

Early-life intestinal epithelial development is controlled by the epigenetic modifier LSD1.
Abstract
Intestinal epithelial homeostasis is maintained by adult intestinal stem cells, which, alongside Paneth cells, appear after birth in the neonatal period. We aimed to identify regulators of neonatal intestinal epithelial development by testing a small library of epigenetic modifier inhibitors in Paneth cell–skewed organoid cultures. We found that lysine-specific demethylase 1A (Kdm1a/Lsd1) is absolutely required for Paneth cell differentiation. Lsd1-deficient crypts, devoid of Paneth cells, are still able to form organoids without a requirement of exogenous or endogenous Wnt. Mechanistically, we find that LSD1 enzymatically represses genes that are normally expressed only in fetal and neonatal epithelium. This gene profile is similar to what is seen in repairing epithelium, and we find that Lsd1-deficient epithelium has superior regenerative capacities after irradiation injury. In summary, we found an important regulator of neonatal intestinal development and identified a druggable target to reprogram intestinal epithelium toward a reparative state.
INTRODUCTION
The intestinal epithelium undergoes a marked change during the neonatal period. Crypt formation occurs after birth together with the appearance of Paneth cells (PCs) and the development of adult intestinal stem cells (ISCs). Adult ISCs rely on niche factors such as Wnt ligands. In vivo, mesenchymal cells are important sources of Wnt to support ISC maintenance (1), whereas in vitro, it is PCs that are required to supply the necessary Wnt (2–5). In contrast, Wnt ligands or PCs are dispensable for fetal organoid homeostasis (6, 7). Thus, ISCs undergo a fetal-to-adult transition that includes a change in Wnt dependency.
Recently, the existence of bona fide fetal ISCs has been challenged by the finding that any fetal epithelial cell can be or become an adult ISC as long as the appropriate environment is supplied (8). This model fits nicely with studies showing that after injury, the intestinal epithelium is temporarily reprogrammed into a fetal-like state that is needed for proper repair (9–11). This, in turn, complements work specifying that adult intestinal epithelial lineages can dedifferentiate to give rise to new ISCs to rebuild the epithelium after injury (12, 13). In hindsight, these high levels of cell fate reversion make sense because the intestine is a common site for chemical and mechanical challenges as well as the host for many putative pathogens. Nonetheless, it is not yet fully understood how the fetal-to-adult ISC transition, or its reversal upon injury, is mediated, and whether epithelial reprogramming can be targeted therapeutically.
Adult ISCs give rise to all intestinal epithelial subtypes, and ISCs have a strikingly similar chromatin landscape compared to enterocytes (12, 14). In contrast, the epigenetic makeup of secretory progenitors is distinguishable from ISCs and enterocytes (12, 15), which was reversed upon irradiation when these secretory precursors dedifferentiate into ISCs (12). Three groups separately identified a crucial role for the polycomb repressive complex 2 (PRC2) in maintaining crypt physiology (16–18). In summary, although it is clear that there is an important role for certain epigenetic modifiers in intestinal epithelial biology, the role and importance of others remain undefined. Here, we exploit the availability of chemical probes targeting epigenetic modifiers (19), and combine it with intestinal organoid cultures (20), to identify the demethylase LSD1 (lysine-specific demethylase 1) as a critical regulator of postnatal epithelial maturation including PC differentiation.
RESULTS
Identification of LSD1 as a regulator of PCs
The intestinal epithelium undergoes a marked change during the neonatal period including the appearance of PCs. To study PC differentiation in organoids, we developed a differentiation protocol [adapted from (21)] using CHIR [glycogen synthase kinase 3 (GSK3) inhibitor] and DAPT (γ-secretase inhibitor) to activate Wnt and block Notch signaling, respectively (fig. S1A). CHIR-DAPT (CD) treatment led to a robust enrichment of lysozyme+ PCs by confocal microscopy, mRNA, and protein expression compared to standard EGF (epidermal growth factor), Noggin, and R-spondin 1 (ENR) organoid growing conditions (Fig. 1A and fig. S1B). Next, we tested chemical probes targeting 18 methyltransferases and demethylases and identified the LSD1 inhibitor GSK-LSD1 to consistently repress PC differentiation (Fig. 1, B and C; fig. S1, C and D; and table S1). In support, GSK-LSD1 similarly affected PC differentiation in organoids grown in ENR conditions (Fig. 1D and fig. S1E), and use of a different LSD1 inhibitor (ORY-1001) led to a similar near loss of PCs (fig. S1F). Consistent with the irreversible binding nature of GSK-LSD1 to its target (22), we found that upon withdrawal of GSK-LSD1, PCs reappeared after two organoid splitting events (Fig. 1E and fig. S1G).
Fig. 1. Inhibition of LSD1 blocks PC differentiation and allows niche-independent expansion of ISCs.
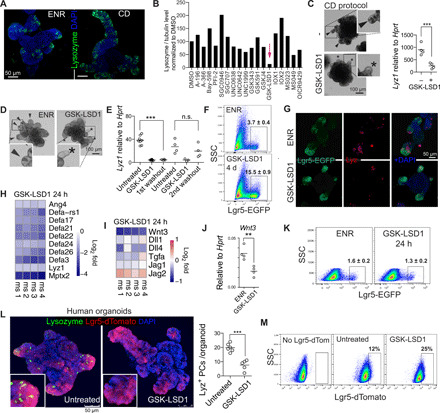
(A) Confocal images of lysozyme and DAPI staining of ENR-grown and CHIR-DAPT (CD)–grown (CD protocol as in fig. S1A) organoids. (B) Quantification of lysozyme/tubulin levels by Western blot of organoids treated with indicated inhibitors in CD conditions. Concentrations used can be found in table S1. Data are mean from two independent experiments (see fig. S1, C and D, for raw data). (C) Bright-field images and inserts of CD-grown organoids with and without GSK-LSD1 (1 μM). Lyz1 expression relative to Hprt by qPCR. (C and D) Arrows indicate PC+ crypts based on the presence of granular cells, and asterisks indicate PC− crypts. (D) Bright-field images and inserts of ENR-grown organoids with or without GSK-LSD1 (1 μM). (E) Lyz1 expression relative to Hprt by qPCR. n.s., not significant. (F and K) Flow cytometry of Lgr5-EGFP organoids. Representative plot; n = 3 different mouse lines; mean ± SEM is noted. (G) Images of Lgr5-EGFP (anti-GFP) and lysozyme (LYZ) staining of organoids. (H and I) Heatmaps of indicated genes from RNA-seq of 24-hour GSK-LSD1–treated organoids. N = 4 different mouse organoid lines, and expression is relative to each individual control. (J) Wnt3 expression relative to Hprt by qPCR. (L) Images of human LGR5-dTomato organoids, additionally stained for lysozyme. Quantified number of PCs per human organoid from two different experiments (n = 1 patient). (M) Flow cytometry of LGR5-dTomato–treated human organoids with 1 μM (n = 1 patient). **P < 0.01 and ***P < 0.001.
Inhibition of LSD1 renders Lgr5+ cells independent of niche-providing PCs in vitro
PCs are crucial for adult small intestinal organoids as they supply niche factors to retain a stem cell population, and normally, PC-devoid organoids only sustain growth upon Wnt supplementation (2–5). To test the role of LSD1 in ISCs, we used Lgr5–enhanced green fluorescent protein (EGFP)–derived organoids and treated them with GSK-LSD1 in normal ENR medium. We found that GSK-LSD1 treatment resulted in a three- to fivefold increase in percentage of LGR5+ cells (Fig. 1, F and G). We define LGR5+ cells by GFPHIGH, as it was previously shown that these cells, but not GFPLOW cells, have clonal capacity (20). To test whether only a selection or all PC genes are down-regulated upon LSD1 inhibition, we performed RNA sequencing (RNA-seq) on organoids treated with GSK-LSD1 for 24 hours, between day 1 and 2 after splitting, which we anticipate is right when PCs develop. We found a robust down-regulation of all PC-specific genes [gene set from (23)] (Fig. 1H). Thus, GSK-LSD1 treatment expands the LGR5+ population and renders these cells independent of PC-derived Wnt3 (Fig. 1, I and J). In contrast to Wnt3 expression, we did not observe a lack of other niche factors including Notch ligands such as Dll1 (Fig. 1I). Notch ligands are not uniquely expressed by PCs and also can be found in enteroendocrine cells (24). We further found that GSK-LSD1 treatment led to down-regulation of genes associated with goblet cells (GCs) but not enteroendocrine cells (fig. S1H). In addition, 24-hour GSK-LSD1 treatment did not lead to an expansion of the LGR5+ population (Fig. 1K). Thus, the expansion of LGR5+ cells upon LSD1 inhibition does not precede or outcompete PC loss and may be a separate event. Nevertheless, using a recently described culture condition that allows PC differentiation in human organoids (25), we found that GSK-LSD1 also blocks PC differentiation in human organoids while simultaneously expanding the LGR5+ population (Fig. 1, L and M).
LSD1 is required for PCs but not goblet or enteroendocrine cells in vivo
To test the role of LSD1 in vivo, we crossed Lsd1f/f (26) with Villin-Cre (27) mice to delete Lsd1 in intestinal epithelial cells specifically [knockout (KO) mice]. Although these mice appear normal, we found that KO mice lack PCs throughout the small intestine (Fig. 2A and fig. S1I). We did observe that KO mice had “escaper” crypts still expressing LSD1, and LSD1+ crypts were positive for lysozyme (fig. S1J). Thus, these mice do not completely lack PCs. Now, two genes are known to be absolutely required for PC differentiation in vivo: Sox9 and Atoh1 (28, 29). We did not observe differences in SOX9 expression (Fig. 2A), and although we found fewer Atoh1+ cells in KO epithelium (Fig. 2A), reduction of Atoh1+ cells unlikely causes a complete lack of PCs. We reasoned that perhaps KO crypts are filled with PC precursors expressing Wnt3; however, similar to GSK-LSD1–treated organoids, Wnt3 was markedly reduced in KO mice crypts (Fig. 2A). Next, we examined other intestinal secretory lineages and found a reduction of GCs, but equal numbers of enteroendocrine cells, comparing adult wild-type (WT) and KO littermates (Fig. 2B). When we examined fetal and postnatal intestines of WT and KO littermates, we found that the reduction in GCs emerges after developmental stage P7, similar to when PCs start to develop (Fig. 2, C and D, and fig. S1K). These results suggest that LSD1 KO epithelium maintains neonatal characteristics into adulthood, including the absence of PCs and fewer GCs.
Fig. 2. LSD1 is required for crypt maturation in vivo and Wnt dependency of organoids.
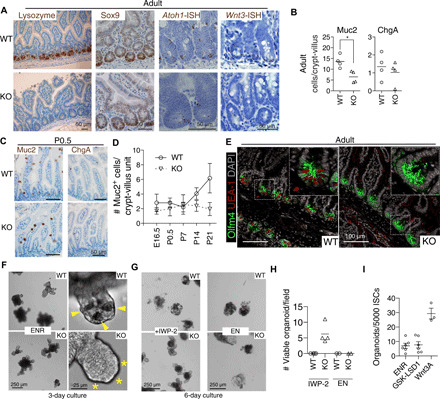
(A) Representative images of antibody (lysozyme and SOX9) and in situ hybridization (ISH) (Atoh1 and Wnt3) staining of WT and KO small intestinal tissue. (B) Quantification of MUC2+ goblet and CHGA+ enteroendocrine cells in adult duodenum intestine. N = 4 mice; *P < 0.05. (C) Representative images of MUC2 and CHGA antibody staining at P0.5. (D) Quantifications of MUC2 (mouse protein) GCs throughout development. N ≥ 3 mice; mean ± SEM is shown. (E) Representative image of OLFM4 (mouse protein) antibody and UEA-1 staining of adult WT and KO tissue. (F to H) Bright-field images of WT and KO organoids with indicated treatments; wells from two different experiments (n = 2 mice) are quantified. (I) Organoid outgrowth from single sorted Lgr5HIGH ISCs; each dot represents a mouse; data pooled from three independent experiments. Mean and SEM are shown.
Mice lacking LSD1 sustain crypt-bottom ISCs independent of PCs, and KO organoids grow independent of endogenous Wnt
To test the role of LSD1 in ISCs in vivo, we used in situ hybridization (ISH) and antibody-based detection of the ISC marker Olfm4 in tissues of WT and KO mice. We found OLFM4+ cells completely filling the bottom of crypts in KO mice compared to the standard PC/ISC pattern observed in WT crypts (Fig. 2E and fig. S1L). In addition, all crypt-base cells in KO mice are Ki67+, suggesting that these OLFM4+ cells are proliferating (fig. S1L). Atoh1−/− mice lack PCs, and Atoh1−/− crypts do not sustain organoid growth without Wnt supplementation (3). In contrast, Lsd1 WT and KO crypts were equally able to form organoids, even in the absence of PCs in KO organoids (Fig. 2F). This led us to hypothesize that KO organoids do not rely on endogenous Wnt. Blockage of Wnt signaling by the porcupine inhibitor IWP-2 showed that treated KO organoids sustained growth unlike WT organoids (Fig. 2, G and H). IWP-2 distinctively reduced growth rate in KO organoids, which makes long-term expansion unfeasible; yet, after splitting, there were still surviving KO organoids under continuous IWP-2 treatment, and LSD1 inhibitor treatment greatly increased splitting efficiency (fig. S1, M and N). In contrast, both KO and WT organoids could not sustain growth in medium lacking R-spondin 1 (Fig. 2, G and H). Thus, loss of LSD1 activity in ISCs renders them not requiring (endogenous) Wnt for growth and even leads to expansion of this population (Fig. 1). However, GSK-LSD1 is not able to replace Wnt3A in the ability to form organoids from single ISCs (Fig. 1I). Although KO organoids resemble aspects of those derived from fetal epithelium that also lack PCs (6, 7), there are some key distinctions. Unlike fetal organoids, KO organoids cannot grow without R-spondin 1. In addition, KO organoids form crypts, whereas fetal or Wnt-supplemented organoids mainly grow as spheroids. It is possible that enteroendocrine progenitors that express Notch ligands such as Dll1/4 are sufficient to allow crypt formation in KO organoids.
Our data thus suggest that adult KO epithelium is in between fetal and adult: KO organoids do not rely on Wnt yet are unable to grow without R-spondin 1, and KO epithelium in vivo lacks PCs and has reduced GC numbers yet with crypts that have OLFM4+ ISCs.
LSD1 represses fetal and neonatal genes that allows PC differentiation independent of YAP/TAZ
Next, we sought to find the mechanism by which LSD1 controls intestinal epithelial biology. We performed RNA-seq on fluorescence-activated cell sorting (FACS)–sorted EPCAM+ small intestinal crypt cells from WT and KO mice. We found 1658 up-regulated and 527 down-regulated genes (Padj < 0.01) in KO cells compared to WT cells (Fig. 3A). In support of our findings that there are Atoh1+ cells in KO crypts and the differential ability of Atoh1-KO and Lsd1-KO crypts to form organoids, we found no shift toward an Atoh1−/− gene signature in the KO transcriptional profile (fig. S2A) (30). To verify our gene expression profile, we tested a PC-specific gene signature (23) and, expectedly, found that this is repressed in KO crypts (Fig. 3B).
Fig. 3. Deletion of LSD1 renders intestinal epithelium fetal-like.
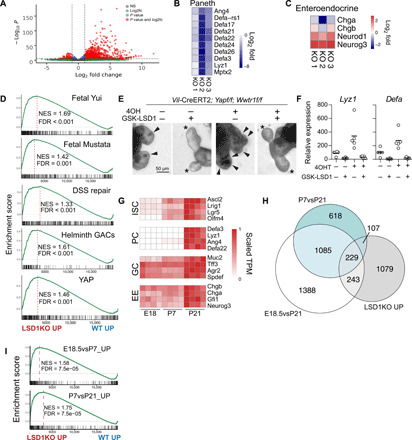
(A) Volcano plot of RNA-seq data comparing WT and KO crypt cells (n = 3). (B) Heatmap of PC-specific genes. (C) Heatmaps of enteroendocrine-associated genes. (D and I) LSD1KO versus WT RNA-seq data were analyzed by gene set enrichment analysis (GSEA) for indicated gene signatures. Normalized enrichment score (NES) and false discovery rates (FDRs) are indicated. (E) Bright-field images of organoids with indicated treatments. Arrows indicate granular PCs, and asterisks indicate crypts devoid of PCs. (F) Expression of Lyz1 and Defa by qPCR relative to Actb; each dot represents a well; data pooled from four independent experiments. (G) Heatmap of genes associated with ISCs, PCs, GCs, and enteroendocrine cells (EE) during development. Expression relative to the highest TPM for that gene, which is set to 1, is shown. (H) Venn diagram of genes significantly higher expressed in LSD1KO compared with genes that are up in P7vsP21 and E18vsP21.
A study reported that an LSD1-GFI1 complex was rapidly disturbed by LSD1 inhibitors and that the scaffolding role of LSD1 rather than its enzyme activity was most crucial (31). Gfi1−/− mice have a near loss of PCs and ISCs, reduced GCs, but more enteroendocrine cells (2, 32). When we examined genes known to be repressed by GFI1 in intestinal epithelium, we found rapid and robust increase of Neurog3 and Neurod1 but not that of mature enteroendocrine cell markers Chga and Chgb both by comparing KO with WT crypts and organoids treated with GSK-LSD1 for 24 hours (Fig. 3C and fig. S1H). This is in accordance with equal enteroendocrine cells in vivo in WT and KO mice (Fig. 2B and fig. S1K). Together, this suggests that inhibition of LSD1 rapidly leads to enzyme-independent derepression of GFI1-targeted enteroendocrine-progenitor markers that, in turn, can limit PC differentiation (32). Furthermore, these enteroendocrine progenitors may take over the role of PCs in Notch-dependent crypt formation (24). However, it does not explain the expansion of ISCs, the complete loss of PCs, or the lack of expansion of mature ChgA+ cells that we observe in KO epithelium, which thus contradict a primarily GFI1-mediated mechanism.
Over the years, there have been various different ISC and stem cell–like populations described, either using genetic markers such as Lgr5 or Bmi1 or by techniques such as label-retaining capacity and single-cell RNA sequencing (scRNA-seq) (13, 33–36). To test whether there was enrichment for a certain stem cell type, we analyzed expression of defining genes for each population (fig. S2, B and C); however, we did not find enrichment for any stem cell subtype including the classical Lgr5+ ISC signature. In addition, we found very limited overlap of an ISC-unique gene set with those up-regulated in KO crypts (fig. 2SD). Instead, we find enrichment of two fetal gene sets in the KO transcriptional profile (Fig. 3D) (7, 9). These fetal genes are normally not expressed in ISCs or any other adult intestinal epithelial cell types and instead are aberrantly expressed in adult KO crypts. We conclude that up-regulation of fetal genes in our bulk RNA-seq is thus not due to the enrichment of ISCs in KO crypts. Recently, two groups elegantly identified and characterized a cellular repair state in the intestinal epithelium that is fetal-like (9, 10). Two repair gene signatures from these studies were also enriched in the KO transcriptional profile (Fig. 3D). Yui et al. (9) revealed that the reparative state was mediated by YAP/TAZ. A separate YAP gene signature was enriched in our KO transcriptional profile (Fig. 3D) (11). To test whether YAP/TAZ is required for LSD1-mediated PC differentiation, we deleted YAP/TAZ in an inducible manner (Vil-CreERT2;Yapf/f;Wwtr1f/f organoids). Tamoxifen treatment led to near undetectable levels of both Yap and Wwtr1 (fig. S2E), impaired survival, and led to an increase in PCs (Fig. 3, E and F), in accordance with previous results (11, 37). GSK-LSD1 treatment completely abrogated PC differentiation independently of YAP/TAZ (Fig. 3, E and F). We also treated organoids derived from mice lacking both Yap and the gene encoding for TAZ Wwtr1 (Vil-Cre;Yapf/f;Wwtr1f/f) with GSK-LSD1 and found that PC differentiation was equally impaired in WT and mutant organoids (fig. S2F). However, we noted that approximately half of these organoids contained Yap and Wwtr1 based on quantitative polymerase chain reaction (qPCR) (fig. S2G).
To distinguish fetal from postnatal intestinal epithelial development, we performed RNA-seq on embryonic (E) day 18.5, as well as postnatal (P) day 7 and P21 intestinal epithelium (fig. S2, H and I). Figure 3G shows how established cell lineage markers behave during development. As expected, we observed a stepwise increase in ISC markers, an abrupt appearance of PC genes at P21, and goblet and enteroendocrine gene expression at E18.5 and P7 stages that only increases slightly at P21 (Fig. 3G). This supports our hypothesis that KO mice have intestinal epithelium that is “stuck” at a P7 stage, lacking PCs, with reduced GC and immature enteroendocrine cells, but with crypts containing ISC-like cells. When we compared genes up-regulated in KO crypts to different developmental stages, we see overlap with both E18.5 and P7 stages compared to P21 (Fig. 3H). In a separate test, we found enrichment by gene set enrichment analysis (GSEA) for our own fetal (E18vsP7) and neonatal (P7vsP21) gene sets (Fig. 3I).
LSD1 controls H3K4me1 levels of genes associated with a fetal-like profile
LSD1 controls embryonic development by repressing enhancers to allow embryonic stem cell differentiation (38). We did not observe major differences in global H3K4me1/2 levels by immunohistochemistry comparing escaper LSD1+ crypts in KO tissue (fig. S3A). To assess whether LSD1 mediates H3K4 demethylation, we performed chromatin immunoprecipitation sequencing (ChIP-seq) for H3K4me1 and H3K4me2 comparing sorted WT and KO crypt cells. Analysis of ChIP-seq for H3K4me1 identified 2059 sites with associated altered methylation levels (Padj < 0.01), of which the majority (1622) were enriched in KO crypts (Fig. 4A). ChIP-seq of H3K4me2 revealed a very similar pattern, and the large majority of genes affected by LSD1 with gain of H3K4me2 were also significant in the analysis for H3K4me1 (Fig. 3, A and B). A previous study found that although ATAC-seq (assay for transposase-accessible chromatin using sequencing) allowed a separation of enterocyte progenitors and secretory progenitors in their chromatin state, H3K4me2 levels at these sites were indistinguishable (12). This suggests that the differences that we observed in our H3K4me1/2 ChIP-seq analysis are not due to differences in adult cell populations between WT and KO crypts. Nevertheless, most of these peaks are not in close proximity to transcriptional start sites (TSSs) (fig. S3B). The top 300 genes associated with increased H3K4me1 levels in the KO are overall enriched in the KO transcriptional profile (Fig. 4C). Furthermore, using a high stringency (Padj < 0.01), we combined our RNA-seq and H3K4me1 ChIP-seq data to establish a core list of 228 genes that are mediated by the enzymatic activity of LSD1 (table S2). Eighty-four percent of the increased H3K4me1 peaks associated with these 228 genes are located outside the 2-kb surrounding the TSS. This suggests that LSD1 would drive enhancer-mediated regulation of these genes, which fits with a role generally associated with LSD1 (38). The LSD1 core signature is enriched in a transcriptional profile comparing fetal with adult organoids (Fig. 4D) (9). Thus, we propose that LSD1 enzymatically represses genes that are required for maturation of intestinal epithelium.
Fig. 4. LSD1 controls H3K4me1/2 levels of fetal-like genes.
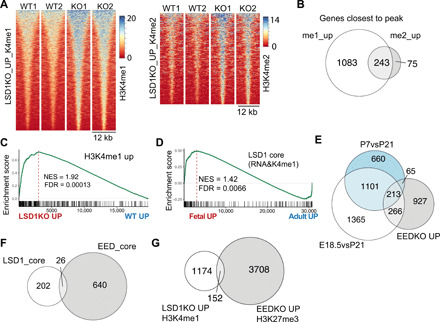
(A) Heatmaps of H3K4me1 and H3K4me2 sites that are significantly up in KO crypts compared to WT crypts. (B) Venn diagram comparing genes associated with H3K4me1 and H3K4me2 peaks that were significantly higher in KO crypts compared to WT crypts. (C) RNA-seq analysis by GSEA of KO versus WT transcriptional profile on a gene set consisting of genes with associated increased H3K4me1 levels in KO crypts. (D) GSEA of LSD1 core gene list (228 genes) on a transcriptome dataset that compares fetal to adult organoids (9). (E) Venn diagram of genes significantly higher expressed in EEDKO compared with genes that are up in P7vsP21 and E18vsP21. (F and G) Venn diagrams comparing the LSD1 core and the EED core (genes up in EED KO crypts AND have H3K27me3 associated peak) as well as the genes associated with increased methylation levels in each separate group.
LSD1 controls genes separately from PRC2
Several groups have shown that embryonic ectoderm development (EED), an essential component of the PRC2, is essential for maintaining adult ISCs and crypts, likely by repressing fetal and embryonic genes (16–18). EED-controlled genes show a remarkable similar overlap with fetal and neonatal genes as LSD1-controlled genes (Fig. 4, E and H). However, when EED KO epithelium returns to a fetal and even embryonic state and mice become moribund (16), LSD1 KO mice retain an early-life postnatal stage and appear normal up to at least 1 year. Fittingly, comparing the LSD1 core with the EED core [genes up in EEDKO crypts and associated with H3K27me3 peaks (17)] revealed little overlap between regulated genes (Fig. 4F). Further analysis also confirmed that most of the putative LSD1-controlled H3K4me1 genes are not “co-repressed” by the PRC2-mediated H3K27me3 (Fig. 4G). Thus, this suggests that both LSD1 and PRC2 control fetal-like genes but in an unrelated fashion.
Developmental H3K4me1 profiles suggest no determining role for LSD1 in demethylation of fetal- or postnatal-specific sites
We found LSD1 to robustly mediate fetal and postnatal genes (Fig. 3), and LSD1 putatively does so by regulating H3K4me1/2 levels at or near those genes (Fig. 4). We next performed H3K4me1 ChIP-seq on FACS-sorted epithelium at different developmental stages. As an example, we find a stepwise increase of H3K4me1 levels near the ISC marker gene Olfm4, whereas another ISC marker, Lgr5, has mostly unchanged H3K4me1 levels (Fig. 5A). When we systemically assess differential sites, we find that there is an abrupt loss of H3K4me1 at P21 from sites that are significantly up in E18.5 and P7 (Fig. 5B). In contrast, ChIP levels that are low in E18.5 or P7 display a stepwise increase that culminates at P21 (Fig. 5B). There are fewer peak changes from E18.5 versus P7, compared to P7 versus P21 or E18.5 versus P21 (3700, 15,522, and 26,942 significant sites, respectively, Padj < 0.01) (Fig. 5B and fig. S3C). This suggests that the major H3K4me1 transition occurs after P7, which would fit very well with our hypothesis that LSD1 controls this transition specifically. However, when we take these sites and display the H3K4me1 ChIP-seq data of WT versus KO crypts, we find a complete lack of regulation of sites lost at P21 (Fig. 5B). This suggests that LSD1 is not involved at all in the observed reduction of H3K4me1 sites at P21. In contrast, we observe a modest increase of H3K4me1 levels in KO crypts of the sites that normally culminate at P21 (Fig. 5B and fig. S3C). In support, if we do the reverse, by taking the significant KO sites from Fig. 4A, and display the different developmental stages, we see a stepwise increase of those peaks in time (Fig. 5C). This unexpected finding suggests that LSD1 broadly modulates H3K4me1 levels of peaks that appear during development, but that LSD1 is not involved in H3K4me1 sites that are lost during the different developmental stages in intestinal epithelium.
Fig. 5. LSD1 does not control fetal- and neonatal-specific H3K4me1 levels.
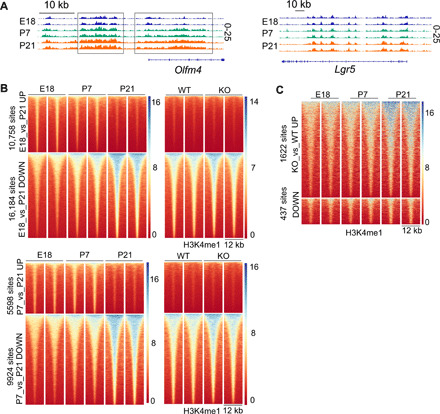
(A) H3K4me1 profiles of Olfm4 and Lgr5 loci at indicated developmental stages. (B) Heatmaps of H3K4me1 sites that were identified to be significantly different between E18 and P21 (top) and P7 versus P21 (bottom). In addition, sites identified between developmental stages were selected to make a heatmap of adult (8 to 12 weeks old) WT and KO H3K4me1 profiles. (C) Significant H3K4me1 sites between KO and WT crypts were selected, and E18, P7, and P21 H3K4me1 data are displayed.
LSD1 expression is down-regulated during repair
We conclude that LSD1 represses fetal/neonatal genes in adult epithelium. A similar gene set is reactivated after damage during the repair phase. Thus, we hypothesized that during repair, derepression of LSD1-controlled fetal/neonatal genes would require LSD1 inactivation. Therefore, we studied LSD1 expression after whole-body irradiation (10 Gy) and found that LSD1 is lower expressed in large crypts that appear 3 days after irradiation, compared to normal-sized crypts of naïve mice (Fig. 6A). This is opposite of the Hippo-transducer YAP expression pattern (fig. S4A). In support, we assessed Lsd1 expression in a recently described scRNA-seq experiment comparing crypt cells during homeostasis and during active repair (35) and found a clear reduction in number of crypt cells expressing Lsd1 (Fig. 6B). In summary, Lsd1 is actively down-regulated in the repair phase of intestinal epithelium.
Fig. 6. LSD1 levels are actively reduced during repair, and Lsd1-deficient epithelium repairs better than WT tissue.
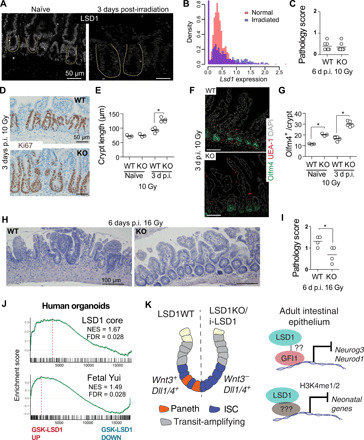
(A) Images of LSD1 antibody staining of naïve and irradiated (10 Gy) intestines. (B) Lsd1 expression from scRNA-seq data from (35) in normal and irradiated crypts. (C) Pathology score of indicated intestines 6 days post-irradiation (p.i.) with 10 Gy. (D) Images of KI67 antibody staining of WT and KO tissue 3 days p.i. (E) Crypt length as quantified from images shown in (D) and fig. S1L; crypt length is determined by KI67+ cells. (F) Images of OLFM4, UEA-1, and DAPI-stained WT and KO intestines 3 days p.i. at 10 Gy. (G) Quantifications of OLFM4+ cells per crypt quantified from images such as shown in (F) and Fig. 2E. (H) Hematoxylin and eosin staining of WT and KO intestinal tissue 6 days p.i. with 16 Gy. (I) Pathology score of indicated intestines 6 days p.i. with 16 Gy. (J) GSEA of indicated signatures on transcriptome data generated from control and GSK-LSD1–treated human organoids. *P < 0.05. (K) Graphical overview of descriptive and mechanistic work from this study.
Lsd1-deficient epithelium has superior reparative capacity
The reparative-like features of KO epithelium prompted us to test whether this would be beneficial upon injury. We irradiated WT and KO mice with 10 Gy, and we did not observe pathological differences 6 days after treatment (Fig. 6C and fig. S4B). However, we did find an increase in crypt length 3 days after injury, as measured by KI67 (mouse protein) cells (Fig. 6, D and E). We found no evidence of appearing PCs after injury in KO tissue (Fig. 6F), but we did observe that the OLFM4+ ISC zone similarly expanded as the KI67+ crypts (Fig. 6, F and G). This suggests that KO epithelium may have better reparative capacities, but with this level of irradiation, WT epithelium is also able to recover after 6 days. Thus, we next irradiated mice with 16 Gy, when WT mice are unable to recover, and in marked contrast, KO epithelium regained crypt-villus structures and had lower pathology scores compared to WT epithelium (Fig. 6, H and I, and fig. S4, C and D). Thus, KO epithelial tissue, which has a preexisting repairing profile, enhances repair in vivo after radiation injury. To test the translational potential of our findings, we performed a gene expression array of GSK-LSD1–treated human organoids and found that the LSD1 core and a fetal signature were enriched in GSK-LSD1–treated human organoids (Fig. 6J).
In conclusion, we have found that LSD1 is required for PC differentiation and that Lsd1-deficient ISCs do not rely on PC-derived Wnt3 to retain stemness (Fig. 6K). Mechanistically, we find that LSD1 normally represses Neurod1 and Neurog3, possibly in concert with GFI1 and independent of its demethylase activity, but that LSD1 represses “neonatal” genes by mediating H3K4me1/2 levels at putative enhancer sites (Fig. 6K).
DISCUSSION
Here, we find that LSD1 is required for postnatal intestinal epithelial maturation. The role we find for LSD1 in suppressing fetal/neonatal genes is near opposite of what recently was found in skin (39). In skin epithelium, inhibition of LSD1 leads to the increase of fate-determining transcription factors and subsequent differentiation, and this enhanced differentiation led to inhibition of skin cancer progression (39). Similarly, in other cancer types including intestinal tumors in zebrafish, LSD1 inhibition leads to differentiation and reduced tumor development (31, 40). In the intestine, this would fit with our finding of the rapid increase in enteroendocrine progenitor genes Neurog3 and Neurod1 likely in a GFI1-mediated manner (Fig. 3C and fig. S1H). However, unlike in GFI1KO mice, LSD1KO epithelium does not have a robust expansion of mature enteroendocrine cells, nor does it have a near loss of ISCs (2). We propose that the H3K4me1-mediated control of fetal/postnatal genes overrules the ability of enteroendocrine progenitors to mature, and it allows the expansion of ISC-like cells at the bottom of crypts and in organoids.
Intestinal epithelial repair is crucial to prevent chronic disease, and YAP/TAZ are established initiators and critical regulators of this process (9, 11). YAP/TAZ are sensors of mechanical signals, so it is plausible that upon damage and subsequent extracellular matrix cues, YAP/TAZ are activated by control of their protein levels. We found a clear reduction of LSD1 levels after irradiation, which coincides with an increase in YAP levels. Although it is unclear how LSD1 levels are controlled after regeneration, in glioblastoma tumor cells GSK3β stabilizes LSD1 by direct phosphorylation (41). Thus, activation of the Wnt pathway, which occurs during the regenerating phase, could lead to fast reduction of LSD1 levels by Wnt-controlled inhibition of GSK3. We further found that LSD1 represses a reparative gene expression profile, similar to that activated by YAP. However, GSK-LSD1–mediated depletion of PCs was completely independent of YAP/TAZ. In combination, after damage, reducing levels of the repressor LSD1 and inducing levels of activators YAP/TAZ can together mediate a robust reparative response.
In summary, we provide evidence that inhibition of LSD1 may be a viable target for the reprogramming of intestinal epithelium into a reparative state that is beneficial after injury, such as inflicted by radiation therapy.
MATERIALS AND METHODS
Animal work
Lgr5-EGFP-IRES-CreERT2 (stock no. 008875) mouse strain was obtained from The Jackson Laboratory. Villin-Cre and Villin-CreERT2 (27) were a gift from S. Robine, Lsd1f/f mice were a gift from S. Orkin (26). Mice were housed and maintained at the Comparative Medicine Core Facility (CoMed), and experiments were ethically approved by the Norwegian Food Safety Authority (FOTS ID 11842). Mice were lethally irradiated (10 or 16 Gy), and small intestinal repair was assessed 3 and 6 days after irradiation. Yapf/f;Wwtr1f/f animals (37) were a gift from S. Piccolo and were crossed with Villin-Cre and Villin-CreERT2 at the University of Copenhagen under the approval of the National Animal Ethics Committee in Denmark.
Crypt, IEC, and ISC isolation and mouse organoid cultures
Adult crypt isolation
Crypt, intestinal epithelial cell (IEC), and ISC isolation, as well as organoid culture, were essentially done as described. For adult crypt isolation, duodenum tissue was rinsed with ice-cold phosphate-buffered saline (PBS) and cut open longitudinally, and villi were scraped off. Tissue was cut in ~2-mm pieces and washed five times with ice-cold PBS. Tissue pieces were incubated in 2 mM EDTA in ice-cold PBS for 30 to 60 min. Crypts were isolated from up to 10 fractions after pipetting up and down five times with PBS. To isolate single cells for sorting, crypts were incubated in TrypLE (Thermo Fisher Scientific) at 37°C for 20 to 45 min. Single cells were stained and sorted, and 4′,6-diamidino-2-phenylindole (DAPI)–negative and Epcam-positive cells were used for RNA-seq and ChIP-seq experiments. ISCs for clonal organoid outgrowth experiments were isolated by sorting DAPI-negative, GFP-high (top 5%) cells from Lgr5-EGFP mice.
E18.5/P7/P21 IEC isolation
Whole (E18.5) or proximal 10-cm (P7 and P21) small intestines were isolated and flushed with ice-cold PBS when possible (P21). Small intestines were opened longitudinally (P21 and P7) and cut into small pieces that were washed with ice-cold PBS and incubated with 2 mM EDTA in ice-cold PBS for 30 min. Whole epithelium was isolated by collecting all fractions, which was used directly for RNA isolation; for ChIP, fractions were made single cell using TrypLE (Thermo Fisher Scientific) and sorted as described.
Organoid cultures
Organoids were grown and maintained in “basal crypt medium” [advanced DMEM/F12 medium supplemented with penicillin/streptomycin, 10 mM Hepes, 2 mM GlutaMax, N2 (Thermo Fisher Scientific, 17502048), and B-27 (Thermo Fisher Scientific, 17504044)] supplemented with N-acetyl-l-cysteine (Sigma-Aldrich, A7250), murine EGF (50 ng/ml; Thermo Fisher Scientific, PMG8041), 20% R-spondin 1s conditioned medium (CM) (gift from C. Kuo), and 10% Noggin-CM (gift from H. Clevers). For ISC clonal experiments, in the first 48 hours after seeding, the medium was supplemented with Rock inhibitor (Y-27632) and Jagged-1 peptide [amino acid sequence CDDYYYGFGCNKFCRPR, made in house, peptide synthesized as described (42)], and 33% WNT3-CM (gift from H. Clevers) served as control. Medium was renewed every other day. For passaging, organoid cultures were washed, and Matrigel and organoids were disrupted mechanically by strong pipetting, centrifuged at 200g, 5 min at 4°C, and resuspended in Matrigel to replate. Imaging of live organoids was done using EVOS FL Auto 2. Structural Genomics Consortium supplied the inhibitors for the screen (www.thesgc.org), all of which are commercially available; concentrations are listed in table S1. In addition, CHIR99021 (3 μM), IWP-2 (2 μM), valproic acid (VPA) (1 mM), and DAPT (10 μM) were used.
Vil-Cre;Yapf/f/Wwtr1f/f and Vil-CreERT2;Yapf/f;Wwtr1f/f organoids were cultured in basal medium supplemented with N-acetyl-l-cysteine (NAC), B27, human EGF (50 ng/ml; PeproTech, AF-100-15) and murine Noggin (100 ng/ml; PeproTech, 250-38), and either mouse RSPO1 (500 ng/ml; R&D Systems, 3474-RS) or 10% RSPO1-CM. Established Vil-CreERT2;Yapf/f;Wwtr1f/f organoids were cultured in the presence of 1 μM 4-OH-tamoxifen (Sigma-Aldrich) for 72 hours before plating in the absence or presence of GSK-LSD1.
Human organoids, culture, and staining
Human small intestine samples were obtained from patients undergoing elective surgery at the Tokyo University Hospital with written informed consent. This was approved by the ethical committee [no. G3553-(7)]. Crypts were isolated by physically removing the stroma, and the remaining epithelium was cut into 1-mm3 pieces, washed at least five times in ice-cold PBS, and incubated in 2.5 mM EDTA in ice-cold PBS for 1 hour. Isolated crypts were then suspended in Matrigel and seeded in 48-well plates. Domes of polymerized Matrigel were given the refined medium consisting of basal crypt medium (see above) supplemented with 10 nM gastrin I (Sigma-Aldrich), 1 mM N-acetylcysteine (Sigma-Aldrich), recombinant mouse Noggin (100 ng/ml; PeproTech), recombinant mouse EGF (50 ng/ml; Thermo Fisher Scientific), recombinant human insulin-like growth factor 1 (IGF-1) (100 ng/ml; BioLegend), recombinant human fibroblast growth factor (FGF) basic (FGF-2) (50 ng/ml; PeproTech), recombinant human R-spondin1 (1 mg/ml; R&D Systems), 500 nM A83-01 (Tocris), and 50% Afamin-Wnt-3A serum-free CM. LGR5-iCaspase9-tdTomato organoids were made previously (25). For staining, organoids were isolated from Matrigel using Cell Recovery Solution (Corning) and fixed in 4% paraformaldehyde for 20 min at room temperature. Next, organoids were washed with PBS and permeabilized with 0.2% Triton X-100 in PBS for 20 min at room temperature. Blocking was done using Power Block Universal Blocking Reagent (BioGenex) for 20 min at room temperature, and rabbit anti-lysozyme antibody [1:1000; Dako, A0099 (Fig. 1H); 1:200; GeneTex, GTX72913 (Fig. 2F)] and anti–red fluorescent protein (RFP) (1:500; Rockland, 600-401-379) were incubated overnight at 4°C. Organoids were washed three times with PBS, and secondary antibody incubation was done for 30 min at room temperature. Nuclear counterstaining was done simultaneously with secondary antibody incubation using Hoechst 33342 (Thermo Fisher Scientific). Stained organoids were suspended in 1 drop of ProLong Diamond Antifade Mountant (Thermo Fisher Scientific) and mounted onto a 35-mm glass bottom dish. Images were captured using a confocal microscope (SP8, Leica).
Immunohistochemical staining of intestinal tissue
For immunohistochemical staining and imaging, tissues were harvested and fixed in swiss rolls. After fixation in formalin, tissues were embedded in paraffin and cut in 4-μm sections. Paraffin sections were rehydrated, and peroxidase activity was blocked in 3% hydrogen peroxide. Antigen retrieval was performed in citrate buffer (pH 6). Sections were stained overnight with primary antibodies against KI67 (1:500; Thermo Fisher Scientific, MA5-14520), lysozyme (1:750; Dako, A0099), SOX9 (1:200; Millipore), LSD1 (1:200; Cell Signaling Technology, 2184S), H3K4me1 (1:100; Cell Signaling Technology, 9723), and H3K4me2 (1:1500; Cell Signaling Technology, 9725). The sections were washed in tris-buffered saline (TBS) and Tween 20 and stained for 1 hour with horseradish peroxidase (HRP)–labeled secondary antibody (Dako, K4003). The staining was developed with diaminobenzidine chromogenic substrate (Dako, K5007) and mounted with Glycergel mounting medium (Dako, C056330). Tissues were imaged using a Nikon Eclipse Ci-L microscope.
Immunofluorescence staining of intestinal tissue and organoids
For immunofluorescence labeling and imaging, tissues (first 5 cm of duodenum) were harvested and fixed in swiss rolls. After fixation in formalin, tissues were paraffin-embedded and cut in 4-μm sections. Briefly, paraffin sections were treated as before for immunohistochemistry (IHC) and, after antigen retrieval, were blocked and permeabilized in PBS with 0.2% Triton X-100, 2% normal goat serum (NGS), 1% bovine serum albumin (BSA), and 0.05% Tween 20. Sections were then stained overnight in the same blocking buffer with primary antibodies against GFP (1:2000; Abcam, 13970), YAP (1:200; Cell Signaling Technology, 14074S), OLFM4 (1:200; Cell Signaling Technology, 39141S), or LSD1 (1:200; Cell Signaling Technology, 2184S). Tissues were then incubated with the corresponding secondary antibodies for 3 hours (1:500; Alexa Fluor), rhodamine-labeled UEA-1 (5 μg/ml; Thermo Fisher Scientific, NC9290135), and Hoechst 33342 (1:10,000). Washes were performed with PBS + 0.1% Tween 20.
For organoid staining, organoids were grown in Matrigel on eight-chamber μ-slides (ibidi, 80826) and fixed after exposition to the specific treatments in PBS containing 4% paraformaldehyde (pH 7.4) and 2% sucrose for 20 to 30 min, permeabilized (PBS, 0.2% Triton X-100), and blocked (PBS–0.2% Triton X-100, 2% NGS, 1% BSA). Primary antibodies against the following antigens were used and diluted in the same blocking buffer: lysozyme (1:500; Dako, A0099), GFP (1:2000; Abcam, 13970), and LSD1 (1:400; Cell Signaling Technology, 2184S) overnight at 4°C with slow agitation. Rhodamine-labeled UEA-1 (5 μg/ml; Thermo Fisher Scientific, NC9290135) and Hoechst 33342 (1:10,000) were used to stain secretory cells and nuclei, respectively, together with the corresponding secondary antibodies (1:500; Alexa Fluor) and incubated overnight in PBS with 0.2% Triton X-100, 1% NGS, and 0.5% BSA at 4°C. Tissue sections and organoids were both mounted using Fluoromount-G (Thermo Fisher Scientific, 00-4958-02) and imaged with a Zeiss 510 Meta Live or a Zeiss LSM880 confocal microscope, using 20× and 40× objective lens.
In situ hybridization
ISH was performed on formalin-fixed, paraffin-embedded (FFPE) tissues using RNAscope 2.5 HD BROWN reagent kit [Advanced Cell Diagnostics (ACD), 322371]. Tissue sections (4 μm) were deparaffinized with Neoclear and 100% ethanol. The slides were pretreated with hydrogen peroxide for 10 min, target antigen retrieval reagent for 15 min, and protease plus reagent for 30 min (ACD, 322300 and 322000). The sections were hybridized with probes for Mm-Wnt3 (ACD, 312241), Mm-Olfm4 (ACD, 311831), Mm-Atoh1 (ACD, 408791), positive control Mm-Ppib (ACD, 313911), and negative control Mm-DapB (ACD, 310043). For amplification and chromogenic detection, the 2.5 HD Detection Reagents BROWN Kit (ACD, 322310) was used. The slides were counterstained with hematoxylin, dehydrated, and mounted with Neo-Mount (Merck, 109016). Tissues were imaged using a Nikon Eclipse Ci-L microscope.
Flow cytometry analysis of organoids
Organoids were mechanically disrupted, centrifuged at 2000 rpm, and incubated with TrypLE (Thermo Fisher Scientific) at 37°C for 50 min for dissociation into single cells. Cells were incubated with DAPI and analyzed using a flow cytometer (FACSCanto II; BD). Stem cell populations were gated as DAPI negative and GFP high and analyzed using FlowJo software.
Western blot
Organoids were harvested in lysis buffer (1% NP-40, 0.02% SDS in 1× TBS) on ice for 30 min. Debris was pelleted by spinning down at 14,000 rpm for 30 min. Supernatant was diluted in 4× NuPAGE sample buffer with 100 mM dithiothreitol, and samples were run using precast 4 to 12% gels using the NuPAGE system and blotted using iBlot 2 (all Thermo Fisher Scientific). Membranes were incubated with antibodies against lysozyme (Dako, A0099) and tubulin (Abcam, Ab6046). Secondary antibodies (HRP linked) were swine anti-rabbit (P039901-2) and goat anti-mouse (P044701-2) (Dako). Imaging was done using SuperSignal West Femto (Thermo Fisher Scientific) on a Lycor machine. Bands were quantified using Image Studio software.
Quantitative polymerase chain reaction
RNA from organoids was isolated using either an RNeasy kit (Qiagen) or Quick-RNA kit (Zymo). Reverse transcription was carried out by using the High-Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific). qPCR was performed using the QuantiFast SYBR Green PCR Kit (Qiagen) using primers for Hprt (forward, cctcctcagaccgcttttt; reverse, aacctggttcatcatcgctaa), Actb (forward, actaatggcaacgagcggttc; reverse, ggatgccagaggattccatacc), Lyz1 (forward, ggcaaaaccccaagatctaa; reverse, tctctcaccaccctctttgc), Lyz1 (forward, gccaaggtctacaatcgttgtgagttg; reverse, cagtcagccagcttgacaccacg), Defa (forward, aatcctcctctctgccctcg; reverse, accagatctctcaatgattcctct), Yap1 (forward, tggccaagacatcttctggt; reverse, caggaacgttcagttgcgaa), Wwtr1 (forward, tggggttagggtgctacagt; reverse, ggattgacggtcatgggtgt), Gapdh (forward, tgttcctacccccaatgtgt; reverse, tgtgagggagatgctcagtg), Olfm4 (forward, ggatcctgaacttttggtgct; reverse, acgccaccatgactacagc), and Wnt3 (forward, ctcgctggctacccaattt; reverse, gaggccagagatgtgtactgc). Samples were commonly analyzed in duplicate, and RNA expression was calculated either normalized to reference gene or additionally normalized to control conditions.
RNA-seq preparation
RNA for WT and LSD1KO crypts was isolated by sorting IECs (DAPI−, Epcam+) in 2× RNA shield buffer (Zymo) and RNA isolation using the Quick-RNA Microprep Kit (Zymo). Library preparation was done using the Illumina TruSeq Stranded protocol, and samples were sequenced at 75 × 2–base pair (bp) paired-end (PE) reads on an Illumina NS500 MO flow cell. Sequencing was performed by the Genomics Core Facility (NTNU). IECs from E18.5, P7, and E18.5 were directly dissolved in RNA isolation buffer, and RNA was isolated using the Quick-RNA Microprep Kit (Zymo). Library preparation was done using the NEBNext Ultra RNA Library Prep Kit. Sequencing was performed by Novogene (UK) Co.
RNA-seq analysis
Sequenced reads were aligned with STAR to the Mus musculus genome build mm10 (43, 44). The number of reads that uniquely aligned to the exon region of each gene in GENCODE annotation M18 of the mouse genome was then counted using featureCounts (45). Genes that had a total count less than 10 were filtered out. Differential expression was then determined with DESeq2 using default settings (46). Interesting differential genes were plotted with a volcano plot using the R package EnhancedVolcano. Heatmaps were generated using the R package pheatmap. Count values for each gene were transformed to rates per base pair by dividing the count for a gene by the length of the total exon region for that gene. Rates per base pair were then converted to transcripts per million (TPM) by dividing the rate per base pair for each gene by the sum of rates per base pair for all the genes in that sample and multiplying with 1 million. Principal components analysis (PCA) was performed using the function sklearn.decomposition.PCA in scikit-learn. GSEA was done by sorting the output from DESeq2 by log2 fold change and with the log2 fold change as weights. GSEA was run with the R package clusterProfiler using 10,000 permutations and otherwise default settings. Gene sets were generated from published datasets including (47) and can be found in table S3.
Microarray analysis
Gene expression in human small intestinal organoids was analyzed using the PrimeView Human Gene Expression Array. Raw expression data were normalized with the rma function in the R/Bioconductor package affy, and the normalized values were used to calculate log fold change (48). For each gene, the probe with the highest absolute log fold change was used. GSEA was run on this list of genes as described for the RNA-seq analysis.
Chromatin immunoprecipitation sequencing
DAPI-negative, Epcam-positive IECs were sorted in PBS containing 20 mM sodium butyrate and cross-linked by incubation in 1% formaldehyde for 8 min. Glycine was added to a final concentration of 125 mM and incubated for 5 min at room temperature. Using a swing-out rotor, cells were washed three times in ice-cold PBS with 20 mM sodium butyrate. After washing, cells were snap-frozen in liquid nitrogen and stored in −80°C. The ChIP-seq was carried out similarly to previously described protocols (49).
Binding of antibodies to paramagnetic beads
The stock of paramagnetic Dynabeads Protein A was vortexed thoroughly to ensure a homogenous suspension before pipetting. Dynabeads stock solution [5 μl per immunoprecipitation (IP)] was transferred into a 1.5-ml tube, which was placed on a magnetic rack, and the beads were captured on the tube wall. The buffer was discarded, and the beads were washed twice with 200 μl of standard radioimmunoprecipitation assay (RIPA) buffer [10 mM tris-HCl (pH 8.0), 140 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% SDS, 0.1% Na-deoxycholate] and resuspended in standard RIPA buffer to a final volume of 100 μl per IP. Ninety-nine microliters of this was aliquoted into each 0.6-ml tube on ice, and antibody [1.2 μg of anti-H3K4me1 (Diagenode, C15410194, lot A1862D) or 4 μl of anti-H3K4me2 (Cell Signaling Technology, #9725, lot 9)] was added per 0.6-ml tube. Tubes were then incubated at 40 rpm on a “head-over-tail” tube rotator for at least 16 hours at 4°C.
Chromatin preparation, Lsd1 cre+/cre−
Cross-linked cell pellets containing 335,000 to 500,000 cells were thawed on ice. The 6- to 10-μl pellets were added to lysis buffer [50 mM tris-HCl (pH 8.0), 10 mM EDTA (pH 8.0), 1% SDS, 20 mM sodium butyrate, 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail] to a total of 160 μl and incubated on ice for 10 min. The samples were sonicated for 8 × 30 s using the UP100H Ultrasonic Processor (Hielscher) fitted with a 2-mm probe. We allowed 30-s pauses on ice between each 30-s session, using pulse settings with 0.5-s cycles and 27% power. After the final sonication, 340 μl of standard RIPA (with 20 mM sodium butyrate, 1 mM PMSF, and protease inhibitor cocktail) was added to the tube while washing the probe, followed by thorough mixing by pipetting. Twenty microliters was removed as input, and the remaining solution was diluted further with 1 ml of standard RIPA buffer (with 20 mM sodium butyrate, 1 mM PMSF, and protease inhibitor cocktail). The samples and inputs were centrifuged at 12,000g in a swinging-bucket rotor for 10 min at 4°C, and the supernatants were transferred to a 1.5-ml tube on ice. Sixty-six thousand to 100,000 cells were used per IP.
Chromatin preparation, E18.5/P7/P21
The H3K4me1 ChIP-seq for the different developmental stages was slightly modified at the chromatin preparation step. Cross-linked cell pellets containing 50,000 to 300,000 cells were thawed on ice. The 10-μl pellets were added to a modified lysis buffer [50 mM tris-HCl (pH 8.0), 10 mM EDTA (pH 8.0), 0.8% SDS, 20 mM sodium butyrate, 1 mM PMSF, and protease inhibitor cocktail] to a total of 120 μl and incubated on ice for 10 min, followed by addition of 30 μl of PBS with 20 mM sodium butyrate. The samples were sonicated as described above. After the final sonication, 360 μl of RIPA without SDS (with 20 mM sodium butyrate, 1 mM PMSF, and protease inhibitor cocktail) was added to the tube while washing the probe, followed by thorough mixing by pipetting. The sample and input were centrifuged at 12,000g in a swinging-bucket rotor for 10 min at 4°C, and the supernatants were transferred to a 1.5-ml tube on ice. Chromatin corresponding to 1000 to 2000 cells was removed from each sample to be decross-linked and sequenced as inputs. For the ChIPs, chromatin corresponding to 50,000 cells was used for E18.5 Replicate A, while chromatin from 100,000 cells was used for the remaining samples.
Immunoprecipitation and washes
Preincubated antibody-bead complexes were washed twice in 200 μl of standard RIPA buffer by vortexing roughly. The tubes were centrifuged in a mini-centrifuge to bring down any solution trapped in the lid, and antibody-bead complexes were captured in a magnetic rack. After removal of RIPA, 177 to 500 μl of chromatin (equivalent of 50,000 to 100,000 cells per ChIP) were added to each tube and then incubated at 4°C, 40 rpm on a head-over-tail rotator for at least 16 hours. For H3K4me1 ChIPs, the chromatin-antibody-bead complexes were washed three times in 100 μl of ice-cold standard RIPA buffer. For H3K4me2 ChIPs, the reactions were washed once in standard RIPA, once in RIPA with increased salt and SDS (300 mM NaCl and 0.20% SDS), once in RIPA with increased salt and SDS (300 mM NaCl and 0.23% SDS), and once in standard RIPA. All washing steps were performed with 100 μl of ice-cold buffer supplemented with 20 mM sodium butyrate, 1 mM PMSF, and protease inhibitor cocktail. Each wash involved rough vortexing at full speed, repeated twice with pauses on ice in between. Next, a wash in 100 μl of TE and tube shift was carried out.
DNA isolation and purification
Tris-EDTA buffer (TE) was removed, and 150 μl of ChIP elution buffer was added [20 mM tris-HCl (pH 7.5), 50 mM NaCl, 5 mM EDTA, 1% SDS, 30 μg of ribonuclease (RNase) A] and incubated at 37°C, 1 hour at 1200 rpm on a thermomixer. The input samples were added to ChIP elution buffer up to 150 μl and incubated similarly. Proteinase K (1 μl; 20 mg/ml stock) was added to each ChIP or input tube and incubated at 68°C, 4 hours at 1250 rpm. The ChIP eluates were transferred to a 1.5-ml tube. Then, a second elution with 150 μl was performed for 5 min and pooled with the first supernatant. The ChIP and input DNA were purified by phenol-chloroform isoamyl alcohol extraction, ethanol-precipitated with 11 μl of acrylamide carrier, and dissolved in 10 to 15 μl of elution buffer (EB) (10 mM tris-HCl).
Library preparation and sequencing
ChIP and input library preparations were performed according to the QIAseq Ultralow Input Library Kit procedure. Sequencing procedures were carried out according to Illumina protocols, on a NextSeq 500 instrument, with 75-bp single-end reads using high-output reagents. The sequencing service was provided by the Norwegian Sequencing Centre (www.sequencing.uio.no).
ChIP-seq analysis
Sequence reads were deduplicated with BBMaps clumpify tool and then aligned with STAR to the M. musculus genome build mm10 (43, 44). Peaks were identified using model-based analysis of ChIP-seq 2 (MACS2) with peak type set to broad and genome size 2652783500 (50). Input files were supplied for H3K4me1/2. Peaks from all samples that were compared in differential expression were merged with BEDTools to create a union set, and featureCounts was used to count the number of reads, including multimappers, for each sample in the union set of peaks (50). Differential peaks was determined from the counts with DESeq2 (46) using default settings. Peak locations were associated with the gene that has the closest TSS with the closest command in BEDtools (51). Ties were resolved by only reporting the first hit. TSSs were downloaded from biomart for GRCm38.p6. The peaks were grouped on the distance to the TSS, and the size of each group was plotted. The list of differential peaks with associated genes was grouped by gene and sorted on the differential peak that had the smallest P value for each gene. Each gene was determined to be either up or down in signal based on whether the total change was above or below zero, where total change is defined as the average of log fold change multiplied by peak length of all peaks associated with that gene. Venn diagrams were created with the R package Euler (52). BigWig files describing the score across the genome were created with deepTools2 and scaled to the count of the sample with the least aligned reads for each group (e.g., H3K4me1 and H3K4me2) (53). Heatmaps of regions of interest were created with deepTools2. Chip-seq profiles were created in integrative genomics viewer (54).
Fetal RNA-seq (from Yui et al.)
Published microarray raw data were downloaded from ArrayExpress under the accession number “E-MTAB-5246” and normalized with “neqc” function in the R package limma, and then log2 fold change was calculated from the normalized expression values (9, 55). GSEA was performed as described in the RNA-seq methods.
scRNA-seq analysis
Preprocessed and normalized scRNA-seq data were downloaded from GSE117783 (35). The control treated cells were randomly subsampled so the two groups had equal number of cells and the density of LSD1-expressing cells was plotted in base R. Y axis is shortened to show distribution of cells that has an expression larger than zero.
Statistical analysis
Statistical significance was determined using either Student’s t test or one-way analysis of variance (ANOVA) with Tukey’s post hoc test, or when n < 10, nonparametric testing (Mann-Whitney test) was done. Significance levels are indicated in figure legends.
Supplementary Material
Acknowledgments
We thank S. Orkin, S. Robine, and S. Piccolo for sharing mouse strains. We thank C. Zaph for the initial support of this study and critical reading of the manuscript. We thank U. Nonstad for assistance with cell sorting. We are indebted to A. Marthinsen for performing the irradiation after working hours and to the Department of Radiology and Nuclear Medicine (St. Olavs Hospital) for allowing the use of their instruments. We thank the imaging (CMIC) and animal care (CoMed) core facilities that assisted in this work (NTNU). Funding: The WT KO crypt RNA-seq was done by the Genomics Core Facility at NTNU, which receives funding from the Faculty of Medicine and Health Sciences and Central Norway Regional Health Authority. The ChIP sequencing was done at the Norwegian Sequencing Centre (www.sequencing.uio.no), a national technology platform hosted by the University of Oslo and supported by the “Functional Genomics” and “Infrastructure” programs of the Research Council of Norway and the Southeastern Regional Health Authorities. Funding of this work was provided by the Norwegian Research Council (Centre of Excellence grant 223255/F50 and “Young Research Talent” 274760 to M.J.O.) and the Norwegian Cancer Society (182767 to M.J.O.). M.M.-A. is the recipient of a Marie Skłodowska-Curie IF (DLV-794391). This work was also supported by the South-Eastern Norway Regional Health Authority, Early Career Grant 2016058, and the Research Council of Norway “Young Research Talent” grant to J.A.D. M.F. is supported by the Norwegian Research Council (grant no. 275286). The SGC is a registered charity (number 1097737) that receives funds from AbbVie; Bayer Pharma AG; Boehringer Ingelheim; Canada Foundation for Innovation; Eshelman Institute for Innovation; Genome Canada through Ontario Genomics Institute (OGI-055); Innovative Medicines Initiative (EU/EFPIA) (ULTRA-DD grant no. 115766); Janssen; Merck KGaA, Darmstadt, Germany; MSD; Novartis Pharma AG; Ontario Ministry of Research, Innovation and Science (MRIS); Pfizer; São Paulo Research Foundation-FAPESP; Takeda; and Wellcome. This project also received funding from the European Union’s Horizon 2020 research and innovation programme (grant agreement INTENS 668294 to M.T.P. and K.B.J.). The Novo Nordisk Foundation Center for Stem Cell Biology is supported by Novo Nordisk Foundation grant number NNF17CC0027852 (K.B.J.). Author contributions: R.T.Z., H.T.L., M.F., M.T.P., Y.O., A.D.-S., M.M.-A., J.O., M.M., N.P., E.K., R.R.S., and M.J.O. performed experiments. K.N. and C.A. provided essential reagents. M.R., F.D., C.A., J.A.D., K.B.J., T.S., and M.J.O. supervised and/or provided conceptual insight. M.J.O. wrote a first draft with subsequent input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All raw data are available through ArrayExpress: 24h GSK-LSD1 organoids RNA-seq: E-MTAB-9077, WT/KO Crypt RNA-seq: E-MTAB-7862, WT/KO Crypt ChIP-seq: E-MTAB-7871, E18/P7/P21 RNA-seq: E-MTAB-8713, E18/P7/P21 H3K4me1 ChIP-seq: E-MTAB-8710, and Human microarray: E-MTAB-7871.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/37/eabc0367/DC1
REFERENCES AND NOTES
- 1.Shoshkes-Carmel M., Wang Y. J., Wangensteen K. J., Tóth B., Kondo A., Massassa E. E., Itzkovitz S., Kaestner K. H., Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 557, 242–246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sato T., van Es J. H., Snippert H. J., Stange D. E., Vries R. G., van den Born M., Barker N., Shroyer N. F., van de Wetering M., Clevers H., Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415–418 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durand A., Donahue B., Peignon G., Letourneur F., Cagnard N., Slomianny C., Perret C., Shroyer N. F., Romagnolo B., Functional intestinal stem cells after Paneth cell ablation induced by the loss of transcription factor Math1 (Atoh1). Proc. Natl. Acad. Sci. U.S.A. 109, 8965–8970 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim T.-H., Escudero S., Shivdasani R. A., Intact function of Lgr5 receptor-expressing intestinal stem cells in the absence of Paneth cells. Proc. Natl. Acad. Sci. U.S.A. 109, 3932–3937 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farin H. F., van Es J. H., Clevers H., Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology 143, 1518–1529.e7 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Fordham R. P., Yui S., Hannan N. R. F., Soendergaard C., Madgwick A., Schweiger P. J., Nielsen O. H., Vallier L., Pedersen R. A., Nakamura T., Watanabe M., Jensen K. B., Transplantation of expanded fetal intestinal progenitors contributes to colon regeneration after injury. Cell Stem Cell 13, 734–744 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mustata R. C., Vasile G., Fernandez-Vallone V., Strollo S., Lefort A., Libert F., Monteyne D., Pérez-Morga D., Vassart G., Garcia M.-I., Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep. 5, 421–432 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Guiu J., Hannezo E., Yui S., Demharter S., Ulyanchenko S., Maimets M., Jørgensen A., Perlman S., Lundvall L., Mamsen L. S., Larsen A., Olesen R. H., Andersen C. Y., Thuesen L. L., Hare K. J., Pers T. H., Khodosevich K., Simons B. D., Jensen K. B., Tracing the origin of adult intestinal stem cells. Nature 570, 107–111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yui S., Azzolin L., Maimets M., Pedersen M. T., Fordham R. P., Hansen S. L., Larsen H. L., Guiu J., Alves M. R. P., Rundsten C. F., Johansen J. V., Li Y., Madsen C. D., Madsen C. D., Nakamura T., Watanabe M., Nielsen O. H., Schweiger P. J., Piccolo S., Jensen K. B., YAP/TAZ-dependent reprogramming of colonic epithelium Links ECM remodeling to tissue regeneration. Cell Stem Cell 22, 35–49.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nusse Y. M., Savage A. K., Marangoni P., Rosendahl-Huber A. K. M., Landman T. A., de Sauvage F. J., Locksley R. M., Klein O. D., Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature 559, 109–113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gregorieff A., Liu Y., Inanlou M. R., Khomchuk Y., Wrana J. L., Yap-dependent reprogramming of Lgr5+ stem cells drives intestinal regeneration and cancer. Nature 526, 715–718 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Jadhav U., Saxena M., O’Neill N. K., Saadatpour A., Yuan G.-C., Herbert Z., Murata K., Shivdasani R. A., Dynamic reorganization of chromatin accessibility signatures during dedifferentiation of secretory precursors into Lgr5+ intestinal stem cells. Cell Stem Cell 21, 65–77.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buczacki S. J. A., Zecchini H. I., Nicholson A. M., Russell R., Vermeulen L., Kemp R., Winton D. J., Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 495, 65–69 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Kazakevych J., Sayols S., Messner B., Krienke C., Soshnikova N., Dynamic changes in chromatin states during specification and differentiation of adult intestinal stem cells. Nucleic Acids Res. 45, 5770–5784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raab J. R., Tulasi D. Y., Wager K. E., Morowitz J. M., Magness S. T., Gracz A. D., Quantitative classification of chromatin dynamics reveals regulators of intestinal stem cell differentiation. Development 147, dev181966 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jadhav U., Cavazza A., Banerjee K. K., Xie H., O’Neill N. K., Saenz-Vash V., Herbert Z., Madha S., Orkin S. H., Zhai H., Shivdasani R. A., Extensive recovery of embryonic enhancer and gene memory stored in hypomethylated enhancer DNA. Mol Cell 74, 542–554.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koppens M. A. J., Bounova G., Gargiulo G., Tanger E., Janssen H., Cornelissen-Steijger P., Blom M., Song J.-Y., Wessels L. F. A., van Lohuizen M., Deletion of polycomb repressive complex 2 from mouse intestine causes loss of stem cells. Gastroenterology 151, 684–697.e12 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Chiacchiera F., Rossi A., Jammula S., Zanotti M., Pasini D., PRC2 preserves intestinal progenitors and restricts secretory lineage commitment. EMBO J. 35, 2301–2314 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheer S., Ackloo S., Medina T. S., Schapira M., Li F., Ward J. A., Lewis A. M., Northrop J. P., Richardson P. L., Kaniskan H. Ü., Shen Y., Liu J., Smil D., McLeod D., Zepeda-Velazquez C. A., Luo M., Jin J., Barsyte-Lovejoy D., Huber K. V. M., Carvalho D. D., Vedadi M., Zaph C., Brown P. J., Arrowsmith C. H., A chemical biology toolbox to study protein methyltransferases and epigenetic signaling. Nat. Commun. 10, 19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato T., Vries R. G., Snippert H. J., van de Wetering M., Barker N., Stange D. E., van Es J. H., Abo A., Kujala P., Peters P. J., Clevers H., Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Yin X., Farin H. F., van Es J. H., Clevers H., Langer R., Karp J. M., Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat. Methods 11, 106–112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohammad H. P., Smitheman K. N., Kamat C. D., Soong D., Federowicz K. E., Van Aller G. S., Schneck J. L., Carson J. D., Liu Y., Butticello M., Bonnette W. G., Gorman S. A., Degenhardt Y., Bai Y., McCabe M. T., Pappalardi M. B., Kasparec J., Tian X., McNulty K. C., Rouse M., McDevitt P., Ho T., Crouthamel M., Hart T. K., Concha N. O., McHugh C. F., Miller W. H., Dhanak D., Tummino P. J., Carpenter C. L., Johnson N. W., Hann C. L., Kruger R. G., A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 28, 57–69 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Haber A. L., Biton M., Rogel N., Herbst R. H., Shekhar K., Smillie C., Burgin G., Delorey T. M., Howitt M. R., Katz Y., Tirosh I., Beyaz S., Dionne D., Zhang M., Raychowdhury R., Garrett W. S., Rozenblatt-Rosen O., Shi H. N., Yilmaz O., Xavier R. J., Regev A., A single-cell survey of the small intestinal epithelium. Nature 551, 333–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Es J. H., Wiebrands K., López-Iglesias C., van de Wetering M., Zeinstra L., van den Born M., Korving J., Sasaki N., Peters P. J., van Oudenaarden A., Clevers H., Enteroendocrine and tuft cells support Lgr5 stem cells on Paneth cell depletion. Proc. Natl. Acad. Sci. U.S.A. 31, 26599–26605 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujii M., Matano M., Toshimitsu K., Takano A., Mikami Y., Nishikori S., Sugimoto S., Sato T., Human intestinal organoids maintain self-renewal capacity and cellular diversity in niche-inspired culture condition. Cell Stem Cell 23, 787–793.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Kerenyi M. A., Shao Z., Hsu Y.-J., Guo G., Luc S., O’Brien K., Fujiwara Y., Peng C., Nguyen M., Orkin S. H., Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. eLife 2, e00633 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.el Marjou F., Janssen K.-P., Chang B. H.-J., Li M., Hindie V., Chan L., Louvard D., Chambon P., Metzger D., Robine S., Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Bastide P., Darido C., Pannequin J., Kist R., Robine S., Marty-Double C., Bibeau F., Scherer G., Joubert D., Hollande F., Blache P., Jay P., Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J. Cell Biol. 178, 635–648 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Q., Bermingham N. A., Finegold M. J., Zoghbi H. Y., Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 294, 2155–2158 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Kim T.-H., Li F., Ferreiro-Neira I., Ho L.-L., Luyten A., Nalapareddy K., Long H., Verzi M., Shivdasani R. A., Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nature 506, 511–515 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maiques-Diaz A., Spencer G. J., Lynch J. T., Ciceri F., Williams E. L., Amaral F. M. R., Wiseman D. H., Harris W. J., Li Y., Sahoo S., Hitchin J. R., Mould D. P., Fairweather E. E., Waszkowycz B., Jordan A. M., Smith D. L., Somervaille T. C. P., Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. 22, 3641–3659 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shroyer N. F., Wallis D., Venken K. J. T., Bellen H. J., Zoghbi H. Y., Gfi1 functions downstream of Math1 to control intestinal secretory cell subtype allocation and differentiation. Genes Dev. 19, 2412–2417 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan K. S., Gevaert O., Zheng G. X. Y., Anchang B., Probert C. S., Larkin K. A., Davies P. S., Cheng Z.-F., Kaddis J. S., Han A., Roelf K., Calderon R. I., Cynn E., Hu X., Mandleywala K., Wilhelmy J., Grimes S. M., Corney D. C., Boutet S. C., Terry J. M., Belgrader P., Ziraldo S. B., Mikkelsen T. S., Wang F., von Furstenberg R. J., Smith N. R., Chandrakesan P., May R., Chrissy M. A. S., Jain R., Cartwright C. A., Niland J. C., Hong Y.-K., Carrington J., Breault D. T., Epstein J., Houchen C. W., Lynch J. P., Martin M. G., Plevritis S. K., Curtis C., Ji H. P., Li L., Henning S. J., Wong M. H., Kuo C. J., Intestinal enteroendocrine lineage cells possess homeostatic and injury-inducible stem cell activity. Cell Stem Cell 21, 78–90.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Es J. H., Sato T., van de Wetering M., Lyubimova A., Yee Nee A. N., Gregorieff A., Sasaki N., Zeinstra L., van den Born M., Korving J., Martens A. C. M., Barker N., van Oudenaarden A., Clevers H., Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat. Cell Biol. 14, 1099–1104 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ayyaz A., Kumar S., Sangiorgi B., Ghoshal B., Gosio J., Ouladan S., Fink M., Barutcu S., Trcka D., Shen J., Chan K., Wrana J. L., Gregorieff A., Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569, 121–125 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Muñoz J., Stange D. E., Schepers A. G., van de Wetering M., Koo B.-K., Itzkovitz S., Volckmann R., Kung K. S., Koster J., Radulescu S., Myant K., Versteeg R., Sansom O. J., van Es J. H., Barker N., van Oudenaarden A., Mohammed S., Heck A. J. R., Clevers H., The Lgr5 intestinal stem cell signature: Robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 31, 3079–3091 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Azzolin L., Panciera T., Soligo S., Enzo E., Bicciato S., Dupont S., Bresolin S., Frasson C., Basso G., Guzzardo V., Fassina A., Cordenonsi M., Piccolo S., YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 158, 157–170 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Whyte W. A., Bilodeau S., Orlando D. A., Hoke H. A., Frampton G. M., Foster C. T., Cowley S. M., Young R. A., Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482, 221–225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egolf S., Aubert Y., Doepner M., Anderson A., Maldonado-Lopez A., Pacella G., Lee J., Ko E. K., Zou J., Lan Y., Simpson C. L., Ridky T., Capell B. C., LSD1 inhibition promotes epithelial differentiation through derepression of fate-determining transcription factors. Cell Rep. 28, 1981–1992.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rai K., Sarkar S., Broadbent T. J., Voas M., Grossmann K. F., Nadauld L. D., Dehghanizadeh S., Hagos F. T., Li Y., Toth R. K., Chidester S., Bahr T. M., Johnson W. E., Sklow B., Burt R., Cairns B. R., Jones D. A., DNA demethylase activity maintains intestinal cells in an undifferentiated state following loss of APC. Cell 142, 930–942 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou A., Lin K., Zhang S., Chen Y., Zhang N., Xue J., Wang Z., Aldape K. D., Xie K., Woodgett J. R., Huang S., Nuclear GSK3β promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat. Cell Biol. 18, 954–966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolscher J. G. M., Oudhoff M. J., Nazmi K., Antos J. M., Guimaraes C. P., Spooner E., Haney E. F., Garcia Vallejo J. J., Vogel H. J., van’t Hof W., Ploegh H. L., Veerman E. C. I., Sortase A as a tool for high-yield histatin cyclization. FASEB J. 25, 2650–2658 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J. M., Sisu C., Wright J., Armstrong J., Barnes I., Berry A., Bignell A., Sala S. C., Chrast J., Cunningham F., Di Domenico T., Donaldson S., Fiddes I. T., García Girón C., Gonzalez J. M., Grego T., Hardy M., Hourlier T., Hunt T., Izuogu O. G., Lagarde J., Martin F. J., Martínez L., Mohanan S., Muir P., Navarro F. C. P., Parker A., Pei B., Pozo F., Ruffier M., Schmitt B. M., Stapleton E., Suner M.-M., Sycheva I., Uszczynska-Ratajczak B., Xu J., Yates A., Zerbino D., Zhang Y., Aken B., Choudhary J. S., Gerstein M., Guigó R., Hubbard T. J. P., Kellis M., Paten B., Reymond A., Tress M. L., Flicek P., GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J. P., Tamayo P., The molecular signatures database hallmark gene set collection. Cell Syst. 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gautier L., Cope L., Bolstad B. M., Irizarry R. A., affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Dahl J. A., Collas P., A rapid micro chromatin immunoprecipitation assay (ChIP). Nat. Protoc. 3, 1032–1045 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.J. Larsson, eulerr: Area-proportional Euler and Venn diagrams with ellipses (2018).
- 53.Ramírez F., Ryan D. P., Grüning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dündar F., Manke T., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., Mesirov J. P., Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K., limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/37/eabc0367/DC1