

Abstract
Inhalation exposures to polycyclic aromatic hydrocarbons (PAHs) have been associated with various adverse health effects, including chronic lung diseases and cancer. Using human bronchial epithelial cell line HBE1, we investigated the effects of structurally different PAHs on tissue homeostatic processes, namely gap junctional intercellular communication (GJIC) and MAPKs activity. Rapid (<1 h) and sustained (up to 24 h) inhibition of GJIC was induced by low/middle molecular weight (MW) PAHs, particularly by those with a bay- or bay-like region (1- and 9-methylanthracene, fluoranthene), but also by fluorene and pyrene. In contrast, linear low MW (anthracene, 2-methylanthracene) or higher MW (chrysene) PAHs did not affect GJIC. Fluoranthene, 1- and 9-methylanthracene induced strong and sustained activation of MAPK ERK1/2, whereas MAPK p38 was activated rather nonspecifically by all tested PAHs. Low/middle MW PAHs can disrupt tissue homeostasis in human airway epithelium via structure-dependent nongenotoxic mechanisms, which can contribute to their human health hazards.
Keywords: gap junctional intercellular communication, polycyclic aromatic hydrocarbons, methylated anthracenes, mitogen-activated protein kinases, human bronchial epithelial cell line, nongenotoxic mechanisms
Graphical Abstract
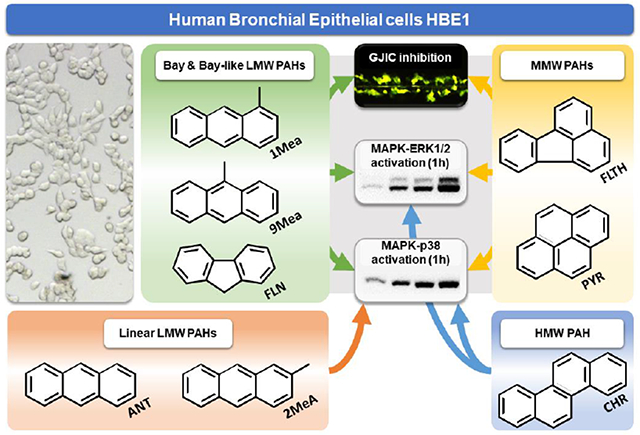
1. INTRODUCTION
Atmospheric pollution is a serious environmental health issue predicted to increase in the future (Oeder et al., 2015). Polycyclic aromatic hydrocarbons (PAHs) represent a prevalent group of airborne pollutants exposing human airways, especially in industrialized and urban areas, where PAHs are anthropogenically produced by incomplete combustion of fossil fuels, vegetative matter or synthetic chemicals (Boström et al., 2002; Kendall et al., 2002). Based on their structure, unsubstituted PAHs can be grouped into low molecular weight (LMW, 2-3 fused aromatic rings, <202 g/mol), middle molecular weight (MMW, ) 4 rings, <228 g/mol), and higher molecular weight (HMW, ≥4 rings, >228 g/mol) (US HHS, 1995).
Increased risks of lung cancer and other respiratory diseases have been associated with environmental or occupational exposures to complex PAHs mixtures, including air pollution, diesel exhaust, coal and coke production or cigarette smoke, which are currently categorized as Group 1 carcinogens (IA RC, 2018). Carcinogenic, mutagenic and nuclear receptor-activating effects of HMW PAHs have been intensely investigated, with benzo[a]pyrene (B[a]P) classified as a human carcinogen (Group 1), and several other HMW PAHs as probable or possible (Group 2A or 2B) carcinogens (IA RC, 2018). However, L/MMW PAHs, including their methylated isoforms, are also highly abundant in complex carcinogenic PAH mixtures such as cigarette smoke, diesel exhaust or coal tar, often significantly exceeding levels of HMW PAHs (Bauer et al., 2018; Boström et al., 2002; Stogiannidis and Laane, 2015), but less information is available regarding toxicity mechanisms and carcinogenicity of L/MMW and/or substituted PAHs (Bauer et al., 2018; Vondráĉek et al., 2007). Even though L/MMW PAHs are mostly non-mutagenic, weakly interacting with nuclear receptors and currently not classifiable as carcinogens (Gad and Gad, 2014; Vondráĉek et al., 2017), some of them have been shown to elicit cellular events that can contribute to tumor-promotion and other pathologies (Bauer et al., 2018). These nongenotoxic effects induced by L/MMW PAHs included dysregulation of gap junctional intercellular communication (GJIC), i.e. biochemical and electrical coupling between adjacent cells mediated via connexin (Cx) protein channels (Loewenstein and Kanno, 1966; Wong et al., 2017). GJIC is a crucial process for homeostatic control of the cell growth, differentiation and apoptosis, required for coordinated cellular and transcriptomic responses across the tissue (Nahta et al., 2015; Trosko and Ruch, 1998). Altered Cx expression and GJIC function have been identified as critical steps in lung cancer promotion and neoplastic transformation (Nahta et al., 2015; Ruch, 2019), and also in development of other lung diseases (Freund-Michel et al., 2016; Wong et al., 2017). In mammals, there are 21 different Cx isoforms (Söhl and Willecke, 2004), which are expressed in a cell-type and tissue-specific manner. Cx43 represents a major GJIC protein expressed in most tissues, and it is also dominant in lower airways and its epithelial cells (Freund-Michel et al., 2016; Wilgenbus et al., 1992).
Inhibition of Cx43-mediated GJIC has been extensively studied for numerous LMW PAHs in a rat liver epithelial in vitro model (Blaha et al., 2002; Sovadinova et al., 2015; Upham et al., 2008, 1998, 1996; Weis et al., 1998). PAH effects on GJIC were structure-dependent, when L/MMW PAHs with a so-called ‘bay’ or ‘bay-like’ region, such as 1- and 9-methylanthracenes (1MeA, 9MeA) or fluoranthene (FLT), rapidly inhibited GJIC (within ≤30 min of exposure). Comparable effects were also induced by L/MMW PAHs fluorene (FLN) or pyrene (PYR). However, linearly arranged LMW PAHs (e.g. anthracene, ANT; 2-methylanthracene, 2MeA), or HMW PAHs (e.g. B[a]P; chrysene, CHR), elicited none or only weak GJIC inhibiting activity (Blaha et al., 2002; Sovadinova et al.,2015; Upham et al.,2008, 1998, 1996; Weis et al., 1998). Moreover, GJIC-inhibiting LMW PAHs specifically activated intracellular signaling pathways mitogen-activated protein kinases (MAPKs) ERK1/2 and p38, and phospholipase A2 (PLA2) and phosphatidylcholine-specific phospholipase C (PC-PLC) (Sovadinova et al., 2015; Upham et al., 2008). LMW PAHs 1MeA, FLT, or their binary mixtures, were found to rapidly dysregulate GJIC and activate MAPK ERK1/2 and p38 in a non-cancerous, Cx43-expressing, mouse epithelial alveolar type II cell line C10. These early signaling events were also associated with induction of inflammatory cytokines and pathways (cyclooxygenase 2, PLA2) and transcriptional changes related to tissue-remodeling (Osgood et al., 2017; Romo et al., 2019; Siegrist et al., 2019). Importantly, these early (<8 h) signaling events in C10 cells were induced by the parental compounds since PAH metabolization was not detected (Siegrist et al., 2019). Apparently, dysregulation of GJIC, intracellular signal transduction and inflammatory responses induced in airway epithelial cells by L/MMW PAHs represent important nongenotoxic mechanisms by which air pollution or cigarette smoke might contribute to the development of chronic toxicities, neoplastic and inflammatory lung diseases (Bauer et al., 2018; Osgood et al., 2017, 2013; Romo et al., 2019; Siegrist et al., 2019). However, the effects of L/MMW PAHs on GJIC and related signaling events have not been studied in human airway cells, although alterations of GJIC by PAHs can be cell type- and species-specific (Osgood et al., 2013; Sovadinova et al., 2015; Kubincova et al., 2019). Therefore, we utilized for the first time the in vitro model of a non-cancerous immortalized human bronchial epithelial cell line (HBE1) to investigate the effects of selected LMW, MMW and HMW PAHs (Table 1) on GJIC and associated signaling events (Cx43 expression, intracellular signaling via MAPK-ERK1/2 and -p38), which might be related to dysregulation of tissue homeostasis and development of lung diseases, such as cancer.
Table 1.
Effects of selected PAHs representatives on molecular and signaling events in HBE1 cells
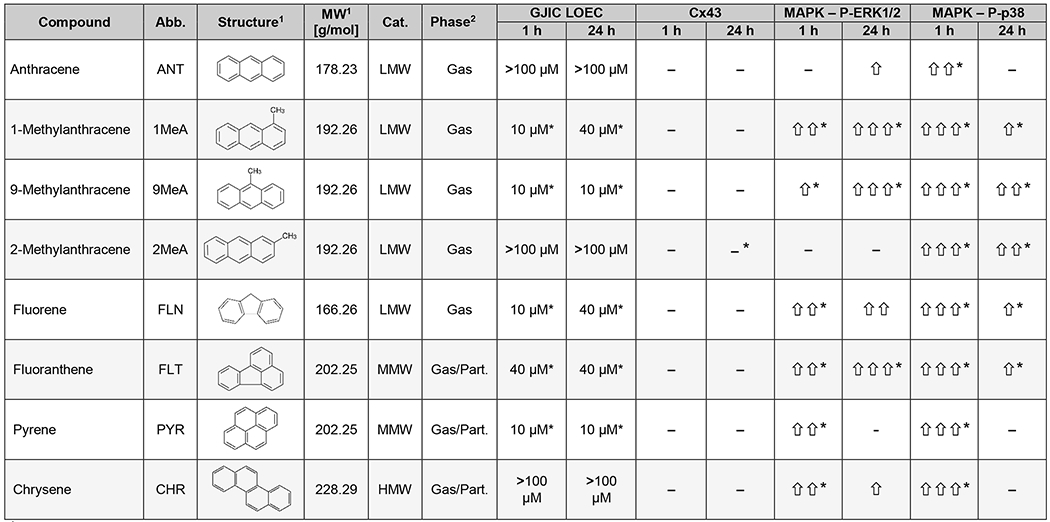 |
Information and chemical structures obtained from chemspider.com
Predicted values of gas/particle partitioning using EPISuite™ octanol/air model; values obtained from chemspider.com Changes in levels of specific proteins in response to 100 μM PAHs were detected by Western blotting: <1.5-fold change in FOC value (−); 1.5–1.99-fold change (⇧); 2.00–2.99-fold change (⇧⇧); >3.00-fold change (⇧⇧⇧). Statistically significant differences from the control are indicated by asterisks (*).
MW, molecular weight; LMW, low molecular weight; MMW, middle molecular weight; HMW, high molecular weight; Cat., category; Part., particle; GJIC, gap junctional intercellular communication; LOEC, lowest observable effect concentration; Cx43, connexin43; MAPK, mitogen-activated protein kinase; P-ERK1/2, phosphorylated extracellular signal-responsive kinases 1 and 2; P-p38, phosphorylated p38 kinase.
2. MATERIALS AND METHODS
2.1. Chemicals
1-Methylanthracene (1MeA, #333573), 2-methylanthracene (2MeA, #M29401), 9-methylanthracene (9MeA, #M29606), fluorene (FLN, #128333), fluoranthene (FLT, #F807), pyrene (PYR, #185515), chrysene (CHR, #245196) and 12-O-tetradecanolyphorbol-13-acetate (TPA, #P8139) were purchased from Sigma-Aldrich (Prague, Czech Republic). 5-Carboxyfluorescein diacetate acetoxymethyl ester (CFDA-AM, #C1354) and Alamar blue® (AB, #DAL1100) were obtained from ThermoFisher Scientific (Pardubice, Czech Republic). Neutral red solution (NR, #N6264) and Lucifer Yellow dilitihium salt (#L0259) were provided by Sigma-Aldrich. Recombinant human insulin (hINS, #I3536), dexamethasone (#D4902), transferrin (#T8158), recombinant human epidermal growth factor (hEGF, #E5036), cholera toxin (#C8052), bovine serum albumin (#A2058) and phenol red-free Dulbecco’s Modified Eagle Medium Nutrient Mixture F-12 (DMEM/F12, #D6434) were obtained from Sigma-Aldrich. DMEM/F12 with phenol red (#L00094) was obtained from Biowest (Nuaillé, France). Endothelial cell growth supplement (#C-30160) was provided by Promocell (Heidelberg, Germany), Plasmocin™ (#ant-mpt) by Invivogen (San Diego, CA). L-glutamine (#XC-T1715), trypsin-EDTA (#LM-T1705) and fetal bovine serum (FBS, #FB-1001) were purchased from Biosera (Nuaillé, France). Acetonitrile (MeCN, #20041-AT0), ethanol (EtOH, #20025-A96), acetic acid (#10047-A80), methanol (MeOH, #20025-A96) and formaldehyde (#40052-A48) were provided by Lach-ner (Neratovice, Czech Republic). Acrylamide/bis 40 % (w/v, 37.5:1, #1068103) and Tris base (#3719202) were obtained from Serva (Heidelberg, Germany), sodium dodecyl sulfate (SDS, #S1377) from Duchefa Biochemie (Haarlem, The Netherlands), dithiothreitol (#D9779) from Sigma-Aldrich, and tetramethylethylene-diamine (#17919) was purchased from ThermoFisher. 1MeA, 2MeA, 9MeA, FLN, FLT, PYR and CHR were dissolved in MeCN, TPA in EtOH, Lucifer Yellow and cholera toxin in phosphate-buffered saline (PBS), hINS in 100 mM acetic acid with 5 % (w/v) bovine serum albumin, hEGF, transferrin and dexamethasone in distilled water.
2.2. Cell culture
Non-cancerous human bronchial epithelial cell line HBE1 originates from non-cystic fibrosis lung tissue of a 60-year old woman. Cells were immortalized with Human Papilloma Virus-18 oncogenes E6 and E7 by Yankaskas et al. (1993). Cells were grown in a serum-free DMEM/F12 (with phenol red, 1.2 g/L sodium bicarbonate and 3.57 g/L HEPES, #L00094, Biowest) supplemented with 4 μg/mL hINS, 5 μg/mL transferrin, 10 ng/mL hEGF, 0.1 μM dexamethasone, 20 ng/mL cholera toxin, 2.5 μg/mL Plasmocin™, 2.5 mM L-glutamine and 0.1 % (v/v) endothelial cell growth supplement. After adding all the supplements, pH was adjusted to 7.40 ± 0.05 and the medium was sterile-filtered using 0.2 μm top-bottle polyethersulfone filter (TPP, Prague, Czech Republic, #99255) into sterile glass bottles. Cells were routinely cultured in 25 cm2 (#90026, TPP) or 75 cm2 (#90076, TPP) tissue culture-treated polystyrene flasks in humidified 5 % CO2 atmosphere at 37 °C. Cells were passaged twice a week using 0.5 mL trypsin-EDTA per 25 cm2 of growth area (GA). After 5 min of incubation (37 °C), trypsin activity was inhibited by the addition of 2.5 mL of 5 % (v/v) FBS in PBS per 25 cm2 GA. The cell suspension was mixed with 5 mL of tempered (37 °C) cultivation media per 25 cm2 GA and centrifuged at 50 g, room temperature for 3 min using sterile centrifuge tubes (TPP). The pellet was resuspended in 5 mL of tempered cultivation media. Cell concentration was measured using Cellometer Auto T4 (Nexcelom Bioscience, Lawrence, MA) prior to seeding cells into a new plasticware.
2.3. Experimental design
All experiments were conducted using the HBE1 cell line between passages 24 and 30. The concentration of cell suspension was adjusted to seed the cells at the density 8-9*104 cells/cm2. 200 μL/well of cell suspension was seeded into the 96-well black Cellstar® microplate with clear bottom (#655090, Greiner Bio-One, Kremsmünster, Austria) for viability assays, 1 mL/well into 24-well clear microplate Cellstar® (#662160, Greiner Bio-One) for GJIC assessment by scrape-load/dye transfer method (SLDT), and 2 mL/dish into 35 mm BioLite cell culture dishes (#130180, Thermo Scientific) for Western blot analyses. The cells were grown to confluency for 48 h prior to the addition of the tested chemicals. Tested chemicals were pipetted directly from stock solutions into each experimental well or dish, and the cells were exposed for 1 h or 24 h. Concentrations of PAHs were selected in the range of 10-100 μM (~2-20 μg/mL), which have been previously shown to have GJIC-inhibiting effects in different rodent cell lines (Blaha et al., 2002; Kubincová et al., 2019; Osgood et al., 2013; Weis et al., 1998). The color of phenol red pH indicator was visually monitored throughout the experiments to assure that pH values were within the physiological range, and no significant differences between the negative control and experimental treatments were observed. The concentration of solvent (vehicle) did not exceed 1 % (v/v), and corresponding vehicle control was run in parallel with negative control (non-treated cells) in each individual experiment. The results of each experiment were compared with negative control and expressed as a fraction of control (FOC). All the experiments were carried out in triplicates and repeated at least three times independently.
2.4. Cell viability assay
We utilized an assay combining three different dyes to evaluate impairment of different vital cellular processes: the integrity of plasma membrane and esterase activity by CFDA-AM, dehydrogenase activity by AB, and lysosomal uptake of NR, conducted according to Kubincová et al. (2019). Briefly, exposed cells were washed with PBS and incubated for 30 min in 100 μL of unsupplemented, phenol red-free DMEM/F12 medium (#D6434, Sigma Aldrich) with 5 % (v/v) AB and 4 μM CFDA-AM. After incubation, the cells were rinsed with PBS and analyzed using FLUOstar Optima (BMG Labtech, Ortenberg, Germany) by measuring fluorescence at 530/590 nm (ex/em) and 485/520 nm (ex/em), respectively. Afterward, the cells were washed with PBS prior to 90 min incubation in 100 μL phenol red-free DMEM/F12 medium with 50 μg/mL NR. After incubation, the cells were rinsed with PBS and NR accumulated by the viable cells was extracted by 1 % (v/v) acetic acid in 50 % (v/v) EtOH. NR uptake (NRU) was quantified by measurement of absorbance using POLARstar Optima (BMG Labtech) at 540 nm and reference wavelength 690 nm.
2.5. Assessment of GJIC
Original SLDT assay (El-Fouly et al., 1987) to assess was modified according to recently published protocols (Babica et al., 2016a; Upham et al., 2016). Shortly, exposed cells were rinsed by PBS supplemented with 0.68 mM calcium chloride and 0.49 mM magnesium chloride (CaMgPBS). The solution of 1 mg/mL Lucifer Yellow dilithium salt in PBS was added to the cells and introduced to the monolayer by three parallel cuts with a stainless steel blade (#17, Proedge Blades, Paterson, NJ). After 6 min of incubation, the cells were washed with CaMgPBS and fixed with 4 % (v/v) formaldehyde solution in PBS. Images from Z1 Axio Observer microscope (Carl Zeiss, Jena, Germany) were obtained with 10× objective using ZEN 2.3. blue edition software (Carl Zeiss). The spread of Lucifer Yellow was evaluated using ImageJ (Schneider et al., 2012) as a diffusion area of the fluorescent dye. The model inhibitor of GJIC, TPA, was used as a positive control.
2.6. Protein extraction and Western blotting
After exposure, the cells were rinsed with PBS and proteins extracted with 150 μL of 20 mM Tris base with 4 % (w/v) SDS and 1 mM dithiothreitol (pH adjusted to 7.6 with hydrochloric acid). Lysed cells were scraped and homogenized using Sonopuls Mini20 sonicator (Bandelin, Berlin, Germany). Protein concentration was measured using the Bio-Rad DC™ protein assay (#5000111, Bio-Rad Laboratories, Prague, Czech Republic,). Protein lysates were subsequently diluted to concentration 1 mg/mL with lysis buffer and 4× Laemmli sample buffer (#1610747, Bio-Rad). Afterward, proteins (10 μg) were separated by SDS polyacrylamide gel electrophoresis (SDS/PAGE) on 12.5 % polyacrylamide gels and electrophoretically transferred to Immobilon-P membrane (Merck Millipore, Prague, Czech Republic) using Mini-Protean® Electrophoresis system (Bio-Rad). Membranes were incubated with the blocking solution (5 %, w/v, low-fat dry milk in Tris-buffered saline with 0.1 %, v/v, Tween 20 [TBS-T]) and subsequently incubated overnight at 4 °C with primary antibodies diluted in blocking solution. All primary and secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA), if not specified otherwise. Following primary antibodies and dilution factors were used – 1:1000 diluted polyclonal rabbit antibody Phospho-p44/42 MAPK ERK1/2 (#4370S), Phospho-p38 MAPK (#4511S), p44/42 MAPK ERK1/2 (#4695), p38 MAPK (#8690S), and 1:4000 diluted polyclonal rabbit antibody against Cx43 (Sigma-Aldrich, #C6219). After the washing step, membranes were incubated for 1 h at room temperature with 1:2500 diluted goat polyclonal horseradish peroxidase-conjugated Anti-Rabbit IgG (#7074S). Secondary antibodies were detected using Clarity™ Western Blotting ECL Substrate (Bio-Rad, #170-5060) and MF-ChemiBis 3.2 documentation system with GelCapture software (DNR Bio-Imaging Systems, Neve Yamin, Israel). Membranes were then washed in TBS-T and re-probed (overnight, 4 °C) with the 1:2000 diluted monoclonal mouse antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Merck Millipore, #MAB374). Membranes were subsequently incubated in 1:2500 diluted horse polyclonal radish peroxidase-conjugated Anti-Mouse IgG (#7076S) secondary antibody for 1 h at room temperature and quantified. WB-F344 epithelial liver cell line w/ or w/o TPA treatment was used as a positive control in each experiment. Densitometric evaluation of Western blot bands was done using ImageJ software. Prior FOC calculation, densitometric data for each band were normalized to GAPDH signal (i.e. the loading control) from the corresponding lane.
2.7. Statistical analyses
FOC data obtained from at least three times independently repeated experiments were combined to calculate the mean ± standard error of the mean (SEM) values, which are presented in the graphs. For data with a normal distribution (Shapiro-Wilk’s Test) and equal variances (Equal Variance Test), statistical differences from the vehicle control were determined by ANOVA followed by Dunnet’s post-hoc test (multiple comparisons) or t-test (two groups comparison). Data with unequal variances were evaluated using the non-parametric Mann-Whitney U test. P values <0.05 were considered significantly different. All calculations were done in SigmaPlot 11.0 (Systat Software, Erkrath, Germany).
3. RESULTS
3.1. Effects of PAHs on cell viability
Potential cytotoxic effects of PAHs at selected concentration range (10-100 μM) were evaluated after 24-h exposure using a combination of three viability dyes, CFDA-AM, AB and NR (Fig. 1). According to the CFDA-AM and AB assays, tested PAHs caused none or only a minor decrease in cell viability, with the results being within +/− 20% of the control and mostly statistically insignificant (Fig. 1A–B). According to NRU results, the exposure to PAHs caused some increases in the cellular uptake of NR, which became the most pronounced (23-31% increase) in response to the treatment by 2MeA (10-40 μM) and FLN (10 μM; Fig. 1C). Concentrations of PAHs used for further experiments thus induced only slight variations in cell viability without a clear concentration- or structure-dependent pattern that would be consistently observable across the three evaluated viability endpoints.
Fig. 1.
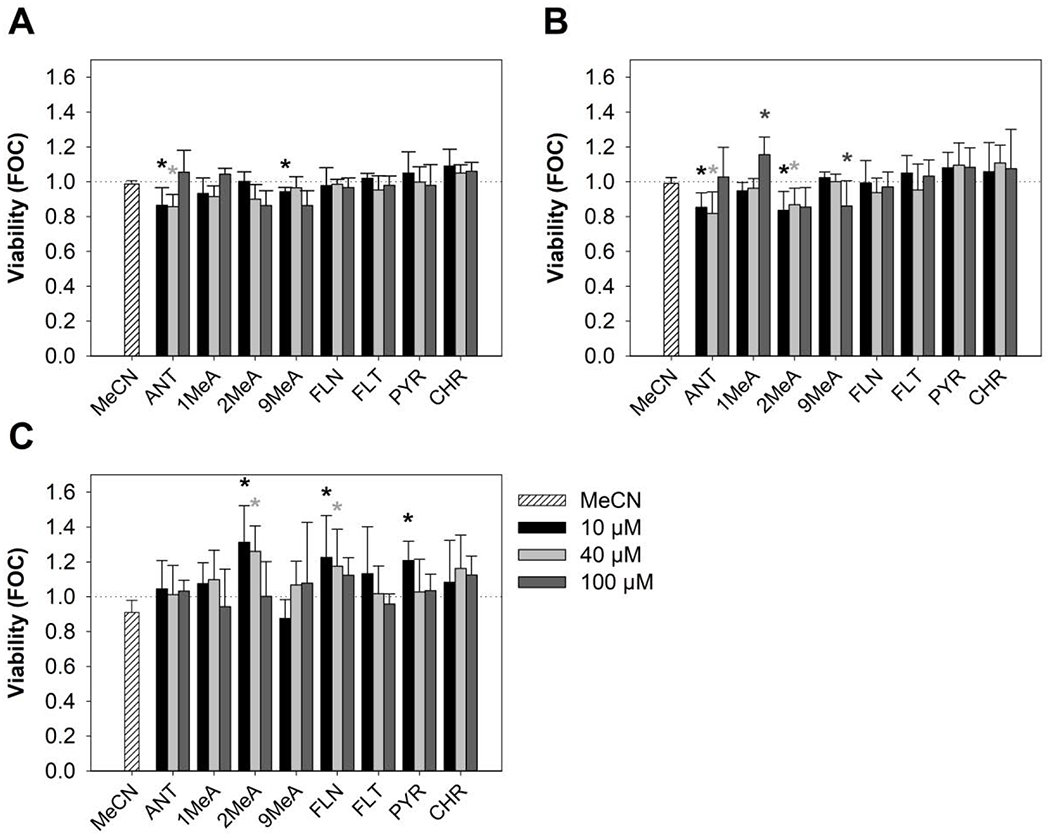
Effects of polycyclic aromatic hydrocarbons (PAHs) on the viability of HBE1 cells. The combination of three different viability dyes was used after 24-h exposure to different concentrations (10 μM, 40 μM, 100 μM) of selected toxicants - (A) 5-carboxyfluorescein diacetate acetoxymethyl ester (CFDA-AM), (B) Alamar blue® (AB) and (C) Neutral red uptake (NRU). Values are plotted as a fraction of negative control (FOC=1, indicated by the dotted line); bars represent mean values ± standard error of the mean of at least 3 independent experiments conducted in triplicate. Asterisks indicate significantly different values from negative control (ANOVA followed by Dunnett’s post-hoc test, p < 0.05). MeCN, acetonitrile vehicle control; ANT, anthracene; 1MeA, 1-methylanthracene; 2MeA, 2-methylanthracene; 9MeA, 9-methylanthracene; FLN, fluorene; FLT, fluoranthene; PYR, pyrene; CHR, chrysene.
3.2. Inhibition of GJIC
The reference tumor promoter and GJIC inhibitor, phorbol ester TPA, showed a characteristic concentration-dependent inhibitory effect on GJIC after both 1-h and 24-h exposure (Appendix Fig. A.1). Following 1 h exposure, 10 nM TPA elicited slight (app. 20%), but statistically significant GJIC inhibition, and represented the lowest observed effect concentration (LOEC). The effects of TPA became more pronounced with the increasing concentration up to 200 nM, where the levels of GJIC were reduced to ~0.5 FOC. The inhibitory effects of TPA increased with exposure prolongation. After 24 h, a statistically significant decrease in cell-cell communication to ~0.5 FOC was observed even at a low concentration of 5 nM (LOEC). Concentrations >10 nM induced almost complete GJIC reduction corresponding to FOC values ~0.2-0.3.
1MeA, 9MeA, FLN, FLT and PYR induced a statistically significant decrease of GJIC after 1-h exposure in a concentration-dependent manner (Fig. 2). Following 1-h exposure, GJIC-inhibiting L/MMW PAHs elicited a statistically significant decrease in cell-cell communication at concentrations ≥ 10 μM (LOEC=10-40 μM), with GJIC reduced to ≤0.8 FOC. The effects became more pronounced with increasing concentration until a plateau was observed at 100 μM with GJIC levels reduced to ~0.5 FOC (Fig. 2A and B). In contrast, linear LMW PAHs without bay or bay-like regions (ANT, 2MeA) or HMW CHR did not induce any major and statistically significant effects on GJIC (LOEC>100 μM; Fig. 2), neither after 1-h nor 24-h exposure. Concentrations >100 μM did not elicit greater inhibition of GJIC for any of the studied PAHs (data not shown). The effects of GJIC-inhibiting PAHs after 24-h exposure were sustained and mostly comparable to 1-h exposure (Fig. 2C and 2D). Only 9MeA was found to induce significantly stronger inhibition of GJIC after prolonged exposure, with GJIC levels reduced to 0.2 FOC after 24 h. Representative photos from GJIC evaluation after 1 h and 24-h exposure are shown in Fig. 2E and F, LOEC values are summarized in Table 1.
Fig. 2.
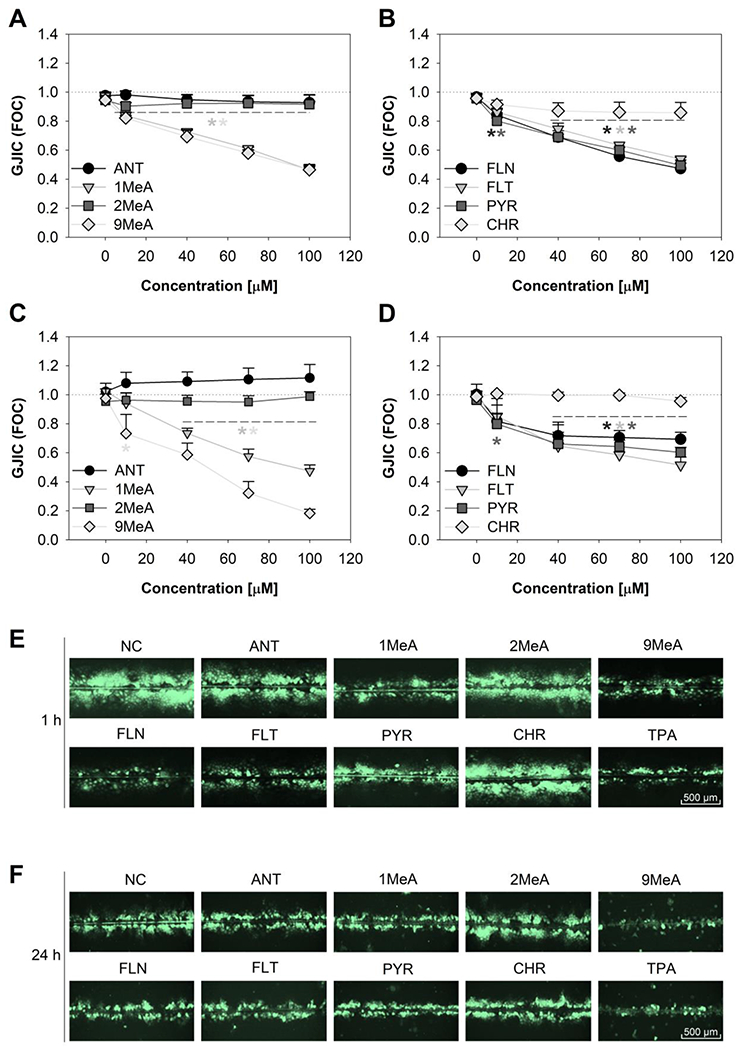
Time- and concentration-dependent inhibition of gap junctional intercellular communication (GJIC) in HBE1 cells after exposure to polycyclic aromatic hydrocarbons (PAHs). Scrape-load/dye transfer method was conducted after (A, B) 1-h and (C, D) 24-h exposure to different concentrations (10 μM, 40 μM, 70 μM, 100 μM) of selected toxicants. Representative pictures of communication cells after (E) 1-h and (F) 24-h exposure to 100 μM of PAHs or 200 nM of TPA. Values are plotted as a fraction of negative control (FOC=1); data represent mean values ± standard error of the mean of at least 3 independent experiments conducted in triplicate. Asterisks indicate significantly different values from the vehicle control (ANOVA followed by Dunnett’s post-hoc test, p < 0.05). GJIC, gap junctional intercellular communication; FOC, fraction of control; NC, negative control; ANT, anthracene; 1MeA, 1-methylanthracene; 2MeA, 2-methylanthracene; 9MeA, 9-methylanthracene; FLN, fluorene; FLT, fluoranthene; PYR, pyrene; CHR, chrysene.
3.3. Effects of PAHs at the molecular level
3.3.1. Cx43 phosphorylation and protein levels
Due to the observed inhibition of GJIC by PAHs, the effects of both 1-h and 24-h exposure on the levels of the major gap junctional protein connexin43 (Cx43) were examined using the Western blot technique (Fig. 3). The concentration of 100 μM, where the most significant effects on GJIC were observed without inducing cytotoxicity, was selected for the experiments. No statistically significant effects of vehicles (MeCN, EtOH) were observed following both 1-h and 24-h exposures (Appendix Fig. A.2). Cx43 was detected as multiple bands around molecular weight 43 kDa representing differentially phosphorylated Cx molecules. GJIC inhibitor TPA is known to induce hyperphosphorylation of Cx43, which typically leads to a shift in the electrophoretic mobility of Cx43 bands detectable by Western blot analysis, as demonstrated in rat liver epithelial cells WB-F344 (Appendix Fig. A.3). Interestingly, no apparent changes in the phosphorylation status of Cx43 in HBE1 or any major changes of total levels of Cx43 were detected in response to PAHs (Fig. 3), while TPA induced only a slight non-significant decrease of Cx43 protein levels after prolonged 24-h exposure.
Fig. 3.
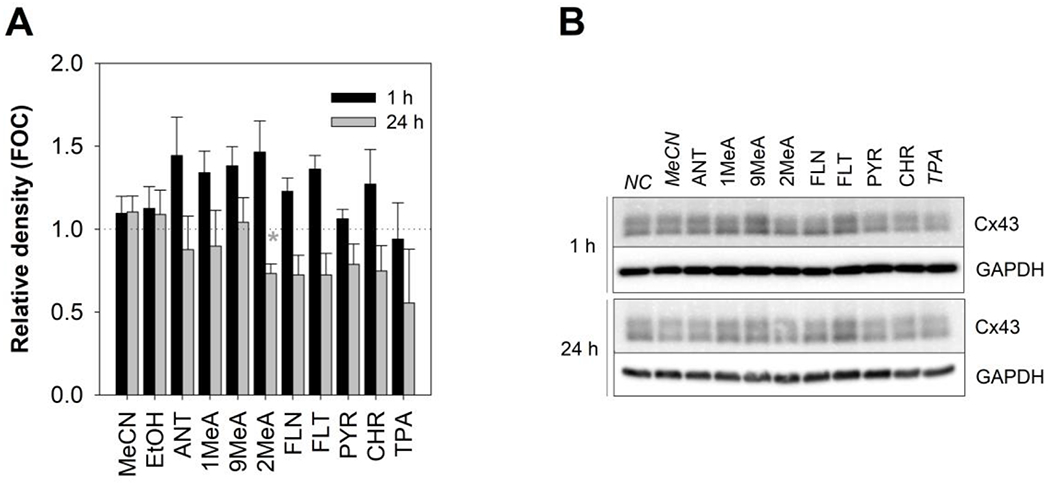
Effects of polycyclic aromatic hydrocarbons (PAHs) on gap junctional protein connexin43 (Cx43) in HBE1 cells. Cx43 levels were evaluated by Western blotting conducted after 1-h and 24-h exposure to 100 μM concentration of selected PAHs and 200 nM TPA (model inhibitor of GJIC) respectively. (A) Graph presents the results of densitometric evaluation of the intensity of Cx43 bands; (B) representative immunoblots are shown on the right. The intensity of Cx43 bands was normalized to the protein loading control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), compared to the negative control (NC) and expressed as a fraction of negative control (FOC =1, indicated by the dotted line). Bars represent mean FOC values ± standard error of the mean of at least 3 independent experiments. Asterisks indicate significantly different values from the vehicle control (t-test or Mann-Whitney test, p < 0.05). FOC, fraction of control; NC, negative control; MeCN, acetonitrile vehicle control; EtOH, ethanol vehicle control; ANT, anthracene; 1MeA, 1-methylanthracene; 2MeA, 2-methylanthracene; 9MeA, 9-methylanthracene; FLN, fluorene; FLT, fluoranthene; PYR, pyrene; CHR, chrysene; TPA, 12-O-tetradecanolyphorbol-13-acetate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Cx43, connexin43.
3.3.2. MAPK-(phospho)ERK1/2 and MAPK-(phospho)p38
Since GJIC inhibition by L/MMW PAHs has been previously associated with the activation of MAPKs (Osgood et al., 2013; Upham et al., 2008), we investigated the effect of PAH exposure (100 μM) on the levels of both phosphorylated (i.e. activated) MAPK-ERK1/2 (P-ERK1/2) and MAPK-p38 kinases (P-p38; Fig. 4). No statistically significant effects of vehicles (MeCN, EtOH) on both phosphorylated and unphosphorylated ERK1/2 and p38 were observed following both 1-h and 24-h exposure (Appendix Fig. A.4). None of the treatment had a major effect on total-ERK1/2 and total-p38 (Fig. 4), thus changes in the levels of phosphorylated, i.e. activated, kinases resulted specifically from altered phosphorylation. Both phosphorylated and unphosphorylated ERK1/2 were detected in HBE1 cells as a major band at molecular weight 42 kDa, and a relatively faint band of 44 kDa respectively. GJIC inhibitor TPA known to induce hyperphosphorylation of ERK1/2, as demonstrated in rat liver epithelial cells WB-F344 (Appendix Fig. A.5) induced similar effect in HBE1 cells.
Fig. 4.
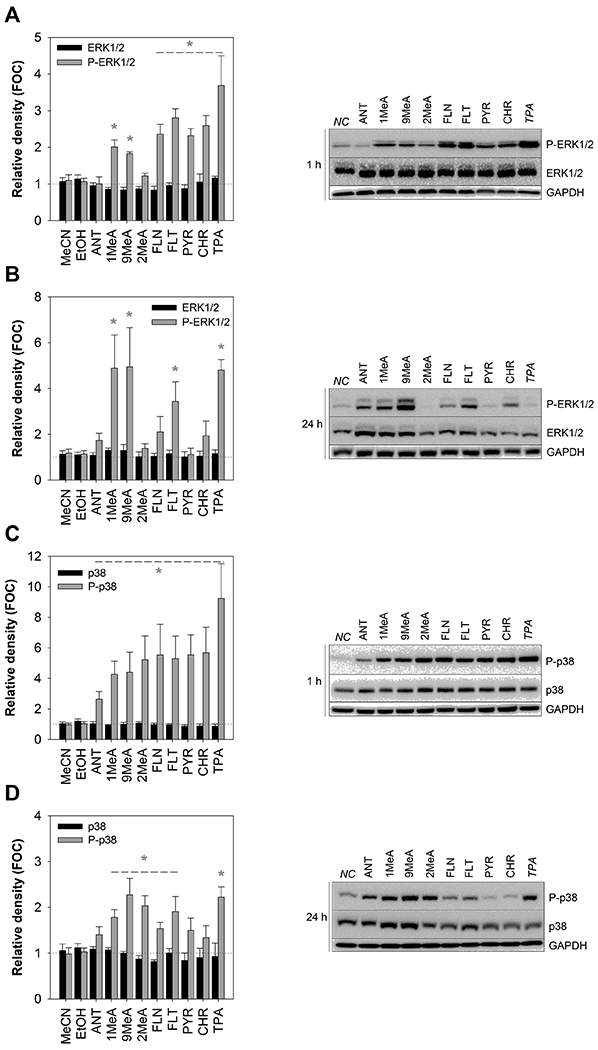
Effects of polycyclic aromatic hydrocarbons (PAHs) on the activation of mitogen-activated protein kinases - extracellular signal-responsive kinases 1/2 (MAPK-ERK1/2) and p38 (MAPK-p38) in the HBE1 cells. Western blotting method was conducted after 1-h and 24-h exposure to 100 μM concentration of selected PAHs and 200 nM TPA to assess the phosphorylation of ERK1/2 after (A) 1-h and (B) 24-h exposure, as well as p38 after (C) 1-h and (D) 24-h exposure. Chemiluminescence-based bands show phosphorylated/total-ERK1/2 (P-/ERK1/2; A-B) and phosphorylated/total-p38 (P-/p38; C-D) after both 1-h and 24-h exposure as well as a protein loading control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Values are plotted as a fraction of negative control (FOC=1); bars represent mean values ± standard error of the mean of at least 3 independent experiments. Asterisks indicate significantly different values from the vehicle control (t-test or Mann-Whitney test, p < 0.05). FOC, fraction of control; NC, negative control; MeCN, acetonitrile vehicle control; EtOH, ethanol vehicle control; ANT, anthracene; 1MeA, 1-methylanthracene; 2MeA, 2-methylanthracene; 9MeA, 9-methylanthracene; FLN, fluorene; FLT, fluoranthene; PYR, pyrene; CHR, chrysene; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TPA, 12-O-tetradecanolyphorbol-13-acetate; P-p38, phosphorylated-p38 kinase; P-ERK1/2, phosphorylated extracellular signal-responsive kinases 1/2.
After 1-h exposure, increased phosphorylation of ERK1/2 was also observable following the treatment with GJIC-inhibiting PAHs 1MeA, 9MeA, FLN, FLT and PYR, but also CHR. On the other hand, ANT and 2MeA, did not induce major effects on ERK activation after 1-h (Fig 4A). Following 24-h exposure, the activation of ERK1/2 by 1MeA, 9MeA and FLT further increased (Fig. 4B), whereas effects of FLN, PYR and CHR on ERK1/2 activity rather decreased or diminished. Significant activation of p38 was observed after a 1-h exposure to PAHs and TPA (Fig. 4C). A relatively weaker response was induced by ANT, while more pronounced effects of the other tested PAHs were comparable, regardless of their structure or GJIC-inhibiting activity. After 24-h of exposure, the effects of PAHs on p38 activation were less apparent, with the strongest responses observed to methylated anthracenes and FLT (Fig. 4D).
4. DISCUSSION
Inhalation of air contaminated with combustion products, such as diesel exhaust fumes or cigarette smoke, represent a major human exposure route to PAH mixtures (Boström et al., 2002; Snook et al., 1976; Gad and Gad, 2014; Stogiannidis and Laane, 2015). L/MMW PAHs are higly prevalent in these mixtures, with two- and three-ringed LMW PAHs occuring mostly in a gas phase, four-ringed MMW PAHs present in both particulate and gas phases, while less abundant HMW PAHs with ≥4 rings found mostly adsorbed to particulate matter (Baek et al., 1991).
Although relatively less researched in comparison with genotoxic HMW PAHs (Lewtas, 2007), L/MMW PAHs have been previously found to inhibit GJIC and activate MAPKs in various rodent cell lines (Blaha et al., 2002; Kubincová et al., 2019), including mouse lung epithelial cells C10 (Bauer et al., 2018; Osgood et al., 2017, 2013; Romo et al., 2019; Siegrist et al., 2019).GJIC is a crucial process for maintaining cell and tissue homeostasis, and GJIC inhibition has been identified as one of the key events in tumor-promotion phase of cancer and one of the main modes of action of nongenotoxic carcinogens (Nahta et al., 2015; Trosko and Ruch, 1998). Mice heterozygous for a Cx43 deficiency developed more lung tumors than their wildtype counterparts (Avanzo et al., 2004). Aberrant Cx43 expression was observed in non-small cell lung cancer patients (Chen et al., 2003). In human and rodent lung cancer cells, GJIC levels and Cx expression is typically reduced, while ectopic Cx expression restores not only cell-cell coupling but also more normal phenotype (Ruch, 2019). In addition to the regulation of the cell growth and differentiation, GJIC in the lung epithelium participates in the coordination of ciliary beating, regulates pulmonary surfactant, physiological Ca2+ signaling, maintains homeostasis of air surface liquid volume, and alters the lung inflammatory response (Johnson and Koval, 2009; Parthasarathi et al., 2006; Scheckenbach et al., 2011; Willebrords et al. 2016). Decreased expression of Cx43 and gap junction activity was observed in lung fibroblasts of idiopathic pulmonary fibrosis patients (Trovato-Salinaro et al., 2006). Additionally, dysregulation of GJIC and/or Cx expression in human airway system has been found to negatively correlate with various diseases, such as airway inflammation (Ram et al., 2009; Willebrords et al. 2016), pulmonary arterial hypertension (Freund-Michel et al., 2016), acute lung injury (Zhang et al., 2010) and chronic pulmonary diseases (Wong et al., 2017).
Although dysregulation of GJIC by inhaled PAHs could potentially impair several physiological processes contributing to acute and chronic respiratory diseases, including cancer, PAHs effects on GJIC in human airway epithelial cells have not been previously characterized. In our study, we found that selected environmentally relevant L/MMW PAHs induced rapid inhibition of GJIC in human, non-cancerous immortalized bronchial epithelial cell line, HBE1. L/MMW PAHs 1MeA 9MeA, FLN, FLT and PYR, but not 2MeA and ANT, elicited statistically significant GJIC inhibition in HBE1 cells at concentrations ≥ 10 μM within 1-exposure, and it lasted for at least 24 h, which is mostly consistent with the previous findings from rodent cell lines, e.g. rat liver epithelial WB-F344 cells, mouse lung epithelial C10 cells, or mouse Leydig TM3 cells (Blaha et al., 2002; Kubincová et al., 2019; Osgood et al., 2013).
Similar structure-dependent activity was observed in WB-F344 cells, where L/MMW PAHs possessing bay or bay-like structural region (1MeA, 9MeA, FLT), as well as FLN and PYR, reached the maximal levels of GJIC inhibition (~0.25-0.45 FOC) within 30 min at concentrations of 30-80 μM, while linearly arranged LMW without a bay/bay-like region (2MeA, ANT) did not affect GJIC (Blaha et al., 2002; Rummel et al., 1999; Upham et al., 1998, 1996; Weis et al., 1998). HMW PAHs with a higher molecular weight and higher lipophilicity, such as CHR, had only weak and transient effect on GJIC in WB-F344 cells (Blaha et al., 2002), which was also observed in our study in HBE1 cells. These structure-dependent effects of PAHs are also supported by Osgood et al. (2017, 2013) who reported inhibition of GJIC in mouse lung epithelial C10 cells by 0.5-24-h exposure to 1MeA and FLT (≥10 μM), but not by 2MeA. Similarly, GJIC was inhibited by 1MeA, 9MeA, FLN and FLT (≥12.5 μM, 0.5-24 h) in mouse Leydig TM3 cells, whereas none or a weak effect was induced by ≤200 μM ANT, 2MeA or B[a]P (Kubincová et al., 2019). However, the inhibition of GJIC induced by FLN in TM3 cells was only transient in contrast to the other GJIC inhibiting PAHs. Moreover, no inhibition of GJIC (≤200 μM, 0.5-24 h) was induced by the same PAHs in the mouse Sertoli TM4 cell line (Kubincová et al., 2019). In addition, B[a]P was found to be a weak inhibitor of GJIC also in WB-F344 (Blaha et al., 2002), but potently inhibited GJIC in C10 cells (≥1-10 μM) after prolonged 4-24 h exposures (Bauer et al., 2018). Apparently, the effects of L/MMW PAHs on GJIC might be not only structure-specific but also cell type-specific and possibly species-specific.
Importantly, these concentrations of L/MMW PAHs inducing GJIC inhibition and/or MAPK activation in HBE1 cells (10-100 μM, i.e. ~2-20 μg/mL) are in the range used in other in vitro studies with human bronchial cells, where toxic PAH effects were reported for concentrations between 1-500 μg/mL or 1-200 μM (Abbas et al., 2019; Cervena et al., 2017; Chang et al., 2019). In agreement with previous studies (Blaha et al., 2002; Kubincová et al., 2019; Zhou et al., 2016), no major cytotoxic responses were detected up to 100 μM concentrations of the tested PAHs in HBE1, which indicates that GJIC inhibition was probably not caused by general cellular response to membrane damage or cytotoxic stress. Since membrane and intracellular accumulation of PAHs by Cx43-expressing TM3 and TM4 cells did not correlate with the inhibition of GJIC (Kubincová et al., 2019), the effects of PAHs on GJIC do not seem to be caused by their direct interaction with Cx proteins. Moreover, GJIC dysregulation by selected PAHs has been shown to depend on activation of several signal transduction pathways in different cell lines (Kubincová et al., 2019; Osgood et al., 2013; Upham et al., 2008), which indicates that the effects on GJIC are mediated rather indirectly by modulation of cellular processes and pathways regulating GJIC. GJIC is regulated at different levels, including quality and quantity of Cx channels (via control of Cx transcription, translation, maturation of Cx proteins, their membrane localization, and degradation), and also by regulation of Cx channel permeability, e.g. by voltage-dependent gating, chemical gating due to interactions between Cx proteins and other molecules, or by post-translational modifications of Cxs (Nielsen et al., 2012; Axelsen et al., 2013). In our study, rapid dysregulation of GJIC occurred most likely through gating mechanisms, while 24-h exposure could involve other levels of GJIC control. The lack of significant reductions of Cx43 protein levels by PAHs does not suggest major changes in transcription, translation and degradation rate of Cx43, but it cannot be excluded that longer PAH exposures could affect Cx fate without changes in its steady-state protein levels, e.g. by alterations of its maturation, trafficking and turnover rates (Kubincova et al., 2019). Phosphorylation of Cx43 at multiple phosphosites, which are targeted by intracellular protein kinases (e.g. MAPK/ERK1/2, protein kinase C, src), represents major post-translational modification and well-recognized mechanism controlling both channel gating and Cx43 fate (Husøy et al., 2001; Musil and Goodenough, 1991; Sirnes et al., 2009). For example, TPA activates protein kinase C and MAPK ERK1/2 which hyperphosphorylate Cx43, as indicated by a shift in the electrophoretic mobility of Cx bands, leading to GJIC inhibition, Cx internalization and degradation (Husøy et al., 2001). Such TPA-induced GJIC-inhibition associated with hyperphosphorylation of Cx43 was detected in WB-F344 (Fig A.3) or C10 cells (Osgood et al., 2013), while no significant shift of Cx43 bands and only a weak reduction of total Cx43 levels was caused by TPA in HBE1 cells. A similar response was recently reported also for TM3 cells, where TPA rapidly inhibited GJIC without changes in the Cx43 phosphorylation pattern, with a reduction of total Cx43 levels occurring after 24-h treatment (Kubincová et al., 2019). Similarly, GJIC-inhibiting PAHs did not affect Cx43 levels or electrophoretic mobility in HBE1 cells, as well as in WB-F344 cells, C10 cells or TM3 cells (Kubincová et al., 2019; Osgood et al., 2013; Upham et al., 2008). A reduction of Cx43 protein levels in response to PAHs was reported only during longer (≥2 h) exposures of C10 (Bauer et al., 2018; Osgood et al., 2017, 2013), or TM3 cells (Kubincová et al., 2019). Considering the fact that GJIC-inhibiting L/MMW PAHs also rapidly activated MAPK ERK1/2 and p38 in HBE1 cells, future research should evaluate the possibility that protein kinases activated by L/MMW PAHs could alter one or a few specific phosphosites of Cx43 protein and thus affect GJIC function without any detectable shifts in Cx43 electrophoretic mobility and/or decrease of Cx43 protein levels. In previous studies, L/MMW PAHs were not found to activate ERK1/2 and p38 in TM3 cells (Kubincova et al., 2019), however, ERK1/2 and p38 were specifically activated by GJIC inhibiting L/MMW PAHs and not by 2MeA or ANT in WB-F344 or C10 cells (Osgood et al., 2017, 2013; Rummel et al., 1999; Upham et al., 2008). Similarly in HBE1, the pattern of ERK1/2 activation mostly corresponded to GJIC inhibiting activity, as the most pronounced and sustained ERK1/2 activation was caused by the most potent GJIC inhibitors with the bay or bay-like region (i.e. 1MeA, 9MeA, and FLT), while non-inhibiting PAHs (ANT, 2MeA) did not significantly activate ERK1/2. However, the activation of p38 in HBE1 cells occurred without a clear relationship of the PAHs to their GJIC-inhibiting potency, in contrast to WB-F344 or C10 cells (Osgood et al., 2017, 2013; Rummel et al., 1999; Upham et al., 2008). Collectively, the effects of PAHs at a cellular and molecular level can be tissue-, cell type- and possibly species-specific, which also applies to the role of MAPKs in PAH-induced dysregulation of GJIC. For example, blocking the activity of ERK1/2 in TM3 cells attenuated PAH effects on GJIC (Kubincova et al., 2019). In contrast, L/MMW PAH-induced inhibition of GJIC was independent of both ERK1/2 or p38 activity in WB-F344 cells, but it was dependent on phospholipase PC-PLC and partially PLA2 (Upham et al., 2008; Sovadinova et al., 2015). MAPK-independent activation of PC-PLC was found to be the key regulator of GJIC in response to several other environmental toxicants (Babica et al., 2016b; Sovadinova et al., 2015). Inhibitors of PLA2, p38, and an anti-inflammatory agent, were found to attenuate dysregulation of GJIC by 1-MeA, FLT, or their binary mixture, in C10 cells (Osgood et al., 2017, 2013; Romo et al., 2019; Siegrist et al., 2019). Obviously, additional mechanisms of GJIC control, other than MAPK-dependent Cx phosphorylation, can participate in PAH-induced dysregulation of GJIC, probably in a cell type-specific manner. Thus, future mechanistic studies will be needed to decipher the exact mechanism(s) of GJIC dysregulation in the response of human bronchial epithelial cells to PAH exposures, including the role of specific kinases and phosphosites, other post-translational modifications of Cx proteins (e.g. nitrosylation, hydroxylation, acetylation, methylation, γ-carboxyglutamation, ubiquitination or SUMOylation), as well as alterations of other signaling pathways and cellular processes known to regulate Cx and GJIC, such as changes in calcium concentrations, membrane potential and fluidity, intracellular pH, or interactions of Cxs with other proteins (Nielsen et al. 2012; Axelsen et al., 2013). Importantly, connections between PAH-induced oxidative stress and GJIC inhibition should be examined, as PAH are known to induce oxidative stress and related responses in airway epithelial cells (Abbas et al., 2019; Osgood et al., 2017), and alterations in redox potential are known to affect Cxs and GJIC (Nielsen et al. 2012; Axelsen et al., 2013). Moreover, eventual effects of PAHs on other Cxs should be investigated, since e.g. Cx26 or Cx32 have been also detected in human bronchial epithelial tissue along with the most prominent Cx43 (Losa et al., 2011; Willebrords et al., 2016).
In addition to GJIC, the activation of MAPKs observed in human bronchial epithelial cells indicate another important nongenotoxic mechanism which might contribute to PAH-induced respiratory toxicities and diseases. MAPKs ERK1/2, p38 or JNK can phosphorylate, depending on the conditions and cell type, up to several hundreds of downstream intracellular protein targets, localized in different cellular compartments, such as cytoplasm (e.g. MAP Activated Protein Kinases), cytoplasmic organelles, various membranes (e.g. Cxs), and particularly in the nucleus, where they modulate the activity of transcription factors and chromatin remodeling enzymes (Plotnikov et al., 2011). MAPK pathways ERK1/2, p38 and JNK play a complex role in the control of cellular processes and behavior, including proliferation, differentiation, apoptosis and stress responses (Plotnikov et al., 2011). Thus, dysregulation of these pathways is involved in the induction and progression of various chronic diseases, including lung cancer, asthma or chronic obstructive pulmonary disease (Qian et al., 2016), which have been also linked to PAH exposures (Crotty Alexander et al., 2015; Goldizen et al., 2016; Yang et al., 2017). In rodent experimental models, increased levels of MAPK activity were involved in cigarette smoke-induced lung epithelial-mesenchymal transition (Liang et al., 2018), epithelial hyperplasia and squamous metaplasia (Du et al., 2017; Zhong et al., 2005), inflammation, oxidative stress and injury (Li et al., 2018), and mucus hypersecretion (Wang et al., 2018). Activation of MAPKs was also associated with tumor-promoting, inflammatory and stress responses in various human bronchial or alveolar in vitro models exposed to complex PAH mixtures, such as cigarette smoke (Luppi et al., 2005; Xu et al., 2015; Yu et al., 2011), diesel exhaust extracts (Bonvallot et al., 2001; Dornhof et al., 2017; Kawasaki et al., 2001; Libalova et al., 2016; Totlandsdal et al., 2015) or fine particulate matter from the combustion (Corsini et al., 2013; Dornhof et al., 2017). However, the contribution of individual PAHs present in the complex mixtures to activation of MAPKs in human bronchial or pulmonary cells is not clear, as most of the studies focused on MAPK-activating effects of HMW PAH B[a]P and its metabolites (Chuang et al., 2008; Ding et al., 2009; Ko et al., 2008; Vázquez-Gómez et al., 2018).
Previous studies reported MAPK activation by L/MMW PAHs in various rodent cell lines, including mouse lung epithelial cells C10 (Osgood et al., 2013, 2017; Siegrist et al., 2019). Our study newly found that GJIC-inhibiting L/MMW PAHs activate ERK1/2, and all studied 2-4-ring PAHs activate p38 in human bronchial epithelial cells HBE1. As discussed above, the structure-dependent effects of L/MMW PAHs on GJIC as well as MAPKs appear to be cell type-, tissue- or species-specific, and interspecific differences in cell biology have been recognized in general as a factor that can limit the translation of biological perturbations observed in rodent airway epithelial cells to humans (Rhrissorrakrai et al., 2015). Thus it is critical to use in vitro models providing the desired biological context in terms of target species, tissue and cell type. Since the effects of L/MMW PAHs on GJIC and/or MAPKs have been so far only rarely studied in human cells (Tai et al., 2007), our study brings important information that these cellular responses are relevant also for human airway epithelial cells, which underlines the need to further investigate human health effects and risks related to inhalation exposures to L/MMW PAHs.
5. CONCLUSIONS
Our study demonstrates that processes connected to neoplastic and inflammatory diseases, such as inhibition of GJIC and activation of MAPKs, can be altered within a human airway epithelium by L/MMW PAHs. Even though evaluation of these critical cellular events in an immortalized human cell line can serve as a valuable tool for hazard identification and toxicity assessment, future studies should further address tumor-promoting, stress and inflammatory events downstream of GJIC inhibition and MAPK activation in response to L/MMW PAHs. This should include validation of these effects, for example, by using primary cells or more advanced 3D cultures or co-cultures of an airway epithelium (Chang et al., 2019; Klein et al., 2013; Miller and Spence, 2017). Less is known about L/MMW PAHs due to their lack of or weak genotoxicity, but they represent a highly relevant and abundant fraction in the complex, toxic and carcinogenic PAH mixtures, such as polluted air, cigarette smoke or diesel exhaust fumes (Bauer et al., 2018; Birrer et al., 1992; Nesnow et al., 1985; Stogiannidis and Laane, 2015; Weisburger and Williams, 1983). Our results provide further evidence that L/MMW PAHs represent a human health hazard and that the inhalation exposures to these ubiquitous contaminants could, via nongenotoxic and pro-inflammatory mechanisms, contribute to the tumor-promoting phase of carcinogenesis or other human lung diseases, such as chronic obstructive pulmonary diseases (Bauer et al., 2018; Osgood et al., 2017, 2013a; Romo et al., 2019; Siegrist et al., 2019). Thus, our study supports the need to further address the effects of L/MMW PAHs and their mixtures in order to improve human health risk assessment of PAH exposures.
Supplementary Material
HIGHLIGHTS.
Airborne PAHs altered cellular signalling in human airway epithelial cells HBE1
Gap junctional intercellular communication was rapidly inhibited (<60 min) by PAHs
Signal transduction pathways MAPK ERK1/2 and p38 were activated
Potent effects induced by environmentally prevalent low molecular weight PAHs
PAHs can disrupt human airway epithelial homeostasis via nongenotoxic mechanisms
Acknowledgments
6. FUNDING INFORMATION
This work was supported by CETOCOEN EXCELLENCE Teaming 2 project supported by Horizon2020 (Grant Number 857560), the Czech Ministry of Education, Youth and Sports (02.1.01/0.0/0.0/18_046/0015975), RECETOX Research Infrastructure (LM2018121), by the Flight Attendant Medical Research Institute (CIA130022, AKB), and the National Institute for Environmental Health Sciences at the National Institutes of Health (R15ES024893, AKB).
ABBREVIATIONS
- 1MeA
1-methylanthracene
- 2MeA
2-methylanthracene
- 9MeA
9-methylanthracene
- FLN
fluorene
- FLT
fluoranthene
- PYR
pyrene
- CHR
chrysene
- B[a]P
benzo[a]pyrene
- TPA
12-O-tetradecanolyphorbol-13-acetate
- GJIC
gap junctional intercellular communication
- LMW
low molecular weight
- PAHs
polycyclic aromatic hydrocarbons
- MAPKs
mitogen-activated protein kinases
- ERKs
extracellular signal-responsive kinases
- P-ERK1/2
phosphorylated extracellular signal-responsive kinases
- P-p38
phosphorylated p38 kinase
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
- SLDT
scrape-loading/dye transfer
- PBS
phosphate buffered saline
- MeCN
acetonitrile
- EtOH
ethanol
- NR
Neutral red
- NRU
neutral red uptake
- AB
Alamar blue®
- CFDA-AM
5-carboxyfluorescein diacetate acetoxymethyl ester
- MeOH
methanol
- hINS
recombinant human insulin
- hEGF
recombinant human epidermal growth factor
- SEM
standard error of the mean
- FOC
fraction of control
- CaMgPBS
calcium- and magnesium-supplemented PBS
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- TBS-T
Tris-buffed saline with 0.1 % (v/v) Tween 20
- LMW
low molecular weight
- HMW
high molecular weight
- MMW
middle molecular weight
- Cx
connexin
- GA
growth area
- LOEC
lowest observable effect concentration
- PLA2
phospholipase A2
- PC-PLC
phosphatidylcholine-specific phospholipase C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
7. REFERENCES
- Abbas I, Badran G, Verdin A, Ledoux F, Roumie M, Lo Guidice J-M, Courcot D, & Garçon G, 2019. In vitro evaluation of organic extractable matter from ambient PM2.5 using human bronchial epithelial BEAS-2B cells: Cytotoxicity, oxidative stress, pro-inflammatory response, genotoxicity, and cell cycle deregulation. Environ. Res 171, 510–522. [DOI] [PubMed] [Google Scholar]
- Axelsen LN, Calloe K, Holstein-Rathlou NH, Nielsen MS, 2013. Managing the complexity of communication: regulation of gap junctions by post-translational modification. Front. Pharmacol 4, 130 10.3389/fphar.2013.00130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzo JL, Mesnil M, Hernandez-Blazquez FJ, Mackowiak II, Cabrera Mori CM, da Silva TC, Salgado Oloris SC, Gárate AP, Gomes Massironi SM, Yamasaki H, Zaidan Dagli ML, 2004. Increased susceptibility to urethane-induced lung tumors in mice with decreased expression of connexin43. Carcinogenesis 25, 1973–1982. 10.1093/carcin/bgh193 [DOI] [PubMed] [Google Scholar]
- Babica P, Sovadinová I, Upham BL, 2016a. Scrape Loading/Dye Transfer Assay BT - Gap Junction Protocols, in: Vinken M, Johnstone SR (Eds.), . Springer New York, New York, NY, pp. 133–144. 10.1007/978-1-4939-3664-9_9 [DOI] [Google Scholar]
- Babica P, Zurabian R, Kumar ER, Chopra R, Mianecki MJ, Park J-S, Jaša L, Trosko JE, Upham BL, 2016b. Methoxychlor and Vinclozolin Induce Rapid Changes in Intercellular and Intracellular Signaling in Liver Progenitor Cells. Toxicol. Sci 153, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek SO, Field RA, Goldstone ME, Kirk PW, Lester JN, Perry R, 1991. A review of atmospheric polycyclic aromatic hydrocarbons: Sources, fate and behavior. Water. Air. Soil Pollut 60, 279–300. 10.1007/BF00282628 [DOI] [Google Scholar]
- Bauer AK, Velmurugan K, Plöttner S, Siegrist KJ, Romo D, Welge P, Brüning T, Xiong KN, Käfferlein HU, 2018. Environmentally prevalent polycyclic aromatic hydrocarbons can elicit co-carcinogenic properties in an in vitro murine lung epithelial cell model. Arch. Toxicol 92, 1311–1322. 10.1007/S00204-017-2124-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrer MJ, Alani R, Cuttitta F, Preis LH, Sabich AL, Sanders DA, Siegfried JM, Szabo E, Brown PH, 1992. Early events in the neoplastic transformation of respiratory epithelium. J. Natl. Cancer Inst. Monogr 31–37. [PubMed] [Google Scholar]
- Blaha L, Kapplova P, Vondracek J, Upham B, Machala M, 2002. Inhibition of gap-junctional intercellular communication by environmentally occurring polycyclic aromatic hydrocarbons. Toxicol. Sci 65, 43–51. 10.1093/toxsci/65.1.43 [DOI] [PubMed] [Google Scholar]
- Bonvallot V, Baeza-Squiban A, Baulig A, Brulant S, Boland S, Muzeau F, Barouki R, Marano F, 2001. Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 expression. Am. J. Respir. Cell Mol. Biol 25, 515–521. 10.1165/ajrcmb.25.4.4515 [DOI] [PubMed] [Google Scholar]
- Boström C-E, Gerde P, Flanberg A, Jernström B, Johansson C, Kyrklund T, Rannug A, Törnqvist M, Victorin K, Westerholm R, 2002. Cancer risk assessment, indicators, and guidelines for polycyclic aromatic hydrocarbons in the ambient air. Environ. Health Perspect 110, 451–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervena T, Rossnerova A, Sikorova J, Beranek V, Vojtisek-Lom M, Ciganek M, Topinka J, Rossner P Jr., 2017. DNA Damage Potential of Engine Emissions Measured In Vitro by Micronucleus Test in Fluman Bronchial Epithelial Cells. Basic. Clin. Pharmacol. Toxicol 121 (S3), 102–108. 10.1111/bcpt.12693 [DOI] [PubMed] [Google Scholar]
- Chang Y, Siddens LK, Heine LK, Sampson DA, Yu Z, Fischer KA, Lohr CV, Tilton SC, 2019. Comparative mechanisms of PAH toxicity by benzo[a]pyrene and dibenzo[def,p]chrysene in primary human bronchial epithelial cells cultured at air-liquid interface. Toxicol. Appl. Pharm 379, 114644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JT, Cheng YW, Chou MC, Sen-Lin T, Lai WW, Ho WL, Lee H, 2003. The correlation between aberrant connexin 43 mRNA expression induced by promoter methylation and nodal micrometastasis in non-small cell lung cancer. Clin. Cancer Res 9, 4200–4204. [PubMed] [Google Scholar]
- Chuang S-M, Wang L-H, Hong J-H, Lin Y-W, 2008. Induction of Rad51 protein levels by p38 MAPK decreases cytotoxicity and mutagenicity in benzo[a]pyrene-exposed human lung cancer cells. Toxicol. Appl. Pharmacol 230, 290–297. 10.1016/j.taap.2008.03.001 [DOI] [PubMed] [Google Scholar]
- Corsini E, Budello S, Marabini L, Galbiati V, Piazzalunga A, Barbieri P, Cozzutto S, Marinovich M, Pitea D, Galli CL, 2013. Comparison of wood smoke PM2.5 obtained from the combustion of FIR and beech pellets on inflammation and DNA damage in A549 and THP-1 human cell lines. Arch. Toxicol 87, 2187–2199. 10.1007/s00204-013-1071-z [DOI] [PubMed] [Google Scholar]
- Crotty Alexander LE, Shin S, Hwang JH, 2015. Inflammatory Diseases of the Lung Induced by Conventional Cigarette Smoke: A Review. Chest 148, 1307–1322. 10.1378/chest.15-0409 [DOI] [PubMed] [Google Scholar]
- Ding J, Ning B, Gong W, Wen W, Wu K, Liang J, He G, Huang S, Sun W, Han T, Huang L, Cao G, Wu M, Xie W, Wang H, 2009. Cyclin D1 induction by benzo[a]pyrene-7,8-diol-9,10-epoxide via the phosphatidylinositol 3 kinase/Akt/MAPK and p70s6k-dependent pathway promotes cell transformation and tumorigenesis. J. Biol. Chem 284, 33311–33319. 10.1074/jbc.M109.046417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornhof R, Maschowski C, Osipova A, Gieré R, Seidl M, Merfort I, Humar M 2017. Stress fibers, autophagy and necrosis by persistent exposure to PM2.5from biomass combustion. PLoS One 12, 1–20. 10.1371/journal.pone.0180291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Lu J, Zhou L, Wu B, Zhou F, Gu L, Xu D, Sun Y, 2017. MAPK/FoxA2-mediated cigarette smoke-induced squamous metaplasia of bronchial epithelial cells. Int. J. COPD 12, 3341–3351. 10.2147/COPD.S143279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- E SM, F SR, C HH, F AR, T CO, 1976. Polynuclear Aromatic Hydrocarbons of Tobacco Smoke: Isolation and Identification. Beiträge zurTab. / Contrib. to Tob. Res 10.2478/cttr-2013-0388 [DOI] [Google Scholar]
- El-Fouly MH, Trosko JE, Chang C-C, 1987. Scrape-loading and dye transfer. Exp. Cell Res 168, 422–430. [DOI] [PubMed] [Google Scholar]
- Freund-Michel V, Muller B, Marthan R, Savineau J-P, Guibert C, 2016. Expression and role of connexin-based gap junctions in pulmonary inflammatory diseases. Pharmacol. Ther 164, 105–119. [DOI] [PubMed] [Google Scholar]
- Gad SC, Gad SE, 2014. Polycyclic Aromatic Hydrocarbons (PAHs). Encycl. Toxicol. Third Ed 1040–1042. 10.1016/B978-0-12-386454-3.00911-8 [DOI] [Google Scholar]
- Goldizen FC, Sly PD, Knibbs LD, 2016. Respiratory effects of air pollution on children. Pediatr. Pulmonol 51, 94–108. 10.1002/ppul.23262 [DOI] [PubMed] [Google Scholar]
- Husøy T, Cruciani V, Sanner T, Mikalsen SO, 2001. Phosphorylation of connexin43 and inhibition of gap junctional communication in 12-O-tetradecanoylphorbol-13-acetate-exposed R6 fibroblasts: minor role of protein kinase C beta I and mu. Carcinogenesis 22, 221–231. 10.1093/carcin/22.2.221 [DOI] [PubMed] [Google Scholar]
- IARC, 2018. Agents Classified by the International Agency for Research On Cancer Monographs. http://monographs.iarc.fr/ENG/Classification/index.php
- Johnson LN, Koval M, 2009. Cross-Talk Between Pulmonary Injury, Oxidant Stress, and Gap Junctional Communication. Antioxid. Redox Signal 11,355–367. 10.1089/ars.2008.2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki S, Takizawa H, Takami K, Desaki M, Okazaki H, Kasama T, Kobayashi K, Yamamoto K, Nakahara K, Tanaka M, Sagai M, Ohtoshi T, 2001. Benzene-extracted components are important for the major activity of diesel exhaust particles: Effect on interleukin-8 gene expression in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol 24, 419–426. 10.1165/ajrcmb.24.4.4085 [DOI] [PubMed] [Google Scholar]
- Kendall M, Duarte A, Rocha-Santos T, Hamilton R, Williams I, 2002. Airborne particulate-associated polyaromatic hydrocarbons, n-alkanes, elemental and organic carbon in three European cities. J. Environ. Monit 4, 890–896. 10.1039/b203888a [DOI] [PubMed] [Google Scholar]
- Klein SG, Serchi T, Hoffmann L, Blömeke B, Gutleb AC, 2013. An improved 3D tetraculture system mimicking the cellular organisation at the alveolar barrier to study the potential toxic effects of particles on the lung. Part. Fibre Toxicol 10, 31 10.1186/1743-8977-10-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J-C, Hong J-H, Wang L-H, Lin Y-W, 2008. The role of repair protein Rad51 in synergistic cytotoxicity and mutagenicity induced by epidermal growth factor receptor inhibitor (Gefitinib, IressaR) and benzo[a]pyrene in human lung cancer. Exp. Cell Res 314, 1881–1891. [DOI] [PubMed] [Google Scholar]
- Kubincová P, Sychrová E, Raška J, et al. Polycyclic Aromatic Hydrocarbons and Endocrine Disruption: Role of Testicular Gap Junctional Intercellular Communication and Connexins. Toxicol Sci 2019;169(1):70–83. [DOI] [PubMed] [Google Scholar]
- Lewtas J, 2007. Air pollution combustion emissions: Characterization of causative agents and mechanisms associated with cancer, reproductive, and cardiovascular effects. Mutat. Res. Mutat. Res 636, 95–133. [DOI] [PubMed] [Google Scholar]
- Li A, Liu Y, Zhu X, Sun X, Feng X, Li D, Zhang J, Zhu M, Zhao Z, 2018. Methylallyl sulfone attenuates inflammation, oxidative stress and lung injury induced by cigarette smoke extract in mice and RAW264.7 cells. Int. Immunopharmacol 59, 369–374. 10.1016/j.intimp.2018.04.028 [DOI] [PubMed] [Google Scholar]
- Liang Z, Wu R, Xie W, Zhu M, Xie C, Li X, Zhu J, Zhu W, Wu J, Geng S, Xu W, Zhong C, Han H, 2018. Curcumin reverses tobacco smoke-induced epithelial-mesenchymal transition by suppressing the MAPK pathway in the lungs of mice. Mol. Med. Rep 17, 2019–2025. 10.3892/mmr.2017.8028 [DOI] [PubMed] [Google Scholar]
- Libalova H, Rossner P, Vrbova K, Brzicova T, Sikorova J, Vojtisek-Lom M, Beranek V, Klema J, Ciganek M, Neca J, Pencikova K, Machala M, Topinka J, 2016. Comparative analysis of toxic responses of organic extracts from diesel and selected alternative fuels engine emissions in human lung BEAS-2B cells. Int. J. Mol. Sci 17, 1–24. 10.3390/ijms17111833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewenst Wr, Kanno Y, 1966. Intercellular communication and control of tissue growth - lack of communication between cells. Nature 209, 1248-, 10.1038/2091248a0 [DOI] [PubMed] [Google Scholar]
- Losa D, Chanson M, Crespin S, 2011. Connexins as therapeutic targets in lung disease. Expert Opin. Ther. Targets 15, 989–1002. 10.1517/14728222.2011.584875 [DOI] [PubMed] [Google Scholar]
- Luppi F, Aarbiou J, van Wetering S, Rahman I, de Boer WI, Rabe KF, Hiemstra PS, 2005. Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: role of glutathione. Respir. Res 6, 140 10.1186/1465-9921-6-140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AJ, Spence JR, 2017. In Vitro Models to Study Human Lung Development, Disease and Homeostasis. Physiology 32, 246–260. 10.1152/physiol.00041.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil LS, Goodenough DA, 1991. Biochemical-analysis ofconnexin43 intracellular-transport, phosphorylation, and assembly into gap junctional plaques. J. Cell Biol 115, 1357–1374. 10.1083/jcb.115.5.1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahta R, Al-Mulla F, Al-Temaimi R, Amedei A, Andrade-Vieira R, Bay SN, Brown DG, Calaf GM, Castellino RC, Cohen-Solal KA, Colacci AM, Cruickshanks N, Dent P, Di Fiore R, Forte S, Goldberg GS, Hamid RA, Krishnan H, Laird DW, Lasfar A, Marignani PA, Memeo L, Mondello C, Naus CC, Ponce-Cusi R, Raju J, Roy D, Roy R, Ryan EP, Salem HK, Scovassi AI, Singh N, Vaccari M, Vento R, Vondracek J, Wade M, Woodrick J, Bisson WH, 2015. Mechanisms of environmental chemicals that enable the cancer hallmark of evasion of growth suppression. Carcinogenesis 36, S2–S18. 10.1093/carcin/bgv028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesnow S, Triplett LL, Slaga TJ, 1985. Studies on the tumor initiating, tumor promoting, and tumor co-initiating properties of respiratory carcinogens. Carcinog. Compr. Surv 8, 257–277. [PubMed] [Google Scholar]
- Nielsen MS, Axelsen LN, Sorgen PL, Verma V, Delmar M, & Holstein-Rathlou NH (2012). Gap junctions. Compr. Physiol 2(3), 1981–2035. 10.1002/cphy.c110051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeder S, Kanashova T, Sippula O, Sapcariu SC, Streibel T, Arteaga-Salas JM, Passig J, Dilger M, Paur H-R, Schlager C, MOlhopt S, Diabate S, Weiss C, Stengel B, Rabe R, Harndorf H, Torvela T, Jokiniemi JK, Hirvonen M-R, Schmidt-Weber C, Traidl-Hoffmann C, BeruBe KA, Wlodarczyk AJ, Prytherch Z, Michalke B, Krebs T, Prevot ASH, Kelbg M, Tiggesbaumker J, Karg E, Jakobi G, Scholtes S, Schnelle-Kreis J, Lintelmann J, Matuschek G, Sklorz M, Klingbeil S, Orasche J, Richthammer P, MCiller L, Elsasser M, Reda A, Groger T, Weggler B, Schwemer T, Czech H, RCiger CP, Abbaszade G, Radischat C, Hiller K, Buters JTM, Dittmar G, Zimmermann R, 2015. Particulate Matter from Both Heavy Fuel Oil and Diesel Fuel Shipping Emissions Show Strong Biological Effects on Human Lung Cells at Realistic and Comparable In Vitro Exposure Conditions. PLoS One 10, e0126536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osgood RS, Upham BL, Hill T, Helms KL, Velmurugan K, Babica P, Bauer AK, 2013. Polycyclic Aromatic Hydrocarbon-Induced Signaling Events Relevant to Inflammation and Tumorigenesis in Lung Cells Are Dependent on Molecular Structure. PLoS One 8, 11 10.1371/journal.pone.0065150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osgood RS, Upham BL, Bushel PR, Velmurugan K, Xiong KN, Bauer AK, 2017. Secondhand smoke-prevalent polycyclic aromatic hydrocarbon binary mixture-induced specific mitogenic and pro-inflammatory cell signaling events in lung epithelial cells. Toxicol. Sci 157, 156–171. 10.1093/toxsci/kfx027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathi K, Ichimura H, Monma E, Lindert J, Quadri S, Issekutz A, Bhattacharyay J, 2006. Connexin 43 mediates spread of Ca2+-dependent proinflammatory responses in lung capillaries. J. Clin. Invest 116, 2193–2200. 10.1172/JCI26605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov A, Zehorai E, Procaccia S, Seger R, 2011. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1813(9), 1619–1633. 10.1016/J.BBAMCR.2010.12.012 [DOI] [PubMed] [Google Scholar]
- Qian F, Deng J, Wang G, Ye RD, Christman JW, 2016. Pivotal Role of Mitogen-Activated Protein Kinase-Activated Protein Kinase 2 in Inflammatory Pulmonary Diseases. Curr. Protein Pept. Sci 17, 332–342. 10.2174/1389203716666150629121324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram A, Singh SK, Singh VP, Kumar S, Ghosh B, 2009. Inhaled carbenoxolone prevents allergic airway inflammation and airway hyperreactivity in a mouse model of asthma. Int. Arch. Allergy Immunol 149, 38–46. 10.1159/000176305 [DOI] [PubMed] [Google Scholar]
- Rhrissorrakrai K, Belcastro V, Bilal E, Norel R, Poussin C, Mathis C, Dulize RHJ, Ivanov NV, Alexopoulos L, Jeremy Rice J, Peitsch MC, Stolovitzky G, Meyer P, Hoeng J, 2015. Understanding the limits of animal models as predictors of human biology: Lessons learned from the sbv IMPROVER Species Translation Challenge. Bioinformatics 31,471–483. 10.1093/bioinformatics/btu611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romo D, Velmurugan K, Upham BL, Dwyer-Nield LD, Bauer AK, 2019. Dysregulation of gap junction function and cytokine production in response to non-genotoxic polycyclic aromatic hydrocarbons in an in vitro lung cell model. Cancers (Basel). 11, 1–19. 10.3390/cancers11040572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruch RJ, 2019. Connexin43 suppresses lung cancer stem cells. Cancers (Basel). 11 10.3390/cancers11020175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rummel AM, Trosko JE, Wilson MR, Upham BL, 1999. Polycyclic aromatic hydrocarbons with bay-like regions inhibited gap junctional intercellular communication and stimulated MAPK activity. Toxicol. Sci 49, 232–240. 10.1093/toxsci/49.2.232 [DOI] [PubMed] [Google Scholar]
- Scheckenbach KEL, Crespin S, Kwak BR, Chanson M, 2011. Connexin channel-dependent signaling pathways in inflammation. J. Vase. Res 48, 91–103. 10.1159/000316942 [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW, 2012: NIH Image to ImageJ: 25 years of image analysis, Nat. Met 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist KJ, Romo D, Upham BL, Armstrong M, Quinn K, Vanderlinden L, Osgood RS, Velmurugan K, Elie M, Manke J, Reinhold D, Reisdorph N, Saba L, Bauer AK, 2019. Early Mechanistic Events Induced by Low Molecular Weight Polycyclic Aromatic Hydrocarbons in Mouse Lung Epithelial Cells: A Role for Eicosanoid Signaling. Toxicol. Sci 169, 180–193. 10.1093/toxsci/kfz030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirnes S, Kjenseth A, Leithe E, Rivedal E, 2009. Interplay between PKC and the MAP kinase pathway in Connexin43 phosphorylation and inhibition of gap junction intercellular communication. Biochem. Biophys. Res. Commun 382, 41–45. 10.1016/j.bbrc.2009.02.141 [DOI] [PubMed] [Google Scholar]
- Söhl G, Willecke K, 2004. Gap junctions and the connexin protein family. Cardiovasc. Res 62, 228–232. 10.1016/j.cardiores.2003.11.013 [DOI] [PubMed] [Google Scholar]
- Sovadinova I, Babica P, Boke H, Kumar E, Wilke A, Park J-S, Trosko JE, Upham BL, 2015. Phosphatidylcholine Specific PLC-Induced Dysregulation of Gap Junctions, a Robust Cellular Response to Environmental Toxicants, and Prevention by Resveratrol in a Rat Liver Cell Model. PLoS One 10, 1–16. 10.1371/journal.pone.0124454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stogiannidis E, Laane R, 2015. Source characterization of polycyclic aromatic hydrocarbons by using their molecular indices: an overview of possibilities. Rev. Environ. Contam. Toxicol 234, 49–133. 10.1007/978-3-319-10638-0_2 [DOI] [PubMed] [Google Scholar]
- Tai M-H, Upham BL, Olson LK, Tsao M-S, Reed DN, Trosko JE, 2007. Cigarette smoke components inhibited intercellular communication and differentiation in human pancreatic ductal epithelial cells. Int. J. cancer 120, 1855–62. 10.1002/ijc.22530 [DOI] [PubMed] [Google Scholar]
- Totlandsdal AI, Låg M, Lilleaas E, Cassee F, Schwarze P, 2015. Differential proinflammatory responses induced by diesel exhaust particles with contrasting PAH and metal content. Environ. Toxicol 30, 188–196. 10.1002/tox.21884 [DOI] [PubMed] [Google Scholar]
- Trosko JE, Ruch RJ, 1998. Cell-cell communication in carcinogenesis. Front. Biosci 10.2741/a275 [DOI] [PubMed] [Google Scholar]
- Trovato-Salinaro A, Trovato-Salinaro E, Failla M, Mastruzzo C, Tomaselli V, Gili E, Crimi N, Condorelli DF, Vancheri C, 2006. Altered intercellular communication in lung fibroblast cultures from patients with idiopathic pulmonary fibrosis. Respir. Res 7, 1–9. 10.1186/1465-9921-7-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upham BL, Blaha L, Babica P, Park J-S, Sovadinova I, Pudrith C, Rummel AM, Weis LM, Sai K, Tithof PK, Guzvic M, Vondracek J, Machala M, Trosko JE, 2008. Tumor promoting properties of a cigarette smoke prevalent polycyclic aromatic hydrocarbon as indicated by the inhibition of gap junctional intercellular communication via phosphatidylcholine-specific phospholipase C. Cancer Sci. 99, 696–705. 10.1111/j.1349-7006.2008.00752.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upham BL, Sovadinova I, Babica P, 2016. Gap Junctional Intercellular Communication: A Functional Biomarker to Assess Adverse Effects of Toxicants and Toxins, and Health Benefits of Natural Products. JoVE e54281 10.3791/54281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upham BL, Weis LM, Rummel AM, Masten SJ, Trosko JE, 1996. The effects of anthracene and methylated anthracenes on gap junctional intercellular communication in rat liver epithelial cells. Fundam. Appl. Toxicol 34, 260–264. 10.1006/faat.1996.0195 [DOI] [PubMed] [Google Scholar]
- Upham BL, Weis LM, Trosko JE, 1998. Modulated gap junctional intercellular communication as a biomarker of PAH epigenetic toxicity: Structure-function relationship. Environ. Health Perspect 106, 975–981. 10.2307/3434140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- US HHS, 1995. Toxicological profile for Polyaromatic Hydrocarbons. US Department of Health and Human Services, ATSDR, https://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=122&tid=25, Version updated on April 8, 2019. [Google Scholar]
- Vázquez-Gómez G, Rocha-Zavaleta L, Rodríguez-Sosa M, Petrosyan P, Rubio-Lightbourn J, 2018. Benzo[a]pyrene activates an AhR/Src/ERK axis that contributes to CYP1A1 induction and stable DNA adducts formation in lung cells. Toxicol. Lett. 289, 54–62. 10.1016/j.toxlet.2018.03.012 [DOI] [PubMed] [Google Scholar]
- Vondráček J, Pěnčíková K, Neča J, Ciganek M, Grycová A, Dvořák Z, Machala M, 2017. Assessment of the aryl hydrocarbon receptor-mediated activities of polycyclic aromatic hydrocarbons in a human cell-based reporter gene assay. Environ. Pollut 220, 307–316. 10.1016/j.envpol.2016.09.064 [DOI] [PubMed] [Google Scholar]
- Vondráĉek J, Ŝvihálková-Ŝindlerová L, Pênĉíková K, Marvanová S, Krĉmáģ P, Ciganek M, Neĉa J, Trosko JE, Upham B, Kozubík A, Machala M, 2007. Concentrations of methylated naphthalenes, anthracenes, and phenanthrenes occurring in Czech river sediments and their effects on toxic events associated with carcinogenesis in rat liver cell lines. Environ. Toxicol. Chem 26, 2308–2316. 10.1897/07-161R.1 [DOI] [PubMed] [Google Scholar]
- Wang H, Yang T, Wang T, Hao N, Shen Y, Wu Y, Yuan Z, Chen L, Wen F, 2018. Phloretin attenuates mucus hypersecretion and airway inflammation induced by cigarette smoke. Int. Immunopharmacol 55, 112–119. 10.1016/j.intimp.2017.12.009 [DOI] [PubMed] [Google Scholar]
- Weis LM, Rummel AM, Masten SJ, Trosko JE, Upham BL, 1998. Bay or baylike regions of polycyclic aromatic hydrocarbons were potent inhibitors of gap junctional intercellular communication. Environ. Health Perspect 106, 17–22. 10.1289/ehp.9810617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisburger JH, Williams GM, 1983. The distinct health risk analyses required for genotoxic carcinogens and promoting agents. Environ. Health Perspect 50, 233–245. 10.2307/3429555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilgenbus KK, Kirkpatrick CJ, Knuechel R, Willecke K, Traub O, 1992. Expression of Cx26, Cx32 AND Cx43 gap junction proteins in normal and neoplastic human tissues. Int. J. Cancer 51, 522–529. 10.1002/ijc.2910510404 [DOI] [PubMed] [Google Scholar]
- Willebrords J, Crespo Yanguas S, Maes M, Decrock E, Wang N, Leybaert L, Kwak BR, Green CR, Cogliati B, Vinken M, 2016. Connexins and their channels in inflammation. Crit. Rev. Biochem. Mol. Biol 51:413–439. doi: 10.1080/10409238.2016.1204980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P, Laxton V, Srivastava S, Chan YWF, Tse G, 2017. The role of gap junctions in inflammatory and neoplastic disorders (Review). Int. J. Mol. Med 39, 498–506. 10.3892/ijmm.2017.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Balsiger R, Tyrrell J, Boyaka PN, Tarran R, Cormet-Boyaka E, 2015. Cigarette smoke exposure reveals a novel role for the MEK/ERK1/2 MAPK pathway in regulation of CFTR. Biochim. Biophys. Acta 1850, 1224–1232. 10.1016/j.bbagen.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang WC, Lung SCC, Sun Z, Chen C, Chen JK, Zou Q, Lin YH, Lin CH, 2017. Polycyclic aromatic hydrocarbons are associated with increased risk of chronic obstructive pulmonary disease during haze events in China. Sci. Total Environ 574, 1649–1658. 10.1016/j.scitotenv.2016.08.211 [DOI] [PubMed] [Google Scholar]
- Yankaskas JR, Haizlip JE, Conrad M, Koval D, Lazarowski E, Paradiso AM, Rinehart CA, Sarkadi B, Schlegel R, Boucher RC, 1993. Papilloma-virus immortalized tracheal epithelial cells retail a well-differentiated phenotype. Am. J. Physiol 264, C1219–C1230. [DOI] [PubMed] [Google Scholar]
- Yu H, Li Q, Kolosov VP, Perelman JM, Zhou X, 2011. Regulation of cigarette smoke-induced mucin expression by neuregulin1β/ErbB3 signalling in human airway epithelial cells. Basic Clin. Pharmacol. Toxicol 109, 63–72. 10.1111/j.1742-7843.2011.00686.x [DOI] [PubMed] [Google Scholar]
- Zhang J, Wang W, Sun J, Li Q, Liu J, Zhu H, Chen T, Wang H, Yu S, Sun G, Chen W, Yi D, 2010. Gap junction channel modulates pulmonary vascular permeability through calcium in acute lung injury: an experimental study. Respiration. 80, 236–245. 10.1159/000274384 [DOI] [PubMed] [Google Scholar]
- Zhong C-Y, Zhou Y-M, Douglas GC, Witschi H, Pinkerton KE, 2005. MAPK/AP-1 signal pathway in tobacco smoke-induced cell proliferation and squamous metaplasia in the lungs of rats. Carcinogenesis 26, 2187–2195. 10.1093/carcin/bgi189 [DOI] [PubMed] [Google Scholar]
- Zhou G, Xiao W, Xu C, Hu Y, Wu Xiaokai, Huang F, Lu X, Shi C, Wu Ximei, 2016. Chemical constituents of tobacco smoke induce the production of interleukin-8 in human bronchial epithelium, 16HBE cells. Tob. Indue. Dis 14, 1–9. 10.1186/s12971-016-0089-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.