

Abstract
Focal adhesion kinase (FAK) has important functions in bone homeostasis but its role in early osteoprogenitor cells is unknown. We show herein that mice lacking FAK in Dermo1-expressing cells exhibited low bone mass and decreased osteoblast number. Mechanistically, FAK-deficient early osteoprogenitor cells had decreased proliferation and significantly reduced mammalian/mechanistic target of rapamycin complex 1 (mTORC1) signaling, a central regulator of cell growth and proliferation. Furthermore, our data showed that the pharmacological inhibition of FAK kinase-dependent function alone was sufficient to decrease the proliferation and compromise the mineralization of early osteoprogenitor cells. In contrast to the Fak deletion in early osteoprogenitor cells, FAK loss in Col3.6 Cre-targeted osteoblasts did not cause bone loss, and Fak deletion in osteoblasts did not affect proliferation, differentiation, and mTORC1 signaling but increased the level of active proline-rich tyrosine kinase 2 (PYK2), which belongs to the same non-receptor tyrosine kinase family as FAK. Importantly, mTORC1 signaling in bone marrow stromal cells (BMSCs) was reduced if FAK kinase was inhibited at the early osteogenic differentiation stage. In contrast, mTORC1 signaling in BMSCs was not affected if FAK kinase was inhibited at a later osteogenic differentiation stage, in which, however, the concomitant inhibition of both FAK kinase and PYK2 kinase reduced mTORC1 signaling. In summary, our data suggest that FAK promotes early osteoprogenitor cell proliferation by enhancing mTORC1 signaling via its kinase-dependent function and the loss of FAK in osteoblasts can be compensated by the upregulated active PYK2.
Keywords: FAK, mTORC1, osteoprogenitor, osteoblast, PYK2
Introduction
Focal adhesion kinase (FAK) is an intracellular non-receptor tyrosine kinase. Since its discovery,(1) numerous studies have shown critical roles of FAK in cell adhesion, migration, proliferation, and differentiation.(2,3) FAK can function through its kinase-dependent and kinase- independent functions in different cellular processes.(4–6)
Osteoblasts from osteoporotic human bone had decreased FAK activation compared to osteoblasts from non-osteoporotic control,(7) suggesting an important role of FAK in human bone health. However, our understanding of the roles and mechanisms of FAK in regulating bone homeostasis is very limited. To circumvent the embryonic lethality of Fak global knockout,(8) Fak has been deleted by different osteoblast-targeted Cre transgenic mice.(9–11) An earlier study showed that Fak deletion in osteoblasts by Col2.3-Cre does not affect mouse bone development.(9) Our recent study showed that Fak deletion in Osterix-expressing committed osteoblast precursor cells leads to osteopenia in mice.(10)
mTORC1 is a major regulator of protein synthesis,(12) and it controls cell growth and proliferation through two downstream effectors including the ribosomal protein S6 kinases (S6K1 and 2) and the eukaryotic translation initiation factor 4E (eIF4E)-binding proteins (4EBP1, 2, and 3).(13) S6K1 is required for 5’-terminal oligopyrimidine (5’-TOP) mRNA translation. Because ribosomal proteins and translation elongation factors are encoded by 5’-TOP mRNAs, signaling through the phosphorylation of S6K1 via mTORC1 activation promotes ribosomal biogenesis and enhance protein biosynthesis. The ribosomal protein S6 is the best known S6K1 substrate and its phosphorylation status faithfully reflects the S6K1 activity. 4EBP1 is a repressor of translation initiation; upon phosphorylation by mTORC1 activation, 4EBP1 releases from eIF4E, allowing eIF4E to bind with eIF4G to initiate cap-dependent translation.(13)
Dermo1 (Twist2)-Cre mouse is one of the commonly used transgenic mice to target early osteoprogenitor cells.(14,15) Inactivation of β1-integrin by Dermo1-Cre leads to severely impaired skeletal development.(16) Importantly, integrin-mediated cell adhesion to extracellular matrix activates FAK, which is the key component of the signal transduction pathways triggered by integrins.(17–19) To directly determine the role of FAK in early osteoprogenitor cells in bone homeostasis, we generated Fak conditional knockout mouse in which Fak was deleted by Dermo1-Cre. In addition, we used Col3.6-Cre(20) to delete Fak in osteoblasts. We showed that Fak deletion by Dermo1-Cre but not by Col3.6-Cre led to bone mass decrease that was accounted for by the decreased cell proliferation and decreased mTORC1 signaling in FAK- deficient early osteoprogenitor cells but not in FAK-deficient osteoblasts.
Materials and Methods
Mice
The floxed Fak (Fakflox/flox), Dermo1-Cre, and Col3.6-Cre, and Rosa26-RFP (Ai14) mice were described previously.(5,20–22) Fakflox/flox, Dermo1-Cre, and Col3.6-Cre mice were backcrossed for at least 8 generations onto a C57BL/6 background. Male Fakflox/flox mice were bred with female Fakflox/+;Dermo1-Cre mice to generate Fakflox/flox (Control), Fakflox/+;Dermo1-Cre (CHet) and Fakflox/flox;Dermo1-Cre (CKODermo1) mice. Male Fakflox/flox;RFP/RFP mice were crossed with female Fakflox/+;Dermo1-Cre mice to generate Fakflox/+;Dermo1-Cre; Ai14 (hereafter, CHet-RFP) and Fakflox/flox;Dermo1-Cre; Ai14 (hereafter, CKODermo1-RFP) mice. Male Fakflox/flox;Col3.6-Cre mice were bred with female Fakflox/flox mice to generate Fakflox/flox (Control) and Fakflox/flox;Col3.6-Cre (CKO3.6) mice. Mice were housed under pathogen-free conditions at 22 ± 2 °C on a 12:12-h light/dark cycle. Mice were euthanized using overdosing carbon dioxide. All the experimental procedures were carried out with the approval of the Institutional Animal Care and Use Committee at the University of Michigan.
MicroCT analysis
Femora and third lumbar vertebra were dissected and analyzed by micro-computed tomography using an eXplore Locus SP (GE Healthcare Pre-Clinical Imaging, London, ON, Canada) as we described previously.(23) The microCT image used for the analysis of vertebra cortical bone area was established 0.63 mm inferior to the end of cranial growth plate (Fig S1). The analyzer was blind to the identification of the samples.
Histomorphometry
Static histomorphometry was performed similarly to what we described previously.(23–25) All parameters were measured according to the Report of the American Society of Bone and Mineral Research Histomorphometry Nomenclature Committee.(26) The analyzer was blind to the identification of the samples.
Bone marrow stromal cells (BMSCs) isolation and culture
BMSCs were collected from 6-week-old mice in the same way as previously described.(10) Briefly, the end of each femur and tibia was cut and cells were flushed using alpha-MEM. Then the cells were cultured in 100-mm dish for 3 days before half change of the basic medium including alpha-MEM medium, 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml of streptomycin, followed by the full medium change on day 7 and every other day afterward. In some experiments, the adherent cells were trypsinized and plated for experiments on day 10.
BMSCs osteogenic differentiation and Histochemical assay
BMSCs isolated from control, CHet and CKODermo1 mice were seeded in 6-well plate at 1.3 × 107 cells/well and cultured as described above. The osteogenic medium containing 50 mg/ml of ascorbic acid and 10 mM of beta glycerophosphate was used on day 7 and changed every other day afterward. The alkaline phosphatase (ALP) staining and Alizarin red (AR) staining were performed on day 10 and day 21 respectively as previously described.(10) Crystal violet staining was performed on day 10. Briefly, cells were fixed with 100% methanol at −20°C for 15 mins and stained with 0.5% crystal violet solution at room temperature for 15 mins. Image J was used to measure the size of the colony after ALP staining and crystal violet staining. BMSCs collected from control and CKOCol3.6 mice were seeded in 12-well plate at 70,000 cells/well after being trypsinized on day 10. After 24 hours, cells were subjected to osteogenic medium that was changed every other day. ALP staining and AR staining were performed on day 7 and day 21 respectively.
FAK kinase inhibitor and FAK/PYK2 dual inhibitor treatment
BMSCs isolated from wild type mice and CKOCol3.6 mice were seeded in 6-well plate at 1.3 × 107 cells/well. FAK kinase inhibitor PF-573,228 (# B1523, Apexbio Technology) or FAK/PYK2 dual inhibitor PF-562,271 (# A8320, Apexbio Technology) was added with different doses at different time points every other day. Cell lysates were collected at indicated time points for western blot analysis. ALP staining and crystal violet staining were performed on day 10. Von Kossa staining was performed on day 21. In brief, cells were fixed using 70% ethanol in room temperature for 30 mins, hydrated to dH2O and then incubated in 5% silver nitrate at 37°C for 1h before being exposed to light for 30 mins.
Proliferation assay
For experiments using control and CKOCol3.6 mice, BMSCs were seeded in 12-well plate at 70,000 cells/well after trypsinizing the mesenchymal colonies on day 10. For the experiments using other mice, BMSCs were directly seeded in 12-well plate at 5× 106 cells/well. Cells were trypsinized and the cell number was counted on day 3 and day 7. Ki67 immunostaining was performed on day 3 and day 7 as previously described.(10) Briefly, cells were fixed with 4% paraformaldehyde and immunostained using anti-Ki67 antibody (# 9129, Cell Signaling Technology).
Western blot
Western blot was performed as previously described.(10) Antibody information is described below. Anti-β-catenin: # 610153 BD Transduction laboratories; anti-Vinculin: # V4505, Sigma- Aldrich; anti-p-PYK2 (Tyr402): # 3291, Cell Signaling Technology; anti-PYK2: # 3292, Cell Signaling Technology; anti-p-FAK (Tyr397): # 3283, Cell Signaling Technology; anti-FAK: #SC-558, Santa Cruz, USA; anti-p-S6 (Ser240/244): # 5364, Cell Signaling Technology; anti-S6: # 2217, Cell Signaling Technology; anti-p-mTOR (Ser2448): # 2971, Cell Signaling Technology; anti-mTOR: # 2972, Cell Signaling Technology; anti-p-4E-BP1 (Thr37/46): # 2855, Cell Signaling Technology; anti-4E-BP1: # 9452, Cell Signaling Technology; anti-p-AKT (Ser473): # 4060, Cell Signaling Technology; anti-AKT: # 4691, Cell Signaling Technology.
Statistics
For the comparison between two groups, Student’s t-test was used. For the comparison in more than two groups, one way ANOVA with Tukey’s multiple comparison tests were performed using GraphPad Prism 8.0. For the comparison of two groups in which have two different variables, two way ANOVA with Sidak’s multiple comparison tests were performed using GraphPad Prism 8.0.
Results
Deletion of Fak in Dermo1-expressing cells leads to osteopenia in mice
To determine the role of FAK in early osteoprogenitor cells, we deleted Fak using Dermo1-Cre. Dermo1-Cre mice were generated by inserting the Cre recombinase gene within the first exon of Dermo1, thus resulting in haploinsufficiency.(21) To determine whether Dermo1 haploinsufficiency affects normal bone development, we analyzed Dermo1-Cre/+ and littermate wild type mice. Our data showed that Dermo1 haploinsufficiency did not affect body weight (Fig S2) and bone mass (Fig S3) in both male and female mice. Fak deletion in Dermo1-expressing cells did not affect the body weight and femoral bone length of male mice and it slightly decreased the body weight without affecting the femoral bone length of female mice at two months of age (Fig S4). To determine the FAK deletion efficiency, western blot was performed using lysates extracted from femurs of 2-month-old mice. Our data showed that FAK was similarly deleted in male and female mice (Fig S5). MicroCT analysis showed that Fak deletion in Dermo1-expressing cells led to a significant decrease in femoral trabecular bone mass (Fig 1A–E) and a significant decrease in femoral cortical bone size (Fig 1F–H) in 2-month-old male mice. Because of the concomitant decrease in both total cross-sectional area and marrow area, there was no change in cortical area and thickness in CKODermo1 male mice (Fig 1I–J). In vertebra, Fak deletion led to a significant decrease in both trabecular and cortical bone mass in male mice (Figure S6). However, Fak deletion in Dermo1-expressing cells did not affect femoral bone mass in 2-month-old female mice although female CKODermo1 mice showed the similar trend in the decrease of trabecular thickness and total cross-sectional area, and a decrease in marrow area (Figure S7). Our data suggested that this gender difference was not due to the difference in FAK deletion efficiency between male and female CKODermo1 mice (Fig S5). The reason underlying this gender difference is unclear.
Figure 1. Fak deletion in Dermo1-expressing cells leads to osteopenia in mice.
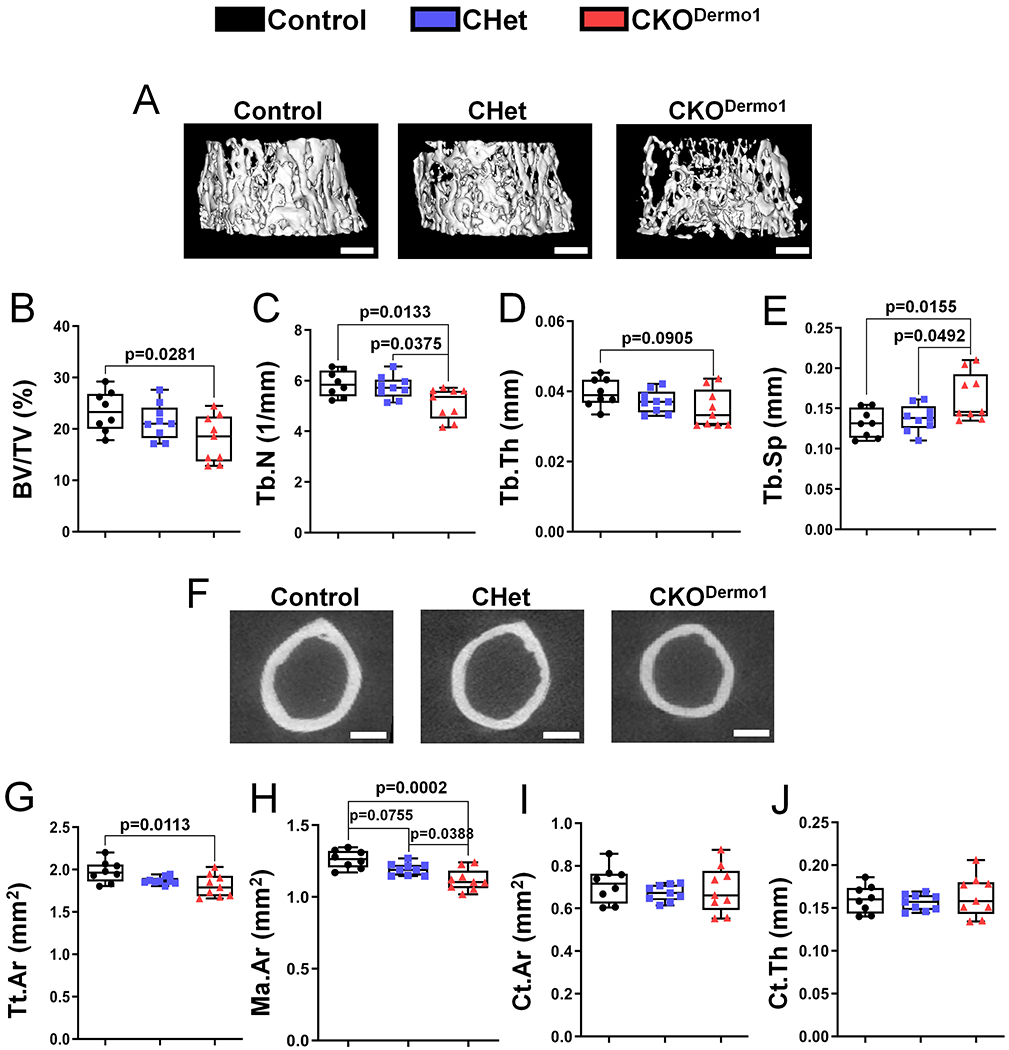
MicroCT analysis were performed in femurs of 2-month-old male Fakflox/flox (Control), Fakflox/+;Dermo1-Cre (CHet) and Fakflox/flox;Dermo1-Cre (CKODermo1) mice. (A-E) Femoral trabecular bone: (A) Representative microCT image, scale bar=500 μm; (B) BV/TV, bone volume fraction; (C) Tb.N, trabecular number; (D) Tb.Th, trabecular thickness; (E) Tb.Sp, trabecular spacing. (F-J) Femoral cortical bone: (F) Representative microCT image, scale bar=300 μm; (G) Tt.Ar, total cross-sectional area inside the periosteal envelope; (H) Ma.Ar, marrow area; (I) Ct.Ar, cortical bone area; (J) Ct.Th, average cortical thickness. Values were presented as median and interquartile range (n=8 for control group, n=9 for CHet group, and n=9 for CKODermo1 group).
Histomorphometry analysis also showed the decrease of femoral trabecular bone mass in CKODermo1 male mice (Fig 2A–E), which was accounted for by a significant decrease in osteoblast number and surface without change in osteoclast number and surface (Fig 2F–I). In addition, Fak deletion in Dermo1-expressing cells did not affect the bone marrow adipocyte number (data not shown).
Figure 2. Fak deletion in Dermo1-expressing cells leads to decreased osteoblast number.
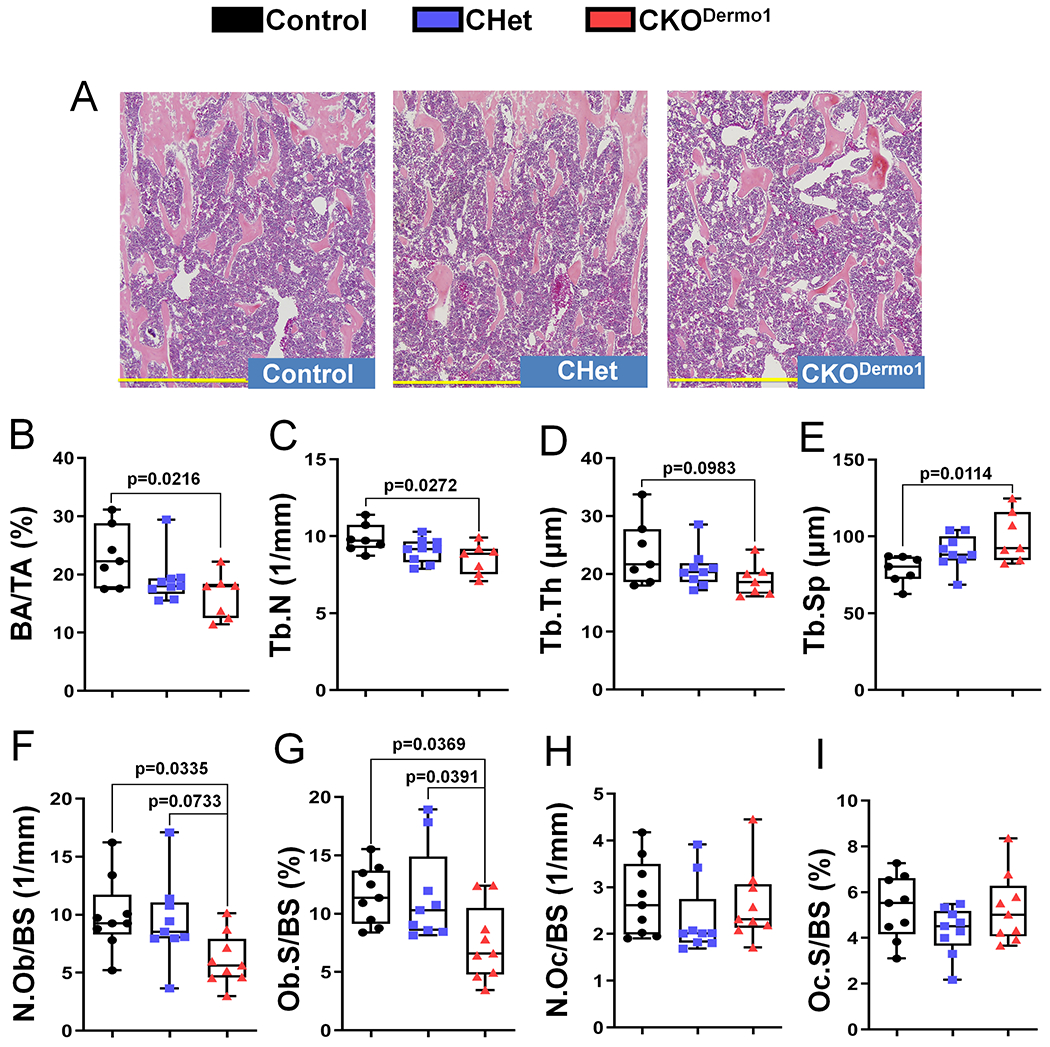
Histomorphometric analysis was performed in femurs of 2-month-old male Fakflox/flox (Control), Fakflox/+;Dermo1-Cre (CHet) and Fakflox/flox;Dermo1-Cre (CKODermo1) mice. (A) Representative histological image, scale bar=0.7 mm. (B) BA/TA, bone area per tissue area. (C) Tb.N, trabecular number. (D) Tb.Th, trabecular thickness. (E) Tb.Sp, trabecular spacing. (F) N.Ob/BS, osteoblast number per bone surface. (G) Ob.S/BS, osteoblast surface per bone surface. (H) N.Oc/BS, osteoclast number per bone surface. (I) Oc.S/BS, osteoclast surface per bone surface. Values were presented as median and interquartile range (n=7 for control group, n=9 for CHet group, and n=7 for CKODermo1 group).
FAK deletion in Dermo1-expressing cells leads to decreased BMSC proliferation
To elucidate the mechanism responsible for the decreased bone mass and decreased osteoblast number in CKODermo1 mice, we determined the effect of Fak deletion on the proliferation of early mesenchymal lineage cells using primary bone marrow stromal cell (BMSC) culture system. Because of the heterogeneity nature of BMSC culture, first we performed lineage mapping experiments by introducing Rosa26-RFP reporter into CHet and CKODermo1 mice. BMSCs were isolated from CHet-RFP and CKODermo1-RFP mice and the percentage of RFP-positive cells within BMSC colonies were analyzed. Our data showed that there was a significant decrease in the percentage of RFP-positive cells in CKODermo1-RFP group compared to CHet-RFP group (Fig 3A–B), suggesting that the Fak-deleted BMSCs had compromised ability to increase their number.
Figure 3. Fak deletion in Dermo1-expressing cells leads to decreased BMSC proliferation.
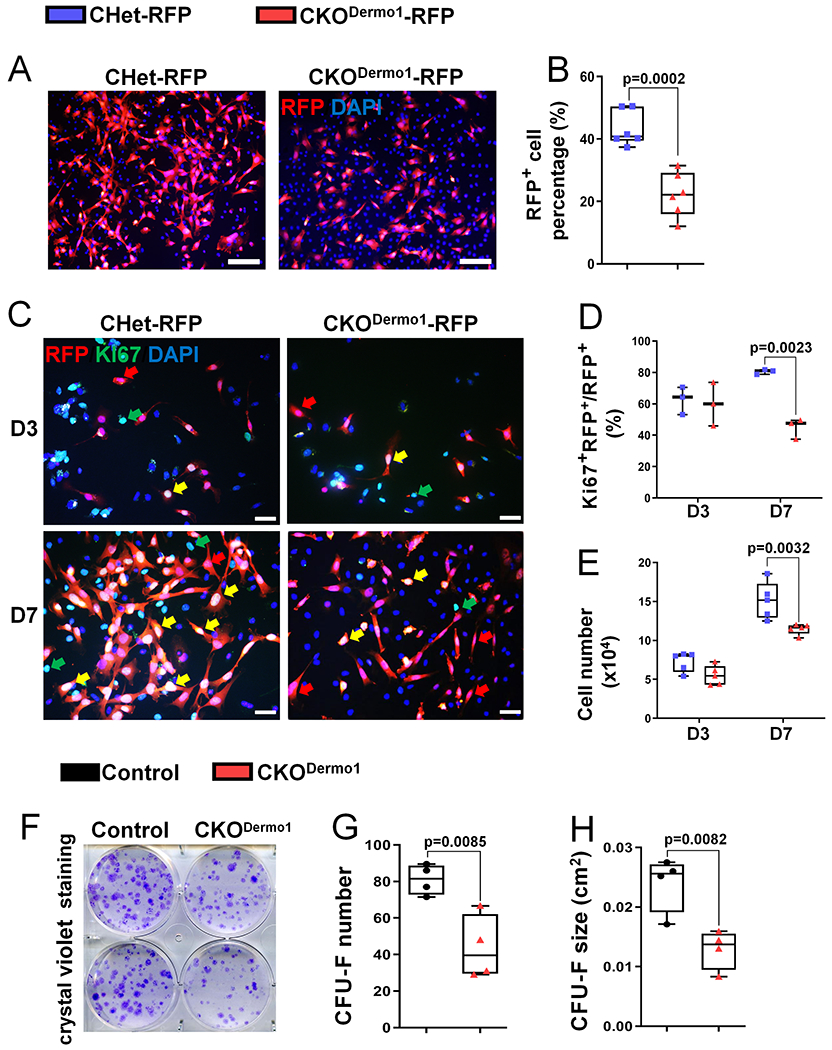
(A-B) Analysis of percentage of RFP-positive cells in clonal BMSCs isolated from CHet-RFP and CKODermo1-RFP mice: (A) Representative fluorescent images. Red indicates RFP-positive signal and blue (Dapi staining) indicates nucleus. Scale bar=100 μm; (B) Quantification of the percentage of RFP-positive cells in day 7 culture illustrated in (A). Data were the average of 6 independent experiments. (C-D) Analysis of Ki67-positive cells in the population of RFP- positive clonal BMSCs isolated from CHet-RFP and CKODermo1-RFP mice: (C) Representative fluorescent images in day 3 (D3) and day 7 (D7) cultures. Yellow arrows indicate the RFP+Ki67+DAPI+cells; red arrows indicate the RFP+Ki67−DAPI+cells; green arrows indicate the RFP−Ki67+DAPI+cells. Scale bar=20 μm; (D) Quantification of the percentage of Ki67 and RFP double positive cells illustrated in (C) (n=3). (E) Quantification of the cell number cultured for 3 and 7 days in the same culture system as (C) (n=5). (F-H) CFU-F (colony forming unit-fibroblast) assay of control and CKODermo1 BMSCs: (F) Representative image of crystal violet staining at day 10; (G) Quantification of the CFU-F number; and (H) Quantification of the CFU-F size described in (F) (n=4). Each data point represents the average of one independent experiment. Values were presented as median and interquartile range.
To determine the mechanism that was accountable for the decrease in the percentage of RFP-positive cells in CKODermo1-RFP group, we performed immunofluorescence staining of Ki67, a proliferation marker. Our data showed that there were significantly less Ki67-positive cells within the RFP-positive cells of CKODermo1-RFP group compared to that of CHet-RFP group in day 7 culture while there was no difference between these two groups at day 3 (Fig 3C– D). Consistent with a decrease in proliferation, BMSCs isolated from CKODermo1 mice had a significant decrease in cell number in day 7 culture (Fig 3E). To determine whether Dermo1 haploinsufficiency itself affects the BMSC proliferation, we performed similar experiments using BMSCs isolated from Dermo1-Cre/+ mice and their wild type littermates. Our data showed that two groups had comparable percentage of Ki67-positive BMSCs and total cell number (Fig S8).
Next, we determined the effect of Fak deletion in Dermo1-expressing cells on the colony-forming units-fibroblastic (CFU-F) formation. Our data showed CKODermo1 group had a significant decrease in both CFU-F number and size (Fig 3F–H). Similarly, CKODermo1 group had a significant decrease in both the number and size of ALP-positive CFU-F (CFU-ALP) (Fig S9A–C). In addition, the CKODermo1 group had significantly compromised mineralization (Fig S9D–E).
FAK kinase inhibition leads to decreased BMSC proliferation
FAK has both kinase–dependent and–independent functions.(4,27) To determine the contribution of the kinase-dependent function of FAK to the BMSC proliferation, we utilized the specific FAK kinase inhibitor PF-573,228,(28) which is a potent ATP-competitive, reversible inhibitor of FAK kinase activity and was developed to treat various cancers.(29,30) It was shown that PF- 573,228 can block >75-80% endogenous FAK activity at the concentration of 0.3-3 μM in different cell types.(28) Thus, we first determined the effect of PF-573,228 on the inhibition of FAK kinase activation at the dose of 0.1 μM to 2 μM. Our data showed that 1 μM PF-573,228 effectively inhibited FAK kinase activation shown by the significantly decreased phospho- FAKTyr397 level (Fig 4A–B). When BMSCs isolated from CHet-RFP mice were treated with 1 μM PF-573,228, there was a significant decrease in the percentage of Ki67-positive cells within the RFP-positive cells (Fig 4C–D) and a significant decrease in total cell number (Fig 4E). Furthermore, PF-573,228 significantly decreased CFU-F number and size (Fig 4F–H), ALP- positive CFU-F number and size (Fig S10A–C) and mineralization (Fig S10D–E) of BMSCs. Thus, our data suggested that the loss of FAK’s kinase-dependent function contributed significantly to the proliferation inhibition in Fak-deleted Dermo1-expressing BMSCs.
Figure 4. FAK kinase inhibition leads to decreased BMSC proliferation.
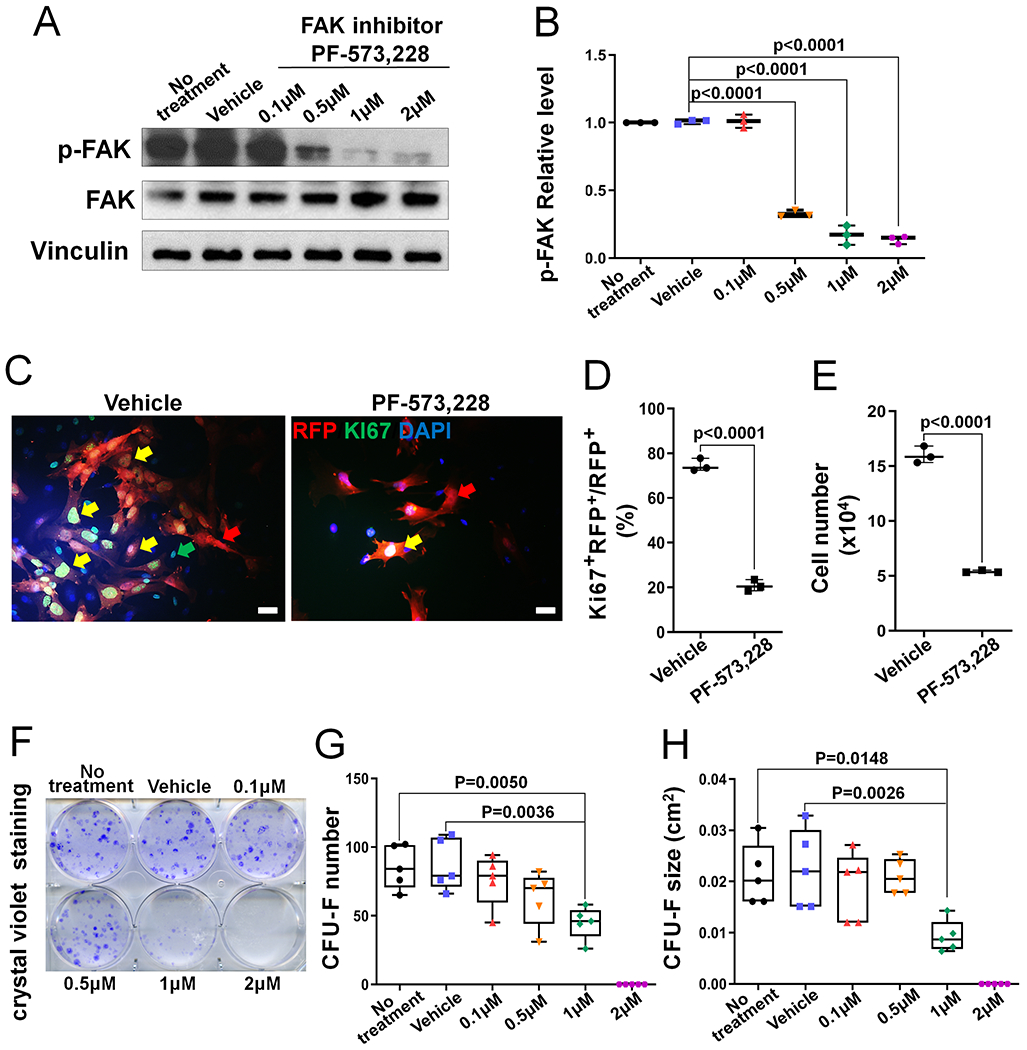
(A) Representative western blot image showing that phospho-FAK (p-FAK) level in BMSCs was dose-dependently suppressed by FAK kinase inhibitor PF-573,228. (B) Quantification of the p-FAK relative level in controls (no treatment and vehicle treatment) and FAK inhibitor-treated BMSCs. p-FAK was normalized to total FAK protein (n=3). (C) Representative fluorescent images of BMSCs isolated from CHet-RFP mice after the treatment with vehicle or FAK inhibitor PF-573,228 for 7 days. Yellow arrows indicate the RFP+Ki67+DAPI+cells; red arrows indicate the RFP+Ki67− DAPI+cells; green arrows indicate the RFP−Ki67+DAPI+cells. Scale bar=20 μm. (D) Quantification of the percentage of Ki67 and RFP double positive cells in the cultures described in (C) (n=3). (E) Quantification of the total cell number in the cultures described in (C) (n=3). (F) Representative image of crystal violet staining in BMSC culture treated with indicated doses of FAK inhibitor PF-573,228 for 10 days. (G) Quantification of the CFU-F number; and (H) Quantification of the CFU-F size in cultures described in (F). Each data point represents the average of one independent experiment (n=5). Values were presented as median and interquartile range.
FAK deficiency in Dermo1-expressing cells leads to decreased mTORC1 signaling
To elucidate the molecular mechanism that was responsible for the decreased proliferation in CKODermo1 BMSCs, we started with determining the effect of Fak deletion in Dermo1-expressing cells on β-catenin level because we previously showed that Fak deletion in Osterix-expressing cells leads to a significant decrease in β-catenin level.(10) First, we determined the FAK deletion efficiency in our BMSC cultures. In most of the experiments (5 out of 9) performed, there was sufficient FAK deletion; however, in some experiments (4 out of 9), FAK was not sufficiently deleted (Fig S11A). For cell signaling study, we focused on the five experiments with sufficient FAK deletion. Our data showed that Fak deletion in Dermo1-expressing cells did not affect β-catenin level (Fig 5A–B). This is unexpected because our previous study showed Fak deletion in Osterix-expressing cells significantly decreases β-catenin level.(10) Next, we examined the effect of Fak deletion in Dermo1-expressing cells on the mTORC1 signaling because our previous study suggested that mTORC1 signaling is an important positive regulator on proliferation of osteoprogenitor cells.(31) Our data showed that Fak deletion in Dermo1-expressing cells significantly inhibited mTORC1 activity demonstrated by the decrease in the level of phospho-mTOR and phospho-S6 (Fig 5A–B). However, there was no change in phospho-4EBP1, the other downstream effector of mTORC1 (Fig 5A–B). In addition, Fak deletion in Dermo1-expressing cells significantly decreased phospho-AKT level and there was no change in phospho-PYK2 level (Fig 5A–B). Of note, in the experiments in which FAK was not sufficiently deleted, the levels of phospho-S6 and phospho-AKT were not decreased (Fig S11B), further supporting the connection between FAK and AKT/mTORC1 signaling. To further determine the regulation of mTORC1 activity by FAK in Dermo1-expressing BMSCs, we performed immunofluorescence staining using antibody against phospho-S6. Our data showed a similar expression pattern between CHet-RFP and CKODermo1-RFP in day 3 culture; however, there was a significant decrease in the percentage of phospho-S6 positive cells within RFP-positive cells in CKODermo1- RFP group in day 7 culture (Fig 5C–D).
Figure 5. Fak deficiency in osteoprogenitor cells leads to decreased mTORC1 signaling.
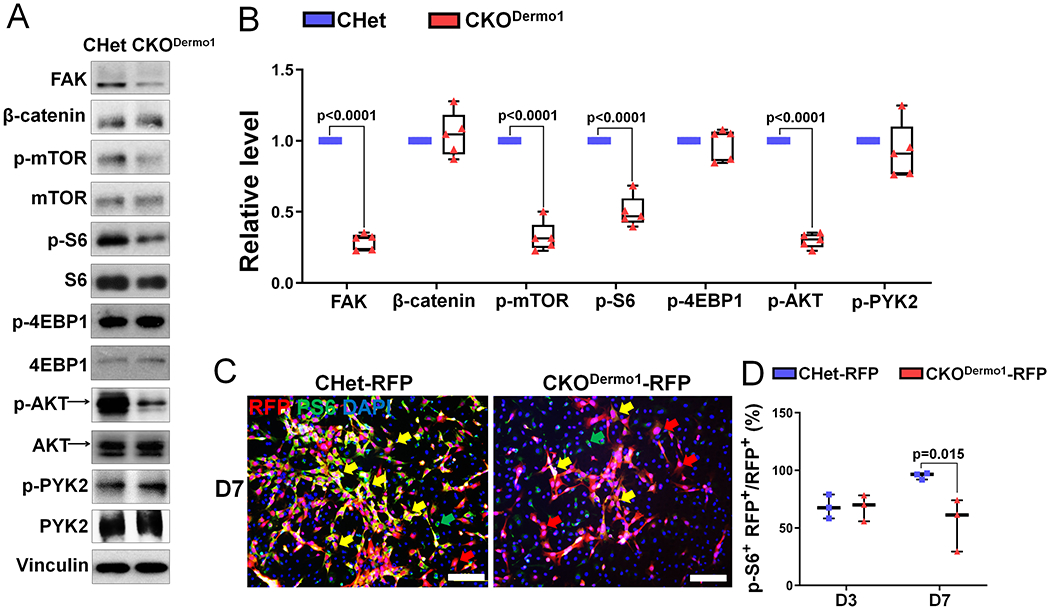
(A-B) BMSCs isolated from CHet and CKODermo1 mice were analyzed by western blot: (A) Representative western blot images using indicated antibodies. (B) Quantification of the relative level of signaling molecules illustrated in (A) (n=5). FAK and β-catenin were normalized to Vinculin and phospho-proteins were normalized to the respective total proteins. (C-D) BMSCs isolated from CHet-RFP and CKODermo1-RFP mice were analyzed by immunofluorescence using anti-p-S6 antibody: (C) Representative fluorescent image in day 7 culture. Yellow arrows indicate the RFP+p-S6+DAPI+cells; red arrows indicate the RFP+p-S6−DAPI+cells; green arrows indicate the p-S6−Ki67+DAPI+cells. Scale bar=100 μm. (D) Quantification of percentage of the p- S6 and RFP double positive cells illustrated in (C) (n=3). Values were presented as median and interquartile range
Fak deletion in osteoblasts does not affect bone development
Our current study and previous study(10) showed that FAK in Dermo1-expressing early osteoprogenitor cells and Osterix-expressing committed osteoblast precursors plays important role in bone development. To determine the effect of Fak deletion in osteoblasts on bone development, we analyzed Fak conditional knockout mice targeted by Col3.6-Cre (CKOCol3.6).
In femoral bone of CKOCol3.6 mice, FAK was efficiently deleted in association with an increase in phospho-PYK2 level (Fig 6A). CKOCol3.6 mice developed normally (Fig 6B). CKOCol3.6 male mice had a transient small but statistically significant decrease in body weight during early postnatal growth with similar body length (Fig 6C–D). In addition, Fak deletion in osteoblast did not affect the body weight but slightly decreased the body length in CKOCol3.6 female mice (Fig S12). Furthermore, microCT analysis showed that CKOCol3.6 male mice had similar trabecular (Fig 6E–H) and cortical (Fig 6I–L) bone parameters compared with control mice. Histomorphometric analysis showed that there were no changes in both osteoblast and osteoclast parameters in CKOCol3.6 male mice compared with the controls (data not shown).
Figure 6. Fak deletion in osteoblasts does not affect bone development.
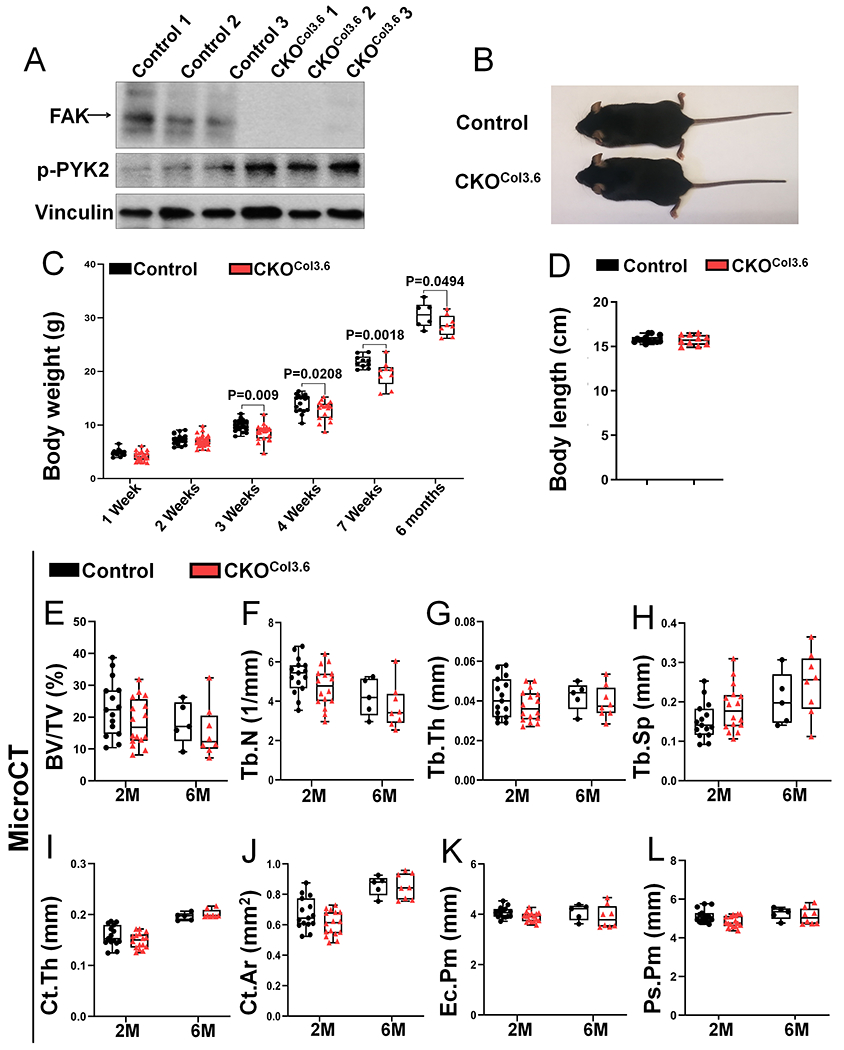
(A) FAK and phospho-PYK2 (p-PYK2) protein levels were analyzed by western blot in cortical bone isolated from femur and tibia of 1-month-old Fakflox/flox (Control) and Fakflox/flox;Col3.6-Cre (CKOCol3.6) male mice. (B) Representative image of 7-week-old control and CKOCol3.6 mice. (C) Body weight of male control and CKOCol3.6 mice at indicated ages (n=16, 26, 22, 17, 10, 6 in 1 week, 2 weeks, 3 weeks, 4 weeks, 7 weeks and 6 months groups respectively for control mice; and n=22, 34, 23, 17, 9, 8 in 1 week, 2 weeks, 3 weeks, 4 weeks, 7 weeks and 6 months groups respectively for CKOCol3.6 mice). (D) Body length of 7-week-old male control and CKOCol3.6 mice (n=14 for control mice, n=10 for CKOCol3.6 mice). (E-L) MicroCT analysis of femoral trabecular and cortical bone in 2-month-old (2M) and 6-month-old (6M) male mice: (E) BV/TV, bone volume fraction; (F) Tb.N, trabecular number; (G) Tb.Th, trabecular thickness; (H) Tb.Sp, trabecular spacing; (I) Ct.Th, average cortical thickness; (J) Ct.Ar, cortical bone area; (K) Ec.Pm, endocortical perimeter; (L) Ps.Pm, periosteal perimeter (n=15 and 5 in 2M and 6M groups respectively for control mice, n=16 and 8 in 2M and 6M groups respectively for CKOCol3.6 mice). Values were presented as median and interquartile range.
Fak deletion in osteoblasts does not affect BMSC proliferation and mTORC1 activity
To further determine the extent that FAK regulates osteoblast function, we performed cellular and signaling studies using BMSCs isolated from CKOCol3.6 and control mice. First, our data showed that Fak deletion in osteoblasts did not affect the BMSC proliferation evidenced by the similar percentage of Ki67-positive cells (Fig 7A–B) and similar cell number (Fig 7C) in CKOCol3.6 group compared to control group. Second, Fak deletion in osteoblasts did not compromise the osteogenic differentiation and mineralization of BMSCs (Fig 7D–E). Third, despite the effective FAK deletion, there was no change in the levels of phospho-S6 and β- catenin in CKOCol3.6 BMSCs (Fig 7F–G). Notably, there was a significant increase in phospho- PYK2 level in CKOCol3.6 group (Fig 7F–G).
Figure 7. Fak deletion in osteoblasts does not affect proliferation and mTORC1 activity.
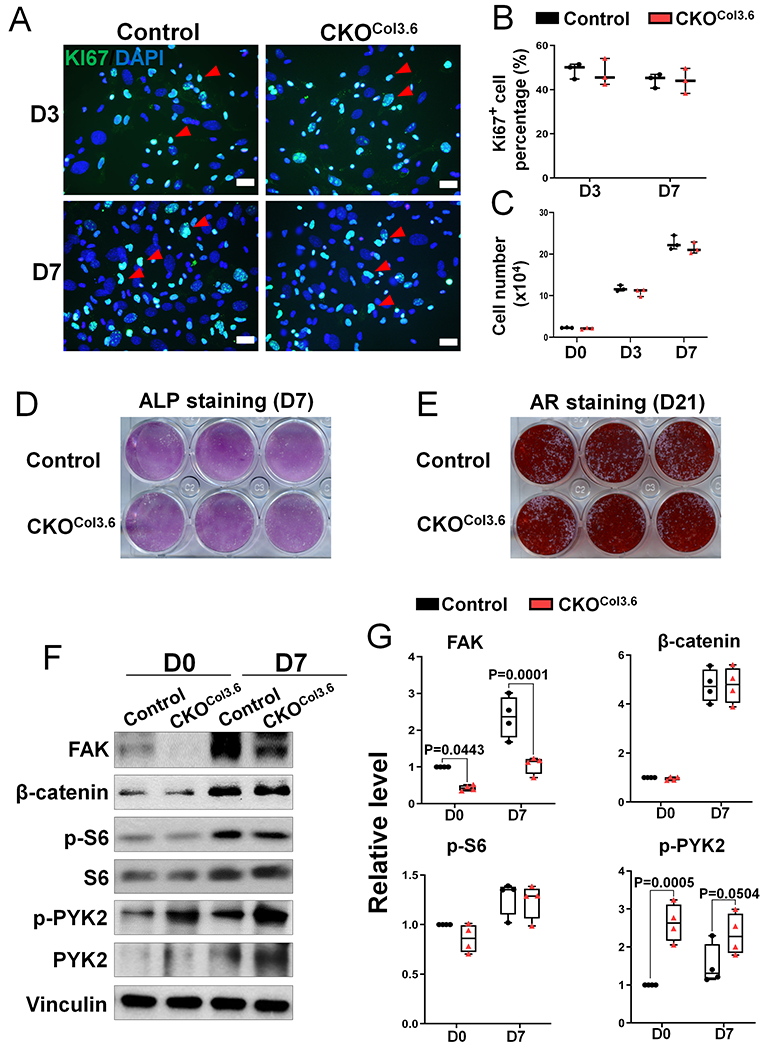
(A) Representative fluorescent images of Ki67 (green) and Dapi (blue) staining in Fakflox/flox (Control) and Fakflox/flox;Col3.6-Cre (CKOCol3.6) BMSCs after 3 and 7 days culture. Red arrow heads point to the Ki67+DAPI+cells. Scale bar=20 μm. (B) Quantification of the percentage of Ki67-positive BMSCs described in (A) (n=3). (C) Quantification of BMSC cell number in the same culture as (A) (n=3). (D) Alkaline phosphatase (ALP) staining was performed after 7 days osteogenic culture of control and CKOCol3.6 BMSCs. Image was the representative of 6 independent experiments. (E) Alizarin red (AR) staining was performed after 21 days osteogenic culture of control and CKOCol3.6 BMSCs. Image was the representative of 6 independent experiments. (F-G) Western blot was performed on lysates of BMSCs isolated from Fakflox/flox (Control) and Fakflox/flox;Col3.6-Cre (CKOCol3.6) mice: (F) Representative western blot image using indicated antibodies in day 0 (first day after plating) and day 7 cultures; (G) Quantification of the relative protein levels as illustrated in (E). FAK and β-catenin were normalized to Vinculin and phospho-proteins were normalized to the respective total proteins (n=4). Values were presented as median and interquartile range.
The inhibition of mTORC1 signaling by FAK kinase inhibitor is cell differentiation stage-dependent
To elucidate the underlying mechanisms by which FAK was differentially required to support the proliferation and mTORC1 signaling in early osteoprogenitor cells vs. osteoblasts, we turned to the strategy of treating BMSCs with FAK kinase inhibitor at different differentiation stages. First, we characterized the osteogenic differentiation status of BMSCs in our culture model at different time points. ALP staining showed negative staining at day 3 but wide-spread alkaline phosphatase-positive colonies in day 7 and day 10 cultures (Fig 8A). In addition, we found nearly all cells were negative for Osterix, a marker of committed osteoblast precursor, at the first two days of culture; there were increasingly more and more Osterix-positive cells starting from day 3 and nearly all of them were Osterix-positive at day 7 (data not shown). Next, we designed a treatment regimen using FAK kinase inhibitor to further determine the effect of FAK kinase inhibition on Wnt/β-catenin and mTORC1 signaling at different differentiation stages (Fig 8B). Our data showed that β-catenin level was only consistently inhibited if FAK kinase inhibitor was added when cells started to express Osterix, which was around day 3 in our culture system; on the other hand, AKT and mTORC1 signaling was consistently inhibited when FAK kinase inhibitor was added at earlier culture in cohort I but not later culture in cohort II (Fig 8C–E). Importantly, our data showed that there was a significant increase in phospho-PYK2 level when FAK kinase inhibitor was added in later culture but not in early culture (Fig 8C–E), which was similar to the increase in phospho-PYK2 level observed in BMSCs of CKOCol3.6 mice (Fig 7F–G). This suggested that the lack of effect of FAK inhibition in osteoblasts on AKT and mTORC1 signaling was likely due to the compensatory increase in PYK2 activity. Finally, we determined the effect of FAK kinase inhibition at different differentiation stages on BMSC mineralization by von Kossa staining (Fig S13A). Our data showed that early FAK kinase inhibition starting from day 1 or day 3 led to significantly decreased mineralized nodule numbers; however FAK kinase inhibition starting from day 7 did not decrease BMSC mineralization. Interestingly, the transient FAK kinase inhibition from day 1 to day 7 was sufficient to compromise the later mineralization (Fig S13B–C). Taken together, our data indicated that FAK kinase activity has an important role in early osteoblast lineage cells but not in late osteoblasts.
Figure 8. FAK kinase inhibition decreases mTORC1 signaling of BMSCs in a cell differentiation stage-dependent manner.
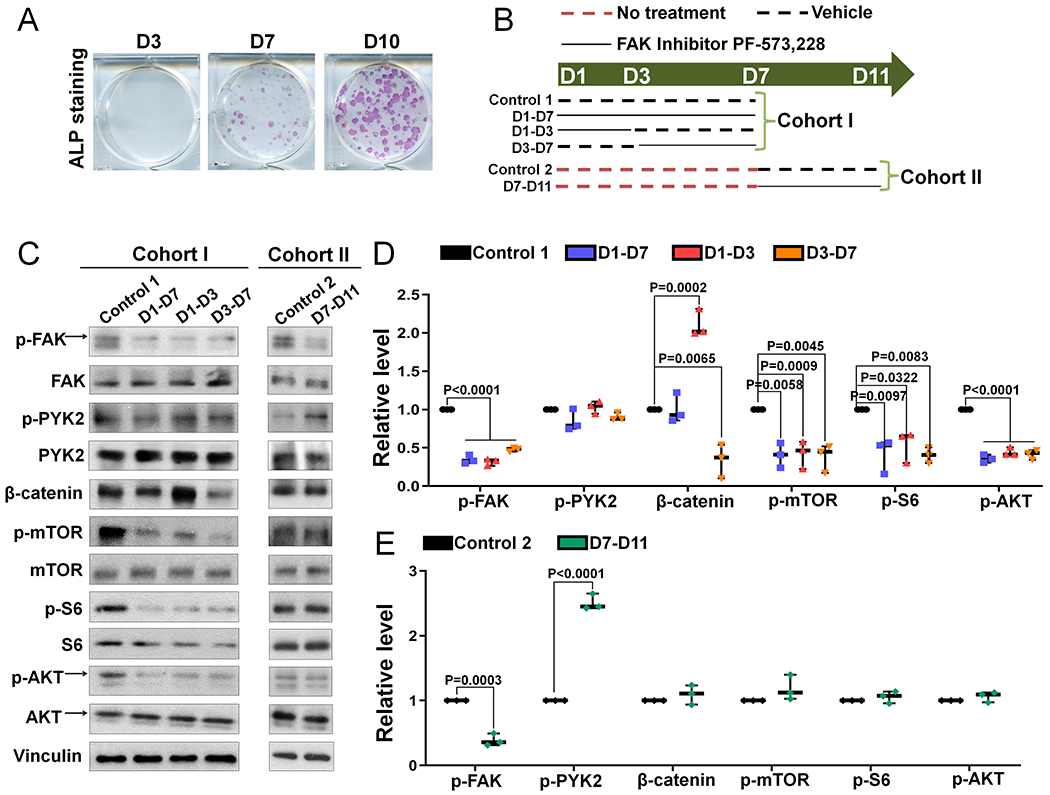
(A) Representative images of alkaline phosphatase (ALP) staining at indicated days of culture (n=3). (B) Cartoon showing the experimental design of FAK inhibitor PF-573,228 treatment used in (C-D). Dashed red line represented no treatment, dashed black line represented vehicle treatment and solid black line represented 1 μM FAK inhibitor PF-573,228 treatment at indicated culture time in different groups. There were two cohorts of culture: 7 days culture and 11 days culture. In cohort I, D1-D7 means FAK inhibitor was added from day 1 to day 7; D1-D3 means FAK inhibitor was added from day 1 to day 3; D3-D7 means FAK inhibitor was added from day 3 to day 7. In cohort II, D7-D11 means FAK inhibitor was added from day 7 to day 11. (C) At the end of culture in each cohort, BMSCs were analyzed with indicated antibodies. (D) Quantification of the relative level of the signaling molecules illustrated in Cohort I (n=3). (E) Quantification of the relative level of the signaling molecules illustrated in Cohort II (n=3). Phospho-proteins were normalized to the respective total proteins and β-catenin was normalized to Vinculin. Values were presented as median and interquartile range.
Dual inhibition of FAK kinase and PYK2 kinase decreases mTORC1 signaling and compromises osteogenic differentiation of BMSCs at later differentiation stage
To test the hypothesis that PYK2 compensates for the loss of FAK at later differentiation stage of BMSCs, we determined the effect of dual inhibition of FAK kinase and PYK2 kinase on mTORC1 signaling by using FAK/PYK2 dual inhibitor PF-562,271(32) at a later differentiation stage of BMSCs. First, our data showed that 1 μM FAK/PYK2 dual inhibitor effectively inhibited the levels of both phospho-FAKTyr397 and phospho-PYK2Tyr402 (Fig 9A–C) in wild type BMSCs. Next, the addition of FAK/PYK2 dual inhibitor starting from day 7 BMSC culture significantly decreased the mTORC1 signaling and the level of phospho-AKT in wild type BMSCs (Fig 9D–F). Finally, our data showed that FAK/PYK2 dual inhibitor treatment starting from day 7 culture inhibited osteogenic differentiation and mineralization (Fig S14). This inhibitory effect was absent in FAK inhibitor treatment (Fig S13, D7-D21 group), highlighting the significant difference between these two inhibitors.
Figure 9. Dual inhibition of FAK kinase and PYK2 kinase decreases mTORC1 signaling of BMSCs at later differentiation stage.
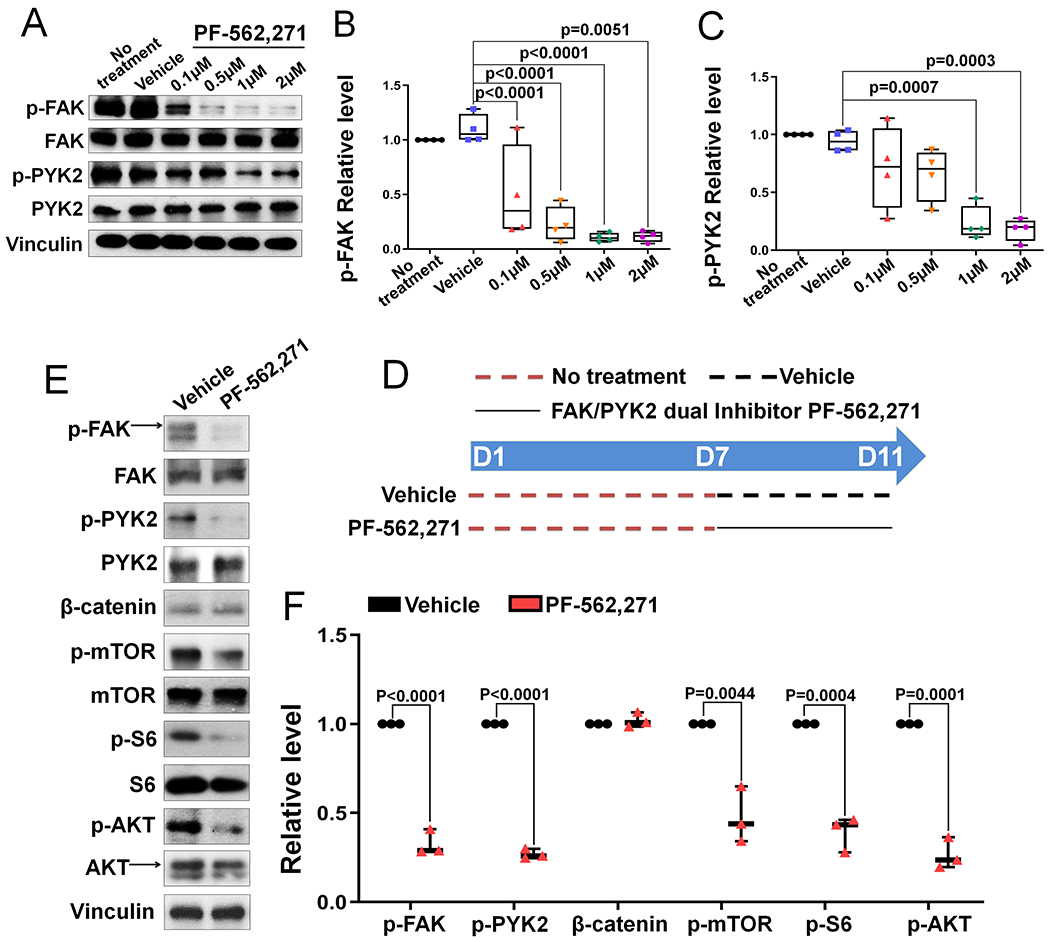
(A) Representative western blot image showing that phospho-FAK (p-FAK) and phospho-PYK2 (p-PYK2) levels in BMSCs were dose-dependently suppressed by FAK/PYK2 dual inhibitor PF-562,271. (B-C) Quantification of the relative p-FAK level (B) and p-PYK2 level (C) illustrated in (A) (n=4). p-FAK and p-PYK2 levels were normalized by their respective total protein levels. (D) Cartoon showing the experimental design of FAK/PYK2 dual inhibitor PF-562,271 treatment used in (E-F). Dashed red line indicates no treatment, dashed black line indicates vehicle treatment and solid black line indicates 1 μM PF-562,271 treatment. D7-D11 means FAK/PYK2 dual inhibitor was added from day 7 to day 11. E) BMSCs were collected at day 11 and extracted proteins were analyzed by western blot using indicated antibodies. (F) Quantification of the relative protein level illustrated in (E). Phospho-proteins were normalized to the respective total proteins and β-catenin was normalized to Vinculin (n=3). Values were presented as median and interquartile range.
To further determine the potential compensatory role of PYK2 when FAK is absent at later differentiation stage of BMSCs, we used the BMSCs isolated from CKOCol3.6 mice because our data showed that PYK2 activity was increased and there was no change in the proliferation and differentiation in CKOCol3.6 BMSCs compared to controls (Fig 7). As expected, the addition of FAK/PYK2 dual inhibitor starting from day 7 significantly decreased the p-PYK2 level in CKOCol3.6 BMSCs (Fig S15B). Importantly, dual inhibitor treatment decreased the mTORC1 signaling and the level of phospho-AKT in CKOCol3.6 BMSCs (Fig S15C–D). Lastly, although CKOCol3.6 BMSCs had comparable osteogenic differentiation and mineralization potential compared to control (Fig S16A–B), FAK/PYK2 dual inhibitor treatment starting from day 7 culture inhibited osteogenic differentiation and mineralization in CKOCol3.6 BMSCs (Fig S16C– G).
Altogether, our data showed that FAK regulates Wnt/β-catenin signaling and mTORC1 signaling of osteoblast lineage cells in a differentiation stage-dependent manner. FAK kinase inhibition in early osteoprogenitor cells compromised mTORC1 signaling but its inhibition in osteoblasts did not compromise mTORC1 signaling or in vitro mineralization because of the compensation by upregulated active PYK2.
Discussion
We previously showed that Fak deletion by Osterix-Cre leads to enhanced bone marrow adipogenesis in association with a significant decrease in β-catenin level.(10) In this study we showed that Fak deletion by Dermo1-Cre or Col3.6-Cre did not increase bone marrow adipogenesis nor decreased β-catenin level, indicating the regulation of β-catenin level by Fak is cell differentiation stage-dependent. To support this notion, our data showed a significant decrease in β-catenin level when FAK kinase activity was inhibited by FAK specific kinase inhibitor in BMSCs around the start of Osterix expression but the earlier or later inhibition of FAK kinase activity did not affect β-catenin level. Moreover, an early transient FAK kinase inhibition surprisingly increased β-catenin level; suggesting the complex regulatory mechanism on β-catenin level by FAK. Our previous report also showed that while Fak deletion in Osterix-expressing cells significantly decreases the β-catenin level in BMSCs but it does not decrease the β-catenin level in more mature calvarial osteoblasts.(10) The underlying mechanism for the differential effects of FAK inhibition at different differentiation stages on β-catenin level remains to be determined.
Through both genetic and pharmacological approaches, we identified a novel regulatory function of FAK on mTORC1 signaling in early osteoprogenitor cells. Our data also showed that FAK positively regulated the phospho-AKT level in these cells. The serine/threonine protein kinase AKT is a downstream effector of PI3K. Because FAK positively regulates PI3K/AKT pathway,(33) and Fak overexpression can promote proliferation partly through PI3K/AKT pathway,(34) the proliferation defect in Fak-deficient early osteoprogenitor cells is likely contributed by the decrease in AKT signaling. Furthermore, AKT can promote mTORC1 activity by inactivating TSC2,(35,36) and AKT deficiency impairs cell proliferation in mTORC1- dependent manner.(37) Our data showed a concomitant decrease in both AKT and mTORC1 signaling in FAK-deficient cells. Thus, the proliferation defect in the mutant cells is potentially mediated through AKT/mTORC1 axis. AKT can phosphorylate mTOR at Thr2446 and Ser2448;(13) our data showed that FAK inhibition decreased Ser2448 phosphorylation of mTOR, further supporting the notion that FAK may regulate mTORC1 through AKT pathway. It is generally believed that AKT-mediated mTORC1 regulation applies to both S6K1 and 4EBP1 pathways because both S6K1 phosphorylation and 4EBP1 phosphorylation are significantly decreased in the cells lacking AKT1 and AKT2.(38) However, it has also been reported that knockdown of Akt1, 2, and 3 in bladder cancer cell lines specifically decreases S6K1 phosphorylation but not 4EBP1 phosphorylation level.(39) In addition, silencing or pharmacological inhibition of mTOR suppresses S6K1 but not 4EBP1 phosphorylation in the same cancer cell lines.(39) Thus, it was proposed that 4EBP1 phosphorylation is regulated independent of AKT/mTORC1 axis. In our study, FAK-deficient early osteoprogenitor cells had decreased phosphorylation of AKT and mTOR, and mTORC1 signaling was only defective in S6K1 branch but not 4EBP1 branch. This suggests that similar regulatory mechanisms exist in early osteoprogenitor cells compared to the reported bladder cancer cell lines. However, the mechanism of FAK’s selective regulation on S6K1 pathway in early osteoprogenitor cells remains to be determined.
In this study, we used Col3.6-Cre transgenic mice to target osteoblasts. We previously characterized its expression and activity.(20) A recent study by Rajshankar et al. used this same transgenic mouse to delete Fak in osteoblasts and showed very different phenotype from ours. In that study, it was shown that the Fak conditional knockout mice are much shorter, have severely crooked and much shorter tails, and have a significant decrease in bone mass.(11) None of these phenotypes was observed in our Fak conditional knockout mice using the same Col3.6-Cre. There are two potential explanations for these discrepancies. First is the difference in genetic background. The mice used in Rajshankar et al.’s study were B6/CD-1 mixed background and ours had been backcrossed for at least eight generations onto a C57BL/6 background. Second is the difference in the Fak floxed mouse line. Rajshankar et al. used the Fak floxed mice generated by Drs. Beggs and Reichardt’s group in which LoxP sites flank the second kinase domain exon.(40) In this study, we used the Fak floxed mice that we previously generated by flanking the third coding exon which is located at its FERM domain.(5) Cre-mediated deletion of exon 3 leads to a frame-shift mutation and produces a small truncated and nonfunctional peptide.
In this report, we showed that Fak deletion in Dermo1-expressing early osteoprogenitor cells had defective proliferation due to a decrease in mTORC1 activity. In contrast, Fak deletion in osteoblasts had no significant effect on proliferation and differentiation as well as mTORC1 activity. Our data suggested that FAK is dispensable in mature osteoblasts and this is likely due to the compensatory role of PYK2, which belongs to the same non-receptor tyrosine kinase family as FAK(41). We draw this conclusion because: 1) our data and another study(9) showed an upregulation of PYK2 activity in FAK-deficient osteoblasts; 2) our data showed that FAK/PYK2 dual inhibitor but not FAK inhibitor decreased mTORC1 signaling and inhibited the osteogenic differentiation of BSMCs if added at a later differentiation stage (after the expression of Osterix); and 3) our data showed that Fak deletion in osteoblasts using Col3.6-Cre did not affect the mTORC1 signaling and differentiation of BMSCs, but the additional inhibition of PYK2 kinase decreased mTORC1 signaling and compromised BMSC differentiation. In addition, a previous study showed that PYK2 knockdown reduces S6K1 expression and phosphorylation in prostate cancer cells.(42)
In summary, our study identified a novel FAK-mTORC1 signaling axis mediated by the kinase-dependent function of FAK in regulating cell proliferation; and this signaling cascade specifically operates in the early osteoprogenitor cells but not in osteoblasts. As a result, Fak deletion in early osteoprogenitor cells led to osteopenia in mice but its deletion in osteoblasts did not affect bone development. Altogether, our studies suggested that FAK plays differential roles at different differentiation stages of osteoblast lineage cells: in early osteoprogenitor cells, it promotes the proliferation via positively regulating mTORC1 signaling; in committed osteoblast precursors, it promotes osteoblast proliferation and differentiation via positively regulating both mTORC1 and Wnt/β-catenin signaling; and in osteoblasts, the loss of FAK can be compensated by upregulated active PYK2 (Fig 10).
Figure 10. Schematic model of the differential roles of FAK in the cells of osteoblast lineage.
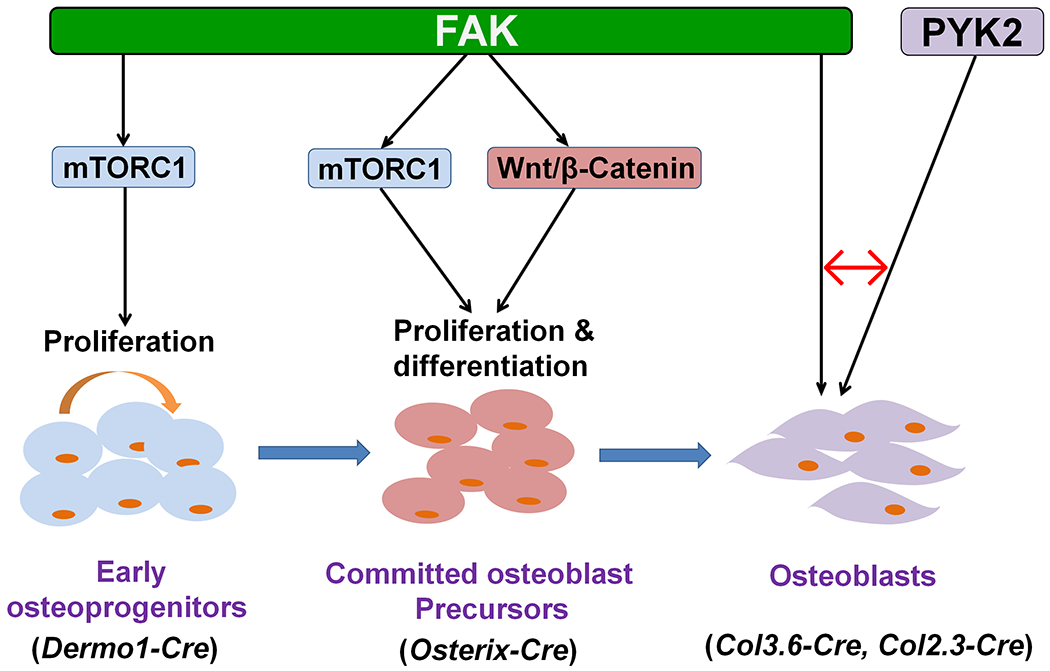
The model depicts the mechanisms of FAK action at three distinct stages of osteoblast lineage in which the roles of FAK have been addressed by genetic and pharmacological approaches as well as the respective Cre transgenes used to delete Fak, including Dermo1-Cre (this study), Osterix-Cre,(10) Col3.6-Cre (this study) and Col2.3-Cre.(9) Red ↔ indicates that the loss of FAK in osteoblasts can be compensated by the upregulated active PYK2.
Supplementary Material
Acknowledgments
We thank Ms. Andrea Clark for microCT scanning and reconstruction, and Dr. Laurie McCauley for valuable discussion. MicroCT work was partly supported by P30 Core Center award to University of Michigan from NIH/NIAMS (AR 69620). FL was supported by Department of Biologic and Materials Sciences & Prosthodontics, University of Michigan School of Dentistry.
Footnotes
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1002/jbmr.4029
Supplemental data have been included with the submission
Disclosure
All authors state that they have no conflict of interest.
References:
- 1.Guan JL, Shalloway D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature. August 20 1992;358(6388):690–2. Epub 1992/08/20. [DOI] [PubMed] [Google Scholar]
- 2.Luo M, Guan JL. Focal adhesion kinase: a prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. March 28 2010;289(2):127–39. Epub 2009/08/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaller MD. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci. April 01 2010;123(Pt 7):1007–13. [DOI] [PubMed] [Google Scholar]
- 4.Zhao X, Peng X, Sun S, Park AY, Guan JL. Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J Cell Biol. June 14 2010;189(6):955–65. Epub 2010/06/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen TL, Park AY, Alcaraz A, Peng X, Jang I, Koni P, et al. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol. June 20 2005;169(6):941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. January 18 2008;29(1):9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perinpanayagam H, Zaharias R, Stanford C, Brand R, Keller J, Schneider G. Early cell adhesion events differ between osteoporotic and non-osteoporotic osteoblasts. J Orthop Res. November 2001;19(6):993–1000. [DOI] [PubMed] [Google Scholar]
- 8.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. October 12 1995;377(6549):539–44. [DOI] [PubMed] [Google Scholar]
- 9.Kim JB, Leucht P, Luppen CA, Park YJ, Beggs HE, Damsky CH, et al. Reconciling the roles of FAK in osteoblast differentiation, osteoclast remodeling, and bone regeneration. Bone. July 2007;41(1):39–51. Epub 2007/04/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun C, Yuan H, Wang L, Wei X, Williams L, Krebsbach PH, et al. FAK Promotes Osteoblast Progenitor Cell Proliferation and Differentiation by Enhancing Wnt Signaling. J Bone Miner Res. December 2016;31(12):2227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajshankar D, Wang Y, McCulloch CA. Osteogenesis requires FAK-dependent collagen synthesis by fibroblasts and osteoblasts. FASEB J. March 2017;31(3):937–53. [DOI] [PubMed] [Google Scholar]
- 12.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. April 13 2012;149(2):274–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. August 15 2004;18(16):1926–45. [DOI] [PubMed] [Google Scholar]
- 14.Elefteriou F, Yang X. Genetic mouse models for bone studies--strengths and limitations. Bone. December 2011;49(6):1242–54. Epub 2011/09/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.VanKoevering KK, Williams BO. Transgenic mouse strains for conditional gene deletion during skeletal development IBMS BoneKEy. 2008;5(5):151–70. [Google Scholar]
- 16.Shekaran A, Shoemaker JT, Kavanaugh TE, Lin AS, LaPlaca MC, Fan Y, et al. The effect of conditional inactivation of beta 1 integrins using twist 2 Cre, Osterix Cre and osteocalcin Cre lines on skeletal phenotype. Bone. November 2014;68:131–41. Epub 2014/09/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan JL. Integrin signaling through FAK in the regulation of mammary stem cells and breast cancer. IUBMB Life. April 2010;62(4):268–76. Epub 2010/01/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guan JL. Focal adhesion kinase in integrin signaling. Matrix Biol. October 1997;16(4):195–200. Epub 1997/12/24. [DOI] [PubMed] [Google Scholar]
- 19.Cary LA, Guan JL. Focal adhesion kinase in integrin-mediated signaling. Front Biosci. January 15 1999;4:D102–13. Epub 1999/01/16. [DOI] [PubMed] [Google Scholar]
- 20.Liu F, Woitge HW, Braut A, Kronenberg MS, Lichtler AC, Mina M, et al. Expression and activity of osteoblast-targeted Cre recombinase transgenes in murine skeletal tissues. Int J Dev Biol. September 2004;48(7):645–53. Epub 2004/10/08. [DOI] [PubMed] [Google Scholar]
- 21.Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, et al. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. July 2003;130(13):3063–74. [DOI] [PubMed] [Google Scholar]
- 22.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. January 2010;13(1):133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu F, Fang F, Yuan H, Yang D, Chen Y, Williams L, et al. Suppression of autophagy by FIP200 deletion leads to osteopenia in mice through the inhibition of osteoblast terminal differentiation. J Bone Miner Res. November 2013;28(11):2414–30. Epub 2013/05/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandhoke TK, Huang YF, Liu F, Gronowicz GA, Adams DJ, Harrison JR, et al. Osteopenia in transgenic mice with osteoblast-targeted expression of the inducible cAMP early repressor. Bone. July 2008;43(1):101–9. Epub 2008/05/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi HK, Yuan H, Fang F, Wei X, Liu L, Li Q, et al. Tsc1 Regulates the Balance Between Osteoblast and Adipocyte Differentiation Through Autophagy/Notch1/beta-Catenin Cascade. J Bone Miner Res. November 2018;33(11):2021–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. January 2013;28(1):2–17. Epub 2012/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo M, Zhao X, Chen S, Liu S, Wicha MS, Guan JL. Distinct FAK activities determine progenitor and mammary stem cell characteristics. Cancer Res. September 1 2013;73(17):5591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. May 18 2007;282(20):14845–52. Epub 2007/03/31. [DOI] [PubMed] [Google Scholar]
- 29.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. September 2014;14(9):598–610. Epub 2014/08/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golubovskaya VM. Targeting FAK in human cancer: from finding to first clinical trials. Front Biosci (Landmark Ed). 2014;19:687–706. Epub 2014/01/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang F, Sun S, Wang L, Guan JL, Giovannini M, Zhu Y, et al. Neural Crest-Specific TSC1 Deletion in Mice Leads to Sclerotic Craniofacial Bone Lesion. J Bone Miner Res. July 2015;30(7):1195–205. Epub 2015/02/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. November 2011;10(11):2135–45. Epub 2011/09/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sonoda Y, Watanabe S, Matsumoto Y, Aizu-Yokota E, Kasahara T. FAK is the upstream signal protein of the phosphatidylinositol 3-kinase-Akt survival pathway in hydrogen peroxide-induced apoptosis of a human glioblastoma cell line. J Biol Chem. April 9 1999;274(15):10566–70. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto D, Sonoda Y, Hasegawa M, Funakoshi-Tago M, Aizu-Yokota E, Kasahara T. FAK overexpression upregulates cyclin D3 and enhances cell proliferation via the PKC and PI3-kinase-Akt pathways. Cell Signal. June 2003;15(6):575–83. [DOI] [PubMed] [Google Scholar]
- 35.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. September 2002;4(9):648–57. [DOI] [PubMed] [Google Scholar]
- 36.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. July 2002;10(1):151–62. [DOI] [PubMed] [Google Scholar]
- 37.Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, et al. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell. October 2006;10(4):269–80. [DOI] [PubMed] [Google Scholar]
- 38.Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. June 1 2003;17(11):1352–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nawroth R, Stellwagen F, Schulz WA, Stoehr R, Hartmann A, Krause BJ, et al. S6K1 and 4E-BP1 are independent regulated and control cellular growth in bladder cancer. PLoS One. 2011;6(11):e27509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, et al. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. October 30 2003;40(3):501–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71(3-4):435–78. Epub 1999/06/04. [DOI] [PubMed] [Google Scholar]
- 42.Hsiao YH, Huang YT, Hung CY, Kuo TC, Luo FJ, Yuan TC. PYK2 via S6K1 regulates the function of androgen receptors and the growth of prostate cancer cells. Endocr Relat Cancer. August 2016;23(8):651–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.