

Bioluminescence-induced proteinaceous photodynamic therapy enables cancer treatment by self-luminescent ROS generation.
Abstract
Despite the potential of photodynamic therapy (PDT), its comprehensive use in cancer treatment has not been achieved because of the nondegradable risks of photosensitizing drugs and limits of light penetration and instrumentation. Here, we present bioluminescence (BL)–induced proteinaceous PDT (BLiP-PDT), through the combination of luciferase and a reactive oxygen species (ROS)–generating protein (Luc-RGP), which is self-luminescent and degradable. After exposure to coelenterazine-h as a substrate for luciferase without external light irradiation, Luc-RGP fused with a small lead peptide–induced breast cancer cell death through the generation of BL-sensitive ROS in the plasma membrane. Even with extremely low light energy, BLiP-PDT exhibited targeted effects in primary breast cancer cells from patients and in in vivo tumor xenograft mouse models. These findings suggest that BLiP-PDT is immediately useful as a promising theranostic approach against various cancers.
INTRODUCTION
Photodynamic therapy (PDT), a form of clinically approved phototherapy, has emerged as an alternative treatment for cancers and malignant diseases (1). A light-activated photosensitizer (PS) and oxygen are considered the major components of PDT (2), in which the generation of reactive oxygen species (ROS) is induced through two types of oxidative photoreactions, leading to localized cell death and tissue devastation. In contrast to classical radiation or chemical therapy, PDT is characterized by the selective ablation of malignant cells, high tumor specificity, and minimal invasiveness. To expand its usefulness, many studies on PDT have explored the development of PS drugs alone or in combination with nanoparticle hybrids (3, 4), immunostimulants (5, 6), and/or other active targeting ligands (7, 8). However, despite these achievements, several major challenges posed by the inherent properties of chemical PS drugs, such as in vivo accumulation and light penetration issues, limit their clinical effectiveness. For example, the Food and Drug Administration–approved tetrapyrrole drugs (e.g., porphyrin or chlorin), comprising the largest group of PS drugs, may cause severe pain or sunlight-induced photodermatosis (9). Even the use of relatively safe prodrugs (e.g., aminolevulinic acid) to endogenously create protoporphyrin IX via the heme biosynthetic pathway often results in hepatic porphyria, renal failure, or hyperthermia owing to its high blood accumulation and slow degradation (10, 11). Furthermore, in addition to the expensive and complex light-illuminating instrumentation in classical PDT, the light penetration depth in the visible and near-infrared regions (400 to 900 nm) is limited to 1 to 2 mm beneath the skin, due to light scattering (12), which substantially restricts the wider applications of PDT in vivo.
To circumvent these limitations of PDT, the combination of bioluminescence (BL) with a chemical PS-conjugated quantum dot was recently demonstrated (13, 14); it achieved ROS-induced cell death via internal activation of the PS. However, this approach still retains the risks for chemical (or nanomaterial) toxicity. In another approach, a panel of PS protein-encoding genes (15–17) or their luciferase-fused genes (18) were constructed to kill cancer cells by light via gene transfection into mammalian cells. However, this approach relies on viral gene delivery, which carries another risk for genotoxicity in targeted cancer therapy.
Here, we report BL-induced proteinaceous PDT (denoted BLiP-PDT) as a novel PDT method that requires neither a chemical PS nor gene transfection. BLiP-PDT is based on a protein biosensor, tailored to exert self-luminescence, ROS generation, target specificity, and theranostic functions. These abilities are achieved by the use of luciferase and an ROS-generating protein, named Luc-RGP, which is further linked to a lead peptide (LP) to enable targeted cancer therapy. With the aim of solving the intrinsic issues of classical PDT, we demonstrate the manner in which the BL-sensitive protein probes specifically induce cancer cell death without an external light source and describe how this phenomenon is elicited in breast cancer (BC) cells, primary BC cells from patients, and an in vivo tumor xenograft mouse model. It should also be noted that, although, in nature, luciferase and its substrate may have resulted from evolutionary pressure to protect organisms as an antioxidant via luciferin-ROS or luciferase-ROS interactions (19), we have exploited a new role of luciferase as an ROS facilitator, which is divergent from its original function. Similar ROS-generating phenomena have been found in light-activated pigments or channels in multiple branches of living organisms (20, 21).
RESULTS
Luc-RGP functions as a protein probe for BLiP-PDT
To establish BLiP-PDT without chemical PS or gene transfection, we prepared two different Luc-RGPs comprising two Renilla luciferase variants [Rluc8.6 (22) or Rluc8 (23)] and two representative RGPs [KillerRed (KR) (15) or MiniSOG (MS) (24)]: Rluc8.6-KR and Rluc8-MS. To endow target specificity to the Luc-RGPs, the cancer cell–targeting LP (WLEAAYQRFL) was fused at their C termini (Fig. 1, A and B) and purified with affinity tags after bacterial protein expression (fig. S1, A and B). In the case of KR-fused protein, monomeric and dimeric protein constructs were mixed after purification, but only the dimeric protein construct emitted fluorescence (FL) (fig. S1A). This is primarily due to the intrinsic dimeric nature of KR (fig. S1, C to F). The LP target ligand was chosen as it was reported to have higher uptake efficiency by cancer cells compared with a commonly used tumor-homing tripeptide, Arg-Gly-Asp (RGD) (25). The peptide sequence was previously identified through peptide arrays from an originally selected phage-displaying peptide (p160) (26) and reported to have a high-binding affinity for some BC cells and neuroblastoma cells (25, 26). Owing to the large spectral overlap of emission/excitation wavelengths between the two proteins (Luc and RGP), high BL resonance energy transfer (BRET) efficiency was observed upon the addition of the synthetic derivative Rluc substrate, coelenterazine-h (Co-h): ~41.3% for Rluc8.6-KR and ~25.0% for Rluc8-MS (fig. S2, A to F). The photochemical properties of RGPs or model PS [Rose Bengal (RB)] are summarized in table S1. RGPs and Luc-RGPs showed similar FL colors at corresponding excitation wavelengths, whereas BL imaging of Luc-RGPs showed longer emission characteristics than Luc alone owing to the BRET phenomenon (Fig. 1C).
Fig. 1. Design and characterization of BL-sensitizing protein probes in vitro.
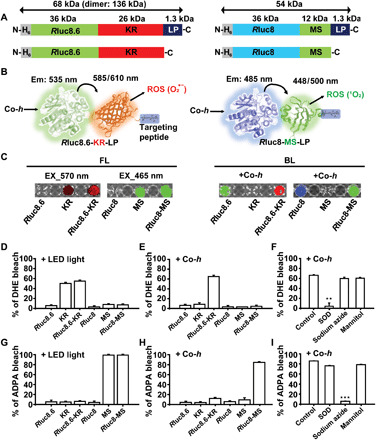
(A) Schematic of the primary structure of the BL-sensitizing probes from the N to the C termini. H6, hexahistidine; Rluc8.6 (with green emission) and Rluc8 (blue emission), Renilla luciferase mutants; MS, MiniSOG. (B) Schematic of two photodynamic protein probes: Rluc8.6-KR-LP (left) and Rluc8-MS-LP (right). Co-h, coelenterazine-h (a substrate for luciferase). The arrow indicates the BRET phenomenon from the emission (em) of Rluc8.6 (or Rluc8) to the excitation (ex) of KR (or MS), which leads to the generation of ROS, such as superoxide (O2•-) or singlet oxygen (1O2). The blue box indicates the chemical structure of the LP. (C) FL (left) and BL (right) images of Luc (Rluc8.6 or Rluc8), RGP (KR or MS), or Luc-RGP fusion protein on a microplate. FL was measured with filters for excitation/emission wavelengths of 570/620 nm (or 465/520 nm), and BL was measured with emission filters for blue (500 nm), green (520 nm), or red (620 nm) wavelengths after Co-h treatment using an IVIS Spectrum. (D to I) Detection of ROS (D to F, superoxide using DHE; G to I, singlet oxygen using ADPA) generated from various proteins upon light irradiation or Co-h addition. The effects of different ROS scavengers were tested after mixing with Rluc8.6-KR (F) or Rluc8-MS (I). The error bars indicate SD values from triplicate experiments; the significance of the difference in ROS generation between control and test samples were evaluated (**P < 0.01, ***P < 0.001, n = 3 replicates, Student’s t test).
To examine whether Luc-RGPs could generate ROS by enzyme-substrate reaction or light irradiation, the levels of superoxide (Fig. 1, D to F) and singlet oxygen (Fig. 1, G to I) were evaluated by measuring photobleaching effect of the ROS-responsive dyes, dihydroethidium (DHE) and anthracene-9,10-dipropionic acid (ADPA), respectively. Light-emitting diode (LED) light (10 mW cm−2 for 10 min)–irradiated KR (or its conjugate) yielded predominantly superoxide (O2•-), whereas LED light irradiation of MS (or its conjugate) only yielded singlet oxygen (1O2) (Fig. 1, D and G), which was consistent with the previous report (24). Notably, only Luc-RGPs yielded considerable ROS levels in the presence of Co-h without light irradiation, while neither Luc nor RGP generated ROS under the same conditions (Fig. 1, E and H). The induction of ROS by Luc-RGPs was dependent on Co-h concentration and the reaction time (fig. S3, A to D). In addition, we confirmed that the Luc-RGP–medicated induction of ROS was markedly inhibited by specific ROS scavengers [superoxide dismutase (SOD) or sodium azide] (Fig. 1, F and I). Collectively, these findings indicate that self-luminescent Luc-RGPs, induced by the substrate, serve as ROS generators in the absence of external light irradiation.
BC cell death by BLiP-PDT
We assessed the ability of Luc-RGP-LPs (Rluc8.6-KR-LP) to target and kill malignant BC MCF-7 cells to determine their potential as BLiP-PDT agents (Fig. 2A). For effective discrimination of cell death and protein probe–binding events, reagents for live cell staining [4′,6-diamidino-2-phenylindole (DAPI), shown in blue] and dead cell staining [SYTOX Green (SG) dye, shown in green] were added to the cell culture medium after the cells were treated with the protein probes. KR (shown in red) was also imaged to confirm the presence of Luc-RGP-LPs in the cell membranes. In Fig. 2B, cell death was not observed after treatment with the various protein probes in the absence of light (left); in contrast, in the presence of Co-h–stimulated BL (right), Rluc8.6-KR-LP notably induced cell death as shown by the absence of DAPI staining and the strong SG staining. Cell surface–binding events were only observed for Rluc8.6-KR-LP (in red from KR) in both cases. Unexpectedly, control cells (without protein treatment) and cells treated with control protein probes (KR and Rluc8.6-KR) showed neither protein binding nor cell death even in the presence of BL. Quantitative data assessed by fluorescence-activated cell sorting (FACS) showed that the proportion of Rluc8.6-KR-LP–treated cells was drastically increased to 87.5% of the total cells, compared with the untreated control (1.2%) or Rluc8.6-KR–treated cells (3.4%); the subsequent addition of Co-h caused a significant increase in SG-stained dead cells and a substantial decrease in DAPI-stained viable cells with Rluc8.6-KR-LP treatment (Fig. 2C). These quantitative data were comparable with the values of mean FL intensity in confocal images (fig. S3, E and F). We showed that a higher Co-h concentration and a longer incubation time for Co-h resulted in greater cytotoxic effect of Rluc8.6-KR-LP (Fig. 2D). Similar to Rluc8.6-KR-LP, Rluc8-MS-LP also considerably induced cell death in the presence of BL and was dependent on the substrate concentration (fig. S4). The BL- or LED-mediated induction of cell death was confirmed by the MTT [3-(4,5dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (fig. S5). When we investigated whether the protein probe worked in serum-containing medium, no significant difference was observed in the cytotoxic effects in serum-free and serum-containing medium at >8 hours after protein treatment (Fig. 2E); effects were apparent for Rluc8.6-KR-LP treatment concentrations above 1 μM (fig. S6). These results clearly indicated that Luc-RGP-LPs had a higher binding specificity to MCF-7 cells and successfully induced cytotoxic effects through BLiP-PDT reactions.
Fig. 2. Cytotoxic effect of BLiP-PDT in MCF-7 cells.
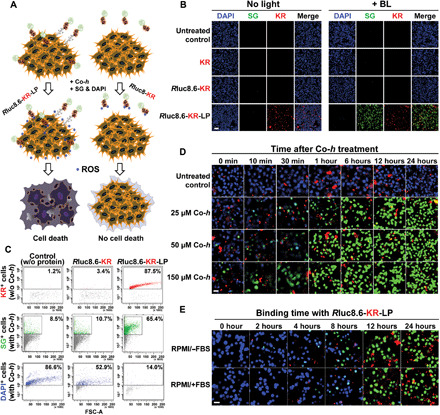
(A) Schematic of targeted cancer therapy using BLiP-PDT with Rluc8.6-KR-LP (left) or Rluc8-KR (right). (B) Fluorescence confocal images of protein-untreated or protein-treated cells without light and with Co-h for BL. Images were obtained after sequential treatment with SG and DAPI. Multiple colors were obtained from DAPI [blue; ex, 360 nm], SG (green; ex, 504 nm), and KR (red; ex, 585 nm). Scale bar, 100 μm. (C) Quantitative FACS analysis of KR-binding cell populations (top) and dead (SG-positive, center) and live (DAPI-positive, bottom) cell populations. All conditions were similar to those of (B). (D) Fluorescence confocal images of MCF-7 cells with 10 μM Rluc8.6-KR-LP at 37°C for 24 hours over time (0 to 24 hours) after Co-h treatment at different concentrations (final, 0 to 150 μM). Images at various incubation times were obtained after sequential treatment with SG and DAPI. Scale bar, 20 μm. (E) Fluorescence confocal images of MCF-7 cells over time (0 to 24 hours) after binding with 10 μM Rluc8.6-KR-LP in FBS-free (top) and FBS-containing (bottom) RPMI medium. Co-h (final, 150 μM) was added to the protein-treated cells and incubated for 30 min at 37°C to ensure ROS generation. Images were obtained after sequential treatment with SG and DAPI. Scale bar, 20 μm.
Next, we investigated whether BLiP-PDT could effectively impair a panel of known BC cell lines (Fig. 3, A to D) and primary triple-negative BC cells (TNBCs) from patients (Fig. 3, E and F). The cytotoxic effect of Rluc8.6-KR-LP and Rluc8-MS-LP with Co-h was assessed; each cell line was found to respond differently. MCF-7, MDA-MB-435, and MDA-MB-231 cells were similarly damaged by both protein probes; however, there was no effect on BT-474, SK-BR-3, and MCF-10A cells (Fig. 3, A to D). Notably, the cell line–dependent and light-activated cytotoxic effects observed were similar upon LED light illumination (fig. S7). Neither partial binding events nor marginal cytotoxic effects were observed in the three unaffected cell lines, even after 2 hours of Co-h treatment. Although BLiP-PDT shows selective treatment of some BC cell lines, Luc-RGP-LP–targeted cells were not correlated with the known receptors in peptide-binding outcomes [i.e., KRT1 (keratin 1), ER (estrogen receptor), PR (progesterone receptor), HER2 (human epidermal growth factor receptor 2), AR (androgen receptor), and EGFR (epidermal growth factor receptor); table S2]. For example, two basal-like TNBC cell lines (MDA-MB-231 and MCF-10A cells) and even AR-expressing cells (i.e., all tested cell lines except MCF-10A) responded heterogeneously. Notably, mRNA and protein expressions of integrin β1 (ITGB1) were highly correlated with cytotoxicity by Luc-RGP-LP, as compared with those of KRT1 (table S2 and fig. S8). It is likely that the peptide sequence may bind to integrin subunits, based on previous reports representing the biodistribution similar to RGD in tumor-bearing mice (27) and high expression levels of integrin β1, β5, and ανβ5 in the three targeted BC cell lines (28). Although no further exploration of the peptide-binding target was conducted in this study, this strategy is advantageous for BC therapy because this probe could simultaneously target a variety of intrinsic subtypes at different stages of BC, as defined by gene expression profiling and clinical outcomes among highly heterogeneous BC (29). To determine its feasibility, we examined the cytotoxic effect of Luc-RGP-LPs in three primary TNBCs from patients with malignant BC (table S3). Compared with the untreated control cells (no LED and no BL), BL resulted in a greater proportion of cell death than LED with Rluc8.6-KR-LP in primary TNBCs but not with Rluc8.6-KR (Fig. 3E). Unlike BC cell lines, the KR- and SG-stained areas in the three primary TNBCs cell lines did not overlap perfectly; the SG-stained area was more extensive than the KR-stained areas (Fig. 3F). We assumed that KR was not detectable at very low concentrations but could still emit ROS. This result indicates that BLiP-PDT using Luc-RGP-LPs was able to impair primary human breast tumors with heterogeneous subtypes exhibiting different phenotypic features.
Fig. 3. Cytotoxic effect of BLiP-PDT in various BC cell lines and primary TNBC cells.
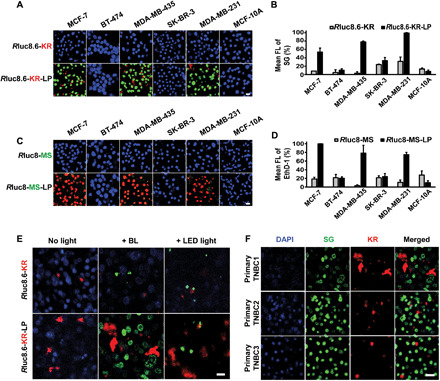
(A) Representative fluorescence confocal images and (B) quantitative graphs in FL intensity of six BC cell lines treated with Rluc8.6-KR (top) or Rluc8.6-KR-LP (bottom). Blue, green, and red colors were obtained from DAPI, SG, and KR, respectively, as described in Fig. 2B. Scale bar, 20 μm. (C) Representative fluorescence confocal images and (D) quantitative graphs in FL intensity of six BC cell lines treated with Rluc8-MS (top) or Rluc8-MS-LP (bottom). Blue, green, and red colors were obtained from DAPI, MS, and EthD-1, respectively. Scale bar, 20 μm. (E) Fluorescence confocal images of patient primary TNBC cells treated with Rluc8.6-KR (top) or Rluc8.6-KR-LP (bottom) without and with light irradiation (BL or LED right). Blue, green, and red colors were obtained from DAPI, SG, and KR, respectively. Scale bar, 20 μm. (F) Fluorescence confocal images of three primary TNBC cells from patients after BLiP-PDT with Rluc8.6-KR-LP. Multiple colors were obtained from DAPI (blue), SG (green), and KR (red). Scale bar, 20 μm.
Real-time monitoring of cell death by BLiP-PDT
To gain an insight into the mechanism of cell death, we monitored the cytotoxic effect of the BL-induced photodynamic protein probe in MCF-7 cells using real-time optical diffraction tomography (ODT). Unlike classical phase-contrast microscopy and differential interference microscopy, this tomographic technique provides label-free and quantitative three-dimensional (3D) mapping of the complex refractive index (RI) in live cells (30); thus, RI distribution with additional FL imaging of SG in MCF-7 cells (targeted cells with Rluc8.6-KR-LP) was clearly distinguished from the untargeted cells (naïve and Rluc8.6-KR–treated MCF-7 cells; Fig. 4, and also, see movies S1 to S3). Upon the addition of Co-h, rapid diffusion of the SG dye proceeded (within a few minutes), and the presence of swollen microsized vesicles (or blebs) from the plasma membrane was only observed in the MCF-7 cells treated with the targeted probe (from 30 min after Co-h treatment) (Fig. 4A). The dynamic fluctuations in dry mass (Fig. 4B) and volume (Fig. 4C) were quantitatively measured from multiple cells (n = 30) based on the average density of RI tomograms: control MCF-7 (M/M0: 0.975 ± 0.040 and V/V0: 1.028 ± 0.077 at 60 min), MCF-7 with Rluc8.6-KR (M/M0: 0.880 ± 0.083 and V/V0: 1.003 ± 0.055 at 60 min), and MCF-7 with Rluc8.6-KR-LP (M/M0: 0.691 ± 0.110 and V/V0: 1.133 ± 0.163 at 60 min). These findings supported the fact that a greater decrease in dry mass and a larger volume fluctuation occurred in the targeted cells (by the destruction of condensed organelle structures) compared with the control cells. These reconstructed 3D RI tomograms of MCF-7 cells, unlike classical diffraction-limited images, precisely and quantitatively provide structural and biophysical information on the manner in which MCF-7 cells are ruptured by the BLiP-PDT. In contrast to a previous report about dye-labeled peptides (27), the peptide-conjugated protein (Rluc8.6-KR-LP), which has a high molecular weight (approximately 68.2 kDa), was not easily internalized through the cell membrane; instead, it appeared to remain outside the cell membrane until the cell burst, as further evidenced by the fluorescent 3D images of KR detected using ODT (Fig. 4D). The membrane-bound probe triggered the rapid membrane rupture via light-activated ROS, subsequently inducing a marked increase in intracellular ROS in organelles (endoplasmic reticulum or mitochondria) and the cytosol, as shown by the live cell–permeable ROS-responsible dyes (fig. S9). These findings strongly support that, in the presence of Co-h, targeted BLiP-PDT initially induces cell necrosis through the loss of cell membrane integrity, which agreed with data obtained using compromised plasma membrane–permeable dyes, such as SG (Fig. 2, B to E) and EthD-1 (fig. S4). We speculated that the rapid and irreversible damage of the cell membrane may be predominantly necrotic rather than apoptotic; however, the necroptosis- or pyroptosis-mediated pathways could not be excluded, as observed for other plasma membrane–targeted cell death events (31, 32).
Fig. 4. 3D ODT images by RI mapping in BL-sensitizing MCF-7 cells.
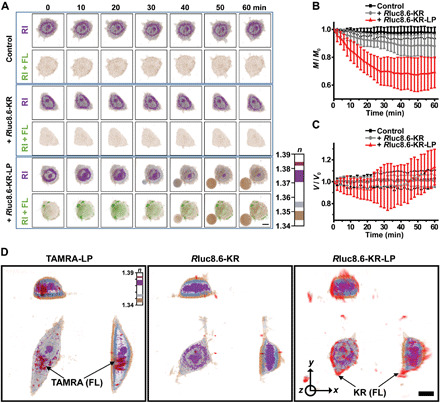
(A) ODT images of live MCF-7 cells showing 3D morphological changes by RI distribution in controls (no protein probe, top), Rluc8.6-KR–treated (center), or Rluc8.6-KR-LP–treated cells (bottom). Cells were incubated with protein probes in serum-containing medium at 37°C for 2 hours, and time-lapse images were obtained at 10-min intervals immediately after sequential treatment with Co-h and SG. Each RI image was overlaid with the corresponding FL image of SG and shown below (RI + FL). Scale bar, 10 μm. See also movies S1 to S3. (B) Dynamic changes in dry mass (M/M0) and (C) volume (V/V0) based on 3D RI distribution in live cells (n = 30). Measurements were taken at 2-min intervals, and error bars indicate SD values. (D) FL-combined 3D RI images of live MCF-7 cells incubated with TAMRA-LP, Rluc8.6-KR, or Rluc8.6-KR-LP in serum-containing medium at 37°C for 2 hours. FL images were obtained at an excitation wavelength of 575 nm; the RI color bar indicates the RI values in each cell. Scale bar, 10 μm.
In vivo effect of BLiP-PDT on tumor xenograft mouse model
We next sought to evaluate in vivo efficacy of BLiP-PDT in a mouse model xenografted with breast tumor cells (Fig. 5). Despite using MCF-7 as a standard BC cell line, invasive and triple-negative MDA-MB-231 cells, known to be insensitive to antiestrogen treatment (33), were subcutaneously implanted in NOD SCID-γ immunocompromised mice. The protein probe Rluc8.6-KR-LP (denoted RKL) was intratumorally injected into MDA-MB-231–bearing mice, followed by subcutaneous spread of Co-h; tumor growth was monitored over time (Fig. 5A). With the identification of tumor-growing sites in three individual mice (Fig. 5B), we observed a strong FL intensity at tumor sites treated with the RKL probe (top image in Fig. 5C), whereas a strong BL intensity was observed only when the RKL probe and the substrate (Co-h) were simultaneously administered (bottom image in Fig. 5C), confirming the target binding and substrate-dependent BL generation. We found that consecutive infusions of RKL and Co-h significantly inhibited the growth of the tumor, which exhibited a smaller size (127 mm3) at day 34, compared with the two control groups (523 and 868 mm3, in Fig. 5, D and E). In the histological analysis, hematoxylin and eosin (H&E) and Ki-67–expressed immunohistochemistry (IHC) images from tissue sections of protein or substrate-untreated tumors indicated malignant cell proliferation and extensive Ki-67 expression, which are characteristic of breast tumors (34). In contrast, tissue section images of protein/substrate-treated tumor sections indicated considerable inhibition of malignant cell proliferation, with low expression of Ki-67 (Fig. 5F). To further demonstrate in vivo therapeutic effect, BLiP-PDT was compared with laser light–irradiated proteinaceous RKL probe and chemical PS (RB) that has similar excitation wavelength to KR (fig. S10, A to D). When three repeated injections of probes into the tumor-growing sites of MDA-MB-231–bearing mice with the subsequent light irradiations at 530 nm were performed, RKL- and RB-based PDTs did not cause a significant reduction in tumor growth despite intense light intensity (1440 W for 15 min), indicating limited light transmittance and ROS generation at a given light irradiation. In contrast, BLiP-PDT, similar to that in Fig. 5, showed more significant inhibition in tumor growth than other groups including phosphate-buffered saline (PBS)–treated and untreated controls. In addition, RB-based cytotoxicity was observed nonspecifically at relatively high concentrations and prolonged irradiation time (fig. S10, E to G), compared with that (fig. S5) by protein probe. Thus, the suppression of BC tumors by a BL-sensitizing protein probe in the tumor xenograft mouse model suggested the significant potential of BLiP-PDT for targeted therapy of BCs in vivo.
Fig. 5. In vivo effect of BLiP-PDT in tumor xenograft mouse model.
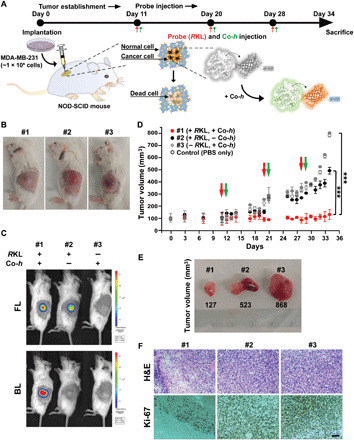
(A) Experimental timeline for the in vivo studies. The xenograft mouse model was established by subcutaneous injection of 1 × 106 MDA-MB-231 cells into NOD-SCID-γ immunocompromised mice. After 10 days, 50 μM Rluc8.6-KR-LP (RKL; final, 1 nmol) and 245 μM Co-h (final, 5 μg) were simultaneously injected into mice three times. (B) Light-field optical images of mice bearing breast tumors after implantation. A representative image from each group (n = 5) is presented (photo credit: Y.K.K., Asan Medical Center). (C) In vivo FL (top) and BL (center) images of mice bearing breast tumors immediately after the injection of RKL probe and Co-h (#1). Negative controls, #2 (without Co-h) and #3 (without RKL probe), were compared. (D) Tumor growth curves of #1 to #3 mice. The injection time points for the RKL probe and Co-h are indicated in red and green, respectively. Error bars indicate SD values (n = 5 per group); the significance of the difference in tumor volume between the control (closed gray circle) and test mice (red or black circles) was evaluated (***P < 0.001, Student’s t test). (E) Representative tumor resection optical image is displayed from #1 to #3 after the completion of BLiP-PDT (photo credit: Y.K.K., Asan Medical Center). (F) H&E-stained (top) and IHC-stained (bottom) images from tumors shown in (C). IHC was performed using anti–Ki-67 (cell-dividing marker) antibody. Scale bar, 200 μm.
DISCUSSION
We demonstrated the novel application of BL in PDT, using Luc-RGP-LPs, to impair BC in vitro and in vivo. The self-luminescent BL, which does not require external light irradiation, promoted ROS generation (superoxide or singlet oxygen) through a BRET process with RGPs. Owing to the specific binding capability of LP to BC cells, BLiP-PDT exhibited the BC-targeted cytotoxicity, thus presenting a significant advance in therapy by overcoming two major limitations of conventional PDT technology: (i) the risk of harm from photosensitizing chemicals and (ii) the requirement of a light source and expensive device configurations. Although PDT has a long history of use for the treatment of various cancers, special precautions regarding the use of chemical PS prodrugs must be taken until the PS drugs have been eliminated from the human body, because their aberrant accumulation and light-activated reactions may cause severe side effects (10, 11, 35). They also passively accumulate around tumor sites owing to the enhanced permeability retention effects in leaky vasculature and tortuous blood vessels or the overexpression of low-density lipoproteins in tumor cells (36); thus, this may increase nonspecific accretion at high concentrations and excessive ROS generation upon irradiation.
In contrast, our BLiP-PDT system relies on a protein probe with high target specificity, self-luminescence, and low phototoxicity. First, the protein probe with an LP target ligand did not influence normal or untargeted cells at all; in addition, the LP-deficient protein probe did not damage target cells upon light illumination (Figs. 2 and 3). This indicates that target specificity of BLiP-PDT to cancer cells was achieved by LP, unlike chemical PDT using RB (fig. S10, E to G). Second, considering an average emission value of 1.5 × 1022 photons s−1 per mole of Rluc (37), the total photon emission from BL using 5 nmol Rluc was 4.5 × 1016 photons over 10 min, which was ~3.7 × 102–fold lower (only 0.26%) than that (1.7 × 1019 photons over 10 min) of LED light irradiation (10 mW cm−2; see more details in note S1). The superior effect of BLiP-PDT on in vivo tumor therapy under the intratumoral injection condition was demonstrated by comparison with proteinaceous or chemical PDT by laser light irradiation (fig. S10). These results indicated that the extremely low BL intensity generated the ROS by BRET transiently and locally at close proximity to targeted regions (approximately <20 nm), enabling the cancer treatment by BL-activated ROS generation. This observation also makes it possible to use this method as an alternative to classical optogenetic control, as reported earlier (38). Moreover, as RGPs have a lower phototoxicity than chemical PSs (15), the combination of Luc and RGP for BLiP-PDT is particularly advantageous for minimizing photodamage to normal tissues. This potential was identified in MCF-10A cells (Fig. 3, A and C), which are considered a normal and nontransformed control in many BC studies (39). BLiP-PDT does not encounter any light penetration issue in vivo, providing a simple therapeutic platform without an external light source, which is not yet possible in classical PDT methods. Last, this BLiP-PDT method can be also combined with immune therapy with the release of intracellular damage-associated molecular patterns via the inflammatory necrotic pathway (40).
In terms of protein stability and degradability, the protein probe used showed high stability in the serum for 48 hours in vitro despite a short lifetime of BL (fig. S11); in contrast, only a weak FL signal was observed at the tumor sites in the mouse model at 24 hours after the intratumoral injection of protein probe (fig. S12), which was presumably due to rapid proteolysis in the in vivo environment. This property may preclude the use of protein probes for long-term PDT to suppress tumor recurrence. As frequently observed in protein delivery studies in vivo, this protein probe administered intravenously was not successful in tumor targeting in tumor xenograft mouse models. Although further development and optimization should be addressed, this may be attributed to its slow intravenous infusion by large size, its short biological half-life by rapid degradation, or dispersion by high expression of ITGB1 in organs. In addition, at this time, this strategy requires repeated injections of fusion protein and Co-h to achieve tumoricidal effect, which may be a limitation for clinical use. Nevertheless, we believe that these limitations will be overcome and outweighed by the obvious advantages of BLiP-PDT as a unique and alternative method to significantly diminish the side effects in classical PDTs. For example, protein engineering or combination strategy with antibody fragments or other polymers may allow the development of improved BLiP-PDT probes that achieve higher targeting precision in vivo. It is noteworthy that Luc-RGP not only served as a proteinaceous PDT material that does not require gene delivery but also functioned at relatively lower concentrations and with a shorter lifetime than previously reported chemical PSs or hybrid nanomaterials (3, 4).
In conclusion, we demonstrated the BLiP-PDT method as a new type of PDT using luciferase-fused RGP. By taking advantage of self-triggered ROS production, we showed that BLiP-PDT selectively induced damage to breast tumors by destroying their plasma membranes without affecting untargeted cells. The use of various combinations of luciferases and RGPs to broaden the scope and versatility of BLiP-PDT will render this approach suitable for immediate use as a promising theranostic method against various cancers. In addition, our developed method will potentiate many opportunities for further study, including BL-activated perturbation and differentiation of target cells, with enhanced simplicity and specificity.
MATERIALS AND METHODS
Protein expression, purification, and in vitro characterization
The KR gene was amplified by pCS2-NXE + mem-KR (Addgene, no. 45761 plasmid) using two primers (forward, 5′-CGG AAT TCG GAG GAG GAA TGC TGT GCT GTA TGA GAA G-3′; reverse, 5′-CCC AAG CTT CTA ATC CTC GTC GCT ACC GAT G-3′) and cloned into the pRSETB-Rluc8.6 plasmid containing an N-terminal His6-tag in the laboratory, with Eco RI and Hind III (Takara) restriction sites used to create pRSETB-Rluc8.6-KR. An LP sequence (Trp-Leu-Glu-Ala-Ala-Tyr-Gln-Arg-Phe-Leu) was sequentially cloned into the C terminus of pRSETB-Rluc8.6-KR using a forward primer (5′-CGG AAT TCA TGG GAG GAG GAC TGT GCT GTA TGA GAA G-3′) and three reverse primers (first, 5′-CGG CCT CCA GCC ATC CTC CTC CAT CCT C-3′; second, 5′-AAG CGC TGG TAG GCG GCC TCC AGC CAT C-3′; and third, 5′-CCC AAG CTT CTA CAG GAA GCG CTG GTA GGC G-3′). The Eco RI site was flanked with the KR gene on the upstream (5′), while Hind III was flanked with the LP gene on the downstream (3′). The PCR product was digested with Eco RI and Hind III and ligated into pRSETB-Rluc8.6-KR digested by the same enzymes to create pRSETB-Rluc8.6-KR-LP. Because the N terminus of the KR within a dimer is sufficiently separated than C terminus, fusion of Rluc8.6 at the N terminus and a short LP at the C terminus of KR can avoid structural constraint in the dimeric protein. The MiniSOG (MS) gene was amplified by mCherry-miniSOG-N1 (Addgene, no. 54803 plasmid) using two primers (forward, 5′-CGG AAT TCG GAG GAG GAA TGG AGA AGA GCT TCG TG-3′; reverse, 5′-CCC AAG CTT CTA GCC GTC CAG CTG CAC G-3′) and cloned into the pRSETB-RLuc8 plasmid containing an N-terminal His6-tag in the laboratory, with the Eco RI and Hind III restriction sites used to create pRSETB-Rluc8-MS. An LP with the sequence was sequentially cloned into the C terminus of pRSETB-Rluc8-MS using a forward primer (5′-CGG AAT TCA TGG GAG GAG GAC TGT GCT GTA TGA GAA G-3′) and three reverse primers (first, 5′-GGC CTC CAG CCA TCC TCC TCC GCC G-3′; second, 5′-AGC GCT GGT AGG CGG CCT CCA GCC ATC-3′; and third, 5′-CCC AAG CTT CTA CAG GAA GCG CTG GTA GGC G-3′). Eco RI site was flanked with the MS gene on the upstream (5′), while Hind III was flanked with the LP gene on the downstream (3′). The PCR product was digested with Eco RI and Hind III and ligated into pRSETB-Rluc8-MS digested by the same enzymes to create pRSETB-Rluc8-MS-LP. Three glycines (GGG) were inserted as linker sequences between the independent protein domains. For protein expression and purification, 500 ml of Luria broth supplemented with ampicillin (100 mg ml−1) was inoculated with Escherichia coli BL21 harboring plasmids. The culture was grown at 37°C until the optical density at 600 nm (OD600) reached 0.6 to 0.8. Expression was conducted at 20°C for 20 hours and induced using 1 mM isopropyl β-d-thiogalactoside (IPTG). The cells were then harvested by centrifugation at 7800 rpm for 20 min at 4°C and resuspended in 20 ml of buffer A [50 mM sodium phosphate containing 300 mM NaCl and 10 mM imidazole (pH 8.0)]. The resuspended pellet was lysed by sonication in the presence of 40 mg of lysozyme, followed by sequential centrifugations of 7800 rpm for 20 min at 4°C and 14,000 rpm for 15 min at 4°C. The His6-tagged expressed proteins were isolated from the supernatant using an Ni2+-NTA agarose-filled column (Pierce) equilibrated with buffer A. The column was washed thrice with 5 ml of buffer B [50 mM sodium phosphate containing 300 mM NaCl and 20 mM imidazole (pH 8.0)]. The bound protein was eluted from the column by 5 ml of buffer C [50 mM sodium phosphate containing 300 mM NaCl and 250 mM imidazole (pH 8.0)]. The buffer in the eluent proteins was replaced by 1× PBS (pH 7.4) using a PD-10 desalting column (GE Healthcare). For large amounts, protein purification was performed with a fast protein liquid chromatography (AKTA, Pharmacia Biotech) system using a HisTrap His-tag protein purification column (GE Healthcare) followed by gel filtration on a Superdex 200 HR 10/30 column (Pharmacia Biotech). The concentration of the purified fusion protein was estimated from the measurement of the absorbance at 280 nm based on calculated extinction coefficient values. The protein product was loaded onto 5 to 12% acrylamide gel under reducing or nonreducing conditions and estimated from the intensities of bands obtained from sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). For denaturation, proteins were incubated at 95°C for 5 min and loaded onto the SDS-PAGE gel. The gel was examined by FL imaging using a custom-built transilluminator equipped with an LED lamp; bands in the gel were visualized by Coomassie Brilliant Blue staining. Native PAGE (10% polyacrylamide) was also performed to analyze nondenaturating proteins in the absence of SDS. To characterize the photochemical properties of the proteins, the absorbance spectra of 10 μM of single or fusion proteins dissolved in 1× PBS (pH 7.4) were measured using a ultraviolet-visible spectrophotometer (Cary 60, Agilent Technologies); FL and BL spectra were measured using a microplate reader (Varioskan, Thermo Fisher Scientific). For BL measurement, luciferase-fused proteins were analyzed immediately after treatment with 50 μM Co-h (Nanolight) dissolved in 1× PBS (pH 7.4). For the stability test of Rluc8 (or Rluc8.6) or its fusion proteins, 10 μM protein was incubated in 1× PBS (pH 7.4) or undiluted normal mouse serum (Jackson ImmunoResearch, no. 015-000-001) for different time intervals (1, 2, 4, 8, 12, 24, and 48 hours) at 37°C. The BL intensity of each reaction was measured using a GLOMAX Luminometer (Promega) immediately after treatment with 150 μM Co-h. The FL and BL images of proteins on a microplate were obtained using an IVIS Spectrum (PerkinElmer). FL images were collected with an excitation filter of 465 to 570 nm and an emission filter of 520 to 620 nm; BL images were obtained using appropriate emission filters (500 nm for blue, 520 nm for green, and 630 nm for red) without an excitation filter immediately after Co-h injection.
Matrix-assisted laser desorption/ionization–mass spectrometry analysis
The desalting and purification of protein digests were performed using ZipTip C4 microcolumns. Briefly, the ZipTip C4 was wetted five times with 10 μl of 100% acetonitrile and equilibrated with 10 μl of 0.1% TFA in pure water. The protein solution was slowly aspirated and dispensed 10 times to bind the proteins to the microcolumns. The desalting was done by aspirating and dispensing 10 μl of 0.1% TFA through the ZipTic C4 columns five times. Last, the proteins were eluted with 10 μl of the matrix solution [10 mg of sinapinic acid (SA) in 1 ml of 70% acetonitrile in water]. A 1-μl aliquot of the purified protein was dropped on the target plate and mixed with SA as a matrix. SA was dissolved in 70:30 (v/v) acetonitrile/water at a concentration of 10 mg ml−1. The MS spectra were obtained in a linear mode using a Bruker UltrafleXtreme matrix-assisted laser desorption/ionization (MALDI) MS instrument (Bremen, Germany) equipped with a SmartBeam II laser.
ROS measurement
For in vitro ROS analysis, superoxide and singlet oxygen were measured using DHE (Thermo Fisher Scientific, no. D7008) and ADPA (Abcam, no. ab145414), respectively. First, 10 μM purified protein was mixed with 100 μM DHE or 50 μM ADPA in an ROS reaction buffer [50 mM Hepes-KOH (pH 7.4)] for a final volume of 100 μl. The mixture was irradiated with LED light (10 mW cm−2, 30 min) or Co-h (final, 150 μM) treatment. Unless otherwise stated, MS or its conjugate was preincubated with flavin mononucleotide (FMN; Sigma-Aldrich, no. F6750) before use. The ROS level was analyzed before and after LED light or BL by measuring the decrease in FL intensity of DHE or ADPA. The change in FL was measured using a microplate reader set to an excitation wavelength of 370 nm and emission wavelength of 420 nm for DHE, or an excitation wavelength of 380 nm and emission wavelength of 430 nm for ADPA. This excitation wavelength had a minimal effect on the excitation of KR or MS. To measure the ROS-scavenging effect, three representative scavengers were used: SOD (superoxide anion–specific scavenger, Sigma-Aldrich, no. S8160), sodium azide (NaN3, singlet oxygen-specific scavenger, Sigma-Aldrich, no. S8032), and d-mannitol (hydroxyl radical–specific scavenger, Sigma-Aldrich, no. M4125). Each scavenger (final, 100 mM) was initially mixed with different proteins (final, 10 μM) in the ROS reaction buffer, and then DHE (final, 100 μM) or ADPA (final, 50 μM) was added to the mixture. The change in FL of DHE or ADPA was measured before and after irradiation by LED light or Co-h–stimulated BL light in the control group (no scavenger). For imaging of intracellular ROS stress in live cells, MCF-7 cells treated with protein probes were incubated with 10 μM CellROX Green (Thermo Fisher Scientific, no. C10444) or 10 μM CellROX Deep Red (Thermo Fisher Scientific, no. C10422) in serum-free medium for 30 min at 37°C. After treatment without or with Co-h (final, 150 μM) for 30 min, the cells were imaged using a confocal microscope (Nikon C2si+, Japan) at 485-nm excitation and 520-nm emission for CellROX Green or at 640-nm excitation and 665-nm emission for CellROX Deep Red. To avoid overlapping the FL colors, CellROX Green was used for Rluc8.6-KR-LP and CellROX Deep Red for Rluc8-MS-LP. The mean FL intensity in each well was quantified using a microplate reader.
Cell culture and FL imaging
Human breast cell lines MCF-7, SK-BR-3, MDA-MB-231, and BT-474 were obtained from the Korean Cell Line Bank. MDA-MB-435 and MCF-10A cells were from the American Type Culture Collection. Primary BC cells from patients were obtained from Asan Medical Center, and the study was approved by the institutional review boards (IRBs) of Asan Medical Center (IRB approval number 2014-0800). Cells were propagated in either RPMI 1640 (HyClone, no. SH30027; for MCF-7, SK-BR-3, and BT-474 cells) or Dulbecco’s modified Eagle’s medium (DMEM; HyClone, no. SH30243; for MDA-MB-231 and MDA-MB-435 cells) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1× penicillin-streptomycin (Thermo Fisher Scientific). Noncancerous MCF-10A cells were cultured in Mammary Epithelial Cell Base Medium (Lonza, no. CC-3150) supplemented with additives. All cells were inoculated in cell culture flasks (T-75, SPL, no. 70075) and dissociated from the flasks using trypsin-EDTA (Gibco, no. 25200-072) and centrifugation (1000 rpm, 3 min). Subsequently, 100 μl of the solution containing the appropriate concentration of cells was transferred to each well of a polystyrene, flat-bottomed, 96-well tissue culture plate (SPL, no. 30096) using a multichannel pipette to yield an initial confluency of 40 to 50%. The cells were grown to ~5 × 105 cells ml−1 in a humidified 5% CO2 incubator at 37°C. When cell confluency reached 80 to 90%, all control and test protein probes were added to the culture medium in the presence or absence of FBS, and the cells were incubated for 24 hours. After the cells were washed and the medium was refreshed, the culture plate was subjected to BL (by the addition of Co-h) or LED light irradiation for an appropriate time. The cells were incubated with serum-free medium containing 261 nM SYTOX Green Nucleic Acid Stain (SG, Invitrogen, no. S7020) or 1× ethidium homodimer (EthD-1, Cell BioLabs, no. 108003) for 30 min at 37°C to monitor and quantify dead cells. Cells were then treated with DAPI for 5 min by dropping Vectashield mounting medium with DAPI (Vector Laboratories, no. H-1200) to monitor and quantify live cells. All images were collected using a confocal microscope at different time points (e.g., time after treatment with proteins, Co-h, or LED light) or different concentrations (e.g., proteins or Co-h). The excitation and emission wavelengths for KR, MS, SG, EthD-1, and DAPI images were at 585/610, 448/500, 504/523, 525/590, and 360/460 nm, respectively. To avoid overlapping the FL colors, SG was used for Rluc8.6-KR-LP and EthD-1 for Rluc8-MS-LP. For external light irradiation, a cell-cultured microplate was placed on a custom-built transilluminator equipped with an LED lamp, and cells were excited at a light power density of 10 mW cm−2 using an appropriate filter set. The data were analyzed using ImageJ (National Institutes of Health) or NIS-Elements AR (ver. 4.40, Advanced Research). All SG or EthD-1 positively stained areas were normalized to the untreated control to represent the cytotoxic effects of BLiP-PDT. In addition, for colorimetric cell viability test, the cells were incubated with fresh medium containing MTT (0.5 mg ml−1; Cell Biolabs, no. 125201) for 2 hours at 37°C. After the culture medium was removed, 100 μl of dimethyl sulfoxide (DMSO; Sigma-Aldrich) was added to each well to induce the reduction of MTT by mitochondrial dehydrogenases in living cells. The optical density in each well was measured at 570 nm using a microplate reader; the stained cells were imaged using a bright-field microscope (Olympus CKX41-A32PHP).
Flow cytometric analysis
MCF-7 cells were incubated with 10 μM Rluc8.6-KR or 10 μM Rluc8.6-KR-LP in serum-free RPMI for 24 hours at 37°C in 96-well round-bottomed microplates (SPL, no. 34069). To assess the binding effect of the protein probe on the cells, protein-untreated (control), Rluc8.6-KR–treated, and Rluc8.6-KR-LP–treated cells were analyzed using a flow cytometer (FACSCanto, BD Biosciences) equipped with 405-, 488-, and 633-nm lasers (BD Biosciences). To monitor the cytotoxic effects, these three cell types were also treated with Co-h (final, 150 μM) and incubated for 24 hours at 37°C, followed by the sequential addition of SG and DAPI. Before FACS analysis, the cells cultured on microplates were dissociated from the wells using trypsin-EDTA and centrifuged at 1000 rpm for 3 min. The cells were filtered through a 100-μm cell strainer and resuspended in 400 μl 1× DPBS containing 5% FBS. We collected 10,000 to 50,000 events using a forward scatter threshold of 10,000. The data were collected for pulse height, area, and width parameters for each channel. For KR, data were collected with the 633-nm laser and 660/20-nm bandpass filter. For SG, data were collected with the 488-nm laser and 530/30-nm bandpass filter. For DAPI, data were collected with the 405-nm laser and 450/50-nm bandpass filter. Flow cytometry data were analyzed using FlowJo Software (TreeStar). For histogram analysis, flow cytometry data in .FCS format were exported into a text-based format using FlowJo software (ver. 10.6.1., FlowJo, LLC) and analyzed in Microsoft Excel.
Optical diffraction tomography
MCF-7 cells (1 × 104) were subcultured in a TomoDish (50 mm by 10 mm, Tomocube) and maintained in RPMI 1640 for 24 hours; subsequently, fresh medium containing Rluc8.6-KR or Rluc8.6-KR-LP (final, 10 μM) was added, followed by incubation for 2 hours at 37°C in humidified 5% CO2. The cells were washed twice with 1× PBS (pH 7.4), and then, the medium was replaced with RPMI 1640 containing Co-h (final, 150 μM) and SG (final, 261 nM). 3D cell images with RI distribution were acquired using ODT, as described previously (30), with a Mach Zehnder interferometric microscope. A commercial microscope (HT-2H, Tomocube) was used to construct a 3D RI map of cells through the reconstruction of a 2D RI distribution of the isosurface. The visualizations of 3D RI distribution were rendered using Tomostudio (HT-2H, Tomocube). The cell quantitative changes in volume and dry mass were calculated by the built-in Tomostudio. The data were presented as the means ± SD.
In vivo BLiP-PDT in tumor xenograft
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Asan Medical Center and performed in compliance with the institutional guidelines (IACUC approval number 2018-12-194). MDA-MB-231 cells were grown in DMEM supplemented with 5% FBS and 1% penicillin and streptomycin. The cultured cells were suspended in DMEM at a density of 1 × 106. Then, 50 μl of cells was mixed with 50 μl of Matrigel (Corning, no. 354248); the total solution (100 μl) was subcutaneously inoculated into the pad of 7-week-old nonobese diabetic–severe combined immunodeficient (NOD-SCID) mice. When the tumor volume reached close to approximately 100 mm3 after 10 days, the mice were intratumorally injected with 20 μl of Matrigel-containing protein (50 μM Rluc8.6-KR-LP; final, 1 nmol) suspended in 1× PBS three times for 34 days (n = 5). On the basis of the sufficient concentration of protein probe in serum-containing culture media (10 μM at 100 μl equivalent to final 1 nmol; fig. S6), its molecular weight (1 nmol corresponds to 68 μg at 68 kDa), and the typical average mouse weight of 25 g, we determined the amount of weight-adjusted single dose (~2.7 mg/kg), which is within a reasonable range for clinical use. At 1 day after protein injection, 50 μl of 245 μM Co-h solution (final, 12.25 nmol equivalent to 5 μg) suspended in 1× PBS was subcutaneously injected. The sequential injection (protein probe and Co-h) at 1-day intervals was performed three times on days 11/12, 20/21, and 28/29 for 34 days. Mice injected with PBS only and mice injected with either protein probe or Co-h were used as control groups (n = 5 for each group). The tumor size was measured every day using a digital caliper (GemRed, no. AP952910); the tumor volume (mm3) was expressed as (length × width2)/2, where length represents the largest tumor diameter (mm) and width represents the perpendicular tumor diameter (mm). After the mice were euthanized at 5 weeks of age, the tumor masses were removed, weighed, and fixed in 5% neutral buffered formaldehyde solution for histological analysis or preserved at −70°C. For in vivo imaging, the mice were anesthetized with 2% isoflurane and imaged using an IVIS Spectrum. Mouse BL images were obtained without an excitation/emission filter immediately after Co-h injection; FL images were taken using an excitation filter at 570 nm (from 540 to 590 nm) and an emission filter at 620 nm (from 550 to 650 nm) with an exposure time of 5 s. For comparison of Rluc8.6-KR-LP and RB (Sigma-Aldrich, no. 330000) under light irradiation condition, an in vivo experiment was performed in a similar manner except for light irradiation. After MDA-MB-231 cells at a density of 1 × 106 were subcutaneously inoculated into the mice and grown for 11 days, the mice were intratumorally injected with 20 μl of 50 μM Rluc8.6-KR-LP (final, 1 nmol) or 20 μl of 50 μM RB (final, 1 nmol) suspended in 1× PBS three times on days 12, 21, and 28 for 33 days (n = 4 for each group). At 1 day after protein or RB injection, subcutaneous injection of Co-h solution (final, 5 μg) or laser light irradiation (for 15 min) was subsequently performed on days 13, 22, and 29. The green light irradiation at 530 nm (1.6 J cm−2) was performed using an IVIS spectrum system. Mice injected with PBS only and mice without any injection were used as control groups (n = 4 for each group).
Histological imaging and Western blot
Breast tumor tissues were fixed in 4% paraformaldehyde (PFA), dehydrated, embedded in paraffin, sectioned, and stained with standard H&E. For IHC staining, the samples were incubated with primary antibodies including anti-Ki67 antibody (1:200 dilutions, Abcam, no. ab 16667). The sections were subsequently incubated with secondary antibodies (1:500 dilutions, Ventana, no. 760-4311) and visualized using Universal DAB Detection kit (Ventana, no. 760-500). The images were acquired using a Motic BA400 microscope. Western blots were performed using lysates from six BC cell lines (MCF-7, BT-474, MDA-MB-435, SK-BR-3, MDA-MB-231, and MCF-10A). Samples were subjected to SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were incubated with primary anti-ITGB1 (Santa Cruz, no. sc-374429) and anti-KRT1 (Santa Cruz, no. sc-376224) antibodies. Signals were developed by enhanced chemiluminescence (Thermo Fisher Scientific, no. 32106) using a secondary antibody [anti-mouse immunoglobulin G (IgG) antibody linked with horseradish peroxidase (HRP); Cell Signaling, no. 7076S] and analyzed using an ImageQuant LAS 4000 biomolecular imager (GE Healthcare Life Sciences, Waukesha, WI) and a bundled Multi Gauge 3.0 software. Western blot values in the figures represent the relative densities of the bands normalized to that of using anti–β-actin antibody (Santa Cruz, no. sc-47778).
Statistical analysis
The data are presented as means with SD, as noted in each case. All values of n are provided. For comparisons between two data sets, paired or unpaired two-sided Student’s t tests were used. P < 0.05 was considered to indicate a statistically significant difference; P < 0.05, P < 0.01, and P < 0.001 are indicated with single, double, and triple asterisks, respectively.
Supplementary Material
Acknowledgments
We would like to thank K. Cho and J.H. Choi (KBSI, Ochang, Center of Research Equipment) for the MALDI-TOF/TOF mass spectrometer analysis. Funding: This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (no. 2019R1A2C2004858 to Y.-P.K.), Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) (no. HI18C1241 to S.L.) funded by the Ministry of Health and Welfare, Commercializations Promotion Agency for R&D Outcomes (COMPA) (no. 2018-jdh-3-sb2-2 to S.L.) funded by the Ministry of Science and ICT, and partially supported by the Basic Science Research Program (no. 2012R1A6A1029029 to Y.-P.K.) through the NRF funded by the Ministry of Education. This work was also supported by the KBSI Research Fund (D010720 to S.L.). Author contributions: E.H.K., S.P., Y.K.K., M.M., and J.P. performed the experiments and analyzed the data; K.J.L. and Y.-P.K. designed primary patient breast cancer and in vivo experiments; S.L. and Y.-P.K. designed FL imaging and ODT experiments; and K.J.L., S.L., and Y.-P.K. conceived the study and drafted the manuscript. All authors discussed and approved the final manuscript. Competing interests: E.H.K. and Y.-P.K. are inventors on patent applications related to a composition and a BLiP-PDT method for cancer cell death, filed by Industry-University Foundation, Hanyang University Cooperation (IUF-HUC) (KR patent application no. KR1020190131285, 22 October 2019; PCT patent application no. PCT/KR2019/013900, 22 October 2019). K.J.L. and Y.-P.K. are inventors on a patent application related to a BLiP-PDT method for inhibiting cancer metastasis, filed by Asan Foundation, Ulsan University, and IUF-HUC (KR patent application no. KR1020200022983, 25 February 2020). All the other authors declare that they have no competing interest. Data and materials availability: The authors declare that the data supporting the findings of the present study are available in the paper and/or the Supplementary Materials. or from the corresponding authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/37/eaba3009/DC1
REFERENCES AND NOTES
- 1.Dolmans D. E. J. G. J., Fukumura D., Jain R. K., Photodynamic therapy for cancer. Nat. Rev. Cancer 3, 380–387 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Zhou Z., Song J., Nie L., Chen X., Reactive oxygen species generating systems meeting challenges of photodynamic cancer therapy. Chem. Soc. Rev. 45, 6597–6626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jang B., Park J. Y., Tung C. H., Kim I. H., Choi Y., Gold nanorod-photosensitizer complex for near-infrared fluorescence imaging and photodynamic/photothermal therapy in vivo. ACS Nano 5, 1086–1094 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Lucky S. S., Soo K. C., Zhang Y., Nanoparticles in photodynamic therapy. Chem. Rev. 115, 1990–2042 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Xia Y. M., Gupta G. K., Castano A. P., Mroz P., Avci P., Hamblin M. R., CpG oligodeoxynucleotide as immune adjuvant enhances photodynamic therapy response in murine metastatic breast cancer. J. Biophotonics 7, 897–905 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Q., Xu L. G., Liang C., Wang C., Peng R., Liu Z., Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat. Commun. 7, 13193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakajima T., Sano K., Choyke P. L., Kobayashi H., Improving the efficacy of photoimmunotherapy (PIT) using a cocktail of antibody conjugates in a multiple antigen tumor model. Theranostics 3, 357–365 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim J., Park W., Kim D., Lee E. S., Lee D. H., Jeong S., Park J. M., Na K., Tumor-specific aptamer-conjugated polymeric photosensitizer for effective endo-laparoscopic photodynamic therapy. Adv. Funct. Mater. 29, 1900084 (2019). [Google Scholar]
- 9.Borgia F., Giuffrida R., Caradonna E., Vaccaro M., Guarneri F., Cannavo S. P., Early and late onset side effects of photodynamic therapy. Biomedicines 6, 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sylantiev C., Schoenfeld N., Mamet R., Groozman G. B., Drory V. E., Acute neuropathy mimicking porphyria induced by aminolevulinic acid during photodynamic therapy. Muscle Nerve 31, 390–393 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Pallet N., Karras A., Thervet E., Gouya L., Karim Z., Puy H., Porphyria and kidney diseases. Clin. Kidney J. 11, 191–197 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hong G., Diao S., Chang J., Antaris A. L., Chen C., Zhang B., Zhao S., Atochin D. N., Huang P. L., Andreasson K. I., Kuo C. J., Dai H., Through-skull fluorescence imaging of the brain in a new near-infrared window. Nat. Photonics 8, 723–730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu C.-Y., Chen C.-W., Yu H.-P., Lin Y.-F., Lai P.-S., Bioluminescence resonance energy transfer using luciferase-immobilized quantum dots for self-illuminated photodynamic therapy. Biomaterials 34, 1204–1212 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Kim Y. R., Kim S., Choi J. W., Choi S. Y., Lee S.-H., Kim H., Hahn S. K., Koh G. Y., Yun S. H., Bioluminescence-activated deep-tissue photodynamic therapy of cancer. Theranostics 5, 805–817 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bulina M. E., Chudakov D. M., Britanova O. V., Yanushevich Y. G., Staroverov D. B., Chepurnykh T. V., Merzlyak E. M., Shkrob M. A., Lukyanov S., Lukyanov K. A., A genetically encoded photosensitizer. Nat. Biotechnol. 24, 95–99 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Liao Z.-X., Li Y.-C., Lu H.-M., Sung H.-W., A genetically-encoded KillerRed protein as an intrinsically generated photosensitizer for photodynamic therapy. Biomaterials 35, 500–508 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Ryumina A. P., Serebrovskaya E. O., Shirmanova M. V., Snopova L. B., Kuznetsova M. M., Turchin I. V., Ignatova N. I., Klementieva N. V., Fradkov A. F., Shakhov B. E., Zagaynova E. V., Lukyanov K. A., Lukyanov S. A., Flavoprotein miniSOG as a genetically encoded photosensitizer for cancer cells. Biochim. Biophys. Acta 1830, 5059–5067 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Proshkina G. M., Shramova E. I., Shilova O. N., Ryabova A. V., Deyev S. M., Phototoxicity of flavoprotein miniSOG induced by bioluminescence resonance energy transfer in genetically encoded system NanoLuc-miniSOG is comparable with its LED-excited phototoxicity. J. Photoch. Photobio. B 188, 107–115 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Widder E. A., Bioluminescence in the ocean: Origins of biological, chemical, and ecological diversity. Science 328, 704–708 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Béjà O., Aravind L., Koonin E. V., Suzuki M. T., Hadd A., Nguyen L. P., Jovanovich S. B., Gates C. M., Feldman R. A., Spudich J. L., Spudich E. N., De Long E. F., Bacterial rhodopsin: Evidence for a new type of phototrophy in the sea. Science 289, 1902–1906 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Stoeckenius W., The rhodopsin-like pigments of halobacteria: Light-energy and signal transducers in an Archaebacterium. Trends Biochem. Sci. 10, 483–486 (1985). [DOI] [PubMed] [Google Scholar]
- 22.Loening A. M., Wu A. M., Gambhir S. S., Red-shifted Renilla reniformis luciferase variants for imaging in living subjects. Nat. Methods 4, 641–643 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Loening A. M., Fenn T. D., Wu A. M., Gambhir S. S., Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng. Des. Sel. 19, 391–400 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Shu X., Lev-Ram V., Deerinck T. J., Qi Y., Ramko E. B., Davidson M. W., Jin Y., Ellisman M. H., Tsien R. Y., A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLOS Biol. 9, e1001041 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmed S., Mathews A. S., Byeon N., Lavasanifar A., Kaur K., Peptide arrays for screening cancer specific peptides. Anal. Chem. 82, 7533–7541 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Zhang J., Spring H., Schwab M., Neuroblastoma tumor cell-binding peptides identified through random peptide phage display. Cancer Lett. 171, 153–164 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Askoxylakis V., Zitzmann S., Mier W., Graham K., Kramer S., von Wegner F., Fink R. H. A., Schwab M., Eisenhut M., Haberkorn U., Preclinical evaluation of the breast cancer cell-binding peptide, p160. Clin. Cancer Res. 11, 6705–6712 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Taherian A., Li X., Liu Y., Haas T. A., Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer 11, 293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subik K., Lee J.-F., Baxter L., Strzepek T., Costello D., Crowley P., Xing L., Hung M.-C., Bonfiglio T., Hicks D. G., Tang P., The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer 4, 35–41 (2010). [PMC free article] [PubMed] [Google Scholar]
- 30.Park Y., Depeursinge C., Popescu G., Quantitative phase imaging in biomedicine. Nat. Photonics 12, 578–589 (2018). [Google Scholar]
- 31.Zhang Y., Chen X., Gueydan C., Han J., Plasma membrane changes during programmed cell deaths. Cell Res. 28, 9–21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakajima K., Takakura H., Shimizu Y., Ogawa M., Changes in plasma membrane damage inducing cell death after treatment with near-infrared photoimmunotherapy. Cancer Sci. 109, 2889–2896 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osborne C. K., Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 339, 1609–1618 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Cuzick J., Dowsett M., Pineda S., Wale C., Salter J., Quinn E., Zabaglo L., Mallon E., Green A. R., Ellis I. O., Howell A., Buzdar A. U., Forbes J. F., Prognostic value of a combined estrogen receptor, progesterone receptor, Ki-67, and human epidermal growth factor receptor 2 immunohistochemical score and comparison with the genomic health recurrence score in early breast cancer. J. Clin. Oncol. 29, 4273–4278 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Tetard M.-C., Vermandel M., Mordon S., Lejeune J.-P., Reyns N., Experimental use of photodynamic therapy in high grade gliomas: A review focused on 5-aminolevulinic acid. Photodiagniosis Photodyn. Ther. 11, 319–330 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Kwiatkowski S., Knap B., Przystupski D., Saczko J., Kędzierska E., Knap-Czop K., Kotlińska J., Michel O., Kotowski K., Kulbacka J., Photodynamic therapy – mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 106, 1098–1107 (2018). [DOI] [PubMed] [Google Scholar]
- 37.So M.-K., Xu C., Loening A. M., Gambhir S. S., Rao J., Self-illuminating quantum dot conjugates for in vivo imaging. Nat. Biotechnol. 24, 339–343 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Wojtovich A. P., Foster T. H., Optogenetic control of ROS production. Redox Biol. 2, 368–376 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neve R. M., Chin K., Fridlyand J., Yeh J., Baehner F. L., Fevr T., Clark L., Bayani N., Coppe J.-P., Tong F., Speed T., Spellman P. T., DeVries S., Lapuk A., Wang N. J., Kuo W.-L., Stilwell J. L., Pinkel D., Albertson D. G., Waldman F. M., McCormick F., Dickson R. B., Johnson M. D., Lippman M., Ethier S., Gazdar A., Gray J. W., A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castano A. P., Mroz P., Hamblin M. R., Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 6, 535–545 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/37/eaba3009/DC1