

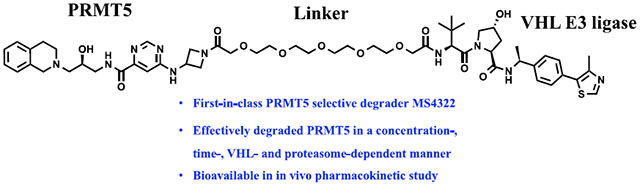
Abstract
The aberrant expression of protein arginine methyltransferase 5 (PRMT5) has been associated with multiple cancers. Using the proteolysis targeting chimera technology, we discovered a first-in-class PRMT5 degrader 15 (MS4322). Here, we report the design, synthesis, and characterization of compound 15 and two structurally similar controls 17 (MS4370) and 21 (MS4369), with impaired binding to the von Hippel-Lindau E3 ligase and PRMT5, respectively. Compound 15, but not 17 and 21, effectively reduced the PRMT5 protein level in MCF-7 cells. Our mechanism studies indicate that compound 15 degraded PRMT5 in an E3 ligase- and proteasome-dependent manner. Compound 15 also effectively reduced the PRMT5 protein level and inhibited growth in multiple cancer cell lines. Moreover, compound 15 was highly selective for PRMT5 in a global proteomic study and exhibited good plasma exposure in mice. Collectively, compound 15 and its two controls 17 and 21 are valuable chemical tools for exploring the PRMT5 functions in health and disease.
Graphical Abstract
INTRODUCTION
As the predominant member of type II protein arginine methyltransferases (PRMTs), protein arginine methyltransferase 5 (PRMT5) catalyzes monomethylation and symmetric dimethylation of arginine residues of its histone substrates, including H3R2, H3R8, and H4R3, and its nonhistone substrates, including p53, EGFR, N-MYC, SmD3, and RNA polymerase II, when interacting with its binding partner MEP50.1–5 Through methylation of these substrates, PRMT5 regulates multiple biological processes, such as chromatin remodeling, gene expression, mRNA splicing, DNA replication and repair, and cell cycle regulation.6–8 The aberrant expression of PRMT5 has been associated with infectious disease, heart disease, and cancers, including breast cancer, lung cancer, and hepatocellular cancer (HCC).9–11 Genetic knockdown of PRMT5 was effective to suppress tumor growth in vitro and in vivo.12 For example, PRMT5 knockdown decreased the proliferation, invasion, and migration of HCC cell lines.13 The knockdown of PRMT5 in AR-positive LNCaP prostate cancer cells completely suppressed tumor growth in xenograft mouse models.14 In addition, the knockdown of PRMT5 drastically prolonged the survival in a murine model of BCR-ABL-driven chronic myelogenous leukemia15,16 and of MYC-driven B cell lymphoma.17 Moreover, the PRMT5 knockdown effectively suppressed the growth of patient-derived xenograft glioblastoma (GBM) tumors in vivo.18 Collectively, these findings suggest that PRMT5 down-regulation may provide a potential therapeutic strategy for treating cancer.
Multiple small-molecule PRMT5 inhibitors have been reported.19–29 Among them, GSK3326595 and JNJ64619178 are currently being evaluated in the clinic. These compounds potently inhibited the methyltransferase activity of PRMT5, and some of them displayed promising tumor growth inhibition effects in animal models. However, different from PRMT5 genetic knockdown, these catalytic inhibitors cannot eliminate the potential scaffolding functions of PRMT5.30 Recently, proteolysis targeting chimera (PROTAC) has emerged as a powerful technology to reduce the protein levels of the target of interest by hijacking the cellular ubiquitin–proteasome system.30–34 PROTACs are expected to phenocopy the effects of genetic knockdown or knockout as they not only inhibit the target proteins as traditional inhibitors do but also more importantly eliminate the other functions (e.g., scaffolding function) of the protein targets. Another potential advantage of PROTACs over occupancy-driven small-molecule inhibitors is that PROTACs may potentially overcome the resistance to small-molecule inhibitor treatments.35 To the best of our knowledge, PRMT5 PROTAC degraders have not been reported to date.
In this study, we report the design, synthesis, and characterization of first-in-class PRMT5 degraders. Through a brief structure–activity relationship (SAR) study, a potent PRMT5 degrader, compound 15 (MS4322), was discovered by linking the PRMT5 inhibitor EPZ01566619 to a von Hippel-Lindau (VHL) E3 ligase ligand, (S,R,S)-AHPC-Me (VHL-2). Compound 15 effectively reduced PRMT5 protein levels in multiple cell lines. In particular, it reduced the PRMT5 protein level in MCF7 cells in a concentration-, time-, VHL- and proteasome-dependent manner. In addition, compound 15 inhibited cell growth at least as effectively as EPZ015666 in multiple cancer cell lines and exhibited good plasma exposure in a mouse pharmacokinetic (PK) study. Furthermore, the results from our mass spectrometry (MS)-based global proteomics analysis indicate that compound 15 is a highly selective PRMT5 degrader. To the best of our knowledge, compound 15 is also the first degrader of any PRMTs, a class of important epigenetic enzymes. Moreover, we developed two structurally similar control compounds, 21 (MS4369) and 17 (MS4370), which displayed an impaired binding to PRMT5 and VHL, respectively, and could not reduce PRMT5 protein levels in cells. Overall, we provide compound 15 and its two control compounds 17 and 21 to the research community as useful chemical tools for further investigating the physiological and pathophysiological functions of PRMT5.
RESULTS AND DISCUSSION
Design, Synthesis, and Evaluation of an Initial Set of Putative PRMT5 Degraders.
EPZ015666 is the first potent and selective PRMT5 inhibitor, and the X-ray cocrystal structure of PRMT5:MEP50 in complex with EPZ015666 and the cofactor S-adenosyl-l-methionine (SAM) revealed that EPZ015666 binds in the substrate-binding site of PRMT5.19 Importantly, the oxetane moiety of EPZ015666 is solvent-exposed and does not interact with any PRMT5 residues (Figure 1A). We therefore hypothesized that the addition of a linker to this moiety would have a minimal effect on the binding of the resulting compounds to PRMT5. To facilitate the installation of a linker, we replaced the oxetane group with an azetidine group, leading to compound 3. Five putative PRMT5 degraders 9–13 were designed by linking compound 3 to the E3 ligase VHL binder VHL-1 with five different linkers (Figure 1B).
Figure 1.
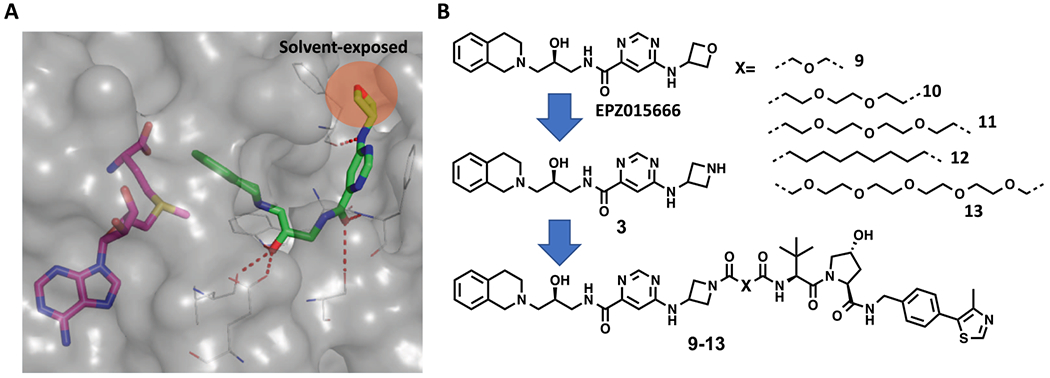
Design of PRMT5 putative degraders 9, 10, 11, 12, and 13. (A) Crystal structure of PRMT5:MEP50 (gray) in complex with SAM (magenta) and EPZ015666 (green) (PDB: 4X61). The highlighted oxetane moiety of EPZ015666 is solvent-exposed. Key H-bond interactions between the EPZ015666 and PRMT5 residues are highlighted by red dashed lines. (B) Chemical structures of EPZ015666, compound 3, and the five putative PRMT5 degraders 9–13.
We developed a concise synthetic route to prepare bivalent compounds 9–13. Commercially available 1,2,3,4-tetrahydroi-soquinoline and tert-butyl (S)-(oxiran-2-ylmethyl)carbamate underwent epoxide opening, deprotection, and amide coupling with 6-chloropyrimidine-4-carboxylic acid, yielding compound 1 but not compound 1a (Scheme 1). This is most likely due to the fact that the chloro group on the pyrimidine ring was substituted by 1-hydroxy-7-aza-benzo-triazole (HOAt) to yield 1 under the amide coupling reaction condition. Fortunately, compound 1 can be readily converted to 2 using tert-butyl 3-aminoazetidine-1-carboxylate as a nucleophilic substitution reagent. Deprotection of intermediate 2 under the acidic condition afforded intermediate 3. The amide coupling reactions between the intermediate 3 and linker-attached VHL-1 derivatives 4–8 resulted in the desired compounds 9–13.
Scheme 1. Synthesis of PRMT5 Putative Degraders 9–13a.
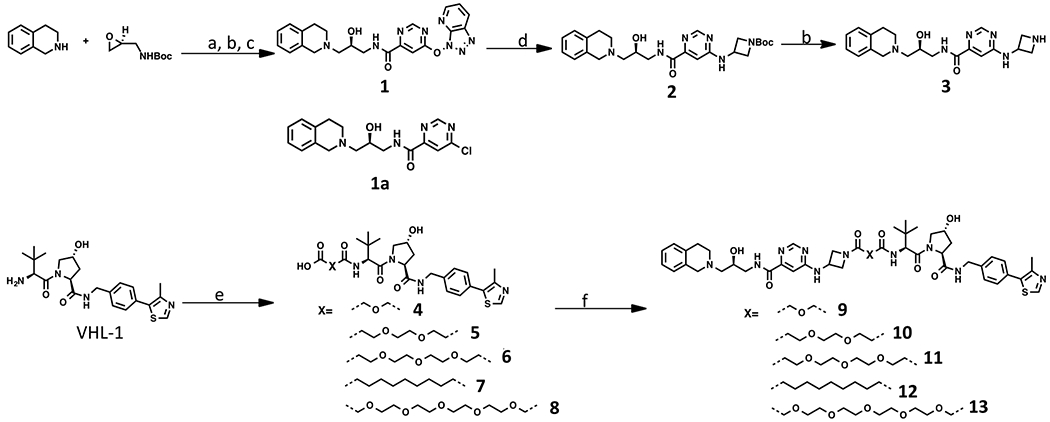
aReagents and conditions: (a) i-PrOH, reflux; (b) TFA, DCM; (c) EDCI·HCl, HOAt, NMM, 6-chloropyrimidine-4-carboxylic acid, DMSO, 18% over three steps; (d) tert-butyl 3-aminoazetidine-1-carboxylate, NMP, 90%; (e) dicarboxylic acids, EDCI·HCl, HOAt, NMM, DMSO, 54–78%; (f) 3, EDCI·HCl, HOAt, NMM, DMSO, 36–60%.
Previous studies have shown that MCF-7 cells, an estrogen receptor (ER) + breast cancer cell line, have sufficient PRMT5 and MEP50 protein levels.36 In addition, MCF-7 cells were successfully used for assessing the VHL-recruiting ER degrader ERD-308.37 These results indicate that MCF-7 cells are suitable for evaluating the effect of our putative PRMT5 degraders on reducing the PRMT5 protein level. We therefore treated MCF-7 cells with compounds 9–13 at three different concentrations (0.2, 1, and 5 μM). As illustrated by Figure 2, although compounds 9–11, which contain relatively short polyethylene glycol (PEG) linkers, and compound 12, which contains an all carbon linker, did not significantly reduce PRMT5 protein levels, compound 13, which contains a longer PEG linker, reduced the PRMT5 protein level at 1 μM and more effectively at 5 μM.
Figure 2.

Effects of compounds 9–13 on reducing the PRMT5 protein level in MCF-7 cells. MCF-7 cells were treated with compounds 9–13 at the indicated concentrations (0.2, 1, and 5 μM) for 6 d. Cell lysates were collected, and the PRMT5 protein levels were detected by western blots. The results are representative of at least two independent experiments.
Design and Synthesis of PRMT5 Degrader 15 and Its Control Compounds 17 and 21.
It has been reported that the VHL binder (S,R,S)-AHPC-Me (VHL-2) (Scheme 2) can result in more effective degraders than the ones derived from VHL-1.38,39 For example, compared with VHL-1-based MEK1/2 degraders, VHL-2-based degraders displayed a higher potency in reducing the MEK1/2 protein levels.3 Therefore, we designed compound 15 by replacing the VHL-1 moiety of degrader 13, the most effective compound from our initial SAR study, with VHL-2 (Figure 3). In addition, we designed two close analogues, compounds 17 and 21 (Figure 3), as controls for compound 15. Compound 17 is a diastereoisomer of compound 15 and was designed to diminish its binding to the E3 ligase VHL, based on the previous reports that the diastereoisomer of VHL-2 led to impaired binding to VHL,38 while possessing the same linker and PRMT5 binding moiety, thus maintaining a similar inhibitory potency for PRMT5 as compound 15. It was previously reported that the replacement of the 1,2,3,4-tetrahydroisoquinoline moiety of EPZ015666 with the piperidine group led to a significantly diminished PRMT5 inhibition activity.19 We therefore designed the control compound 21 by replacing the 1,2,3,4-tetrahydroisoquinoline moiety of compound 15 with the piperidine group to reduce its inhibitory potency for PRMT5, while maintaining the same binding affinity to VHL as compound 15. Because of their high structural similarity, compounds 15, 17, and 21 are expected to have similar physicochemical properties.
Scheme 2. Synthesis of Compounds 15, 17, and 21a.
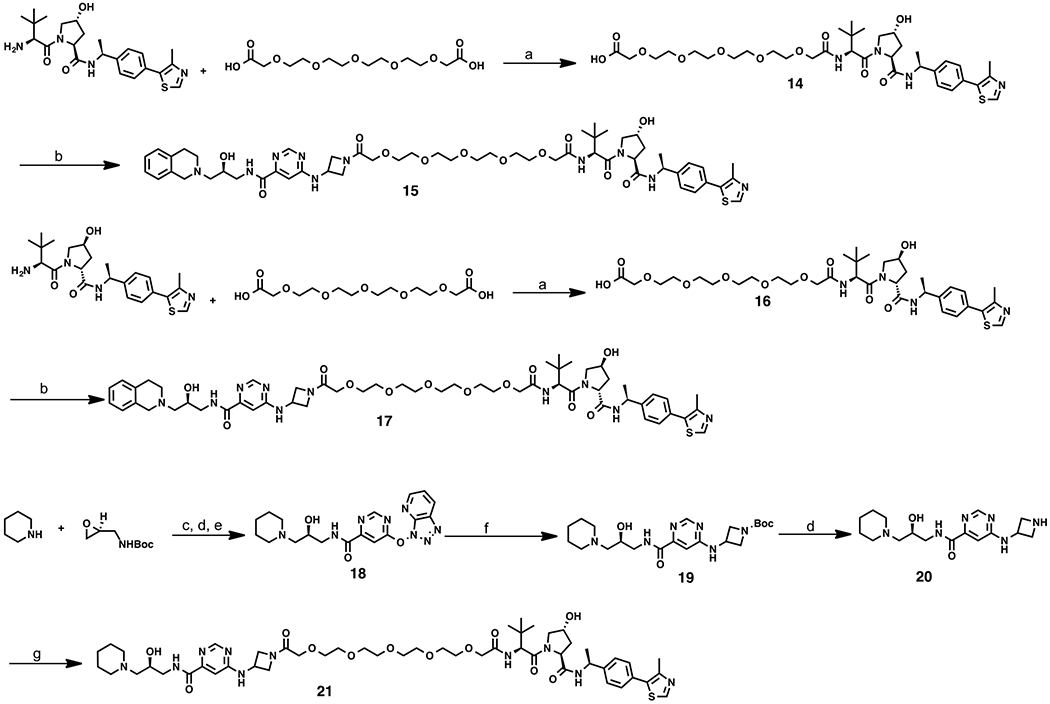
aReagents and conditions: (a) EDCI·HCl, HOAt, NMM, DMSO, 50%; (b) 3, EDCI·HCl, HOAt, NMM, DMSO, 35–53%; (c) i-PrOH, reflux; (d) TFA, DCM; (e) EDCI·HCl, HOAt, NMM, 6-chloropyrimidine-4-carboxylic acid, DMSO; (f) tert-butyl 3-aminoazetidine-1-carboxylate, NMP, 13% over four steps; (g) 14, EDCI·HCl, HOAt, NMM, DMSO, 49%.
Figure 3.
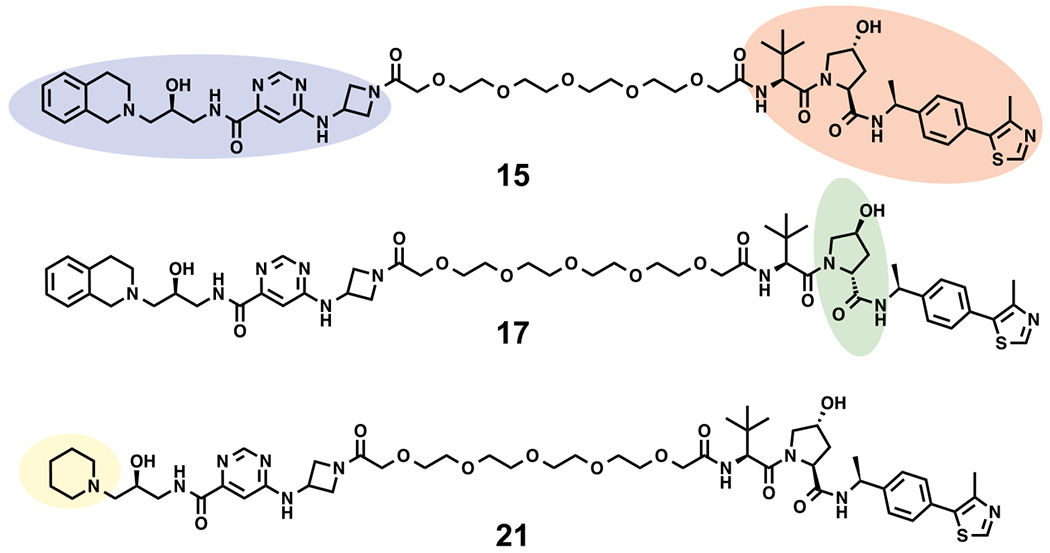
Design of the PRMT5 degrader 15 and its two control compounds 17 and 21.
Compounds 15, 17, and 21 were prepared using the synthetic routes outlined in Scheme 2. Starting from VHL-238 and commercially available 3,6,9,12,15-pentaoxaheptadecanedioic acid, intermediate 14 was produced under an amide coupling condition. Under the same amide coupling condition, degrader 15 was prepared from carboxylic acid 14 and amine 3. Using the same synthetic sequence, compound 17 was synthesized from a diastereoisomer of VHL-2, which was prepared according to the published procedures.38 To synthesize compound 21, intermediate 19 was prepared using the same synthetic route as preparing intermediate 3 from piperidine and tert-butyl (S)-(oxiran-2-ylmethyl)-carbamate, via epoxide opening, deprotection, amide coupling, and nucleophilic aromatic substitution (SNAr) reactions. Deprotection of intermediate 19 provided intermediate 20. An amide coupling reaction between amine 20 and acid 14 yielded compound 21.
Inhibition of PRMT5 Methyltransferase Activity in Biochemical Assays.
We first evaluated the effect of compounds 15, 17, and 21 on inhibiting the methyltransferase activity of PRMT5 in a human PRMT5/MEP50 radioactive biochemical assay using tritiated SAM (3H-SAM) as the methyl donor. Compared with the PRMT5 inhibitor EPZ015666 (IC50 = 30 ± 3 nM), compound 15 (IC50 = 18 ± 1 nM), which contains a linked VHL-2 moiety, and compound 17 (IC50 = 12 ± 1 nM), which contains a diastereoisomer of VHL-2, maintained high potency in inhibiting the PRMT5 enzymatic activity (Figure 4). As expected, compound 21, which comprises a piperidinyl group instead of the tetrahydroisoquinolinyl group on the PRMT5 binding moiety, indeed displayed much reduced potency in inhibiting the PRMT5 methyltransferase activity.
Figure 4.
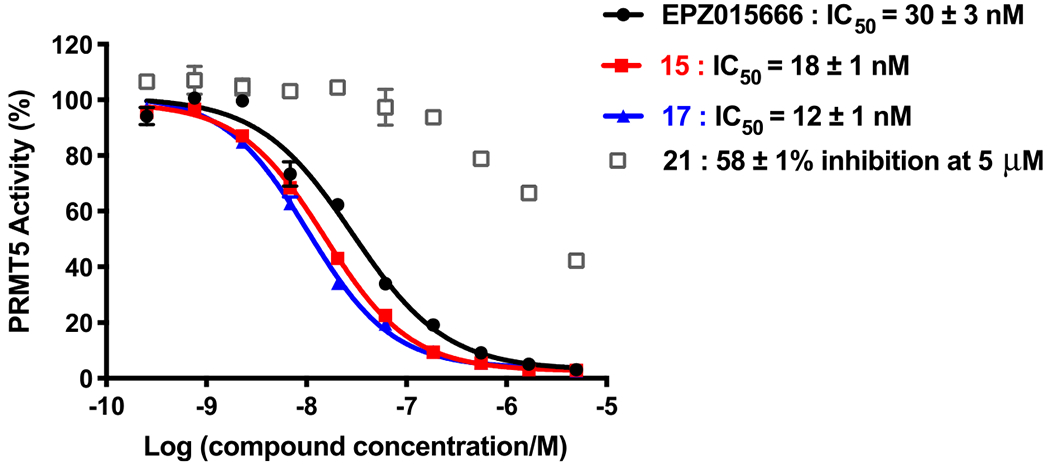
Effects of compounds 15, 17, and 21 on inhibiting the PRMT5 methyltransferase activity in a radioactive biochemical assay. Substrate: H4 (1–15)-biotin (400 nM). Cofactor: 3H-SAM (1 μM). EPZ015666 was used as a positive control. IC50 determination experiments were performed in duplicate, and the values are presented as mean ± SD.
PRMT5 Degradation and Mechanism of Action Studies in MCF-7 Cells.
We next assessed the PRMT5 degradation effects of compounds 15, 17, and 21 in MCF-7 cells. We were pleased to find that compound 15 effectively reduced the PRMT5 protein level in a concentration-dependent manner, with a DC50 value of 1.1 ± 0.6 μM and Dmax = 74 ± 10% (Figure 5A,B), and compound 15 was more effective than compound 13 at degrading PRMT5 (Figure 2). In addition, the two control compounds, 17 and 21, and the PRMT5 inhibitor EPZ015666 did not reduce PRMT5 protein levels at 5 μM (Figure 5A). Thus, compounds 17 and 21 are good negative controls for the PRMT5 degradation activity of compound 15. Next, we conducted a time course study for determining the effect of compound 15 on PRMT5 degradation. MCF-7 cells were treated with compound 15 at 5 μM for 0–8 d. Significant PRMT5 degradation was observed after a 2 d treatment, and the degradation effect was gradually increased to the maximum after a 6 to 8 d treatment (Figure 5C). It is worth to note that the degradation kinetics by compound 15 is significantly slower than that by typical degraders targeting kinases.39–43 It is unclear why PRMT5 degradation induced by compound 15 is slower. This warrants further investigation. We also compared the effect of compound 15 and EPZ015666 on inhibiting global arginine symmetric dimethylation (SDMA) (Figure 5D). In MCF-7 cells treated with 0.1, 0.3, 1, and 3.0 μM of these two compounds for 6 d, both the PRMT5 degrader 15 and the inhibitor EPZ015666 concentration-dependently decreased global SDMA, and compound 15 appeared slightly more effective than EPZ015666.
Figure 5.
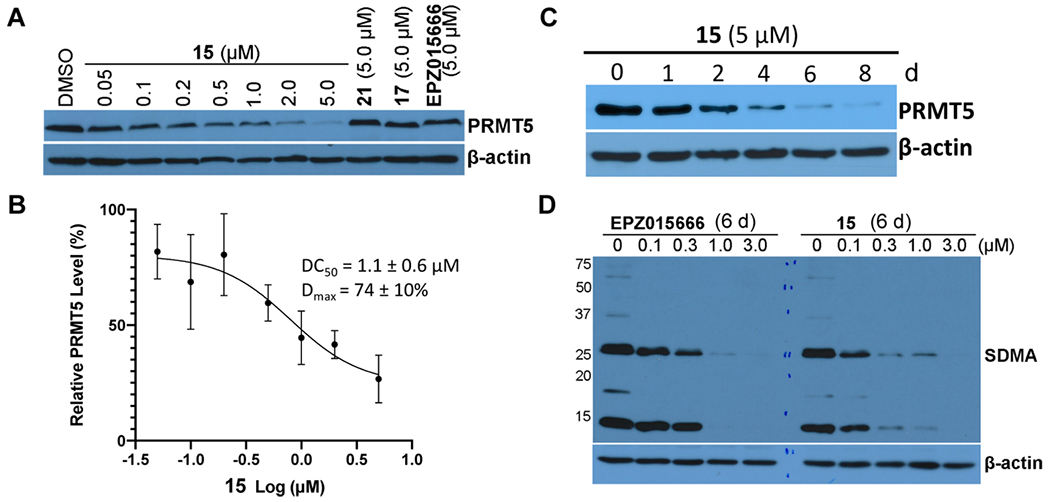
Effects of compound 15 on reducing the PRMT5 protein level and inhibiting SDMA in MCF-7 cells. (A,B) Compound 15 concentration-dependently reduced the PRMT5 protein level, whereas compounds 17 and 21 and EPZ015666 did not. MCF-7 cells were treated with the indicated concentration of 15, 17, 21, or EPZ015666 for 6 d. Cell lysates were collected, and the protein levels of PRMT5 were detected by western blotting. The relative strength of the PRMT5 signal on the western blots was measured by densitometry. DC50 and Dmax values are presented as mean ± SD from three independent experiments. (C) Compound 15 reduced the PRMT5 protein level in a time-dependent manner. MCF-7 cells were treated with 5 μM of compound 15 for 0, 2, 4, 6, or 8 d. The PRMT5 protein levels were detected by western blotting. (D) Compound 15 and EPZ015666 inhibited SDMA. MCF-7 cells were treated with the indicated concentration of EPZ015666 or compound 15 for 6 d. The SDMA levels were detected by western blotting. Western blot results are representative of at least two independent experiments.
To demonstrate that the observed PRMT5 degradation induced by compound 15 is dependent on the ubiquitin proteasome system, we perform a set of rescue experiments. We co-treated MCF-7 cells with 5 μM of compound 15 and 30 μM of PRMT5 inhibitor EPZ015666, 100 μM of VHL E3 ligase ligand VH-298,44 2 μM of neddylation inhibitor MLN4924,45 or 30 μM of proteasome inhibitor MG-132. As illustrated in Figure 6A, the co-treatment with EPZ015666, which competes with compound 15 at the PRMT5 substrate binding site, or VH-298, which competes with compound 15 at the VHL binding site, significantly rescued the reduction of the PRMT5 protein level compared to the dimethyl sulfoxide (DMSO) control (Figure 6A, lane 2). Similarly, co-treatment with the proteasome inhibitor MG-132 or the cullin-RING E3 ubiquitin ligase (CRL) neddylation inhibitor MLN4924 also partially restored the PRMT5 protein level in MCF-7 cells. These results, together with the lack of degradation effects by the control compounds 17 and 21, indicate that compound 15 degrades the PRMT5 protein through a PRMT5-, VHL-, and proteasome-dependent mechanism.
Figure 6.
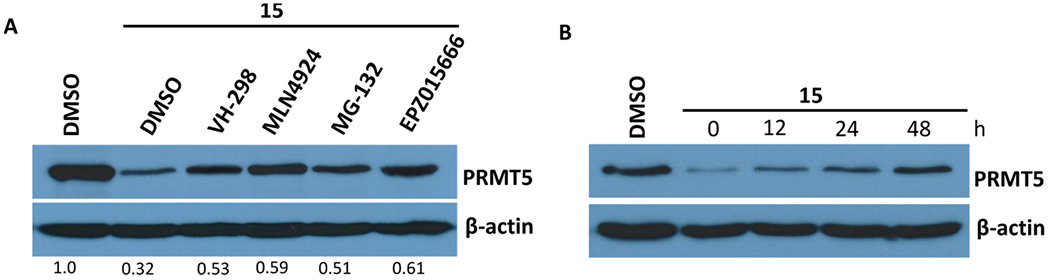
(A) Compound 15 reduced the PRMT5 protein level in MCF-7 cells in a PRMT5-, E3 ligase VHL-, and proteasome-dependent manner. MCF-7 cells were treated with 5 μM of compound 15 for 7 d. During the last 24 h, the cells were co-treated with 100 μM VH-298, 2 μM MLN4924, 30 μM MG-132, or 30 μM EPZ015666. The protein levels of PRMT5 were detected by western blotting. (B) PRMT5 degradation induced by compound 15 was reversible. MCF-7 cells were treated with 5 μM of compound 15 for 6 d, and compound 15 was then washed out. The PRMT5 protein levels were detected by western blotting at 0, 12, 24, and 48 h post the washout.
It has been reported that PROTAC-induced protein degradation does not impair the resynthesis of the protein.46 We therefore evaluated whether the PRMT5 degradation induced by compound 15 can be rescued by using a washout assay (Figure 6B). We treated MCF-7 cells with 5 μM of compound 15 for 6 d and then washed out compound 15 and found that the PRMT5 protein level was gradually recovered over 12, 24, and 48 h. These results suggest that the cellular PRMT5 protein level can be recovered after the removal of compound 15.
Effects of PRMT5 Degrader 15 on Cell Growth Inhibition.
We next evaluated the antiproliferative effect of degrader 15 in MCF-7 cells. As illustrated in Figure 7, compound 15 potently inhibited MCF-7 cell proliferation in a concentration-dependent manner. It was at least as effective as the positive control EPZ015666. This result is consistent with the effect of compound 15 and EPZ015666 on inhibiting global SDMA (Figure 5D). As expected, the negative control compound 21, with much reduced PRMT5 inhibitory activity (Figure 4), did not display any antiproliferative effect on MCF-7 cells. Importantly, the control compound 17, which exhibited similar PRMT5 inhibitory activity as compound 15 (Figure 4) but did not degrade PRMT5 (Figure 5A), displayed much weaker antiproliferative activity than compound 15. Collectively, these results suggest that the observed antiproliferative effect of compound 15 is mainly because of its PRMT5 degradation activity but not its PRMT5 inhibitory activity.
Figure 7.
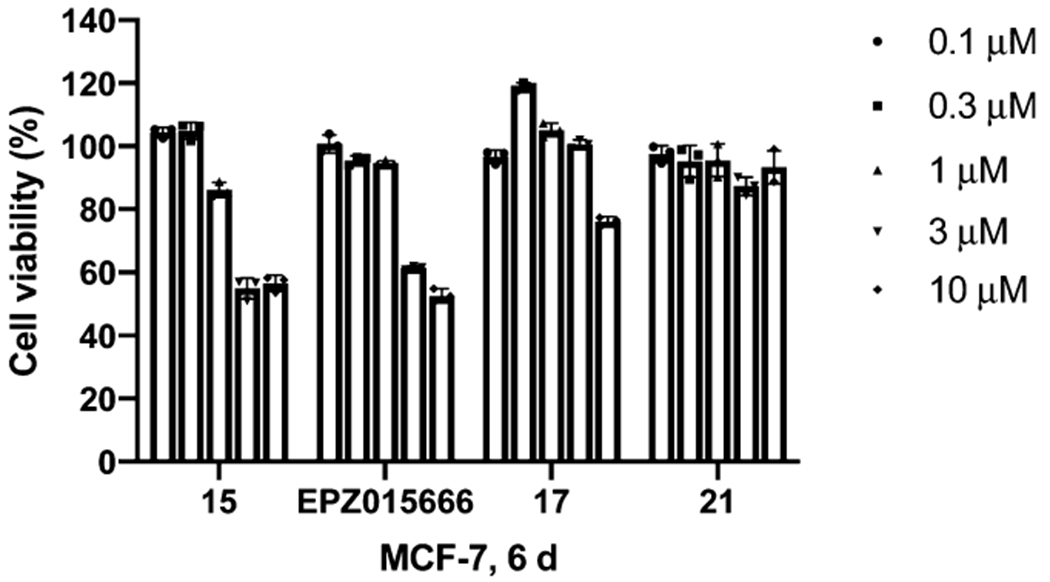
Effects of compounds 15, 17, 21, and EPZ015666 on inhibiting the proliferation of MCF-7 cells. MCF-7 cells were treated with the indicated concentration of compounds 15, 17, 21, or EPZ015666 for 6 d. The relative cell viabilities to the untreated group are shown as mean ± SD (n = 3).
Compound 15 is a Highly Selective PRMT5 Degrader.
To assess the selectivity of compound 15 across the proteome, we performed MS-based label-free quantitative (LFQ) global proteomics analysis of MCF-7 cells treated with compound 15 or DMSO (Figure 8). The samples for MS analysis were prepared by treating MCF-7 cells with compound 15 (at 5 μM) or DMSO for 5 d. The compound concentration and treatment time were selected based on the above concentration range and time course studies (Figure 5A–C). Out of the more than 5000 proteins identified, PRMT5, its binding partner WDR77 (MEP50), and AGRN (an unrelated protein) are the only proteins whose levels were reduced by compound 15, as indicated by the volcano plot (Figure 8). These results strongly suggest that compound 15 is a highly selective PRMT5 degrader.
Figure 8.
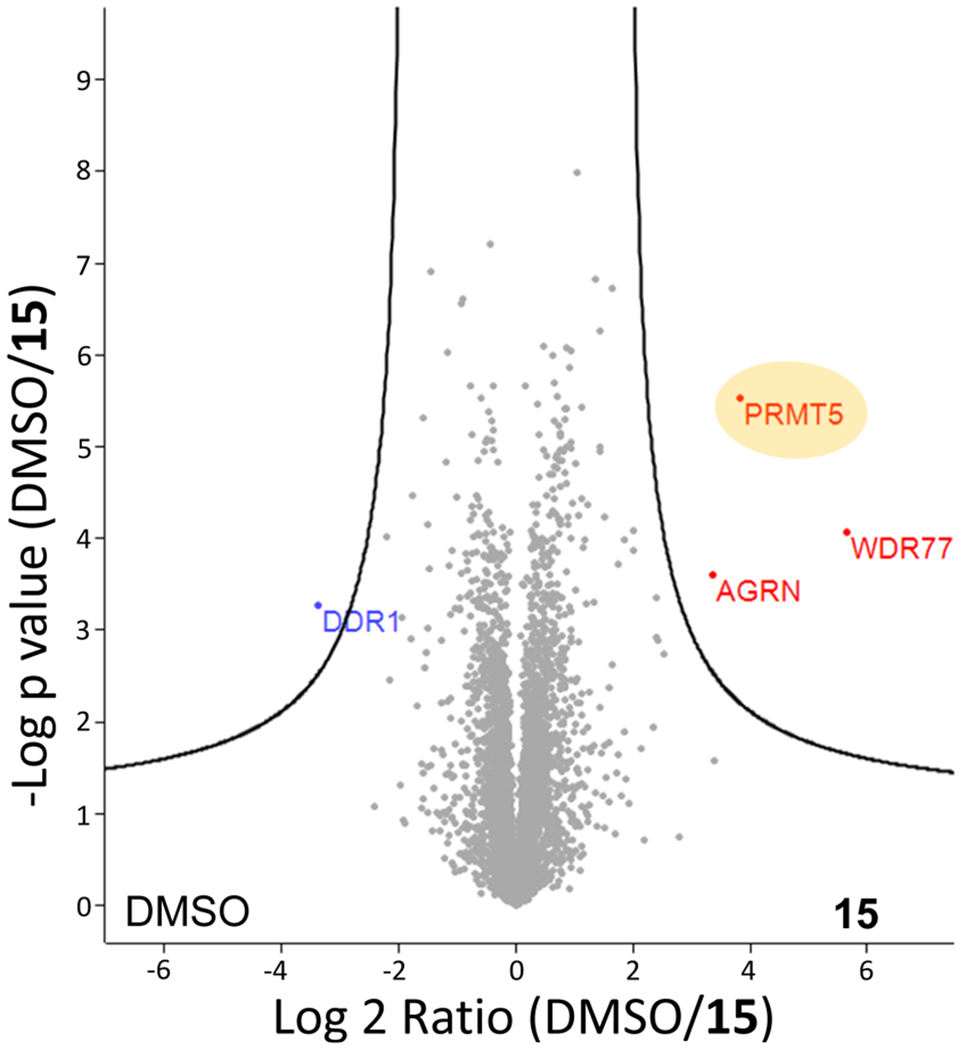
Global proteomic analysis indicates that compound 15 is a highly selective PRMT5 degrader. MCF-7 cells were treated with compound 15 at 5 μM or DMSO for 5 d and then harvested and lysed for liquid chromatography–MS (LC–MS) analysis. LFQ was used to calculate the peptide intensity; the relative abundance of PRMT5 (LFQ intensity value) between the compound 15-treated and DMSO-treated groups is shown.
We next evaluated the effects of compound 15 on PRMT5 degradation and cell growth inhibition in other cancer cell lines. We were pleased to find that the PRMT5 degradation induced by compound 15 is not limited to MCF-7 cells. The PRMT5 protein levels in Hela (cervical cancer), A549 (lung adenocarcinoma), A172 (GBM), and Jurkat (leukemia) cells were significantly reduced after the cells were treated with compound 15 at 5 μM for 6 d (Figure 9). Furthermore, compound 15 concentration-dependently inhibited the proliferation of Hela, A549, A172, and Jurkat cells, with a similar potency as the PRMT5 inhibitor EPZ015666 in Hela, A549, and Jurkat cells, and a slightly better potency than EPZ015666 in A172 cells (Figure 10).
Figure 9.
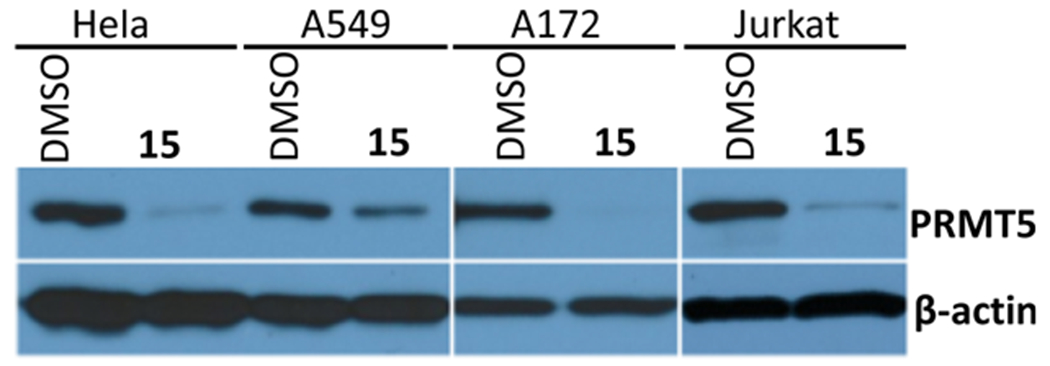
Compound 15 reduced the PRMT5 protein levels in HeLa, A549, A172, and Jurkat cells. The cells were treated with 5 μM of compound 15 for 6 d. The protein levels of PRMT5 were detected by western blotting. The western blotting results are representative of two independent experiments.
Figure 10.
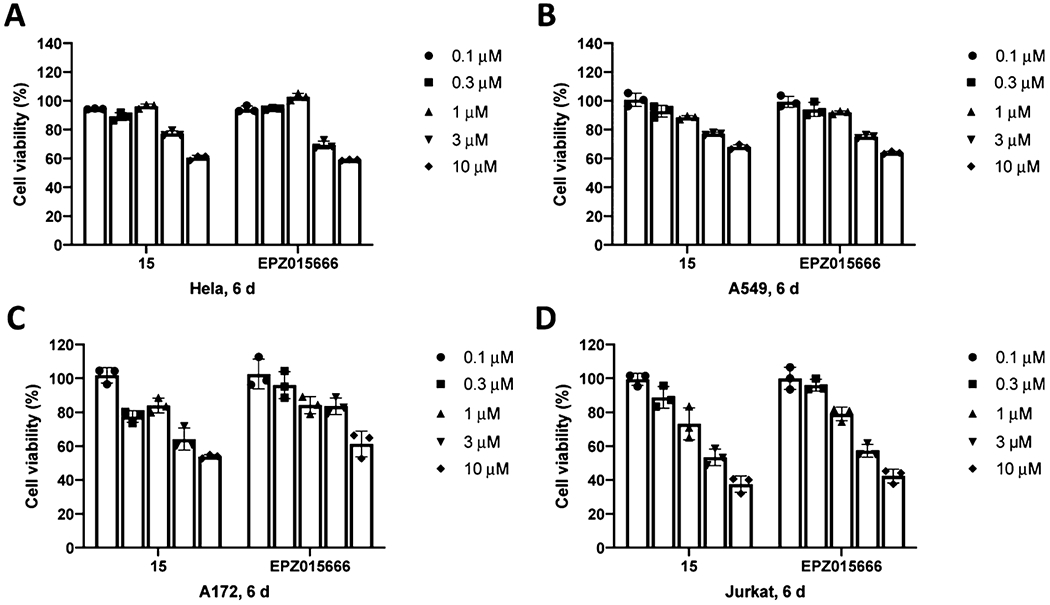
Compound 15 and EPZ015666 inhibited the proliferation of HeLa (A), A549 (B), A172 (C), and Jurkat (D) cells. The cells were treated with 15 or EPZ015666 at the indicated concentrations for 6 d. The relative cell viabilities to the untreated group are shown as mean ± SD (n = 3).
In Vivo Mouse PK Study.
Lastly, we assessed the in vivo mouse PK properties of compound 15. Compound concentrations in plasma from male Swiss albino mice were determined following a single intraperitoneal (IP) administration of compound 15 at 150 mg/kg. We were pleased to find that a single IP injection resulted in good exposure levels in plasma (Figure 11). In particular, a very high plasma concentration (14 ± 2 μM) was achieved at 2 h post dosing. Even after 12 h, the concentration of compound 15 was still above 100 nM. In addition, compound 15 was well tolerated by the treated mice, and no clinical signs were observed in the PK study. Taken together, these results suggest that compound 15 could be a valuable chemical tool for investigating the effects of PRMT5 degradation in vivo.
Figure 11.
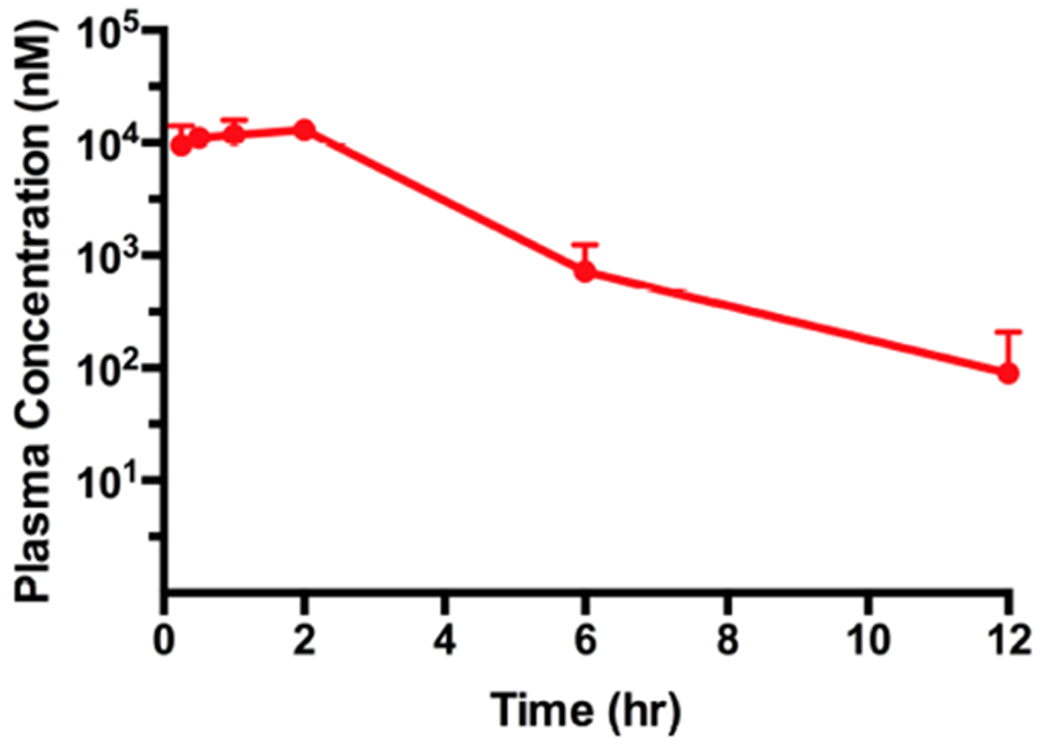
Plasma concentrations of compound 15 over 12 h, following a single 150 mg/kg IP injection in male Swiss albino mice. The compound concentration shown at each time point is the mean ± SD from three test mice.
CONCLUSIONS
Although the PROTAC technology has been successfully applied to the generation of numerous small-molecule degraders for a number of kinases,32,40,41,47 including ALK, MEK/1/2, and EGFR degraders recently discovered by us,39,42,43 very few small-molecule degraders of histone methyltransferases (HMTs, also known as protein methyltransferases) have been reported.48 Through a brief SAR study, we discovered a first-in-class PRMT5 degrader, compound 15, which is also the first degrader of any PRMTs, an important subclass of HMTs. Compound 15 is an E3 ligase VHL-recruiting bifunctional small molecule, effectively reducing the PRMT5 protein level in MCF-7 cells in a concentration-, time-, PRMT5-, VHL-, and proteasome-dependent manner. The results from our MS-based quantitative global proteomics analysis indicate that compound 15 is a highly selective PRMT5 degrader. Compound 15 was at least as effective as the parent PRMT5 inhibitor EPZ015666 at inhibiting global SDMA and proliferation in MCF-7 cells. In addition, compound 15 was able to significantly reduce PRMT5 protein levels in other cancer lines such as Hela, A549, A172, and Jurkat cells and inhibit the proliferation of these cells. Furthermore, compound 15 displayed good plasma exposure in a mouse PK study. Moreover, we developed two structurally similar control compounds, 17, which exhibited similar PRMT5 inhibitory activity as compound 15 but did not degrade PRMT5, and 21, which displayed much reduced inhibitory activity for PRMT5 and did not degrade PRMT5. Collectively, compound 15 and its controls 17 and 21 are a set of valuable chemical tools for further exploring PRMT5 functions in physiology and pathophysiology.
EXPERIMENTAL SECTION
Chemistry General Procedure.
All commercially available chemical reagents were directly used without further purification. A Teledyne ISCO CombiFlash Rf+ instrument equipped with a variable wavelength UV detector and a fraction collector was used to conduct flash column chromatography. RediSepRf HP C18 and flash silica columns were used for purification. High-performnace liquid chromatography (HPLC) spectra for all compounds were acquired using an Agilent 1200 series system with a diode array detector. Chromatography was performed on a 2.1 × 150 mm Zorbax 300SB-C18 5 μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 mL/min. The gradient program was as follows: 1% B (0–1 min), 1–99% B (1–4 min), and 99% B (4–8 min). High-resolution mass spectrometry (HRMS) data were acquired in positive ion mode using an Agilent G1969A API-TOF system with an electrospray ionization (ESI) source. Nuclear magnetic resonance (NMR) spectra were acquired on either a Bruker DRX-600 spectrometer (800, 600 MHz 1H NMR) or a Bruker Avance-III 800 MHz spectrometer (201 MHz 13C NMR). Chemical shifts are reported in ppm (δ). Preparative HPLC was performed on Agilent Prep 1200 series with a UV detector set to 254 nm or 220 nM. Samples were injected into a Phenomenex Luna 5 μM C18 column, with dimensions of 250 × 30 mm, at room temperature. The flow rate was 40 mL/min. A linear gradient was used with 10% of MeOH (A) in H2O (with 0.1% trifluoroacetic acid (TFA)) (B) to 100% of MeOH (A). HPLC was used to establish the purity of target compounds. All final compounds had >95% purity using the HPLC methods described above. All final compounds are characterized by the salt form of trifluoroacetic acid.
(S)-6-((3H-[1,2,3]Triazolo[4,5-b]pyridin-3-yl)oxy)-N-(3-(3,4-dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)pyrimidine-4-carboxamide (1).
To the solution of tert-butyl (S)-(oxiran-2-ylmethyl)-carbamate (1 g, 5.77 mmol) in isopropanol (10 mL) was added 1,2,3,4-tetrahydroisoquinoline (770 mg, 5.77 mmol). The resulting solution was heated at reflux for 6 h, before the volatile was removed under reduced pressure. The resulting residue was dissolved in dichloromethane (DCM, 5 mL). After the resulting solution was treated with TFA (5 mL) for 1 h, the volatiles were evaporated under reduced pressure. The resulting brown oil was used in the next step without further purification. The residue was added to a solution of 6-chloropyrimidine-4-carboxylic acid (951 mg, 6 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI, 1.66 g, 8.67 mmol), HOAt (1.9 g, 14.45 mmol), and N-methylmorpholine (NMM, 1.12 g, 11.12 mmol) in DMSO (30 mL), and the solution was stirred for 6 h. Reverse-phase chromatography was used to purify the mixture without additional workup to give intermediate 1 (580 mg, 18%). 1H NMR (600 MHz, methanol-d4): δ 8.75 (dd, J = 4.4, 1.5 Hz, 1H), 8.61 (dd, J = 8.5, 1.5 Hz, 1H), 8.37 (d, J = 1.2 Hz, 1H), 7.99 (d, J = 1.2 Hz, 1H), 7.61 (dd, J = 8.5, 4.5 Hz, 1H), 7.10–7.02 (m, 3H), 6.98 (d, J = 7.3 Hz, 1H), 4.17–4.08 (m, 1H), 3.73 (s, 2H), 3.65–3.51 (m, 2H), 2.97–2.80 (m, 4H), 2.75–2.65 (m, 2H). MS (ESI) m/z: 447.2 [M + H]+.
tert-Butyl (S)-3-((6-((3-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)carbamoyl) pyrimidin-4-yl)amino)azetidine-1-carboxylate (2).
To a solution of 1 (580 mg, 1 mmol) in N-methyl-2-pyrrolidone (NMP, 10 mL) was added tert-butyl 3-aminoazetidine-1-carboxylate (1 g, 5.81 mmol). The resulting mixture was stirred overnight before being quenched with water (50 mL). The mixture was extracted with ethyl acetate (3 × 50 mL). The combined organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel flash chromatography (methanol/DCM, 0–10%) to afford intermediate 2 (434 mg, yield 90%). 1H NMR (600 MHz, methanol-d4): δ 8.25 (s, 1H), 7.14–7.02 (m, 4H), 6.99 (d, J = 7.3 Hz, 1H), 4.71–4.62 (m, 1H), 4.33–4.20 (m, 2H), 4.10–4.03 (m, 1H), 3.86–3.76 (m, 2H), 3.71 (s, 2H), 3.56–3.44 (m, 2H), 2.92 (t, J = 5.9 Hz, 2H), 2.88–2.76 (m, 2H), 2.65 (d, J = 6.1 Hz, 2H), 1.44 (s, 9H). MS (ESI) m/z: 483.2 [M + H]+.
(S)-6-(Azetidin-3-ylamino)-N-(3-(3,4-dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)pyrimidine-4-carboxamide (3).
To a solution of 2 (434 mg, 0.9 mmol) in DCM (5 mL) was added TFA (5 mL). After the reaction was stirred for 30 min, the volatiles were removed under reduced pressure to give intermediate 3 as a TFA salt (560 mg, yield 100%). 1H NMR (600 MHz, methanol-d4): δ 8.60 (s, 1H), 7.36–7.14 (m, 5H), 4.67–4.57 (m, 1H), 4.46–4.31 (m, 4H), 4.27–4.19 (m, 2H), 3.94–3.80 (m, 1H), 3.61–3.07 (m, 8H). MS (ESI) m/z: 383.2 [M + H]+.
Synthesis of VHL-1 PEG Linkers (4–8).
To a solution of diacid (4 mmol) in dimethylformamide (10 mL) and DCM (250 mL) was added NMM (10 mmol), VHL-1 (2 mmol), HOAt (2.4 mmol), and EDCI (2.4 mmol) at 0 °C. The resulting reaction solution was stirred at 0 °C for 6 h before being stirred at room temperature (rt) overnight. The progress of the reaction was monitored by LC–MS. After VHL-1 was totally consumed, the reaction was concentrated. The resulting residue was purified by reverse-phase chromatography to yield the desired product.
2-(2-(((S)-1-((2S,4R)-4-Hydroxy-2-((4-(4-methylthiazol-5-yl)-benzyl)carbamoyl) pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-amino)-2-oxoethoxy)acetic Acid (4).
(810 mg, yield 69%) as white solid. 1H NMR (600 MHz, CD3OD): δ 8.97 (s, 1H), 7.47 (d, J = 8.2 Hz, 2H), 7.43 (d, J = 8.1 Hz, 2H), 4.69 (s, 1H), 4.60–4.47 (m, 3H), 4.36 (d, J = 15.5 Hz, 1H), 4.27–4.17 (m, 2H), 4.16–4.07 (m, 2H), 3.89 (d, J = 11.0 Hz, 1H), 3.81 (dd, J = 11.0, 3.8 Hz, 1H), 2.48 (s, 3H), 2.22 (dd, J = 13.1, 7.6 Hz, 1H), 2.13–2.06 (m, 1H), 1.05 (s, 9H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H35N4O7S, 547.2221; found, 547.2230.
3-(2-(3-(((S)-1-((2S,4R)-4-Hydroxy-2-((4-(4-methylthiazol-5-yl)-benzyl) carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-amino)-3-oxopropoxy)ethoxy) propanoic Acid (5).
(680 mg, yield 64%) as white solid. 1H NMR (600 MHz, CD3OD): δ 8.98 (d, J = 20.1 Hz, 1H), 7.48 (d, J = 8.0 Hz, 2H), 7.43 (d, J = 8.1 Hz, 2H), 4.64 (s, 1H), 4.59–4.51 (m, 2H), 4.49 (s, 1H), 4.35 (d, J = 15.5 Hz, 1H), 3.89 (d, J = 11.0 Hz, 1H), 3.80 (dd, J = 10.9, 3.8 Hz, 1H), 3.76–3.67 (m, 4H), 3.63–3.55 (m, 4H), 2.60–2.43 (m, 7H), 2.21 (dd, J = 13.1, 7.6 Hz, 1H), 2.08 (ddd, J = 13.2, 9.1, 4.5 Hz, 1H), 1.04 (s, 9H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C30H43N4O8S, 619.2796; found, 619.2800.
(S)-15-((2S,4R)-4-Hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)-carbamoyl) pyrrolidine-1-carbonyl)-16,16-dimethyl-13-oxo-4,7,10-trioxa-14-azaheptadecanoic Acid (6).
(677 mg, yield 57%) as white solid. 1H NMR (600 MHz, CD3OD): δ 8.95 (s, 1H), 7.47 (d, J = 8.1 Hz, 2H), 7.42 (d, J = 8.1 Hz, 2H), 4.65 (s, 1H), 4.59–4.51 (m, 2H), 4.49 (s, 1H), 4.35 (d, J = 15.5 Hz, 1H), 3.89 (d, J = 11.1 Hz, 1H), 3.80 (dd, J = 10.9, 3.9 Hz, 1H), 3.76–3.67 (m, 4H), 3.66–3.54 (m, 8H), 2.60–2.50 (m, 3H), 2.50–2.43 (m, 4H), 2.21 (dd, J = 13.1, 7.6 Hz, 1H), 2.13–2.08 (m, 1H), 1.04 (s, 9H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C32H47N4O9S, 663.3058; found, 663.3059.
11-(((S)-1-((2S,4R)-4-Hydroxy-2-((4-(4-methylthiazol-5-yl)-benzyl)carbamoyl) pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-amino)-11-oxoundecanoic Acid (7).
(930 mg, yield 78%) as white solid. 1H NMR (600 MHz CD3OD): δ 8.95 (s, 1H), 7.49 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 7.8 Hz, 2H), 4.66 (s, 1H), 4.61–4.50 (m, 3H), 4.38 (d, J = 15.6 Hz, 1H), 3.93 (d, J = 9.6 Hz, 1H), 3.82 (dd, J = 11.4, 3.6 Hz, 1H), 2.50 (s, 3H), 2.35–2.21 (m, 5H), 2.12–2.07 (m, 1H), 1.66–1.57 (m, 4H), 1.37–1.29 (m, 10H), 1.06 (s, 9H). HRMS (ESI-TOF) calcd for C33H49N4O6S, 629.3367; found, 629.3368.
(S)-19-((2S,4R)-4-Hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)-carbamoyl) pyrrolidine-1-carbonyl)-20,20-dimethyl-17-oxo-3,6,9,12,15-pentaoxa-18-azahenicosanoic Acid (8).
(496 mg, yield 54%) as white solid. 1H NMR (600 MHz, CD3OD): δ 8.89 (s, 1H), 7.47 (d, J = 8.1 Hz, 2H), 7.42 (d, J = 8.1 Hz, 2H), 4.69 (s, 1H), 4.59–4.46 (m, 3H), 4.36 (d, J = 15.5 Hz, 1H), 4.16–4.00 (m, 4H), 3.87 (d, J = 11.0 Hz, 1H), 3.80 (dd, J = 11.0, 3.7 Hz, 1H), 3.76–3.53 (m, 16H), 2.48 (s, 3H), 2.22 (dd, J = 13.1, 7.6 Hz, 1H), 2.12–2.07 (m, 1H), 1.04 (s, 9H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C34H51N4O11S, 723.3270; found, 723.3269.
N-((S)-3-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)-6-((1-(2-(2-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)-benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-amino)-2-oxoethoxy)acetyl)azetidin-3-yl)amino)pyrimidine-4-carboxamide (9).
To a solution of 3 (10 mg, 0.016 mmol), 4 (10 mg, 0.02 mmol), EDCI (6 mg, 0.03 mmol), and HOAt (4 mg, 0.03 mmol) in DMSO (1 mL) was added NMM (15 mg, 0.15 mmol). After stirring overnight at room temperature, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford 9 as a white solid (6 mg, yield 36%). 1H NMR (600 MHz, methanol-d4): δ 8.96 (s, 1H), 8.52 (s, 1H), 7.48–7.41 (m, 4H), 7.34–7.16 (m, 5H), 4.71–4.47 (m, 4H), 4.44–4.33 (m, 4H), 4.25–4.05 (m, 9H), 3.98–3.95 (m, 1H), 3.91–3.76 (m, 5H), 3.57–3.49 (m, 2H), 3.44–3.35 (m, 2H), 2.47 (s, 3H), 2.26–2.21 (m, 1H), 2.12–2.06 (m, 1H), 1.04 (s, 9H). 13C NMR (151 MHz, MeOD): δ 172.96, 170.44, 169.89, 169.63, 157.07, 151.71, 147.17, 146.45, 139.01, 130.65, 129.87, 128.98, 128.50, 128.11, 127.60, 126.91, 126.45, 69.91, 69.68, 68.55, 64.24, 59.47, 57.47, 57.07, 56.73, 54.98, 43.16, 42.31, 41.75, 39.09, 37.55, 36.26, 35.65, 35.59, 25.57, 24.53, 14.22. HPLC 95% pure, tR = 3.97 min; HRMS (ESI-TOF) m/z: [M + H]+ for C46H59N10O8S+, 911.4233; found, 911.4233.
N-((S)-3-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)-6-((1 -(3-(2-(3-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)-benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-amino)-3-oxopropoxy)ethoxy)propanoyl)azetidin-3-yl)amino)-pyrimidine-4-carboxamide (10).
To a solution of 3 (10 mg, 0.016 mmol), 5 (10 mg, 0.02 mmol), EDCI (6 mg, 0.03 mmol), and HOAt (4 mg, 0.03 mmol) in DMSO (1 mL) was added NMM (15 mg, 0.15 mmol). After stirring overnight at rt, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford 10 as a white solid (8 mg, yield 48%). 1H NMR (600 MHz, methanol-d4): δ 8.95 (s, 1H), 8.54 (s, 1H), 7.49–7.34 (m, 4H), 7.33–7.14 (m, 5H), 4.79–4.74 (m, 1H), 4.66–4.44 (m, 7H), 4.44–4.28 (m, 5H), 4.16–4.10 (m, 1H), 3.90–3.77 (m, 5H), 3.75–3.67 (m, 4H), 3.61–3.49 (m, 6H), 3.44–3.37 (m, 2H), 2.56–2.38 (m, 7H), 2.25–2.20 (m, 1H), 2.11–2.03 (m, 1H), 1.03 (s, 9H). 13C NMR (151 MHz, MeOD): δ 173.11, 172.51, 172.35, 170.73, 156.98, 151.75, 147.12, 139.07, 132.33, 130.65, 129.84, 128.98, 128.50, 128.11, 127.63, 126.91, 126.45, 70.12, 70.10, 70.02, 69.69, 66.91, 66.68, 59.46, 57.53, 57.29, 56.62, 54.26, 43.17, 42.29, 42.18, 40.56, 37.57, 35.98, 35.39, 31.90, 25.59, 24.53, 14.21. HPLC 95% pure, tR = 3.98 min; HRMS (ESI-TOF) m/z: [M + H]+ for C50H67N10O9S+, 983.4808; found, 983.4795.
N-((S)-3-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)-6-((1-((S)-15-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)-carbamoyl)pyrrolidine-1-carbonyl)-16,16-dimethyl-13-oxo-4,7,10-trioxa-14-azaheptadecanoyl)azetidin-3-yl)amino)pyrimidine-4-carboxamide (11).
To a solution of 3 (10 mg, 0.016 mmol), 6 (10 mg, 0.016 mmol), EDCI (6 mg, 0.03 mmol), and HOAt (4 mg, 0.03 mmol) in DMSO (1 mL) was added NMM (15 mg, 0.15 mmol). After stirring overnight at rt, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford 11 as a white solid (10 mg, yield 60%). 1H NMR (600 MHz, methanol-d4): δ 8.96 (s, 1H), 8.55 (s, 1H), 7.50–7.39 (m, 4H), 7.33–7.14 (m, 5H), 4.81–4.76 (m, 1H), 4.66–4.48 (m, 7H), 4.44–4.31 (m, 5H), 4.17–4.14 (m, 1H), 3.91–3.78 (m, 5H), 3.73–3.69 (m, 4H), 3.64–3.48 (m, 10H), 3.41–3.35 (m, 2H), 2.58–2.38 (m, 7H), 2.25–2.20 (m, 1H), 2.11–2.07 (m, 1H), 1.03 (s, 9H). 13C NMR (151 MHz, MeOD): δ 173.09, 172.52, 172.34, 170.74, 151.78, 147.10, 139.09, 132.36, 130.66, 129.82, 128.97, 128.50, 128.11, 127.63, 126.91, 126.46, 70.13, 70.10, 70.06, 70.04, 70.02, 69.96, 69.82, 69.69, 69.68, 66.88, 66.61, 59.45, 57.56, 57.20, 56.60, 54.29, 43.20, 42.90, 42.29, 40.60, 39.03, 37.57, 36.09, 35.94, 35.87, 35.39, 31.88, 25.65, 25.64, 25.60, 24.53, 14.20. HPLC 95% pure, tR = 4.00 min; HRMS (ESI-TOF) m/z: [M + H]+ for C52H71N10O10S+, 1027.5070; found, 127.5066.
N-((S)-3-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)-6-((1-(11-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)-benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-amino)-11-oxoundecanoyl)azetidin-3-yl)amino)pyrimidine-4-carboxamide (12).
To a solution of 3 (20 mg, 0.032 mmol), 7 (16 mg, 0.025 mmol), EDCI (12 mg, 0.06 mmol), and HOAt (8 mg, 0.06 mmol) in DMSO (1 mL) was added NMM (30 mg, 0.30 mmol). After stirring overnight at rt, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford 12 as a white solid (11 mg, yield 39%). 1H NMR (600 MHz, methanol-d4): δ 8.92 (s, 1H), 8.53 (s, 1H), 7.49–7.39 (m, 4H), 7.34–7.18 (m, 5H), 4.79–4.73 (m, 1H), 4.65–4.48 (m, 7H), 4.43–4.32 (m, 5H), 4.09–4.05 (m, 1H), 3.92–3.79 (m, 5H), 3.55–3.50 (m, 2H), 3.42–3.37 (m, 2H), 2.47 (s, 3H), 2.26–2.08 (m, 6H), 1.59 (s, 4H), 1.32 (s, 10H), 1.03 (s, 9H). 13C NMR (151 MHz, MeOD): δ 174.66, 174.56, 173.08, 170.95, 151.68, 139.04, 130.65, 129.90, 128.97, 128.50, 128.11, 127.61, 126.92, 126.45, 69.68, 59.43, 57.55, 57.10, 56.62, 54.29, 43.14, 42.29, 40.53, 37.53, 35.25, 35.18, 30.87, 29.00, 28.94, 28.89, 28.84, 25.63, 25.59, 25.59, 24.54, 24.52, 14.23. HPLC 95% pure, tR = 4.00 min; HRMS (ESI-TOF) m/z: [M + H]+ for C53H73N10O7S+, 993.5379; found, 993.5378.
N-((S)-3-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)-6-((1-((S)-19-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)-carbamoyl)pyrrolidine-1-carbonyl)-20,20-dimethyl-17-oxo-3,6,9,12,15-pentaoxa-18-azahenicosanoyl)azetidin-3-yl)amino)-pyrimidine-4-carboxamide (13).
To a solution of 3 (20 mg, 0.032 mmol), 8 (16 mg, 0.02 mmol), EDCI (6 mg, 0.03 mmol), and HOAt (4 mg, 0.03 mmol) in DMSO (1 mL) was added NMM (30 mg, 0.30 mmol). After stirring overnight at rt, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford 13 as a white solid (10 mg, yield 38%). 1H NMR (600 MHz, methanol-d4): δ 8.90 (s, 1H), 8.52 (s, 1H), 7.48–7.41 (m, 4H), 7.35–7.16 (m, 5H), 4.81–4.66 (m, 6H), 4.62–4.46 (m, 6H), 4.42–4.32 (m, 5H), 4.25–4.21 (m, 1H), 4.10–4.02 (m, 4H), 3.89–3.67 (m, 4H), 3.70–3.43 (m, 17H), 2.47 (s, 3H), 2.26–2.21 (m, 1H), 2.08 (t, J = 11.2 Hz, 1H), 1.03 (s, 9H). 13C NMR (151 MHz, MeOD): δ 172.99, 170.70, 170.50, 170.30, 157.14, 151.71, 147.22, 139.02, 132.26, 130.66, 129.90, 129.13, 128.99, 128.50, 128.11, 127.61, 126.91, 126.45, 70.84, 70.32, 70.21, 70.11, 70.04, 70.02, 69.67, 69.23, 59.43, 57.93, 56.77, 56.72, 54.97, 43.15, 42.31, 41.78, 37.60, 35.70, 25.58, 25.55, 24.54, 14.26. HPLC 95% pure, tR = 3.78 min; HRMS (ESI-TOF) m/z: [M + H]+ for C54H75N10O12S, 1087.5281; found, 1087.5261.
(S)-19-((2S,4R)-4-Hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)-phenyl)ethyl)carbamoyl)pyrrolidine-1-carbonyl)-20,20-dimethyl-17-oxo-3,6,9,12,15-pentaoxa-18-azahenicosanoic Acid (14).
To a solution of (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (VHL-2) (33.6 mg, 0.06 mmol), 3,6,9,12,15-pentaox-aheptadecanedioic acid (31.03 mg, 0.1 mmol), EDCI (12 mg, 0.06 mmol), and HOAt ((8 mg, 0.06 mmol) in DMSO (2 mL) was added NMM (30 mg, 0.3 mmol). After being stirred overnight at rt, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford 14 as an oil (22 mg, yield 50%). 1H NMR (600 MHz, methanol-d4): δ 9.25 (s, 1H), 7.51–7.38 (m, 4H), 5.04–4.97 (m, 2H), 4.68 (s, 1H), 4.60–4.54 (m, 1H), 4.44 (s, 1H), 4.13 (s, 1H), 4.06–4.04 (m, 1H), 3.85 (d, J = 11.1 Hz, 1H), 3.76–3.61 (m, 17H), 3.35–3.30 (m, 1H), 2.52 (s, 3H), 2.24–2.20 (m, 1H), 1.99–1.93 (m, 1H), 1.51 (d, J = 7.0 Hz, 3H), 1.05 (s, 9H). MS (ESI) m/z: 737.3 [M + H]+.
N-((S)-3-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)-6-((1-((S)-19-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)-phenyl)ethyl)carbamoyl)pyrrolidine-1-carbonyl)-20,20-dimethyl-17-oxo-3,6,9,12,15-pentaoxa-18-azahenicosanoyl)azetidin-3-yl)-amino)pyrimidine-4-carboxamide (15).
To a solution of 3 (10 mg, 0.016 mmol), 14 (10 mg, 0.014 mmol), EDCI (6 mg, 0.03 mmol), and HOAt (4 mg, 0.03 mmol) in DMSO (1 mL) was added NMM (15 mg, 0.15 mmol). After being stirred overnight at rt, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford 15 as a white solid (6 mg, yield 35%). 1H NMR (800 MHz, methanol-d4): δ 9.03 (s, 1H), 8.59 (s, 1H), 7.50–7.43 (m, 4H), 7.34 (t, J = 7.4 Hz, 1H), 7.31–7.27 (m, 2H), 7.26–7.20 (m, 2H), 5.07–5.01 (m, 1H), 4.75 (t, J = 8.8 Hz, 1H), 4.70 (s, 1H), 4.68–4.57 (m, 2H), 4.49–4.34 (m, 4H), 4.30–4.23 (m, 1H), 4.18–4.11 (m, 2H), 4.11–4.03 (m, 2H), 4.00–3.95 (m, 1H), 3.87 (d, J = 11.0 Hz, 2H), 3.78–3.61 (m, 18H), 3.61–3.39 (m, 5H), 3.30–3.11 (m, 2H), 2.51 (s, 3H), 2.24 (dd, J = 13.2, 7.6 Hz, 1H), 2.04–1.92 (m, 1H), 1.53 (d, J = 7.2 Hz, 3H), 1.07 (s, 9H). 13C NMR (201 MHz, MeOD): δ 171.76, 170.68, 170.51, 170.29, 164.14, 163.01, 156.76, 151.88, 146.93, 144.48, 132.41, 130.66, 129.75, 129.11, 128.50, 128.12, 126.92, 126.45, 126.30, 126.10, 104.05, 70.81, 70.32, 70.20, 70.10, 70.04, 69.67, 69.57, 69.17, 64.26, 59.23, 57.81, 56.83, 56.69, 54.96, 48.77, 42.84, 41.88, 37.46, 35.66, 25.60, 24.53, 21.00, 14.08. HPLC 95% pure, tR = 3.83 min; HRMS (ESI-TOF) m/z: [M + H]+ for C55H77N10O12S+, 1101.5438; found, 1101.5427.
(S)-19-((2R,4S)-4-Hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)-phenyl)ethyl)carbamoyl)pyrrolidine-1-carbonyl)-20,20-dimethyl-17-oxo-3,6,9,12,15-pentaoxa-18-azahenicosanoic Acid (16).
To a solution of (2R,4S)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (100 mg, 0.2 mmol), 3,6,9,12,15-pentaoxaheptadecanedioic acid (100 mg, 0.3 mmol), EDCI (76 mg, 0.4 mmol), and HOAt (53 mg, 0.4 mmol) in DMSO (2 mL) was added NMM (101 mg, 1 mmol). After being stirred overnight at rt, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford 16 as an oil (66 mg, yield 50%). 1H NMR (600 MHz, methanol-d4): δ 9.34 (s, 1H), 7.58 (d, J = 8.2 Hz, 2H), 7.52 (d, J = 8.2 Hz, 2H), 5.04 (q, J = 7.1 Hz, 1H), 4.66–4.62 (m, 1H), 4.59 (t, J = 7.5 Hz, 1H), 4.53–4.46 (m, 1H), 4.23–3.98 (m, 4H), 3.97–3.92 (m, 1H), 3.79–3.55 (m, 17H), 2.57 (s, 3H), 2.29–2.19 (m, 1H), 2.17–2.09 (m, 1H), 1.48 (d, J = 7.1 Hz, 3H), 1.08 (s, 9H). MS (ESI) m/z: 737.1 [M + H]+.
N-((S)-3-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)-6-((1-((S)-19-((2R,4S)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)-phenyl)ethyl)carbamoyl)pyrrolidine-1-carbonyl)-20,20-dimethyl-17-oxo-3,6,9,12,15-pentaoxa-18-azahenicosanoyl)azetidin-3-yl)-amino)pyrimidine-4-carboxamide (17).
To a solution of 3 (10 mg, 0.016 mmol), 16 (10 mg, 0.014 mmol), EDCI (6 mg, 0.03 mmol), and HOAt (4 mg, 0.03 mmol) in DMSO (1 mL) was added NMM (15 mg, 0.15 mmol). After being stirred overnight at rt, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford 17 as a white solid (9 mg, yield 53%). 1H NMR (800 MHz, methanol-d4): δ 9.13 (s, 1H), 8.58 (d, J = 9.5 Hz, 1H), 7.59–7.41 (m, 4H), 7.38–7.26 (m, 3H), 7.25–7.14 (m, 2H), 5.07–5.01 (m, 1H), 4.79–4.72 (m, 1H), 4.71–4.56 (m, 3H), 4.55–4.35 (m, 4H), 4.33–4.24 (m, 2H), 4.13 (s, 2H), 4.06–3.84 (m, 5H), 3.78–3.40 (m, 23H), 3.33–3.08 (m, 1H), 2.54 (s, 3H), 2.30–2.19 (m, 1H), 2.19–2.10 (m, 1H), 1.48 (d, J = 6.8 Hz, 3H), 1.09 (s, 9H). 13C NMR (201 MHz, MeOD): δ 172.14, 171.01, 170.52, 170.21, 163.07, 159.78, 151.75, 146.63, 144.43, 132.62, 130.69, 129.44, 129.29, 129.02, 128.51, 128.11, 126.91, 126.47, 104.13, 70.77, 70.36, 70.16, 70.13, 70.05, 69.98, 69.94, 69.66, 69.28, 69.19, 64.23, 59.39, 57.80, 57.19, 55.66, 54.81, 48.59, 43.38, 42.04, 37.66, 34.74, 25.65, 25.45, 24.53, 21.22, 14.13. HPLC 95% pure, tR = 3.78 min; HRMS (ESI-TOF) m/z: [M + H]+ for C55H77N10O12S+, 1101.5438; found, 1101.5425.
tert-Butyl (S)-3-((6-((2-hydroxy-3-(piperidin-1-yl)propyl)-carbamoyl)pyrimidin-4-yl)amino)azetidine-1-carboxylate (19).
Intermediate 19 (220 mg, yield 13% over four steps) was prepared according to the same procedure used for preparing intermediate 2 from piperidine (255 mg, 3 mmol) and tert-butyl (S)-(oxiran-2-ylmethyl)carbamate (519 mg, 3 mmol). 1H NMR (500 MHz, methanol-d4): δ 8.57 (s, 1H), 7.22 (s, 1H), 4.50–4.13 (m, 4H), 3.89–3.81 (m, 2H), 3.66–3.39 (m, 4H), 3.29–2.89 (m, 4H), 2.12–1.70 (m, 5H), 1.67–1.23 (m, 10H). MS (ESI) m/z: 435.2 [M + H]+.
(S)-6-(Azetidin-3-ylamino)-N-(2-hydroxy-3-(piperidin-1-yl)-propyl)pyrimidine-4-carboxamide (20).
A solution of 19 (220 mg, 0.4 mmol) in DCM (2 mL) was treated with TFA (2 mL) for 30 min. The volatiles were removed under reduced pressure to give compound 20 (260 mg, yield 100%). 1H NMR (600 MHz, methanol-d4): δ 8.91 (s, 1H), 7.66 (s, 1H), 5.06–4.95 (m, 1H), 4.72 (dd, J = 12.9, 6.9 Hz, 1H), 4.43–4.36 (m, 1H), 4.24 (s, 2H), 3.64–3.37 (m, 4H), 3.25–3.18 (m, 4H), 3.12–2.99 (m, 2H), 2.98–2.88 (m, 1H), 1.98–1.63 (m, 4H), 1.61–1.42 (m, 1H). MS (ESI) m/z: 335.3 [M + H]+.
6-((1-((S)-19-((2S,4R)-4-Hydroxy-2-(((S)-1 -(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidine-1-carbonyl)-20,20-dimethyl-17-oxo-3,6,9,12,15-pentaoxa-18-azahenicosanoyl)azetidin-3-yl)-amino)-N-((S)-2-hydroxy-3-(piperidin-1-yl)propyl)pyrimidine-4-carboxamide (21).
To a solution of 20 (10 mg, 0.02 mmol), 14 (10 mg, 0.014 mmol), EDCI (6 mg, 0.03 mmol), and HOAt (4 mg, 0.03 mmol) in DMSO (1 mL) was added NMM (15 mg, 0.15 mmol). After being stirred overnight at rt, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford 21 as a white solid (8 mg, 49%). 1H NMR (800 MHz, methanol-d4): δ 9.08 (s, 1H), 8.60 (s, 1H), 7.54–7.42 (m, 4H), 7.25 (s, 1H), 5.06–5.01 (m, 1H), 4.78–4.72 (m, 1H), 4.71 (s, 1H), 4.65–4.56 (m, 1H), 4.51–4.39 (m, 2H), 4.34–4.24 (m, 2H), 4.19–4.12 (m, 2H), 4.11–4.03 (m, 2H), 4.02–3.94 (m, 1H), 3.87 (d, J = 11.0 Hz, 1H), 3.83–3.45 (m, 23H), 3.27–3.20 (m, 1H), 3.12 (dd, J = 13.3, 10.5 Hz, 1H), 3.05 (td, J = 12.4, 3.5 Hz, 1H), 2.98 (td, J = 12.3, 3.2 Hz, 1H), 2.52 (s, 3H), 2.24 (dd, J = 13.3, 7.7 Hz, 1H), 2.02–1.75 (m, 6H), 1.54 (d, J = 7.1 Hz, 3H), 1.07 (s, 9H). 13C NMR (201 MHz, methanol-d4): δ 171.77, 170.68, 170.53, 170.29, 163.81, 163.05, 160.20, 159.43, 152.02, 151.44, 146.75, 144.56, 132.58, 129.63, 129.12, 126.32, 126.12, 104.07, 70.85, 70.35, 70.27, 70.23, 70.15, 70.13, 70.07, 70.05, 69.69, 69.57, 69.25, 63.91, 59.34, 59.23, 57.80, 56.82, 56.70, 54.93, 54.80, 51.89, 48.78, 43.30, 41.95, 37.44, 35.66, 25.60, 22.42, 21.24, 21.00, 14.00. HPLC 95% pure, tR = 3.76 min; HRMS (ESI-TOF) m/z: [M + H]+ for C51H77N10O12S+, 1053.5438; found, 1053.5429.
Cell Culture and Cell Viability Assay.
MCF-7, HeLa, A549, and A172 cells were cultured in Dulbecco’s modified Eagle medium, supplemented with 10% fetal bovine serum (FBS), in 5% CO2 at 37 °C. Jurkat cells were cultured in RPMI 1640 medium, supplemented with 10% FBS, in 5% CO2 at 37 °C. A certain number of different cells were seeded and then treated with the indicated compounds for certain days (the medium plus compounds was replaced every other day). The cells were then harvested using Promega CellTiter-Glo (Promega) to count the cell titer, according to the manufacturer’s instructions. The ratio of the cell titer of the experiment group to that of the control group (DMSO-treated) was calculated as the normalized viability.
Western Blot and Antibodies.
Western blotting was performed with a standard protocol. Briefly, cells were harvested and washed with ice-cold phosphate-buffered saline (PBS) and lysed with radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 0.1% sodium dodecyl sulfate, 1% sodium deoxycholate, 5 mM ethylenediaminetetraacetic acid), supplemented with protease inhibitor cocktail (Roche). After being loaded for polyacrylamide gel electrophoresis, the total proteins were transferred to the polyvinylidene difluoride membrane and sequentially incubated in 5% fat-free milk in PBST (PBS + 0.05% Tween 20), primary antibody diluted in PBST, and secondary antibody diluted in PBST. Then, the membrane was incubated with enhanced chemiluminescent reagents and developed with X-ray films. The antibodies used in this study included anti-PRMT5 (active Motif), anti-SDMA (Cell Signaling Technologies), and anti-β-actin (Sigma).
PRMT5/MEP50 Methyltransferase Inhibition Assay.
In this biochemical assay (FlashPlate), which monitors the transfer of a 3H-labeled methyl group from the cofactor SAM to the substrate H4 (1–15), human PRMT5/MEP50 was used as the enzyme (5 nM). The concentration of the substrate core histone was 0.04 μM, and the concentration of the cofactor SAM was 1 μM. Four compounds were tested in a 10-concentration IC50 mode with threefold serial dilution, in duplicate, starting at 5 μM. Compound pre-incubation was performed for 20 min at rt in a mixture of enzyme and substrate.
MS Assay.
Sample Preparation.
MCF7 cells were treated by compound 15 and DMSO, respectively, at a concentration of 5 μM for 5 d. The medium was replaced by a fresh medium with compound 15 and DMSO every 2 d (48 h). Each proteomics experiment result was obtained from two biological replicates, and each sample subjected to LS–MS was validated by repetitive western blot experiments with two different PRMT5 antibodies (Santacruz; A-11; CST 2252S). The harvested cells were lysed in a lysis buffer (8 M urea, 50 mM Tris-HCl pH 8.0). Sonication (5 s on 5 s off, 2 × 30 s) was performed to shear genomic DNA. The lysates were centrifugated for 30 min at 10,000g at 4 °C, and the supernatant was transferred to a clean tube. The protein concentration was determined (bicinchoninic acid assay), and the protein was reduced with 5 mM dithiothreitol, alkylated with 15 mM iodoacetamide in the dark, and then diluted with 3× volume of 25 mM Tris (pH 8.0) buffer and 1 mM CaCl2. The final urea concentration is 2 M. Trypsin was added into the protein solution with 1:100 ratio (trypsin/protein) and digested in 12–16 h or overnight at rt.
MS Analysis.
Peptides were cleaned up by C18 stage tips, and the concentration was determined (peptide assay, Thermo 23275). The cleaned peptides were dissolved in 0.1% formic acid and analyzed on a Q-Exactive HF-X system coupled with an Easy nanoLC 1200 (Thermo Fisher Scientific, San Jose, CA). Peptides (0.5 μg) were loaded on to an Acclain PepMap RSLC C18 column (250 mm × 75 μm ID, C18, 2 μm, Thermo Fisher). The analytical separation of all peptides was achieved with a 130 min gradient. A linear gradient of 5–30% buffer B over 110 min was executed at a 300 nL/min flow rate followed by a ramp to 100% B in 5 min, and 15 min wash with 100% B, where buffer A was aqueous 0.1% formic acid and buffer B was 80% acetonitrile and 0.1% formic acid. LC–MS experiments were performed in a data-dependent mode with full MS (externally calibrated to a mass accuracy of <5 ppm and a resolution of 60,000 at m/z 200), followed by high-energy collision-activated dissociation-tandem mass spectrometry (MS/MS) of the top 20 most intense ions with a resolution of 15,000 at m/z 200. High-energy collision-activated dissociation-MS/MS was used to dissociate peptides at a normalized collision energy of 27 eV in the presence of nitrogen bath gas atoms. Dynamic exclusion was 30.0 s. There were two biological replicates for one treatment, and each sample was subjected to two technical LC–MS replicates.
MS Data Analysis.
Mass spectra processing and peptide identification were performed on the Andromeda search engine in MaxQuant software (version 1.6.10.43) against a human UniProt database (UP000005640). All searches were conducted with a defined modification of cysteine carbamidomethylation, with methionine oxidation and protein amino-terminal acetylation as dynamic modifications. Peptides were confidently identified using a target-decoy approach, with a peptide false discovery rate (FDR) of 1% and a protein FDR of 1%. A minimum peptide length of 7 amino acids was required; maximally two missed cleavages were allowed; the initial mass deviation for the precursor ion was up to 7 ppm, and the maximum allowed mass deviation for fragment ions was 0.5 Da. Data processing and statistical analysis were performed on Perseus (version1. 6.10.50). Protein quantitation was performed on biological replicate runs, and a two-sample t test statistics was used to report statistically significant expression fold changes.
PK Assay.
Six male Swiss albino mice were administered intraperitoneally, with 15 formulated in normal saline at a single 150 mg/kg dose. Blood samples (approximately 60 μL) were collected under light isoflurane anesthesia at 0.25, 0.5, 1, 2, 6, and 12 h. At each time point, blood samples were collected from three mice. Immediately after collection, plasma was harvested by centrifugation and stored at −70 °C until analysis. All samples were processed for analysis by protein precipitation using acetonitrile and analyzed with the fit-for-purpose LC–MS/MS method (LLOQ = 9.25 ng/mL for plasma).
Supplementary Material
ACKNOWLEDGMENTS
The research described here was supported in part by an endowed professorship (to J.J.) from the Icahn School of Medicine at Mount Sinai and the grants R01GM122749 (to J.J.) and R01GM126421 (to M.T.B.) from the U.S. National Institutes of Health.
ABBREVIATIONS
- PRMT5
protein arginine methyltransferase 5
- SAM
S-adenosylmethionine
- MTA
methylthioadenosine
- MTAP
methylthioadenosine phosphorylase
- VHL
von Hippel-Lindau
- PROTAC
proteolysis targeting chimera
- SAR
structure–activity relationship
- VHL-2
(S,R,S)-AHPC-Me
- PEG
poly-(ethylene glycol)
- SDMA
arginine symmetric dimethylation
- PK
pharmacokinetic
- TFA
trifluoroacetic acid
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HOAt
1-hydroxy-7-aza-benzo-triazole
- NMM
N-methylmorpholine
- NMP
N-methyl-2-pyrrolidone
- DMSO
dimethyl sulfoxide
- ESI
electrospray ionization
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01111.
1H NMR, 13C NMR, and HPLC spectra of compounds 15, 17, and 21; raw data of compounds 15, 17, 21, and EPZ015666 inhibiting the PRMT5 methyltransferase activity in the PRMT5 biochemical assay; raw data of compounds 15, 17, 21, and EPZ015666 in cell viability assays (PDF)
Molecular formula strings and biological data (CSV)
Contributor Information
Yudao Shen, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Guozhen Gao, Department of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center, Smithville, Texas 78957, United States.
Xufen Yu, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Huensuk Kim, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Li Wang, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Ling Xie, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Megan Schwarz, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Xian Chen, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Ernesto Guccione, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Jing Liu, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological, Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Mark T. Bedford, Department of Epigenetics and Molecular, Carcinogenesis, The University of Texas MD Anderson Cancer, Center, Smithville, Texas 78957, United States.
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological, Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
REFERENCES
- (1).Branscombe TL; Frankel A; Lee J-H; Cook JR; Yang Z.-h.; Pestka S; Clarke S. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J. Biol. Chem. 2001, 276, 32971−32976. [DOI] [PubMed] [Google Scholar]
- (2).Pal S; Vishwanath SN; Erdjument-Bromage H; Tempst P; Sif S Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004, 24, 9630−9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Pal S; Baiocchi RA; Byrd JC; Grever MR; Jacob ST; Sif S Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007, 26, 3558−3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Migliori V; Müller J; Phalke S; Low D; Bezzi M; Mok WC; Sahu SK; Gunaratne J; Capasso P; Bassi C; Cecatiello V; De Marco A; Blackstock W; Kuznetsov V; Amati B; Mapelli M; Guccione E. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol. 2012, 19, 136−144. [DOI] [PubMed] [Google Scholar]
- (5).Musiani D; Bok J; Massignani E; Wu L; Tabaglio T; Ippolito MR; Cuomo A; Ozbek U; Zorgati H; Ghoshdastider U; Robinson RC; Guccione E; Bonaldi T Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci. Signaling 2019, 12, No. eaat8388. [DOI] [PubMed] [Google Scholar]
- (6).Karkhanis V; Hu Y-J; Baiocchi RA; Imbalzano AN; Sif S Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 2011, 36, 633−641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Blanc RS; Richard S Arginine methylation: the coming of age. Mol. Cell 2017, 65, 8−24. [DOI] [PubMed] [Google Scholar]
- (8).Fong JY; Pignata L; Goy P-A; Kawabata KC; Lee SC-W; Koh CM; Musiani D; Massignani E; Kotini AG; Penson A; Wun CM; Shen Y; Schwarz M; Low DH; Rialdi A; Ki M; Wollmann H; Mzoughi S; Gay F; Thompson C; Hart T; Barbash O; Luciani GM; Szewczyk MM; Wouters BJ; Delwel R; Papapetrou EP; Barsyte-Lovejoy D; Arrowsmith CH; Minden MD; Jin J; Melnick A; Bonaldi T; Abdel-Wahab O; Guccione E Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell 2019, 36, 194–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yang Y; Bedford MT Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37−50. [DOI] [PubMed] [Google Scholar]
- (10).Stopa N; Krebs JE; Shechter D The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell. Mol. Life Sci. 2015, 72, 2041−2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Richters A Targeting protein arginine methyltransferase 5 in disease. Future Med. Chem. 2017, 9, 2081−2098. [DOI] [PubMed] [Google Scholar]
- (12).McCorvy JD; Wacker D; Wang S; Agegnehu B; Liu J; Lansu K; Tribo AR; Olsen RHJ; Che T; Jin J; Roth BL Structural determinants of 5-HT2B receptor activation and biased agonism. Nat. Struct. Mol. Biol. 2018, 25, 787−796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shimizu D; Kanda M; Sugimoto H; Shibata M; Tanaka H; Takami H; Iwata N; Hayashi M; Tanaka C; Kobayashi D; Yamada S; Nakayama G; Koike M; Fujiwara M; Fujii T; Kodera Y The protein arginine methyltransferase 5 promotes malignant phenotype of hepatocellular carcinoma cells and is associated with adverse patient outcomes after curative hepatectomy. Int. J. Oncol. 2017, 50, 381−386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Deng X; Shao G; Zhang H-T; Li C; Zhang D; Cheng L; Elzey BD; Pili R; Ratliff TL; Huang J; Hu C-D Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene 2017, 36, 1223−1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jin Y; Zhou J; Xu F; Jin B; Cui L; Wang Y; Du X; Li J; Li P; Ren R; Pan J Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J. Clin. Invest. 2016, 126, 3961−3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhu F; Guo H; Bates PD; Zhang S; Zhang H; Nomie KJ; Li Y; Lu L; Seibold KR; Wang F; Rumball I; Cameron H; Hoang NM; Yang DT; Xu W; Zhang L; Wang M; Capitini CM; Rui L PRMT5 is upregulated by B-cell receptor signaling and forms a positive-feedback loop with PI3K/AKT in lymphoma cells. Leukemia 2019, 33, 2898−2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Koh CM; Bezzi M; Low DHP; Ang WX; Teo SX; Gay FPH; Al-Haddawi M; Tan SY; Osato M; Saboò A; Amati B; Wee KB; Guccione E. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96−100. [DOI] [PubMed] [Google Scholar]
- (18).Yan F; Alinari L; Lustberg ME; Katherine Martin L; Cordero-Nieves HM; Banasavadi-Siddegowda Y; Virk S; Barnholtz-Sloan J; Bell EH; Wojton J; Jacob NK; Chakravarti A; Nowicki MO; Wu X; Lapalombella R; Datta J; Yu B; Gordon K; Haseley A; Patton JT; Smith PL; Ryu J; Zhang X; Mo X; Marcucci G; Nuovo G; Kwon C-H; Byrd JC; Chiocca EA; Li C; Sif S; Jacob S; Lawler S; Kaur B; Baiocchi RA Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res. 2014, 74, 1752−1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Chan-Penebre E; Kuplast KG; Majer CR; Boriack-Sjodin PA; Wigle TJ; Johnston LD; Rioux N; Munchhof MJ; Jin L; Jacques SL; West KA; Lingaraj T; Stickland K; Ribich SA; Raimondi A; Scott MP; Waters NJ; Pollock RM; Smith JJ; Barbash O; Pappalardi M; Ho TF; Nurse K; Oza KP; Gallagher KT; Kruger R; Moyer MP; Copeland RA; Chesworth R; Duncan KW A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432−437. [DOI] [PubMed] [Google Scholar]
- (20).Mao R; Shao J; Zhu K; Zhang Y; Ding H; Zhang C; Shi Z; Jiang H; Sun D; Duan W; Luo C Potent, selective, and cell active protein arginine methyltransferase 5 (PRMT5) inhibitor developed by structure-based virtual screening and hit optimization. J. Med. Chem. 2017, 60, 6289−6304. [DOI] [PubMed] [Google Scholar]
- (21).Ye Y; Zhang B; Mao R; Zhang C; Wang Y; Xing J; Liu Y-C; Luo X; Ding H; Yang Y; Zhou B; Jiang H; Chen K; Luo C; Zheng M Discovery and optimization of selective inhibitors of protein arginine methyltransferase 5 by docking-based virtual screening. Org. Biomol. Chem. 2017, 15, 3648−3661. [DOI] [PubMed] [Google Scholar]
- (22).Duncan KW; Rioux N; Boriack-Sjodin PA; Munchhof MJ; Reiter LA; Majer CR; Jin L; Johnston LD; Chan-Penebre E; Kuplast KG; Porter Scott M; Pollock RM; Waters NJ; Smith JJ; Moyer MP; Copeland RA; Chesworth R Structure and property guided design in the identification of PRMT5 tool compound EPZ015666. ACS Med. Chem. Lett. 2016, 7, 162−166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Smil D; Eram MS; Li F; Kennedy S; Szewczyk MM; Brown PJ; Barsyte-Lovejoy D; Arrowsmith CH; Vedadi M; Schapira M Discovery of a dual PRMT5-PRMT7 inhibitor. ACS Med. Chem. Lett. 2015, 6, 408−412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Prabhu L; Chen L; Wei H; Demir O.; Safa A; Zeng L; Amaro RE; O’Neil BH; Zhang Z-Y.; Lu T. Development of an AlphaLISA high throughput technique to screen for small molecule inhibitors targeting protein arginine methyltransferases. Mol. BioSyst. 2017, 13, 2509−2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kong G-M; Yu M; Gu Z; Chen Z; Xu R-M; O’Bryant D; Wang Z Selective small-chemical inhibitors of protein arginine methyltransferase 5 with anti-lung cancer activity. PLoS One 2017, 12, No. e0181601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bonday ZQ; Cortez GS; Grogan MJ; Antonysamy S; Weichert K; Bocchinfuso WP; Li F; Kennedy S; Li B; Mader MM; Arrowsmith CH; Brown PJ; Eram MS; Szewczyk MM; Barsyte-Lovejoy D; Vedadi M; Guccione E; Campbell RM LLY-283, a potent and selective inhibitor of arginine methyltransferase 5, PRMT5, with antitumor activity. ACS Med. Chem. Lett. 2018, 9, 612−617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhu K; Song J-L; Tao H-R; Cheng Z-Q; Jiang C-S; Zhang H Discovery of new potent protein arginine methyltransferase 5 (PRMT5) inhibitors by assembly of key pharmacophores from known inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 3693−3699. [DOI] [PubMed] [Google Scholar]
- (28).Zhu K; Tao H; Song J-L; Jin L; Zhang Y; Liu J; Chen Z; Jiang C-S; Luo C; Zhang H Identification of 5-benzylidene-2-phenylthiazolones as potent PRMT5 inhibitors by virtual screening, structural optimization and biological evaluations. Bioorg. Chem. 2018, 81, 289−298. [DOI] [PubMed] [Google Scholar]
- (29).Lin H; Wang M; Zhang YW; Tong S; Leal RA; Shetty R; Vaddi K; Luengo JI Discovery of potent and selective covalent protein arginine methyltransferase 5 (PRMT5) inhibitors. ACS Med. Chem. Lett. 2019, 10, 1033−1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sun X; Gao H; Yang Y; He M; Wu Y; Song Y; Tong Y; Rao Y PROTACs: great opportunities for academia and industry. Signal Transduction Targeted Ther. 2019, 4, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lai AC; Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16, 101−114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Schapira M; Calabrese MF; Bullock AN; Crews CM Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discovery 2019, 18, 949−963. [DOI] [PubMed] [Google Scholar]
- (33).Ohoka N; Shibata N; Hattori T; Naito M Protein knockdown technology: application of ubiquitin ligase to cancer therapy. Curr. Cancer Drug Targets 2016, 16, 136−146. [DOI] [PubMed] [Google Scholar]
- (34).Churcher I Protac-induced protein degradation in drug discovery: breaking the rules or just making new ones? J. Med. Chem. 2018, 61, 444−452. [DOI] [PubMed] [Google Scholar]
- (35).Gao H; Sun X; Rao Y PROTAC technology: opportunities and challenges. ACS Med. Chem. Lett. 2020, 11, 237−240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chiang K; Zielinska AE; Shaaban AM; Sanchez-Bailon MP; Jarrold J; Clarke TL; Zhang J; Francis A; Jones LJ; Smith S; Barbash O; Guccione E; Farnie G; Smalley MJ; Davies CC PRMT5 is a critical regulator of breast cancer stem cell function via histone methylation and FOXP1 expression. Cell Rep 2017, 21, 3498−3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hu J; Hu B; Wang M; Xu F; Miao B; Yang C-Y; Wang M; Liu Z; Hayes DF; Chinnaswamy K; Delproposto J; Stuckey J; Wang S Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J. Med. Chem. 2019, 62, 1420−1442. [DOI] [PubMed] [Google Scholar]
- (38).Raina K; Lu J; Qian Y; Altieri M; Gordon D; Rossi AMK; Wang J; Chen X; Dong H; Siu K; Winkler JD; Crew AP; Crews CM; Coleman KG PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 7124−7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wei J; Hu J; Wang L; Xie L; Jin MS; Chen X; Liu J; Jin J Discovery of a first-in-class mitogen-activated protein kinase kinase 1/2 degrader. J. Med. Chem. 2019, 62, 10897−10911. [DOI] [PubMed] [Google Scholar]
- (40).Su S; Yang Z; Gao H; Yang H; Zhu S; An Z; Wang J; Li Q; Chandarlapaty S; Deng H; Wu W; Rao Y Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J. Med. Chem. 2019, 62, 7575−7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Brand M; Jiang B; Bauer S; Donovan KA; Liang Y; Wang ES; Nowak RP; Yuan JC; Zhang T; Kwiatkowski N; Müller AC; Fischer ES; Gray NS; Winter GE. Homologselective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300−306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhang C; Han X-R; Yang X; Jiang B; Liu J; Xiong Y; Jin J Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK). Eur. J. Med. Chem. 2018, 151, 304−314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cheng M; Yu X; Lu K; Xie L; Wang L; Meng F; Han X; Chen X; Liu J; Xiong Y; Jin J Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. J. Med. Chem. 2020, 63, 1216−1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Frost J; Galdeano C; Soares P; Gadd MS; Grzes KM; Ellis L; Epemolu O; Shimamura S; Bantscheff M; Grandi P; Read KD; Cantrell DA; Rocha S; Ciulli A Potent and selective chemical probe of hypoxic signalling downstream of HIF-alpha hydroxylation via VHL inhibition. Nat. Commun 2016, 7, 13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Tong S; Si Y; Yu H; Zhang L; Xie P; Jiang W MLN4924 (Pevonedistat), a protein neddylation inhibitor, suppresses proliferation and migration of human clear cell renal cell carcinoma. Sci. Rep. 2017, 7, 5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Bondeson DP; Mares A; Smith IED; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611−617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Tovell H; Testa A; Zhou H; Shpiro N; Crafter C; Ciulli A; Alessi DR Design and characterization of SGK3-PROTAC1, an isoform specific SGK3 kinase PROTAC degrader. ACS Chem. Biol. 2019, 14, 2024−2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ma A; Stratikopoulos E; Park K-S; Wei J; Martin TC; Yang X; Schwarz M; Leshchenko V; Rialdi A; Dale B; Lagana A; Guccione E; Parekh S; Parsons R; Jin J Discovery of a first-inclass EZH2 selective degrader. Nat. Chem. Biol. 2020, 16, 214−222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.