

Integrative single-cell analysis de-constructs the heterogeneity of human reprogramming and uncovers its stage-specific GRNs.
Abstract
Cellular reprogramming suffers from low efficiency especially for the human cells. To deconstruct the heterogeneity and unravel the mechanisms for successful reprogramming, we adopted single-cell RNA sequencing (scRNA-Seq) and single-cell assay for transposase-accessible chromatin (scATAC-Seq) to profile reprogramming cells across various time points. Our analysis revealed that reprogramming cells proceed in an asynchronous trajectory and diversify into heterogeneous subpopulations. We identified fluorescent probes and surface markers to enrich for the early reprogrammed human cells. Furthermore, combinatory usage of the surface markers enabled the fine segregation of the early-intermediate cells with diverse reprogramming propensities. scATAC-Seq analysis further uncovered the genomic partitions and transcription factors responsible for the regulatory phasing of reprogramming process. Binary choice between a FOSL1 and a TEAD4-centric regulatory network determines the outcome of a successful reprogramming. Together, our study illuminates the multitude of diverse routes transversed by individual reprogramming cells and presents an integrative roadmap for identifying the mechanistic part list of the reprogramming machinery.
INTRODUCTION
Somatic cells can be reverted to pluripotency by inducing the expression of four transcription factors (TFs), namely OCT4, SOX2, KLF4, and MYC (OSKM), in a process known as cellular reprogramming (1). Discovery of this phenomenon has raised the hopes for advancing the field of regenerative medicine (2). However, cellular reprogramming suffers from extremely low efficiency especially for the human cells, resulting in a heterogeneous population in which few cells can be characterized as pluripotent (3, 4). Although a handful of studies analyzed bulk population to understand the reprogramming mechanisms (5–8), ensemble measurement of the heterogeneous population impedes the discerning of transcriptomic and epigenetic changes taking place in the minority of cells undergoing the route toward successful reprogramming. Single-cell sequencing technologies provide tools to decipher the types of cells present in a heterogeneous mixture (9). In the present study, we adopted the parallel genome-wide single-cell assays including single-cell RNA sequencing (scRNA-Seq) and single-cell assay for transposase-accessible chromatin (scATAC-Seq) (10, 11) to profile the transcriptome and chromatin accessibility of human reprogramming cells across various stages. We identified cellular diversification and trajectories, where an individual cell displays different kinetics and potential for reprogramming. In addition, with a set of cell surface markers and a fluorescent probe BDD2-C8, we were able to enrich the early-intermediary cells undergoing the route toward successful reprogramming. Moreover, we identified the modulators driving the changes in chromatin accessibility and gene regulatory networks accessibility, as cells advanced toward the diverse reprogramming trajectories. Notably, the pivot from FOSL1 (FOS-like 1, AP-1 transcription factor subunit) to TEAD4 (TEA domain transcription factor 4)–centric regulatory networks is essential for the acquisition of the pluripotent state.
RESULTS
Single-cell profiling of human cell fate reprogramming
To study the heterogeneity of human reprogramming, we analyzed a total of 33,468 scRNA-Seq and scATAC-Seq libraries prepared for day 0 (BJ), day 2 (D2), day 8 (D8), day 12 (D12), and day 16 (D16) OSKMinduced reprogramming cells (Fig. 1A). On D16, cells were sorted using TRA-1-60, a pluripotent stem-cell-specific surface marker, to distinguish the successfully reprogrammed (D16+) from the non-reprogrammed (D16−) cells (Fig. 1A and fig. S1, A and B). The generated induced pluripotent stem cells (iPSCs) were characterized with immunostaining, DNA methylation, and teratoma assay (fig. S1, C to E). In this study, we used two distinct approaches for scRNA-Seq library preparation (Fig. 1A). Microfluidic cell capture–based assay (Fluidigm C1) reads the full-length transcripts from hundreds of cells with high resolution, whereas the droplet-based assay (10X Genomics) probes the 3′ end of the transcripts from thousands of cells albeit at a relatively low genomic coverage. scATAC-Seq libraries were prepared on capture-based microfluidic chips (Fluidigm C1) (Fig. 1A). In addition, we have screened for fluorescence probes to distinguish early-intermediate cells poised for successful reprogramming (Fig. 1A). On top, we have also prepared chromatin immunoprecipitation sequencing (ChIP-Seq) libraries for the upstream TFs to dissect their regulatory networks (Fig. 1A). The cumulative data enable us to characterize the subpopulations in-depth and to construct a trajectory map of human reprogramming.
Fig. 1. Single-cell systems used for deconvoluting the heterogeneity in human cellular reprogramming.
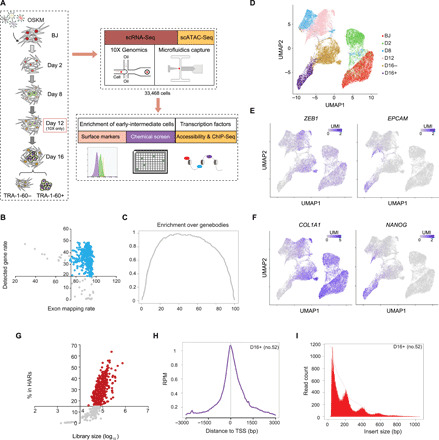
(A) Overview of the prepared single-cell NGS libraries across various time points of human cellular reprogramming. The microfluidic platform was used to prepare 439 scRNA-Seq and 891 scATAC-Seq libraries (duplicates) of good quality. 10X Genomics platform was utilized to prepare 32,138 scRNA-Seq libraries of good quality. (B) QC of microfluidic capture–based scRNA-Seq libraries. Dotplot demonstrates the exon mapping percentage (x axis) of each scRNA-Seq library, along with its corresponding detected gene rate (y axis). Blue dots represent libraries passing the QC filters. (C) Average enrichment of capture-based scRNA-Seq libraries over genebodies. (D) UMAP plot for the prepared 10X scRNA-Seq libraries. (E and F) Superimposition of the expression levels for MET genes (E) and fibroblast and pluripotent genes (F). (G) QC of scATAC-Seq libraries. Dotplot demonstrates the library size (x axis) of each scATAC-Seq library, along with its contribution to the respective time point’s HARs (y axis). Red dots represent the libraries passing the QC filters. (H) Average enrichment profile of a D16+ scATAC-Seq library around transcription start sites (TSS) of the genome with a window of −3000 bp to 3000 bp. (I) Histogram of insert size metric of a D16+ scATAC-Seq library revealing nucleosomal pattern.
The majority of the capture-based scRNA-Seq libraries demonstrated high exon mapping percentage (≥75%) and gene detection rate (≥20%), with even distribution over the genebodies (Fig. 1, B and C). Furthermore, epithelial and pluripotency genes were progressively expressed with the advancement of reprogramming, as opposed to the mesenchymal and fibroblast genes (fig. S1F). Similarly, the majority of the 10X libraries were of good quality (fig. S1, G and H). UMAP clustering revealed a dynamic transcriptomic transition from the parental BJ to D16+ cells (Fig. 1D). Expectedly, ZEB1 (mesenchymal) and COL1A1 (somatic) were abundantly expressed in the early time points and non-reprogrammed cells (Fig. 1, E and F). On the contrary, EPCAM (epithelial) and NANOG and LIN28A (pluripotent) were expressed highly in the successfully reprogrammed cells (Fig. 1, E and F, and fig. S1I). Likewise, most of the scATAC-Seq libraries passed the previously reported quality control (QC) indices (10) and exhibited enrichment over transcription start site (TSS) regions and nucleosomal distributions (Fig. 1, G to I, and fig. S1, J and K). Collectively, we generated reliable single-cell libraries for tens of thousands of reprogramming cells, providing a rich resource to decipher its deep molecular mechanisms.
Identification of heterogeneous subgroups with diverse reprogramming potentials
To determine the diverse populations present at each reprogramming time point, we clustered scRNA-Seq libraries using reference component analysis (RCA) (12). RCA clustering was reported to demonstrate higher accuracy and less technical bias than the existing algorithms for capture-based scRNA-Seq libraries (12). Briefly, RCA projects scRNA-Seq libraries to a reference-guided transcriptomic space and clusters cells based on their similarity to various cell types of different lineage origins in the RCA global panel (12). BJ cells correlated substantially to the smooth muscle lineage, which was also observed across the published BJ RNA-Seq libraries (fig. S2, A and B). D2 cells were marked by four distinct subgroups (G1 to G4), among which G1-2 cells displayed lower correlation to the fibroblasts and mesenchymal stem cells (MSCs) (Fig. 2A and fig. S2C). MSCs are multipotent stromal cells with the capacity to differentiate into mesodermal lineages such as osteocytes, chondrocytes, myocytes, and adipocytes (13). Correlation of reprogramming cells to MSCs might possibly be associated with the mesoderm germ layer origin of BJ fibroblast, the starting cells induced for reprogramming in this study. On the other hand, D8 cells were distributed across three discrete subgroups (Fig. 2A and fig. S2C). D8 G1 cells corresponded to the fibroblasts, smooth muscles, myocytes, and MSC lineages, while G3 cells displayed substantial similarity to the PSCs. D8 G2 cells represented the intermediate state. Likewise, two subpopulations were present in D16+ cells, among which G2 cells were highly associated with PSCs, while G1 cells maintained detectable correlation to the MSCs, adipose cells, and endothelial cells, other than PSCs (Fig. 2A and fig. S2C). In summary, RCA analysis indicates that the reprogramming cells are highly diverse, some of which may deviate from the route to pluripotency and acquire alternative lineage cell fates.
Fig. 2. Identification of diverse reprogramming subgroups and construction of reprogramming trajectories.
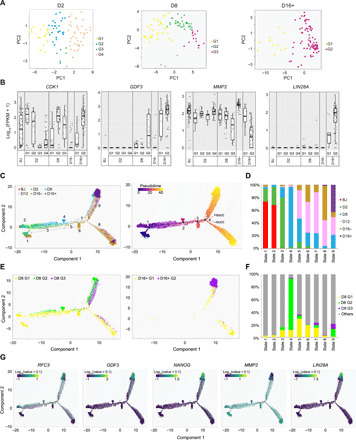
(A) PCAs showing subgroups present in D2 (left), D8 (middle), and D16+ (right) cells determined by RCA, based on their correlation to cells of various lineages in the RCA panel. Each color represents a subgroup. Gray color indicates the minority outlier cells, which do not belong to the indicated subgroups. (B) Boxplots showing single-cell expression of the differentially expressed genes CDK1 (D2), GDF3 (D8), MMP2 (D16+), and LIN28A (D16+) across the time points and their respective subgroups. Lines represent the median expression. (C) Left: Trajectory of reprogramming cells constructed from the 10X scRNA-Seq libraries based on DDRTree dimension reduction. Colors represent time points. Right: Pseudotime calculated by Monocle. Color indicates pseudotime. (D) Stacked columns indicating the distribution of reprogramming time points across the pseudotemporal states. Colors represent time points. (E) Superimposition of D8 subgroups (left) and D16+ subgroups (right) on the trajectory of reprogramming. Colors represent subgroups. (F) Stacked columns revealing the distribution of D8 subgroups across the pseudotemporal states. Colors represent D8 subgroups, and gray color indicates cells of the other time points. (G) Superimposition of expression of the D8 subgroup-specific genes (RFC3 and GDF3) and the D16+ subgroup–specific genes (NANOG, MMP2, and LIN28A) on the reprogramming trajectories.
We then performed differential gene expression (DGE) analysis to evaluate the subgroup-specific genes (fig. S2D and table S1). Among D2 subgroups, G3 cells had the most distinct transcriptomic profile with exclusive expression of a remarkable number of genes (fig. S2D and table S1). Whereas the majority of D2 G1-2 genes were expressed highly in D16+ G2 but not in D16- cells, suggestive of their higher reprogramming propensity (fig. S2D and table S1). It includes cell cycle–associated genes, such as CDK1, implicating the importance of cell cycle at the early stage of reprogramming, which is in agreement with the previous study (Fig. 2B) (14). Among D8 subgroups, G1- and G1-2–specific genes were expressed highly in BJ and D16− but not in D16+ cells, including genes associated with extracellular matrix organization and collagen catabolic process—such as JUNB, LUM, COL1A1, and COL6A3—which were reported as barriers of reprogramming (fig. S2D and table S1) (15). Whereas the opposite trend was observed for D8 G2-3– and G3-specific genes (fig. S2D and table S1). For instance, DNA replication factor RFC3 was vastly expressed in D8 G2-3 cells, and pluripotent marker GDF3 was specifically expressed in the D8 G3 cells (Fig. 2B and table S1). Supporting this notion, stemness analysis revealed that D8 G1-2–specific genes significantly associated with the differentiated lineages, whereas D8 G3-specific genes exhibited a high stemness score for PSCs (fig. S2E). Likewise, in agreement with the RCA correlation, D16+ G1-specific genes were also abundantly expressed in the D16− cells, containing genes involved in extracellular matrix organization such as MMP2, suggesting that D16+ G1 cells were at most partially reprogrammed (Fig. 2B, fig. S2D, and table S1). On the other hand, epithelial genes and pluripotent genes including CDH1, NANOG, and LIN28A were specifically expressed in D16+ G2 cells (Fig. 2B and table S1). In line with this, stemness analysis uncovered the association of D16+ G1-specific genes to the differentiated lineages and D16+ G2 genes to pluripotency (fig. S2F).
To test the diverse reprogramming potentials of D8 subgroups, we then correlated them with the subgroups of various time points (fig. S2G). D8 G3 highly correlated with D16+ G2, whereas D8 G1 cells strongly correlated with D16− cells and cells of early time points (BJ and D2). On the other hand, D8 G2 cells represented an intermediate state, which moderately correlated with both cells of early time points and D16+/−.
Pseudotemporal trajectory of the reprogramming process
We next analyzed 10X libraries to construct the trajectory (16, 17) of cellular reprogramming, which consisted of nine states and four branching events (Fig. 2C). Notably, pseudotime highly correlated with the actual reprogramming time points (Fig. 2, C and D). We then asked how RCA subgroups distribute across the pseudotemporal trajectory. To answer, we identified the RCA subgroups from 10X libraries in the same manner as Fluidigm scRNA-Seq libraries and superimposed the RCA subgroup identities on the trajectory map (Fig. 2E and fig. S3A). RCA subgroups identified from two datasets not only shared the correlation patterns to various lineages but also displayed statistical similarity to each other (fig. S3B). Notably, the majority of the D2 G1-3 cells were found in state 3 (95% of G1, 65% of G2, and 80% of G3), whereas G4 cells scattered across the early states (fig. S3C). Combined RCA analysis demonstrated that D2 G1-2 cells clustered closer to the D8 G2 cells and correlated stronger to the embryonic stem cell (ESC) fate than the other D2 subgroups, further substantiating their higher reprogramming propensity (fig. S3, D and E). As for D8 subgroups, G1 cells enriched across the states other than state 9 (successfully reprogrammed), whereas G3 cells were mostly found in state 9 (Fig. 2, E and F). On the other hand, D8 G2 cells mainly belonged to states 4 and 9 (Fig. 2, E and F). Intriguingly, state 4 (627 cells) composed almost entirely of D8 G2 (544 cells) (Fig. 2, E and F). Expectedly, D16+ G2 cells were the major constituents of state 9, whereas cells of D16+ G1 were enriched in both state 8 (non-reprogrammed) and state 9, corroborating their partial or non-reprogrammed identities (Fig. 2E and fig. S3F). Further, subgroup-specific markers were expressed differentially along the pseudotime axis (Fig. 2G). RFC3 (D8 G2-3), GDF3 (D8 G3), and NANOG and LIN28A (D16+ G2) were expressed highly in the cells on the successful reprogramming trajectory, whereas MMP2 (D16+ G1) showed the opposite trend (Fig. 2G).
In addition to RCA, we have also examined the lineages reported for mouse reprogramming cells in our system (fig. S3G) (18). To do so, we first determined the signature genes for the corresponding lineages by mining the related human studies and databases (19–23) (http://biocc.hrbmu.edu.cn/CellMarker/). On the basis of expression of the signature genes, reprogramming cells were then annotated with lineage identities. Similar to mouse reprogramming, fibroblast identity fainted as human reprogramming progressed, whereas pluripotent identity arose at the late stage (fig. S3G). Notably, during early stage of mouse reprogramming, mesenchymal-to-epithelial transition (MET) event caused the bifurcation of early population into stromal and epithelial cells (18). However, because of the differences in kinetics and mechanisms, MET is not an early event in human reprogramming (19). Supporting this, we also observed down-regulation of mesenchymal genes at the early stage, whereas most of epithelial genes were expressed only at the late stage of human reprogramming (fig. S1F). Consistent with this notion, epithelial lineage was only enriched in states 8 and 9 comprising of late time points (D12 and D16) in human reprogramming (fig. S3G). In addition, trophoblast and neural lineages appeared at the intermediate-late stage of mouse reprogramming (18). Likewise, we have also observed enrichment of trophoblast and neural lineages in cells of states 8 and 9 (D12 and D16−) (fig. S3G). Intriguingly, we found that some of state 8 cells (D12 and D16-) resembled immune lineage (fig. S3G). Together, these analyses provide a plethora of data, allowing to identify the cell fates and the crucial modulators affecting the reprogramming trajectory at an unprecedented single-cell resolution.
Fluorescent probes screen for the early-intermediate reprogramming cells
To enrich for the intermediate cells with high reprogramming potential, we conducted a screen for a library of 34 fluorescent dyes generated using a diversity-oriented fluorescence library approach (DOFLA) (fig. S4A) (24). Briefly, staining signals of fluorescent probes were measured in the intermediate D8 cells cultured with reprogramming medium supplemented with either dimethyl sulfoxide (DMSO) (control) or transforming growth factor–β (TGF-β) inhibitor (A83-01), which was shown to markedly accelerate the kinetics of the reprogramming process (25). Because of the inherently low reprogramming efficiency of BJ fibroblast, staining signals of the fluorescent probes were expected to be weak in the D8 cells cultured with the control medium, otherwise, the probes were considered to display unspecific staining. In addition to that, dyes with differential staining signals between the reprogramming cells cultured with the control and A83-01 medium were identified to be capable of distinguishing early reprogrammed cells. Among the top-ranked dyes, we selected three representative dyes for functional validations, namely, BDD1-A2, BDD2-A6, and BDD2-C8 (fig. S4, A to C). Their staining signals colocalized with TRA-1-60 (fig. S4B). Indeed, the top 10% of D8 cells stained with the candidate probes gave rise to a higher number of TRA-1-60+ colonies than that of the bottom 10% of cells (fig. S4D). Among these dyes, BDD2-C8 consistently distinguished the early reprogrammed cells induced from both BJ and MRC5 fibroblasts (fig. S4, D and E). In addition, BDD2-C8 also precisely captured changes in reprogramming efficiency upon depletion of the key modulators (fig. S4F) (6).
We next prepared 192 capture-based scRNA-Seq libraries for the top 10% and bottom 10% of D8 cells stained with BDD2-C8 (D8BDD2-C8 high and D8BDD2-C8 low). In the ensuing RCA analysis, D8BDD2-C8 high and D8BDD2-C8 low cells clustered close to the D8 G2-3 and G1, respectively, which was further substantiated by Pearson correlation analysis (fig. S4, G to I). Gene Ontology (GO) terms enriched by D8BDD2-C8 high-specific genes were related to cell cycle, embryo development, and stem cell population maintenance; whereas D8BDD2-C8 low genes were predominantly represented by epithelial to mesenchymal transition (EMT), extracellular matrix organization, and development processes (fig. S4J and table S1). In terms of structural complexes, D8BDD2-C8 low genes were specifically enriched in the endoplasmic reticulum (ER) lumen and Golgi (fig. S4K and table S1). Notably, BDD2-C8 localized in the ER and Golgi (fig. S4L). On top, secretory genes were found to be highly expressed in the D8BDD2-C8 low cells implicating its active ER-Golgi secretion pathway (fig. S4M). Depletion of these genes indeed resulted in the retention of BDD2-C8 (fig. S4, N and O). Together, BDD2-C8 may be actively effluxed from BJ and the non-reprogrammed cells (D8 G1) but retained in the pluripotent cells and intermediate cells with high reprogramming potential, due to their differential ER-Golgi secretion activities.
Surface markers to enrich for the early-intermediate cells with diverse reprogramming potentials
Through the analysis of scRNA-Seq libraries, we have also identified surface markers to enrich for the intermediate cells with varying reprogramming potentials. The following criteria were applied to shortlist the surface markers: (i) differentially expressed in D8 subgroups; (ii) highly expressed in at least one of the D8 subgroups, for the ease of downstream validations; (iii) representatives of each category with similar expression dynamics across the reprogramming time points and the respective subgroups. Accordingly, we shortlisted CD13, CD44, and CD201 for further validations in the following study.
In general, the selected surface markers displayed reduced expression with the progression of reprogramming, except for PROCR (CD201) which demonstrated the trend in the capture-based scRNA-Seq libraries exclusively (Fig. 3A and fig. S5, A and B). This is an example indicating the importance of using both scRNA-Seq strategies for deciphering the molecular changes in reprogramming. Notably, the surface markers exhibited higher expression in D8 G1/G2 and D16− cells than in D8 G3 and D16+ cells, respectively (fig. S5A). Expression dynamics of the surface markers were validated by time-course fluorescence-activated cell sorting (FACS) analysis (Fig. 3B). We then examined the correlation between BDD2-C8 and the identified surface markers. Expectedly, D8BDD2-C8 high cells demonstrated lower levels of CD13, CD44, and CD201 (Fig. 3C and fig. S5C). Notably, CD201 was expressed in the BDD2-C8–sorted cells with a subtler difference, which could possibly be explained by the high expression of CD201 in the majority of top 10% BDD2-C8 cells (G2-like) and the bottom 10% BDD2-C8 cells (G1-like). Furthermore, D8 cells negatively stained for CD13 and CD44 exhibited lower expression of mesenchymal markers but higher epithelial and pluripotency genes, including the D8 G3 marker GDF3 (Fig. 3D). Different from than in CD13- and CD44-sorted cells, the majority of MET genes expressed at comparable levels in the CD201-sorted cells, in line with the distinct observations seen earlier for the correlation between the surface markers and BDD2-C8 staining signals (Fig. 3D). Nonetheless, D8 populations negatively stained for all the shortlisted surface markers gave rise to more TRA-1-60+ colonies, indicating their capability in isolating the early reprogrammed cells with high stemness feature (Fig. 3E).
Fig. 3. Identification of surface markers for the early-intermediate reprogramming cells.
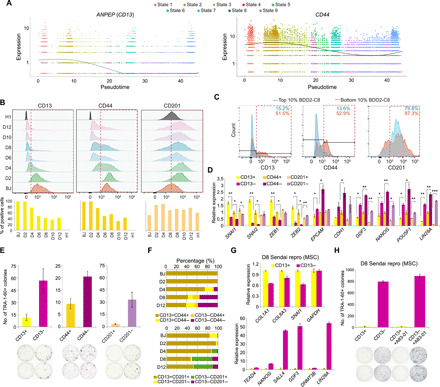
(A) Dotplots indicating the expression of ANPEP and CD44 along the pseudotime. Smooth lines are composed of multiple dots representing the mean expression level at each pseudotime, regardless of the state. (B) Stacked histograms (top) showing the fluorescence intensities (x axis) of the surface markers in the cells indicated on the left. Red dotted boxes highlight the positively stained populations. Quantifications are shown below. (C) Overlaid histograms showing the staining signals of the surface markers in the top 10% and bottom 10% BDD2-C8–stained cells. Red dotted boxes highlight the positively stained populations. The numbers on top indicate the percentages of positively stained cells. (D) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) measuring the relative expression levels in the D8-sorted cells. n = 2; error bar indicates SD. * indicates P < 0.05; ** indicates P < 0.005; *** indicates P < 0.0005. (E) Quantification of TRA-1-60+ colonies yielded from the D8-sorted cells. Representative images are shown below. n = 2; error bar indicates SD. (F) Bar charts showing the distribution of costaining signals of the surface markers across the cells of various reprogramming time points. (G) qRT-PCR exhibiting the relative expression of the collagen/mesenchymal genes (top) and pluripotent genes (bottom) in the D8 CD13-sorted cells induced from MSC using Sendai virus. n = 2; error bar indicates SD. (H) Quantification of TRA-1-60+ colonies yielded from D8 CD13-sorted cells induced from MSC using Sendai virus (top). Representative images are shown below. n = 2; error bar indicates SD.
To verify the earlier assumption for whether the different patterns observed for CD201, we performed time-course costaining for the identified surface markers and analyzed using FACS. In line with the earlier results, CD13 and CD44 markers showed extensive overlaps across the time points (Fig. 3F and fig. S5D). Proportion of population with double-negative staining for CD13 and CD44 increased with the advance of reprogramming, whereas the reverse trend was observed for the population with the double-positive staining profile (Fig. 3F and fig. S5D). On the other hand, a substantial number of reprogramming cells exhibited inconsistent staining signals for CD13 and CD201 (CD13−CD201+), possibly representing D8 G2-like cells, the percentage of which rose as reprogramming progressed (Fig. 3F and fig. S5E). Together, other than the population labeled by CD13 and CD44, CD201 also marks a distinct population of intermediate cells with lower reprogramming propensities.
We have also tested the applicability of the surface markers in an alternative reprogramming system induced from MSCs using Sendai viruses. As the kinetics of Sendai virus induction is similar to that of lentivirus, experiments were conducted in the D8 intermediate reprogramming cells (Fig. 3, G and H, and fig. S5, F to H). FACS analysis indicated that CD13, CD44, and CD201 were abundantly expressed in both the parental MSCs and D8 cells (fig. S5F). However, few cells showed negative staining for CD201, indicating its inability to distinguish the early reprogrammed cells induced by Sendai virus (fig. S5F). Similar to the lentivirus-mediated BJ reprogramming, costaining of CD13 and CD44 in D8 MSCs demonstrated substantial overlapping signals (fig. S5G). Notably, D8 CD13− MSCs exhibited up to 60-fold higher expression of GDF3 and other pluripotency genes than the CD13+ cells (Fig. 3G and fig. S5H). Moreover, D8 CD13− MSCs resulted in a remarkably higher reprogramming efficiency with or without the aid of TGF-β inhibition (Fig. 3H and fig. S5H). In summary, expression patterns of CD13 and CD44 were reproduced in the alternative Sendai reprogramming system induced from MSCs. Furthermore, CD13 effectively marked the cells with a high reprogramming efficiency.
Refined classification of the intermediate reprogramming population
To further decipher the heterogeneity within the intermediate reprogramming cells, we prepared 10X libraries for D8 cells sorted with CD13. Most of the libraries passed the QC thresholds (fig. S6A). Expectedly, clustering showed two distinct groups with differential CD13 expression (Fig. 4A and fig. S6B). The majority of the genes that were highly expressed in CD13+ cells were correspondingly expressed in the D8BDD2-C8 low cells and vice versa, which is in agreement with the FACS correlation between the staining signals of BDD2-C8 and CD13 (fig. S6C). To finely characterize the subpopulations, we performed Seurat analysis for 10X libraries of D8 CD13-sorted cells and identified eight clusters, among which clusters 5 to 8 were mainly composed of CD13+ cells (Fig. 4B and fig. S6D). We then performed RCA analysis for the CD13-sorted 10X libraries (Fig. 4C). In general, CD13 clusters exhibited similar correlation patterns to the D8 RCA subgroups, in terms of the mesenchymal lineages and pluripotency (Fig. 4C). For instance, distinctly separate CD13+ clusters (5 to 8) demonstrated high correlation to the mesenchymal lineages, but not to ESCs, resembling the correlation pattern of D8 G1 cells (Fig. 4C). On the contrary, closely clustered CD13− clusters (1, 3, and 4) showed the opposite pattern, similar to that of D8 G3 (Fig. 4C). On the other hand, cells of cluster 2 exhibited a transitional profile (Fig. 4C). In addition, we have also performed mathematical imputation using MAGIC (26) for a pairwise comparison between CD13 and GDF3, the D8 G3 marker. A strongly negative correlation was observed between the expression of CD13 and GDF3 in D8 cells (Fig. 4D). Notably, cells of cluster 7 lowly expressed both CD13 and GDF3, therefore, were found at the bridge of the correlation plot, suggesting their intermediary profiles (fig. S6, D and E).
Fig. 4. Refined classification and enrichment of early-intermediate reprogramming cells.
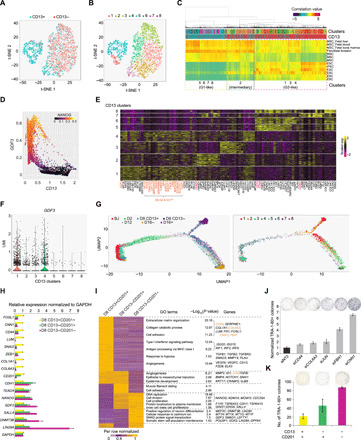
(A and B) t-SNE plots indicating the CD13 antigen profiles (A) and Seurat clusters (B) of the D8 CD13-sorted 10X libraries. (C) RCA clustering of the D8 CD13-sorted 10X libraries. (D) MAGIC plot showing the correlation between CD13 and GDF3. Colors represent the expression levels of NANOG. (E) Heatmap showing the DEGs of CD13 clusters. Genes highlighted in orange are expressed highly in D8 G2 and G3. (F) Violin plot demonstrating the expression of GDF3 across the clusters. (G) Left: Trajectory constructed by the 10X scRNA-Seq libraries of various time points and D8 CD13-sorted cells. Right: Superimposition of the CD13 clusters. (H) qRT-PCR showing the relative expression in the D8 CD13 & CD201-sorted cells. n = 2; error bar indicates SD. (I) Left: Heatmap showing the DEGs of D8 CD13 & CD201-sorted cells determined from their bulk RNA-Seq libraries. GO terms and the associated genes are indicated on the right. (J) Normalized TRA-1-60+ colonies upon knockdown of genes highly expressed in CD13+CD201+ cells at D5 of reprogramming. Representative images are shown above. n = 3; error bar indicates SD. (K) Quantification of TRA-1-60+ colonies yielded from the D8 CD13 & CD201-sorted cells. Representative images are shown above. n = 2; error bar indicates SD. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; MHC, major histocompatibility complex.
RCA and MAGIC findings were corroborated by DGE analysis for CD13 clusters (Fig. 4E). Briefly, CD13+ clusters 5, 6, and 8 extensively shared DEGs, including mesenchymal gene SNAI2 and genes associated with collagen catabolic process, whereas CD13− clusters 1, 3, and 4 highly expressed D8 G2-3–specific genes, among which clusters 1 and 4 exhibited the highest expression of D8 G3 marker GDF3 (Fig. 4, E and F). Furthermore, the pseudotemporal analysis for 10X libraries of D8 CD13-sorted cells and cells of the other time points revealed the localization of CD13+ clusters 5, 6, and 8 and CD13− clusters 1, 3, and 4 at the early and late pseudotime, respectively (Fig. 4G). In line with their high GDF3 expression, cells of clusters 1 and 4 concentrated at the branch shared by D16+ cells, whereas cells of cluster 3 were found at the unsuccessful reprogramming branch (Fig. 4G). On the other hand, clusters 2 and 7 shared DEGs, including the somatic gene JUNB and surface marker CD201 (Fig. 4E). Consistent with the FACS correlation results, MAGIC analysis indicated the extensive correlation between CD13 and CD44 across the clusters but not between CD13 and CD201 (Fig. 4E and fig. S6, F to H). The inconsistency between CD13 and CD201 was mostly contributed by the intermediary clusters 2 and 7, which displayed low CD13 but high CD201 expression (Fig. 4E and fig. S6, D and I). Based on the evidences gathered thus far, we hypothesized that the dual sorting with CD13 and CD201 surface markers would allow us to enrich for the successfully reprogrammed early-intermediate cells with higher purity.
To verify, we then categorized D8 cells into double-negative (CD13−CD201−), double-positive (CD13+CD201+), and intermediary (CD13−CD201+) cells, which were then subjected to the transcriptomic analysis. CD13+CD201+ cells highly expressed the fibroblast-associated genes, mesenchymal genes, and genes associated with extracellular matrix and cell adhesion (Fig. 4, H and I, and table S1). On the contrary, CD13−CD201− cells highly expressed genes related to pluripotency, epithelial lineage, cell division, and stem cell population maintenance (Fig. 4, H and I, and table S1). However, CD13−CD201+ cells exhibited an intermediate transcriptional profile (Fig. 4, H and I, and table S1). Apart from this, genes highly expressed in CD13+CD201+ cells were mostly enriched in the D16− but not in the D16+ cells, whereas the opposite was observed for CD13−CD201− specific genes (fig. S6K and table S1). Moreover, depletion of genes highly expressed in the CD13+CD201+ population resulted in higher numbers of reprogrammed colonies (Fig. 4J). Notably, D8 CD13−CD201− population gave rise to the highest number of TRA-1-60+ colonies, followed by CD13−CD201+ (intermediate) and CD13+CD201+ cells (lowest) (Fig. 4K). The presence and reprogramming potentials of the three distinct D8 populations were validated in an alternative BJ reprogramming system induced using Sendai viruses (fig. S6, L to N). Together, concurrent use of CD13 and CD201 antibodies enabled us to dissect the precise intermediate populations differentially poised for successful reprogramming.
Stage-specific regulatory networks of cellular reprogramming
TFs define the cell-selective regulatory networks underlying the cellular identities and functions (27). However, the stage-specific core regulatory networks of the human cellular reprogramming remain elusive. To this end, we performed DGE analysis for TFs across the pseudotemporal states, which were then categorized as Early Silenced, Late Silenced, Transient, Early Expressed, and Late Expressed (Fig. 5A, fig. S7A, and table S1). Notably, many TFs exhibited similar expression trends in the D8 RCA subgroups and the D8 BDD2-C8– and CD13 & CD201-sorted cells. For example, Early Silenced TFs (e.g., FOSL1, CREB3L1, AHRR, DRAP1, and ELL2) were mostly silenced in D8 BDD2-C8+ and D8 CD13− cells, including CD13−CD201+ and CD13−CD201− cells (Fig. 5A and fig. S7B). Comparatively, most of Late Silenced TFs were still highly expressed in the D8 BDD2-C8+ and not differentially expressed among D8 CD13 & CD201-sorted populations (Fig. 5A and fig. S7B). The majority of Transient TFs exhibited higher expression in the D8 CD13+CD201+ and the D8 CD13−CD201+ populations, whereas some lineage-associated factors, such as, HAND1 (mesoderm) and ASXL3 and NEUROG2 (neuroectoderm) were found to be highly expressed in the D8 CD13-CD201+ or the D8 CD13-CD201- cells (Fig. 5A and fig. S7C). Contrastingly, Early Expressed TFs demonstrated higher expression in the D8 CD13−CD201− and the D8 CD13−CD201+ (Fig. 5A and fig. S7D). Notably, the D8 CD13−CD201− cells expressed the highest levels of Late Expressed TFs, such as PRDM14, DNMT3B, and LHX6 (Fig. 5A and fig. S7D).
Fig. 5. Stage-specific TF regulatory networks of reprogramming.
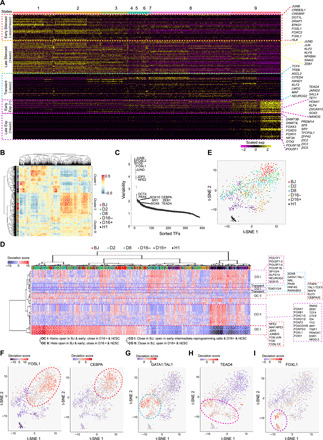
(A) Heatmap showing the TFs’ expression across the pseudotime states. Color code on top represents the pseudotime states. Representative TFs of each category are listed on the right. (B) Correlation between scATAC-Seq libraries based on the calculated JASPAR motif deviations in the HARs. Side color bar indicates time points of the scATAC-Seq libraries. (C) Plot indicating the significantly variable motifs in terms of accessibility in the scATAC-Seq libraries. y axis represents the variability score assigned to each JASPAR motif, whereas x axis represents the motif rank. (D) scATAC-Seq heatmap based on the deviation scores of the significantly variable JASPAR motifs. Color code on top represents time points. Motifs were classified to three major types according to the dynamics of accessibility across the time points. (E) t-SNE plot of scATAC-Seq libraries based on the deviation scores of JASPAR motifs. (F to I) Superimposition of motif enrichment scores for OC motifs FOSL1 and CEBPA (F), Transient motif GATA1:TAL1 (G), CO motifs: type I-TEAD4 (H); type II-FOXL1 (I) on the t-SNE plot. Colors indicate the motif accessibility levels.
To investigate how the regulatory TFs accessed their genomic targets, we then analyzed time-course scATAC-Seq libraries of reprogramming cells (Fig. 1A). Both batches of libraries showed similar promoter accessibility profiles (fig. S7E). To integrate scATAC-Seq and scRNA-Seq datasets, we applied Seurat, which predicts the corresponding scRNA-Seq cluster for each scATAC-Seq library based on the cluster-specific gene activities and co-embeds both datasets in the same low-dimensional space for visualization (fig. S7F) (28). In general, scATAC-Seq and scRNA-Seq libraries were well blended, and the predicted time points for scATAC-Seq libraries demonstrated a good correlation with the actual time points (fig. S7F). Next, correlation of scATAC-Seq libraries among themselves resulted in three major clusters (Fig. 5B). The early cells consisting mostly of BJ and D2 clustered together (cluster II), while D8, D16+, and H1 cells shared a similar accessibility profile (cluster I) (Fig. 5B). A third cluster composed mainly of D8 and D16− cells (cluster III) (Fig. 5B). We then used chromVAR (29) to identify the TFs determining the variable epigenome accessibility (Fig. 5C). Notably, chromatins with motif sequences of TFs—such as FOSL1 and its partners, CEBPA, ZEB1, PAX6, SOX8, SOX10, OCT4, and TEAD4—were found to be the most heterogeneous in terms of accessibility across the various time points of reprogramming cells and human embryonic stem cells (hESCs) (Fig. 5C and table S1).
According to the dynamics of motif accessibility, TFs were then categorized to OC (Open in BJ but Close in D16+ and hESC), Transient, and CO (Close in BJ but Open in D16+ and hESC) (Fig. 5D). In particular, OC TFs belonged mostly to the FOS-JUN-AP1 complex, such as FOSL1 and JDP2 (Jun dimerization protein 2) (Fig. 5, D to F, and fig. S7G). Intriguingly, motifs of lineage specifiers [mesendoderm (ME): GATA1, SOX8, and HNF4G; ectoderm (ECT): PAX6, NRL, and RXR) were found to be accessible only in the intermediate reprogramming cells but not in the hESCs (Fig. 5, D, E, and G, and fig. S7H]. This observation corroborated the model of counteracting lineage specification networks underlying the induction of pluripotency (30). On the contrary, CO type I TFs (e.g., TEAD4 and POU5F1) exhibited accessibility starting from the D8 reprogramming cells, while CO type II TFs (e.g., FOXL1, TCF4, and YY2) demonstrated accessibility in D16+ cells and ESCs exclusively (Fig. 5, D, E, H, and I; and fig. S7, I to K). Notably, TFs of FOX family and YY1 were previously reported to be important for reprogramming (31, 32). Independently, we used single-cell regulatory network inference and clustering (SCENIC) analysis to infer the regulon activities of TFs, based on the coexpression of TFs and the potential target genes containing TF motif sequences (33). Corroborating our earlier results, Silenced and OC TFs showed decrease in their regulon activity as reprogramming progressed, whereas most of Expressed and CO TFs displayed the opposite dynamics (fig. S7L). Likewise, regulon activity of the Transient TFs was observed only in the intermediate cells. Together, these data represent the compendium of TFs, which regulate the networks of downstream key modulators in the cellular reprogramming process.
Identification of key regulators for the intermediate stage of reprogramming
To deduce TFs essential for the intermediate cells to acquire pluripotency, we analyzed D8 scATAC-Seq libraries individually (fig. S8A). Intriguingly, the most variable motifs of D8 cells belonged to the FOS-JUN-AP1 and TEAD families (Fig. 6A and table S1). Notably, D8 cells were either accessible for FOSL1-JUN-AP1 or TEAD4 motifs (Fig. 6B and fig. S8B). In addition, FOSL1 and TEAD4 displayed a contrasting expression pattern and regulon activity during reprogramming (Fig. 6, C and D, and fig. S8, C and D). In D8, FOSL1 and TEAD4 were exclusively expressed in the CD13+ and CD13− cells, respectively (Fig. 6C).
Fig. 6. TFs contributing to the heterogeneity in chromatin accessibility of the intermediate reprogramming cells.
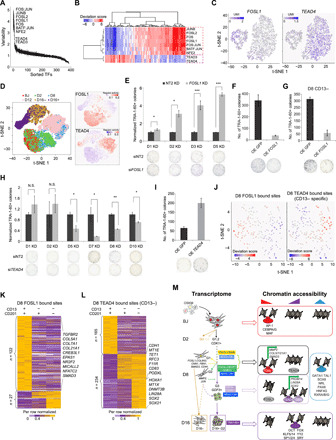
(A) Plot indicating the significantly variable motifs in terms of accessibility in D8 cells. (B) Clustering of D8 scATAC-Seq libraries based on the accessibility of variable motifs. (C) Expression of FOSL1 and TEAD4 in the D8 CD13-sorted cells. (D) t-SNE plot based on the regulon activity matrix (left) and superimposition of regulon activities for FOSL1 and TEAD4 (right). (E) The number of normalized TRA-1-60+ colonies upon knockdown (KD) of FOSL1 at the indicated reprogramming time points. Representative images are shown below, n = 3. Error bar indicates SD. * indicates P < 0.05; *** indicates P < 0.0005. (F and G) Quantification of TRA-1-60+ colonies upon overexpression (OE) of FOSL1 in D5 cells (F) and D8 CD13− cells (G). Representative images are shown below, n = 3. Error bar indicates SD. (H) The number of normalized TRA-1-60+ colonies upon knockdown of TEAD4 at the indicated reprogramming time points. Representative images are shown below, n = 2. Error bar indicates SD. * indicates P < 0.05; ** indicates P < 0.005; N.S. indicates not significant. (I) Quantification of TRA-1-60+ colonies upon overexpression of TEAD4. Representative images are shown below, n = 3. Error bar indicates SD. (J) Clustering of D8 scATAC-Seq libraries based on the accessibility of FOSL1 and TEAD4 bound sites. (K and L) Heatmaps showing the expression of functional FOSL1 (K) and TEAD4 targets (L) in the D8 CD13 & CD201-sorted cells. (M) Proposed model of the study.
Given its expression and motif accessibility at the early stage of reprogramming, FOSL1 was hypothesized to act as a roadblock. To verify, reprogramming efficiency was assessed upon the depletion of FOSL1 by small interfering RNA (siRNA), which showed slight effects to cell proliferation and lasted for about 4 days (fig. S8, E to G). Depletion of FOSL1 at day 1, 2, 3, and 5 post-OSKM induction resulted in an increased reprogramming efficiency (Fig. 6E). Specificity of the FOSL1 siRNA construct was validated using a mutant construct, which showed no phenotypic effects (fig. S8, H and I). Depletion of FOSL1 in an alternative Sendai virus reprogramming cells reproduced the phenotypic change (fig. S8J). Conversely, ectopic expression induced a drastic reduction in the number of reprogrammed colonies (Fig. 6F). Noteworthy, high reprogramming propensity of CD13− cells was negated by the elevated FOSL1 level (Fig. 6G).
Contrastingly, we posit that TEAD4 might serve as an effector. To prove, efficiency and specificity of siTEAD4 were first tested. Knockdown effect of siTEAD4 lasted for around 5 days in hESCs (fig. S8K). When knockdown was performed on D5 cells, TEAD4 expression did not restore after siRNA transfection, which could be due to the perpetuations of the non-reprogrammed state introduced by siTEAD4 (fig. S8L). Notably, depletion of TEAD4 only from D5 onward resulted in reduced number of reprogrammed colonies (Fig. 6H). This established the vital role of TEAD4 at the intermediate-late stages of reprogramming. Specificity of siTEAD4 was affirmed by the mutant construct showing no phenotypic change (fig. S8, M and N). In contrast, overexpression of TEAD4 resulted in a notable increase in the number of reprogrammed colonies (Fig. 6I). Depletion of TEAD4 revoked the reprogramming potential of D8 CD13− cells (fig. S8O). In addition, phenotypic changes of siTEAD4 in the reprogramming cells was not through demolishing pluripotency (fig. S8, P to R). Collectively, these analyses illustrate that the pivot from FOSL1 to TEAD4-directed regulatory network is essential for successful reprogramming.
Mechanistic roles of FOSL1 and TEAD4 in cellular reprogramming
To find the direct targets of FOSL1, we prepared ChIP-Seq libraries for D8 cells, which demonstrated the expected genomic distribution profile and enriched with FOSL1 motif and its regulatory partners (fig. S9, A and B). We also investigated the genomic binding profile of TEAD4 in the D8 CD13-sorted cells (fig. S9, C and D). CD13+ TEAD4 bound sites (53.6%) were shared by CD13− cells, whereas CD13− ChIP-Seq libraries detected 1.7-fold higher numbers of TEAD4 bound loci than that of CD13+ cells (18,550 versus 10,986 sites) (fig. S9D). In addition, CD13− specific sites showed greater enrichment of TEAD4 motif and exclusive enrichment of pluripotency-associated OCT4-SOX2-TCF-NANOG motif, indicating the differential regulatory role of TEAD4 in the D8 CD13-sorted cells (fig. S9E).
We next clustered BJ, D16−, and D16+ scATAC-Seq libraries, based on the accessibility of FOSL1 and TEAD4 bound sites (fig. S9F). Notably, FOSL1-specific sites exhibited higher accessibility in BJ and D16− cells, whereas CD13− specific and CD13 common TEAD4 bound sites were mostly accessible in D16+ cells (fig. S9, F and G). These differential accessible sites were annotated as the functional FOSL1 and TEAD4 targets (table S1). Similar to the motif enrichment pattern, most of the D8 cells were accessible for either FOSL1 (“FOSL1 ChIP only” cells) or CD13− TEAD4 bound sites (“TEAD4 ChIP only” cells) (Fig. 6J and fig. S9, H to J). We next asked whether their binding has any consequence to the transcription of target genes. To this end, we analyzed the expression of functional FOSL1 and TEAD4 targets in the D8 CD13 & CD201-sorted cells. Most of the D8 FOSL1 targets (e.g., TGFBR2, SMAD3, and COL5/7/21A1) were expressed highly in the CD13+CD201+ cells (Fig. 6K). In contrast, D8 CD13−CD201− cells demonstrated high expression of the TEAD4 targets, including key pluripotent genes such as DNMT3B, LIN28A, SOX2, and PODXL, which codes for TRA-1-60 (Fig. 6L). This was further substantiated at the single-cell resolution by the coupled NMF (nonnegative matrix factorizations) analysis for D8 scRNA-Seq and scATAC-Seq libraries, which established regulatory connectivity between the accessible chromatin regions and expression of target genes (fig. S9K). Notably, cluster 1 composed of TEAD4 ChIP-only cells (scATAC-Seq) and D8 RCA G3 cells (scRNA-Seq), while cluster 2 composed of FOSL1 ChIP-only cells (scATAC-Seq) and D8 RCA G1 (scRNA-Seq) cells (fig. S9K). Differential analysis demonstrated that TEAD4 and its targets, such as DNA methyltransferase TET1 and epithelial gene CDH1, were highly accessible and expressed in the cluster 2 cells, whereas FOSL1 and its targets, such as MMP2 and SMAD3, displayed the opposite trends (fig. S9, K and L). Besides, interactome analysis revealed that cluster 2 genes related to ER-Golgi transport and extracellular matrix organization, including FOSL1 targets MMP2 and several collagen genes (fig. S9M). Notably, downstream targets of FOSL1 and TEAD4 were themselves modulators of reprogramming. For instance, knockdown of FOSL1 bound genes, MMP2 and SMAD3, resulted in higher numbers of reprogrammed colonies (fig. S9N). Conversely, depletion of TEAD4 targets, PRDM14 and SOX2, showed the opposite effect (fig. S9O).
DISCUSSION
In this study, we presented the single-cell roadmap of the human cellular reprogramming process, which reveals the diverse cell fate trajectory of individual reprogramming cells (Fig. 6M). D2 cells consist of four subgroups, out of which CDK1+ cells showed greater propensity for reprogramming. Among the three subgroups of D8 cells, G1 represents the unsuccessful reprogramming cells, whereas G3 cells with GDF3 expression are highly primed for successful reprogramming. GDF3 was previously shown to be expressed in pluripotent stem cells and played a role in regulating cell fate via bone morphogenetic protein signaling inhibition (34). G2 is the intermediary group between G1 and G3. In-house–developed fluorescent probe (BDD2-C8) and the identified surface markers (CD13, CD44, and CD201) enable the segregation of the heterogeneous population based on their reprogramming potential. Noteworthy, combinatorial use of the surface markers enables a more refined segregation. The toolkits to decipher the intermediate cells with different stemness capacity will help deepen our understanding of the mechanisms of the reprogramming process. In addition, the ability to enrich for early reprogramming cells will help increase the success rate of iPSC generation from the cell lines or patient-derived primary cells, which are refractory to reprogramming.
Moreover, TF analysis reveals the stage-specific regulatory networks of reprogramming. Accessible regions with the motifs of FOS-JUN-AP1 and CEBPA are rapidly closed upon induction, which were reported to act as repressors in mouse reprogramming (35). An earlier report showed that FOSL1 lost binding from many of the genomic regions as early as day 2 in mouse reprogramming (35). Our study reveals that, in the human system, FOSL1 regulates myriad of genes, which serve as barriers of reprogramming, including MMP2, SMAD3, TGFBR2, and collagen genes. Notably, chromatin regions with the motifs of lineage TFs are open in the intermediate cells, which might be due to the induction of ME and ECT lineages driven by OCT4 and SOX2 (36). This is further supported by the replacement of OCT4 and SOX2 with the ME and ECT lineage specifiers, for both mouse and human reprogramming (30, 37). We unravel the transitory epigenetic accessibility directed by the lineage TFs, which contribute extensively to the diverse cell fate potentials observed during cellular reprogramming. We describe the crucial switch from a FOSL1 to a TEAD4-centric expression, which collectively regulates genomic accessibility, cell lineage transcription program, and network of functional downstream modulators favoring the acquisition of the pluripotent state.
MATERIALS AND METHODS
Experimental design
To deconstruct the heterogeneity, reprogramming cells were collected at various time points after induction and subjected for single-cell next-generation sequencing (NGS) library preparation. Reprogramming was induced from BJ fibroblast with polycistronic O2S, K2M vectors delivered using lentivirus. BJ, D2, D8, D16 TRA-1-60+, and D16 TRA-1-60− were harvested for scRNA-Seq and scATAC-Seq library preparation on the Fluidigm C1 platform. The same series of time points, with the addition of D12, was collected for scRNA-Seq library preparation using the droplet-based 10X Genomics.
scRNA-Seq library construction (Fluidigm)
The scRNA-Seq libraries were prepared following the published protocol with minor modifications (11). Briefly, cells undergoing reprogramming were trypsinized into single cells at each time point. Cells were washed three times with C1 DNA-seq Cell Wash Buffer (Fluidigm). Cells at a concentration of 300 to 600 cells/μl were mixed with C1 Cell Suspension Reagent at a ratio of 3:2. Next, the cell mixture was loaded into C1 Single-Cell Auto Prep IFC (integrated fluidic circuits) microfluidic chips according to the “STRT seq, 1862× (10-17 μm diameter cells)” protocol. Cells were captured at the 96 capture sites in the microfluidic chip and stained using a green fluorescent calcein-AM dye (LIVE/DEAD cell viability assay, Life Technologies) and imaged by a Leica CTR 6000 microscope. mRNA-Seq libraries were prepared following the Generate complementary DNA (cDNA) libraries with the C1 Single-Cell mRNA Seq HT IFC and Reagent Kit V2 manual.
scRNA-Seq library construction (10X Genomics)
The 10X Genomics scRNA-Seq libraries were prepared following the Single-cell 3′ Reagents Kits v2 User Guide. Briefly, cell suspensions (~6000 cells) were loaded in a C1 Chromium Instrument (10X Genomics) to generate single-cell gel beads in emulsion (GEMs). scRNA-Seq libraries were prepared using the Chromium Single Cell 3′ Library, Gel Bead Kit v2, and Chromium i7 Multiplex Kit (10X Genomics). Sequencing was performed on Illumina HiSeq 4000 platform using 98 base pairs (bp) pair-end sequencing parameter
scATAC-Seq library construction (Fluidigm)
The scATAC-Seq libraries were prepared following the published protocol with minor modifications (10). Briefly, cells undergoing reprogramming at different time points were trypsinized into single cells and washed three times with the C1 DNA-seq Cell Wash Buffer (Fluidigm). Cells at a concentration of 300 to 600 cells/μl were combined mixed with the C1 Cell Suspension Reagent at a ratio of 3:2. The mixture of cells was then loaded into the C1 Single-Cell Auto Prep IFC microfluidic chips according to the “ATACseq: Cell Load and Stain (1862×)” protocol. Cells were captured at the 96 capture sites in the chip and stained using the green fluorescent calcein-AM dye (LIVE/DEAD cell viability assay, Life Technologies) and imaged by the Leica CTR 6000 microscope.
On the IFC, the lysis and transposition reaction took place at 37°C for 30 min, followed by inactivation of Tn5 using EDTA at 50°C for 30 min. Next, excess EDTA was quenched by MgCl2 at room temperature. The digested accessible regions were then amplified for 8 cycles. The 96 scATAC-Seq libraries were harvested from microfluidic chips and transferred to a 96-well plate for incorporating cell-specific indexes by additional 14 cycles of polymerase chain reaction (PCR). Following that, the 96 scATAC-Seq libraries were pooled and purified by a Qiagen PCR purification kit, and the quality of the libraries was assessed by an Agilent 2100 bioanalyzer.
Cell lines and reagents
The human neonatal fibroblast cell line BJ (Stemgent, Cambridge, MA) and the human lung fibroblast cell line MRC-5 (American Type Culture Collection (ATCC) CCL-171) were maintained in the medium, which is composed of Dulbecco’s Modified Eagles Medium (DMEM) with High Glucose (4500 mg/l) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich), minimum essential medium (MEM) nonessential amino acid solution (100X), 200 mM l-glutamine (100X), and penicillin-streptomycin (10,000 U/ml) (100X). hESCs and iPSCs were grown in mTeSR medium (STEMCELL Technologies). The mentioned reagents were purchased from Life Technologies, unless otherwise specified.
Cellular reprogramming (lentivirus)
At one day prior to the induction, BJ was seeded at a density of 25,000 cells/ml onto a 12-well plate. At D0, BJ cells were infected with polycistronic O2S, K2M lentivirus (Addgene) in the presence of polybrene (4 mg/ml; Sigma-Aldrich). These cells were maintained with BJ medium until D6. At D5, cells were trypsinized, replated at a density of 50,000 cells/ml onto a Matrigel (Corning)–coated 12-well plate. From D6 to D12, reprogramming cells were cultured with hESC medium, composed of 20% knockout serum replacement, MEM nonessential amino acid solution (100X), 200 mM l-glutamine (100X), penicillin-streptomycin (10,000 U/ml) (100X), basic fibroblast growth factor (8 ng/ml), and 0.1 mM β-mercaptoethanol in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12). From day 12 onward, medium was changed to a mixture of mouse embryonic fibroblast (MEF)–conditioned hESC medium and mTeSR (STEMCELL Technologies) in a 1:1 ratio.
Production of lentiviral supernatants
293T cells were plated at a density of 1.0 × 107 cells per 15-cm dish. The next day, cells were transfected with 9 μg of overexpression vector, 9 μg of psPAX2, and 0.9 μg of pMD2.G plasmid diluted with OptiMEM at a total of 225ul by mixing with 54 μl of TransIT LT1 (Mirus Bio) diluted with 696 μl of OptiMEM (Invitrogen) per plate. The supernatant was collected at 48 and 72 hours after transfection, filtered through 0.45-μm pore size filters, and concentrated using ultracentrifugation for 1.5 hours at 23,000 rpm.
Cellular reprogramming (Sendai virus)
At two days prior to the induction, 25,000 BJ or MSC cells were plated onto a 12-well plate. At D0, cells were infected with KOS, MYC, and KLF4 Sendai virus (CytoTune-iPS 2.0 Sendai Reprogramming Kit) at multiplicities of infection of 10:10:6. On the next day, medium was changed to reduce the toxicity effects of the Sendai virus. Cells were cultured in BJ and MSC medium for the first 4 to 5 days. At D4 or D5, cells were passaged and transferred onto Matrigel (Corning)–coated plates. Reprogramming cells were then cultured with medium, composed of BJ/MSC medium and mTeSR medium (STEMCELL Technologies) in a 1:1 ratio. A83-01 (STEMCELL Technologies) was supplemented to the reprogramming medium to increase the reprogramming efficiency.
Magnetic-activated cell sorting
Enrichment of cells by magnetic-activated cell sorting (MACS) was performed according to the manufacturer’s instructions. Briefly, cells were trypsinized to single cells and resuspended in 1% FBS (Sigma-Aldrich) at a concentration of 20 million/ml. Single-cell suspensions were incubated with anti-human TRA-1-60-PE (phycoerythrin) antibody (Miltenyi Biotec), anti-human CD13-FITC (fluorescein isothiocyanate) antibody (Miltenyi Biotec), and anti-human CD201-PE antibody (Miltenyi Biotec) in a 1:11 dilution, in the fridge for 10 min. Cells were then incubated with anti-PE MicroBeads (Miltenyi Biotec) in a 1:5 dilution, in the fridge for 15 min. Next, cells were separated into TRA-1-60+ and TRA-1-60− (flow-through) cells using the magnetic sorter.
Staining and imaging in 96-well plates (fluorescent probes screen)
A preprepared library of fluorescent probes was used for screen. For staining, cells were washed with phosphate-buffered saline (PBS) and incubated in cell culture medium supplemented with an optimal concentration of fluorescent probes for 1 hour at 37°C. It was followed by three washes with PBS and destaining with culture medium for 3 hours at 37°C. Hoechst 3342 (1:20,000; Invitrogen) was added to each well and stained for 30 min. The cells were then washed once with PBS.
Cells were imaged with an IXU ultra plate-scanning confocal microscope (Molecular Devices) at ×10 magnification, and nine pictures were taken per well. Granule area, integrated fluorescent intensity, and nuclei number were quantified using MetaXpress Image Acquisition and Analysis software V2.
BDD2-C8 dye live staining and FACS sorting
D8 reprogramming cells were incubated with hESC medium containing BDD2-C8 at a final concentration of 0.05 μM at 37°C for 1 hour. It was followed by three washes with PBS and destaining with hESC medium for 3 hours at 37°C. During destaining steps, fresh hESC medium was changed every 1 hour.
BDD2-C8–stained cells were trypsinized and subjected for sorting with flow cytometry (MoFlo XDP Cell Sorter, Beckman Coulter). According to the staining intensity of BDD2-C8, the top 10% and bottom 10% of D8 reprogramming cells were enriched for single-cell real-time quantitative reverse transcription PCR (qRT-PCR), scRNA-Seq library preparation, and replating onto a 12-well plate for TRA-1-60+ colony counting assay.
Organelle probe and fluorescent probe costaining
Cells were incubated with organelle-specific probes (diluted with culture media) at 37°C for 30 min and washed three times with PBS. Cells were then stained with fluorescent probes at 37°C for 1 hour, followed by destaining at 37°C for 3 hours. The information of organelle-specific probes is as follows: ER-specific probe: ER-Tracker Red, 500 nM; Golgi-specific probe: BODIPY FL C5-ceramide, 5 μM; mitochondria-specific probe: MitoTracker Green FM, 250 nM.
Costaining of cell surface markers and BDD2-C8
Cells were stained with BDD2-C8 for 1 hour, followed by 3 hours of destaining. Next, cells were trypsinized and resuspended with 1% FBS (Sigma-Aldrich) at a concentration of 1.5 million/ml. Single-cell suspensions were incubated with anti-human CD13-FITC, anti-human CD201(EPCR)-APC-Vio770 antibody (at a dilution of 1:11), and anti-human CD44-VioBlue (at a dilution of 1:51) (Miltenyi Biotec), in the fridge for 10 min. Cells were then subjected for flow cytometry analysis.
siRNA transfection in 12-well plates
siRNA (50 μl of 500 nM siGENOME, Dharmacon) was prepared and frozen at −20°C before use. For reverse transfection, a master mix of 0.8 μl of DharmaFECT1 (Dharmacon) transfection reagent and 150 μl of OptiMEM (Invitrogen) mix was added to the tubes containing the siRNA. The DharmaFECT1 and siRNA mix was incubated at room temperature for 20 min before transferring onto 80,000 cells in 800 μl medium in a well of 12-well plate.
TRA-1-60 immunohistochemical staining
Cells were fixed with 4% paraformaldehyde for 20 min at room temperature. After three PBS washes, cells were blocked with 7% FBS (Gibco) for 30 min at room temperature. Cells were then incubated with anti-human TTRA-1-60-biotin (eBioscience, cat. no. 13-8863-82) diluted with 7% FBS at 1:300 at 4°C overnight. After three PBS washes, cells were next stained with streptavidin-horseradish peroxidase antibody (BioLegend, cat. no. 405210) diluted with 7% FBS at 1:500 at room temperature for 1 hour. DAB Substrate Kit (Vector Laboratories, cat. no. SK-4100) was applied for visualizing the TRA-1-60+ colonies. Plate was coated with milk and scanned by an Epson Perfection 4490 scanner. The image was analyzed by Cell Profiler.
Immunostaining
Cells were washed with PBS and fixed with 4% paraformaldehyde for 20 min at room temperature. After three PBS washes, cells were permeabilized by 0.25% Triton X-100. Next, plates were washed with PBS three times and blocked using 7% FBS (Gibco) for 30 min at room temperature. Cells were then incubated with the primary antibody at 4°C overnight. After three PBS washes, cells were stained with secondary antibody for 1 hour at room temperature. Hoechst 3342 (1:20,000; Invitrogen) was added to and stained for 30 min.
RNA extraction, reverse transcription, and real-time qPCR
Total RNA was extracted using the TRIzol reagent (Invitrogen). Contaminant DNA was removed by deoxyribonuclease I (Ambion) treatment, and the RNA was further purified using the QIAGEN RNeasy Kit. First-strand cDNA was synthesized using the iScript cDNA synthesis Kit (Bio-Rad) according to the manufacturer’s instructions. Quantitative real-time PCR was performed on the CXF384 Real-time System (Bio-Rad), using a Kapa SYBR Fast qPCR kit (Kapa Biosystems). The expression level of each gene was normalized to that of β-actin, unless otherwise stated.
Bulk RNA-Seq
RNA-seq libraries were prepared using the TruSeq Stranded mRNA Library Prep Kit following the manual guide of the kit. Briefly, mRNA was enriched by the beads conjugated with Poly-T oligo, fragmentized, and reverse-transcribed to cDNA. After ligating the sequencing adaptors, cDNA was amplified for 15 cycles of PCR. The library was sequenced on a HiSeq 4000 sequencer with a 101 base pairs (bp) pair-end sequencing parameter.
Chromatin immunoprecipitation (ChIP)
ChIP was performed according to a previous study (38). Briefly, 10 million cells were cross-linked using 1% formaldehyde for 10 min at room temperature, followed by quenching using 200 mM glycine. After that, cells were lysed using lysis buffer consisting of 10 mM Tris-HCl (pH 8), 100 mM NaCl, 10 mM EDTA, 0.25% Triton X-100, and protease inhibitor cocktail (Roche). Cells were then resuspended in 1% SDS lysis buffer composed of 50 mM Hepes-KOH (pH 7.5), 150 mM NaCl, 1% SDS, 2 mM EDTA, 1% Triton X-100, 0.1% NaDOC, and protease inhibitor cocktail. The suspension was nutated for 15 min at 4°C before spinning down to collect the chromatin pellet. The pellet was then washed two times with 0.1% SDS lysis buffer containing 50 mM Hepes-KOH (pH 7.5), 150 mM NaCl, 0.1% SDS, 2 mM EDTA, 1% Triton X-100, 0.1% NaDOC, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail. Chromatin was then fragmented by sonication, which was conducted with cells on ice for 14 cycles, power amplitude of 35%, and 30-s pulses on with 59.9-s pulses off (Branson Sonifier 250). The chromatin solution was clarified by centrifugation at 20,000g at 4°C for 45 min and then precleared with Dynabeads protein A (Life Technologies) for 2 hours at 4°C. The precleared chromatin sample was incubated with 50 μl of Dynabeads protein A loaded with 5 μg of antibody overnight at 4°C. The beads were washed three times with 0.1% SDS lysis buffer, once with 0.1% SDS lysis buffer/0.35 M NaCl, once with 10 mM Tris-HCl (pH 8), 1 mM EDTA, 0.5% NP-40, 0.25 LiCl, and 0.5% NaDOC, and once with Tris-EDTA (TE) buffer (pH 8.0). The immunoprecipitated material was eluted from the beads by heating for 45 min at 68°C in 50 mM Tris-HCl (pH 7.5), 10 mM EDTA, and 1% SDS. Reverse cross-linking was carried out by incubating the samples with Pronase (1.5 μg/ml) at 42°C for 2 hours followed by a 6-hour incubation at 67°C. DNA was then purified using a MinElute PCR Purification kit (Qiagen). Purified ChIP DNA was further processed for library preparation following the manufacturer’s instruction (Illumina). Fold enrichment of each primer is calculated by normalizing to a negative control primer.
Computational analysis
scRNA-Seq libraries analysis (Fluidigm)
Libraries were mapped to hg19 assembly using STAR aligner (version 2.5.3a), allowing up to two mismatches and removing reads mapping to more than one region. Cuffquant (version 2.2.1) was used to count reads belonging to each gene in the genome with the option -u and -b included in the used script. Cuffnorm (version 2.2.1) was used to normalize the counts and calculate the fragments per kilobase of transcript per million fragments mapped (FPKM) among libraries belonging to the same time point.
Filtering scRNA-Seq libraries (Fluidigm)
Percentage of genes measured and exon mapping percentage were determined using the RNA-Seq QC options of SeqMonk software (version 1.40.1) (www.bioinformatics.babraham.ac.uk/projects/seqmonk/). Libraries with mapping to exon rate below 75% were filtered out. Moreover, libraries with below 20% of measured genes were also filtered out.
Distribution of reads over transcripts (Fluidigm scRNA-Seq)
The determination of distribution of reads over the transcripts of housekeeping genes was performed using the geneBody_coverage.py module of RseQC python package (version 2.6.4).
Reference component analysis
FPKM counts of all high-quality scRNA-Seq libraries were uploaded to R for RCA (version 1.0) analyses (12). The expression data were projected into the reference component space using the “Global Panel” method as indicated in the software’s vignette.
Differential expression analysis (Fluidigm scRNA-Seq)
The differentially expressed genes were calculated among the subgroups of each time point. If the gene (i) has average FPKM value more than 5, (ii) was expressed in more than 50% of the cells, and (iii) was expressed at more than threefold higher in average expression in the subgroup, it is considered to be highly expressed in the respective subgroup.
GO analysis
A web-accessible program, DAVID (database for annotation, visualization, and integrated discovery) (https://david.ncifcrf.gov/), was used for performing GO analysis. The GO terms were extracted from GOTERM_BP_DIRECT under the Functional Annotation tab.
Boxplot
Boxplots were generated by “ggplot2” (version 2.2.1) package in R, based on the log10 (FPKM + 1) values of specified genes in all the time points and the respective subgroups.
Pearson correlation
Pearson correlation between D8 subgroups and cells of all the other time points was performed using Excel, based on the differentially expressed genes of D8 subgroups.
Stemness analysis
Stemness score was obtained by inputting the highly expressed genes of each subgroup in StemChecker (http://stemchecker.sysbiolab.eu/).
QC of 10X Genomics scRNA-Seq libraries analysis
Fastqs were generated from the 10X raw data using the mkfastq module of cellranger (version 2.1.1). The 10X libraries belonging to each reprogramming time point were quantified using the cell ranger count module. Then, the libraries were aggregated using cellranger aggr script with disabled normalization by setting the option --normalize=NONE. Next, the aggregated libraries were fed to Seurat package (version 3.0) using the Read10X function. The QC was performed using VlnPlot function of Seurat package. Mitochondrial DNA percentage in each library was measured by grepping genes starting with MT and summing the values using colSums. The correlation between the number of genes and unique molecular identifiers (UMI) or between the percentage of mitochondrial DNA and UMI was performed using the GenePlot function of Seurat. Cells with detected genes below 2500 or above 6500 were filtered out. Cells with mitochondrial content above 10% were removed.
Seurat analysis [10X Genomics scRNA-Seq libraries of reprogramming cells (time points)]
After filtering low-quality cells, we normalized and scaled the cells using “sctransform.” This was followed by calculating principal components analysis (PCA) and generating the Elbow Plot. UMAP (Uniform Manifold Approximation and Projection) was generated using RunUMAP. The lineage scores were calculated using “AddModuleScore” of Seurat and superimposed on the UMAPs using FeaturePlot using min.cutoff = “q75.”
Pseudotemporal analysis (reprogramming cells of various time points)
The output folder of cellranger aggr (without normalization) was fed into Monocle (Version 2.6.4) using the newCellDataSet function. The “negbinomal.size” expression family was chosen. Afterwards the size factors and dispersions of the libraries were estimated. Genes that were expressed in minimum of 10 cells were considered for subsequent analyses. Differential gene expression analysis was performed using “differentialGeneTest” while using the size factors for correction of the estimations. Genes that are differentially expressed with q value < 0.01 were considered for the tree constructions. Dimension reduction was performed using DDRTree methods, and the trajectory was plotted using the plot_cell_trajectory. For superimposition of gene expression on the measured cell trajectories, the marker option of plot_cell_trajectory option was used, and use_color_gradient was set as TRUE.
Seurat analysis (10X Genomics scRNA-Seq libraries of D8 CD13-sorted cells)
CD13+ and CD13− libraries were aggregated using the cellranger aggr script with disabled normalization by setting the option --normalize=NONE. Next, the aggregated libraries were fed to Seurat package (version 3.0) using the Read10X function. The QC was performed using the VlnPlot function of Seurat package. Mitochondrial DNA percentage in each library was measured by grepping genes starting with MT and summing the values using colSums. The percentage of mitochondrial DNA was then added as a MetaData. Cells that passed QC had genes detected between 3000 and 6000, UMI count between 10,000 and 32,500, and mitochondrial contamination below 0.08. Variable genes were detected using the following parameters in “FindVariableGenes” function: mean.function = ExpMean, dispersion.function = LogVMR, x.low.cutoff = 0.0125, x.high.cutoff = 3, y.cutoff = 0.5.
Libraries were then normalized using LogNormalize method with 10,000 scale. After that, the libraries were scaled to UMI and percentage of mitochondrial DNA. CD13− cells that demonstrated expression of CD13 were considered as sorting error and filtered out. PCA was determined using variable genes, and t-SNE (t-distributed stochastic neighbor embedding) was performed using dims 1:9. Clusters were determined using FindClusters with 0.6 resolution.
Pseudotemporal analysis (reprogramming cells of various time points with D8 CD13-sorted cells)
BJ, D2, D12, D16−, D16+, D8 CD13−, and D8 CD13+ 10X libraries were aggregated using cellranger “aggr” without normalization. Next, the aggregated libraries were fed to Seurat package using the Read10X function. Cells with genes detected below 1875 or above 7000 were filtered out. In addition, cells with mitochondrial DNA above 0.1 were filtered out. The Seurat object was then fed into Monocle (version 3 alpha) using “importCDs.” Then, “estimateSizeFactors” and “estimateDispersions” were performed. This was followed by “preprocessCDS” with 20 dimensions. Then, dimensions were reduced using UMAP (39). Cells were then portioned using “PartitionCells” with default parameters. Last, “learnGraph” was used, “RGE_method” set to DDRTree, and trajectory was plotted using “plot_cell_trajectory.”
Mathematical imputation of 10X Genomics scRNA-Seq libraries
Raw expression matrix was generated using the mat2csv function of cellranger. The matrix was then transposed using R. This matrix was then used as input for MAGIC (version 1.4.0) (26). We filtered genes that had below 10 UMI and filtered cells with total UMI < 7000. The matrix was then normalized using “library.size.normalize” function of MAGIC and then transformed using “sqrt” function. Imputation was then performed using “magic” function with default parameters.
Determination of regulatory TFs
Differentially expressed genes across the states were determined using “differentialGeneTest” function of Monocle (Version 2.6.4). The “fullModelFormulaStr” was set to states. The genes that were significant were then annotated (Molecular Function) using Metascape (http://metascape.org/gp/index.html). Significant TFs were then visually inspected one by one to determine the significant regulators across the pseudotime states. After filtering low-quality cells as indicated above, the states to which each reprogramming cell belonged to in the pseudotime trajectory were added using the addMetaData function to Seurat reprogramming object. The TFs heatmap was generated using the “DoHeatmap” function and grouping the cells by states.
Mapping of scATAC-Seq libraries
Libraries were mapped to hg19 genome assembly using STAR aligner (version 2.5.3a). Reads that contain more than two mismatches and/or mapped to more than one position in the genome were filtered out. The option --alignIntronMax was set to 1 to disable the splice awareness of the aligner. The option -alignEndsType was set to EndToEnd to make STAR compatible with DNA reads.
Determination of highly accessible regions (HARs)
Mapped files of libraries belonging to the same time point were merged using samtools. After that, the MarkDuplicates module of PICARD tools (http://broadinstitute.github.io/picard) was used to remove duplicates. Then, MACS2 (Version 2.1.0) was used to call peaks. The --nomodel --nolambda --keep-dup all --call-summits options were used. ChrM and all ambiguous chromosomes were excluded from this analysis. The top 50,000 peaks in the peaks summit file were extended to ±250 bp. These were then considered to be the highly accessible regions (HARs) in each respective time point.
Filtering scATAC-Seq libraries
Duplicates were removed from each scATAC-Seq as mentioned above. This was followed by the addition of group information using the AddOrReplaceReadGroups module of PICARD (version 2.17.6). A sequence dictionary for hg19 genomes lacking ChrM and other ambiguous chromosomes was generated using CreateSequenceDictionary module of PICARD. Reads in each library were sorted lexicographically by using the ReorderSam module of PICARD tools. The option ALLOW_INCOMPLETE_DICT_CONCORDANCE was set to TRUE, which helps in removing ChrM and ambiguous mapped reads from the libraries. The generated bam files were then indexed using samtools. The coverage of each scATAC-Seq over the HARs of their corresponding time point was measured using the DepthOfCoverage module of GATK (Genome Analysis Toolkit) tools v3.5. The option –countType was set to COUNT_FRAGMENTS to ensure reliable estimates. Libraries should have coverage over a minimum 15% of the HAR coordinates to be considered for further analysis. The coverage values are recorded in the total_cvg columns present in the interval_summary output of GATK. In addition, library size of each library was determined using the MarkDuplicates of PICARD tools. Any library having a size less than 10,000 was discarded.
Average enrichment profile generation
The processed scATAC-Seq libraries, in which ChrM and ambiguously mapped reads were removed, were used to generate the average enrichment profile plots. The software ngs.plot was used (version 2.61). The options -R was set to tss and -L was set to 3000 to measure the average enrichment of reads on ±3000 bp around the TSS regions.
Nucleosomal pattern determination
The CollectInsertSizeMetrics module of PICARD (version 2.17.6) tool was used to generate the insert size histograms. The libraries were free from reads mapping to ChrM or other ambiguous chromosomes before running the scripts.
Prediction of promoters
First, the HARs were annotated using annotatePeaks.pl script. Then, regions that were 3000 bp upstream to 200 bp downstream from the nearest gene were considered to be putative promoters. We ensured that these regions do not fall in the genebody of any gene. The coverage of the scATAC-Seq libraries over these putative promoters was measured using GATK tools as mentioned above. The values were normalized by dividing each value present in the total_cvg column by the respective library size and multiplying that by 10,000. These normalized values were then uploaded to R, and PCA plots were generated using “FactoMineR.”
ChromVAR analysis of scATAC-Seq libraries
Deduplicated mapped scATAC-Seq files and the narrowPeaks HARs were uploaded to chromVAR software (version 1.0.2), and the coverage of the libraries over the HARs was measured using getCounts function of chromVAR. Libraries were filtered to remove dead or double cell libraries using filterSamples option. The HARs were annotated with JASPAR motifs using matchMotifs option. Variability of JASPAR motifs across all cells was quantified using the computeDeviations function. This was followed by computing the variability using computeVariability option. Then, the scATAC-Seq libraries were correlated on the basis of the computed deviations using the “getSampleCorrelation” module.
Clustering of scATAC-Seq libraries based on deviation scores that were quantified was measured using “deviationsTSNE” of chromVAR with thresholds and perplexities ensuring that the results were not due to batch effect. Superimposition of motif enrichment scores for FOSL1, OCT4, and TEAD4 on the scATAC-Seq clusters. These genes were considered in the annotation field in the “plotDeviationsTSNE” script of chromVAR.
Differential accessibility analysis
The differential accessibility analysis was performed using DESeq2 (version 1.18.1). The unnormalized coverage values over regions of interest were fed to R. The time points of the libraries were considered as conditions. The size factors were estimated using the “poscounts” method. This was followed by normalization and differential accessibility analysis using the “DESeq” function. The reported regions underwent independent hypothesis weighting to ensure the removal of false-positive hits by using the filterFun=ihw in the “results” option of DESeq2. The MA (log ratio versus abundance) plot was generated using the plotMA function of DESeq2.
ChIP-Seq analysis
Libraries were mapped to hg19 genome using STAR (version 2.5.3) as performed for the scATAC-Seq libraries (mentioned above). Subsequently, tag directories were created for each mapped file using makeTagDirectory script of Homer (version 4.9). This was followed with peak calling using GEM (version 3.4) (40). The --k_min 6 --k_max 13 --t 10 --outHOMER –outBED were the additional options passed to GEM. The default read distribution file of GEM was used.
Genomic distribution plots
The identified binding sites of FOSL1 were uploaded to PAVIS (peak annotation and visualization) (https://manticore.niehs.nih.gov/pavis2/). The hg19 known Genes Assembly was used for annotating the peaks. For other parameters, the default values were used.
Motif analysis
Motifs enriched in regions of interest were determined using the findMotifsGenome.pl script of HOMER (version 4.9). The option “size” was set to “given” in the script.
Statistical analysis
Two-tailed Student’s t test was performed for calculating statistical significance.
Supplementary Material
Acknowledgments
We are grateful to Y.F. Seah and Y.Y. Zeng for helpful discussion. Funding: H.L. is supported by the NIH (AG056318, AG61796, and CA208517), the Glenn Foundation for Medical Research, Mayo Clinic Center for Biomedical Discovery, Center for Individualized Medicine, Mayo Clinic Cancer Center, and the David F. and Margaret T. Grohne Cancer Immunology and Immunotherapy Program. L.F.Z. is supported by the Singapore Ministry of Education Academic Research Fund (MOE2015-T2-1-093) and Singapore National Research Foundation under its Cooperative Basic Research Grant administered by the Singapore Ministry of Health’s National Medical Research Council (NMRC/CBRG/0092/2015). N.Y.K. and Y.T.C. are supported by the JCO Development Programme Grant (1334k00083). Y.H.L. is supported by the NRF Investigatorship award (NRFI2018-02 grant), JCO Development Programme Grant (1534n00153 and 1334k00083), and the Singapore National Research Foundation under its Cooperative Basic Research Grant administered by the Singapore Ministry of Health’s National Medical Research Council (NMRC/CBRG/0092/2015). We are grateful to the Biomedical Research Council, Agency for Science, Technology and Research, Singapore for research funding. Author contributions: Q.R.X. and C.A.E.F. designed and performed research, analyzed data, and wrote the paper. P.G., Y.S.C., T.W., and C.X.D.T. conducted research. N.Y.K., S.S., Y.T.C., J.X., J.J.C., G.Q.D., H.L., and L.F.Z. analyzed data. Y.H.L. designed research, analyzed data, and wrote the paper. Competing interests: During the conduct of this study, G.Q.D. held equity in and received consulting fees from iPierian Inc., True North Therapeutics, and MPM Capital. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Raw NGS sequencing data are deposited to GEO under the accession number GSE100345. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/37/eaba1190/DC1
REFERENCES AND NOTES
- 1.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S., Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Seah Y. F. S., El Farran C. A., Warrier T., Xu J., Loh Y.-H., Induced pluripotency and gene editing in disease modelling: Perspectives and challenges. Int. J. Mol. Sci. 16, 28614–28634 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheow L. F., Courtois E. T., Tan Y., Viswanathan R., Xing Q., Tan R. Z., Tan D. S. W., Robson P., Loh Y.-H., Quake S. R., Burkholder W. F., Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat. Methods 13, 833–836 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Stadtfeld M., Hochedlinger K., Induced pluripotency: History, mechanisms, and applications. Genes Dev. 24, 2239–2263 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin H., Diaz A., Blouin L., Lebbink R. J., Patena W., Tanbun P., LeProust E. M., McManus M. T., Song J. S., Ramalho-Santos M., Systematic identification of barriers to human iPSC generation. Cell 158, 449–461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toh C.-X. D., Chan J.-W., Chong Z.-S., Wang H. F., Guo H. C., Satapathy S., Ma D., Goh G. Y. L., Khattar E., Yang L., Tergaonkar V., Chang Y.-T., Collins J. J., Daley G. Q., Wee K. B., Farran C. A. E., Li H., Lim Y.-P., Bard F. A., Loh Y.-H., RNAi reveals phase-specific global regulators of human somatic cell reprogramming. Cell Rep. 15, 2597–2607 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Yang C.-S., Chang K.-Y., Rana T. M., Genome-wide functional analysis reveals factors needed at the transition steps of induced reprogramming. Cell Rep. 8, 327–337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang H.-T., El Farran C. A., Xing Q. R., Zhang L.-F., Li H., Lim B., Loh Y.-H., Global H3.3 dynamic deposition defines its bimodal role in cell fate transition. Nat. Commun. 9, 1537 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gawad C., Koh W., Quake S. R., Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 17, 175–188 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Buenrostro J. D., Wu B., Litzenburger U. M., Ruff D., Gonzales M. L., Snyder M. P., Chang H. Y., Greenleaf W. J., Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramsköld D., Luo S., Wang Y.-C., Li R., Deng Q., Faridani O. R., Daniels G. A., Khrebtukova I., Loring J. F., Laurent L. C., Schroth G. P., Sandberg R., Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 30, 777–782 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H., Courtois E. T., Sengupta D., Tan Y., Chen K. H., Goh J. J. L., Kong S. L., Chua C., Hon L. K., Tan W. S., Wong M., Choi P. J., Wee L. J. K., Hillmer A. M., Tan I. B., Robson P., Prabhakar S., Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 49, 708–718 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Ullah I., Subbarao R. B., Rho G. J., Human mesenchymal stem cells - Current trends and future prospective. Biosci. Rep. 35, e00191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruiz S., Panopoulos A. D., Herrerías A., Bissig K. D., Lutz M., Berggren W. T., Verma I. M., Izpisua Belmonte J. C., A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Curr. Biol. 21, 45–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiao J., Dang Y., Yang Y., Gao R., Zhang Y., Kou Z., Sun X.-F., Gao S., Promoting reprogramming by FGF2 reveals that the extracellular matrix is a barrier for reprogramming fibroblasts to pluripotency. Stem Cells 31, 729–740 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N. J., Livak K. J., Mikkelsen T. S., Rinn J. L., The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiu X., Mao Q., Tang Y., Wang L., Chawla R., Pliner H. A., Trapnell C., Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schiebinger G., Shu J., Tabaka M., Cleary B., Subramanian V., Solomon A., Gould J., Liu S., Lin S., Berube P., Lee L., Chen J., Brumbaugh J., Rigollet P., Hochedlinger K., Jaenisch R., Regev A., Lander E. S., Optimal-transport analysis of single-cell gene expression identifies developmental trajectories in reprogramming. Cell 176, 928–943.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cacchiarelli D., Trapnell C., Ziller M. J., Soumillon M., Cesana M., Karnik R., Donaghey J., Smith Z. D., Ratanasirintrawoot S., Zhang X., Ho Sui S. J., Wu Z., Akopian V., Gifford C. A., Doench J., Rinn J. L., Daley G. Q., Meissner A., Lander E. S., Mikkelsen T. S., Integrative analyses of human reprogramming reveal dynamic nature of induced pluripotency. Cell 162, 412–424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe K., Liu Y., Noguchi S., Murray M., Chang J.-C., Kishima M., Nishimura H., Hashimoto K., Minoda A., Suzuki H., OVOL2 induces mesenchymal-to-epithelial transition in fibroblasts and enhances cell-state reprogramming towards epithelial lineages. Sci. Rep. 9, 6490 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takaishi M., Tarutani M., Takeda J., Sano S., Mesenchymal to epithelial transition induced by reprogramming factors attenuates the malignancy of cancer cells. PLOS ONE 11, e0156904 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanton S., Boyle M. J., He Z., Santel M., Weigert A., Sanchís-Calleja F., Guijarro P., Sidow L., Fleck J. S., Han D., Qian Z., Heide M., Huttner W. B., Khaitovich P., Pääbo S., Treutlein B., Camp J. G., Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Petropoulos S., Edsgärd D., Reinius B., Deng Q., Panula S. P., Codeluppi S., Plaza Reyes A., Linnarsson S., Sandberg R., Lanner F., Single-cell RNA-seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell 165, 1012–1026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yun S.-W., Leong C., Zhai D., Tan Y. L., Lim L., Bi X., Lee J.-J., Kim H. J., Kang N.-Y., Ng S. H., Stanton L. W., Chang Y.-T., Neural stem cell specific fluorescent chemical probe binding to FABP7. Proc. Natl. Acad. Sci. U.S.A. 109, 10214–10217 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan F., Qian C., Tang K., Abd-Allah S. M., Jing N., Inhibition of transforming growth factor β (TGF-β) signaling can substitute for Oct4 protein in reprogramming and maintain pluripotency. J. Biol. Chem. 290, 4500–4511 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Dijk D., Sharma R., Nainys J., Yim K., Kathail P., Carr A. J., Burdziak C., Moon K. R., Chaffer C. L., Pattabiraman D., Bierie B., Mazutis L., Wolf G., Krishnaswamy S., Pe’er D., Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729.e27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neph S., Stergachis A. B., Reynolds A., Sandstrom R., Borenstein E., Stamatoyannopoulos J. A., Circuitry and dynamics of human transcription factor regulatory networks. Cell 150, 1274–1286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R., Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schep A. N., Wu B., Buenrostro J. D., Greenleaf W. J., ChromVAR: Inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shu J., Wu C., Wu Y., Li Z., Shao S., Zhao W., Tang X., Yang H., Shen L., Zuo X., Yang W., Shi Y., Chi X., Zhang H., Gao G., Shu Y., Yuan K., He W., Tang C., Zhao Y., Deng H., Induction of pluripotency in mouse somatic cells with lineage specifiers. Cell 153, 963–975 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takahashi K., Tanabe K., Ohnuki M., Narita M., Sasaki A., Yamamoto M., Nakamura M., Sutou K., Osafune K., Yamanaka S., Induction of pluripotency in human somatic cells via a transient state resembling primitive streak-like mesendoderm. Nat. Commun. 5, 3678 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Gabut M., Samavarchi-Tehrani P., Wang X., Slobodeniuc V., O’Hanlon D., Sung H.-K., Alvarez M., Talukder S., Pan Q., Mazzoni E. O., Nedelec S., Wichterle H., Woltjen K., Hughes T. R., Zandstra P. W., Nagy A., Wrana J. L., Blencowe B. J., An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 147, 132–146 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Aibar S., González-Blas C. B., Moerman T., Huynh-Thu V. A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., van den Oord J., Atak Z. K., Wouters J., Aerts S., SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine A. J., Brivanlou A. H., GDF3, a BMP inhibitor, regulates cell fate in stem cells and early embryos. Development 133, 209–216 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Chronis C., Fiziev P., Papp B., Butz S., Bonora G., Sabri S., Ernst J., Plath K., Cooperative binding of transcription factors orchestrates reprogramming. Cell 168, 442–459.e20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loh K. M., Lim B., A precarious balance: Pluripotency factors as lineage specifiers. Cell Stem Cell 8, 363–369 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Montserrat N., Nivet E., Sancho-Martinez I., Hishida T., Kumar S., Miquel L., Cortina C., Hishida Y., Xia Y., Esteban C. R., Izpisua Belmonte J. C., Reprogramming of human fibroblasts to pluripotency with lineage specifiers. Cell Stem Cell 13, 341–350 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Yang B. X., El Farran C. A., Guo H. C., Yu T., Fang H. T., Wang H. F., Schlesinger S., Seah Y. F. S., Goh G. Y. L., Neo S. P., Li Y., Lorincz M. C., Tergaonkar V., Lim T.-M., Chen L., Gunaratne J., Collins J. J., Goff S. P., Daley G. Q., Li H., Bard F. A., Loh Y.-H., Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 163, 230–245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McInnes L., Healy J., Saul N., Großberger L., UMAP: Uniform manifold approximation and projection. J. Open Source Softw. 3, 861 (2018). [Google Scholar]
- 40.Guo Y., Mahony S., Gifford D. K., High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLOS Comput. Biol. 8, e1002638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/37/eaba1190/DC1