Scholar]
- 24. Zhang, J. & Machado, S.G. Statistical issues including design and sample size calculation in thorough QT/QTc studies. J. Biopharm. Stat. 18, 451–467 (2008). [DOI] [PubMed] [Google Scholar]
- 25. Hohmann, N. , Haefeli, W.E. & Mikus, G. CYP3A activity: towards dose adaptation to the individual. Expert Opin. Drug Metab. Toxicol. 12, 479–497 (2016). [DOI] [PubMed] [Google Scholar]
- 26. Garnett, C.E. et al Concentration‐QT relationships play a key role in the evaluation of proarrhythmic risk during regulatory review. J. Clin. Pharmacol. 48, 13–18 (2008). [DOI] [PubMed] [Google Scholar]
- 27. Malik, M. Facts, fancies and follies of drug‐induced QT/QTc interval shortening. Br. J. Pharmacol. 159, 70–76 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Upon request, and subject to certain criteria, conditions, and exceptions, BeiGene will provide access to individual de‐identified participant data from BeiGene‐sponsored global interventional clinical studies conducted for medicines (i) for indications that have been approved or (ii) in programs that have been terminated. BeiGene will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data requests may be submitted to medicalinformation@beigene.com.