

Abstract
Determining factors that contribute to interindividual and intra‐individual variability in pharmacokinetics (PKs) and drug metabolism is essential for the optimal use of drugs in humans. Intestinal microbes are important contributors to variability; however, such gut microbe‐drug interactions and the clinical significance of these interactions are still being elucidated. Traditional PKs can be complemented by untargeted mass spectrometry coupled with molecular networking to study the intricacies of drug metabolism. To show the utility of molecular networking on metabolism we investigated the impact of a 7‐day course of cefprozil on cytochrome P450 (CYP) activity using a modified Cooperstown cocktail and assessed plasma, urine, and fecal data by targeted and untargeted metabolomics and molecular networking in healthy volunteers. This prospective study revealed that cefprozil decreased the activities of CYP1A2, CYP2C19, and CYP3A, decreased alpha diversity and increased interindividual microbiome variability. We further demonstrate a relationship between the loss of microbiome alpha diversity caused by cefprozil and increased drug and metabolite formation in fecal samples. Untargeted metabolomics/molecular networking revealed several omeprazole metabolites that we hypothesize may be metabolized by both CYP2C19 and bacteria from the gut microbiome. Our observations are consistent with the hypothesis that factors that perturb the gut microbiome, such as antibiotics, alter drug metabolism and ultimately drug efficacy and toxicity but that these effects are most strongly revealed on a per individual basis.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ There are few human studies investigating the clinical effect and mechanism of the microbiome modulating drug metabolism. Animal studies suggest that bacteria present in the gut may modulate regulatory elements in cytochrome P450 (CYP) genes leading to alterations in activity.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study addressed the following questions: (i) Does the antibiotic cefprozil modify the microbiome and cause significant changes in drug metabolizing enzyme activity? (ii) Can untargeted metabolomics with molecular networking provide additional insight into drug metabolism by the host and the microbiome?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This study showed that the antibiotic cefprozil decreased CYP1A2, CYP2C19, and CYP3A activity in humans, increased beta diversity, and decreased alpha diversity, which correlated with increased drug and drug metabolites. In addition, this study illustrates the important use of untargeted metabolomics and molecular networking together with targeted pharmacokinetics (PKs) to differentiate host and bacterial drug metabolism.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This study is at the interface of translational science by asking a clinically important question: Does altering the microbiome with antibiotics affect drug metabolism leading to important changes in drug action? We investigate this question using a novel method of untargeted metabolomics and molecular networking tools combined with traditional PK analysis. This methodology may enhance future drug metabolism studies, particularly involving the microbiome.
Identifying factors that contribute to the variability of drug action forms the basis for precision medicine. Drug metabolism is influenced by a variety of factors, including genetics, ontogeny, diet, xenobiotics, and circadian rhythms, is dynamic and occurs in multiple locations in the human body (e.g., gut lumen and liver). The impact of the gut microbiota on drug metabolism has been of recent interest leading to the burgeoning field of pharmacomicrobiomics 1 with implications for toxicity 2 , 3 and efficacy. 4 , 5 Gut microbes interact directly with drugs and indirectly with the human intestinal drug metabolizing enzymes and transporters. Recent investigations have shown that bacteria may generate metabolites previously unreported by traditional methods. 6 Additionally, animal and human studies suggest that microbes can alter the activities of cytochrome P450 enzymes (CYP) in the intestine leading to alterations in drug action. 7 , 8 , 9 , 10
A traditional approach to studying drug metabolism in humans involves investigating the pharmacokinetics (PKs) of probe compounds that serve as biomarkers for enzyme or transporter activity. A probe compound is chosen and validated based upon its high specificity for the enzyme or transporter of interest, its safety profile, and availability. Cocktail studies involving the simultaneous administration of multiple noninteracting drug probes enable multiple enzymes to be probed with improved efficiency. The Cooperstown cocktail has been validated 11 and used to study CYP1A2 (caffeine), CYP2D6 (dextromethorphan), CYP2C9 (warfarin), CYP2C19 (omeprazole), and CYP3A4 (midazolam). Enzyme phenotypes are assessed by PK parameters, such as oral clearance or parent:metabolite area under the curve (AUC) ratios.
Mass spectrometry (MS) is a common technology used in measuring drugs and their transformation in biofluids and tissues; MS is used for targeted PK methods, which provide accurate quantification with low limits of detection as well as untargeted methods, including those used for metabolomics, and provide a broad characterization of the chemicals. 12 MS detects the mass‐to‐charge (m/z) of ions and charged versions of neutral chemicals, thus providing information on the intrinsic mass of the chemical as well as a corresponding abundance, which is related to but not directly proportional to concentration. Further, MS can provide structural information of chemicals via product ion scans (one type of MS2) that contain interpretable fragments of a chemical structure when supplied with an excess of internal energy (such as higher‐energy collision dissociation). Often, MS is coupled to chemical separation, such as liquid chromatography (LC) or gas chromatography, prior to mass analysis to facilitate analysis of complex samples. Targeted MS methods, such as those used for PKs, are very sensitive (often able to detect chemicals in the low part‐per‐billion range) and specific as the chemicals to be detected are determined a priori, but are rarely used to monitor tens to hundreds of chemicals concurrently. Untargeted MS methods are more akin to a screening approach in which the aim is to detect all chemicals present in a given sample without any prior knowledge. With respect to the understanding of drug metabolism, targeted PKs and untargeted MS provide complementary information.
One challenge of the untargeted MS approach is the overwhelming amount of data generated. A solution is to use molecular networking, which connects chemicals with similar molecular structures based on MS2 fragmentation patterns via the Global Natural Products Social Molecular Networking (GNPS) platform. 13 , 14 The MS2 spectra are also used to provide putative annotations for their corresponding MS1 feature (i.e., m/z and retention time). Chemical annotations are generated by comparing measured MS2 spectra with that of reference MS2 spectra in GNPS. GNPS annotations are considered level 2 (putative annotation based on spectral library similarity) or level 3 (putatively characterized compound class based on spectral similarity to known compounds of a chemical class) by the 2007 metabolomics standard initiative. 15 Feature‐based molecular networking provides the ability to link the MS2 data processed in GNPS with the MS1 features making qualitative assessments of PKs possible.
Our approach to improving understanding of the influence of the intestinal microbiome on human drug metabolism is reported in this proof‐of‐concept investigation. Traditional targeted PK analysis was conducted using a modified Cooperstown cocktail to simultaneously assess CYP1A2, CYP2C19, CYP2D6, and CYP3A activities in healthy volunteers before and after a 7‐day course of oral cefprozil—an antibiotic with no known direct effect on CYP‐mediated drug metabolism. This was paired with untargeted MS coupled with molecular networking to detect and qualitatively measure drug metabolites in plasma, urine, and feces. Finally, 16S rRNA analysis of the microbiome was used to investigate changes in microbial composition and diversity after antibiotic therapy. Utilizing this methodology allowed us to more deeply interrogate gut microbial and host drug metabolism interactions and the effects of antibiotic perturbation of the gut microbiome on CYP drug metabolism.
Methods
Clinical protocol
This prospective, two‐period, crossover study was approved by the UC San Diego Human Research Protections Program (Protocol #161940). All procedures were conducted upholding the ethical standards according to the Declaration of Helsinki. All subjects provided written informed consent.
Fourteen healthy human subjects aged ≥ 18 years who met the inclusion/exclusion criteria were enrolled. Subjects were excluded if they were or had household contact within 2 weeks of the following: healthcare facility worker, farm worker, slaughterhouse worker, animal care worker; or were a vegetarian, smoker, consumed moderate amounts of alcohol chronically, on antibiotic therapy, hospitalized within the last 12 months, living with someone or an animal that had been on antibiotic therapy in the last month, had any gastrointestinal illness, any chronic illness, any infection requiring chemotherapy or antibiotics, diarrhea in the past 3 months, taking medications affecting the gastrointestinal system, CYP3A, CYP1A2, CYP2D6, CYP2C19 enzyme systems, immunomodulatory medications, allergy to beta‐lactams, pregnant or lactating, taking any herbal, dietary, or naturally derived supplements, or had an abnormal body mass index.
In order to phenotype multiple drug metabolizing enzymes simultaneously, we used a modified and validated Cooperstown cocktail. 16 , 17 The Cooperstown cocktail is a panel of four drugs used in PK studies to determine drug metabolizing enzyme activity. We used the following drug probes: caffeine for CYP1A2, 18 omeprazole for CYP2C19, 19 dextromethorphan for CYP2D6, 20 and midazolam for CYP3A. 21 We excluded warfarin and the evaluation of CYP2C9. We genotyped subjects for common allelic variants of CYP2C19, CYP2D6, CYP3A4, and CYP3A5.
Subjects were admitted to the outpatient unit of the Clinical and Translational Research Institute at UC San Diego. On study days 1 and 9, simultaneous single oral doses of caffeine 2 mg/kg, omeprazole 40 mg, dextromethorphan 30 mg, and midazolam 0.075 mg/kg were administered. On study days 2–8, subjects took the antibiotic cefprozil 500 mg orally twice daily. The cefprozil dosing regimen was chosen based upon its therapeutic use and a previous study demonstrating a reproducible effect on the gut microbiome. 22 Blood (5 mL) samples were taken prior to and 5 minutes, 0.5, 1, 2, 4, 5, 6, and 8 hours after drug administration on study days 1 and 9. Urine was sampled every 2 hours during the 8 hours after drug administration to dynamically assess metabolomics. Duplicate aliquots were obtained for metabolomic analysis. Fecal samples were obtained prior to study day 1 as a baseline, daily during antibiotic therapy, and on study days 1 and 9, as well as days 15 and 38, which corresponded to 7‐day and 30‐day postantibiotic therapy. Subjects fasted overnight, including avoidance of alcohol or caffeine, and were given standardized meals and snacks on study days 1 and 9 (Figure 1 ).
Figure 1.
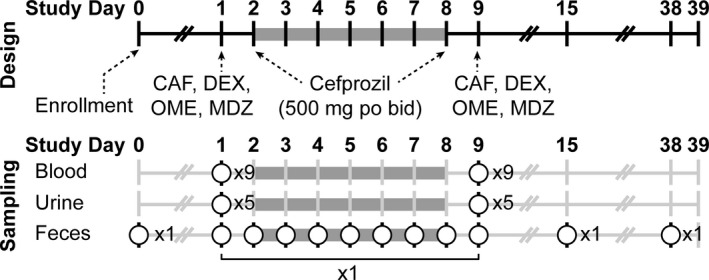
Clinical protocol design and sampling schema. Subjects received the Cooperstown cocktail on study days 1 and 9. Cefprozil was taken study days 2–8. Nine blood samples, five urine samples, and fecal samples were obtained on study days 1 and 9. Fecal samples were taken each day during cefprozil therapy and on study days 15 and 38. CAF, caffeine; DEX, dextromethorphan; MDZ, midazolam; OME, omeprazole.
Pharmacokinetic analysis
Caffeine, omeprazole, 5‐hydroxyomeprazole, dextromethorphan, dextrorphan, and midazolam PKs were determined by noncompartmental analysis using Phoenix version 8.1 (Pharsight, Cary, NC, USA). The AUC from time zero to infinity (AUC0–∞) was calculated as the sum of AUC from time zero to the last measurable concentration (AUC0–last) plus the ratio of the last measurable concentration and the elimination rate constant. Midazolam and caffeine apparent oral clearances were calculated as F*Dose/AUC0‐∞. A log‐linear trapezoidal method was used to calculate AUC0‐last. Additional PK parameters calculated included maximum plasma concentration (Cmax), the volume of distribution, elimination rate constant, and half‐life.
Statistical analysis
Fourteen subjects were randomized to detect a 25% difference (80% power) with an alpha = 0.05 based upon the change in midazolam oral clearance using previously published mean and SD data. 23 Data were log‐transformed prior to analyses. Based on January 2020 Food and Drug Administration Guidance for Clinical Drug Interaction Studies: Study Design, Data Analysis, and Clinical Implications (https://www.fda.gov/media/134581/download), analyses of variance and a general linear model were performed that included subject and treatment effects as factors and calculated least squares geometric mean ratios (LS‐GMRs). Ninety percent confidence intervals (CIs) were calculated and expressed as a percentage relative to the LS‐GMR of the pre‐antibiotic cocktail phase. If the 90% CI is within the 0.8 to 1.25 interval, it is concluded that there is no significant drug interaction. In contrast, values outside the 0.8 to 1.25 interval are indicative of an interaction between cefprozil and the specific drug‐metabolizing enzyme. 24 This statistical approach has been used in previous phenotyping cocktail studies. 25 , 26 Statistical analyses were performed using SAS version 9.3 (SAS Institute, Cary, NC, USA).
Pharmacogenomic analysis
Genotyping of the genes and variants listed below was performed by using polymerase chain reaction (PCR) and TaqMan allele discrimination in a custom designed microarray. TaqMan reagents consisted of a pair of unlabeled PCR primers and a TaqMan probe with a FAM or VIC dye label on the 5′ end and minor groove binder and nonfluorescent quencher on the 3′ end. TaqMan probes were designed to anneal within a DNA region amplified by a specific set of primers. As the Taq polymerase extended the primer and synthesized the nascent strand, the 5' to 3' exonuclease activity of the polymerase degraded the probe that annealed to the template. Degradation of the probe released the fluorophore from it and broke the close proximity to the quencher, thus relieving the quenching effect and allowing fluorescence of the fluorophore. The fluorescence detected in the quantitative PCR thermal cycler was directly proportional to the fluorophore released and the amount of DNA template present in the PCR. Using TaqMan probes for both the normal and variant allele enabled genotyping. Each variant was tested individually or within a custom array. For copy number analysis, gene specific (CYP2C19, CYP2D6, and CYP3A) and a reference gene were compared. Individual samples were run in quadruplicate. Each replicate was normalized to the reference gene to obtain a ΔCt (FAM dye Ct, VIC dye Ct), and then an average ΔCt for each sample (from the 4 replicates) was calculated. All samples were then normalized to a calibrator sample to determine ΔΔCt. The relative quantity was 2‐ΔΔCt, and copy number was 2 X relative quantity.
Targeted mass spectrometry
Quantitative simultaneous determination of caffeine, omeprazole, 5‐hydroxy (5‐OH) omeprazole, and midazolam in human plasma and dextromethorphan, dextrorphan, and dextrorphan‐O‐glucuronide in human urine was accomplished by the use of high‐performance liquid chromatography with tandem MS detection. Caffeine, omeprazole, 5‐OH omeprazole, and midazolam were precipitated from 50 µL of plasma with 100 µL of 100% acetonitrile (ACN). Twenty µl of supernatant was injected directly onto a C‐18 reversed phase high‐performance liquid chromatography (HPLC) column (MacMod Ace‐5, 2.1 × 150 mm). The LC mobile phase consisted of HPLC grade water with 0.1% formic acid (elute A) and ACN with 0.1% formic acid (elute B), and eluted with a gradient program of 0.5–3 minutes/90% B; 3.25–10 minutes/10% B at a flow rate of 0.4 mL/min. MS/MS detection was made in positive electrospray ionization mode, at mass transitions of 196 → 137 m/z (caffeine), 346 → 198 m/z (omeprazole), 362 → 214 m/z (5‐OH omeprazole), and 326 → 291 m/z (midazolam). The method had a dynamic range of 2–25,000 ng/mL. Dextromethorphan, dextrorphan, and dextrorphan‐O‐glucuronide were precipitated from 100 µL of urine with 200 µL of 100% ACN. Twenty µL of supernatant was injected directly onto a C‐18 reversed phase HPLC column (MacMod Ace‐5, 2.1 × 150 mm). The LC mobile phase consisted of HPLC grade water with 0.1% formic acid (elute A) and ACN with 0.1% formic acid (elute B), and eluted with a gradient program of 0.5–3 minutes/100% B; 3.50–10 minutes/0% B at a flow rate of 0.3 mL/minutes. MS/MS detection was made in positive electrospray ionization mode, at mass transitions of 272 → 171 m/z (dextromethorphan), 261 → 156 m/z (dextrorphan), and 434 → 258 m/z (5‐OH dextrorphan‐O‐glucuronide). The method had a dynamic range of 0.98–500 ng/mL. For analytes in both plasma and urine, calibration standards were used to generate a curve using a linear regression algorithm to plot the peak area ratio vs. concentration with 1/× weighting, over the full dynamic range of analyte concentrations. Dextromethorphan to dextrorphan molar ratios were evaluated for CYP2D6 activity; however, as many samples were below the lower limit of quantitation of the assay (0.98 ng/mL), the results were not interpretable.
Untargeted mass spectrometry
Sample preparation: Plasma
Three hundred µL of MeOH (100%) was added to each well of the 96‐well plate Phree Phospholipid Removal Kit and centrifuged at 500 g for 5 minutes, 3 times prior to sample addition; the MeOH was discarded in the laboratory hazardous waste. Blood plasma was stored at −80°C prior to extraction in 1.5 mL microtubes. The blood plasma microtubes were thawed at room temperature prior to extraction. Blood plasma samples were placed into one of four Phree Phospholipid Removal Kit 96‐well plates randomly. The thawed blood plasma samples were vortexed for 5 seconds and centrifuged for 1 minute at 5,000 rpm prior to pipetting 50 µL of each sample into the 96‐well Phree Phospholipid Removal Kit. Two hundred µL of MeOH (100%) was added to each well using a multichannel pipette; the solution was aspirated and dispensed 5 times to mix the blood plasma and organic solvent. A 96‐well plate (Eppendorf Microplate 96/U‐PP) was placed under the Phree Phospholipid Removal Kit to collect the sample and centrifuged at 500 g for 5 minutes. The Phree Phospholipid Removal Kit portion was discarded in the solid biohazardous waste and the sample‐containing 96‐well plate was evaporated until dry using a CentriVap Benchtop Vacuum Concentrator (Labconco, Kansas City, MO). The 96‐well plate containing the dried extract was covered (Storage Mat III 3080) and stored at −80°C prior to analysis. Immediately prior to analysis, the dried extract material was resuspended in 200 µL of MeOH‐water (1:1), sonicated for 5 minutes, centrifuged for 5 minutes at 500 g, and covered with a plate‐sealing film (Zone‐Free Sealing Films).
Sample preparation: Urine
Urine samples collected using BD Vacutainer tubes were stored at −80°C prior to extraction. Urine samples were thawed at room temperature. The Vacutainer tubes were vortexed for 5 seconds and centrifuged at 600 g for 5 minutes. The Vacutainer tube cap for each sample was carefully removed and 20 µL was pipetted into a well in the 96‐well plate (Eppendorf Microplate 96/U‐PP). Five µL of an internal standard solution (20 µg/mL solution of omeprazole‐d3, caffeine‐d3, midazolam‐d4, dextromethorphan‐d3, and dextrorphan‐d3) was added to the sample as well as 175 µL of ACN‐water (1:174). The 96‐well plate was covered with a plate sealing film (Zone‐Free Sealing Films) for analysis.
Sample preparation: Feces
Fecal samples were stored at −80°C prior to extraction. The swab tip of the BD Falcon SWUBE Collection and Transport System swabs were cut into Nunc 96‐Well Polypropylene DeepWell Storage Plates. Sample barcodes were scanned using a barcode scanner and saved into a Google Sheets spreadsheet generating a record of which sample was positioned in each well of the plate. Three hundred µL of MeOH‐water (1:1) was added to each well using a multichannel pipette. The deep well plate was covered with a storage mat and floated in an ultrasonic bath for 5 minutes. The samples were placed in a 4°C refrigerator overnight to extract. Subsequently, the swabs were removed from each well using tweezers, rinsing in between with nanopure water. The swab tips were disposed of in the solid biohazardous waste. Extracts were evaporated until dry using a CentriVap Benchtop Vacuum Concentrator (Labconco, Kansas City, MO, USA). The 96‐well plates containing the dried extract were covered (96‐deep well plate mats, Nunc 96 Well Caps for 1.0 mL Polystyrene DeepWell Plates) and stored at −80°C prior to analysis. Immediately prior to analysis, the dried extract material was resuspended in 300 µL of MeOH‐water (1:1), sonicated for 5 minutes, and centrifuged for 5 minutes at 500 g. One hundred µL of extract from each well was transferred into a 96‐well plate (Eppendorf Microplate 96/U‐PP) and diluted twofold using MeOH‐water (1:1), and covered with a plate‐sealing film (Zone‐Free Sealing Films).
Data acquisition
Blood plasma, urine, and fecal samples were analyzed using an ultra‐high‐performance LC (UltiMate 3000; Thermo) coupled to a quadrupole time‐of‐flight MS (maXis Impact, Bruker). Chromatographic separation was carried out on the analytical C18 column with corresponding C18 guard cartridge maintained at 40°C during separation. Then, 5.0 µL of the extract was injected per sample. Mobile phase composition was as follows: A, water with 0.1% formic acid (v/v) and B, ACN with 0.1% formic acid (v/v). Gradient elution was performed as follows: 0.0–0.8 minutes, 3% B; 0.8–5.0 minutes, 100% B; 5.0–6.5 minutes, 100% B; and 6.6 minutes–7.6 minutes, 3% B. MS data were collected using data dependent acquisition. An MS1 scan from m/z 50–1,500 at 3 Hz was followed by MS2 scans. Fragmentation was produced by stepped collision induced dissociation, of the five most abundant ions in the prior MS1 scan. Electrospray ionization, positive mode, was used to convert solution phase molecules into gas‐phase ions for MS analysis using the following source parameters: drying gas, 9.0 L min‐1; dry gas heater, 200 °C; capillary voltage, +4.5 kV; end plate offset, −0.5 kV; and nebulizer, 2.0 bar. Hexakis (2,2‐difluoroethoxy) phosphazene, lock mass standard, was sublimed in the ionization source; the lock mass standard was added such that a signal of ~ 1 × 105 signal was observed.
Data processing and data analysis
Lock mass corrected.mzXML files were generated from the raw qToF file (.d) using DataAnalysis (Bruker). MZmine2 was used to perform feature finding 27 (file with all parameters available in MassIVE MSV000082493), yielding a data matrix of MS1 features (i.e., m/z and retention time) and associated peak area. Feature‐based molecular networking outputs were generated from MZmine2 using the “export to GNPS” module, which generates a “quant.csv” which contains the MS1 feature information and a corresponding .mgf file, which contains MS2 information linked to the MS1 features when an MS2 was detected.
Molecular networking (GNPS)
A molecular network was created with the feature based molecular networking workflow (https://ccms‐ucsd.github.io/GNPSDocumentation/featurebasedmolecularnetworking/) on GNPS (https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=b3966445abad4658a3cdc63c8198a666). The data were filtered by removing all MS2 product ions within ± 17 m/z of the precursor m/z. MS2 spectra were window filtered by choosing only the top 6 fragment ions in the ± 50 m/z window throughout the spectrum. The precursor m/z tolerance was set to 0.02 m/z and a MS2 product ion m/z tolerance of 0.02 m/z. A network was then created where edges were filtered to have a cosine score above 0.7 and more than 4 matched peaks. Further, edges between two nodes were kept in the network if and only if each of the nodes appeared in each other’s respective top 10 most similar nodes. Finally, the maximum size of a molecular family (i.e., network component) was set to 100, and the lowest scoring edges were removed from molecular families until the molecular family size was below this threshold. The spectra in the network were then searched against GNPS spectral libraries. The library spectra were filtered in the same manner as the input data. All matches kept between network spectra and library spectra were required to have a score above 0.7 and at least 4 matched peaks.
Curves were generated in blood plasma, urine, and feces samples using code (R language) available at GitHub (see below). The MS1 features corresponding to the drug and drug metabolites were subsetted from the MS1 feature table (“quant.csv”) generated for feature‐based molecular networking by linking the chemical annotations from GNPS (based on MS2 spectral matching) or the monoisotopic mass (confirmed by manual interpretation of the MS2 product ion spectra). MS1 features detected in urine were normalized by the peak area of creatinine to compensate for variable concentration in random urine collection. MS1 features detected in fecal samples were normalized by the peak area of stercobilin, a heme catabolite responsible for the brown color of feces, to compensate for variable amounts in material extracted. Blood samples were analyzed without normalization as a fixed amount of volume was extracted in each sample. The results were subsequently plotted using the ggplot2 library in R. Note, nonfinite values were dropped when applying log10‐scaling to the y‐axis to facilitate interpretation as the peak areas for the drug and drug metabolites span multiple orders of magnitude.
Metabolomics data and code availability
All MS data (.d and .mzXML files) are publically available via GNPS/MassIVE (massive.ucsd.edu), a public MS data repository, under the accession number MSV000082493. All other code and materials (R) used to process data and generate plots are freely available at GitHub (github.com/alan‐jarmusch/UntargetedMS‐DrugMetabolism).
Microbiome
Sample and data processing
The Earth Microbiome Project 28 DNA extraction and 16S sequencing protocol was used for sample processing. In brief, stool sample DNA was extracted from swabs using the 96‐well MoBio Powersoil DNA kit, and barcoded 515F‐806R primers targeting the V4 region of the 16S rRNA gene were used for 16S amplification. The resulting V4 amplicons were sequenced at UCSD Institute for Genomic Medicine on an Illumina MiSeq.
Raw 16S sequencing data were uploaded to Qiita, 29 where it was demultiplexed, trimmed to 150 bp reads, and processed to suboperational taxonomic units (sOTU)s using Deblur. 30 The resulting feature table and representative sequences were downloaded and further analyzed with QIIME 2 31 to perform rarefaction, taxonomic assignments, phylogenetic tree generation, and differential abundance testing, and to calculate alpha and beta diversity. Taxonomic assignments used the naive bayes sklearn classifier 32 in QIIME 2 trained on the 515F/806R region of Greengenes 13_8 99% operational taxonomic units (OTUs). Phylogenetic tree generation was performed by inserting representative sequences into the Greengenes 13_8 99% tree with SEPP 33 in QIIME 2. The feature table was rarefied to 5,000 reads per sample before performing alpha diversity (observed OTUs) and beta diversity (unweighted UniFrac) calculations in QIIME 2. Statistical significance of alpha diversity comparisons between days was determined with the alpha‐group‐significance command (Kruskal–Wallis) in QIIME 2. Differentially abundant features (microbes) between study days 1 and 9 were calculated with discrete false discovery rate 34 on the rarified feature table collapsed to species‐level and on the noncollapsed table, implemented in QIIME 2. Repeated measures correlations were calculated using the python package Pingouin, 35 which implements repeated measures correlation (rmcorr) 36 in python and were run individually for each metabolite. Unweighted UniFrac distances used in repeated measures correlations were the unweighted UniFrac distances for each individual subject at study days 2–9 compared with their study day 0 (baseline) sample. Comparisons of beta diversity variability between subjects by day were calculated by taking the unweighted UniFrac distances between all subjects for each day and performing pairwise Kruskal–Wallis tests with Scipy 37 between the study day 0 distances and all other days.
Data visualizations were generated with the python packages seaborn (seaborn: statistical data visualization — seaborn 0.9.0 documentation. http://seaborn.pydata.org/) and matplotlib. 38
Microbiome data availability
Sequencing data are publicly available in Qiita at https://qiita.ucsd.edu/study/description/11766 and the EBI accession number is ERP117671. Jupyter notebooks with data analysis code are available at https://github.com/afvrbanac/Tsunoda‐Drug‐Metabolism.
Results
Cefprozil decreased activity of multiple CYP isoforms
After a 7‐day course of oral cefprozil at therapeutic doses (500 mg twice daily), the LS‐GMR of the PK parameter for caffeine, omeprazole, and midazolam decreased (Table 1 ). Furthermore, the 90% CI of the LS‐GMR for caffeine, omeprazole, and midazolam were all outside the 0.8 to 1.25 interval. Consequently, the decrease in the LS‐GMRs and accompanying 90% CIs suggest a decrease in CYP1A2, CYP2C19, and CYP3A4 activities. Due to uninterpretable results, dextromethorphan (CYP2D6) data are not reported. No trends could be explained from sex, race, or the genetic variation in the CYPs (Table S1 ).
Table 1.
Pharmacokinetic measurement of CYP activity prior to and after cefprozil
| CYP | Parameter | Geometric mean period 1 (no antibiotic) | Geometric mean period 2 (after antibiotic) | LS‐GMR | 90% CI |
|---|---|---|---|---|---|
| CYP1A2 | Caffeine (CL/F; mL/hour) | 24.9 | 23.5 | 0.93 | 0.53–1.33 a |
| CYP2C19 | OME/5‐OH OME (AUC) | 1.4 | 1.2 | 0.89 | 0.63–1.17 a |
| CYP3A4 | Midazolam (CL/F; mL/hour) | 9.7 | 9.6 | 0.98 | 0.78–1.19 a |
AUC, area under the curve; CI, confidence interval; CL/F, total clearance; CYP, cytochrome P450; LS‐GMR, least squares geometric mean ratio; OME, omeprazole; 5‐OH OME, 5‐hydroxy omeprazole.
90% CI is outside the 0.8–1.25 interval.
Cefprozil modified the gut microbiome
Microbiome alpha diversity (i.e., the number of observed OTUs per sample, remained similar between enrollment (study day 0) and the pre‐antibiotic Cooperstown cocktail administration (study day 1). The antibiotic cefprozil was taken orally beginning on study day 2, which resulted in an observed decrease in alpha diversity over the week‐long course (Figure 2a ), and alpha diversity was significantly lower on study days 5, 7, 8, and 9 (postantibiotic Cooperstown cocktail administration) compared with study day 1. Alpha diversity recovered to pre‐antibiotic levels by study day 15 after cessation of antibiotics on study day 8. Although cefprozil decreased alpha diversity in subjects, the induced changes in the microbiome of subjects were individualistic. Unweighted UniFrac distances, a measure of beta diversity between subjects were significantly higher on study days 3–9 and 15 compared with enrollment (study day 0), Figure S1 . Cefprozil was qualitatively detected using untargeted metabolomics in the fecal samples beginning on study day 3 through study day 9, affirming subject adherence, Figure S2 .
Figure 2.
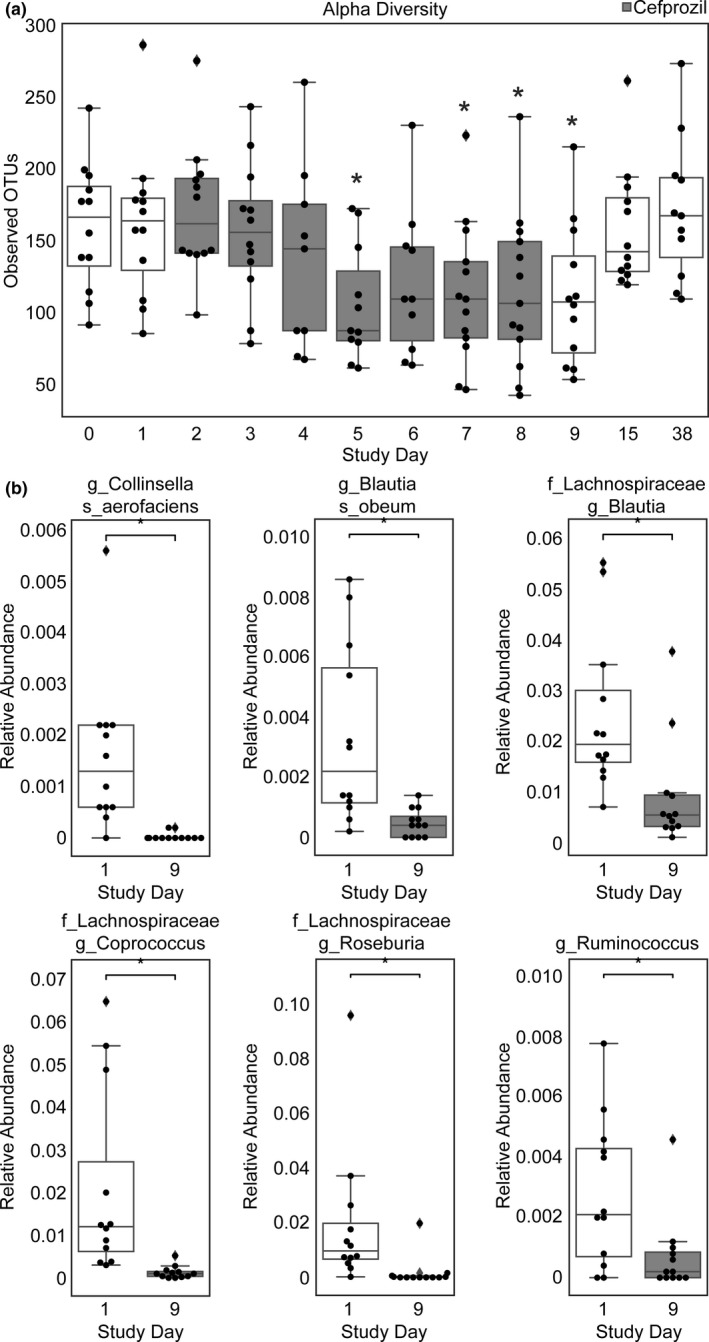
Box and whisker plots displaying changes in fecal microbiota. (a) Alpha diversity was measured by observed operational taxonomic units (OTUs). Boxes shaded in grey indicate the period of time in which oral cefprozil was administered. Asterisks indicate statistical significance (Kruskal‐Wallis) compared with alpha diversity on study day 1. (b) Differentially abundant bacteria between study days 1 and 9. Statistical significance was assessed with dsFDR(49) on the rarified feature table collapsed to species‐level. Plots are displayed as relative abundance for ease of interpretability. The boxes represent the 25%, 50%, and 75% quartile and the whiskers extend ±1.5 times the interquartile range.
Despite an increase in microbiome variability (i.e., beta diversity) between subjects with cefprozil treatment, changes in certain bacteria were consistent across subjects. Collinsella aerofaciens, Lachnospiraceae Blautia, Blautia obeum, Coprococcus, Roseburia, and Ruminococcus were significantly decreased in the study day 9 fecal samples compared with study day 1 (Figure 2b ). Although the Lachnospiraceae family overall was significantly lower in study day 9 samples, differential abundance testing at the sOTU level (individual 16S sequences) revealed multiple bacteria in the Lachnospiraceae family that increased with antibiotic treatment, including a Lachnospiraceae Clostridium sOTU and a Clostridium lavalense sOTU (Table S2 ).
Untargeted metabolomics and molecular networking enhance characterization of drug metabolism
Metabolomics data collected using untargeted MS were analyzed using molecular networking. Molecular networking links similar chemicals based on similar MS/MS fragmentation patterns. Every unique MS/MS spectrum is displayed as a node (ellipse) in Figure 3 and colored by annotation status (green annotated or grey unannotated). The majority of nodes, a proxy for how many different chemicals were detected, were unannotated. The CYP probe drugs, midazolam (components 1 and 3), omeprazole (components 2 and 5), and dextromethorphan (component 4) were molecular networked. Midazolam metabolites generated by CYP3A4 and glucuronosyltransferase (1‐hydroxymidazolam, 1‐hydroxymidazolam‐O‐glucuronide, and dihydroxymidazolam glucuronide) were detected in component 3. Omeprazole metabolites generated by CYP2C19, CYP3A4, and glucuronosyltransferase were detected in component 2 (5‐hydroxyomeprazole, 5‐carboxyomeprazole, and 5‐hydroxyomeprazole‐O‐glucuronide, respectively). Component 4 contained the CYP2D6‐derived metabolite dextrorphan. Caffeine was detected in the samples but is not included in the network as it falls below the networking criteria for the minimum number of fragment peaks (i.e., product ions).
Figure 3.
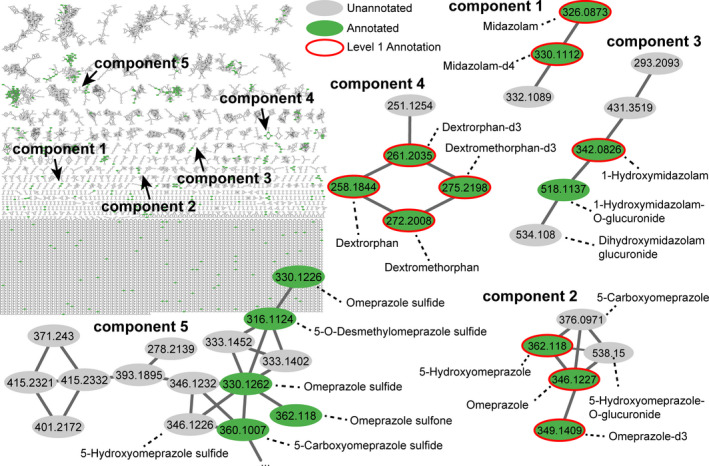
Molecular networking via Global Natural Products Social Molecular Networking for all samples with nodes (ellipse) representing unique tandem mass spectrometry (MS/MS) consensus spectra and edge (lines) indicating similarity in MS/MS fragmentation. Annotation is concurrently performed via MS/MS spectral library matching (level 2) and is indicated in grey (unannotated) or green (annotated). Authentic chemical standards improve confidence in annotation (level 1), red node outline. Illustrative molecular networking components (i.e., chemical families) are enlarged to highlight the detection and networking of CYP drug probes as well as metabolites.
Component 5 of the molecular network (Figure 3 ) included additional omeprazole metabolites, including omeprazole sulfide (a reported bacterial metabolite) and omeprazole sulfone (a reported CYP3A metabolite). 16 , 17 , 18 An unannotated node with a measured m/z of 346.1226 was linked to omeprazole sulfide, and was tentatively annotated as hydroxyomeprazole sulfide (presumed to be the 5‐hydroxy metabolite) upon the manual interpretation of the MS/MS spectra (Figure S3 and S4 ). Characteristic product ions of m/z 313.1422 (neutral loss of ‐SH), 198.0571, and m/z 149.0684 were noted. The neutral loss of 18.0110 (‐H2O), 32.9803 (‐HS), 63.9620 (‐SO2) was observed in omeprazole, omeprazole sulfide, and omeprazole sulfone, respectively. The neutral loss corresponding to the substituted benzimidazole ring (m/z 148.0652), also detected as a product ion of m/z 149.0684, was observed in omeprazole, omeprazole sulfide, and omeprazole sulfone, and the respective product ions of m/z 198.0581, 182.0619, and 214.0521. The detection of m/z 198.0571 corresponding to the charged substituted pyridine ring (and neutral loss of the substituted benzimidazole ring) matched that of omeprazole. The fragments suggested the presence of a chemical, isobaric with omeprazole with the same molecular formula, but differing in the location of oxygen atoms. The tentatively annotated hydroxyomeprazole sulfide shared a product ion of m/z 167.0922 with that of 5‐hydroxyomeprazole indicating hydroxylation. In summary, the link between molecular networking indicated a chemical similar to omeprazole sulfide, a bacterial metabolite, with an MS/MS spectra indicative of hydroxylation of the substituted pyridine ring, which we presume is the known 5‐hydroxymetabolite produced by CYP2C19.
The metabolomics data provided a global view of drug metabolism when analyzed via multivariate statistics (i.e., principal component analysis (PCA)). The urine data were relatively similar in chemical composition indicated by the grouping and a large amount of overlap in the PCA score plots in Figure 4a , b for pre‐antibiotic and postantibiotic study days. Data along the principal component (PC1) axis, displayed in the density plot (inset), indicate a shift associated with time after drug administration. The median in samples PC1 values shift in the positive PC1 direction after 0 minutes (pre‐administration of drugs) and continues to do so until ~ 240 or 360 minutes before beginning shifting in the negative direction on the PC1 axis at 480 minutes. The high‐level view of metabolism indicated that chemical shifts following the metabolism of the drugs is detectable, whereas modest differences are noted reflecting the minor effect of adding a few drugs to the complex chemical composition of urine. Untargeted metabolomics data provided semiquantitative information about the temporal changes in plasma, urine, and fecal samples of known drug and drug metabolites, such as omeprazole, hydroxyomeprazole, and omeprazole sulfide (Figure 4c – e ), as well as putatively annotated drug metabolites resulting from molecular networking (e.g., hydroxyomeprazole sulfide). Assuming similar ionization efficiency (i.e., the rate of converting neutral molecules to charged forms for detection via MS) based on the structural similarity of omeprazole and plotted metabolites, one could interpret the relative levels of the metabolites. In plasma, Figure 4c , omeprazole, 5‐hydroxyomeprazole, and hydroxyomeprazole sulfide were similar in abundance, whereas omeprazole sulfide was less abundant. The level of omeprazole began to increase ~ 30 minutes after dosage. Hydroxyomeprazole levels began increasing ~ 60 minutes after dosage, quickly increasing to the peak level at 120 minutes. Similar trends in omeprazole sulfide and hydroxyomeprazole sulfide were observed with a more gradual increase to peak levels (~ 120 to 240 minutes) beginning at 30 to 60 minutes. In urine, omeprazole and omeprazole sulfide were less abundant than hydroxyomeprazole sulfide and 5‐hydroxyomperazole. Similar to the blood levels, hydroxyomeprazole sulfide levels more gradually increased over time compared with hydroxyomeprazole. The values for study day 1 and study day 9 are displayed in different colors revealing trends, but the changes were not statistically significant in plasma or urine. The fecal data, Figure 4e , indicated that during the period of time after the initial dose of omeprazole, the parent drug, and its metabolites were detected in fecal samples over the following week.
Figure 4.
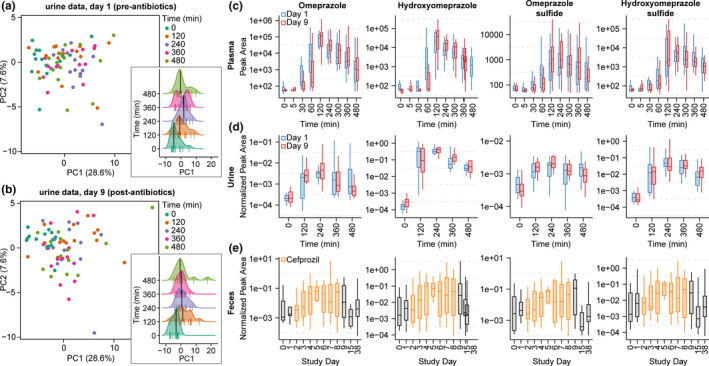
Principal component analysis of untargeted metabolomics urine data for (a) study day 1 (pre‐antibiotics) and (b) study day 9 (postantibiotics). Points represent urine samples and are colored by the time in which they were collected after drug consumption. Density plots for principal component (PC)1 values are inset. Lines at the bottom of each distribution reflect individual points, the median value is indicated (black line), and the distributions are offset by time. (c) Plot of time vs. peak area (log‐scaled) for omeprazole, hydroxyomeprazole, omeprazole sulfide, and hydroxyomeprazole sulfide in plasma samples. (d) Plot of time vs. peak area normalized by creatinine (log‐scaled) for omeprazole, hydroxyomeprazole, omeprazole sulfide, and hydroxyomeprazole sulfide in urine samples. (e) Plot of study day vs. peak area normalized to stercobilin (log‐scaled) for omeprazole, hydroxyomeprazole, omeprazole sulfide, and hydroxyomeprazole sulfide in fecal samples obtained daily. The period of antibiotic use is indicated in orange. The boxes represent the 25%, 50%, and 75% quantile and the whiskers extend ± 1.5 times the interquartile range. Note, values are log10‐scaled to facilitate interpretation and points with nonfinite values are not displayed; however, statistical analyses were performed using all points.
Decreased microbiome alpha diversity correlated with increased formation of drug and drug metabolites in feces
Microbiome perturbation was associated with elevated drug and drug metabolites in fecal samples. Using a repeated measures correlation 36 to account for interindividual variation, we found microbiome alpha diversity was anticorrelated with both levels of omeprazole and omeprazole metabolites (Figure 5 ), independent of time (Figure S5 ). Additionally, the unweighted UniFrac distance for each individual’s sample to their study day 0 (baseline) sample was positively correlated with omeprazole and omeprazole metabolites (Figure 5 ). Taken together, this indicates that the magnitude of microbiome disruption, measured by both microbiome alpha diversity changes and individual deviation from baseline (beta diversity), was associated with omeprazole and omeprazole metabolite accumulation in feces. A similar trend was observed for other drugs, including caffeine and midazolam (Figure S6 ), although midazolam accumulation in feces was significantly associated with time (Figure S5 ).
Figure 5.
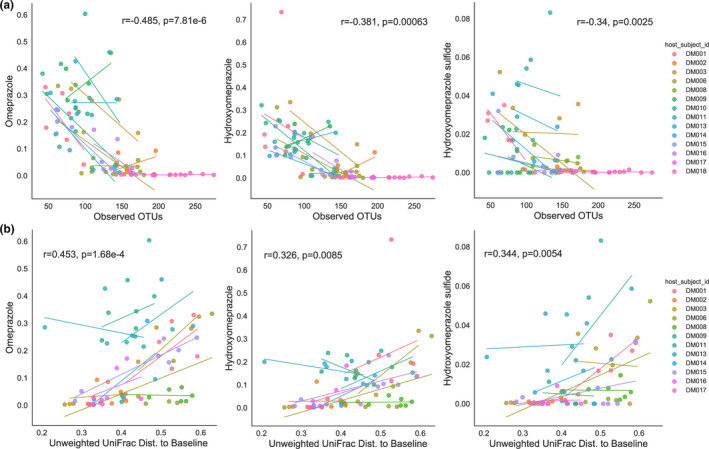
Correlations among fecal omeprazole (OME), 5‐hydroxy (5‐OH) OME, and 5‐OH OME sulfide levels, and (a) alpha diversity (observed operational taxonomic unit (OTUs)) or (b) unweighted UniFrac distance to each subject’s baseline sample. Plots display regression lines for each individual subject and statistics displayed are from the repeated measures correlation using days 2–9. Subjects DM10 and DM18 were omitted as their study day 0 (baseline) samples were either below the sequencing quality threshold or not collected.
Discussion
After a 7‐day course of the oral antibiotic cefprozil, the phenotyping parameters for caffeine, omeprazole, and midazolam were altered reflecting decreased activities of CYP1A2, CYP2C19, and CYP3A, respectively. Cefprozil had a greater effect on CYP2C19 and modest effects on CYP3A4 and CYP1A2. The mechanism for this effect is not likely due to cefprozil directly affecting CYP‐mediated metabolism as the majority of the drug is excreted unchanged in the urine; hepatic metabolism is thought to be minimal, and no CYP‐mediated drug‐drug interactions have been reported with these probe compounds. 39 Cefprozil is a second‐generation cephalosporin that has activity against gram positive bacteria (e.g. Streptococci, Haemophilus influenza) as well as gram negative bacteria (e.g., Escherichia coli); and affects specific organisms in the gut microbiome. 22 Therefore, we propose that cefprozil is modifying the gut microbiome and affecting CYP activity. It is possible that other antibiotics that more dramatically alter gut microorganisms may have different effects on CYP‐mediated metabolism.
Cefprozil decreased microbiome alpha diversity, which significantly recovered by study day 15. Similar to a previous study, 22 there was significant interindividual variability in the gut microbial changes after cefprozil (Figure S1 ). Most of the gut bacteria that decreased with cefprozil treatment (Lachnospiraceae, Blautia, Coprococcus, Roseburia, and Ruminococcus) were from the Firmicutes phyla, which together with Bacteroides composes the majority of the human gut microbiome. Although none of these bacteria have been previously associated with changes in drug metabolism, interesting associations with human disease and drug activity have been reported. Increased Lachnospiraceae Blautia was associated with survival in graft‐vs.‐host disease. 40 Coprococcus has been shown to be depleted in depression. 41 Roseburia has been associated with several diseases suggesting that it may be a biomarker for a healthy microbiome. 42 Collinsella aerofaciens from the actinobacteria phylum was significantly decreased in our study. Interestingly, C. aerofaciens has been associated with inflammatory bowel diseases, aging, and drug activity. A recent in vitro drug screen suggests that C. aerofaciens may contribute to the metabolism of 16 different drugs 43 and an increased abundance was associated with greater response to anti‐PD‐1 therapy in patients with metastatic melanoma. 44
Recent investigations have drawn attention to the role of Faecalibacterium prausnitzii in the disposition of the CYP3A substrate and immunosuppressive agent tacrolimus. The abundance of F. prausnitzii in feces was positively correlated to tacrolimus dosing in patients who underwent kidney transplantation 10 ; and, upon further investigation, F. prausnitzii was shown to form a tacrolimus metabolite. 6 We did not observe a change in F. prausnitzii in our data, although it was shown to be present throughout individuals over time (Figure S7 ). Cefprozil did not significantly affect this particular organism in our cohort.
Metabolomics performed using untargeted MS provided information on drug metabolites via molecular networking, an overall picture of drug metabolism via PCA, and qualitative information on the temporal dynamics of drug metabolites. Molecular networking was able to connect parent and metabolite drugs, including known CYP biotransformation products, such as 1‐hydroxymidazolam (CYP3A4), dextrorphan (CYP2D6), and 5‐hydroxyomeprazole (CYP2C19) across blood plasma, urine, and feces. Deuterated chemical standards of the parent drugs and select metabolites networked with their respective parent drugs increased confidence in chemical annotations provided via spectral library matching in GNPS. Phase II drug metabolites were concurrently detected (e.g., glucuronidation). The drug metabolites that were not assigned a chemical annotation were more often than not due to the unavailability of a reference MS2 spectrum in the GNPS (a compilation of nearly all public MS2 reference spectra, including MoNA, MassBank, MassBank EU, etc.); however, connections to known metabolites could be established using molecular networking facilitating annotation de novo.
Omeprazole sulfide was detected in the blood, urine, and fecal samples in support of previous reports in human blood plasma, 45 human urine, 46 and wastewater samples. 47 The exact mechanism or location in the human body at which omeprazole sulfide is produced cannot be conclusively determined in this study; however, there is prior evidence that omeprazole sulfide can be produced by the intestinal microbiome of rat feces ex vivo as well as in the blood of rats after oral ingestion and intrarectal dosage. 48 Further, human and rabbit feces incubated with sulphinpyrazone and sulindac resulted in sulfoxide reduction, which is also observed in rabbits in vivo. 49 Biochemically, the reduction of the sulfoxide functional group may serve as an electron acceptor in anaerobic respiration. We cannot discount the role of chemical reduction, particularly in acidic conditions, that may occur. Regardless of the mechanism and location of production, our study affirms that omeprazole sulfide can be detected in blood plasma, urine, and feces in humans. Further, molecular networking indicated the presence of a chemical, connected to omeprazole sulfide, which was tentatively identified as hydroxyomeprazole sulfide based on MS/MS and exact mass (2.02 ppm mass error). This metabolite was reported previously in wastewater samples 47 ; but a source was never identified. We postulate that this metabolite is the combined product of both host CYP and intestinal gut microbiota metabolism, although we did not test this directly.
With respect to the omeprazole metabolites, we investigated the changes in alpha diversity and beta diversity, and the formation of omeprazole and its metabolites over study days 2–9. We found alpha diversity was negatively correlated with omeprazole and omeprazole metabolites, associating a loss of microbial diversity with the accumulation of omeprazole, 5‐OH omeprazole, and 5‐OH omeprazole sulfide in feces. This was not simply an artifact of time, neither omeprazole nor its metabolites were significantly correlated with day (Figure S5 ). We also quantified the magnitude of microbiome changes for each individual over the course of the study by looking at the unweighted UniFrac distances from the study day 0 baseline sample to study days 2–9. An increase in UniFrac distance to baseline, indicating a greater change in microbiome composition, was significantly correlated with omeprazole, 5‐OH omeprazole, and 5‐OH omeprazole sulfide accumulation in feces. Although we cannot directly link these results to bioavailability of the drug, disruption of the microbiome seems to be associated with drug and drug metabolite content in feces. Notably, we did not observe a significant correlation with changes in CYP activity and alpha or beta diversity, although unlike fecal metabolite correlations, we were limited to data from only study days 1 and 9 where CYP activity was measured.
In conclusion, using the tools of targeted MS and untargeted MS/molecular networking and microbiome analysis, we demonstrated a relationship between perturbation of the gut microbiome and drug metabolism. Untargeted MS data analyzed by molecular working via GNPS were useful in connecting drugs with their known CYP and phase II drug metabolites, suggesting the presence of uncharacterized drug metabolites, which could be confirmed using exact masses, molecular formulae, and interpretation of MS2 product ion spectra; and that qualitative assessment of PK levels in multiple human biofluids is possible. We affirm the presence of omeprazole sulfide in human blood plasma, urine, and fecal samples. We report the presence of 5‐hydroxyomeprazole sulfide, 5‐carboxyomeprazole sulfide, and 4‐hydroxyomeprazole sulfide‐O‐glucuronide in human blood plasma and urine, and hypothesize they are produced through the combination of human CYP and phase II metabolism and that of intestinal microbiota. Enhanced interrogation of drug metabolism with our methodology provides insight into the chemical modifications, which may be previously unknown and allows differentiation between host and microbe. Alteration of drug metabolizing enzymes by antibiotics may be an important mechanism to consider in the variability of drug PKs, drug action, and pharmacodynamics. Future studies to identify gut microbes and their products and the molecular mechanisms of this interaction will be important. Other factors that change the gut microbiome may also significantly impact intestinal and hepatic drug metabolism. Understanding these contributions to overall drug bioavailability brings us closer to optimal drug utilization and discovery.
Funding
Funding for this project was provided by the National Institutes of Health (UL1TR001442, Tsunoda; U01 AI124316‐01, P.C.D. and R.K. P41 GM103484, Center for Computational Mass Spectrometry; GMS10RR029121, P.C.D.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. A.K.J. thanks the American Society for Mass Spectrometry for their support. S.M.T. received a CTRI Pilot Project grant.
Conflict of Interest
Pieter C. Dorrestein is a scientific advisory board member for Sirenas. All other authors declared no other competing interests for this work.
Author Contributions
A.K.J., A.V., and S.M.T. wrote the manuscript. S.M.T., A.K.J., and P.C.D. designed the research. S.M.T., A.K.J., A.V., and J.D.M. performed the research. A.K.J., P.C.D., A.V., R.K., M.A., M.L., J.D.M., J.D.M., and S.M.T. analyzed the data. P.C.D. and R.K. contributed new reagents/analytical tools.
Supporting information
Acknowledgments
The authors acknowledge David Ashley, Mark Wallace, MD, and Tina Misel, RN, NP, for their coordination, enrollment, and recruitment assistance of the healthy volunteers; and Victoria M. Pratt, PhD, for the pharmacogenomic analysis.
Contributor Information
Rob Knight, Email: robknight@health.ucsd.edu.
Pieter C. Dorrestein, Email: pdorrestein@health.ucsd.edu.
Shirley M. Tsunoda, Email: smtsunoda@health.ucsd.edu.
References
- 1. Sharma, A. , Buschmann, M.M. & Gilbert, J.A. Pharmacomicrobiomics: the holy grail to variability in drug response? Clin. Pharmacol. Ther. 106, 317–328 (2019). [DOI] [PubMed] [Google Scholar]
- 2. Nakayama, H. , Kinouchi, T. , Kataoka, K. , Akimoto, S. , Matsuda, Y. & Ohnishi, Y. Intestinal anaerobic bacteria hydrolyse sorivudine, producing the high blood concentration of 5‐(E)‐(2‐bromovinyl)uracil that increases the level and toxicity of 5‐fluorouracil. Pharmacogenetics 7, 35–43 (1997). [DOI] [PubMed] [Google Scholar]
- 3. Okuda, H. , Ogura, K. , Kato, A. , Takubo, H. & Watabe, T. A possible mechanism of eighteen patient deaths caused by interactions of sorivudine, a new antiviral drug, with oral 5‐fluorouracil prodrugs. J. Pharmacol. Exp. Ther. 287, 791–799 (1998). [PubMed] [Google Scholar]
- 4. Maini Rekdal, V. , Bess, E.N. , Bisanz, J.E. , Turnbaugh, P.J. & Balskus, E.P. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364, eaau6323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gopalakrishnan, V. et al Gut microbiome modulates response to anti‐PD‐1 immunotherapy in melanoma patients. Science 10, 97–103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guo, Y. et al Commensal gut bacteria convert the immunosuppressant tacrolimus to less potent metabolites. Drug Metab. Dispos. 47, 194–202 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fu, Z.D. , Selwyn, F.P. , Cui, J.Y. & Klaassen, C.D. RNA‐Seq profiling of intestinal expression of xenobiotic processing genes in germ‐free mice. Drug Metab. Dispos. 45, 1225–1238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Selwyn, F.P. , Cheng, S.L. , Klaassen, C.D. & Cui, J.Y. Regulation of hepatic drug‐metabolizing enzymes in germ‐free mice by conventionalization and probiotics. Drug Metab. Dispos. 44, 262–274 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bjorkholm, B. , Bok, C.M. , Lundin, A. , Rafter, J. , Hibberd, M.L. & Pettersson, S. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS ONE 4, e6958 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee, J.R. et al Gut microbiota and tacrolimus dosing in kidney transplantation. PLoS ONE 10, e012239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Streetman, D.S. et al Combined phenotypic assessment of CYP1A2, CYP2C19, CYP2D6, CYP3A, N‐acetyltransferase‐2 and xanthine oxidase with the "Cooperstown cocktail". Clin. Pharmacol. Ther. 68, 375–383 (2000). [DOI] [PubMed] [Google Scholar]
- 12. Schrimpe‐Rutledge, A.C. , Codreanu, S.G. , Sherrod, S.D. & McLean, J.A. Untargeted metabolomics strategies‐challenges and emerging directions. J. Am. Soc. Mass Spectrom. 27, 1897–1905 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang, M. et al Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 34, 828–837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Quinn, R.A. , Nothias, L.F. , Vining, O. , Meehan, M. , Esquenazi, E. & Dorrestein, P.C. Molecular networking as a drug discovery, drug metabolism, and precision medicine strategy. Trends Pharmacol. Sci. 38, 143–154 (2017). [DOI] [PubMed] [Google Scholar]
- 15. Sumner, L.W. et al Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 3, 211–221 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chainuvati, S. et al Combined phenotypic assessment of cytochrome P450 1A2, 2C9, 2C19, 2D6, and 3A, N‐acetyltransferase‐2, xanthine oxidase activities with the "Cooperstown 5+1 cocktail". Clin. Pharmacol. Ther. 74, 437–447 (2003). [DOI] [PubMed] [Google Scholar]
- 17. Ma, J.D. , Nafziger, A.N. , Villano, S.A. , Gaedigk, A. & Bertino, J.S. Jr. Maribavir pharmacokinetics and the effects of multiple‐dose maribavir on cytochrome P450 (CYP) 1A2, CYP 2C9, CYP 2C19, CYP 2D6, CYP 3A, N‐acetyltransferase‐2, and xanthine oxidase activities in healthy adults. Antimicrob. Agents Chemother. 50, 1130–1135 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gu, L. , Gonzalez, F.J. , Kalow, W. & Tang, B.K. Biotransformation of caffeine, paraxanthine, theobromine and theophylline by cDNA‐expressed human CYP1A2 and CYP2E1. Pharmacogenetics 2, 73–77 (1992). [DOI] [PubMed] [Google Scholar]
- 19. Abelo, A. , Andersson, T.B. , Antonsson, M. , Naudot, A.K. , Skanberg, I. & Weidolf, L. Stereoselective metabolism of omeprazole by human cytochrome P450 enzymes. Drug Metab. Dispos. 28, 966–972 (2000). [PubMed] [Google Scholar]
- 20. Yu, A. & Haining, R.L. Comparative contribution to dextromethorphan metabolism by cytochrome P450 isoforms in vitro: can dextromethorphan be used as a dual probe for both CTP2D6 and CYP3A activities? Drug Metab. Dispos 29, 1514–1520 (2001). [PubMed] [Google Scholar]
- 21. Patki, K.C. , Von Moltke, L.L. & Greenblatt, D.J. In vitro metabolism of midazolam, triazolam, nifedipine, and testosterone by human liver microsomes and recombinant cytochromes p450: role of cyp3a4 and cyp3a5. Drug Metab. Dispos 31, 938–944 (2003). [DOI] [PubMed] [Google Scholar]
- 22. Raymond, F. et al The initial state of the human gut microbiome determines its reshaping by antibiotics. ISME J. 10, 707–720 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsunoda, S.M. , Velez, R.L. , von Moltke, L.L. & Greenblatt, D.J. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect of ketoconazole. Clin. Pharmacol. Ther. 66, 461–471 (1999). [DOI] [PubMed] [Google Scholar]
- 24. Williams, R.L. , Chen, M.L. & Hauck, W.W. Equivalence approaches. Clin. Pharmacol. Ther. 72, 229–237 (2002). [DOI] [PubMed] [Google Scholar]
- 25. Stopfer, P. et al Optimization of a drug transporter probe cocktail: potential screening tool for transporter‐mediated drug‐drug interactions. Br. J. Clin. Pharmacol. 84, 1941–1949 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wyen, C. et al Effect of an antiretroviral regimen containing ritonavir boosted lopinavir on intestinal and hepatic CYP3A, CYP2D6 and P‐glycoprotein in HIV‐infected patients. Clin. Pharmacol. Ther. 84, 75–82 (2008). [DOI] [PubMed] [Google Scholar]
- 27. Pluskal, T. , Castillo, S. , Villar‐Briones, A. & Oresic, M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry‐based molecular profile data. BMC Bioinformatics 11, 395 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thompson, L.R. et al A communal catalogue reveals Earth's multiscale microbial diversity. Nature 551, 457–463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gonzalez, A. et al Qiita: rapid, web‐enabled microbiome meta‐analysis. Nat. Methods 15, 796–798 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Amir, A. et al Deblur rapidly resolves single‐nucleotide community sequence patterns. mSystems 2, 1–7, pii: e00191‐16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bolyen, E. et al Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abraham, A. et al Machine learning for neuroimaging with scikit‐learn. Front. Neuroinform. 8, 14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Janssen, S. et al Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems 3, 1–14, pii: e00021‐18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiang, L. , Amir, A. , Morton, J.T. , Heller, R. , Arias‐Castro, E. & Knight, R. Discrete false‐discovery rate improves identification of differentially abundant microbes. mSystems 2, 1–15, pii: e000092‐17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vallat, R. Pingouin: statistics in Python. J. Open Source Softw. 3, 1026 (2018). [Google Scholar]
- 36. Bakdash, J.Z. & Marusich, L.R. Repeated measures correlation. Front. Psychol. 8, 456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Virtanen, P. et al SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hunter, J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007). [Google Scholar]
- 39. Barriere, S.L. Review of in vitro activity, pharmacokinetic characteristics, safety, and clinical efficacy of cefprozil, a new oral cephalosporin. Ann. Pharmacother. 27, 1082–1089 (1993). [DOI] [PubMed] [Google Scholar]
- 40. Jenq, R.R. et al Intestinal blautia is associated with reduced death from graft‐versus‐host disease. Biol. Blood Marrow Transplant. 21, 1373–1383 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Valles‐Colomer, M. et al The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 4, 623–632 (2019). [DOI] [PubMed] [Google Scholar]
- 42. Machiels, K. et al A decrease of the butyrate‐producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63, 1275–1283 (2014). [DOI] [PubMed] [Google Scholar]
- 43. Zimmermann, M. , Zimmermann‐Kogadeeva, M. , Wegmann, R. & Goodman, A.L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570, 462–467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matson, V. et al The commensal microbiome is associated with anti‐PD‐1 efficacy in metastatic melanoma patients. Science 359, 104–108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Regardh, C.G. , Gabrielsson, M. , Hoffman, K.J. , Lofberg, I. & Skanberg, I. Pharmacokinetics and metabolism of omeprazole in animals and man–an overview. Scand. J. Gastroenterol. Suppl. 108, 79–94 (1985). [DOI] [PubMed] [Google Scholar]
- 46. Nevado, J.J. , Penalvo, G.C. , Dorado, R.M. & Robledo, V.R. Simultaneous determination of omeprazole and their main metabolites in human urine samples by capillary electrophoresis using electrospray ionization‐mass spectrometry detection. J. Pharm. Biomed. Anal. 92, 211–219 (2014). [DOI] [PubMed] [Google Scholar]
- 47. Boix, C. , Ibanez, M. , Zamora, T. , Sancho, J.V. , Niessen, W.M. & Hernandez, F. Identification of new omeprazole metabolites in wastewaters and surface waters. Sci. Total Environ. 468–469, 706–714 (2014). [DOI] [PubMed] [Google Scholar]
- 48. Watanabe, K. , Furuno, K. , Eto, K. , Oishi, R. & Gomita, Y. First‐pass metabolism of omeprazole in rats. J. Pharm. Sci. 83, 1131–1134 (1994). [DOI] [PubMed] [Google Scholar]
- 49. Strong, H.A. , Renwick, A.G. , George, C.F. , Liu, Y.F. & Hill, M.J. The reduction of sulphinpyrazone and sulindac by intestinal bacteria. Xenobiotica 17, 685–696 (1987). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All MS data (.d and .mzXML files) are publically available via GNPS/MassIVE (massive.ucsd.edu), a public MS data repository, under the accession number MSV000082493. All other code and materials (R) used to process data and generate plots are freely available at GitHub (github.com/alan‐jarmusch/UntargetedMS‐DrugMetabolism).