

Summary
CRISPR/Cas9 is a promising technology for gene correction. However, the edition is often biallelic, and uncontrolled small insertions and deletions (indels) concomitant to precise correction are created. Mutation-specific guide RNAs were recently tested to correct dominant inherited diseases, sparing the wild-type allele. We tested an original approach to correct compound heterozygous recessive mutations. We compared editing efficiency and genotoxicity by biallelic guide RNA versus mutant allele-specific guide RNA in iPSCs derived from a congenital erythropoietic porphyria patient carrying compound heterozygous mutations resulting in UROS gene invalidation. We obtained UROS function rescue and metabolic correction with both guides with the potential of use for porphyria clinical intervention. However, unlike the biallelic one, the mutant allele-specific guide was free of on-target collateral damage. We recommend this design to avoid genotoxicity and to obtain on-target scarless gene correction for recessive disease with frequent cases of compound heterozygous mutations.
Keywords: CRISPR/Cas9, gene therapy, iPSCs, precise genome editing, congenital erythropoietic porphyria, compound heterozygous recessive disease, mutation specific
Graphical Abstract
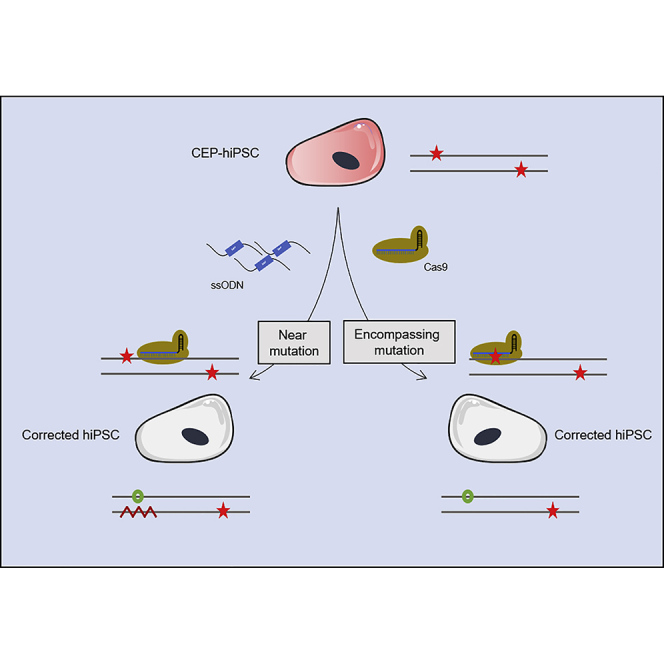
Highlights
-
•
CRISPR editing allows erythropoietic porphyria full metabolic correction in hiPSCs
-
•
Mutation-specific gRNA keeps intact WT allele in cases of heterozygous compound
Bedel and colleagues correct congenital erythropoietic porphyria in human iPSCs. They recommend mutation-specific gRNA design to correct heterozygous compound diseases by CRISPR/Cas9 to protect the WT allele and limit genotoxicity.
Introduction
Congenital erythropoietic porphyria (CEP) is an autosomal recessive disorder caused by a profound deficiency in the enzymatic activity of uroporphyrinogen III synthase (UROS) (EC 4.2.1.75), the fourth enzyme of the heme biosynthetic pathway (Erwin and Desnick, 2019; Ged et al., 2009; Richard et al., 2008). This defect leads to the accumulation of the fluorescent type I porphyrin isomers, causing dermatological lesions and hemolytic anemia. The clinical severity of the disease and the lack of specific treatment, besides bone marrow transplantation (Harada et al., 2001; Lagarde et al., 1998; Peinado et al., 2013; Shaw et al., 2001; Tezcan et al., 1998; Thomas et al., 1996), are strong arguments for gene therapy (Richard et al., 2008). Additive gene therapies are successful in treating monogenic hematopoietic disorders (Fischer et al., 2015; Morris et al., 2017; Ribeil et al., 2017). We and others (Bishop et al., 2006; Ged et al., 2006; Yasuda and Desnick, 2019) have generated an animal model of CEP (Urosc.744C>A/c.744C>A-knockin mice [p.Pro248Gln]) to evaluate the efficacy of gene therapy. Lentiviral additive gene therapy with UROS cDNA into hematopoietic stem cells resulted in the complete and long-term enzymatic, metabolic, and phenotypic correction of the disease, with a better survival of the corrected red blood cells (Robert-Richard et al., 2008). These data were a proof of concept of a successful gene therapy for this disease. However, reports of proviral insertional leukemogenesis (Hacein-Bey-Abina et al., 2008) underscore the need for safer methods. The discovery of key transcription factors enabling reprogramming of a somatic cell into a pluripotent stem cell, called an induced pluripotent stem cell (iPSC) (Takahashi et al., 2007), has provoked an exciting rebound in gene therapy research. Indeed, these cells allow clonal selection of corrected cells with safe provirus integration and further expansion for autologous grafting.
We successfully performed additive lentiviral gene therapy in hiPSCs (human iPSCs) from a CEP patient without risk of insertional oncogenesis by selection of corrected hiPSC clones with only one integration in a genomic safe harbor (Bedel et al., 2012).
However, exogenous sequence integration in the genome may carry unknown side effects and the inserted transgene is under the control of a non-physiological promotor. The CRISPR/Cas9 system is a seducing alternative to additive gene therapy (Doudna and Charpentier, 2014). Editing a gene at its endogenous locus by removing or correcting deleterious mutations rather than adding a new transgene has the potential to solve insertional mutagenesis and non-physiological gene regulation (Cong et al., 2013; Kohn and Kuo, 2017). CRISPR/Cas9 is an RNA-guided DNA endonuclease system targeting a specific genomic sequence complementary to a single-guide RNA (sgRNA) (Cong et al., 2013; Doudna and Charpentier, 2014; Hsu et al., 2013; Jinek et al., 2012). Most publications report the use of engineered Cas9-nucleases to invalidate genes. Cas9 produces double-strand breaks (DSBs) at sites of interest mainly solved by the error-prone non-conservative non-homologous end-joining (NHEJ) repair pathway, which introduces insertions and deletions (indels) leading to disrupted targeted sequences (Boch et al., 2009; Porteus and Baltimore, 2003; Zhang et al., 2011). It is also possible to edit genes by homology direct repair (HDR) if a DNA template is provided (Yang et al., 2014). However, simultaneous to HDR, NHEJ occurs (Liu et al., 2019). The on-target NHEJ activated in response to DNA DSBs has often been underestimated. Precise genome editing (PGE) ratio measures needed HDR versus competitive unwanted indels. For example, in the CEP model and for UROS targeting, we recently demonstrated that uncontrolled DNA sequence modifications induced by the NHEJ are very frequent in HEK293T, with a PGE ratio of 0.53 (Cullot et al., 2019). In any on-target site, repair introducing indels is twice as frequent as precise HDR. This PGE ratio is even lower in hiPSCs (Li et al., 2018; Liang et al., 2017). This underlines the need to find new tools to improve the PGE ratio.
Several approaches have been proposed to improve the PGE ratio. It is possible to insert a positive selection cassette in the donor template. However, this carries a genotoxic risk by transgene integration and is susceptible to immune response against the exogenous protein. Other techniques have been described, such as the inhibition of NHEJ by suppressing DNA ligase IV (Hu et al., 2018b; Maruyama et al., 2015), KU70 (Chu et al., 2015), 53BP1 (Canny et al., 2018; Jayavaradhan et al., 2019; Nambiar et al., 2019), and polymerase Ɵ (Saito et al., 2017; Zelensky et al., 2017); alternatively, HDR activation using the RAD51 agonist (Song et al., 2016) or synchronizing the cells in S-G2 phases (Janssen et al., 2019; Zhang et al., 2017) promotes HDR.
Interestingly, it is also possible to play on the design of the guide RNA (gRNA). Indeed, a mutant allele-specific gRNA invalidated only mutant alleles in dominant diseases in vitro (Burnight et al., 2017; Giannelli et al., 2018; Monteys et al., 2017; Yamamoto et al., 2017) and in vivo (Xie et al., 2016). This approach was used to correct a dominant mutant allele using a template with (Smith et al., 2015) or without (Rabai et al., 2019) a selection cassette.
We hypothesized that a mutant allele-specific gRNA could be precisely corrected by HDR in recessive diseases with compound heterozygous mutations. Avoiding DSB in the wild-type (WT) allele is the only way to prevent indels, preserving WT allele integrity and obtaining a perfect PGE ratio.
In this work, we propose to correct hiPSCs derived from a CEP patient carrying two heterozygous mutations for the UROS gene by genome editing using CRISPR/Cas9. We compared editing efficiency and genotoxicity by biallelic gRNA versus mutant allele-specific gRNA. We showed that a mutant allele-specific guide was mandatory to perfectly correct cells without on-target indels and obtain efficient correction of CEP recessive disease with compound heterozygous mutations, which is interesting for future clinical applications.
Results
CEP-hiPSC Characterization
To evaluate the mutant allele-specific approach to correct CEP, we used hiPSCs previously reprogrammed keratinocytes from a CEP patient in our lab (Bedel et al., 2012). After cell expansion, the genotype was confirmed by PCR and Sanger sequencing (Figure 1A). The patient is compound heterozygous for the UROS gene with one mutation in exon 4 (c.217C > T) and another in exon 10 (c.683C > T). Although these combined mutations lead to a dramatic decrease of UROS enzymatic activity (0.15 ± 0.07 versus 19.3 ± 1.7 for WT cells, n = 3) (Figure 1B), CEP-hiPSCs do not spontaneously accumulate type I porphyrins detectable by flow cytometry (Figure 1C, left). By contrast, the exposure to the heme biosynthesis precursor (δ-amino-levulinic acid [ALA]) induced appearance of specific fluorocytes due to type I porphyrin accumulation in the mutated cells (Figure 1C, right).
Figure 1.

CEP-hiPSC Characterization
(A) CEP-hiPSC UROS genotyping. The CEP-hiPSC is composite heterozygous, with one allele mutated in exon 4 (c.217T > C) and the other one on the exon 10 (c.683C > T).
(B) WT-hiPSC and CEP-hiPSC UROS enzymatic activity. Quantification of UROS enzymatic activity by high-performance liquid chromatography (HPLC) of WT or CEP-hiPSC. Data are represented as mean ± SEM. n ≥ 5 independent experiments for each IPSC lines.
(C) WT-hiPSC and CEP-hiPSC metabolic activity. Type I fluorocyte accumulation (PE-Cy5-A) by flow cytometry: illustrative results of WT (top) and CEP-hiPSC (bottom) before and after δ-aminolevulinic acid (ALA) addition (5 mM, 12 h).
(D) Experimental workflow for UROS gene editing and result analysis. CEP-hiPSCs were transfected by nucleofection with a fluorescent ssODN template, a ZsGreen-nuclease plasmid, and an RNA guide plasmid targeting exon 4 or 10. Then polyclonal and further monoclonal analysis were performed: HDR was quantified by RFLP, and confirmed by ICE and NGS. The indels were quantified by ICE and NGS. The UROS functionality was assessed by quantifying UROS-specific activity by HPLC and type I porphyrin accumulation by flow cytometry. For the subclones, chromosomal integrity was checked by FISH (analysis only performed for subclones).
Correction of the UROS Mutations Comparing Two-gRNA Design Strategy
We aimed to compare the efficacy of the mutant allele-specific gRNA approach to a classic biallelic gRNA. We designed a biallelic gRNA-10 for exon 10, targeting both alleles (mutant and WT) close to the c.683C > T mutation (Figures 1D and 2A). For exon 4, the gRNA-4 includes the mutation c.217C > T and is mutant allele specific (Figures 1D and 5A).
Figure 2.

Polyclonal Analysis of UROS Exon 10 Editing by CRISPR/Cas9
(A) Top, schematic UROS locus on chromosome 10 with UROS gene overview and the exon 10 targeted region. Middle, detailed view of exon 10 region close to the c.683T mutation. CRISPR-mediated HDR design using an sgRNA targeting the sequence just next to the mutation (sgRNA-10 highlighted in blue) and a 75nt-ssODN-A647 carrying a silent restriction site ApaI (highlight in yellow) and the correction of the mutation (c.683C in red). Right, scheme of ApaI-digested PCR products obtained for alleles with or without HDR and an illustrative RFLP analysis. Bottom, exon 10 gene editing workflow. The CEP-hiPSCs were transfected with a ZsGreen-nuclease plasmid, the sgRNA-10 plasmid, and a 75nt-ssODN-A647 template. The cells were sorted by cytometry to get back only the cells transfected with the ZsGreen-nuclease and the 75nt-ssODN-A647 template.
(B) Flow cytometry illustration of the double-positive ZsGreen+/A647+ sorted cells (left) and their targeting region analysis: Sanger sequence (top), ICE (middle), and RFLP (bottom).
Figure 5.

Polyclonal Analysis of UROS Exon 4 Editing by CRISPR/Cas9
(A) Top, schematic UROS locus on chromosome 10 with UROS gene overview and the targeted exon 4. Bottom, detailed view of the exon 4 region and CRISPR-mediated HDR design using c.217C-targeting sgRNA (sgRNA-4 highlighted in blue) and an 80nt-ssODN-A647 carrying a silent restriction site SacI (highlight in yellow) and the correction of the mutation (c.217T in red). Right, scheme of SacI-digested PCR products obtained for alleles with or without HDR and an illustrative RFLP analysis.
(B) Flow cytometry illustration of the double-positive ZsGreen+/A647+ sorted cells (top left) and their targeting region analysis: Sanger sequence (top), ICE (middle), NGS (bottom left), and RFLP (bottom).
(C) Indel sequence and their contribution percentage given by ICE software.
Because editing is low in primary stem cells, we designed tools allowing transfected cell sorting, using two plasmids expressing Cas9-ZsGreen or the gRNA, with fluorescent Alexa 647-ssODN HDR templates (75 or 80 bp). These templates bear silent mutations introducing restriction sites allowing HDR quantification by restriction fragment length polymorphism (RFLP) (Figure 1D). Indels and HDR rates were confirmed by sequencing and ICE/TIDER (Inference of CRISPR edits/Tracking of Insertions, Deletions and Recombination events) software analyses. We characterized enzymatic and metabolic activities of the corrected cells and their subclones (UROS enzymatic activity rescue, porphyrin disappearance) (Figure 1D).
Biallelic gRNA for Exon 10 UROS Editing
After transfection with the Cas9-ZsGreen, gRNA-10 plasmids, and the 75nt-ssODN-A647 template, cells were sorted for ZsGreen and Alexa Fluor 647 staining (Figures 2A and 2B). Sanger sequencing revealed indels from the cut site indicating a DSB. This result was confirmed by ICE and TIDER analyses (83% of indels) (Figures 2B and S1A, left). These techniques also indicated 2% of HDR, which is confirmed by RFLP (2.8% of alleles with an ApaI site), showing a low frequency of precise editing (Figure 2B).
To sort the corrected cells, which no longer accumulate fluorescent porphyrins, we exposed the cells to ALA, and sorted the non-fluorescent cells (PE-Cy5 negative = porphyrin negative) (Figure 3A). As expected in the non-fluorescent cells, the mutated T reverted to C, and the two expected silent mutations (ApaI) were introduced. HDR was quantified by ICE (38%) and TIDER (22%), and confirmed by RFLP (41%) (Figures 3A and S1A, right). These data suggest that only one of the two alleles was edited by HDR in these corrected cells. We obtained a 24-fold increase of UROS activity compared with isogenic CEP-hiPSCs (3.65 ± 0.33 versus 0.15 ± 0.07 for CEP-hiPSCs before editing). This enzymatic rescue prevented porphyrin accumulation, but was partial compared with non-isogenic WT-hiPSCs from a healthy patient (3.65 ± 0.33 versus 19.3 ± 1.7 U/mg protein for WT cells) (Figure 3B). However, Sanger sequence chromatography and ICE/TIDER analyses revealed that this correction was concomitant with a high indel rate (53% and 23%, respectively) (Figure 3A). These indels are two symmetric 25-base deletions in favor of microhomology-mediated end joining repair (Figure 3C). Most indels seemed to be the same deletion due to clonal correction or clonal expansion of these corrected cells: the PGE ratio (HDR/indels) ranked between 0.7 and 0.9 with ICE or TIDER results, confirming the predominant NHEJ repair pathway. We then cultured corrected CEP-hiPSCs under clonal conditions to find one perfectly corrected subclone without indels (HDR/WT or HDR/HDR). All six analyzed subclones contained the ApaI site and the corrected c.683C base, but also indels (Figure 4A), while UROS enzymatic activity was restored as for the polyclonal cell population (3.8 ± 0.4 U/mg protein, n = 6 clones) (Figure 4B). In conclusion, in all the analyzed corrected cells, both specific correction of the mutated allele and indels on the homologous allele occurred because the gRNA targeted both alleles.
Figure 3.

Molecular and Metabolic Analysis of UROS Exon 10-Corrected Cells
(A) Top left, cytometry sorting of corrected cells, with disappearance of porphyrins after ALA exposure (5 mM, 12 h) (porphneg cells, PE-Cy5-A). Top right, CEP-hiPSCs and porphyrin-negative sorted cells exon 10 genotyping. The targeted mutation and its correction are highlight in red, and the inserted silent restriction site ApaI in yellow. Middle right, ICE analysis with the indels and HDR percentages. Bottom right, RFLP analysis of the targeted region.
(B) Quantification of UROS enzymatic activity in the sorted population. Data are represented as mean ± SEM. n ≥ 4 independent experiments for each hiPSC line.
(C) Indel sequences and their contribution percentage given by ICE software.
Figure 4.

UROS Exon 10 Subclone Genotyping
(A) Sanger sequence of six porphyrin-negative subclones. The corrected mutation is highlighted in red and the silent restriction site ApaI in yellow. HDR and indel analysis by RFLP and ICE are indicated.
(B) UROS enzymatic activity of polyclonal porphyrin-negative sorted population and corrected subclones. Data are represented as mean ± SEM, ∗∗∗p < 0.001 versus CEP-hIPSC lines, n ≥ 4 independent experiments for CEP and polyclonal porphyrin-negative (corrected) cell lines. n = 6 corrected subclones.
Mutant Allele-Specific gRNA for Exon 4 UROS Editing
Unlike exon 10, we used a mutant allele-specific RNA guide (gRNA-4) that targeted only the mutated allele for the editing of exon 4 (Figure 5A). Using the same strategy, we transfected the CEP-hiPSCs with the ZsGreen-nuclease plasmid, the gRNA-4 plasmid with an ssODN template (80pb-ssODN-A647) bearing a silent mutation introducing a SacI restriction site (Figure 5A). After sorting of the double-positive ZsGreen+/A647+ cells, we obtained corrected cells (4.7% by RFLP, 5% by deep sequencing, undetectable by ICE/TIDER analyses) (Figures 5B and S1B, left). As shown by Sanger sequencing, correction events were concomitant with indels: 37%, 33%, and 30.9% by ICE, TIDER and deep sequencing analyses, respectively (Figures 5B and S1B, left). Indel sequences consisted of five short deletions (1–5 bases) and one base insertion (Figure 5C), probably due to NHEJ repair. We sorted, in the presence of ALA, the porphyrin-negative cells, supposedly the corrected cells. Sanger chromatography confirmed a perfect mutation correction, c.217T (Figure 6A). Molecular analysis confirmed C to T editing with a high HDR rate by ICE (70%), TIDER (45%), RFLP (39%), and deep sequencing (44.4%) (Figures 6A and S1B, right). Although no indel events were detectable on the chromatogram and by ICE/TIDER analyses (Figures 6A, 6B, and S1B, right), deep sequencing revealed around 7% of alleles with indels in the polyclonal mix. Cells demonstrated phenotypic correction with restoration of metabolic activity by high-performance liquid chromatography (3.54 ± 0.4 U/mg protein), corresponding to a 24-fold increase compared with the isogenic CEP-hiPSCs (Figure 6C). To verify absence of indels in the corrected cells, we cultured seven “porphyrin-negative” subclones. Six out of the seven contained the corrected mutation and the SacI digestion site without indels (Figure 7A), resulting in a perfect PGE ratio. One was not corrected, but carried indels, probably because of imperfect cell sorting, and in agreement with deep sequencing results (7% of alleles with indels). The six corrected subclones restored UROS metabolic activity (3.95 ± 0.3 U/mg protein, n = 6) (Figure 7B) similar to the polyclonal cell population. Together these data showed similar enzymatic correction between UROS exon 10- and UROS exon 4-corrected cells, but without genotoxicity on the opposite allele thanks to a gRNA encompassing the mutation.
Figure 6.

Molecular and Metabolic Analysis of UROS Exon 4-Corrected Cells
(A) Top left, cytometry sorting of corrected cells, with disappearance of porphyrins after ALA exposure (5 mM, 12 h) (porphneg cells, PE-Cy5-A). Top right, CEP-hiPSCs and porphyrin-negative sorted cell exon 4 genotyping. The targeted mutation and its correction are highlighted in red, and the inserted silent restriction site SacI in yellow. Middle right, ICE analysis with the indel and HDR percentages. Bottom right, RFLP analysis of the targeted region. Bottom left, NGS analyses of allelic outcome.
(B) Sequences and their contribution percentage given by ICE software.
(C) Quantification of UROS enzymatic activity in the sorted population by HPLC. Data are represented as mean ± SEM, n ≥ 6 independent experiments for each hIPSC line.
Figure 7.

UROS Exon 4 Subclone Correction without Karyotypic Instability
(A) Exon 4 subclone genotyping. The corrected mutation is highlighted in red and the silent restriction site SacI in yellow. HDR and indel analysis by RFLP and ICE are indicated. Loq, limit of quantification 2%.
(B) UROS enzymatic activity of polyclonal porphyrin-negative sorted population and six corrected subclones. Data are represented as mean ± SEM, ∗p < 0.05, ∗∗p < 0.01 versus CEP-hIPSC lines, n ≥ 6 independent experiments for CEP and polyclonal porphyrin-negative (corrected) cell lines. n = 6 corrected subclones.
(C) DNA-FISH assay using UROS-framing probes (top) (−4.6 Mb upstream and +4.4 Mb downstream of the UROS locus, respectively, green [G] and orange [O]). Bottom, illustrative DNA-FISH results for the exon 4 subclones (magnification factor ×630).
(D) Illustrative schema of mutation-specific gRNA importance for heterozygous composite gene editing. The mutation is represented by a blue cross, the gRNA by a red line. The black dotted lines are the cut site of nuclease Cas9. Green dots represent the corrected mutation by HDR, and the red triangles are indels created by NHEJ repair of DSB.
To check putative off-target damages by CRISPR/Cas9 nuclease and the gRNA-4, we quantified indels in the top 10 off-target sequences predicted by CRISPOR. We observed low indel rates (7% and 5%) in only two of the top 10 ranked loci. The first one is located in an intronic region and the second one in an intergenic region (Figure S2). No off-target site is on chromosome 10 minimizing risks of internal chromosomal rearrangement.
We recently reported that UROS exon 4 targeting by CRISPR/Cas9 nuclease in HEK293T could induce chromosome 10q terminal truncations, with megabase-scale deletion (Cullot et al., 2019). Thus, we revealed that genome integrity in exon 4 perfectly corrected CEP cells. We focused on the UROS locus (10q26.2), which is harbored in iPSC polyclonal populations and subclones. We performed fluorescence in situ hybridization (FISH) analysis using probes framing UROS (a proximal probe at 4.6 Mb upstream of UROS, labeled in green [G], and a distal probe 4.4 Mb downstream of UROS, labeled in orange [O]) (Figure 7C). Polyclonal analysis did not reveal loss of the orange probe, which would identify chromosomal end losses. All the subclones were 2O/2G (Figure 7C), suggesting the absence of megabase-scale deletions and chromosome 10q terminal truncation. In the same way, none of the tested subclones lost the orange signal (0/3 subclones). In addition, we observed amplification of two alleles by Sanger sequences (heterozygous sequences, Figure 7A), suggesting the correct integrity of the diploid clones, without kilobase deletions. To confirm the absence of Cas9-induced chromosomal instability, we performed karyotype analysis after three rounds of Cas9 nuclease nucleofections with the gRNA-4. We did not observe any chromosome abnormality after editing (mitosis with one X chromosome deletion were already present) (Figure S3).
Collectively, these data suggest that the use of a mutant allele-specific gRNA approach to edit exon 4 produced a clean and efficient correction of UROS in hiPSCs, with a perfect PGE ratio (Figure 7D).
Discussion
Here, we succeeded in targeting and correcting by HDR the mutant alleles (in exon 4 and exon 10 of the UROS gene) in hiPSCs from a compound heterozygous CEP patient. To evaluate the genomic correction, we used deep sequencing and ICE/TIDER software that compared Sanger chromatograms. Generally, we obtained good correlations between them and the gold standard deep sequencing. However, low HDR rates are not detected by software analyses (<5%) and we observed some discrepancies between the results from these two pieces of software. These interesting tools, easy-to-use and costless, need to be used with precaution for precise quantification.
We obtained full metabolic correction of CEP-hiPSCs by CRISPR/Cas9, by guide design and with disappearance of porphyrin accumulation. Correction of one mutation, either in exon 4 or in exon 10, led to the same partial UROS enzymatic rescue. Intriguingly, monoallelic corrections did not restore half of the regular hiPSC UROS enzymatic activity with two WT alleles, probably because they are not isogenic and the heterogeneity between clones is high. As UROS activity is partially restored by both approaches (mutant allele specific or biallelic), they could be considered equivalent in editing. However, in mammalian cells, DSBs are predominantly repaired by NHEJ, while HDR is less active (Chu et al., 2015; Li et al., 2018; Liang et al., 2017). In our work, the first correction design consisted of a routine approach, targeting the UROS exon 10 by a biallelic targeting gRNA complementary to a sequence next to the mutation. This always resulted in undesired indels in the WT allele in parallel to correct editing of the other allele. This observation relates to the experimental difficulty in generating specific heterozygous mutations at the cellular level since most CRISPR/Cas9 editing events are biallelic (Wang et al., 2013). The PGE ratio achieved in hiPSCs was the same as that in the HEK293T cells (Cullot et al., 2019), with indels twice as frequent as precise correction. Besides inducing gene KO, on-target indels could lead to the production of abnormal proteins with immunogenic properties, or potentially pathogenic by gain of function. Indeed, mutations in coding regions can promote abnormal interactions, alter the interaction of the mutant protein with its natural binders, and/or promote misfolding/aggregation (Li et al., 2019).
The second correction strategy to achieve allele specificity while not affecting the WT allele, designed a guide sequence encompassing the mutation site. Together with the hiPSC-clonal approach, this design produced scarless editing, without undesired on-target indels and limited potential genotoxicity induced by CRISPR/Cas9 nuclease. Usually, gRNA of endonucleases, such as Streptococcus pyogenes Cas9 (SpCas9) and Staphylococcus aureus (SaCas9), can tolerate mismatches between gRNA and target DNA. In our study, we designed a mutant-specific guide with mutation next to the PAM (protospacer adjacent motif) because this location does not accept mismatch (Hsu et al., 2013). So, we drastically spared the WT allele. This would not be the case if the mutation was in the guide but far from the PAM. In this case, guide would be permissive to this mutation, and would cut the WT allele too. This is in agreement with the fact that the CRISPR/Cas system can be a highly specific genome-editing tool capable of distinguishing alleles differing by a single nucleotide, when this difference is in the seed sequence (Slaymaker et al., 2016).
To our knowledge, this is the first report of a CEP-hiPSC correction by CRISPR/Cas9 but also the first allele-specific correction of a compound heterozygote for a recessive disease. Mutant allele-specific gRNA was already reported to target heterozygous mutation for indel-mediated inactivation in three autosomal dominant diseases in vitro (Monteys et al., 2017; Yamamoto et al., 2017) and in vivo (György et al., 2019; Monteys et al., 2017; Xie et al., 2016). Recently, Rabai et al. (2019) showed elegant editing by HDR of DNM2 using an allele-specific guide, but for the autosomal dominant form of centronuclear myopathy. Importantly, in patients with rare recessive diseases, compound heterozygosity of pathogenic mutations is the most likely inheritance model if the parents are non-consanguineous (Kamphans et al., 2013). In this situation, allele-specific correction may be the most suitable approach. To this end, the Pollard lab developed a new software, AlleleAnalyzer, to facilitate allele-specific gRNA design (Keough et al., 2019).
Despite high versatility and efficacy, the CRISPR/Cas9 nuclease tool has still limitations. First, it bears a risk of off-target effects. The allele-specific guide design has no impact on putative off-target risk. In our model, we observed low percentages of indels in off-target sequences. This flaw could be solved by the new generation higher-fidelity Cas (eSpCas9, spCas9-HF1, and HypaCas9 [Ikeda et al., 2019]). For example, HypaCas9 enabled the discrimination of single-nucleotide polymorphisms and the introduction of monoallelic mutations in mouse zygotes (Ikeda et al., 2019). Second, on-target genotoxicity due to DSB was described recently. CRISPR/Cas9 can promote large deletions from a few kilobases (Adikusuma et al., 2018; Kosicki et al., 2018; Shin et al., 2017) to many megabases (Cullot et al., 2019) after only one cut. These losses cause loss of heterozygosity (LOH). In the case of CEP-hiPSCs, we did not observe any kilobase- or megabase-scale deletions by FISH or PCR. We cannot exclude rare genomic abnormalities (below FISH limit of detection) or a distinct locus genotoxicity. Gorter de Vries et al. (2019) recently warned of the LOH risk not by loss of genetic material but by replacement of the targeted sequence by a copy of the opposite allele. They observed this risk using allele-specific genome editing in diploid yeast, repaired by the break-induced repair (BIR) mechanism. Ma et al. (2017) confirmed this risk in human embryos. In our case, all subclones had the restriction site integration, demonstrating that the repair mechanism involved the template by HDR and not the other homolog chromosome by BIR: the clonal approach with hiPSCs renders possible the careful selection of cells with genomic integrity and scarless editing.
The heterozygous composite proportion in patients with recessive diseases is frequent (e.g., phenylketonuria and Tay-Sachs disease). For example, for cystic fibrosis, which is the more prevalent genetic disease, this proportion is 40%. In these cases, the mutant allele-specific guide approach is the best way to avoid undesired uncontrolled editing of the WT allele and on-target side effects due to the risk of the production of abnormal protein. The fact that the PAM should be localized near the heterozygous mutation may be solved by new-generation Cas9 tools, such as xCas9 with broad PAM eligibility (Hu et al., 2018a). Since unwanted indels are caused by DSB, another solution could be an editing without DSB with the recent prime-editor (Anzalone et al., 2019) or the base editor systems (Molla and Yang, 2019). Indeed, c.217T > C is eligible as a CBE base editor. However, their efficacy and specificity in hiPSCs have still to be improved.
Experimental Procedures
Cell Culture and Transfection
iPSCs from a CEP patient and WT individual were described previously (Bedel et al., 2012) and obtained after informed consent in accordance with the ethical standards of the responsible committee on human experimentation (Centre Hospitalier Universitaire de Bordeaux). hiPSC clones were maintained as undifferentiated cells in cocultures with mitomycined mouse embryonic fibroblasts. The ES medium used was the following: KO-DMEM (Invitrogen, Villebon sur Yvette, France) containing 20% KOSR (Invitrogen) (vol/vol), 10 ng/mL human bFGF (PeproTech), 1 mM GlutaMAX (Invitrogen), 100 mM non-essential amino acids (Invitrogen), 100 mM 2-mercaptoethanol (Sigma-Aldrich, Saint Louis, MO, USA), 50 mg/mL ascorbic acid (Sigma-Aldrich), 0.5 mM sodium butyrate (Sigma-Aldrich), 50 U/mL penicillin, and 50 mg/mL streptomycin (Invitrogen).The ES medium was changed every day. hiPSC subclones were cultured onto a feeder-free Cellartis DEF-CS Culture System (Takara Bio Europe), therefore exhibiting proliferation as monolayer hiPSC lines with continuous passaging twice a week, as described previously (Asplund et al., 2016).
Editing Tools
Cells were transfected by electroporation using the Nucleofector AMAXA 4D electroporation system (Lonza, Bale, Switzerland), using a P3 primary cell kit and CB-150 program. In brief, 800,000 cells were nucleofected with 20 μg of nuclease-containing plasmid, 20 μg gRNA-containing plasmid, and 1.7 μM of specified ssODN. Cells were then seeded onto 12-well plates (Corning, Tewksbury, MA, USA) and cultured as described above. Transfected cells were then positively selected 24 h after transfection by ZsGreen-positive and A647-positive selection by fluorescent-activated cell sorting (FACS) using BD FACSAria.
The nuclease-containing plasmid was a modified version of lentiCRISPRv2 obtained from Addgene (Cambridge, MA, USA) (no. 52961). The ZsGreen sequence was inserted by digesting the lentiCRISPRv2 by BamHI-SacII (New England Biolabs, Ipswich, MA, USA) and replaced this fragment by a BamHI-P2A-ZsGreen-WPRE sequence synthetized by Eurofins Genomics (Germany).
All sgRNAs were designed using the CHOPCHOPv2 algorithm68 (chopchop.cbu.uib.no) and based on a unique sequence with 20 nucleotides. All ssODN templates used in the study were purchased from Integrated DNA Technologies (Coralville, IA, USA). For 80nt-ssODN-A647 and 75nt-ssODN-A647, an Alexa Fluor 647 was chemically linked at the 5' terminal end to ssODN by NHS Ester link (Table S1).
Flow Cytometry for Porphyrin Accumulation
UV-sensitive type I porphyrins were excited at 496 nm and the emitted wavelength was approximately 667 nm, detected by the PE-Cy5-A PMT channel (FACSCanto, BD, Franklin Lakes, NJ, USA). Cells were sorted by BD FACSAria.
Sanger Sequencing and ICE Analysis for Allelic Outcomes
Genomic DNA was extracted using NucleoSpin Tissue (Macherey-Nagel, Duren, Germany) according to the manufacturer's protocol. The genomic region flanking UROS exon 4 (or exon 10) was amplified by PCR (HotStarTaq Plus DNA Polymerase, QIAGEN, Venlo, the Netherlands) with adequate primers (Table S1). PCR products were purified with NucleoSpin Gel and PCR Clean-up (Macherey-Nagel). Sanger sequencing was performed by Eurofins Genomics. ICE and TIDER (Brinkman et al., 2018) were used to determine HDR and indels frequencies. PCR product from non-transfected CEP-hiPSCs was provided as control chromatogram.
NGS Deep Sequencing for Allelic Outcomes
Genomic DNA was extracted using NucleoSpin Tissue (Macherey-Nagel) according to the manufacturer's protocol. The genomic region flanking UROS exon4 was amplified by PCR (KAPA HiFi DNA Polymerase, Kapa Biosystems, Cape Town, South Africa) with adequate primers (Table S1). PCR products were purified with NucleoSpin Gel and PCR Clean-up (Macherey-Nagel). To prepare sequencing libraries, the Illumina Nextera XT Kit (Illumina, San Diego, CA, USA) was used and nested-PCR using Illumina primers was performed on purified PCR products. An Illumina MiSeq instrument (Illumina) was used for high-throughput sequencing. The average depth of each genome analysis was 30,000. Quality of paired-end reads was checked with FastQC (Galaxy, https://usegalaxy.org/). Then, quantification of HDR was performed on the restriction site, and percentage of insertion and deletion and base rates were done at the cut site using Alamut Visual software.
Microhomology Analysis
The sequences around the gRNA-4 and gRN-10 target site of UROS exon 4 or 10, respectively, were uploaded to Microhomology-Predictor of the CRISPR RGEN Tool (http://www.rgenome.net/mich-calculator/) for microhomology sequence analysis. One of the 25-bp deletions for gRNA-10 has corresponding pattern scores of 114.7. The corresponding indel pattern was also identified by ICE analyze.
RFLP for HDR Quantification
UROS exon 4 or exon 10 PCR products were digested, respectively, with SacI or ApaI restriction enzyme (New England Biolabs) for 1 h at 37°C. Then, 5-ng digestion products were loaded into the Agilent 2200 TapeStation (Santa Clara, CA, USA) capillary electrophoresis using D1000 ScreenTape and D1000 reagents according to the manufacturer's protocol. Quality control of enzymatic digestion efficiency is included in each assay.
UROS Enzymatic Activity and Metabolic Correction
UROS activity was determined by an enzyme-coupled assay as described previously (Tsai et al., 1987). In brief, porphobilinogen was first converted to hydroxymethylbilane, the natural substrate for UROS, by hydroxymethylbilane synthase. Then, the uroporphyrinogen reaction products were oxidized to their respective uroporphyrin isomers, which were then resolved and quantitated by reversed-phase high-pressure liquid chromatography. One unit was defined as the amount of enzyme that formed 1 nmol of uroporphyrinogen III per hour at 37°C.
Off-target Analysis
For sgRNA targeting the exon 4 UROS locus, the top 10 predicted off-target sites, identified by CRISPOR software, were amplified in genome-edited corrected CEP-hiPSCs and subjected to Sanger sequencing, followed by comparison with non-transfected cells by ICE analysis.
Primers used for off-target analysis are in Table S1.
Cytogenetic Examination of Chromosome 10
FISH was performed on interphase nuclei, with probes targeting locus 10q26.11 on chromosome 10 (BAC RP11–79M19 probe, labeled in green) (Empire Genomics, Buffalo, NY, USA), and locus 10q26.2 (BAC RP11–31M22 probe, labeled in orange) (Empire Genomics). Preparations were pre-treated as indicated below. In brief, the slides were successively immersed in a 2× saline sodium citrate buffer for 10 min at 37°C, in a 0.01% pepsin solution for10 min at 37°C, in a 1× phosphate-buffered saline (PBS) solution for 5 min, in a 3.7% formaldehyde solution for 5 min, and in a 1× PBS solution for 5 min. FISH probes and DNA were then co-denaturated according to the manufacturers' protocols, and hybridization was performed overnight at 37°C. The slides were then successively immersed in wash solutions and the nucleic acids were counterstained by 4,6-diamidino-2-phenylindole. The slides were then placed under an Axio Imager 2 microscope with an epi-fluorescence source (Carl Zeiss AG, Oberkochen, Germany). The microscope was linked to the Metafer4 software for automated image acquisition and processing (MetaSystems, Altlussheim, Germany). Genomic DNA was extracted with the Wizard Kit (Promega, Madison, USA) following the protocol validated in the laboratory.
Karyotype
After FrdU synchronization followed by a thymidine chase, standard R-banding analysis was performed on metaphase chromosomes obtained with all iPSC clones. At least 20 metaphase chromosomes were fully karyotyped.
Statistics
Statistical significance was inferred when necessary. GraphPad Prism 6 software was used for statistical analysis. Results are presented as mean ± SEM (standard error of the mean). Shapiro-Wilkinson test was done to assess the normality distribution. When it was positive, the two-tailed unpaired t test was done to compare means of two groups and one-way ANOVA, completed with unprotected Fisher's least significant difference test, was used to compare three groups. When normality distribution failed, non-parametric Kruskal-Wallis was used. All comparisons are shown with black bars. Null hypothesis was rejected when p < 0.05. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, non-significant.
Data and Code Availability
All next-generation sequencing datasets have been deposited in the NCBI database under BioProject accession no. PRJNA645083. The accession number for the exon 4 of CEP-hiPSC non-transfected reported in this paper is NCBI database: SAMN15491845.”, for transfected CEP-hiPSC with nuclease and RNA guide plasmids and ssODN is SAMN15491847 (Figure 5), and for corrected cells on exon 4 or porphyrin-negative cells is SAMN15491846 (Figure 6).
Author Contributions
F.P. performed the experiments and cultured iPSCs. G.C. carried out the data analysis. J.T. and J.B. performed FISH analysis and designed the experiments. S.A. analyzed the deep sequencing data. I.L.-G. carried out FACS analysis. M.L. performed UROS enzymatic assay. I.M. cultured iPSCs and mouse embryonic fibroblast cells. J.-M.B., E.R., and S.D. helped in CRISPR/Cas9 discussions and wrote the manuscript. F.M.-G. and A.B. cultured iPSCs, designed and analyzed the data, and wrote the manuscript. All authors acquired the funding and approved the final manuscript.
Acknowledgments
We thank Dr Feng Zhang for sharing plasmids through Addgene (Cambridge, MA, USA) and Vincent Pitard and Atika Zouine from the cytometry platform at Bordeaux University. This work was supported by the Association Française contre les Myopathies grant no. 20957, and Agence de Biomédecine. We thank Florence Lichou, Beatrice Turcq, and Nicolas Sevenet (Inserm U1218, Institut Bergonié, Bordeaux, France) for NGS analysis, and Valérie Prouzet-Mauléon for helpful CRISPR discussion. We thank Catherine Pain and Prof. Alain Taieb for providing CEP patient keratinocytes (Bordeaux Hospital). We thank Katia Obama, Isabelle Lamy, and Sandrine Hamon for administrative assistance and financial management.
Published: August 13, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.07.015.
Supplemental Information
References
- Adikusuma F., Piltz S., Corbett M.A., Turvey M., McColl S.R., Helbig K.J., Beard M.R., Hughes J., Pomerantz R.T., Thomas P.Q. Large deletions induced by Cas9 cleavage. Nature. 2018;560:E8–E9. doi: 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asplund A., Pradip A., van Giezen M., Aspegren A., Choukair H., Rehnström M., Jacobsson S., Ghosheh N., El Hajjam D., Holmgren S. One standardized differentiation procedure robustly generates homogenous hepatocyte cultures displaying metabolic diversity from a large panel of human pluripotent stem cells. Stem Cell Rev. Rep. 2016;12:90–104. doi: 10.1007/s12015-015-9621-9. [DOI] [PubMed] [Google Scholar]
- Bedel A., Taillepierre M., Guyonnet-Duperat V., Lippert E., Dubus P., Dabernat S., Mautuit T., Cardinaud B., Pain C., Rousseau B. Metabolic correction of congenital erythropoietic porphyria with iPSCs free of reprogramming factors. Am. J. Hum. Genet. 2012;91:109–121. doi: 10.1016/j.ajhg.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop D.F., Johansson A., Phelps R., Shady A.A., Ramirez M.C.M., Yasuda M., Caro A., Desnick R.J. Uroporphyrinogen III synthase knock-in mice have the human congenital erythropoietic porphyria phenotype, including the characteristic light-induced cutaneous lesions. Am. J. Hum. Genet. 2006;78:645–658. doi: 10.1086/502667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boch J., Scholze H., Schornack S., Landgraf A., Hahn S., Kay S., Lahaye T., Nickstadt A., Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- Brinkman E.K., Kousholt A.N., Harmsen T., Leemans C., Chen T., Jonkers J., van Steensel B. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 2018;46:e58. doi: 10.1093/nar/gky164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnight E.R., Gupta M., Wiley L.A., Anfinson K.R., Tran A., Triboulet R., Hoffmann J.M., Klaahsen D.L., Andorf J.L., Jiao C. Using CRISPR-Cas9 to generate gene-corrected autologous iPSCs for the treatment of inherited retinal degeneration. Mol. Ther. 2017;25:1999–2013. doi: 10.1016/j.ymthe.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canny M.D., Moatti N., Wan L.C.K., Fradet-Turcotte A., Krasner D., Mateos-Gomez P.A., Zimmermann M., Orthwein A., Juang Y.-C., Zhang W. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR–Cas9 genome-editing efficiency. Nat. Biotechnol. 2018;36:95–102. doi: 10.1038/nbt.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu V.T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K., Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullot G., Boutin J., Toutain J., Prat F., Pennamen P., Rooryck C., Teichmann M., Rousseau E., Lamrissi-Garcia I., Guyonnet-Duperat V. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019;10:1136. doi: 10.1038/s41467-019-09006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna J.A., Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Erwin A.L., Desnick R.J. Congenital erythropoietic porphyria: recent advances. Mol. Genet. Metab. 2019;128:288–297. doi: 10.1016/j.ymgme.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A., Notarangelo L.D., Neven B., Cavazzana M., Puck J.M. Severe combined immunodeficiencies and related disorders. Nat. Rev. Dis. Primers. 2015;1:15061. doi: 10.1038/nrdp.2015.61. [DOI] [PubMed] [Google Scholar]
- Ged C., Mendez M., Robert E., Lalanne M., Lamrissi-Garcia I., Costet P., Daniel J.Y., Dubus P., Mazurier F., Moreau-Gaudry F. A knock-in mouse model of congenital erythropoietic porphyria. Genomics. 2006;87:84–92. doi: 10.1016/j.ygeno.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Ged C., Moreau-Gaudry F., Richard E., Robert-Richard E., de Verneuil H. Congenital erythropoietic porphyria: mutation update and correlations between genotype and phenotype. Cell. Mol. Biol. 2009;55:53–60. [PubMed] [Google Scholar]
- Giannelli S.G., Luoni M., Castoldi V., Massimino L., Cabassi T., Angeloni D., Demontis G.C., Leocani L., Andreazzoli M., Broccoli V. Cas9/sgRNA selective targeting of the P23H rhodopsin mutant allele for treating retinitis pigmentosa by intravitreal AAV9.PHP.B-based delivery. Hum. Mol. Genet. 2018;27:761–779. doi: 10.1093/hmg/ddx438. [DOI] [PubMed] [Google Scholar]
- Gorter de Vries A.R., Couwenberg L.G.F., van den Broek M., de la Torre Cortés P., ter Horst J., Pronk J.T., Daran J.-M.G. Allele-specific genome editing using CRISPR–Cas9 is associated with loss of heterozygosity in diploid yeast. Nucleic Acids Res. 2019;47:1362–1372. doi: 10.1093/nar/gky1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- György B., Nist-Lund C., Pan B., Asai Y., Karavitaki K.D., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Solanes P., Spataro S. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat. Med. 2019;25:1123–1130. doi: 10.1038/s41591-019-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S., Garrigue A., Wang G.P., Soulier J., Lim A., Morillon E., Clappier E., Caccavelli L., Delabesse E., Beldjord K. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada F.A., Shwayder T.A., Desnick R.J., Lim H.W. Treatment of severe congenital erythropoietic porphyria by bone marrow transplantation. J. Am. Acad. Dermatol. 2001;45:279–282. doi: 10.1067/mjd.2001.114730. [DOI] [PubMed] [Google Scholar]
- Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z., Shi Z., Guo X., Jiang B., Wang G., Luo D., Chen Y., Zhu Y.-S. Ligase IV inhibitor SCR7 enhances gene editing directed by CRISPR–Cas9 and ssODN in human cancer cells. Cell Biosci. 2018;8:1–15. doi: 10.1186/s13578-018-0200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda A., Fujii W., Sugiura K., Naito K. High-fidelity endonuclease variant HypaCas9 facilitates accurate allele-specific gene modification in mouse zygotes. Commun. Biol. 2019;2:1–7. doi: 10.1038/s42003-019-0627-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen J.M., Chen X., Liu J., Gonçalves M.A.F.V. The chromatin structure of CRISPR-Cas9 target DNA controls the balance between mutagenic and homology-directed gene-editing events. Mol. Ther. Nucleic Acids. 2019;16:141–154. doi: 10.1016/j.omtn.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayavaradhan R., Pillis D.M., Goodman M., Zhang F., Zhang Y., Andreassen P.R., Malik P. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019;10:2866. doi: 10.1038/s41467-019-10735-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphans T., Sabri P., Zhu N., Heinrich V., Mundlos S., Robinson P.N., Parkhomchuk D., Krawitz P.M. Filtering for compound heterozygous sequence variants in non-consanguineous pedigrees. PLoS One. 2013;8:e70151. doi: 10.1371/journal.pone.0070151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keough K.C., Lyalina S., Olvera M.P., Whalen S., Conklin B.R., Pollard K.S. AlleleAnalyzer: a tool for personalized and allele-specific sgRNA design. Genome Biol. 2019;20:1–9. doi: 10.1186/s13059-019-1783-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn D.B., Kuo C.Y. New frontiers in the therapy of primary immunodeficiency: from gene addition to gene editing. J. Allergy Clin. Immunol. 2017;139:726–732. doi: 10.1016/j.jaci.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarde C., Hamel-Teillac D., De Prost Y., Blanche S., Thomas C., Fischer A., Nordmann Y., Ged C., De Verneuil H. [Allogeneic bone marrow transplantation in congenital erythropoietic porphyria. Gunther’s disease] Ann. Dermatol. Venereol. 1998;125:114–117. [PubMed] [Google Scholar]
- Li X.-L., Li G.-H., Fu J., Fu Y.-W., Zhang L., Chen W., Arakaki C., Zhang J.-P., Wen W., Zhao M. Highly efficient genome editing via CRISPR–Cas9 in human pluripotent stem cells is achieved by transient BCL-XL overexpression. Nucleic Acids Res. 2018;46:10195–10215. doi: 10.1093/nar/gky804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zhang Y., Li X., Yi S., Xu J. Gain-of-function mutations: an emerging advantage for cancer biology. Trends Biochem. Sci. 2019;44:659–674. doi: 10.1016/j.tibs.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X., Potter J., Kumar S., Ravinder N., Chesnut J.D. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 2017;241:136–146. doi: 10.1016/j.jbiotec.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Liu M., Rehman S., Tang X., Gu K., Fan Q., Chen D., Ma W. Methodologies for improving HDR efficiency. Front. Genet. 2019;9:691. doi: 10.3389/fgene.2018.00691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H., Marti-Gutierrez N., Park S.-W., Wu J., Lee Y., Suzuki K., Koski A., Ji D., Hayama T., Ahmed R. Correction of a pathogenic gene mutation in human embryos. Nature. 2017;548:413–419. doi: 10.1038/nature23305. [DOI] [PubMed] [Google Scholar]
- Maruyama T., Dougan S.K., Truttmann M.C., Bilate A.M., Ingram J.R., Ploegh H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla K.A., Yang Y. CRISPR/Cas-mediated base editing: technical considerations and practical applications. Trends Biotechnol. 2019;37:1121–1142. doi: 10.1016/j.tibtech.2019.03.008. [DOI] [PubMed] [Google Scholar]
- Monteys A.M., Ebanks S.A., Keiser M.S., Davidson B.L. CRISPR/Cas9 editing of the mutant huntingtin allele in vitro and in vivo. Mol. Ther. 2017;25:12–23. doi: 10.1016/j.ymthe.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris E.C., Fox T., Chakraverty R., Tendeiro R., Snell K., Rivat C., Grace S., Gilmour K., Workman S., Buckland K. Gene therapy for Wiskott-Aldrich syndrome in a severely affected adult. Blood. 2017;130:1327–1335. doi: 10.1182/blood-2017-04-777136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambiar T.S., Billon P., Diedenhofen G., Hayward S.B., Taglialatela A., Cai K., Huang J.-W., Leuzzi G., Cuella-Martin R., Palacios A. Stimulation of CRISPR-mediated homology-directed repair by an engineered RAD18 variant. Nat. Commun. 2019;10:1–13. doi: 10.1038/s41467-019-11105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado C.M., Heredia C.D.de, To-Figueras J., Arias-Santiago S., Nogueras P., Elorza I., Olivé T., Bádenas C., Moreno M.J., Tercedor J. Successful treatment of congenital erythropoietic porphyria using matched unrelated hematopoietic stem cell transplantation. Pediatr. Dermatol. 2013;30:484–489. doi: 10.1111/pde.12117. [DOI] [PubMed] [Google Scholar]
- Porteus M.H., Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- Rabai A., Reisser L., Reina-San-Martin B., Mamchaoui K., Cowling B.S., Nicot A.-S., Laporte J. Allele-specific CRISPR/Cas9 correction of a heterozygous DNM2 mutation rescues centronuclear myopathy cell phenotypes. Mol. Ther. Nucleic Acids. 2019;16:246–256. doi: 10.1016/j.omtn.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeil J.-A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., El Nemer W. Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- Richard E., Robert-Richard E., Ged C., Verneuil F.M.-G., de H. Erythropoietic porphyrias: animal models and update in gene-based therapies. Curr. Gene Ther. 2008;8:176–186. doi: 10.2174/156652308784746477. [DOI] [PubMed] [Google Scholar]
- Robert-Richard E., Moreau-Gaudry F., Lalanne M., Lamrissi-Garcia I., Cario-André M., Guyonnet-Dupérat V., Taine L., Ged C., Verneuil H.de. Effective gene therapy of mice with congenital erythropoietic porphyria is facilitated by a survival advantage of corrected erythroid cells. Am. J. Hum. Genet. 2008;82:113–124. doi: 10.1016/j.ajhg.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S., Maeda R., Adachi N. Dual loss of human POLQ and LIG4 abolishes random integration. Nat. Commun. 2017;8:1–10. doi: 10.1038/ncomms16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P.H., Mancini A.J., McConnell J.P., Brown D., Kletzel M. Treatment of congenital erythropoietic porphyria in children by allogeneic stem cell transplantation: a case report and review of the literature. Bone Marrow Transplant. 2001;27:101–105. doi: 10.1038/sj.bmt.1702738. [DOI] [PubMed] [Google Scholar]
- Shin H.Y., Wang C., Lee H.K., Yoo K.H., Zeng X., Kuhns T., Yang C.M., Mohr T., Liu C., Hennighausen L. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 2017;8:1–10. doi: 10.1038/ncomms15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C., Abalde-Atristain L., He C., Brodsky B.R., Braunstein E.M., Chaudhari P., Jang Y.-Y., Cheng L., Ye Z. Efficient and allele-specific genome editing of disease loci in human iPSCs. Mol. Ther. 2015;23:570–577. doi: 10.1038/mt.2014.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Yang D., Xu J., Zhu T., Chen Y.E., Zhang J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016;7:1–7. doi: 10.1038/ncomms10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tezcan I., Xu W., Gurgey A., Tuncer M., Cetin M., Öner C., Yetgin S., Ersoy F., Aizencang G., Astrin K.H. Congenital erythropoietic porphyria successfully treated by allogeneic bone marrow transplantation. Blood. 1998;92:4053–4058. [PubMed] [Google Scholar]
- Thomas C., Ged C., Nordmann Y., Verneuil H.de, Pellier I., Fischer A., Blanche S. Correction of congenital erythropoietic porphyria by bone marrow transplantation. J. Pediatr. 1996;129:453–456. doi: 10.1016/s0022-3476(96)70082-3. [DOI] [PubMed] [Google Scholar]
- Tsai S., Bishop D.F., Desnick R.J. Coupled-enzyme and direct assays for uroporphyrinogen III synthase activity in human erythrocytes and cultured lymphoblasts: enzymatic diagnosis of heterozygotes and homozygotes with congenital erythropoietic porphyria. Anal. Biochem. 1987;166:120–133. doi: 10.1016/0003-2697(87)90554-9. [DOI] [PubMed] [Google Scholar]
- Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C., Zhang Y.-P., Song L., Luo J., Qi W., Hu J., Lu D., Yang Z., Zhang J., Xiao J. Genome editing with CRISPR/Cas9 in postnatal mice corrects PRKAG2 cardiac syndrome. Cell Res. 2016;26:1099–1111. doi: 10.1038/cr.2016.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y., Makiyama T., Harita T., Sasaki K., Wuriyanghai Y., Hayano M., Nishiuchi S., Kohjitani H., Hirose S., Chen J. Allele-specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long-QT syndrome with a CALM2 mutation. Hum. Mol. Genet. 2017;26:1670–1677. doi: 10.1093/hmg/ddx073. [DOI] [PubMed] [Google Scholar]
- Yang L., Mali P., Kim-Kiselak C., Church G. CRISPR-Cas-mediated targeted genome editing in human cells. In: Storici F., editor. Gene Correction: Methods and Protocols. Humana Press; 2014. pp. 245–267. [DOI] [PubMed] [Google Scholar]
- Yasuda M., Desnick R.J. Murine models of the human porphyrias: contributions toward understanding disease pathogenesis and the development of new therapies. Mol. Genet. Metab. 2019;128:332–341. doi: 10.1016/j.ymgme.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelensky A.N., Schimmel J., Kool H., Kanaar R., Tijsterman M. Inactivation of Pol θ and C-NHEJ eliminates off-target integration of exogenous DNA. Nat. Commun. 2017;8:1–7. doi: 10.1038/s41467-017-00124-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Cong L., Lodato S., Kosuri S., Church G.M., Arlotta P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 2011;29:149–153. doi: 10.1038/nbt.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-P., Li X.-L., Li G.-H., Chen W., Arakaki C., Botimer G.D., Baylink D., Zhang L., Wen W., Fu Y.-W. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017;18:1–18. doi: 10.1186/s13059-017-1164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All next-generation sequencing datasets have been deposited in the NCBI database under BioProject accession no. PRJNA645083. The accession number for the exon 4 of CEP-hiPSC non-transfected reported in this paper is NCBI database: SAMN15491845.”, for transfected CEP-hiPSC with nuclease and RNA guide plasmids and ssODN is SAMN15491847 (Figure 5), and for corrected cells on exon 4 or porphyrin-negative cells is SAMN15491846 (Figure 6).