

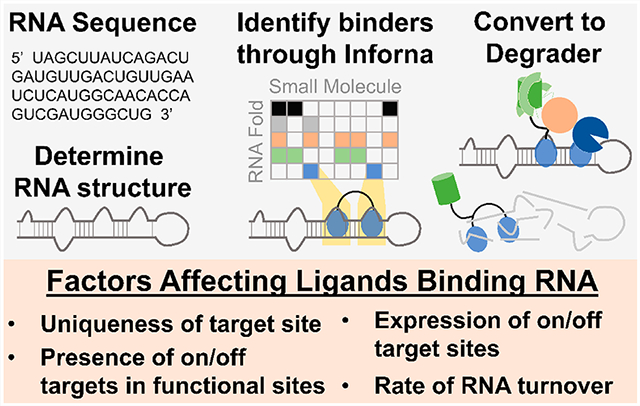
Abstract
RNA offers nearly unlimited potential as a target for small molecule chemical probes and lead medicines. Many RNAs fold into structures that can be selectively targeted with small molecules. This Perspective discusses molecular recognition of RNA by small molecules and highlights key enabling technologies and properties of bioactive interactions. Sequence-based design of ligands targeting RNA has established rules for affecting RNA targets and provided a potentially general platform for the discovery of bioactive small molecules. The RNA targets that contain preferred small molecule binding sites can be identified from sequence, allowing identification of off-targets and prediction of bioactive interactions by nature of ligand recognition of functional sites. Small molecule targeted degradation of RNA targets (ribonuclease-targeted chimeras, RIBOTACs) and direct cleavage by small molecules have also been developed. These growing technologies suggest that the time is right to provide small molecule chemical probes to target functionally relevant RNAs throughout the human transcriptome.
Graphical Abstract
DRUG DISCOVERY IN THE “-OMICS” ERA
In 2001, the dissemination of the draft sequence of the human genome was noted as a crowning scientific achievement.1 Genomics inspired “omics” for nearly every biomolecule such as the proteome, the glycome, and the transcriptome.2–5 Many of these studies have been aimed at associating differential biomolecule expression to disease states.6 Ideally, being armed with an encyclopedia of genome sequence, one could readily identify the basis for understudied diseases and rapidly develop effective, orally bioavailable small molecule treatments.
Because of the focus of using genomics to inform medicinal development, one of the first genome-wide analyses determined the number of open reading frames (ORFs). Surprisingly, these annotations, culminating in the Encyclopedia of DNA Elements (ENCODE) project,7 revealed that canonical ORFs are present in only ⅓ of the genes that humans were previously predicted to have. Additionally, only 2% of human DNA is translated into protein, the most well studied small molecule drug target (Figure 1).8 Interestingly, it was found that 90% of the genome is transcribed into RNA, with the vast majority of these having non-protein-coding functions.8,9 Noncoding RNAs have been subsequently found to have many different functions, and analysis of the noncoding RNA between organisms follows their complexity, in contrast to differences in their respective protein-coding genes (Figure 1).10,11 Historically, protein is the predominant biomolecule considered for small molecule drug targeting. Thus, druggability, or whether a target protein is a member of a family that has been previously targeted with a small molecule, was initially assessed across the genome for protein-coding genes. This analysis revealed that 15% of proteins are considered “druggable”, i.e., within genes that have been targeted with a small molecule (Figure 1A).12,13 Recent studies suggest that this notion may need reassessment, however. Perhaps the language should be changed to “undrugged”, as a wide variety of difficult to target proteins have been targeted by small molecules, including the mutant Kirsten rat sarcoma (KRAS) proto-oncogene.14,15
Figure 1.

RNA as a viable drug target. (A) The conventional binary approach to small molecule drugs is their molecular recognition of proteins. Among the ~20 000 proteins that comprise the proteome, only about 15% are in traditional “druggable” protein families. In turn, this only represents a fraction of the genome that is transcribed, leaving much of the transcriptome underexploited as therapeutic targets. (B) Noncoding genes relate to the complexity of the organism, as the relative number of coding bases remains similar, while the relative number of long noncoding RNAs (lncRNA) significantly increases, indicating that much of the intricacies of human biology and disease are represented among noncoding regions. (C) Due to the importance of coding and noncoding RNA to biology, small molecules interacting with RNA can act on the transcriptome, resulting in varied downstream effects. Importantly, validated activities for small molecules that target human RNA include: (i) changing gene expression by modulating the stability of mRNA by direct binding; (ii) affecting its noncoding RNA effectors; (iii) affecting the epitranscriptome; or (iv) influencing alternative splicing. Affecting the transcriptome with small molecule drugs can rescue disease by modulating the translation of beneficial or detrimental proteins.
A dogma for the development of small molecules against this fraction of the proteome has been centered on some tenants that perhaps need re-evaluation. First, binding sites for a bioactive ligand must be in an active site or an allosteric site to affect function. Second, the occupancy-driven view of pharmacology dictates that high target occupancy is necessary for effective target inhibition. Thus, maintained exposure to the target is the major driver of pharmacodynamic profiles. Third, the proteome is targetable at defined three-dimensional clefts that allow for high surface area recognition of ligands.
Many groups have worked tirelessly to challenge these assertions. While there are no hard and fast rules in drug discovery, pragmatic solutions to important biomedical problems always win out. For example, recent developments on targeting KRAS have shown that ligands can be developed to target important proteins that do not have the traditionally desired ligand binding pockets.16–18 There are a variety of new ligands in the clinic that affect these previously perceived as impossible-to-drug targets. Additionally, work in the targeted protein degradation, or proteolysis targeting chimeras (PROTACs), area has challenged notions of occupancy-driven pharmacology, as well as the necessity for bioactive ligands targeting allosteric or active, i.e., functional, sites.19 For example, the ability to recruit E3-ubiquitin ligases to selectively degrade target proteins bound by selective ligands suggests that small molecules do not require occupancy to enable a pharmacological effect. Additionally, ligand occupancy or “ligandability” has been studied across the proteome and has provided an encyclopedia of information on proteome-wide ligand occupancy.20,21
RNA could be advantageous as a small molecule drug target, following the paradigm of occupancy-driven inhibition of functional sites with defined three-dimensional structure employed for enzymes and receptors. RNA is also an attractive target as it is causative and/or upstream of pathological mechanisms related to disease states (Figure 1).22,23 Importantly, RNAs can be analyzed from tissue or liquid biopsies more easily than proteins because nucleic acids can be amplified and sequenced. This Perspective focuses on our current understanding of the molecular recognition of RNA by small molecules, the design of bioactive small molecules, and tools used to study RNA target validation, engagement, and selectivity. There are many excellent reviews on RNA chemical biology and drug discovery that cover topics outside the scope of this Perspective.24–29
ASSESSING RNA AS A DRUG TARGET
Since the beginning of modern medicine, RNA played an essential role as a small molecule drug target (Figure 2). Streptomycin was found to target the bacterial ribosome and shortly after its discovery, became a first-in-line treatment for Mycobacterium tuberculosis. Prior to this breakthrough discovery, tuberculosis was a devastating disease, and those affected were housed in sanitariums until they succumbed to the infection. Other natural products isolated from species of Streptomyces bacteria are also tried and true antibacterials such as other aminoglycoside antibiotics like neomycin B, the active ingredient in Neosporin. Studies of these compounds and their derivatives quickly expanded to various other aminoglycosides-based antibiotics, which are still in wide use today.30 While these aminoglycosides are effective as broad-spectrum antibiotics, they tend to be promiscuous binders, leading to their limited use unless they are modified.31 However, between their discovery during WWII and now, there have been few ligands that target RNA that have progressed to the clinic (Figure 2).
Figure 2.
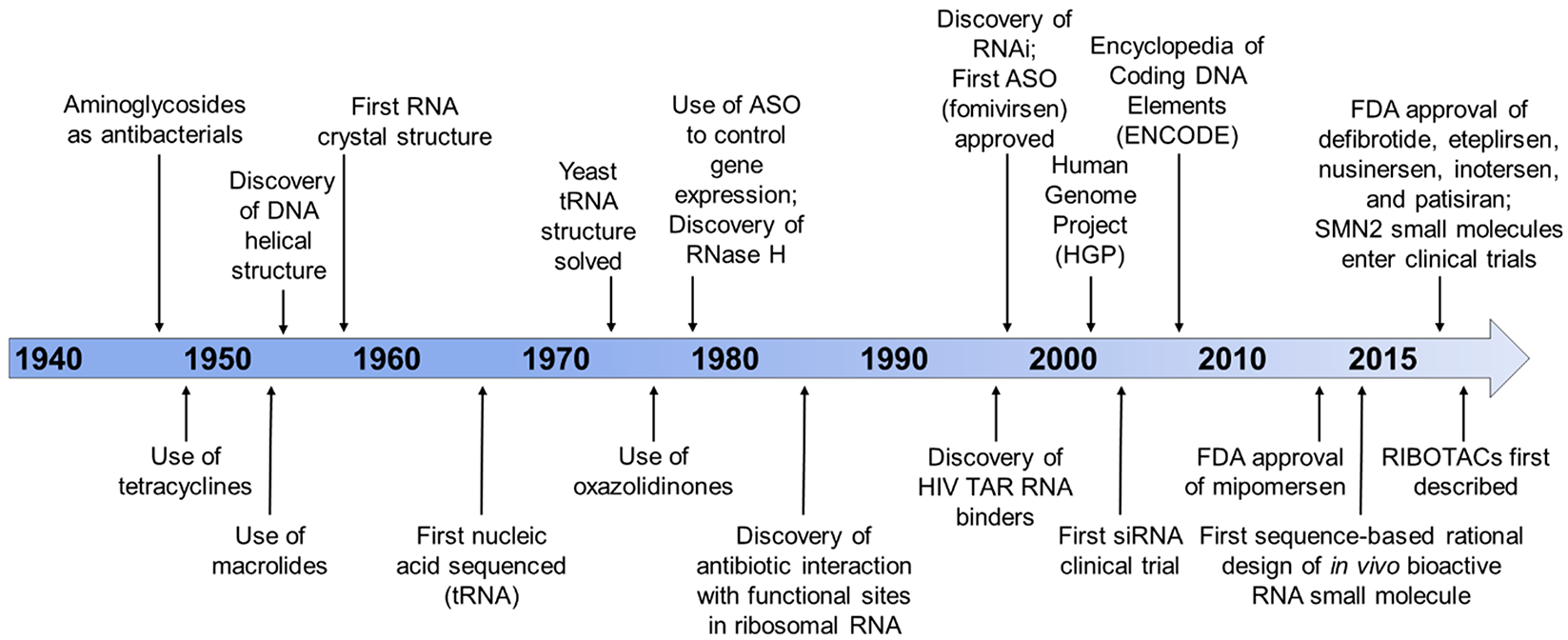
Timeline of major developments in the RNA-targeting field. The history of drugging RNA is tied closely with molecular biology discoveries (DNA/RNA structural determination). Antibacterials that targeted RNA preceded the first investigations into antisense oligonucleotides. However, FDA approvals of antisense oligonucleotides increased upon completion of the human genome project. Recent developments, such as the use of rational design-based approaches, the initiation of clinical trials for small molecule drugs treating spinal muscular atrophy (SMA), and the first report of ribonuclease targeting chimeras (RIBOTACs), demonstrate the rapid development of small molecules targeting RNA. These advancements provide a convincing argument to turn our focus to the druggable transcriptome.
Phenotypic screens have been broadly used in the drug discovery industry beyond screening bacterial lysates to find active antibacterials. These approaches provide compounds that affect a wide variety of pathways. The challenge with phenotypic screens is identifying the engaged targets to understand the mechanism of action, compounded by the fact that target validation tools for these assays were generally designed to probe protein-centric pathways. For example, pooled CRISPR and shRNA hairpin libraries knock down ORFs (protein) or forced expression of the ORF can be used to track down a target for a small molecule.32,33
Although these phenotypic screens do not traditionally consider RNA as a potential target, they have indeed discovered compounds that modulate RNA. Unfortunately, these screens do not necessitate interaction with the target RNA of interest to modulate downstream RNA biology. Discovered serendipitously through phenotypic screening of small molecules interfering with the bacterial riboflavin biosynthetic pathway, Merck reported ribocil, which acts to inhibit the flavin mononucleotide (FMN) riboswitch and subsequently disrupt translation of the downstream mRNA.34 After demonstrating that riboflavin genes (ribA and ribB) are essential for Escherichia coli growth, ~57 000 compounds were screened for inhibition of bacterial growth that could be reversed by riboflavin supplementation. This screen identified that ribocil inhibited cellular production of riboflavin and its metabolites. Compound-resistant E. coli strains were then sequenced, revealing that all mutants contained base pair mutations affecting the aptamer region of the FMN riboswitch, which controls ribB expression. Additional binding and reporter gene assays indicated that ribocil was a competitive inhibitor of FMN and that inhibition was ablated in FMN mutant constructs, validating this RNA as the compound’s target. Although ribocil selectively binds the FMN riboswitch and is structurally distinct from the endogenous FMN riboswitch ligand, the rapid development of bacterial resistance in various species precluded it from progression as a clinical candidate. Overall, the development of ribocil has proven that phenotypic screens can provide small molecules that bind RNA, encouraging investigators to consider RNA modulation as a mechanism of action.
An early adaptor of these phenotypic studies was PTC Therapeutics. Their programs include compounds that perturb readthrough of stop codons and affect pre-mRNA splicing outcomes. Phenotypic screening identified ataluren, a clinical candidate for treatment of Duchenne’s muscular dystrophy (DMD). DMD is caused by a premature stop codon in the dystrophin mRNA, and ataluren is thought to stimulate its readthrough. Although ataluren was conditionally approved for treatment of DMD in Europe, the FDA declined to accept it as a drug based on a clinical trial in which ataluren missed its primary end point.35,36 While exact details of its mechanism are not known, studies revealed that ataluren directly binds and stabilizes firefly luciferase, thus allowing for its identification in the luciferase-based nonsense codon suppression assay.37,38 Therefore, in order to avoid false positives, target activity observed from phenotypic screens must also consider potential interference with downstream assay signals.
Risdaplam is a second compound that emerged from these screens and affects pre-mRNA splicing as a treatment for spinomuscular atrophy (SMA). This autosomal recessive neurodegenerative disease is caused by deficiencies in survival motor neuron (SMN) protein, which is caused by deletion or loss in function of the SMN1 gene.39 SMA is classified by disease severity, which scales with the reduction of SMN protein levels. The loss of functional SMN protein can be compensated by its expression through the SMN2 gene. However, SMN2 typically produces shorter and nonfunctional SMN protein due to a C to T single nucleotide change that causes biased exon 7 skipping, decreasing the half-life of the resulting protein. Phenotypic screens led by Roche/PTC identified compounds that increased inclusion of exon 7 in a luciferase-based cellular assay. Following efficacy studies in patient-derived cells and mouse models and RNA-Seq analysis to verify selectivity, the orally active Risdaplam was identified to convert the SMN2-encoding pre-mRNA into a longer lived version by modifying its splicing outcome.40 In a similar report, phenotypic screening at Novartis identified a compound, Branaplam, that similarly increased production of functional SMN protein through splicing modulation, which is now progressing through clinical development.41
Due to their identification through phenotypic screens, the mechanisms of action of these splicing modulators have not been completely defined.42 Multiple studies have proposed that compounds in the Risdiplam series can achieve such exceptional molecular recognition and specificity by formation of a higher order complex comprising the exonic splicing enhancer (ESE) sequence, the 5′-splice site (5′-ss), and other regulatory splicing proteins.43 Wang and others used several approaches, including chemical cross-linking and isolation by pull-down (Chem-CLIP), in order to determine direct target engagement and interactions of these compounds with RNA.42,44 These Chem-CLIP studies, in addition to a series of genomic and proteomic experiments of this class of compounds, revealed their direct binding to the AGGAAG motif present in exon 7 of the SMN2 pre-mRNA. Small molecule binding promotes conformational changes that result in increased binding of splicing modulators (FUBP1, KHSRP) to enhance SMN2 splicing.42 These studies suggest that compounds can directly bind and alter SMN2 RNA structure, thereby affecting interactions with other splicing modifiers.42,43 Unfortunately, the specificity afforded by these compounds in the context of interacting with specific splicing proteins and RNA motifs in SMA may not be available for all RNA targets.
Collectively, none of these efforts purposefully targeted RNA. While they validate that small molecules can affect several different RNA functions, purposeful targeting of RNA is altogether different. Tools such as Chem-CLIP will therefore by key for exploiting RNA as a drug target, providing direct evidence of target engagement and hence compound mode of action.
RICH STRUCTURE OF RNA
Mammalian RNA encodes both genetic and structural information, exemplified by noncoding transfer (t)RNA, which was the first nucleic acid sequenced (Figure 2).45 The discovery that the stable cloverleaf structure of tRNA interacts with the codon of messenger (m)RNA represents that RNA–RNA interactions function as a physical template for protein synthesis, setting the stage for the central dogma of biology.46 Beyond their fundamental roles in protein synthesis, non-coding RNAs also play essential regulatory roles. Recent research even points to the fact that organismal complexity is directly associated with the number of noncoding RNAs, rather than the size of the genome (Figure 1).10 As an early example, the Steitz group proved in seminal work that noncoding U1 small nuclear RNA (U1 snRNA) recognizes 5′ splice sites in pre-mRNA,47 demonstrating that RNA structural recognition is necessary for accurate splicing and that RNA adopts intricate structures that influence genetic processes.
RNA folds into elaborate structures that enable its essential functions in diverse applications in biology, encompassing regulation of gene expression, ligand sensing, catalysis, and others.48 In order to minimize its free energy, single stranded RNA forms fully base-paired as well as noncanonically paired regions (hairpins, internal loops, bulges, etc.) that determine higher order (tertiary) folding patterns (Figure 3).48 These secondary and tertiary RNA structural elements are highly dynamic and dependent on their environment (protein, salt concentration, small molecules, etc.), which can greatly affect function. Within structured RNAs, over 50% of residues are confined in Watson–Crick base-paired helices, and emerging studies show that subtle changes to secondary structure, or variation in Watson–Crick base pair interactions within RNA helices, significantly impact the formation of tertiary structures.49,50 Notably, not all human RNAs contain long-range interactions between secondary structural elements, or tertiary structure (unlike RNase P RNA, (t)RNAs, and ribosomal (r)RNAs that display well-defined tertiary structures), including coaxial stacking and pseudoknots, among others. Recent investigations into RNA tertiary folding energetics have also suggested that tertiary structure can be defined through a dynamic thermodynamic ensemble of assembled secondary structural elements based on RNA sequence.51 That is, secondary structure limits the number of tertiary interactions available and hence constrains tertiary structure.
Figure 3.
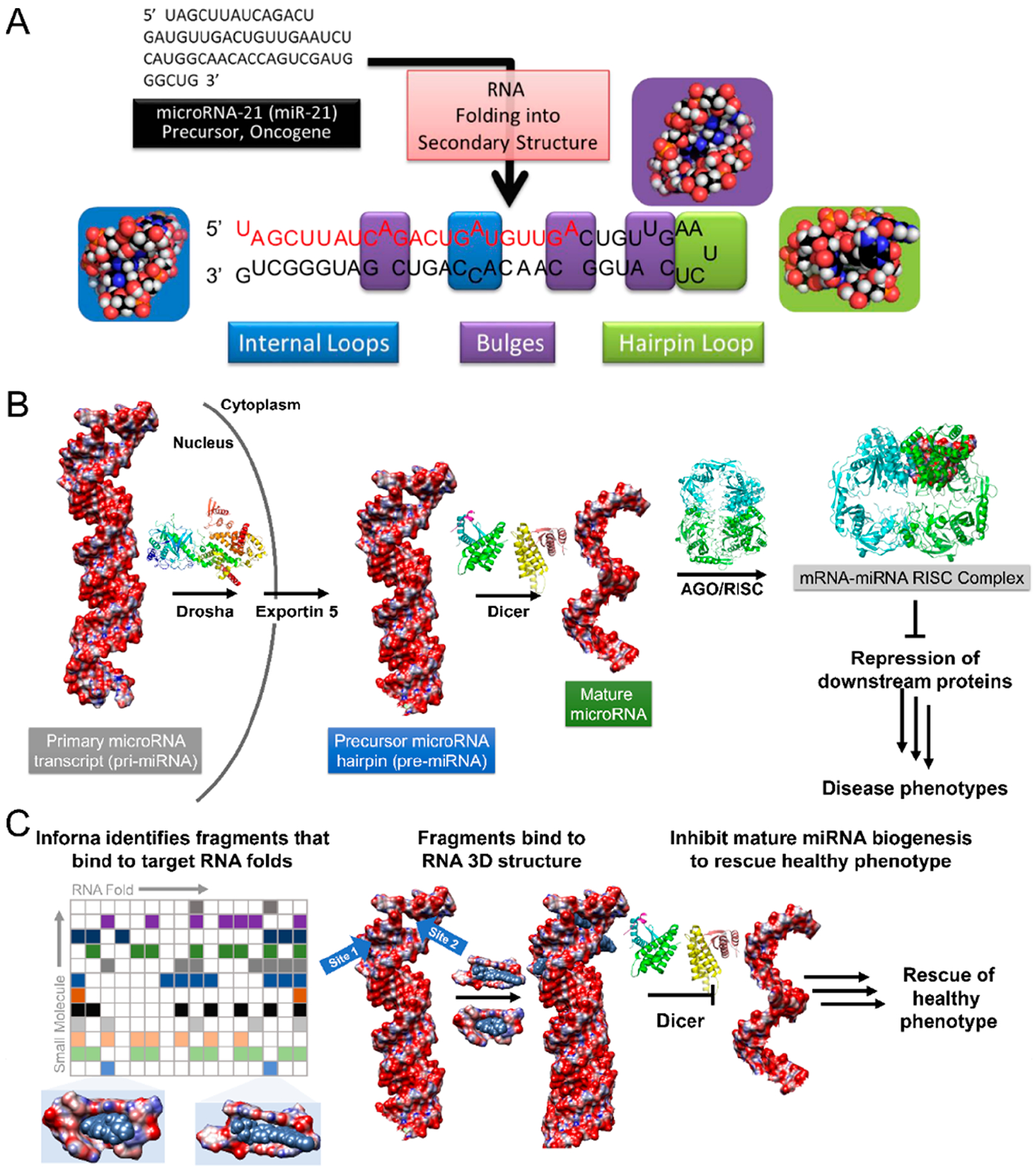
Modular RNA secondary structure motifs form three-dimensional structures. (A) Hierarchical assembly of RNA structure from sequence to secondary structure. Many of these secondary structures can form modular RNA motifs that can allow for small molecule recognition. (B) Structural schematic of microRNA processing. Primary transcripts (pri-miRNAs) are processed by the Drosha endonuclease to yield precursor hairpins (pre-miRNAs), which are exported to the cytoplasm and subsequently processed by the Dicer endonuclease to liberate a mature miRNA. One of the mature strands is then loaded into the argonaute/RNA-induced silencing complex (AGO/RISC), whereupon it acts on RNAs to modulate gene expression. Aberrant miRNA expression can be causative of disease phenotypes. (C) One rational design approach to target RNA is to understand the molecular recognition of structural elements by small molecules, those elements that are preferred by the small molecule and those that are discriminated against. Inforna compares structural elements within an RNA target to a database of these preferred interactions to afford lead small molecules. For example, binding to the pri- or pre-miRNAs at functional Drosha or Dicer sites can prevent their processing to the active, mature strand, thus allowing the rescue of disease-associated phenotypes through the inhibition of biogenesis.
As understanding RNA folding is foundational to deducing its overall structure, much effort has been dedicated toward accurately predicting RNA secondary structure from sequence, including phylogenetic comparison, free energy minimization, and combinations thereof.52–54 For example, entire kingdoms of life have been determined through the conservation of RNA secondary structure and phylogenetic comparison.55 In cases where limited RNA sequences are available, secondary structure prediction through free energy minimization is commonly used.56 These free energy calculations are able to provide both optimal and suboptimal structures.57 This technique is more powerful when incorporating experimental constraints from structural probing, allowing accurate determination of more complicated RNA structures (rRNA, viral RNAs, etc.).58 Use of dimethyl sulfate (DMS) chemical probing has also allowed global monitoring of RNA structure in vivo with single nucleotide resolution, showing that cellular mRNAs are predominantly unfolded.59,60 Optimal integration of chemical probing experimental data and computational tools remains an ongoing challenge in the field of RNA structural determination, as recent quantitative tools to measure covariations in RNA alignments have disputed evidence of proposed secondary structures of various long noncoding RNAs.61 Recently, advanced bioinformatics scanning window models (ScanFold) have allowed generation of high likelihood functional RNA structures.62 These resultant RNA structuromes provide evidence for hubs of structured regions within viral and mammalian RNA.52
RNAs play essential roles in cellular processes; thus their dysregulated expression or misfolding can be causative of disease pathologies.22,23 For example, overexpression of regulatory noncoding RNAs, such as microRNAs (miRNAs) that have defined secondary structural elements but lack tertiary structure, can result in oncogenic phenotypes.63 Improper folding of RNA can also be responsible for disease pathology, as is commonly seen with short tandem repeat, or microsatellite, RNA expansions.64 Due to their misfolding, several pathological mechanisms can cause a wide variety of disease mechanisms. Examples include (i) dysregulation of RNA splicing due to single nucleotide polymorphisms (SNPs), as seen in Tau neurodegenerative disorders; (ii) sequestration of essential proteins that disrupts their normal function, as observed in the sequestration of splicing factors by expanded r(CUG) or r(CCUG) repeats in myotonic dystrophy type 1 (DM1) and type 2 (DM2), respectively; and (iii) the production of toxic or nonfunctional proteins, as observed in r(G4C2) repeat expansions observed in frontotemporal dementia and amyotrophic lateral sclerosis (FTD/ALS).64 Overall, the molecular basis for many diseases is rooted in deregulated RNA function, which is intimately tied to its proper structural folds.
BASICS OF TARGETING RNA
As aberrantly folded RNA structures contribute to disease, it is of utmost importance to provide compounds to correct their dysfunction or to facilitate their study in healthy and disease states. Beyond simple molecular recognition, however, targeting RNA is also a function of a variety of interrelated factors (Figure 4). First of all, drugs to target RNA must be able to access the structural elements present in the target RNA, which may be in prohibitive structural interactions with other biomolecules [i.e., competition with RNA-binding proteins (RBPs)], or the RNA itself may be difficult to access (e.g., blood–brain penetrance). Additionally, selective compounds must be able to disrupt the on-target RNA, while not binding to off-target transcripts, within acceptable limits. Binding to a specific transcript is also related to the expression levels of competing RNA motifs, which is intimately connected to tissue-specific or cellular compartment-specific expression. Furthermore, compounds must be able to elicit observable biological modulations; that is, compounds must interact with RNA at a functional site (Figure 4).
Figure 4.
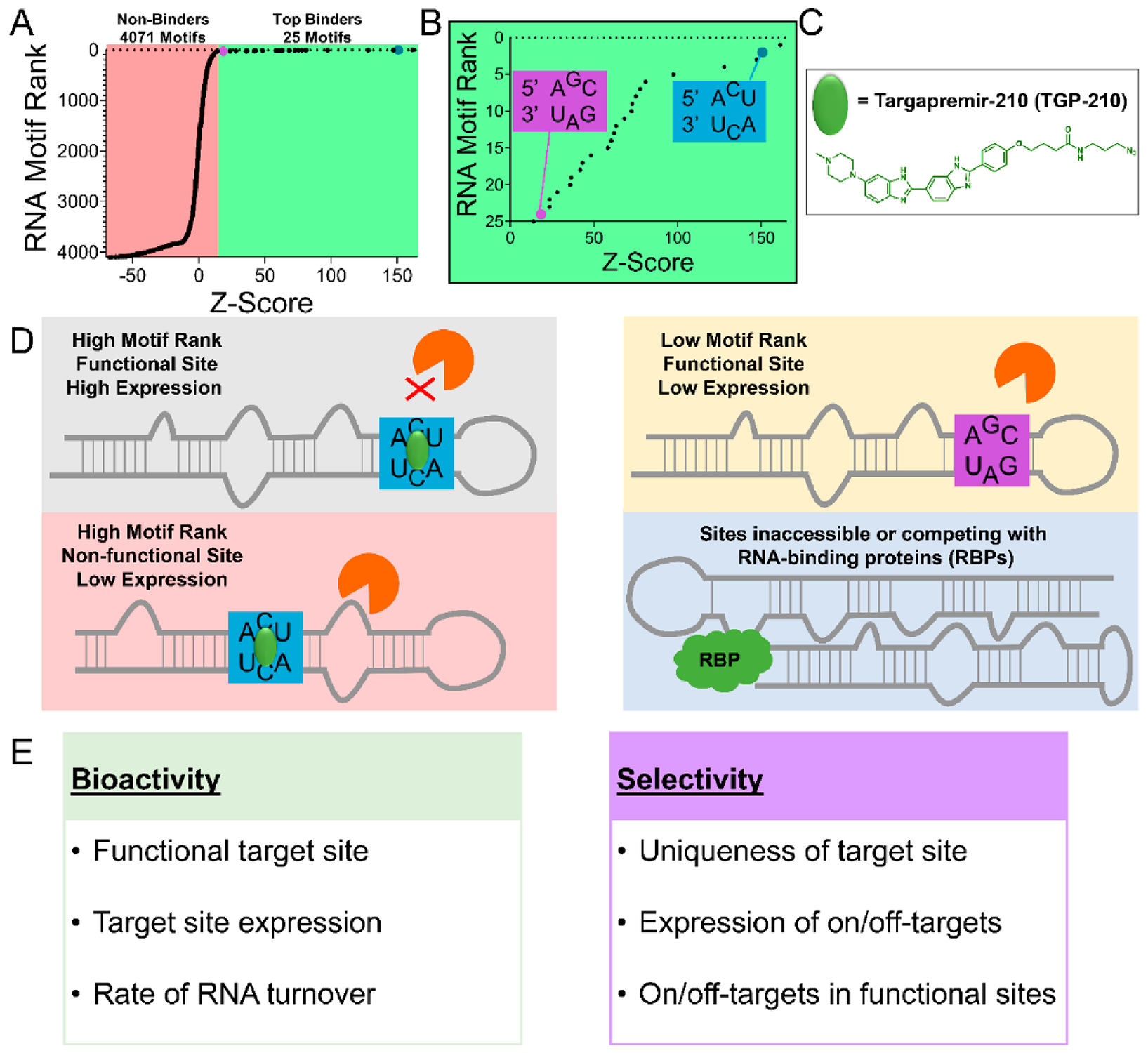
Factors that influence bioactivity and selectivity of RNA-binding ligands. (A) 2DCS selection and HiT-StARTS analysis identifies the top binding RNA motifs and nonbinders to targapremir-210 (TGP-210). Z-Score is a calculated value that represents fitness of the RNA-small molecule interaction. (B) Fitness plot of the top 25 binding motifs are shown. One of the highest fitness RNA motif interactions with TGP-210 is the 5′ ACU/3′ UCA 3 × 3 internal loop (blue box) found in the functional Dicer site of miR-210. The 5′ AGC/3′ UAG RNA motif is a lower affinity interaction (purple box). (C) Structure of TGP-210. (D, E) The bioactivity of TGP-210 to selectively inhibit miR-210 biogenesis is a function of multiple factors, including the fitness of the RNA motif-small molecule interaction, the expression and turnover of the target RNA(s), whether or not occupancy of the target site will result in a functional interaction (i.e., occupying a Dicer/Drosha site), and the accessibility of TGP-210 to off-target sites.
A common approach to affect RNA function is sequence complementarity through antisense oligonucleotide (ASO) hybridization with a target strand. The resultant antiparallel base-paired duplex can either affect the natural folding of the target RNA, thus disrupting its interactions with other biomolecules, or recruit endogenous cellular machinery to cleave the RNA.65 These interactions, however, are dependent on the thermodynamic and kinetic energy barriers necessary for the folding/unfolding/hybridization of the native conformations of both the target RNA and ASO, which can limit their effectiveness against highly structured RNAs.64 Thus, the antisense-based strategy is best applied to weakly structured RNAs. Beyond these difficulties to provide meaningful molecular interactions, oligonucleotides have been known to have inadequate in vivo properties (limited delivery strategies, biodistribution, and tissue penetrance) and exhibit various side effects that include eliciting of an immune response, thrombocytopenia, and hepatotoxicity in patients that have caused clinical trials to be halted.65–68
As the previous FDA standards indicate, ASO drugs are assumed to be selective. Intriguingly, the selectivity of past FDA-approved ASOs have not been thoroughly studied transcriptome-wide or proteome-wide, despite the increasing stringency to provide mode-of-action centric, selective drugs at the molecular level.66,69 Indeed, investigations into the specificity and selectivity of ASOs are more nuanced than previously thought.66 Naively, it might be assumed that selectivity can be enhanced by simply lengthening the oligonucleotide. Herschlag and co-workers found, however, that there is an optimal length of oligonucleotide to achieve selectivity.70 Oligonucleotides that are too long can form thermodynamically stable duplexes containing mismatches with off-targets. Indeed, the sources of hybridization of oligonucleotides with unintended targets are well-studied, and developments in analytical bioinformatics and algorithms have allowed computational screening to determine the maximal on-target transcript effects with an acceptable tolerance of off-targets.66,71 Still, the experimental determination of on-/off-targets through transcriptome-wide profiling of ASOs is still necessary to measure selectivity. Interestingly, there are multiple demonstrations that small molecules that target RNA can rival the selectivity of ASOs or even exhibit enhanced selectivity in certain cases.72–74 Thus, small molecule targeting of RNA structure may overcome the various liabilities of ASOs.
An emerging strategy to selectively target RNA with small molecules is to focus on the molecular recognition of structural elements.75 One approach to rapidly define selective small molecule binders of RNA structural motifs is through a selection-based method that screens a library of RNA motifs vs a library of small molecules embedded in an agarose microarray (Figure 3C).76 Dubbed two-dimensional combinatorial screening (2DCS), the small molecule microarray is incubated with a labeled RNA library that displays discrete structural elements in a randomized region. Weak binders and interactions with elements common to all library members (i.e., outside the randomized region) are competed off with an excess of oligonucleotide competitors (including DNA and regions that mimic the cassette displaying the randomized motif). Upon excision, amplification, and RNA-seq analysis of the bound RNA, the motifs preferred by each small molecule are defined by a statistical method named “high throughput structure–activity relationships through sequencing” (HiT-StARTS).77 HiT-StARTS determines the statistical significance of the enrichment of a given motif in the 2DCS selection vs the starting library. The higher the statistical confidence in this enrichment, the more privileged, or fit, the interaction is. Normalization of the statistical confidence affords a fitness score for all members of an RNA library for a given small molecule. By creating a database of the results from 2DCS and HiT-StARTS, we developed a lead identification strategy for RNA targets, dubbed Inforna (Figure 3C).78,79 That is, Inforna searches RNA targets for structural elements preferred by small molecules, which are chemical leads. Similar to the Watson–Crick base-pairing rules that govern oligonucleotide selectivity to RNA sequence, Inforna provides the small molecule equivalent of experimentally derived RNA fold-small molecule interactions for selective recognition of RNA structure.
While structure-based targeting shows great promise as detailed below, it does have limitations. For example, not all structural elements are functional; thus, even if a small molecule binder is identified, it likely will have no biological consequence. Alternatively, a ligand may not yet have been discovered for a particular functional structure. We have recently developed a strategy dubbed ribonuclease targeting chimeras (RIBOTACs) to overcome these limitations (discussed below). Furthermore, not all structures may be accessible for ligand binding, for example, due to the presence of long-range tertiary interactions or its interaction with RBPs.
SELECTIVE SMALL MOLECULE APPROACHES TO PURPOSEFULLY AFFECT microRNA BIOLOGY
Much of the transcriptome exists in defined hubs of structured RNA folds, and Inforna’s foundation is in querying these highly probable regions of structured RNA elements against experimentally identified and characterized RNA 3D fold-small molecule interactions. Built into Inforna are several strategies to rationally design compounds to purposefully target RNA and affect downstream biology. As selectivity remains the largest concern for small molecule targeting of RNA, Inforna has the ability to predict on- and off-target transcripts (Figure 4) (that is, RNAs with 3D-folds that are optimal (high fitness) or suboptimal for small molecule targeting).80 Additionally, these RNA 3D folds can be limited to parts of RNA that are important for bioactivity, such as the Dicer and Drosha processing sites in miRNAs. By use of the highest fitness and most selective binders from Inforna, small molecules that target oncogenic miRNA precursors have been designed and have shown promising preclinical data in vivo.75,77
Studies have determined that Inforna-defined RNA-small molecule interactions can inform selective targeting of miRNA precursors. These studies have formulated guidelines for targeting RNA with small molecules. The oncogenic miR-210 provides a prototypical example. Aberrant expression of miR-210 is observed in cancer cells that are in low oxygen, hypoxic environments, such as in solid breast cancer tumors.81,82 Inforna identified a compound, Targapremir-210 (TGP-210), that selectively inhibits miR-210 biogenesis by binding to the C/C internal loop displayed in its Dicer processing site.80 Inhibition of miR-210 disrupted the hypoxic circuit, resulting in stimulation of apoptosis in cellular and in vivo models.80,83
Inhibition of miR-210 as a result of selective on-target engagement was confirmed with a Chem-CLIP probe in which TGP-210 was appended to a cross-linking (chlorambucil) module and a purification (biotin) module. Chem-CLIP studies revealed that miR-210 was the most enriched transcript among highly abundant RNAs (rRNAs, tRNAs, mRNAs, etc.) but also among hypoxia-associated miRNAs and miRNAs that contain suboptimal binding sites (Figure 4). As the less expressed miR-497 contained the same C/C internal loop as miR-210, it was also pulled down in the enriched Chem-CLIP fraction. Transcripts like miR-497, or other RNAs that contain predicted interactions with the lead compound, are termed RNA isoforms. Interestingly, TGP-210 bound to pre-miR-497 in a nonfunctional site; thus miR-497 biogenesis remained unaffected.
Competitive Chem-CLIP (C-Chem-CLIP) was also applied by co-treating cells with the TGP-210 Chem-CLIP probe and the parent compound. Here, levels of miR-210 were depleted in the pulled down fraction due to competition for occupancy of target binding sites with TGP-210. These thorough studies elucidated various rules necessary for small molecules targeting RNA structure to selectively affect RNA biology. Importantly, the presence of the C/C internal loop in the functionally relevant Dicer processing site enabled TGP-210 to modulate miR-210’s downstream biology. Despite Chem-CLIP studies indicating target engagement with off-target miR-497, due to its interaction with TGP-210 in a nonfunctional site, its binding remained “silent”. Additionally, abundance of the target plays an important role, as miR-497 expression is 10-fold lower than the overexpressed miR-210 in hypoxia, potentially explaining its lower occupancy by TGP-210 (Figure 4). Giving confidence to target expression being necessary for compound bioactivity is the fact that apoptosis was not triggered in normoxic cells, i.e., in a regular oxygen environment, that do not overexpress miR-210. Overall, target expression levels and occupancy of a biologically relevant site are necessary variables to consider for small molecules that target structured RNAs. Furthermore, target engagement techniques, such as Chem-CLIP, can be used to verify on- and off-targets.
In the case of miR-210, Inforna identified lead RNA motif-small molecule interactions that are uniquely displayed in highly expressed, functional sites. Given the high concentration of bystander RNAs (rRNA, tRNA) and RNA isoforms, there are small molecules that bind motifs found in multiple RNAs.84,85 As an example, Inforna identified a small molecule with overlapping affinity for the functional Drosha sites present in both pri-miR-515 and pri-miR-885.78,86 Selective inhibition of pri-miR-515 was further compounded by the ~2.5-fold increased expression of miR-885 relative to miR-515 (Figure 4). In order to discriminate between these two miRNAs, Inforna queried adjacent structured regions for a lead small molecule, enabling optimization through a modular approach in which multiple motifs can be targeted with the same molecule.73 While many transcripts may contain the specific RNA motif targeted by a small molecule, not all of them will be presented in functional sites, while even fewer RNAs will contain two targetable sites separated by defined, specific distances (Figure 4). By screening a library of RNA-binding modules separated by different linker lengths,87 a dimeric compound, Targaprimir-515 (TGP-515), was developed to selectively inhibit production of miR-515 vs miR-885.73 Treatment with TGP-515 in a nonmigratory breast cancer cell line resulted in the increased biosynthesis of sphingosine 1-phosphate (S1P) through the derepression of sphingosine kinase 1 (SK1), triggering a migratory phenotype. This multivalent approach has been also been applied to target other specific RNAs successfully.88,89
The selectivity of TGP-515 to inhibit miR-515 biogenesis and modulate downstream biology was validated through several approaches. In a similar manner to the miR-210 studies, a Chem-CLIP probe was synthesized, and competition with TGP-515 indicated selective occupancy of only pri-miR-515, and not pri-miR-885. The selectivity of the dimer compound was rigorously confirmed through quantitative polymerase chain reaction (qPCR) profiling, RNA-seq, and global neo-protein studies, demonstrating on-target pathway activation and limited off-target effects in MCF-7 cells (Figure 5). Interestingly, proteomic studies indicated that human epidermal growth factor receptor 2 (HER2) was the most upregulated protein, which is normally not highly abundant in basal MCF-7 cells. Pretreatment with TGP-515 in various HER2-negative cell lines sensitized cells to the anti-HER2 therapies, Herceptin and Kadcyla, proving the hypothesis that increased HER2 production upon TGP-515 treatment can render insensitive cells sensitive to targeted therapies. Generally, the selectivity of a small molecule for its target is one of the first considerations when designing chemical probes. In this study, potential off-targets were identified computationally through Inforna, and suboptimal selectivity was overcome through the use of multivalency. Ultimately, validation and selectivity studies were able to identify a potential precision medicine approach for difficult-to-treat cancers.
Figure 5.
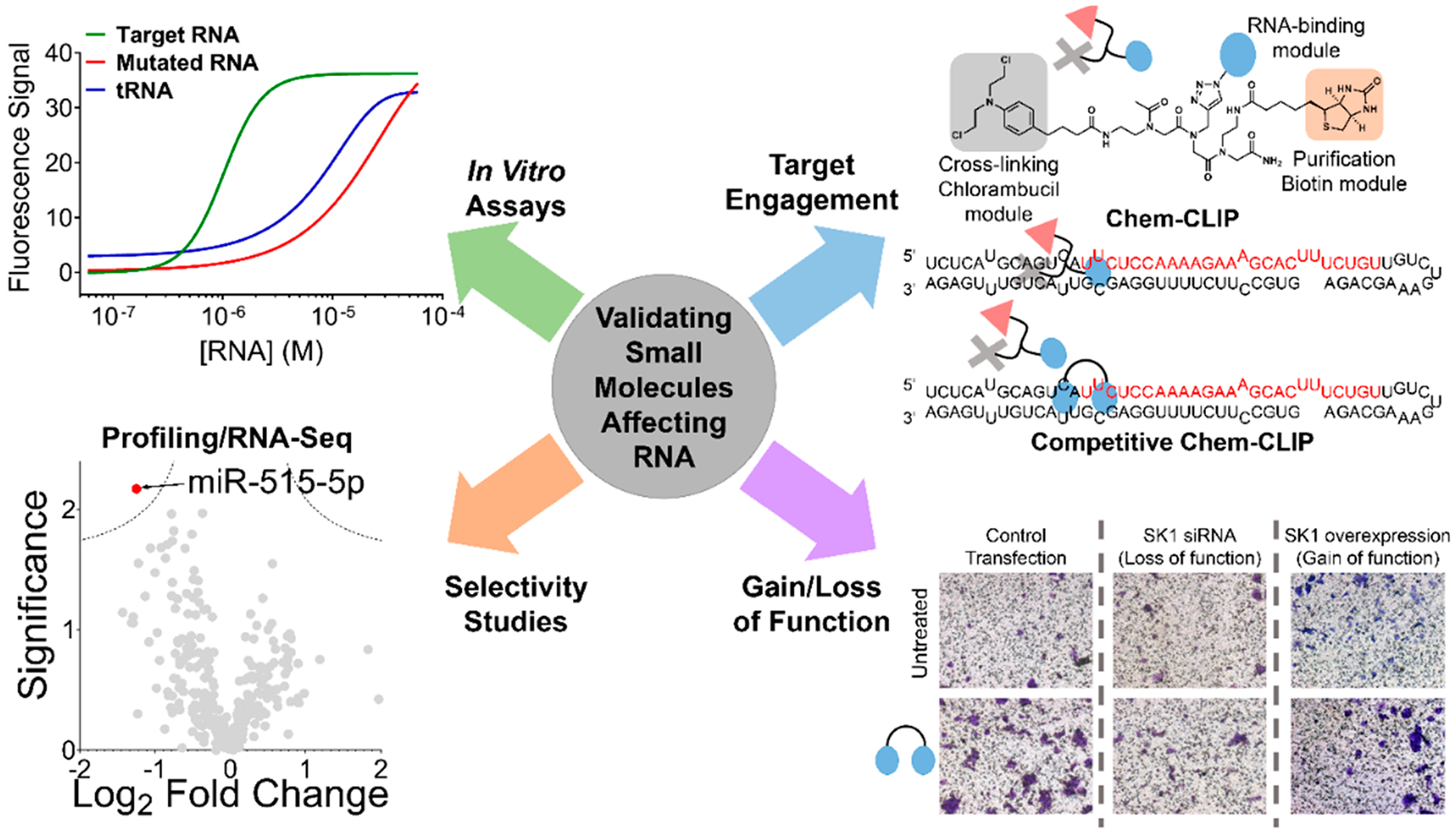
Tools to assess target engagement and selectivity of small molecules targeting RNA. Developing small molecules against RNA starts with identification of a hit, whether through Inforna (Figure 3) or screening approaches (target-based, phenotypic, fragment-based, DNA-encoded, etc.). Considering the factors from Figure 4, the hit must then be validated and optimized, including for in vitro binding affinity to the RNA structural element over RNAs that do not contain the motif and other abundant RNA/DNAs. Further validation in vitro and in cells can be accomplished with target engagement approaches that use chemical probing methods that measure RNA enrichment (Chem-CLIP) or RNA depletion (Competitive (C)-Chem-CLIP), among others. Comprehensively evaluating cellular selectivity on a transcriptome- and proteome-wide scale is also part of the workflow to validate a small molecule RNA target. After demonstration of selective on-target effects, the compound’s functional effect must then be validated in more advanced models, including the effect of a gain or loss in expression of the target.
In a similar case to miR-515, a monomeric compound inhibited the biogenesis of both miR-377 and miR-421 by binding to their common Dicer functional sites.90 Fortuitously, Inforna identified a lead compound that bound to an adjacent RNA motif in the pre-miR-377 hairpin that was not present in pre-miR-421. Upon optimization of the linker spacing between the RNA-binding modules that bound the two sites in premiR-377, the resulting dimeric compound selectively inhibited pre-miR-377, effecting angiogenesis through modulation of VEGFA protein.
Thus, Inforna has proven to be broadly applicable for the purposeful targeting of functional sites in human RNAs with selective, bioactive small molecules, while also providing comprehensive data sets defining RNA landscapes that are key for selective targeting and for identifying chemotypes with RNA-binding capacities. In addition, Inforna can be applied to develop multivalent ligands with improved selectivity and potency for RNAs that display unique targetable RNA motifs.
EXPLOITING RNA STRUCTURE FOR CHEMICAL PROBE DESIGN
Many proteins are difficult to drug directly since they lack traditionally targeted small molecule binding pockets; that is, they have intrinsically disordered domains. Therefore, affecting the coding mRNA upstream of protein production may present a more viable therapeutic option to decrease aberrant protein levels. One aspect of protein folding only recently receiving attention for its possible functional roles is intrinsically disordered regions.91,92 Indeed, only a small fraction of protein crystal structures recorded in the Protein Data Bank (PDB) do not possess disordered regions. These regions could represent a protein–protein interface or potential allosteric site, the folding of which is induced by binding of another protein, metabolite, or small molecule drug. Indeed, the study of molecular recognition of these protein structures by small molecules has provided well-informed paths toward drug design.93
One clinically relevant intrinsically disordered protein is α-synuclein, which is causative of neurodegeneration in Parkinson’s disease due to its aggregation in Lewy bodies.94 While α-synuclein protein is intrinsically disordered, the SNCA mRNA that encodes this pathogenic protein has a highly structured iron responsive element (IRE) in its 5′ untranslated region (UTR) that controls its translation.95 Inforna identified a small molecule, Synucleozid, that directly binds an A bulge in the IRE, inhibiting translation in cells and providing a cytoprotective effect.96 Direct molecular recognition of the RNA by Synucleozid was validated using antisense oligonucleotide ligand binding site mapping (ASO-Bind-Map), a small molecule application of a previously developed technique.97 ASO-Bind-Map profiles binding sites of small molecules in vitro and in cells as the small molecule binds and stabilizes the target RNA’s structural element, thus interfering with ASO binding and subsequent RNase H-mediated cleavage. Targeting α-synuclein upstream of its pathogenic protein demonstrates that targeting RNA elements with extensive structure can inhibit canonical translation. Importantly, affecting disease-causing intrinsically disordered proteins encoded by structured RNAs may not be limited to mRNAs with IRE and could be broadly applied to other proteins that lack binding sites for traditional “drug-like” small molecules.
The biology of RNA repeat expansion disorders presents another notable target for small molecules that bind to structured regions. From a small molecule targeting standpoint, these repeat expansions form stable repeating structural units that can be exploited for targeting, especially with on-site compound synthesis using the disease-causing RNA as a catalyst for inhibitor synthesis.89,98 These repeats are potentially difficult to treat with ASOs that have to disrupt these stable RNA structures. Repeating transcripts are causative of >30 incurable diseases including ALS/FTD, fragile X-associated tremor ataxia syndrome (FXTAS), and DM1 and DM2.99 Disease-causing RNA repeats can be present within coding or noncoding regions, which can affect their downstream biology. In FXTAS and DM, repeats are present in the 5′ and 3′ UTRs, respectively. ASOs sterically blocking protein interactions within the RNA repeat expansions can therefore modulate downstream biology. In the case of FXTAS, however, ASOs can also decrease downstream translation, suggesting that targeting r(CGG) repeat expansions with ASOs could worsen, rather than alleviate, disease. The repeating units, however, are amenable for small molecule targeting, as they can allow for increased specificity through the use of multivalent ligands.
In the case of myotonic dystrophy type 1, the r(CUG) repeats trigger disease by sequestering muscle-blind like protein 1 (MBNL1), which regulates the alternative splicing of various transcripts, including the insulin receptor (IR) and muscle-specific chloride ion channel (Clnc1). This gain of function by the repeats thus results in aberrant splicing products and hence DM1 disease biology. In addition to spliceopathy, other phenotypic complications arise from the expanded repeat such as aggregation of mutant transcripts with RBPs in nuclear foci, activation of stress pathways, haploinsufficiency, alteration of neighboring genes, and dysregulation of translation.100–102 The binding affinity of MBNL1 for r(CUG) repeats has been measured in the nM range (~3–200 nM).103 Therefore, multivalent small molecules that bind the repeats with binding affinities also in the nM range were developed that can compete for binding with MBNL1, freeing it to resume its normal splicing functions. Thus, directly binding RNA to compete with RBPs presents a viable strategy to treat repeat expansion disorders. This strategy is in contrast to small molecules that form a ternary complex with the RNA and protein to treat SMA, as described above.
TOOLS TO ASSESS TARGET ENGAGEMENT AND SELECTIVITY OF RNA-BINDING SMALL MOLECULES
A significant amount of weight in the drug discovery process for any biomolecule is placed on in vitro binding affinity. While important, depending too heavily on binding studies can also be problematic, especially for dynamic RNA targets whose structure can vary in vitro vs in situ. Indeed, studies have generated ligands that interact with RNA with high affinity but display little to no bioactivity. This mismatch between optimized binding affinity and bioactivity could be due to many factors, including in vitro conditions significantly differing from in situ conditions (concentration/expression level, presence of other biomolecules, metal ions, etc.), in vitro models adopting different structures than the cellular RNA, the binding method itself, poor cellular permeability, localization of the small molecule to a cellular compartment where the target is not located, etc. Thus additional methods of analysis are required to validate target engagement (Figures 4 and 5).
Studying the in vitro selectivity of small molecules has been more or less standardized. Typically, binding affinities of the compound to the target RNA are quantified using various biophysical techniques (biolayer interferometry [BLI], surface plasmon resonance [SPR], NMR spectroscopy, microscale thermophoresis [MST], ESI-LC/MS, electrophoretic mobility shift assays [EMSA], etc.), that have been previously used for other biomolecules.104–106 In addition, studying RNA-small molecule interactions has also made the use of ligand or RNA-based fluorescence and fluorescence-based displacement assays.106,107 In these in vitro studies, the selectivity of ligands to the target RNA are measured relative to several controls, including point mutants of the target RNA that change its three-dimensional structure (mutating an internal loop into a base pair, for example), highly expressed RNAs (yeast tRNAs), or bulk DNA (fish sperm DNA). These initial studies provide a starting point to inform the on- and off-rate of the small molecule to bind to a specific region of the RNA in a general sense; however these assays may simplify the complexity of RNA structural dynamics in the cell, which typically exist across a population of states rather than a single explicit structure and where stability of RNA structures can be influenced by RBPs or post-transcriptional modifications.48,54
If we are indeed to develop guidelines for the selective recognition of RNA by small molecules, it is imperative to trace bioactivity to direct engagement of the target. Indeed, even among well-developed protein targeting modalities that have reached clinical trials, the compound action and mechanism of action remains not well-characterized, which can result in off-target effects.108 Until recently, target engagement methods had not been developed for RNA targets. These new methods, which employ cross-linking or cleavage, have been reviewed previously.75 In addition to target engagement, many profiling techniques are available to assess selectivity of small molecules targeting RNA, each with their own experimental advantages and liabilities.109,110 Below, we summarize three such methods.
Quantifying expression differences of RNA transcripts has traditionally been performed through real-time reverse transcription quantitative polymerase chain reaction (RT-qPCR). Indeed, new instrumentation and technologies have allowed for increased throughput while maintaining high sensitivity and resolution, even at the single-cell level.111,112 The use of RT-qPCR has been commonly used to measure differential miRNA expression (Figure 5). RT-qPCR miRNA profiling revealed that small molecule hits, defined by Inforna, that target pri-miR-96 or pri-miR-515 had comparable selectivity to ASOs targeting mature miR-96–5p or mature miR-515–5p, respectively (Figures 5 and 6). Potential liabilities with measuring RNA levels with RT-qPCR include the potential biases in melting temperatures and amplification efficiency, and the labor-intensive process can be prone to user error. RT-qPCR profiling as a tool to measure the transcriptome is still useful to measure a large quantity of genes, but it is used more importantly for validation of known pathways. The method’s reliance on gene specific primers potentially precludes its use as a discovery tool.
Figure 6.
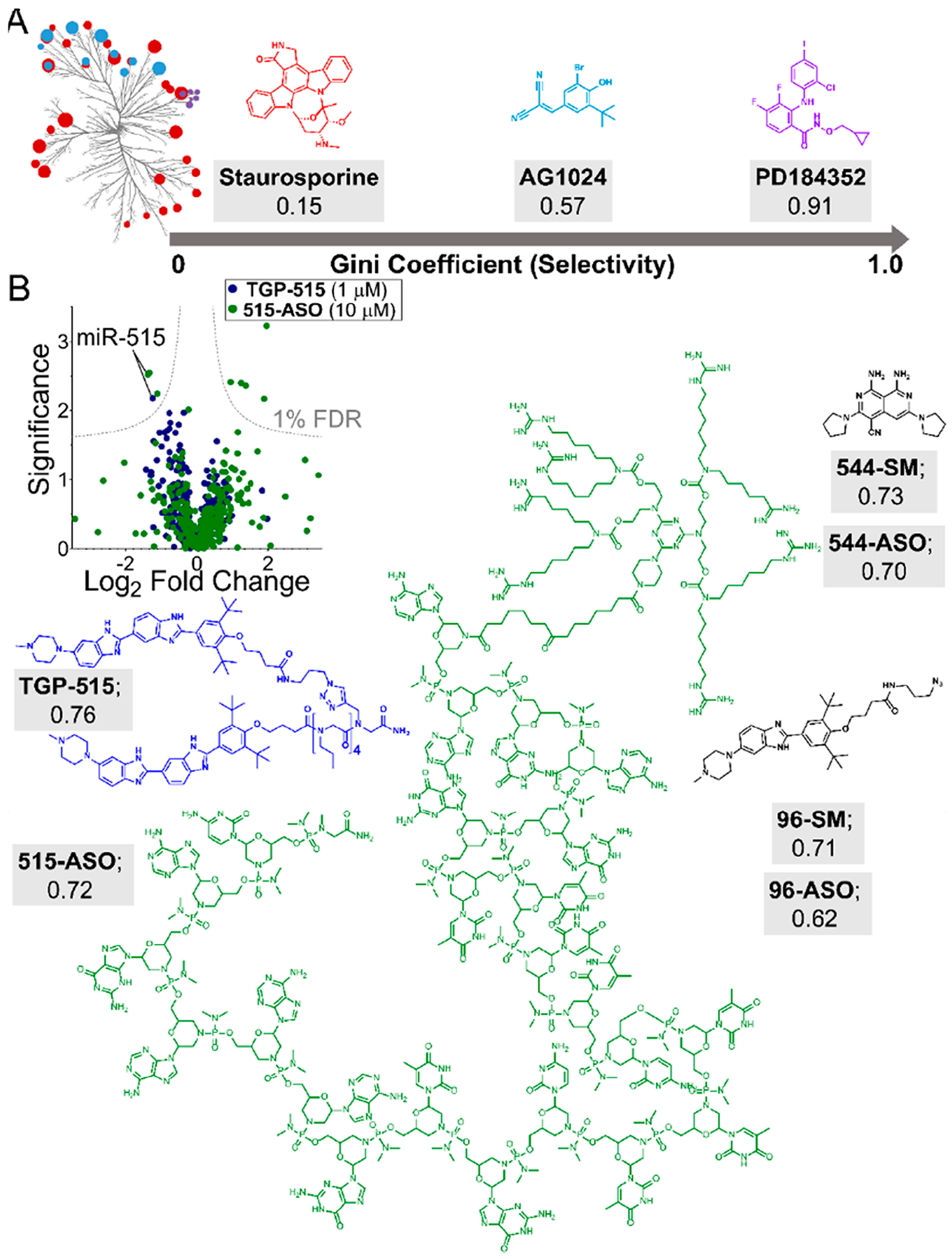
Quantitatively evaluating the selectivity of small molecules targeting RNA. (A) Data from profiling experiments can be used to quantify compound selectivity by calculating a Gini coefficient (GC). GC analysis of kinase inhibitors showed that promiscuous compounds (staurosporine) are characterized by values close to 0, while highly selective compounds exhibit Gini coefficient values close to 1 (PD184352; 0.91), with selective compounds being defined as >0.6. (B) GC analyses can be applied to profiling data (left), such as miRNA qPCR profiling data between small molecules (TGP-515; blue) and ASOs (515-ASO; green). Applying this analysis to various small molecule ligands targeting RNA structure indicates that they demonstrate high selectivity for their targets. Antisense oligonucleotides targeting RNA sequence are also selective for their targets. Shown are the structures and GC values of TGP-515 (blue) and Vivo-Morpholino ASO targeting miR-515 (green), in addition to other small molecule/ASO GC analyses. Overall, Gini coefficients provide a metric to quantitatively define compound selectivity. When applied to RNA targeting, GC analyses demonstrate that small molecules that recognize RNA structure can rival or exceed the selectivity of ASOs designed to bind via Watson–Crick base pairing.
Whole transcriptome, RNA sequencing (RNA-Seq) is the emerging technology to measure transcript abundance in gene expression studies. As RNA-Seq is based on counting reads aligned to reference sequences and does not require specific probe sequences for expression measurements, it overcomes limitations encountered by RT-qPCR profiling. Additionally, the dynamic range of RNA-Seq is only limited by the read count and can be performed with a limited amount of transcript material. A potential limitation with RNA-Seq has been standards for data processing and methods for normalization and statistical analysis, where best practices are still being developed and must be completed on a case-by-case basis. Differential expression analysis from RNA-Seq can also be used to quantitatively define on- and off-targets, as well as to study the downstream effects of compound treatment, i.e., downstream pathway analysis. A recent example includes utilizing RNA-Seq at an early time point in breast cancer cells treated with a compound that inhibited miR-515 biogenesis. This analysis showed the compound’s limited off-target effects on the transcriptome but also the upregulation of downstream proliferative pathways.73
Studying the effects on the downstream proteins takes investigations into small molecule selectivity beyond the transcriptome and can define how the modulation of RNA biology affects the proteome. Although compounds are affecting the transcriptome, proteomics can be used as a readout to measure the change in the protein landscape as a result of affecting RNA. Global proteomics, commonly used to study compounds selectively targeting proteins, or neo-protein synthesis, measuring proteins that are up- or down-regulated after compound treatment, can reveal novel biology. Indeed, the downstream neo-protein synthesis upon inhibition of miR-515 with TGP-515 revealed compound enhancement of HER2 protein expression in a normally HER2 negative cell line. Upon this realization, the levels of HER2 were able to be tuned in order to sensitize their treatment with anti-HER2 precision medicines.
Functional assays are important to verify that the compound is functioning through the predicted RNA-centric mode of action. With TGP-515, important loss- and gain-of-function assays were performed to validate the sensitivity of HER2 negative cell lines affecting miR-515 and SK1 pathways. For example, an siRNA or chemical knockdown of SK1 ablated the migratory phenotype (Figure 5) and sensitivity to HER2 therapies. Similarly, overexpression of SK1 resulted in a similar sensitivity, indicating that derepression of SK1 by miR-515 inhibition was responsible for increased sensitivity to HER2-targeted therapies. In addition to confirmation of target engagement and selectivity through techniques such as Chem-CLIP and profiling, functional assays are necessary to ascertain that the compound is operating through the hypothesized mode of action.
QUANTIFICATION OF SELECTIVITY
Quantification of compound selectivity has previously been measured with kinase inhibitors.113 One particular metric is called a Gini coefficient, which was originally developed as a measure of statistical dispersion of wealth distribution to measure inequality.114 More recently, the Gini coefficient has been used to measure selectivity of kinase inhibitors as it expresses a frequency distribution of differential inhibition among a population of kinases. In brief, Gini coefficients range from 0, indicating a nonselective compound, to 1, indicating selectivity for a single target. For example, a kinase inhibitor that lacks selectivity (inhibits 78/85 kinases tested) had a Gini coefficient of 0.15 while inhibitors considered selective had Gini coefficients ranging from 0.69 to 0.91 (Figure 6).114,115 Applying such an analysis to RT-qPCR profiling data from miRNAs expressed in MCF-7 cells indicated similar selectivity of a small molecule that affects pri-miR-515 and a Vivo-Morpholino modified ASO that targets miR-515–5p (Gini coefficients of 0.75 and 0.72, respectively) (Figure 6). In another analysis, the small molecule and ASO targeting miR-96 displayed comparable levels of selectivity with Gini coefficient values of 0.71 and 0.62, respectively (Figure 6). Besides qPCR profiling data, Gini coefficient analysis can be applied to larger data sets. For example, an analysis on a transcriptome-wide microarray experiment highlighted the broad selectivity of both a small molecule targeting miR-544 (Gini coefficient = 0.73) and an ASO targeting miR-544 (Gini coefficient = 0.70).116 Interestingly, the monomeric miR-544 small molecule has highly drug-like properties, supporting the idea that selective small molecules targeting structured RNA motifs also occupy traditional “drug-like” chemical space. Overall, in order to be able to objectively measure the selectivity of small molecules interacting with RNA, metrics such as the Gini coefficient are useful tools to compare compound selectivity. Importantly, Gini coefficient analyses have shown that the selectivity of small molecules can rival or exceed that of ASOs.
DRUGLIKENESS AND BEYOND THE RULE OF 5
Drug discovery has long used a set of physicochemical parameters to define the chemical space that is most likely to become an orally bioavailable drug. As RNA has previously been considered an “undruggable” target, then perhaps the physicochemical profiles of orally bioactive drugs may not necessarily be the best measure for RNA-binding ligands. Indeed, not all drugs are created for the same purpose, which may inaccurately bias trends toward an average that is not necessarily true for the specific applications of lead compounds.117 For example, many small molecules differ in clearance rates and exposure to the tissue type of interest; i.e., blood–brain barrier penetrant compounds are different from systemic broad spectrum antibiotics. Therefore, the commonly used parameters that make drugs particularly attractive for oral delivery in protein targeting applications may not be appropriate for targeting RNA. In fact, there is an increasing trend of compounds that exist in a space that is extended from the Rule of 5 (eRo5) or even beyond the Rule of 5 (bRo5).118 Indeed, as the methods to analyze the selectivity of compounds has progressively become more rigorous (full proteomics and transcriptomics profiles) and with the greater rigor for compounds with defined modes of action, the landscape of what is considered a “drug” that selectively hits a defined drug target has significantly changed.108 Traditional ASOs, and other biologics such as antibody-drug conjugates, have also seen a rapid rise in approvals as FDA-approved drugs, suggesting that compounds that do not fit into traditional medicinal chemistry parameters are also viable as effective therapeutics.119
In pursuit of more selective and potent compounds, a recent trend has become known as “compound bloat”. That is, drugs and potential drug candidates have been increasing in molecular weight and changing the classic idea of drug-like compounds. As mentioned by others, this “compound bloat” has been in response to decreasing off-target effects that were not previously measurable with technologies available at the time the drug was developed (CRISPR screens, RNA-Seq, proteomics).108 Indeed, targeting multiple “hot sites” of the same target or using binding sites between two different surfaces (i.e., protein–protein interactions (PPI)) has now been implemented.120 However, these innovative techniques have required increased molecular weight and polar surface area, along with an increased number of hydrogen bond donors and acceptors.119
While many drug-like compounds are heavily tested in vitro before in cellulis evaluation, binding contacts are not the only factor that influences bioactivity when concerned with drug-like properties. Furthermore, bioactivity does not necessarily indicate on-target effects, as without proper target engagement and validation, the compound may work in a different mechanism than hypothesized. Indeed, focusing on optimal physicochemical properties is important toward discovering privileged RNA space, but more innovative and novel approaches to the problem of targeting RNA with small molecules may result in compounds that look more “undrug-like” than traditional small molecule therapeutics. More provocatively, chemical matter that affects RNA space most effectively may differ significantly from traditional guidelines used to identify, design, and optimize ligands that affect other biomolecules.
Insight into the physicochemical properties of RNA-binding small molecules can begin to be gleaned from interactions housed in Inforna79 and a recently created catalog named R-BIND.121 Analysis of the compounds in both databases revealed highly similar chemical properties to FDA approved drugs in DrugBank (Figure 7A).122 However, differences are indeed observed. For example, RNA binders on average have more aromatic rings (3.9 ± 1.9 vs 1.4 ± 1.4 in approved drugs) and more H bond donor (5.2 ± 4.5 vs 2.4 ± 0.5.0) and acceptor groups (8.5 ± 6.2 vs 5.1 ± 7.4).
Figure 7.
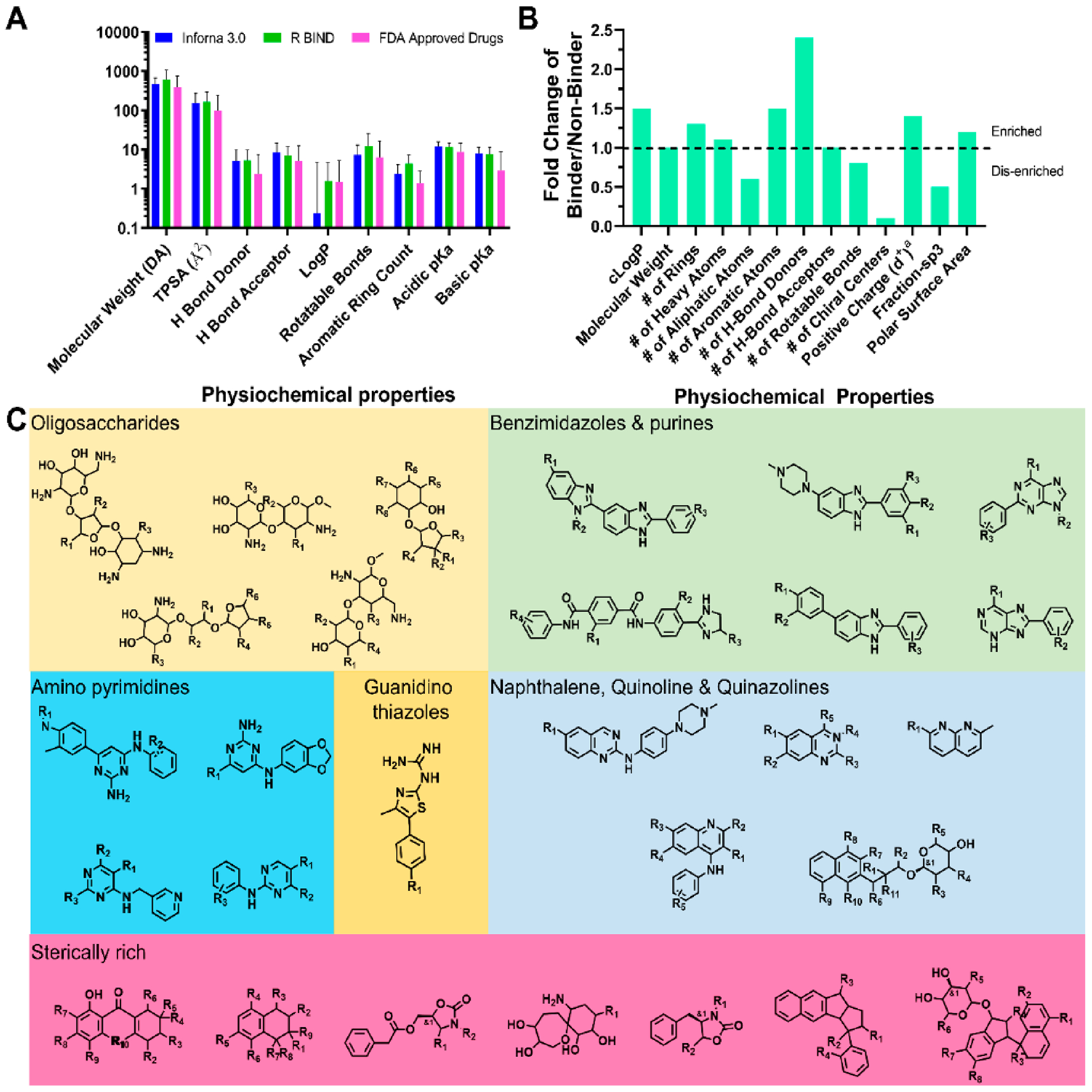
Physiochemical properties of RNA binders and common RNA-binding scaffolds. (A) Physiochemical properties of cataloged RNA binders contained within Inforna and R-BIND show convergence and correlate with properties of FDA approved drugs. FDA approved drugs were taken from DrugBank.122 (B) Properties that are enriched within RNA binders include greater positive charge at pH 7.4, number of H-bond donor and acceptor counts, and total polar surface area (TPSA). RNA binders also exhibit fewer chiral centers, aliphatic atoms, and rotational bonds compared to nonbinders. (C) Scaffolds contained within Inforna and R-BIND that exhibit RNA binding. These include oligosaccharides, benzimidazoles, purines, naphthalenes, quinolines, quinazolines, aminopyrimidines, and quinidine thiazoles. Interestingly, while most data generally point to planar molecules as RNA binders, some sterically rich compounds have been found to bind RNA.
In one large screen to identify novel compounds that bind RNA, a comparison of binding and nonbinding molecules showed that binders had on average 1.5-fold higher cLogP values, 1.3-fold more ring systems, 1.7-fold fewer aliphatic atoms, 1.5-fold more H-bond donors, 2.4-fold more H-bond acceptors, 10-fold fewer chiral centers, and slightly greater positive charge (δ+ of 0.4 vs 0.3 at pH 7.4) (Figure 7B).90 These data indicate a propensity for RNA binders to be structurally rigid and planar, likely driving binding of the RNA via π–π stacking and hydrogen bonding interactions with the nucleobases and/or the phosphate backbone.90
Scaffolds commonly associated with RNA binding that have yielded bioactive ligands include benzimidazoles, bis-benzimidazoles and phenylbenzimidazoles, aminopyrimidines, and quinazolines (Figure 7C). Other scaffolds identified from Inforna and R-BIND include oligosaccharides, naphthalenes, and purines. While the above physiochemical data and previous works suggest a preference for planar molecules, sterically rich compounds have also been identified such as oligosaccharides and those found in Figure 7C including spirocycles and fused ring spirocycles.
EMERGING MODALITIES FOR AFFECTING RNA BIOLOGY
As RNA presents a novel and challenging target, new approaches are needed to affect its downstream biology through its direct targeting. Recent work has demonstrated the use of other modalities to enable the targeted degradation of RNA, such as bleomycin conjugates (reviewed more in-depth previously),69,99,119,123 outside the traditional “drug-like” physicochemical properties.124 Many compounds (described above) have rescued disease-associated phenotypes through a binding mode of action; however, it may be advantageous to expand the mode of action to cleave the RNA target, whether directly or by recruiting endogenous nucleases (Figure 8). These “RNA degraders” can potentially ameliorate disease pathologies in a more potent and selective manner than simple binding compounds.72,89
Figure 8.
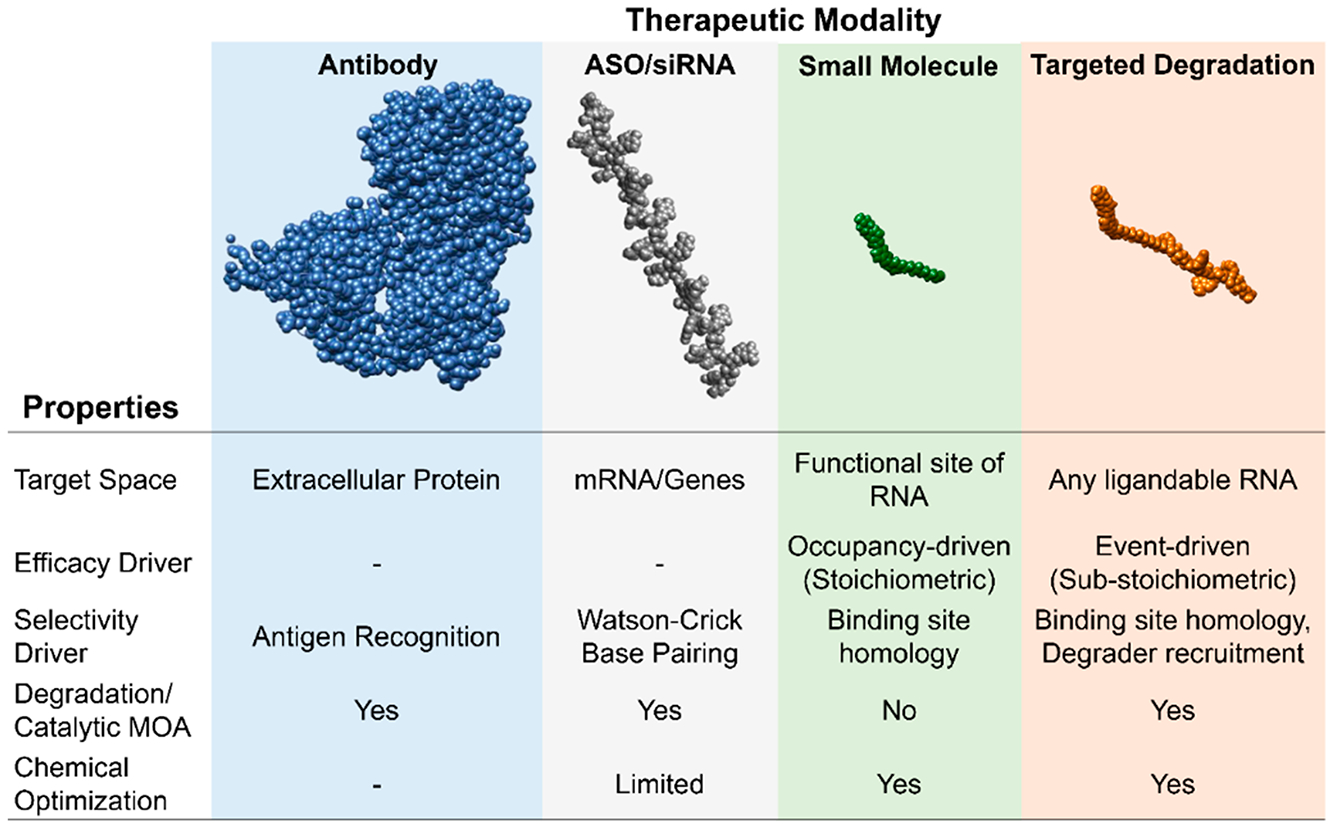
Properties of bioactive therapeutic modalities. Current and emerging strategies to affect downstream biology include antibodies (rituximab, PDB code 4KAQ), ASOs (Nusinersen), small molecules (TGP-210), and targeted degradation approaches (TGP-210 RIBOTAC). Each targets a unique space, but RIBOTACs can affect bioactivity of RNA without binding to a functional site. RIBOTACs also degrade their targets in a catalytic and substoichiometric fashion, thus allowing greater potency. Small-molecule-based modalities are advantageous as their physicochemical properties can be potentially medicinally optimized. ASO, small molecule, and targeted degradation models were made using the Online SMILES Generator (National Cancer Institute).
RNA degraders have been applied extensively to RNA repeat expansion disorders, as many of these highly stable structures can function as scaffolds to sequester proteins or to trigger repeat associated non-ATG (RAN) translation. Recently, Angelbello et al. demonstrated that RNA-degraders rescue DM1 disease pathology through intraperitoneal delivery of Cugamycin (a dimeric r(CUG)-binding compound appended to bleomycin), which selectively cleaves the repeat expansion in a preclinical mouse model.72 In addition to its therapeutic potential, these degrader molecules can also function as effective chemical probes, as described through the development of small molecule nucleic acid profiling by cleavage applied to RNA (RiboSNAP). RiboSNAP uses the cleavage activity of bleomycin to deplete the RNA target’s levels in patient-derived cells, thus demonstrating target engagement. RiboSNAP has also been applied with other noncoding RNAs to map small molecule binding sites,75 thus establishing RNA degraders and RiboSNAP as a new potential class of targeted therapeutic and chemical probing technique.
Recently, our lab has designed small molecules that can recruit a nuclease to a specific transcript, triggering its degradation. RIBOTACs, akin to PROTACs,125 recruit nucleases for the targeted degradation of structured RNA sites (Figure 8). Specifically, RIBOTACs locally recruit endogenous latent ribonuclease (RNase L) to a specific transcript, allowing for assembly of dimeric, active RNase L to selectively degrade the RNA target. This strategy is akin to a small molecule form of CRISPR, without the need for transfection of foreign guide RNA transcripts or proteins. In contrast, RIBOTACs exploit a cell’s endogenous machinery for nucleic acid disposal by bringing together all the “players” in a ternary complex (target RNA:RIBOTAC:RNase L). By locally eliminating aberrantly functioning RNA, rather than simply binding and inhibiting it, RIBOTACs offer specific advantages:
Occupancy vs Event Driven Processes.
Current pharmacological paradigms require small molecules to modulate downstream function by occupying enzyme active sites or blocking receptors, which requires high concentrations of drug at the correct position. Not only can maintaining high concentrations of drug lead to off-target effects, this occupancy-driven model requires RNA with accessible, “druggable” active sites, which may be difficult to target selectively. By eliminating the aberrant RNA, rather than binding and inhibiting them, RIBOTACs circumvent the occupancy-driven model and instead are event-driven: binding enables activation of ribonucleases to initiate degradation (Figures 8 and 9).
Figure 9.
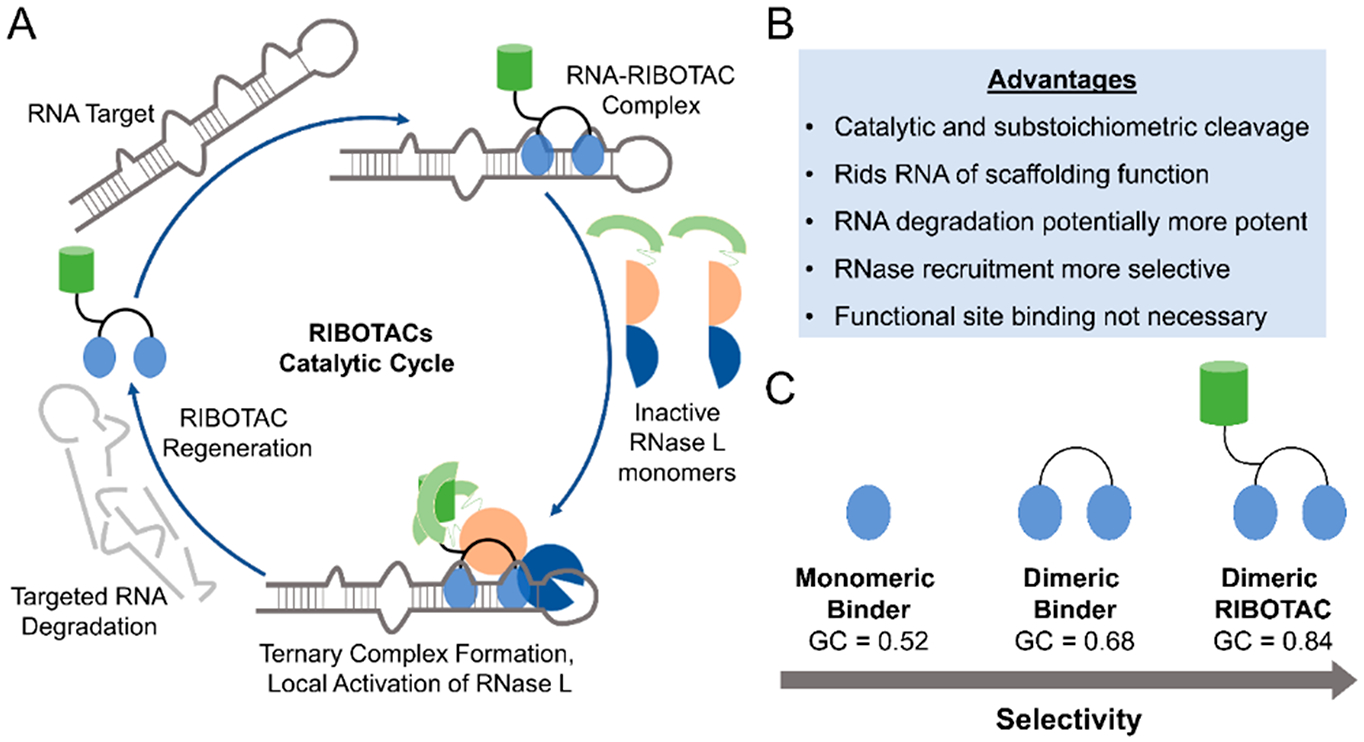
Ribonuclease targeting chimeras (RIBOTACs) as heterobifunctional degraders of RNA. (A) Taking cues from PROTACs and RNase H-based antisense oligonucleotide approaches, RIBOTACs are heterobifunctional compounds that recruit endogenous nucleases to degrade a targeted transcript. RIBOTACs can potentially increase potency of small molecules as they can catalytically and substoichiometrically degrade an RNA target. These RIBOTACs simply need to bind the target (not necessarily at a functional site) and use endogenous ribonuclease pathways to remove the RNA via targeted degradation, which also rids the RNA of any potential scaffolding functions with RBPs. Formation of the ternary complex may also increase selectivity as only meaningful interactions between the RNA:RIBOTAC:RNase L will result in cleavage. While this approach is potentially broadly applicable, development and optimization of both RNA binders and RNase-recruiting modules remain time-consuming, especially as there are a limited number of known RNase activators. Additionally, RIBOTACs that function through RNase L can have less pronounced effects on nuclear RNA, as RNase L is primarily cytoplasmic. (B) Advantages provided by the RIBOTAC approach. (C) Demonstration of increased selectivity of different RNA-binding modules, as indicated by GC analysis. The monomeric RNA-binding module that binds a single functional site on an RNA is less selective than the multivalent ligand targeting the same RNA. Adding an RNase L recruitment module to convert the dimeric compound into a RIBOTAC allows for increased selectivity, potentially due to the requirement of effective ternary complex formation between the RNA, RIBOTAC, and RNase L.
Increased Potency and Catalytic Activity.
Low levels of occupancy of RIBOTACs may be able to maintain a rate of RNA degradation that can provide the desired pharmacological effect. Previous small molecule catalytic activity was seen with Ru(bypy) photocatalysts as singlet oxygen generators,126 and RIBOTACs have demonstrated a similar level of substoichiometric degradation of their targets.127 This catalytic effect also suggests that RIBOTAC concentrations required for degradation may be much lower than those required for levels of inhibition by simple binding. As a catalytic degrader, RIBOTAC action also allows for greater RNA depletion over time; that is, low exposures can lead to meaningful knockdown, reducing potential off-target toxicity.
Avoiding Target Accumulation.
Drug binding can potentially stabilize RNA, thereby increasing its half-life. Once drug exposure drops below a certain inhibitory level, the disease-causing RNA will then persist. Degradation avoids this dilemma entirely, as it eliminates the target RNA completely. Additionally, degradation rather than binding also rids the targeted RNA of their potential disease-causing interactions with other biomolecules, such as RBPs like MBNL1 with r(CUG) expanded repeats. Accumulation of mutations in the drug target can also occur under selective pressure, which may negatively affect drug binding and occupancy, thereby leading to a decrease in efficacy, as observed with ribocil. Interestingly, RIBOTACs do not necessarily have to target an active site to form a ternary complex and degrade the target, as even an allosteric binder can be used. Degradation of the RNA also leads to a lower risk of the target complex evolving a resistant form, but upon formation of resistance, the modularity of RIBOTACs can potentially lead to the simple solution of using an alternative target binder. As ribonucleases are highly conserved and ubiquitous among cells, resistance to RIBOTAC-induced degradation may be much lower than observed for simple binding compounds.
Gain of Specificity.
Achieving good selectivity over off-targets still presents a significant challenge among small molecule inhibitors, especially in RNA targets that contain highly homologous binding sites. RIBOTACs achieve targeted degradation in two steps, first from binding of the RIBOTAC to the target and, second, upon nuclease recruitment to the target RNA (Figure 9). While the first step is mainly controlled by the affinity of the RIBOTAC to form binary complexes with its target RNA, the selectivity of the latter step can be appropriately adjusted for the RNA of interest via linker length or even by modulating the RNase recruiter. Additionally, as described above, compounds typically require binding to active sites within an RNA target to be effective; however RIBOTACs can be designed to bind structural motifs unique to a specific target RNA, thus enabling greater selectivity. Therefore, selectivity can be engineered into each component of the RIBOTAC to modulate the intrinsic binding affinity of the lone RNA-binding module to off-target RNAs. One important factor to consider for future RIBOTACs may be the role of binding kinetics (kon/koff rates). Indeed, with the advent of newer biophysical methods to analyze kinetics, recent studies have shown greater appreciation for on-/off-rates for drugs binding to their targets.128 These properties are further complicated upon accounting for the optimal presentation/rate of recruitment of the RNase recruitment module and the complexities of ternary complex formation with the RNA target and RNA-binding module in the cell, which are required for RIBOTACs to propagate cleavage.
Collectively, our inaugural studies suggest (i) RIBOTACs could be a general strategy broadly applicable to imbue RNA-targeted small molecules with cleavage capability, extending mode of action beyond simple binding and (ii) selectivity, as compared to the RNA-binding compound alone, can be enhanced by conjugation of a degradation module, as also observed with nonspecific kinase inhibitors converted to a PROTAC.129 Additionally, RIBOTACs can use guiding principles from studies into ternary complex formation and the role of kinetics for optimal activity in the PROTACs space when considering RNA-targeted degradation.130,131 Bearing this in mind, future studies to apply the RIBOTAC technology for the druggable transcriptome may require (i) medicinal chemistry of RNA-binding small molecule, linker, and RNase L recruitment modules, especially regarding on-/off-rates; (ii) studies toward the recruitment of other ribonuclease modalities, preferably with favorable physicochemical properties; (iii) new linker chemistries to optimize presentation of the bifunctional ends; and (iv) application of RIBOTACs to in vivo models. Indeed, a recent study has shown that a RIBOTAC using a heterocyclic recruiter of RNase L was able to substoichiometrically degrade pre-miR-21 to impede metastasis of breast cancer to the lung in a mouse model.132 Interestingly, selectivity and Gini coefficient analysis indicated that the RIBOTAC targeting miR-21 was more selective than the parent binding compounds, rivaling or exceeding that of protein-targeted drugs. Further downstream transcriptome and proteome-wide studies demonstrated broad inhibition of oncogenic pathways, as expected upon miR-21 depletion.132
CONCLUSION
As controlled RNA expression manages nearly every function in the human body, modulation of these phenomena represents an integral goal in biomedical research. As a result of chemical probes and small molecule drugs being developed for a tiny fraction of the proteome, a call to develop chemical probes (small molecules, degraders, biologics, etc.) for the entirety of the human proteome was recently announced (Target 2035).133 Indeed, this ambitious aspiration will require many groups willing to work on proteins in the “dark proteome” and the advancement and optimization of new and current technologies (DNA-encoded libraries, massively parallel high throughput screening, functional assays for understudied proteins, etc.).134,135 Another approach to increase coverage of chemical probes would be to study compounds that modulate protein levels by affecting them at the RNA level. Not only could compounds that target RNA effectively inhibit protein expression by binding to their precursor mRNA transcript, but they also could potentially activate proteins by modifying their associated regulatory RNAs (miRNAs, lncRNAs, etc.), thus providing a bilateral approach to affect protein biology.
While effective control of RNA expression using antisense technologies has been demonstrated, these modalities are still limited in their own unique ways, leading researchers to examine if the same level of control and selectivity can be achieved using small molecules. Highlights from this Perspective have illustrated that the selective molecular recognition of RNA with small molecules is possible through the interaction of ligands with structured RNA motif regions. Bioactivity of small molecule ligands requires more than just optimizing for high affinity in vitro binding interactions. Furthermore, the analysis of transcriptome- and downstream proteome-wide selectivity can determine how structural recognition can be exploited for specific RNA modulation. Importantly, target engagement and validation studies are essential to verify RNA-centric modes of action and to rescue disease-associated phenotypes through an expanding toolbox of techniques. Finally, RNA is indeed druggable, although the compounds used may not look like traditional drugs, such as heterobifunctional chimeric compounds that have demonstrated efficacy in selective RNA degradation.
As opportunities arise and challenges are met for RNA-targeting small molecules, it is an exciting time to propose a druggable transcriptome project in order to provide chemical probes for functional RNAs on a transcriptome-wide scale. Developing technologies, such as RIBOTACs, and currently existing methodologies, such as Inforna, can be used in conjunction to study and optimize small molecules targeting all functionally relevant RNAs broadly. As advancements and innovations in the design and discovery small molecules interacting with RNA continue, the use of these techniques can bring about a new paradigm in chemical biology and potentially launch research into new therapeutic modalities and viable medicines.
ACKNOWLEDGMENTS
We thank the members of our laboratory that have helped advance the science of small molecules interacting with RNA, especially human RNAs, over the past 15 years. We thank the funding agencies (National Institutes of Health Grants R01 GM97455, R01 CA249180, P01 NS099114, and DP1 NS096898 to M.D.D.; the Department of Defense Peer Reviewed Medical Research Program Grant W81XWH-18-0718 to M.D.D.; ACS Medicinal Chemistry Predoctoral Fellowship to M.G.C.) that have supported our work in this area.
ABBREVIATIONS USED
- 2′−5′ A
2′−5′ linked oligoadenylate
- 2DCS
two-dimensional combinatorial screening
- AGO/RISC
argonaute/RNA-induced silencing complex
- ASO
antisense oligonucleotide
- ASO-Bind-Map
antisense oligonucleotide ligand binding site mapping
- BLI
biolayer interferometry
- bRo5
beyond rule of 5
- C-Chem-CLIP
competitive cross-linking and isolation by pulldown
- Chem-CLIP
chemical cross-linking and isolation by pulldown
- CRISPR
clustered regularly interspaced short palindromic repeats
- DM1/DM2
myotonic dystrophy type I/II
- DMD
Duchenne’s muscular dystrophy
- EMSA
electrophoretic mobility shift assay
- ENCODE
Encyclopedia of DNA Elements
- eRo5
extended rule of 5
- ESE
exonic splicing enhancer
- ESI-LC/MS
electrospray ionization liquid chromatography/mass spectrometry
- FMN
flavin mononucleotide
- FTD/ALS
frontotemporal dementia/amyotrophic lateral sclerosis
- FXTAS
fragile X-associated tremor ataxia syndrome
- GC
Gini coefficient
- HER2
human epidermal growth factor receptor 2
- HiT-StARTS
high throughput structure–activity relationships through sequencing
- IDP
intrinsically disordered protein
- IRE
iron responsive element
- IRP
iron responsive protein
- KRAS
Kirsten rat sarcoma proto-oncogene
- lncRNA
long noncoding RNA
- MBNL1
muscle-blind like protein 1
- miR
microRNA
- MST
microscale thermophoresis
- PPI
protein–protein interactions
- pre-miR
precursor microRNA hairpin
- pre-mRNA
precursor messenger RNA
- pri-miR
primary microRNA transcript
- PROTAC
proteolysis targeting chimera
- RiboSNAP
small-molecule nucleic acid profiling by cleavage applied to RNA
- RIBOTAC
ribonuclease targeting chimera
- RNase
ribonuclease
- RNA-Seq
RNA sequencing
- RT-qPCR
real time reverse transcription quantitative polymerase chain reaction
- S1P
sphingosine 1-phosphate
- shRNA
short hairpin RNA
- siRNA
short interfering RNA
- SK1
sphingosine kinase 1
- SMA
spinomuscular atrophy
- SMN
survival motor neuron protein
- SPR
surface plasmon resonance
- TGP-210
targapremir-210
- TGP-515
targaprimir-515
- U1 snRNA
U1 small nuclear RNA
- UTR
untranslated region
- VEGFA
vascular endothelial growth factor A
Biographies
Matthew G. Costales completed his B.S. in Chemistry at the University of Maryland, College Park. He spent the following years as an Oak Ridge Institute for Science and Education Fellow in the Immunobiology Group in the Center for Food Safety and Applied Nutrition at the Food and Drug Administration. He then received his Ph.D. in Chemical Biology under the supervision of Prof. Matthew D. Disney at the Scripps Research Institute in Florida for his dissertation research on strategies to selectively target RNA with small molecules. Currently, he is working in the Neuro Discovery group at Dicerna Pharmaceuticals.
Jessica L. Childs-Disney attended Messiah College where she received her B.S. in Chemistry. She then completed her Ph.D. in Biophysical Chemistry, studying how to induce RNA misfolding under the tutelage of Prof. Douglas H. Turner, and postdoctoral studies at the Swiss Federal Institute of Technology (ETH) Zurich. After spending 4 years as an Assistant Professor at Canisius College, she moved to The Scripps Research Institute in Florida where she is a senior staff scientist.
Hafeez S. Haniff finished his B.S. in Chemistry at the University of Central Florida. He is currently completing his Ph.D. in the lab of Prof. Matthew D. Disney at the Scripps Research Institute in Florida. His dissertation research is focused on the development and application of RNA-focused small molecules libraries to modulate the function of RNA-based diseases for regenerative medicine.
Matthew D. Disney received his B.S. in Chemistry from the University of Maryland, College Park. He then completed his M.S. and Ph.D. in Biophysical Chemistry with Prof. Douglas H. Turner. He completed postdoctoral studies in the lab of Prof. Peter H. Seeberger at the Massachusetts Institute of Technology and the Swiss Federal Institute of Technology (ETH) Zurich. He started his independent research career and eventually joined the faculty in the bicoastal Department of Chemistry at The Scripps Research Institute in Florida, where he is Full Professor. His group focuses on developing small molecules to selectively drug human RNAs. His research has also led to the founding of Expansion Therapeutics.
Footnotes
The authors declare the following competing financial interest(s): M.D.D. is a founder of Expansion Therapeutics.
REFERENCES
- (1).Lander ES; Linton LM; Birren B; Nusbaum C; Zody MC; Baldwin J; Devon K; Dewar K; Doyle M; FitzHugh W; Funke R; Gage D; Harris K; Heaford A; Howland J; Kann L; Lehoczky J; LeVine R; McEwan P; McKernan K; Meldrim J; Mesirov JP; Miranda C; Morris W; Naylor J; Raymond C; Rosetti M; Santos R; Sheridan A; Sougnez C; Stange-Thomann Y; Stojanovic N; Subramanian A; Wyman D; Rogers J; Sulston J; Ainscough R; Beck S; Bentley D; Burton J; Clee C; Carter N; Coulson A; Deadman R; Deloukas P; Dunham A; Dunham I; Durbin R; French L; Grafham D; Gregory S; Hubbard T; Humphray S; Hunt A; Jones M; Lloyd C; McMurray A; Matthews L; Mercer S; Milne S; Mullikin JC; Mungall A; Plumb R; Ross M; Shownkeen R; Sims S; Waterston RH; Wilson RK; Hillier LW; McPherson JD; Marra MA; Mardis ER; Fulton LA; Chinwalla AT; Pepin KH; Gish WR; Chissoe SL; Wendl MC; Delehaunty KD; Miner TL; Delehaunty A; Kramer JB; Cook LL; Fulton RS; Johnson DL; Minx PJ; Clifton SW; Hawkins T; Branscomb E; Predki P; Richardson P; Wenning S; Slezak T; Doggett N; Cheng JF; Olsen A; Lucas S; Elkin C; Uberbacher E; Frazier M; Gibbs RA; Muzny DM; Scherer SE; Bouck JB; Sodergren EJ; Worley KC; Rives CM; Gorrell JH; Metzker ML; Naylor SL; Kucherlapati RS; Nelson DL; Weinstock GM; Sakaki Y; Fujiyama A; Hattori M; Yada T; Toyoda A; Itoh T; Kawagoe C; Watanabe H; Totoki Y; Taylor T; Weissenbach J; Heilig R; Saurin W; Artiguenave F; Brottier P; Bruls T; Pelletier E; Robert C; Wincker P; Smith DR; Doucette-Stamm L; Rubenfield M; Weinstock K; Lee HM; Dubois J; Rosenthal A; Platzer M; Nyakatura G; Taudien S; Rump A; Yang H; Yu J; Wang J; Huang G; Gu J; Hood L; Rowen L; Madan A; Qin S; Davis RW; Federspiel NA; Abola AP; Proctor MJ; Myers RM; Schmutz J; Dickson M; Grimwood J; Cox DR; Olson MV; Kaul R; Raymond C; Shimizu N; Kawasaki K; Minoshima S; Evans GA; Athanasiou M; Schultz R; Roe BA; Chen F; Pan H; Ramser J; Lehrach H; Reinhardt R; McCombie WR; de la Bastide M; Dedhia N; Blocker H; Hornischer K; Nordsiek G; Agarwala R; Aravind L; Bailey JA; Bateman A; Batzoglou S; Birney E; Bork P; Brown DG; Burge CB; Cerutti L; Chen HC; Church D; Clamp M; Copley RR; Doerks T; Eddy SR; Eichler EE; Furey TS; Galagan J; Gilbert JG; Harmon C; Hayashizaki Y; Haussler D; Hermjakob H; Hokamp K; Jang W; Johnson LS; Jones TA; Kasif S; Kaspryzk A; Kennedy S; Kent WJ; Kitts P; Koonin EV; Korf I; Kulp D; Lancet D; Lowe TM; McLysaght A; Mikkelsen T; Moran JV; Mulder N; Pollara VJ; Ponting CP; Schuler G; Schultz J; Slater G; Smit AF; Stupka E; Szustakowki J; Thierry-Mieg D; Thierry-Mieg J; Wagner L; Wallis J; Wheeler R; Williams A; Wolf YI; Wolfe KH; Yang SP; Yeh RF; Collins F; Guyer MS; Peterson J; Felsenfeld A; Wetterstrand KA; Patrinos A; Morgan MJ; de Jong P; Catanese JJ; Osoegawa K; Shizuya H; Choi S; Chen YJ; Szustakowki J Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- (2).Anderson NL; Anderson NG Proteome and proteomics: new technologies, new concepts, and new words. Electrophoresis 1998, 19, 1853–1861. [DOI] [PubMed] [Google Scholar]
- (3).Bertozzi CR; Sasisekharan R Glycomics In Essentials of Glycobiology, 2nd ed.; Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, Eds.; Cold Spring Harbor Laboratory Press: New York, 2009. [PubMed] [Google Scholar]
- (4).Ejsing CS; Sampaio JL; Surendranath V; Duchoslav E; Ekroos K; Klemm RW; Simons K; Shevchenko A Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 2136–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).McGettigan PA Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol 2013, 17, 4–11. [DOI] [PubMed] [Google Scholar]
- (6).Yang Y; Adelstein SJ; Kassis AI Target discovery from data mining approaches. Drug Discovery Today 2012, 17, S16–S23. [DOI] [PubMed] [Google Scholar]
- (7).Hangauer MJ; Vaughn IW; McManus MT Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013, 9, No. e1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Djebali S; Davis CA; Merkel A; Dobin A; Lassmann T; Mortazavi A; Tanzer A; Lagarde J; Lin W; Schlesinger F; Xue C; Marinov GK; Khatun J; Williams BA; Zaleski C; Rozowsky J; Röder M; Kokocinski F; Abdelhamid RF; Alioto T; Antoshechkin I; Baer MT; Bar NS; Batut P; Bell K; Bell I; Chakrabortty S; Chen X; Chrast J; Curado J; Derrien T; Drenkow J; Dumais E; Dumais J; Duttagupta R; Falconnet E; Fastuca M; Fejes-Toth K; Ferreira P; Foissac S; Fullwood MJ; Gao H; Gonzalez D; Gordon A; Gunawardena H; Howald C; Jha S; Johnson R; Kapranov P; King B; Kingswood C; Luo OJ; Park E; Persaud K; Preall JB; Ribeca P; Risk B; Robyr D; Sammeth M; Schaffer L; See L-H; Shahab A; Skancke J; Suzuki AM; Takahashi H; Tilgner H; Trout D; Walters N; Wang H; Wrobel J; Yu Y; Ruan X; Hayashizaki Y; Harrow J; Gerstein M; Hubbard T; Reymond A; Antonarakis SE; Hannon G; Giddings MC; Ruan Y; Wold B; Carninci P; Guigó R; Gingeras TR Landscape of transcription in human cells. Nature 2012, 489, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wade JT; Grainger DC Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol 2014, 12, 647–653. [DOI] [PubMed] [Google Scholar]
- (10).Liu G; Mattick JS; Taft RJ A meta-analysis of the genomic and transcriptomic composition of complex life. Cell Cycle 2013, 12, 2061–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Necsulea A; Soumillon M; Warnefors M; Liechti A; Daish T; Zeller U; Baker JC; Grützner F; Kaessmann H The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014, 505, 635–640. [DOI] [PubMed] [Google Scholar]
- (12).Makley LN; Gestwicki JE Expanding the number of “druggable” targets: non-enzymes and protein-protein interactions. Chem. Biol. Drug Des 2013, 81, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Limbird LE The receptor concept: a continuing evolution. Mol. Interventions 2004, 4, 326–336. [DOI] [PubMed] [Google Scholar]
- (14).Lazo JS; Sharlow ER Drugging undruggable molecular cancer targets. Annu. Rev. Pharmacol. Toxicol 2016, 56, 23–40. [DOI] [PubMed] [Google Scholar]
- (15).Dang CV; Reddy EP; Shokat KM; Soucek L Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ostrem JML; Shokat KM Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discovery 2016, 15, 771–785. [DOI] [PubMed] [Google Scholar]
- (17).Kessler D; Gmachl M; Mantoulidis A; Martin LJ; Zoephel A; Mayer M; Gollner A; Covini D; Fischer S; Gerstberger T; Gmaschitz T; Goodwin C; Greb P; Häring D; Hela W; Hoffmann J; Karolyi-Oezguer J; Knesl P; Kornigg S; Koegl M; Kousek R; Lamarre L; Moser F; Munico-Martinez S; Peinsipp C; Phan J; Rinnenthal J; Sai J; Salamon C; Scherbantin Y; Schipany K; Schnitzer R; Schrenk A; Sharps B; Siszler G; Sun Q; Waterson A; Wolkerstorfer B; Zeeb M; Pearson M; Fesik SW; McConnell DB Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 15823–15829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lou K; Steri V; Ge AY; Hwang YC; Yogodzinski CH; Shkedi AR; Choi ALM; Mitchell DC; Swaney DL; Hann B; Gordan JD; Shokat KM; Gilbert LA KRASG12C inhibition produces a driver-limited state revealing collateral dependencies. Sci. Signaling 2019, 12, No. eaaw9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Pettersson M; Crews CM PROteolysis TArgeting Chimeras (PROTACs)—past, present and future. Drug Discovery Today: Technol 2019, 31, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Backus KM; Correia BE; Lum KM; Forli S; Horning BD; Gonzalez-Paez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; Wolan DW; Cravatt BF Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Parker CG; Galmozzi A; Wang Y; Correia BE; Sasaki K; Joslyn CM; Kim AS; Cavallaro CL; Lawrence RM; Johnson SR; Narvaiza I; Saez E; Cravatt BF Ligand and target discovery by fragment-based screening in human cells. Cell 2017, 168, 527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).O’Rourke JR; Swanson MS Mechanisms of RNA-mediated disease. J. Biol. Chem 2009, 284, 7419–7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cooper TA; Wan L; Dreyfuss G RNA and disease. Cell 2009, 136, 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sztuba-Solinska J; Chavez-Calvillo G; Cline SE Unveiling the druggable RNA targets and small molecule therapeutics. Bioorg. Med. Chem 2019, 27, 2149–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ursu A; Vezina-Dawod S; Disney MD Methods to identify and optimize small molecules interacting with RNA (SMIRNAs). Drug Discovery Today 2019, 24, 2002–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rizvi NF; Smith GF RNA as a small molecule druggable target. Bioorg. Med. Chem. Lett 2017, 27, 5083–5088. [DOI] [PubMed] [Google Scholar]
- (27).Di Giorgio A; Duca M Synthetic small-molecule RNA ligands: future prospects as therapeutic agents. MedChemComm 2019, 10, 1242–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Morgan BS; Forte JE; Hargrove AE Insights into the development of chemical probes for RNA. Nucleic Acids Res. 2018, 46, 8025–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Warner KD; Hajdin CE; Weeks KM Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discovery 2018, 17, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Begg EJ; Barclay ML Aminoglycosides - 50 years on. Br. J. Clin. Pharmacol 1995, 39, 597–603. [PMC free article] [PubMed] [Google Scholar]
- (31).Disney MD; Labuda LP; Paul DJ; Poplawski SG; Pushechnikov A; Tran T; Velagapudi SP; Wu M; Childs-Disney JL Two-dimensional combinatorial screening identifies specific aminoglycoside-RNA internal loop partners. J. Am. Chem. Soc 2008, 130, 11185–11194. [DOI] [PubMed] [Google Scholar]
- (32).Datlinger P; Rendeiro AF; Schmidl C; Krausgruber T; Traxler P; Klughammer J; Schuster LC; Kuchler A; Alpar D; Bock C Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Moffat J; Sabatini DM Building mammalian signalling pathways with RNAi screens. Nat. Rev. Mol. Cell Biol 2006, 7, 177–187. [DOI] [PubMed] [Google Scholar]
- (34).Howe JA; Wang H; Fischmann TO; Balibar CJ; Xiao L; Galgoci AM; Malinverni JC; Mayhood T; Villafania A; Nahvi A; Murgolo N; Barbieri CM; Mann PA; Carr D; Xia E; Zuck P; Riley D; Painter RE; Walker SS; Sherborne B; de Jesus R; Pan W; Plotkin MA; Wu J; Rindgen D; Cummings J; Garlisi CG; Zhang R; Sheth PR; Gill CJ; Tang H; Roemer T Selective small-molecule inhibition of an RNA structural element. Nature 2015, 526, 672–677. [DOI] [PubMed] [Google Scholar]
- (35).DeFrancesco L Drug pipeline: 1Q17. Nat. Biotechnol 2017, 35, 400. [DOI] [PubMed] [Google Scholar]
- (36).McDonald CM; Campbell C; Torricelli RE; Finkel RS; Flanigan KM; Goemans N; Heydemann P; Kaminska A; Kirschner J; Muntoni F; Osorio AN; Schara U; Sejersen T; Shieh PB; Sweeney HL; Topaloglu H; Tulinius M; Vilchez JJ; Voit T; Wong B; Elfring G; Kroger H; Luo X; McIntosh J; Ong T; Riebling P; Souza M; Spiegel RJ; Peltz SW; Mercuri E Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [DOI] [PubMed] [Google Scholar]
- (37).Auld DS; Thorne N; Maguire WF; Inglese J Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 3585–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Auld DS; Lovell S; Thorne N; Lea WA; Maloney DJ; Shen M; Rai G; Battaile KP; Thomas CJ; Simeonov A; Hanzlik RP; Inglese J Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 4878–4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Rao VK; Kapp D; Schroth M Gene therapy for spinal muscular atrophy: an emerging treatment option for a devastating disease. J. Manag. Care Spec. Pharm 2018, 24, S3–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cheung AK; Hurley B; Kerrigan R; Shu L; Chin DN; Shen Y; O’Brien G; Sung MJ; Hou Y; Axford J; Cody E; Sun R; Fazal A; Fridrich C; Sanchez CC; Tomlinson RC; Jain M; Deng L; Hoffmaster K; Song C; Van Hoosear M; Shin Y; Servais R; Towler C; Hild M; Curtis D; Dietrich WF; Hamann LG; Briner K; Chen KS; Kobayashi D; Sivasankaran R; Dales NA Discovery of small molecule splicing modulators of survival motor neuron-2 (SMN2) for the treatment of spinal muscular atrophy (SMA). J. Med. Chem 2018, 61, 11021–11036. [DOI] [PubMed] [Google Scholar]
- (41).Charnas L; Voltz E; Pfister C; Peters T; Hartmann A; Berghs-Clairmont C; Praestgaard J; de Raspide M; Deconinck N; Born A; Baranello G; Bertini E; Schara U; Goemans N; Roubenoff R Safety and efficacy findings in the first-in-human trial (FIH) of the oral splice modulator branaplam in type 1 spinal muscular atrophy (SMA): interim results. Neuromuscul. Disord 2017, 27, S207–S208. [Google Scholar]
- (42).Wang J; Schultz PG; Johnson KA Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E4604–E4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sivaramakrishnan M; McCarthy KD; Campagne S; Huber S; Meier S; Augustin A; Heckel T; Meistermann H; Hug MN; Birrer P; Moursy A; Khawaja S; Schmucki R; Berntenis N; Giroud N; Golling S; Tzouros M; Banfai B; Duran-Pacheco G; Lamerz J; Hsiu Liu Y; Luebbers T; Ratni H; Ebeling M; Cléry A; Paushkin S; Krainer AR; Allain FHT; Metzger F Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat. Commun 2017, 8, 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Yang W-Y; Wilson HD; Velagapudi SP; Disney MD Inhibition of non-ATG translational events in cells via covalent small molecules targeting RNA. J. Am. Chem. Soc 2015, 137, 5336–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Holley RW; Apgar J; Everett GA; Madison JT; Marquisee M; Merrill SH; Penswick JR; Zamir A Structure of a ribonucleic acid. Science 1965, 147, 1462–1465. [DOI] [PubMed] [Google Scholar]
- (46).Rich A; RajBhandary UL Transfer RNA: molecular structure, sequence, and properties. Annu. Rev. Biochem 1976, 45, 805–860. [DOI] [PubMed] [Google Scholar]
- (47).Lerner MR; Boyle JA; Mount SM; Wolin SL; Steitz JA Are snRNPs involved in splicing? Nature 1980, 283, 220–224. [DOI] [PubMed] [Google Scholar]
- (48).Ganser LR; Kelly ML; Herschlag D; Al-Hashimi HM The roles of structural dynamics in the cellular functions of RNAs. Nat. Rev. Mol. Cell Biol 2019, 20, 474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Stombaugh J; Zirbel CL; Westhof E; Leontis NB Frequency and isostericity of RNA base pairs. Nucleic Acids Res. 2009, 37, 2294–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Yesselman JD; Denny SK; Bisaria N; Herschlag D; Greenleaf WJ; Das R Sequence-dependent RNA helix conformational preferences predictably impact tertiary structure formation. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 16847–16855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Denny SK; Bisaria N; Yesselman JD; Das R; Herschlag D; Greenleaf WJ High-throughput investigation of diverse junction elements in RNA tertiary folding. Cell 2018, 174, 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Kwok CK; Tang Y; Assmann SM; Bevilacqua PC The RNA structurome: transcriptome-wide structure probing with next-generation sequencing. Trends Biochem. Sci 2015, 40, 221–232. [DOI] [PubMed] [Google Scholar]
- (53).Fallmann J; Will S; Engelhardt J; Grüning B; Backofen R; Stadler PF Recent advances in RNA folding. J. Biotechnol 2017, 261, 97–104. [DOI] [PubMed] [Google Scholar]
- (54).Bevilacqua PC; Ritchey LE; Su Z; Assmann SM Genome-wide analysis of RNA secondary structure. Annu. Rev. Genet 2016, 50, 235–266. [DOI] [PubMed] [Google Scholar]
- (55).Hasegawa M; Iida Y; Yano T -a.; Takaiwa, F.; Iwabuchi, M. Phylogenetic relationships among eukaryotic kingdoms inferred from ribosomal RNA sequences. J. Mol. Evol 1985, 22, 32–38. [DOI] [PubMed] [Google Scholar]
- (56).Turner DH; Mathews DH NNDB: the nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res. 2010, 38, D280–D282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Zuker M On finding all suboptimal foldings of an RNA molecule. Science 1989, 244, 48–52. [DOI] [PubMed] [Google Scholar]
- (58).Mathews DH; Disney MD; Childs JL; Schroeder SJ; Zuker M; Turner DH Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 7287–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Rouskin S; Zubradt M; Washietl S; Kellis M; Weissman JS Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 2014, 505, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Zubradt M; Gupta P; Persad S; Lambowitz AM; Weissman JS; Rouskin S DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 2017, 14, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Rivas E; Clements J; Eddy SR A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat. Methods 2017, 14, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Andrews RJ; Roche J; Moss WN ScanFold: an approach for genome-wide discovery of local RNA structural elements-applications to Zika virus and HIV. PeerJ 2018, 6, No. e6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).He L; Hannon GJ MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet 2004, 5, 522–531. [DOI] [PubMed] [Google Scholar]
- (64).Bernat V; Disney MD RNA structures as mediators of neurological diseases and as drug targets. Neuron 2015, 87, 28–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Bennett CF Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med 2019, 70, 307–321. [DOI] [PubMed] [Google Scholar]
- (66).Hagedorn PH; Hansen BR; Koch T; Lindow M Managing the sequence-specificity of antisense oligonucleotides in drug discovery. Nucleic Acids Res. 2017, 45, 2262–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Chi X; Gatti P; Papoian T Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discovery Today 2017, 22, 823–833. [DOI] [PubMed] [Google Scholar]
- (68).Frazier KS Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist’s perspective. Toxicol. Pathol 2015, 43, 78–89. [DOI] [PubMed] [Google Scholar]
- (69).Valeur E; Jimonet P New modalities, technologies, and partnerships in probe and lead generation: Enabling a mode-of-action centric paradigm. J. Med. Chem 2018, 61, 9004–9029. [DOI] [PubMed] [Google Scholar]
- (70).Herschlag D Implications of ribozyme kinetics for targeting the cleavage of specific RNA molecules in vivo: more isn’t always better. Proc. Natl. Acad. Sci. U. S. A 1991, 88, 6921–6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Pedersen L; Hagedorn PH; Lindholm MW; Lindow M A kinetic model explains why shorter and less affine enzyme-recruiting oligonucleotides can be more potent. Mol. Ther.–Nucleic Acids 2014, 3, No. e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Angelbello AJ; Rzuczek SG; Mckee KK; Chen JL; Olafson H; Cameron MD; Moss WN; Wang ET; Disney MD Precise small molecule cleavage of a r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 7799–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Costales MG; Hoch DG; Abegg D; Childs-Disney JL; Velagapudi SP; Adibekian A; Disney MD A designed small molecule inhibitor of a non-coding RNA sensitizes HER2 negative cancer to Herceptin. J. Am. Chem. Soc 2019, 141, 2960–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Costales MG; Rzuczek SG; Disney MD Comparison of small molecules and oligonucleotides that target a toxic, non-coding RNA. Bioorg. Med. Chem. Lett 2016, 26, 2605–2609. [DOI] [PubMed] [Google Scholar]
- (75).Disney MD; Velagapudi SP; Li Y; Costales MG; Childs-Disney JL Identifying and validating small molecules interacting with RNA (SMIRNAs). Methods Enzymol. 2019, 623, 45–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Velagapudi SP; Costales MG; Vummidi BR; Nakai Y; Angelbello AJ; Tran T; Haniff HS; Matsumoto Y; Wang ZF; Chatterjee AK; Childs-Disney JL; Disney MD Approved anti-cancer drugs target oncogenic non-coding RNAs. Cell Chem. Biol 2018, 25, 1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Velagapudi SP; Luo Y; Tran T; Haniff HS; Nakai Y; Fallahi M; Martinez GJ; Childs-Disney JL; Disney MD Defining RNA-small molecule affinity landscapes enables design of a small molecule inhibitor of an oncogenic noncoding RNA. ACS Cent. Sci 2017, 3, 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Velagapudi SP; Gallo SM; Disney MD Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat. Chem. Biol 2014, 10, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Disney MD; Winkelsas AM; Velagapudi SP; Southern M; Fallahi M; Childs-Disney JL Inforna 2.0: A platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem. Biol 2016, 11, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Costales MG; Haga CL; Velagapudi SP; Childs-Disney JL; Phinney DG; Disney MD Small molecule inhibition of microRNA-210 reprograms an oncogenic hypoxic circuit. J. Am. Chem. Soc 2017, 139, 3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Huang X; Ding L; Bennewith KL; Tong RT; Welford SM; Ang KK; Story M; Le Q-T; Giaccia AJ Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol. Cell 2009, 35, 856–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Qin Q; Furong W; Baosheng L Multiple functions of hypoxia-regulated miR-210 in cancer. J. Exp. Clin. Cancer Res 2014, 33, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Kelly TJ; Souza AL; Clish CB; Puigserver P A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1alpha stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol. Cell. Biol 2011, 31, 2696–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Johnson LF; Abelson HT; Penman S; Green H The relative amounts of the cytoplasmic RNA species in normal, transformed and senescent cultured cell lines. J. Cell. Physiol 1977, 90, 465–470. [DOI] [PubMed] [Google Scholar]
- (85).Liu B; Childs-Disney JL; Znosko BM; Wang D; Fallahi M; Gallo SM; Disney MD Analysis of secondary structural elements in human microRNA hairpin precursors. BMC Bioinf. 2016, 17, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Velagapudi SP; Seedhouse SJ; French J; Disney MD Defining the RNA internal loops preferred by benzimidazole derivatives via 2D combinatorial screening and computational analysis. J. Am. Chem. Soc 2011, 133, 10111–10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Disney MD; Lee MM; Pushechnikov A; Childs-Disney JL The role of flexibility in the rational design of modularly assembled ligands targeting the RNAs that cause the myotonic dystrophies. ChemBioChem 2010, 11, 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Velagapudi SP; Cameron MD; Haga CL; Rosenberg LH; Lafitte M; Duckett DR; Phinney DG; Disney MD Design of a small molecule against an oncogenic noncoding RNA. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 5898–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Rzuczek SG; Colgan LA; Nakai Y; Cameron MD; Furling D; Yasuda R; Disney MD Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol 2017, 13, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Haniff HS; Knerr L; Liu X; Crynen G; Boström J; Abegg D; Adibekian A; Lekah E; Wang K-W; Camerson MD; Yildirm I; Lemurell M; Disney MD Design of a small molecule that stimulates VEGFA informed from an expanded encyclopedia of RNA fold-small molecule interactions. ChemRxiv 2020, DOI: 10.26434/chemrxiv.7967291.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).van der Lee R; Buljan M; Lang B; Weatheritt RJ; Daughdrill GW; Dunker AK; Fuxreiter M; Gough J; Gsponer J; Jones DT; Kim PM; Kriwacki RW; Oldfield CJ; Pappu RV; Tompa P; Uversky VN; Wright PE; Babu MM Classification of intrinsically disordered regions and proteins. Chem. Rev 2014, 114, 6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Uversky VN Intrinsically disordered proteins and their “mysterious” (meta)physics. Front. Phys 2019, 7, 10. [Google Scholar]
- (93).Ruan H; Sun Q; Zhang W; Liu Y; Lai L Targeting intrinsically disordered proteins at the edge of chaos. Drug Discovery Today 2019, 24, 217–227. [DOI] [PubMed] [Google Scholar]
- (94).Kim WS; Kågedal K; Halliday GM Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Res. Ther 2014, 6, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Cahill CM; Lahiri DK; Huang X; Rogers JT Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim. Biophys. Acta, Gen. Subj 2009, 1790, 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Zhang P; Park H-J; Zhang J; Junn E; Andrews RJ; Velagapudi SP; Abegg D; Vishnu K; Costales MG; Childs-Disney JL; Adibekian A; Moss WN; Mouradian MM; Disney MD Translation of the intrinsically disordered protein α-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Zarrinkar PP; Williamson JR Kinetic intermediates in RNA folding. Science 1994, 265, 918–924. [DOI] [PubMed] [Google Scholar]
- (98).Rzuczek SG; Park H; Disney MD A toxic RNA catalyzes the in cellulo synthesis of its own inhibitor. Angew. Chem., Int. Ed. Engl 2014, 53, 10956–10959. [DOI] [PubMed] [Google Scholar]
- (99).Angelbello AJ; Chen JL; Childs-Disney JL; Zhang P; Wang Z-F; Disney MD Using genome sequence to enable the design of medicines and chemical probes. Chem. Rev 2018, 118, 1599–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Mateos-Aierdi AJ; Goicoechea M; Aiastui A; Fernández-Torrón R; Garcia-Puga M; Matheu A; López de Munain A Muscle wasting in myotonic dystrophies: a model of premature aging. Front. Aging Neurosci 2015, 7, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Meola G; Cardani R Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta, Mol. Basis Dis 2015, 1852, 594–606. [DOI] [PubMed] [Google Scholar]
- (102).Udd B; Krahe R The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012, 11, 891–905. [DOI] [PubMed] [Google Scholar]
- (103).Rzuczek SG; Gao Y; Tang Z-Z; Thornton CA; Kodadek T; Disney MD Features of modularly assembled compounds that impart bioactivity against an RNA target. ACS Chem. Biol 2013, 8, 2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Moon MH; Hilimire TA; Sanders AM; Schneekloth JS Measuring RNA-ligand interactions with microscale thermophoresis. Biochemistry 2018, 57, 4638–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Umuhire Juru A; Patwardhan NN; Hargrove AE Understanding the contributions of conformational changes, thermodynamics, and kinetics of RNA-small molecule interactions. ACS Chem. Biol 2019, 14, 824–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Blakeley BD; DePorter SM; Mohan U; Burai R; Tolbert BS; McNaughton BR Methods for identifying and characterizing interactions involving RNA. Tetrahedron 2012, 68, 8837–8855. [Google Scholar]
- (107).Wicks SL; Hargrove AE Fluorescent indicator displacement assays to identify and characterize small molecule interactions with RNA. Methods 2019, 167, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Lin A; Giuliano CJ; Palladino A; John KM; Abramowicz C; Yuan ML; Sausville EL; Lukow DA; Liu L; Chait AR; Galluzzo ZC; Tucker C; Sheltzer JM Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med 2019, 11, No. eaaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Lowe R; Shirley N; Bleackley M; Dolan S; Shafee T Transcriptomics technologies. PLoS Comput. Biol 2017, 13, No. e1005457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Git A; Dvinge H; Salmon-Divon M; Osborne M; Kutter C; Hadfield J; Bertone P; Caldas C Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 2010, 16, 991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Bittker JA High-throughput RT-PCR for small-molecule screening assays. Curr. Protoc. Chem. Biol 2012, 4, 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Citri A; Pang ZP; Südhof TC; Wernig M; Malenka RC Comprehensive qPCR profiling of gene expression in single neuronal cells. Nat. Protoc 2012, 7, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Bosc N; Meyer C; Bonnet P The use of novel selectivity metrics in kinase research. BMC Bioinf. 2017, 18, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Graczyk PP Gini coefficient: a new way to express selectivity of kinase inhibitors against a family of kinases. J. Med. Chem 2007, 50, 5773–5779. [DOI] [PubMed] [Google Scholar]
- (115).Drewry DH; Wells CI; Andrews DM; Angell R; Al-Ali H; Axtman AD; Capuzzi SJ; Elkins JM; Ettmayer P; Frederiksen M; Gileadi O; Gray N; Hooper A; Knapp S; Laufer S; Luecking U; Michaelides M; Muller S; Muratov E; Denny RA; Saikatendu KS; Treiber DK; Zuercher WJ; Willson TM Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS One 2017, 12, No. e0181585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Haga CL; Velagapudi SP; Strivelli JR; Yang WY; Disney MD; Phinney DG Small molecule inhibition of miR-544 biogenesis disrupts adaptive responses to hypoxia by modulating ATM-mTOR signaling. ACS Chem. Biol 2015, 10, 2267–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Kenny PW; Montanari CA Inflation of correlation in the pursuit of drug-likeness. J. Comput.-Aided Mol. Des 2013, 27, 1–13. [DOI] [PubMed] [Google Scholar]
- (118).Shultz MD Two decades under the influence of the Rule of Five and the changing properties of approved oral drugs. J. Med. Chem 2019, 62, 1701–1714. [DOI] [PubMed] [Google Scholar]
- (119).Valeur E; Gueret SM; Adihou H; Gopalakrishnan R; Lemurell M; Waldmann H; Grossmann TN; Plowright AT New modalities for challenging targets in drug discovery. Angew. Chem., Int. Ed. Engl 2017, 56, 10294–10323. [DOI] [PubMed] [Google Scholar]
- (120).Lagorce D; Douguet D; Miteva MA; Villoutreix BO Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep 2017, 7, 46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Morgan BS; Sanaba BG; Donlic A; Karloff DB; Forte JE; Zhang Y; Hargrove AE R-BIND: an interactive database for exploring and developing RNA-targeted chemical probes. ACS Chem. Biol 2019, 14, 2691–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Wishart DS; Knox C; Guo AC; Cheng D; Shrivastava S; Tzur D; Gautam B; Hassanali M DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Valeur E; Narjes F; Ottmann C; Plowright AT Emerging modes-of-action in drug discovery. MedChemComm 2019, 10, 1550–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Lipinski CA Lead- and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technol. 2004, 1, 337–341. [DOI] [PubMed] [Google Scholar]
- (125).Churcher I Protac-induced protein degradation in drug discovery: Breaking the rules or just making new ones? J. Med. Chem 2018, 61, 444–452. [DOI] [PubMed] [Google Scholar]
- (126).Lee J; Udugamasooriya DG; Lim H-S; Kodadek T Potent and selective photo-inactivation of proteins with peptoidruthenium conjugates. Nat. Chem. Biol 2010, 6, 258–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Costales MG; Matsumoto Y; Velagapudi SP; Disney MD Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc 2018, 140, 6741–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Vauquelin G Effects of target binding kinetics on in vivo drug efficacy: koff, kon and rebinding. Br. J. Pharmacol 2016, 173, 2319–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Bondeson DP; Smith BE; Burslem GM; Buhimschi AD; Hines J; Jaime-Figueroa S; Wang J; Hamman BD; Ishchenko A; Crews CM Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol 2018, 25, 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Roy MJ; Winkler S; Hughes SJ; Whitworth C; Galant M; Farnaby W; Rumpel K; Ciulli A SPR-measured dissociation kinetics of PROTAC ternary complexes influence target degradation rate. ACS Chem. Biol 2019, 14, 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Copeland RA The drug-target residence time model: a 10-year retrospective. Nat. Rev. Drug Discovery 2016, 15, 87–95. [DOI] [PubMed] [Google Scholar]
- (132).Costales MG; Aikawa H; Li Y; Childs-Disney JL; Abegg D; Hoch DG; Pradeep Velagapudi S; Nakai Y; Khan T; Wang KW; Yildirim I; Adibekian A; Wang ET; Disney MD Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 2406–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Carter AJ; Kraemer O; Zwick M; Mueller-Fahrnow A; Arrowsmith CH; Edwards AM Target 2035: probing the human proteome. Drug Discovery Today 2019, 24 (11), 2111–2115. [DOI] [PubMed] [Google Scholar]
- (134).Kehe J; Kulesa A; Ortiz A; Ackerman CM; Thakku SG; Sellers D; Kuehn S; Gore J; Friedman J; Blainey PC Massively parallel screening of synthetic microbial communities. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 12804–12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Neri D; Lerner RA DNA-Encoded Chemical Libraries: A selection system based on endowing organic compounds with amplifiable information. Annu. Rev. Biochem 2018, 87, 479–502. [DOI] [PMC free article] [PubMed] [Google Scholar]