

Atomistic molecular dynamics simulation characterizes ebselen binding activity to SARS-CoV-2 main protease.
Abstract
There is an urgent need to repurpose drugs against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Recent computational-experimental screenings have identified several existing drugs that could serve as effective inhibitors of the virus’ main protease, Mpro, which is involved in gene expression and replication. Among these, ebselen (2-phenyl-1,2-benzoselenazol-3-one) appears to be particularly promising. Here, we examine, at a molecular level, the potential of ebselen to decrease Mpro activity. We find that it exhibits a distinct affinity for the catalytic region. Our results reveal a higher-affinity, previously unknown binding site localized between the II and III domains of the protein. A detailed strain analysis indicates that, on such a site, ebselen exerts a pronounced allosteric effect that regulates catalytic site access through surface-loop interactions, thereby inducing a reconfiguration of water hotspots. Together, these findings highlight the promise of ebselen as a repurposed drug against SARS-CoV-2.
INTRODUCTION
A coronavirus of zoonotic origin, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is the etiological agent responsible for the 2019–2020 viral pneumonia COVID-19 outbreak that commenced in Wuhan City, Hubei Province, China (1–4). Currently, targeted therapeutics are limited and, at this point in time, effective treatment options remain tentative and highly restricted.
Conventional drug design and drug development strategies require years of investigations and substantial investments. The repurposing of approved pharmaceutical drugs (and drug candidates already in clinical trials) therefore provides an essential, alternative approach to rapidly identify drugs with clinical potential to help manage emerging infectious diseases for which specific treatments and vaccines are not available. A recent high-throughput screening study considered more than 10,000 compounds against the main protease Mpro of SARS-CoV-2 (5). The main protease is one of the key enzymes in the viral life cycle, as it plays a central role in mediating viral replication and transcription. Its structure is shown in Fig. 1. Along with other, nonstructural proteins (papain-like protease, helicase, and RNA-dependent RNA polymerase) and the spike glycoprotein structural protein, Mpro is essential for interactions between the virus and host cell receptors during viral entry (6), making it an attractive drug target against SARS-CoV-2 (7, 8).
Fig. 1. Structure of Mpro and density maps of ebselen binding.
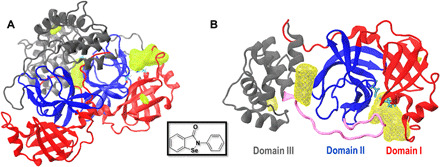
(A) Mpro dimer and (B) protomer A (top view). In both panels, Mpro domains I, II, and III are shown in red, blue, and gray, respectively. Cys145 and His41 (the catalytic dyad) are shown in cyan. Yellow surfaces show the most probable interaction sites between Mpro and ebselen (highest probability density). The loop (residues 185 to 201) connecting domains I and II with domain III is shown in pink (B). The inset shows the chemical structure of the ebselen molecule.
In a recent study by Jin et al. (5), a wide range of approved drugs, drug candidates, and natural products were screened by an enzymatic inhibition assay to identify a possible Mpro inhibitor. Ebselen, an organoselenium compound whose structure is shown in Fig. 1, emerged from that study as a promising drug lead to target this crucial enzyme. It was found to exhibit strong antiviral activity in cell-based assays (5). In previous, pre-COVID-19 work, ebselen was found to have anti-inflammatory, antioxidative, and cytoprotective properties. It has been investigated in the context of multiple diseases, such as bipolar disorders (9) and hearing loss (10, 11). In addition, a recent report indicated that ebselen also showed potent bactericidal activity against multidrug-resistant (MDR) clinical isolates of Staphylococcus aureus (12). Likewise, the use of silver and ebselen in synergistic formulations has been shown to be effective against five, clinically difficult-to-treat MDR Gram-negative bacteria (13). Ebselen has low cytotoxicity (median lethal dose in rats >4600 mg/kg, per os) (14), and its safety in humans has been evaluated in several clinical trials (10, 11, 15). Together, these reports—and the underlying data—underscore the clinical potential of ebselen for CoV-2 treatment.
In an effort to understand the molecular mechanisms through which ebselen interacts with Mpro, in this work, we present results of atomistic molecular simulations that provide useful, previously unknown insights into the Mpro-ebselen complex, which might provide novel avenues to rationally enhance ebselen’s activity. First, we present an analysis of the most probable interaction sites between Mpro-ebselen, as well as absolute binding free energy calculation for a complementary quantitative evaluation. Second, we evaluate the role of different binding sites on molecular stiffness and molecular strain. Last, we examine how ebselen binding modifies the structure and transport of water in Mpro’s catalytic site. We find that ebselen binds to two sites, as opposed to only one at the catalytic site, and we also identify an allosteric mechanism that influences the catalytic site when ebselen is bound at the distant site. Our findings are summarized in a concluding section, where we also provide several suggestions for future experimental work.
RESULTS
Molecular dynamics (MD) simulations of Mpro and ebselen were carried out using the AMBER18 simulation package (see Materials and Methods for details). A total of 3 μs of atomistic MD trajectories of Mpro using ebselen as a molecular probe were organized into 15 replicas of 200 ns each. These trajectories were analyzed to construct probability density maps for the preferred locations of ebselen around the protein. The results are shown in Fig. 1. Two distinct, highly probable binding sites emerged from this analysis. The first is located within the catalytic site, and the second is in a region that is essential for Mpro’s dimerization (16), between the II and III domains. Similar observations were reported in a previous study (17), where simple organic solvents or compounds such as acetonitrile, benzene, dimethyl sulfoxide, methanol, phenol, and urea were used as molecular probes. In addition, the authors considered the potential mutability of residues belonging to the Mpro catalytic site and explained that the development of drug resistance associated with the natural evolution of Mpro could wipe out efforts that target this protein for COVID-19 treatment. Instead, they emphasized the promise of alternative strategies aimed at targeting the region between the II and III domains, which is implicated in dimer formation. The results shown in Fig. 1 are therefore encouraging in that ebselen appears to target both the catalytic site and the dimerization domain.
Figure 2 provides a closer look at ebselen’s binding motifs in both regions (the catalytic site and the intersection between domains II and III). In the catalytic site (Fig. 2A), hydrogen bonding interactions are formed between the carbonyl oxygen of ebselen and the Asn142 and Gln189 side chains. One can also appreciate the hydrophobic contacts between ebselen and Met165, Pro168, Met49, His164, and both residues belonging to the catalytic dyad, His41 and Cys145.
Fig. 2. Binding modes for ebselen-Mpro complexes.
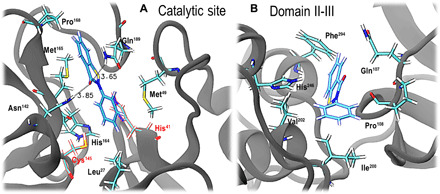
(A) At the catalytic site and (B) at the intersection between domains II and III. In both panels, ebselen, as well as main residues displaying contacts, is shown in sticks.
Figure 2B shows a representative configuration of ebselen at the intersection between domains II and III. There are distinct hydrophobic contacts with Phe294, Pro108, Ile200, Val202, His246, Thr292, Ile249, Pro132, and Ile249 (some residues are not shown for clarity). Highly dynamic hydrogen bonds were observed between ebselen and the Gln107, Gln110, and His246 side chains.
The results shown in Fig. 1 were generated on the basis of direct MD simulations. To arrive at a quantitative estimate of the binding affinity of each site, we used thermodynamic integration (TI) to determine the absolute binding free energy for the catalytic site and the domains II and III site. More specifically, we used particle mesh Ewald molecular dynamics (PMEMD) as implemented in AMBER18 (18) with 11 windows per integration and 10 ns per window. In addition, multiple runs starting from the most probable binding cluster identified in the previous MD simulations were considered. In this way, three independent replicas for each site were used to calculate averages. The results are shown in Table 1.
Table 1. Absolute binding free energy for Mpro-ebselen complexes.
The average and uncertainty energy values were obtained from three independent replicas for each site. Details of the calculation are described in Materials and Methods.
| Binding site |
Absolute binding free energy (kcal/mol) |
| Catalytic site | −5.55 ± 2.28 |
| Domains II and III site | −8.87 ± 1.59 |
Consistent with the probability density maps shown in Fig. 1, the absolute binding free energies corresponding to both sites are negative, serving to underscore the thermodynamic stability of the ebselen-Mpro complex at both sites. Here, we note the binding affinity of ebselen at the second binding site, located between the II and III domains, is, in fact, greater than that at the catalytic site. A previous report suggested that this small molecule could also inhibit Mpro through noncovalent binding, particularly because it has been found to exhibit a stronger inhibition effect than other compounds that were also able to quantitatively modify Cys145 in the catalytic dyad (5). The results presented here could explain ebselen’s high enzymatic inhibition, even when this compound could only partially modify Cys145.
Having identified a distant binding site for ebselen between domains II and III, we examined the role of this binding site, if any, on the catalytic site of the protein. To do so, we determined the local strain induced by the binding throughout the protein. To examine potential synergies of drug binding to both sites, we also considered a third scenario in which both sites (catalytic and distant) are simultaneously occupied by ebselen. Note that local strain provides a measure of local deformation that filters out nontrivial, functional conformational changes from nonfunctional ones, and it is therefore ideally suited to highlight how a perturbation (i.e., ebselen binding) at one site induces conformational changes at other, potentially distant, sites. It is also important to highlight that different measures of strain have recently been used in studies of allostery in proteins (19–21). Here, we used a method originally introduced by our group in the context of local strain in polymeric glasses (22).
Figure 3 shows the shear strain throughout the protein upon binding of ebselen at different sites, alongside the β factor (estimated from the root mean squared fluctuation), which measures thermal fluctuations for each residue. The shear strains are measured relative to the average conformation from all frames of the apo protein trajectory; these strains, therefore, correspond to the deformation upon binding relative to the apo protein. The shear strain for the apo structure is then showing an average internal strain in the protein.
Fig. 3. Strain analysis of ebselen-Mpro complexes.
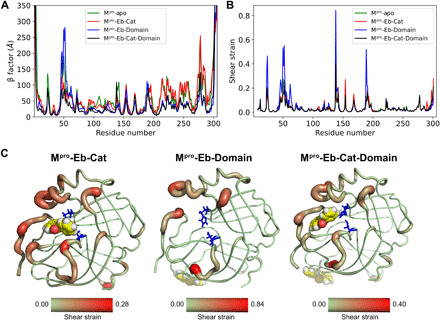
(A) β factor, estimated from the root mean squared fluctuation. (B) Shear strains. In both upper panels, Mpro-apo results are shown in green (Mpro-apo); for ebselen, when bound to the catalytic site (Mpro-Eb-Cat), results are in red. For the domain II–III interface (Mpro-Eb-Domain), results are shown in blue. For two ebselen molecules bound at both sites simultaneously (Mpro-Eb-Cat-Domain), results are shown in black. For the shear strain calculation, only the Cα atoms are included. (C) Shear strains mapped onto the different protein complexes from (B). The redder the region and the larger the radius of the structure, the higher the shear strain. For clarity, only the catalytic site and domains I and II are shown, the drug is shown as spheres, and the catalytic dyad is shown as blue sticks.
From the β factor analysis, it is evident that when ebselen is bound to the domains II and III interface (Fig. 3A, blue lines), the dynamics of the 44–52 loop, which flanks the catalytic site, is notably altered. On the other hand, when ebselen is bound at both sites simultaneously, there is a clear global reduction of the thermal fluctuations among the receptor except for one single region located around residues 137 to 140, where enhanced flexibility is apparent. Apart from these residues, ebselen bound at both sites shows the lowest β factor values; however, at this specific point, this system exhibits as high flexibility as Mpro-apo protein (Fig. 3A, green line).
The strain analysis helps disentangle the effects of functional and nonfunctional fluctuations and provides a more detailed view of the effects of ebselen binding. As seen in Fig. 3A for the β factor analysis, when ebselen is bound to the domain interface, it produces a large strain at the 44–52 loop (Fig. 3B, blue line). Likewise, the 185–201 loop that also flanks the catalytic site, as well as the region comprising residues 137 to 140, exhibits a high strain, which is not immediately apparent in the β factor. When both molecules are bound simultaneously to Mpro (Fig. 3B, black line), a high strain signal is also exhibited around residues 137 to 140, as expected on the basis of the aforementioned β factor results. Last, when ebselen is located in the catalytic site, a much lower strain is showed around all three regions (Fig. 3B, red line). Figure 3C shows that the strain is primarily localized at the two loops flanking the catalytic site (44–52 and 185–201 loops), as well as a loop in catalytic site (residues 22 to 25). A highly strained region also appears around residues 137 to 140. To understand why the 137 to 140 residues show such a large strain when ebselen is bound to both sites, we turn our attention to the molecular images shown in Fig. 4.
Fig. 4. Close-up of the highly strained regions identified from strain analysis.
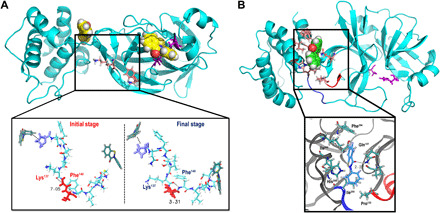
(A) The Mpro structure with ebselen molecules bound to both catalytic site and domains II and III interface simultaneously. In the upper panel, Lys137 and Phe140 are shown as sticks. The lower panel shows the close-up of this region and shows the backbone H-bond between Lys137 and Phe140 at the beginning and end of the simulation. (B) The Mpro structure with ebselen bound to the domains II and III interface. The close-up shows that a hydrogen bonding interaction is formed with Gln107 side chain, and hydrophobic contacts are formed with His246, Val202, Ile249, Phe294, Pro132, and Ile200. This last residue is located at the end of the 185–201 loop, which connects domain III with domains I and II (catalytic site). In both (A) and (B) upper panels, ebselen is shown as spheres and the catalytic dyad as magenta sticks.
In the close-up of the residues shown in Fig. 4A, a specific backbone hydrogen bond forms between Lys137 and Phe140 because of the binding of ebselen between domains II and III; this conformational change induced by the presence of ebselen causes the high strain shown in Fig. 3B. This conformational change takes place in the middle region between the catalytic site (Cys145) and the binding cleft between domains II and III (Pro132), which points to the relevance of these residues.
From these results, it is evident that when ebselen binds between the II and III domains, it exerts a pronounced allosteric effect that affects the loops that regulate access to the catalytic site (44–52 and 185–201 loops). In addition, it affects the residues 137 to 140, where a specific backbone hydrogen bond forms between Lys137 and Phe140. The exact role of this conformational change will be the subject of future studies, but the results presented here show that it serves as a relay between domain III and the catalytic site.
Given that conformational changes in the catalytic site are observed when ebselen binds to Mpro far away from this specific region, we turn our attention to the hydration characteristics of the catalytic site in the Mpro-ebselen complex (when bound to the domains II and III interface) and in the Mpro-apo structure [Protein Data Bank (PDB) code: 6M03] (23). AQUA-DUCT 1.0.5 (24) was used to analyze the water structure and water flux in the Mpro protein, with a time window of 50 ns and sampling every 1 ps. The results are shown in Fig. 5.
Fig. 5. Water analysis of Mpro complexes.
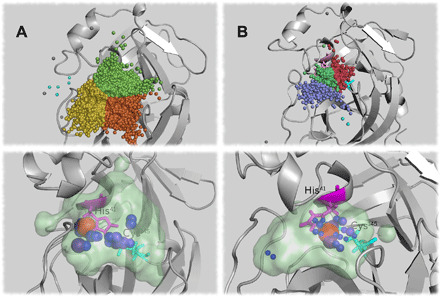
Top: Water inlet clusters for (A) apo protein and (B) ebselen in domains II and III. The biggest cluster in both cases is divided into three parts using K-means (24) and colored for clarity. Bottom: MAV (as light green surface) and hotspots (blue-red color and sphere radius relative to occupation values) for (A) apo and (B) ebselen in domains II and III. The catalytic dyad is represented with sticks (magenta, His41; cyan, Cys145).
Figure 5 (top) shows that ebselen binding between domains II and III leads to fewer water inlets compared with the Mpro-apo state; this is indicative of a water flux reduction in the catalytic site upon ebselen binding, although it binds far from the catalytic site. From the lower panel in Fig. 5, it is evident that a volume reduction of the catalytic site occurs when ebselen is located between domains II and III compared with the apo state; the maximum available volume (MAV) for water in the catalytic region is around 50% smaller in this case. Note that in the apo protein, there is a catalytic water molecule that forms a catalytic triad together with Cys145 and His41 (17, 25), and this catalytic water (red in Fig. 5A, bottom) is preserved and remains close to His41 in the simulations. In contrast, it is clear from Fig. 5B (bottom) that the presence of ebselen induces a displacement and reconfiguration of water hotspots, including the catalytic water (red). These effects could prevent the normal enzymatic function of Mpro, as this catalytic water displacement might damage the catalytic triad required for protein activity (26). Similarly, the observed pocket volume reduction might affect the accessibility of the polyprotein that Mpro cleaves, thereby reducing enzymatic function.
DISCUSSION
As mentioned before, Mpro is an attractive drug target against the COVID-19 virus due to its central role in the viral life cycle. A previous structural and evolutionary investigation suggested that the SARS-CoV-2 Mpro is not a suitable target for de novo development of inhibitors or the repurposing of drugs against the previous SARS coronavirus (17). However, only the flexibility and plasticity of the active sites in Mpro for COVID-19 and the highly similar previous SARS-CoV Mpro were compared. Nevertheless, major differences in both shape and size of the catalytic site were observed, indicating that repurposing SARS drugs for COVID-19 may not be effective. On the basis of their evolutionary analysis, these authors also pointed out that the virus’s mutability will pose further challenges to treatments against the COVID-19 Mpro protein. An alternative to this discouraging scenario, however, would be to target the region between the II and III domains, which is implicated in dimer formation.
Here, we find that there are two highly probable interaction sites between SARS-CoV-2 Mpro and ebselen. One is located within the catalytic cavity and, more importantly, in the region between the II and III domains, which is essential for Mpro dimerization (16). Detailed calculations of the free energy reveal a higher binding affinity of ebselen to domains II and III than to the catalytic site.
The strain analysis reveals that ebselen bound between the II and III domains exert a pronounced allosteric effect that affects the loops regulating access to the catalytic site. In addition, it also affects residues 137 to 140, where a specific backbone hydrogen bond forms between Lys137 and Phe140.
The catalytic site water analysis indicates that the proposed allosteric inhibition by ebselen could occur through a volume reduction of the catalytic pocket and a reconfiguration of water hotspots in that region. Given the catalytic role of water in this enzyme’s activity, these effects could act to prevent the regular enzymatic function of the SARS-CoV-2 Mpro protein.
Together, the results from our comprehensive study of ebselen activity and the underlying data serve to underscore the potential of ebselen for COVID-19 treatment. The discovery of a distant binding site is also encouraging in that it offers potential for development of novel Mpro inhibitors based on the ebselen scaffold; indeed, distant sites could be effective targets for other drugs that might have been deemed futile for catalytic sites. Note that current, massive virtual screening campaigns are focused primarily on targeting the SARS-CoV-2 Mpro catalytic site; our study, however, highlights the potential importance of a different, distant site (27). Further experimental structural characterization will be necessary to validate the predictions presented in this work, particularly those pertaining to the distant binding site. In the next stages of this project, we will seek to investigate the experimental crystal structure of the SARS-CoV-2 Mpro–ebselen complex, to validate our theoretical predictions and help elucidate the inhibitory mechanism. These efforts could also be combined with rational mutagenesis of interacting residues to alanine and binding assays such as surface plasmon resonance, with which to quantify binding and provide a basis for comparisons to theoretical predictions.
A previous evolutionary study (17) showed that mutating a few residues belonging to the catalytic site is energetically unfavorable. Residues such as P39, R40, P52, G143, G146, or L167 could therefore be considered as key anchoring sites for future Mpro inhibitor design. A recent study (5) identified ebselen as a promising repurposed drug against SARS-CoV-2. Our results build on that work by providing a detailed molecular picture of the interactions between ebselen and Mpro. The insights provided here could help facilitate the rational molecular design of new analogs, based on the ebselen scaffold and its binding to a distant site, for future coronaviruses. This could be substantial, particularly given the fact that the SARS-CoV-2 Mpro protein shares a high sequence similarity to the SARS-CoV Mpro (28), and is therefore a likely target for future coronaviruses. This alternative approach, uncovered through the extensive molecular simulations presented here, highlights the need to develop reliable, high-throughput methods to screen drug-protein interactions at the molecular level and that incorporate the role of explicit water molecules. We conclude by pointing out that our focus in this work has been on noncovalent complexes between ebselen-Mpro. In a subsequent stage, we also plan to investigate covalent complexes involving Cys145.
MATERIALS AND METHODS
A total of more than 6 μs of classical MD simulations of SARS-CoV-2 Mpro-apo state and Mpro-ebselen complex were run using the AMBER18 (29) simulation package (3 μs, ebselen as a molecular probe; 2.4 μs, shear strain analysis; 100 ns, water structure and flux analysis; 990 ns, free energy analysis). The receptor initial configuration for the Mpro-ebselen system was taken from a recently reported structure for the Mpro-N3 inhibitor (5) (PDB ID: 6LU7); where the inhibitor and crystallographic water molecules were removed before starting simulations. Force field parameters for ebselen were determined using the Antechamber program and described by the general amber force field (30, 31). The partial atomic charges were determined by the restrained electrostatic potential fitting technique. Those electrostatic potential calculations were performed at the HF/6-31G level with Gaussian 09. For Mpro-apo simulations, we used the recently reported structure (23) (PDB ID: 6M03). Approximately 20,000 TIP3P water model molecules and 4 Na+ ions were added. All simulations were carried out using the ff14SB force field (32). The simulation protocol included a first minimization of 7000 steps, involving 3500 steepest descent steps followed by 3500 steps of conjugate gradient energy minimization, where constraints were applied on the protein heavy atoms (force constant, 500 kcal/mol·Å2) and a second minimization (7000 steps) with no constraints of conjugate gradient energy minimization. Next, during the first equilibration, the temperature was gradually increased from 0 to 300 K over 50 ps using a Langevin thermostat with a temperature coupling constant of 1.0 ps in the canonical ensemble. Density equilibration and production runs were carried out using a constant pressure ensemble (NPT). All simulations were performed using periodic boundary conditions and a 2-fs time step. Long-range electrostatic interactions were calculated using the particle mesh Ewald method with a nonbonded cutoff of 10 Å, and the SHAKE algorithm was used to implement rigid constraints.
Ebselen global density map
To determine the most probable interaction sites between Mpro-ebselen (global hotspot), 15 replicas of 200 ns each were run (a total of 3 μs), where configurations were saved every 20 ps. The CPPTRAJ (33) software was used to process the trajectories, where at the first stage all trajectories were aligned by means of minimizing the distance among protein backbone atoms (C, N, and CA). Grid command was used to track the ebselen molecule and to produce a number density map, where the grid resolution was selected to be 0.5 Å.
Strain analysis
Initial Mpro-ebselen complex configurations were selected from previous global binding affinity study, with ebselen being bound to the catalytic site, to the intersection between domains II and III, and to both of these sites simultaneously. Three replicas of 200 ns for each of these initial setups were run. As a reference simulation, we used the Mpro-apo structure (PDB code: 6M03), and three replicas of 200 ns each were run. In this way, a total of 2400 ns of classical MD were run.
To apply the strain formalism from continuum theory to discrete, atomistic systems, differential operators replace the derivatives (19–21, 34, 35). A radius R around each central atom i containing n other atoms j defines the local neighborhood around the central atom. The instantaneous position of atom i at any timestep in the MD is xi, and the position of the same atom at any timestep of the reference simulation is x0, i. To first order, the distances between atom i and its neighbors j are related through the deformation matrix F by xj − xi = F(x0, j − x0, i), which forms an overdetermined system of linear equations, and an optimized F* is sought by minimizing the difference between the actual distances and the projected distances to an affine deformation: The atomic strain tensor is then found by , and the magnitude is defined as the L2 norm of the shear part of the strain tensor: , since proteins are generally incompressible (19, 21). For this analysis, a radius of 10 Å around each atom is considered (21), and only the Cα atoms are used in the calculations. The reference simulation is the apo protein trajectory, and the strain is then measured using the ebselen simulations to elucidate the effect of binding of ebselen at the different sites. In addition, β factors were estimated over the same trajectories using the atomicfluct command of the CPPTRAJ module of AMBER18.
AQUA-DUCT
To analyze the water structure and water flux in the Mpro protein of SARS-CoV-2, we used AQUA-DUCT 1.0.5 (24). We obtained inlets, MAV, and hotspots for water molecules in the Mpro catalytic region. For calculations, this region was defined as a 5 Å sphere around the center of geometry of the active site residues (H41, C145, H164, and D187) (17). The Mpro protein was studied in two different scenarios, with ebselen in the domains II and III site and the apo protein with no ligand. The time window used in both calculations was 50 ns, sampling every 1 ps. Images were created with open-source PyMOL (36).
Free energy
The absolute binding free energy is defined as follows: ΔGbinding = ΔGL − ΔGRL, where ΔGRL is the free energy change of ebselen annihilation in the Mpro complex and ΔGL is the free energy change of ebselen annihilation in water. To calculate these free energy changes, we use TI implemented in PMEMD for AMBER18. We use the one-step annihilation protocol with soft-core potentials (37). In addition, we adopted a simple approach with multiple runs starting from the most probably binding cluster estimated from previous MD simulations. In this way, three independent replicas for each site were taken into account, as well as three replicas for ebselen solvated in pure water. Eleven equally spaced windows were used (Δλ = 0.1) with 10 ns of simulation time per window. To keep the ligand from wandering in TI calculations, we used a soft restraint of 10 kcal/mol·Å2 (37).
Acknowledgments
We are grateful to the Research Computing Center of the University of Chicago for computational resources. Funding: The authors are grateful to Andrzej Joachimiak and Rick Stevens for helpful discussions. The study of protein-DNA complexes in epigenetics is supported by the NSF, under grant EFRI CEE 1830969, and the development of new DNA models is supported by BIO/MCB 1818328. The study of allostery and its role in triggerable materials is supported by MURI: W911NF-15-1-0568. The development of advanced sampling simulation algorithms and efficient software is supported by the Department of Energy, Office of Science, Basic Energy Sciences, Division of Materials Science and Engineering. The simulations reported here were carried out on the GPU cluster supported by the NSF through grant DMR-1828629. Author contributions: C.A.M. and J.J.d.P. conceived and started the project. C.A.M., F.B., G.R.P.-L., and W.A. ran simulations, calculated properties, and analyzed results. J.J.d.P. guided the project and discussed analysis. All authors contributed to writing. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W.; China Novel Coronavirus Investigating and Research Team , A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Q., Guan X., Wu P., Wang X., Zhou L., Tong Y., Ren R., Leung K. S. M., Lau E. H. Y., Wong J. Y., Xing X., Xiang N., Wu Y., Li C., Chen Q., Li D., Liu T., Zhao J., Liu M., Tu W., Chen C., Jin L., Yang R., Wang Q., Zhou S., Wang R., Liu H., Luo Y., Liu Y., Shao G., Li H., Tao Z., Yang Y., Deng Z., Liu B., Ma Z., Zhang Y., Shi G., Lam T. T. Y., Wu J. T., Gao G. F., Cowling B. J., Yang B., Leung G. M., Feng Z., Early transmission dynamics in wuhan, china, of novel coronavirus–infected pneumonia. N. Engl. J. Med. 382, 1199–1207 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., Chen H.-D., Chen J., Luo Y., Guo H., Jiang R.-D., Liu M.-Q., Chen Y., Shen X.-R., Wang X., Zheng X.-S., Zhao K., Chen Q.-J., Deng F., Liu L.-L., Yan B., Zhan F.-X., Wang Y.-Y., Xiao G.-F., Shi Z.-L., A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., Yuan M.-L., Zhang Y.-L., Dai F.-H., Liu Y., Wang Q.-M., Zheng J.-J., Xu L., Holmes E. C., Zhang Y.-Z., A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L. W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H., Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Zumla A., Chan J. F. W., Azhar E. I., Hui D. S. C., Yuen K.-Y., Coronaviruses — drug discovery and therapeutic options. Nat. Rev. Drug Discov. 15, 327–347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anand K., Palm G. J., Mesters J. R., Siddell S. G., Ziebuhr J., Hilgenfeld R., Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 21, 3213–3224 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G. F., Anand K., Bartlam M., Hilgenfeld R., Rao Z., The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U.S.A. 100, 13190–13195 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh N., Halliday A. C., Thomas J. M., Kuznetsova O. V., Baldwin R., Woon E. C. Y., Aley P. K., Antoniadou I., Sharp T., Vasudevan S. R., Churchill G. C., A safe lithium mimetic for bipolar disorder. Nat. Commun. 4, 1332 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lynch E., Kil J., Development of Ebselen, a glutathione peroxidase mimic, for the prevention and treatment of noise-induced hearing loss. Semin. Hear. 30, 47–55 (2009). [Google Scholar]
- 11.Kil J., Lobarinas E., Spankovich C., Griffiths S. K., Antonelli P. J., Lynch E. D., Le Prell C. G., Safety and efficacy of ebselen for the prevention of noise-induced hearing loss: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 390, 969–979 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Thangamani S., Younis W., Seleem M. N., Repurposing ebselen for treatment of multidrug-resistant staphylococcal infections. Sci. Rep. 5, 11596 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zou L., Lu J., Wang J., Ren X., Zhang L., Gao Y., Rottenberg M. E., Holmgren A., Synergistic antibacterial effect of silver and ebselen against multidrug-resistant Gram-negative bacterial infections. EMBO Mol. Med. 9, 1165–1178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.M. Renson, E. Etschenberg, J. Winkelmann, 2-Phenyl-1,2-benzisoselenazol-3(2H)-one containing pharmaceutical preparations and process for the treatment of rheumatic diseases (1982); https://patents.google.com/patent/US4352799A/en.
- 15.Masaki C., Sharpley A. L., Cooper C. M., Godlewska B. R., Singh N., Vasudevan S. R., Harmer C. J., Churchill G. C., Sharp T., Rogers R. D., Cowen P. J., Effects of the potential lithium-mimetic, ebselen, on impulsivity and emotional processing. Psychopharmacology (Berl) 233, 2655–2661 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li C., Qi Y., Teng X., Yang Z., Wei P., Zhang C., Tan L., Zhou L., Liu Y., Lai L., Maturation mechanism of severe acute respiratory syndrome (SARS) coronavirus 3C-like proteinase. J. Biol. Chem. 285, 28134–28140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bzówka M., Mitusińska K., Raczyńska A., Samol A., Tuszyński J. A., Góra A., Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is an inconvenient target for small-molecule inhibitors design. bioRxiv 2020.02.27.968008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaus J. W., Pierce L. T., Walker R. C., McCammon J. A., Improving the efficiency of free energy calculations in the amber molecular dynamics package. J. Chem. Theory Comput. 9, 4131–4139 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell M. R., Tlusty T., Leibler S., Strain analysis of protein structures and low dimensionality of mechanical allosteric couplings. Proc. Natl. Acad. Sci. U.S.A. 113, E5847–E5855 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gullett P. M., Horstemeyer M. F., Baskes M. I., Fang H., A deformation gradient tensor and strain tensors for atomistic simulations. Model. Simul. Mater. Sci. Eng. 16, 015001 (2007). [Google Scholar]
- 21.Eckmann J.-P., Rougemont J., Tlusty T., Colloquium: Proteins: The physics of amorphous evolving matter. Rev. Mod. Phys. 91, 031001 (2019). [Google Scholar]
- 22.Papakonstantopoulos G. J., Doxastakis M., Nealey P. F., Barrat J.-L., de Pablo J. J., Calculation of local mechanical properties of filled polymers. Phys. Rev. E. 75, 031803 (2007). [DOI] [PubMed] [Google Scholar]
- 23.R. P. D. Bank, RCSB PDB - 6M03: The crystal structure of COVID-19 main protease in apo form; www.rcsb.org/structure/6M03.
- 24.Magdziarz T., Mitusińska K., Bzówka M., Raczyńska A., Stańczak A., Banas M., Bagrowska W., Góra A., AQUA-DUCT 1.0: Structural and functional analysis of macromolecules from an intramolecular voids perspective. Bioinformatics 36, 2599–2601 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anand K., Ziebuhr J., Wadhwani P., Mesters J. R., Hilgenfeld R., Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 300, 1763–1767 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Knight J. D. R., Hamelberg D., McCammon J. A., Kothary R., The role of conserved water molecules in the catalytic domain of protein kinases. Proteins 76, 527–535 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kandeel M., Al-Nazawi M., Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 251, 117627 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu C., Zhou Q., Li Y., Garner L. V., Watkins S. P., Carter L. J., Smoot J., Gregg A. C., Daniels A. D., Jervey S., Albaiu D., Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 6, 315–331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, Y. Huang, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York, P. A. Kollman, AMBER 2018 (University of California, San Francisco, 2018).
- 30.Wang J., Wang W., Kollman P. A., Case D. A., Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 25, 247–260 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A., Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., Simmerling C., ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roe D. R., Cheatham T. E., PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Dube D., Ahalawat N., Khandelia H., Mondal J., Sengupta S., On identifying collective displacements in apo-proteins that reveal eventual binding pathways. PLOS Comput. Biol. 15, e1006665 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Falk M. L., Langer J. S., Dynamics of viscoplastic deformation in amorphous solids. Phys. Rev. E. 57, 7192–7205 (1998). [Google Scholar]
- 36.DeLano W. L., Pymol: An open-source molecular graphics tool. CCP4 Newsletter on protein crystallography. 40, 82–92 (2002). [Google Scholar]
- 37.Steinbrecher T., Joung I., Case D. A., Soft-core potentials in thermodynamic integration: Comparing one- and two-step transformations. J. Comput. Chem. 32, 3253–3263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]