

Abstract
Rationale
Kawasaki disease (KD) is an acute vasculitis of early childhood that can result in permanent coronary artery structural damage. The etiology for this arterial vulnerability in up to 15% of KD patients is unknown. Vascular smooth muscle cell (VSMC) dedifferentiation play a key role in the pathophysiology of medial damage and aneurysm formation, recognized arterial pathology in KD. Platelet hyperreactivity is also a hallmark of KD. We recently demonstrated that uptake of platelets and platelet-derived miRNAs influences VSMC phenotype in vivo.
Objective
We set out to explore whether platelet/vascular smooth muscle cell interactions contribute to coronary pathology in KD.
Methods & Results
We prospectively recruited and studied 242 KD patients, 75 of whom had documented coronary artery pathology. Genome-wide miRNA sequencing and droplet digital PCR (ddPCR) demonstrated that KD patient platelets have significant induction of miR-223 compared to healthy controls. Platelet-derived miR-223 has recently been shown to promote vascular smooth muscle quiescence and resolution of wound healing after vessel injury. Paradoxically, KD patients with the most severe coronary pathology (giant coronary artery aneurysms) exhibited a lack of miR-223 induction. Hyperactive platelets isolated from KD patients are readily taken up by VSMCs, delivering functional miR-223 into the VSMCs promoting VSMC differentiation via downregulation of platelet-derived growth factor receptor β (PDGFRβ). The lack of miR-223 induction in patients with severe coronary pathology leads to persistent VSMC dedifferentiation. In a mouse model of KD (Lactobacillus casei cell wall extract (LCWE) injection), miR-223 knockout (miR-223 KO) mice exhibited increased medial thickening, loss of contractile VSMCs in the media, and fragmentation of medial elastic fibers compared to WT mice, which demonstrated significant miR-223 induction upon LCWE challenge. The excessive arterial damage in the miR-223 knockout could be rescued by adoptive transfer of platelet, administration of miR-223 mimics, or the PDGFRβ inhibitor imatinib mesylate. Interestingly, miR-223 levels progressively increase with age, with the lowest levels found in less than five-year-old. This provides a basis for coronary pathology susceptibility in this very young cohort.
Conclusions
Platelet-derived miR-223 (through PDGFRβ inhibition) promotes VSMC differentiation and resolution of KD induced vascular injury. Lack of miR-223 induction leads to severe coronary pathology characterized by VSMC dedifferentiation and medial damage. Detection of platelet-derived miR-223 in KD patients (at the time of diagnosis) may identify patients at greatest risk of coronary artery pathology. Moreover, targeting platelet miR-223 or VSMC PDGFRβ represents potential therapeutic strategies to alleviate coronary pathology in KD.
Keywords: Kawasaki disease, vascular smooth muscle cell, aneurysm, miR-223, platelets
Subject Terms: Aneurysm, Platelets, Vascular Biology
Graphical Abstract
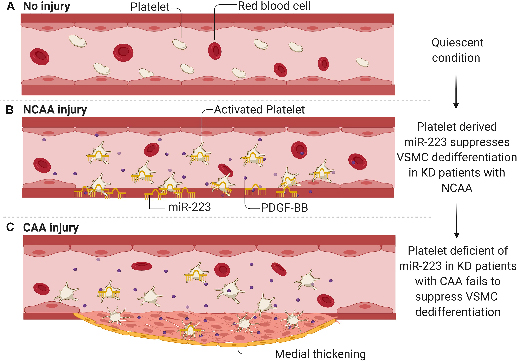
INTRODUCTION
Kawasaki disease (KD) is an acute vasculitis of childhood that can result in permanent structural damage to the coronary arteries, including medial damage, thickening, and aneurysm formation 1. KD is the most common cause of acquired cardiovascular disease in childhood 2. The acute vascular inflammation is characterized by progressive dysfunction and damage to endothelium, extensive activation of platelets, and vascular smooth muscle cell (VSMC) dedifferentiation, potentially leading to pathological vascular remodeling and permanent coronary pathology 3, 4. Standard treatment requires the combination of high-dose intravenous immunoglobulin and oral aspirin 5, primarily focused on the injury process, targeting inflammation, and thrombosis. Despite such therapies, 5 to 15% of KD children develop permanent vascular damage leading to significant morbidity and mortality. New mechanistic insights into the pathogenesis of KD coronary pathology are urgently warranted.
The process leading to arterial damage in KD is well studied. Endothelial damage from vasculitis promotes platelet activation 6–8 and release of factors contributing to VSMC dedifferentiation, e.g. platelet-derived growth factor-BB (PDGF-BB) 9. In response to PDGF-BB, VSMCs undergo a phenotypic switch from a contractile, quiescent differentiated state to a synthetic, dedifferentiated, proliferative inflammatory phenotype 10–12 characterized by loss of smooth muscle cell markers including α-smooth muscle actin (ACTA2), transgelin (TAGLN), and calponin (CNN1) 13. VSMC dedifferentiation has been reported as a unique process of concentric mass formation around the arterial lumen, which contributes to the coronary pathology in KD4, 14. Thus VSMC dedifferentiation plays a critical role in many vascular pathologies including medial damage, thickening, and aneurysm formation 15–17. What is currently lacking is an understanding of the process leading to damage resolution. We recently demonstrated that the horizontal transfer of platelet-derived miRNAs into VSMCs play an important role in arterial damage resolution 18. We hypothesize that aberrant damage resolution may hold the key to persistent and permanent coronary pathology in KD.
We now demonstrate using genome-wide miRNA sequencing and droplet digital PCR (ddPCR) that KD patients with No Coronary Artery Aneurysm (NCAA group) show high miR-223 induction in platelets, whereas KD patients with severe Coronary Artery Aneurysm (CAA group) show low miR-223 induction in platelets, suggesting a role for miR-223 in KD coronary pathology. We further demonstrate that NCAA platelets (high miR-223 expression) deliver miR-223 into VSMCs, thereby inhibiting VSMC dedifferentiation via downregulation of platelet-derived growth factor receptor β (PDGFRβ). Deficiency of miR-223 observed in CAA platelets results in a failure to switch on VSMC quiescence, contributing to continuing pathological repair and severe coronary vascular pathology. In LCWE-injected miR-223 knockout mice, either administration of miR-223 mimic (agomiR-223), platelets with miR-223, or imatinib mesylate (an inhibitor of PDGFRβ) significantly attenuate coronary pathology progression. Thus, platelet miR-223 may serve as a novel biomarker as well as a therapeutic target for KD coronary artery pathology.
METHODS
Details of the experimental methods are available in the Online Appendix.
Please see The Major Resources Table in the Supplemental Materials. Briefly, consecutive consenting patients diagnosed with KD (American Heart Association criteria) 5 and age- and sex- matched healthy controls were recruited to the study (Guangzhou Women and Children’s Medical Center Human Investigation Committee No. 2017102710). The demographic and clinical characteristics of the recruited subjects are presented in Online Table I–III.
Platelets were prepared as previously described 18. To detect the ultrastructural internalization of platelets into cells, and the function of platelet-derived miR-223 in VSMCs, transmission electron microscopy, Ago2 Immunoprecipitation (Ago2-IP) was performed as previously described 19, 20.
L.Casei (American Type Culture Collection, ATCC 11578) cell wall extract (LCWE) was prepared as previously described 20–22. Wild-type C57BL/6, the global miR-223 knockout (miR-223y/-) mice 23 (JAX stock #013198) purchased from Jackson Laboratory (Bar Harbor, USA) and backcrossed to C57BL/6 mice for ≥5 generations were used to establish KD mouse model. The experiments were conducted under the authorization of Animal Care Committee in Guangzhou Medical University (2019–384). The animal procedures were performed by the same operator blinded to the mouse genotype/treatment group, as well as the KD status for the in vivo animal model experiments. Images were assigned a numerical code to ensure that the selection of representative images is carried out in a blinded manner.
Statistical analysis was performed using GraphPad Prism v.8.0.2 (GraphPad, La Jolla, CA, USA). Shapiro-Wilk Normality test was performed to determine data distribution. For normally distributed data, values are presented as mean ± SD. Unpaired two-tailed Student’s t test was used to compare two groups; One-way ANOVA followed by Tukey’s multiple comparisons was performed to determine differences between indicated groups. For data that were not distributed normally (including n≤4), values are presented as medians ± interquartile ranges (IQR). Mann-Whitney test was used for two groups, and Kruskall-Wallis with Dunn’s multiple comparisons test was performed for more than two groups. For experiments set up with more than one variable, repeated measures (RM) Two-way ANOVA with Sidak’s multiple comparisons test was used. For platelet infusion experiment, Wilcoxon matched-pairs signed rank test was used for comparison between percentages of GFP positive cells in circulation before and after platelet infusion. A separate file including all statistics has been added as a supplementary to the manuscript. A difference of P<0.05 was considered significant.
RESULTS
We hypothesized that the underlying mechanism for KD coronary pathology may include not only induction of the vascular wall damage by inflammation, but also inadequate resolution of this damage.
Reduced induction of miR-223 is associated with severe coronary pathology and serves as a disease biomarker (Figure 1)
Figure 1. Kawasaki disease (KD) platelets exhibit differential expression of miR-223 dependent on the severity of coronary pathology.

(A) Heatmap of differentially expressed miRNAs in platelets from healthy control (HC) (n=3), KD patients in non-coronary arterial aneurysm (NCAA) group (n=5) and coronary arterial aneurysm (CAA) group (n=3). (B) CAA patients were divided into three groups, small CAA (SCAA), medium CAA (MCAA), giant CAA (GCAA), according to coronary artery Z-worst. Representative echocardiogram images of corresponding patients with SCAA, MCAA, GCAA or NCAA, were shown. (C) Levels of miR-223 in platelets from HC (n=29), KD patients in NCAA group (n=88) and CAA group (n=28) were determined by droplet digital PCR (ddPCR) and presented as copies per microliter (μl) in each sample (Kruskall-Wallis test and Dunn’s multiple comparisons test). (D) Levels of miR-223 in platelets from each group (SCAA: n=12, MCAA: n=7, GCAA: n=9) were measured by ddPCR (Kruskall-Wallis test and Dunn’s multiple comparisons test). Binary logistic regression analysis was performed, and ROC values were generated to evaluate the ability of platelet miR-223 to distinguish, (E) HC from KD patients with CAA and NCAA, (F) KD patients in NCAA group from CAA group, (G) KD patients with SCAA and MCAA from GCAA. *P<0.05, ***P<0.001, ****P<0.0001.
Genome-wide miRNA sequencing was performed on platelets isolated from healthy controls (HC) and Kawasaki disease (KD) patients. KD patients were further characterized in two groups, NCAA and CAA, according to coronary artery dimension calculated by echocardiography. Supporting the validity of our miRNA profiling, members of the let-7 family accounted for the most miRNAs in platelets from KD patients 33, 34 (Online Figure IA). Among all the miRNAs significantly increased in KD, the top eleven induced miRNAs (unsupervised hierarchical clustering) included miR-223 (Figure 1A highlighted by red box). We recently demonstrated that activated platelets can promote resolution of vascular injury via horizontal transfer of platelet-derived miR-223 into VSMCs 18 where miR-223 is normally undetected 35. Given that miR-223 is increased in NCAA with no increase in CAA, miR-223 may be protective against CAA in KD.
Expression of platelet miR-223 was further analyzed using quantitative Real-Time PCR (RT-qPCR) in KD patients (NCAA n=67, CAA n=35) and HC (n=67) recruited from the cohort study (2017102710). Consistent with the miRNA array, the level of miR-223 was significantly increased in KD with NCAA compared to HC (P<0.0001) and KD patients with coronary artery aneurysms (CAA) (P=0.0016), demonstrating significantly lower miR-223 induction (Online Figure IB). MiR-223 may be differentially expressed dependent on the severity of coronary pathology in KD. KD patients (n=116) and HC patients (n=29) recruited from the cohort study (2017102710) were evaluated for miR-223 using high sensitivity quantitative droplet digital PCR (ddPCR), to accurately determine copy number of platelet-derived miR-223. Blinded to the results, patients with CAA (n=28) were then further divided into three groups, SCAA (small CAA n=12), MCAA (medium CAA n=7), GCAA (giant CAA n=9), using an established clinical index used to quantify the severity of coronary pathology in KD 5 (Figure 1B). Consistent with our initial cohort study, the copy number of platelet miR-223 was significantly increased in KD patients with NCAA (n=88), compared to HC (n=29) or KD patients with CAA (n=28) (Figure 1C). Among the KD patients with CAA, the copy number of platelet miR-223 was significantly reduced dependent on the coronary pathology severity, following the order SCAA > MCAA > GCAA (Figure 1D).
To determine whether platelet miR-223 level can serve as a biomarker for coronary pathology severity in KD, a receiver operating characteristic (ROC) curve was plotted and the area under curve (AUC) was calculated (AUC=1 indicates the highest correlation). The level of platelet miR-223 was able to distinguish KD patients (both NCAA and CAA groups) from HC, with an AUC value of 0.931 (95% confidence interval (CI), 0.889–0.972) (Figure 1E), and the AUC calculated for miR-223 to distinguish the CAA group from NCAA group was 0.677 (95% CI, 0.563–0.791) (Figure 1F). The AUC value obtained for miR-223 to distinguish the SCAA and MCAA group from GCAA group was 0.994 (95% CI, 0.978–1.0) (Figure 1G). Taken together, these results support an induction of miR-223 in KD platelets. The level of induction of miR-223 inversely correlate with coronary artery pathology severity with no significant induction (lowest miR-223 levels) in KD patients with severe coronary artery pathology. The key question arises as to whether lack of miR-223 induction is merely a biomarker for coronary disease pathology or is involved in the coronary pathology process.
KD platelets deliver miR-223 to VSMC (Figure 2 & 3)
Figure 2. Activated KD platelets are internalized into VSMCs delivering miR-223 into VSMCs.

(A) Representative images of VSMCs co-cultured with CMFDA-labeled (green) KD platelets and unstimulated HC platelets, and thrombin stimulated HC platelets for 2, 4, 24 hours. VSMCs stained with ACTA2 (red) and nuclei visualized with Hochest (blue) (n=8). Scale bar: 20μm. (B) Three-dimensional reconstruction of confocal Z-stack images of the whole VSMCs co-cultured with CMFDA-labeled KD (green) platelets were shown at different orientations to demonstrate uptake and perinuclear localization (n=8). Scale bar: 20μm. (C) Transmission electron microscopy imaging of VSMCs co-cultured with KD platelets. The red arrow indicates the entering and internalized platelet. Scale bar: 500 nm. (D) Intracellular miR-223 level in VSMCs after incubation with platelets from unstimulated HC (n=3), thrombin-stimulated HC (n=3) and KD group (n=3) for 0, 2, 4, 8 hours, normalized to U6. The relative level of miR-223 in thrombin-stimulated HC group and KD group was calculated and normalized by HC group at each time point (Kruskall-Wallis test and Dunn’s multiple comparisons test). (E) Intracellular miR-223 level in VSMCs at 4 hours after incubation with increasing concentration of platelets from HC (n=3) and KD patients with NCAA (n=3). Results are normalized to U6. The relative level of miR-223 in KD group was calculated and normalized by HC group at each concentration (Mann Whitney test). Abbreviations: HC, VSMC co-cultured with platelets from HC; KD (NCAA), VSMCs co-cultured with platelets from KD patients with NCAA; HC+thrombin, VSMCs co-cultured with thrombin -stimulated HC platelets. *P<0.05 versus HC group.
Figure 3. KD platelets are internalized into VSMCs delivering miR-223 in lactobacillus casei cell wall extract (LCWE)-induced KD murine model.

The abdominal aorta tissues were collected from day 0 to day 14 after LCWE-injection. The PBS-injected wide type (WT) littermates were used as vehicle control. (A) Representative Hematoxylin and eosin (H & E) -stained sections from LCWE-injected mice were shown. Photomicrographs from I-V show the process of LCWE-induced abdominal aorta lesions (n=30). Scale bar: 50 μm. (B) Relative expression of miR-223 in platelets from PBS-treated WT littermates (n=5), LCWE-treated WT littermates (n=18), PBS-treated miR-223 KO mice (n=5), and LCWE-treated miR-223 KO mice (n=10) at two weeks (Kruskall-Wallis test and Dunn’s multiple comparisons test, **P<0.01, ****P<0.0001 versus LCWE-injected WT littermates). (C) Corresponding immunofluorescence of CD41 and ACTA2 in abdominal aorta sections from PBS (n=6) and LCWE-injected WT littermates (n=6) at two weeks. CD41 stained as green, ACTA2 as red, and nuclei visualized with DAPI (blue). Scale bar: 20 μm. (D) Quantification of CD41 expression in sections was shown (Unpaired t test). (E) Representative images of the sections from PBS (n=6) and LCWE-injected PF4-cre: mT/mG WT mice (n=6). Scale bar: 20 μm. (F) Quantification of the mGFP positive cells in VSMCs of media layer two weeks after LCWE-injection (Unpaired t test). (G) Representative images of miR-223 and PDGFRβ expression in abdominal aorta sections from PBS or LCWE-injected WT littermates (n=4), PBS or LCWE-injected miR-223 knockout (KO) mice (n=4). miR-223 (red), PDGFRβ (green), and DAPI (blue). Scale bar: 20 μm. Quantification of miR-223 (H) and PDGFRβ (I) expression in situ shown (Kruskall-Wallis test and Dunn’s multiple comparisons test). Abbreviations: PBS, PBS-injected mice; LCWE, LCWE-injected mice; Littermates, WT littermates; miR-223 KO, miR-223 KO mice. **P<0.01 versus PBS-injected WT littermates; #P<0.05 versus PBS-injected miR-223 KO mice in G & H.
As we have recently observed, if platelet miR-223 is somehow protective against vascular pathology it needs to be incorporated into VSMCs 18. VSMC dedifferentiation, proliferation, and apoptosis, processes associated with arterial aneurysm formation, have been reported to lead to concentric mass formation around the arterial lumen, contributing to the coronary pathology in KD 4, 14. As earlier described, miR-223 is not normally found in VSMC. Endothelial damage from KD vasculitis promotes platelet activation 6–8 and direct contact between platelets and VSMC. Activated platelets are readily taken up by VSMCs 18. Compared to HC platelets, KD platelets showed higher expression of P-selectin (CD62P), and increased release of platelet factor 4 (PF4), beta-thromboglobulin (β-TG), and CD42b/GPIB alpha, all markers of platelet activation (Online Table IV). CMFDA-labeled HC (not activated), HC activated with thrombin or KD platelets were cocultured with human coronary SMCs and assessed for platelet uptake using high resolution confocal microscopy. Both the HC platelets activated with thrombin and the KD platelets demonstrated significant uptake at 4 and 24 hours compared to the HC platelets (inactivated) (Figure 2A). This is consistent with KD platelets being activated (Online Table IV) even in the absence of agonists such as thrombin. To more precisely localize the uptake of platelets in KD we utilized high resolution confocal microscopy with three-dimensional reconstruction. KD platelets were found to be incorporated into VSMCs, and localized to the cytoplasm at early time points (1–4 hours). After 24 hours of coculture, KD platelets localized to the perinuclear region (Figure 2B), consistent with the previous reports demonstrating activated platelets endocytosed by VSMCs 18. The process of KD platelet internalization in VSMCs was further confirmed by transmission electron microscopy, visualized by the presence of distinct platelet granules that incorporated into VSMCs (Figure 2C I–III). Initially platelets are located at the membrane of VSMCs (Figure 2C I–red arrows show an external platelet), then incorporated into the cell cytoplasm (Figure 2C II–red arrow) and fused with lysosomes (Figure 2C III–red arrow, Online Figure II). We also found that the level of miR-223 was significantly increased in VSMCs cocultured with KD platelets after 2-hour incubation (Figure 2D), and VSMCs cocultured with KD platelets showed induction of miR-223 compared to HC platelets at the same amount of platelet (Figure 2E). In the absence of platelet coculture, no significant miR-223 expression was detected in VSMCs alone (Online Figure III), as observed in previous studies 35. Using purified platelets and a transwell chamber with a filter pore size equal to 1 μm, where only platelet-derived microparticles (PMPs) were allowed to pass through, we found that the induction of miR-223 in VSMCs was mainly contributed by KD platelets rather than PMPs (Online Figure IV A & B). Compared to PMPs and supernatant, VSMCs incubated with KD platelets exhibited significant expression of miR-223 (Online Figure IV C–D). Moreover, VSMC differentiation appeared to be induced by KD platelets but not PMPs (Online Figure IV E–F). These results suggest that activated KD platelets may deliver miR-223 at least in part through activated platelet contact with VSMC and internalization.
To determine whether KD platelets are incorporated by VSMC in vivo, we employed a well-established murine model of KD in which intraperitoneal injection of Lactobacillus casei cell wall extract (LCWE) induces vascular inflammation 21. Consistent with pathology reported in the previous literature 26, our LCWE-induced KD mice developed progressive abdominal aorta arteritis (Figure 3A I–IV, Online Figure V), characterized by the infiltration of immune cells (Figure 3A II, Online Figure VI), disruption of the elastin, necrosis, and medial thickening 36 (Figure 3A III–IV). Corroborating results from KD patients, expression of murine platelet miR-223 increased, two-week after LCWE injection (Figure 3B). Confocal microscopy confirmed localization of platelets (CD41 positive cells) in the medial layer of injured vessels (Figure 3C & D). To further establish platelet uptake in vivo, we employed a platelet-specific Cre recombinase expressing mT/mG mice (PF4-cre: mT/mG) which promotes a switch from tomato red fluorescence to GFP only in platelets. Indeed, two weeks after LCWE injection, GFP positive cells were localized in the medial layer of abdominal aorta, but not in the PBS-injected mice (Figure 3E & F).
Using fluorescent in-situ hybridization (FISH), the expression of miR-223 was detected in the medial VSMCs of only the injured vessel (not in the control uninjured vessel). Expression of miR-223 was not increased in the medial VSMCs of LCWE-injected miR-223 KO mice compared to wild type (WT) littermates (Figure 3G & H). Interestingly, in this experiment expression of PDGFRβ (dedifferentiation marker) was significantly increased in the thickened medial layer of miR-223 KO mice (Figure 3G & I, Online Figure VII negative control). The significance will be apparent later. Taken together, these results provide evidence for the horizontal transfer of platelet-derived miR-223 into VSMCs of injured vessels in the KD mice. This may be in part through a platelet internalization process as described in Figure 2, however miR-223 transfer by other means such as exosomes cannot be completely excluded. We then explored the effect of miR-223 on VSMCs in KD, particularly as miR-223 is normally not found in VSMCs.
KD platelet-derived miR-223 regulate VSMC dedifferentiation via directly targeting PDGFRβ. (Figure 4)
Figure 4. KD platelet-derived miR-223 regulates VSMC dedifferentiation via direct targeting PDGFRβ.

VSMCs were incubated with platelets from HC and KD patients with NCAA for 48 hours, cell proliferation was assessed by, (A) CCK-8 assays (HC: n=17, KD (NCAA): n=16, Mann Whitney test), and (B) BrdU incorporation assays (HC: n=12, KD (NCAA): n=13, Unpaired t test). The expression (C) and quantification (D) of markers for differentiation (ACTA2, CNN1, TAGLN) and dedifferentiation (PCNA, OPN, PDGFRβ) in VSMCs co-cultured with platelets from HC (n=13) and KD patients with NCAA (n=21) were determined by Western blot (Mann Whitney test). Ago2 immunoprecipitation was performed in VSMCs (E) (Ctrl: n=6, KD (NCAA): n=6, Unpaired t test), or VSMCs pre-transfected with miR-223 inhibitor (antagomiR-223) (F) (KD (NCAA)+antagomiRNC: n=6, KD (NCAA)+antagomiR223: n=6, Mann Whitney test), after 24 hours incubation with platelets from KD patients with NCAA, Ago2-associated miR-223 and PDGFRβ mRNA were quantified by quantitative RT-PCR (RT-qPCR). VSMCs transfected with Luc-PDGFRβ (WT) or Luc-PDGFRβ (MT) were co-transfected with miR-223 mimic (agomiR-223) (n=3) or NC-mimic (agomiR-NC) (n=3) in (G), co-cultured with platelets from HC (n=3) or KD patients with NCAA (n=6) in (H), co-transfected with antagomiR-223 (n=6) or NC inhibitor (antagomiR-NC) (n=6) followed by incubation with platelets from KD patients with NCAA as indicated in (I). Luciferase activity was measured with that of Renilla as transfection control (Mann Whitney test or Unpaired t test). Abbreviations: HC, VSMCs co-cultured with platelets from HC; KD (NCAA), VSMCs co-cultured with platelets from KD patients with NCAA. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To understand the role of KD platelet-derived miR-223 in VSMC injury repair, proliferation rate was measured in VSMCs cocultured with platelets containing high miR-223 (KD) versus low miR-223 (HC). VSMC proliferation was significantly reduced after 24 and 48 hours of incubation with NCAA platelets (high miR-223) compared to HC (low miR-223) (Online Figure VIIIA). Importantly, extracellular PDGF-BB (ligand for the receptor) did not differ in VSMC treated with NCAA platelets versus HC platelets (Online Figure VIIIB). With the mouse studies, VSMCs co-cultured with NCAA platelets also exhibited reduced rate of cell proliferation, measured by Cell Counting Kit-8 (CCK-8) and BrdU incorporation assay (Figure 4A & B). Also, the expression of the VSMC differentiation markers (ACTA2, CNN1 and TAGLN) were significantly increased with concomitant reduced expression of VSMC dedifferentiation markers (PCNA, OPN and PDGFRβ) in VSMCs cocultured with NCAA platelets compared to HC platelets (Figure 4C & D). The expression of VSMC differentiation and dedifferentiation markers was also assessed in platelets alone and VSMCs alone, serving as controls (Online Figure IX). These results support a role for platelet-derived miR-223 in suppressing VSMC dedifferentiation.
We then explored the mechanism by which miR-223 inhibits VSMC dedifferentiation. Overexpression of miR-223 using miR-223 mimic (agomiR-223) substantially reduced the level of PDGFRβ protein in VSMCs (Online Figure XA). As miRNAs are incorporated into Argonaute 2 (Ago2) effector complexes to regulate specific mRNAs through translational repression, Ago2 immunoprecipitation was performed in VSMCs cocultured with KD platelets (high miR-223), followed by RT-qPCR detection of miR-223 and PDGFRβ mRNA. We found an increase in the Ago2•miR-223 complexes and PDGFRβ mRNA in VSMCs cocultured with KD platelets, however, such increased association of miR-223 and PDGFRβ with Ago2 was not seen in VSMCs pre-transfected with miR-223 inhibitor (antagomiR-223), indicating that the platelet-derived miR-223 functionally targets PDGFRβ in VSMCs (Figure 4E & F). Bioinformatics analysis further identified that human PDGFRβ harbors a conserved binding site for miR-223–3p within its 3’ untranslated region (3’ UTR) (Online Figure XB). We then measured the activity of a luciferase reporter fused to the wild-type PDGFR-3’ UTR (Luc-PDGFRβ (WT)) or mutant PDGFRβ 3’ UTR (Luc-PDGFRβ (MT)) in which the miR-223 targeting sites were mutated. Administration of agomiR-223 inhibited the luciferase activity in VSMCs transfected with Luc-PDGFRβ (WT), while the inhibitory effect of agomiR-223 on luciferase activity was abrogated in cells transfected with Luc-PDGFRβ (MT) (Figure 4G). Compared to HC platelets, KD platelets (high miR-223) reduced luciferase activity in VSMCs transfected with Luc-PDGFRβ (WT) but not Luc-PDGFRβ (MT) (Figure 4H), and such reduction of luciferase activity induced by KD platelets (high miR-223) was not observed in VSMCs transfected with miR-223 inhibitor (antagomiR-223) (Figure 4I). These results, along with the miR-223 KO mice demonstrating increased PDGFRβ in the thickened medial layer of (Figure 3G & I), support that the horizontal transfer of platelet-derived miR-223 inhibits VSMC dedifferentiation by directly targeting PDGFRβ.
MiR-223 deficient platelets from CAA patients with severe coronary artery pathology fails to suppress VSMC dedifferentiation (Figure 5 & 6)
Figure 5. Platelet deficient of miR-223 in KD patients with CAA fails to suppress VSMC dedifferentiation.

The protein expression (A) and quantification (B) of markers for differentiation (ACTA2, CNN1, TAGLN) and dedifferentiation (PCNA, OPN, PDGFRβ (HC: n=8, NCAA: n=8, CAA: n=15)) in VSMCs after incubation with platelets from HC (n=10), KD patients with CAA (n=20) and NCAA (n=20) were determined by Western blot (One-way ANOVA or Kruskall-Wallis test). VSMCs were pre-treated with agomiR-223 or antagomiR-223, followed by incubation with platelets from HC (n=9), KD patients with NCAA (n=16) or CAA (n=9). The protein expression (C) and quantification (D) of markers for differentiation (ACTA2, CNN1, TAGLN) and dedifferentiation (PCNA, OPN, PDGFRβ (HC: n=8, NCAA: n=9, CAA: n=9)) were measured by Western blot (Unpaired t test or Mann Whitney test). Abbreviations: HC, VSMCs co-cultured with platelets from HC; KD (NCAA), VSMCs co-cultured with platelets from KD patients with NCAA; KD (CAA), VSMCs co-cultured with platelets from KD patients with CAA. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Figure 6. Deficiency of miR-223 aggravates LCWE-induced abdominal aorta lesions, which was resolved by administration with agomiR-223.

One week after LCWE injection, mice were administrated with agomiR-223 via tail intravenous injection. PBS-injected mice were used as vehicle control. The abdominal aorta tissues were collected two weeks after LCWE-injection. Corresponding immunofluorescence of PDGFRβ (A), smoothelin (B), and ACTA2 in abdominal aorta sections were shown. PDGFRβ or smoothelin was stained as green, ACTA2 as red, and nuclei visualized with DAPI (blue). Scale bar: 20 μm. Quantification of PDGFRβ (C) and smoothelin (D) expression in abdominal aorta lesions were shown (n=6 in C & D, One-way ANOVA and Tukey’s multiple comparisons test). (E) Representative H & E-stained sections from PBS or LCWE injected mice were shown. Scale bar: 50μm. (F) The areas of thickened media layer in each group were calculated by ImageJ software and subjected to statistical analysis for significance (PBS-littermates: n=7, LCWE-littermates: n=24, LCWE-littermates + agomiR-223: n=7, PBS-miR-223 KO: n=7, LCWE-miR-223 KO: n=13, LCWE-miR-223 KO + agomiR-223: n=7, One-way ANOVA and Tukey’s multiple comparisons test). Abbreviations: Littermates, WT littermates; miR-223 KO, miR-223 KO mice; PBS, PBS-injected mice; LCWE, LCWE-injected mice; LCWE + agomiR-223, LCWE-injected mice followed by administration with agomiR-223; L, lumen; A, adventitia. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 versus PBS-injected WT littermates; ##P<0.01, ###P<0.001, ####P<0.0001 versus LCWE-injected WT littermates; $ $ $ $P<0.0001 versus LCWE-injected WT littermates followed by administration with agomiR-223; &&&&P<0.0001 versus PBS-injected miR-223 KO mice; ????P<0.001 versus LCWE-injected miR-223 KO mice.
KD patients with significant coronary artery pathology (CAA) had low platelet miR-223 compared to KD patients with little or no coronary pathology (NCAA) (Figure 1). VSMCs cocultured with NCAA platelets exhibited increased VSMC differentiation markers (ACTA2, CNN1 and TAGLN) with concomitant reduction of VSMC dedifferentiation markers (PCNA, OPN and PDGFRβ), but such changes were not found in VSMCs cocultured with CAA platelets (Figure 5A & B). VSMCs transfected with agomiR-223 showed significant increased miR-223 level while VSMCs transfected with antagomiR-223 showed diminished expression of miR-223 (Online Figure XI A & B). In VSMCs incubated with NCAA platelets, the administration of antagomiR-223 significantly reduced the expression of differentiation markers (ACTA2, CNN1 and TAGLN), and increased the expression of dedifferentiation markers (PCNA, OPN and PDGFRβ). In VSMCs incubated with CAA platelets, the administration of agomiR-223 significantly increased the expression of differentiation markers and reduced the expression of dedifferentiation markers (Figure 5C & D).
To demonstrate the role of platelet-derived miR-223 in KD-induced coronary artery pathology in vivo, miR-223 KO mice (analogous to KD patients with low miR-223) were intraperitoneally injected with LCWE. Two weeks after LCWE treatment, the expression of PDGFRβ (a marker of VSMC dedifferentiation) was increased while the expressions of ACTA2 (a marker of VSMC differentiation) and smoothelin (a marker of terminal VSMC differentiation) were decreased in the thickened medial layer of miR-223 KO mice, compared to littermates (Figure 6A & C). In addition, matrix metalloproteinase 9 (MMP9) (a marker of the synthetic state of VSMCs and aneurysm formation) and cleaved caspase-3 (a marker of VSMC apoptosis) were increased in the thickened medial layer of LCWE-injected miR-223 KO mice (Online Figure XII A–D). The altered expression of PDGFRβ, smoothelin, MMP9 and cleaved caspase-3 were attenuated by administration with agomiR-223 in LCWE-injected mice (Figure 6B & D, Online Figure XII A–D). Two weeks after LCWE treatment, the medial thickening was significantly enhanced in the miR-223 KO mice compared to littermates, which was significantly attenuated by agomiR-223 (Figure 6E & F, Online Figure XIII). These results suggested that miR-223 deficiency in KD platelets contributes to persistent and excessive VSMC dedifferentiation, thereby increasing the risk of coronary pathology. MiR-223 may provide a potential therapeutic approach for the coronary artery pathology by applying a brake on excessive VSMC dedifferentiation in KD.
Adoptive transfer of platelet ameliorated the abdominal aorta lesions in LCWE-injected miR-223 KO mice (Figure 7)
Figure 7. Adoptive transfer of platelets ameliorated the abdominal aorta lesions in LCWE-injected miR-223 KO mice.

One week after LCWE injection, washed platelets (0.25×109/200μl per mouse) isolated from PF4-cre: mT/mG mice were intravenously injected, and the abdominal aorta tissues were collected after one week. (A) Representative H & E-stained sections from LCWE injected miR-223 KO mice infused with platelets isolated from PF4-cre: mT/mG mice were shown. PBS-injected mice were used as vehicle control. Scale bar: 50μm. (B) The areas of thickened media layer were calculated by ImageJ software and subjected to statistical analysis for significance (PBS: n=4, LCWE: n=4, LCWE + PLT: n=4, Kruskall-Wallis test and Dunn’s multiple comparisons test). (C) Representative immunofluorescent images of PDGFRβ and ACTA2 in sections from PBS-injected, LCWE-injected, and LCWE-injected miR-223 KO mice followed by platelet infusion. PDGFRβ stained as green, ACTA2 as red, and nuclei visualized with DAPI (blue). Scale bar: 20 μm. (D) Quantification of PDGFRβ in the injured vessels (PBS: n=5, LCWE: n=5, LCWE + PLT: n=5, One-way ANOVA and Tukey’s multiple comparisons test). Abbreviations: PBS, PBS-injected mice; LCWE; LCWE-injected mice; LCWE + PLTs, LCWE-injected mice followed by infusion with platelets; miR-223 KO, miR-223 KO mice. **P<0.01, ****P<0.0001 versus PBS-injected miR-223 KO mice; ####P<0.0001 versus LCWE-injected miR-223 KO mice.
To demonstrate a platelet-specific contribution of miR-223 in suppressing VSMC de-differentiation, washed platelets (0.25× 109 per mouse) 32 isolated from PF4-cre: mT/mG mice (where platelets are GFP positive) were intravenously injected into LCWE-injected miR-223 KO mice and WT littermate controls. Two weeks after LCWE injection, GFP positive platelets were found in the damaged abdominal aorta’s medial layer of both the miR-223 KO mice and the littermate control (Online Figure XIVA). Concurrent flow cytometry analysis confirmed between 3–4% GFP positive platelets remaining in the murine blood samples (Online Figure XIVB). We then compared miR-223 KO mice two weeks post injection with PBS only, or LCWE only, or LCWE and platelets. Compared to PBS-injected miR-223 KO mice, the medial thickening was significantly increased in LCWE-injected miR-223 KO mice, but no significant increase was found in LCWE-injected miR-223 KO mice with adoptive platelet transfer (Figure 7A & B, Online Figure XV). Also, the LCWE-induced induction of PDGFRβ was significantly attenuated by the adoptive platelet transfer (Figure 7C & D). Moreover, level of platelet miR-223 was significantly higher than peripheral blood mononuclear cells (PBMCs) isolated from the same volume of blood (Online Figure XIVC). Taken together, these results support a platelet-specific contribution of miR-223 in suppressing VSMC de-differentiation applying a brake on the repair process.
Targeting miR-223-PDGFRβ axis attenuated LCWE-induced abdominal aorta lesions in KD mice (Figure 8)
Figure 8. Targeting miR-223-PDGFRβ axis attenuated LCWE-induced abdominal aorta lesions in KD mice.

(A) Relative levels of miR-223 in platelets from KD patients with NCAA (n=67), KD patients with GCAA (n=12) versus HC group (n=67) (Kruskall-Wallis test and Dunn’s multiple comparisons test). (B) Relative level of miR-223 in platelets from GCAA patients with (Statin +) (n=12) or without simvastatin (Statin -) (n=12) treatment (Mann Whitney test). (C) Coronary artery Z-worst in GCAA patients at baseline (n=12), at one (1M) (n=12) and six months (6M) (n=9) after treatment with Statin. The data is not applicable for three GCAA patients six months after treatment with Statin. (D) Representative H & E -stained sections from PBS-injected (n=6), LCWE-injected (n=7), LCWE-injected miR-223 KO mice followed by administration with Imatinib mesylate (Imatinib) (n=7), Rapamycin D (n=7), Atorvastatin calcium (Statin) (n=7) were shown. PBS-injected mice were used as vehicle control. Scale bar: 50μm. (E) The areas of thickened media layer were calculated by ImageJ software and subjected to statistical analysis for significance (One-way ANOVA and Tukey’s multiple comparisons test). Abbreviations: PBS, PBS-injected mice; LCWE; LCWE-injected mice; Imatinib, Rapamycin D, Statin indicate LCWE-injected mice followed by administration with imatinib, Rapamycin D, Statin, respectively; miR-223 KO, miR-223 KO mice. **P<0.01 versus PBS-injected miR-223 KO mice; #P<0.05 versus LCWE-injected miR-223 KO mice; &P<0.05 versus LCWE-injected miR-223 KO mice followed by administration with Imatinib mesylate.
Our studies provide important insights into novel therapeutic targets, in particular the miR-223/PDGFRβ/VSMC axis. Despite current state-of-the-art therapies (IVIG targeting the immune system, and aspirin targeting platelets), patients with giant coronary artery aneurysms (GCAA) consistently demonstrate low miR-223 (Figure 8A). Recent clinical studies have suggested statins may be beneficial in KD with protective actions on endothelial damage and inflammation 37. Twelve KD patients with severe coronary pathology (GCAA) recruited from a cohort study (2017102710) were treated with IVIG/aspirin with or without statin. No difference in miR-223 level was found after four weeks of treatment (Figure 8B). Compared to the baseline, no significant reduction of Z-worst was found in the CAA patients treated with combination therapy for one-month or six-month (Figure 8C), with one patient markedly increasing their Z-worst (Figure 8C). We then explored, using the LCWE murine model, whether targeting the miR-223/PDGFRβ/VSMC axis may be beneficial in KD.
To investigate whether platelet miR-223-PDGFRβ axis may provide therapeutic benefit for CAA progression in KD, 1) agomiR-223 (miR-223 mimic, 80mg/kg per mouse) 38 2) Imatinib mesylate (PDGFR α and β inhibitor, 50mg/kg/day per mouse) 28, 3) Rapamycin D (mTORC1 inhibitor recognized to promote VSMC differentiation, 2mg/kg/day per mouse) 31, and 4) Atorvastatin calcium (used in recent clinical trials as an endothelial protectant, 10mg/kg/day per mouse) 27, were introduced to LCWE-induced miR-223 KO mice. MiR-223 KO mice were used to mimic the platelet deficient of miR-223 in KD patients with GCAA. Compared to PBS-treated mice, medial thickening and damage was again evident two weeks after LCWE injection (Figure 8D). Treatment of imatinib mesylate (Imatinib), rapamycin D and agomiR-223 significantly attenuated the medial thickening in the LCWE-injected mice (Figure 8D & E), indicating that PDGFRβ-induced VSMC dedifferentiation plays a crucial role in KD vascular pathology. However, such attenuation of medial thickening was not found in LCWE-injected mice treated with statin, consistent with the results of KD patients with severe coronary pathology (Figure 8D & E, Online Figure XVI). Taken together, both the human studies and mouse model of KD support the importance of miR-223, PDGFRβ and VSMC differentiation in protecting and applying a brake on KD induced injury repair.
The reason why less than 5-year-old children are particularly susceptible to KD and the coronary vascular complications is entirely unknown. The answer may again lie with miR-223. Assessing levels of miR-223 in several age cohorts (<5-year-old n=21, 5–10 year old n=21 and >20 year old n=8) demonstrated the lowest levels in the <5 year old with progressive increase with age (Online Figure XVII). With low miR-223 indicating highest vulnerability to coronary pathology, less than five-year-old would thus be the most at risk cohort for disease. Targeting the miR-223-PDGFRβ axis in KD vascular pathology may provide therapeutic value in this severe childhood disease.
DISCUSSION
Instigation of vascular injury and repair (inflammation, endothelial damage, VSMC dedifferentiation and proliferation) is well studied, but the mechanisms for limiting the repair process, are poorly understood. Based on our current studies, impaired recovery from prolonged inflammatory damage contributes to abnormal remodeling and severe coronary artery pathology in KD. Hyperactive platelets in KD (arising from the inflammation and endothelial damage) are recognized to increase the risk of coronary syndrome 39. Hyperactive platelets deliver miR-223 into VSMCs (miR-223 is not native to VSMC), suppressing VSMC dedifferentiation through PDGFRβ expression inhibition. This brake on VSMCs promotes resolution from arterial injury. Platelet deficient in miR-223 leads to coronary artery pathology as platelet uptake fails to suppress VSMC dedifferentiation (Central illustration). Thus, targeting the miR-223-PDGFRβ axis may provide novel therapeutic option for KD-induced vascular pathologies.
Deficiency of miR-223 in CAA platelets contributes to vascular pathology and the increased risk of aneurysm formation in KD
VSMC dedifferentiation plays an important role in the formation of arterial aneurysms 12, 13, 16. It is known that growth factors released from activated platelets e.g. PDGF induces VSMC dedifferentiation in injured vessels 10, 40. Upon arterial injury in KD, platelets provide an immediate response with activation and release of alpha-granule storage including growth factors such as PDGF-BB. Based on our in vitro and in vivo results, KD platelets also deliver miR-223. In the absence of miR-223, VSMC dedifferentiation proceeds unabated leading to medial thickening, damage, apoptosis and MMP9 release. Increased levels of MMP9 has been found in aneurysmal arteries in KD 36, thought to drive the degradation of vessel wall matrix, promoting CAA formation 41. Our results also showed that the expression of MMP9 was altered in the injured arteries of LCWE-injected mice and was significantly abrogated by administration of agomiR-223 in LCWE-injected miR-223 KO mice. Consistent with our findings, recent studies have suggested that miR-223 may play an important role in endothelial pathology in KD, possibly through a novel endocrine genetic signaling mechanism 33, 42, with high expression level of miR-223 at the acute stage of KD, and relative lower expression in KD with coronary lesions 42, 43.
Measuring platelet miR-223 may allow for identification of patients at high risk of developing GCAA
MiR-223 is one of the most abundantly expressed miRNA in human platelets 44. Diminished platelet miR-223 has been reported to correlate with platelet reactivity and higher risk of coronary artery disease 34. Consistent with previous studies 33, 42, 43, we also found that level of platelet miR-223 was increased at the acute stage of KD, but decreased in coronary lesions of KD. The inflammatory cytokine TNF-alpha (TNF-α) has been found to contribute to altered miR-223 level in cells 33, 45. To better understand the induction of miR-223 in KD, we measured the level of TNF-α in plasma from HC and KD patients, and we found that the level of TNF-α was significantly increased in KD group compared to HC (Online Figure XVIII A). Similar to KD patients, the level of TNF-α in plasma from LCWE-injected mice was also significantly increased compared to the PBS-injected mice group (Online Figure XVIII B). Human or murine platelets incubated with TNF-α showed induction of miR-223 in a time- and concentration-dependent manner (Online Figure XVIII C & D), suggesting that TNF-α contributes to the induction of platelet miR-223 in KD patients and LCWE-injected murine model. Since platelets are anucleate, the expression of platelet miR-223 is regulated by Dicer1-mediated synthesis of mature miRNA from precursor miRNA (pre-miRNA). We therefore measured the platelet expression of Dicer1, mature miR-223, and pre-miR-223 in HC, KD patients with coronary artery aneurysm (NCAA) and KD patients with coronary artery aneurysm (CAA). Compared to HC and CAA group, the levels of mature miR-223 and Dicer1 were significantly higher in NCAA group while the level of pre-miR-223 was significantly lower in NCAA group (Online Figure XVIII E, F & G). Both NCAA and CAA (KD patients) have been induced to produce mature miR-223 (i.e. with TNF-α) in comparison to the HC (control). CAA resembles HC as induction appears to have largely failed (Online Figure XVIII E, F & G). These results at least in part explain why NCAA patients have more platelet miR-223 than CAA patients. As previously described, a recent study demonstrated that TNF-α may contribute to the downregulation of miR-223 33. This was however in human umbilical vein endothelial cells artificially overexpressing miR-223. Inflammation and TNF-α may be damaging/ inducing apoptosis in the endothelial cells, and thus the reduction of miR-223. Such discrepancy on the effect of TNF-α may therefore be attributed to the different cell types (platelet vs endothelial cell) and different experimental treatment (pathophysiological level vs overexpression). Combined, the results are not mutually exclusive and serve to highlight the potential importance of miR-223 transfer from platelets.
It has been reported that cardiac complications, including stenosis, thrombotic occlusion, myocardial infarction, and sudden death occur most often in patients with GCAA 2, it is thus important to identify these patients as early as possible. DdPCR is a recently introduced technology that allows for precise and sensitive absolute quantification of target nucleic acids based on superior sample partitioning, increased signal-to-noise ratio, and Poisson statistics 46. More importantly, it can tolerate wide variations in amplification efficiency without affecting the quantitative results of the target. Based on the statistical analysis of our ddPCR results, the differential expression of miR-223 in KD platelets was dependent on the severity of coronary artery pathology. Importantly we also demonstrate that the lowest levels of miR-223 are observed in less than 5 year old children, the age at highest risk for KD vascular complications. Thus measuring platelet miR-223 may have both diagnostic and prognostic value in KD.
Targeting the miR-223-PDGFRβ axis may serve as an alternative therapy for KD patients with CAA
Prolonged and chronic inflammation induces the vascular damage in KD, which persists even in the whole process of KD 36. Adjunctive therapies such as statins have been introduced to KD with CAA based on their pleiotropic effects of alleviating inflammation and protecting endothelial function 37, 47. However, in our recruited KD patients, twelve GCAA patients with miR-223 deficiency were not responsive to the statins treatment regardless of dose and duration of CAA, suggesting that alternative therapy for coronary lesions is urgently needed for those KD patients with platelet deficient of miR-223.
The important role of PDGF-induced VSMC phenotypic switch in pathogenesis of coronary lesions in KD has been reported previously 4, 14, 48 which suggests that pharmacological inhibition of VSMC dedifferentiation, such as rapamycin D 30 and imatinib 28, 29, may be the potential therapy for CAA progression. Imatinib (STI571; Gleevec® or Glivec®), a tyrosine kinase inhibitor of PDGFRβ, has been reported efficacy in the treatment of systemic rheumatic diseases 49 and pulmonary arterial hypertension 50, but their use has not been described in KD. Based on our results, administration of imatinib attenuated LCWE-induced abdominal aorta lesions in KD mice, supporting the use of imatinib in treating KD vascular lesions. Rapamycin D, known to inhibit PDGF-induced VSMC dedifferentiation 30, attenuated LCWE-induced aortic lesions. Targeting platelet miR-223-PDGFRβ axis may provide alternative therapy for CAA progression in KD patients.
Study limitations
We acknowledge several limitations with our studies. We are unable to directly address the mechanisms in human. However, we have delineated the mechanism in vitro using patient platelets and human coronary vascular smooth muscle cells as well as in vivo using a well-established KD mouse model. Many other mechanisms including miRNAs may also contribute to the coronary pathology of KD however based on the human and animal studies we are confident that miR-223 plays a major role. Larger clinical studies are required to establish miR-223 as a prognostic biomarker as well as for therapies with miR-223 inducers, PDGF inhibitors, and inducers of VSMC differentiation.
Summary
The key finding of this study is that activated KD platelets transfer miR-223 to VSMCs and suppress VSMC dedifferentiation in part through targeting PDGFRβ. Coronary pathology in KD is in part due to lower levels of platelet-derived miR-223 leading to maintenance of the synthetic and proliferative VSMC state. Thus, platelet miR-223 levels can potentially serve as KD diagnostic and prognostic biomarkers. These results not only confirm the pivotal role of activated platelets in maintaining a delicate balance of VSMC response to injury, but also provide a novel mechanism into CAA formation in KD. The mechanism reported here may not only be useful in the early evaluation of prognosis of KD patients but may also provide important insights into miRNA based therapeutic targets for CAA in KD patients.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
The acute vascular inflammation of Kawasaki disease (KD) is characterized by activation of platelets and vascular smooth muscle cell (VSMC) dedifferentiation, potential contributors to KD coronary pathologies.
VSMC dedifferentiation plays a critical role in many vascular pathologies including medial damage, thickening, and aneurysm formation.
Platelet-derived miRNAs play a dual role in vascular injury repair, initiating an immediate repair process and, concurrently, a delayed process to prevent excessive repair.
What New Information Does This Article Contribute?
The level of miR-223 in platelets from KD patients inversely correlates with severity of coronary artery pathology.
Horizontal transfer of KD platelet-derived miR-223 suppresses vascular smooth muscle cell (VSMC) dedifferentiation by inhibiting the expression of PDGFRβ.
Deficiency of miR-223 induction in KD platelets contributes to the increased risk of coronary artery pathology and aneurysm formation.
The miR-223-PDGFRβ axis may provide a therapeutic target that would ameliorate coronary artery pathology in KD.
Platelet hyperreactivity is a hallmark of KD. Despite treatment with intravenous immunoglobulin plus high-dose aspirin, 5 to 15% of KD children develop coronary artery pathology leading to significant morbidity and mortality. VSMC dedifferentiation plays a key role in the pathogenesis of coronary artery pathology and aneurysm formation. Activated platelets have been found to regulate VSMC dedifferentiation via horizontal transfer of platelet-derived miRNAs. We therefore hypothesized that the transfer of platelet-derived miRNAs into VSMCs may promote the resolution of coronary artery pathology in KD. Here we found that KD platelets with low induction of miR-223 had increased risk of severe coronary pathology. The horizontal transfer of KD platelet-derived miR-223 suppresses VSMC dedifferentiation by inhibiting the expression of PDGFRβ. This brake on VSMC dedifferentiation promotes resolution from arterial injury.
Acknowledgments
SOURCES OF FUNDING
This work was supported by the National Natural Science Foundation of China (Grant No. 81670117, 81970437, 81902144, 81903605), the Guangdong Province Science and Technology Project (Grant No.2017A050506008), the Guangzhou science and technology project (Grant No. 201904010483), the Guangdong Outstanding Young Scientist Funding (Grant No. 2016A030306049) and the fund from Guangzhou Women and Children's Medical Center/Guangzhou Institute of Pediatrics (Grant No. IP-2018-022). Further support was also provided by NIH NHLBI grants HL115247 and HL122815.
Nonstandard Abbreviations and Acronyms
- ACTA2
α-smooth muscle actin
- A
adventitia
- AUC
the area under curve
- β-TG
beta-thromboglobulin
- CAA
coronary arterial aneurysm
- CI
confidence interval
- CNN1
calponin
- CD62P
P-selectin
- DdPCR
droplet digital PCR
- FISH
fluorescent in-situ hybridization
- GCAA
giant coronary arterial aneurysm
- HC
healthy control
- KD
Kawasaki disease
- LCWE
lactobacillus casei cell wall extract
- L
lumen
- MCAA
medium coronary arterial aneurysm
- miR-223 KO
miR-223 knockout
- MMP9
matrix metalloproteinase 9
- MFI
mean fluorescence intensity
- NCAA
non-coronary arterial aneurysm
- PDGFRβ
platelet-derived growth factor receptor β
- PDGF-BB
platelet-derived growth factor-BB
- PCNA
proliferating cell nuclear antigen
- PF4
platelet factor 4
- PMP
platelet-derived microparticle
- ROC
receiver operating characteristic
- SCAA
small coronary arterial aneurysm
- TAGLN
transgelin
- TNF-α
TNF-alpha
- VSMC
vascular smooth muscle cell
- WT
wide type
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, Baker AL, Jackson MA, Takahashi M, Shah PB, Kobayashi T, Wu MH, Saji TT, Pahl E. Diagnosis, treatment, and long-term management of kawasaki disease: A scientific statement for health professionals from the american heart association. Circulation. 2017;135:e927–e999 [DOI] [PubMed] [Google Scholar]
- 2.Denby KJ, Clark DE, Markham LW. Management of kawasaki disease in adults. Heart. 2017;103:1760–1769 [DOI] [PubMed] [Google Scholar]
- 3.Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovascular research. 2012;95:194–204 [DOI] [PubMed] [Google Scholar]
- 4.Shulman ST, Rowley AH. Kawasaki disease: Insights into pathogenesis and approaches to treatment. Nat Rev Rheumatol. 2015;11:475–482 [DOI] [PubMed] [Google Scholar]
- 5.Friedman KG, Gauvreau K, Hamaoka-Okamoto A, Tang A, Berry E, Tremoulet AH, Mahavadi VS, Baker A, deFerranti SD, Fulton DR, Burns JC, Newburger JW. Coronary artery aneurysms in kawasaki disease: Risk factors for progressive disease and adverse cardiac events in the us population. J Am Heart Assoc. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burns JC, Glode MP, Clarke SH, Wiggins J, Jr., Hathaway WE. Coagulopathy and platelet activation in kawasaki syndrome: Identification of patients at high risk for development of coronary artery aneurysms. The Journal of pediatrics. 1984;105:206–211 [DOI] [PubMed] [Google Scholar]
- 7.Yahata T, Suzuki C, Yoshioka A, Hamaoka A, Ikeda K. Platelet activation dynamics evaluated using platelet-derived microparticles in kawasaki disease. Circ J. 2014;78:188–193 [DOI] [PubMed] [Google Scholar]
- 8.Taki M, Kobayashi M, Ohi C, Shimizu H, Goto K, Aso K, Murano K. Spontaneous platelet aggregation in kawasaki disease using the particle counting method. Pediatr Int. 2003;45:649–652 [DOI] [PubMed] [Google Scholar]
- 9.Etulain J Platelets in wound healing and regenerative medicine. Platelets. 2018;29:556–568 [DOI] [PubMed] [Google Scholar]
- 10.Risinger GM, Jr.,, Hunt TS, Updike DL, Bullen EC, Howard EW. Matrix metalloproteinase-2 expression by vascular smooth muscle cells is mediated by both stimulatory and inhibitory signals in response to growth factors. J Biol Chem. 2006;281:25915–25925 [DOI] [PubMed] [Google Scholar]
- 11.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang P, Guan Y, Chen J, Li X, McConnell BK, Zhou W, Boini KM, Zhang Y. Contribution of p62/sqstm1 to pdgf-bb-induced myofibroblast-like phenotypic transition in vascular smooth muscle cells lacking smpd1 gene. Cell Death Dis. 2018;9:1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL, Lau CL, Owens GK. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg. 2009;138:1392–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orenstein JM, Shulman ST, Fox LM, Baker SC, Takahashi M, Bhatti TR, Russo PA, Mierau GW, de Chadarevian JP, Perlman EJ, Trevenen C, Rotta AT, Kalelkar MB, Rowley AH. Three linked vasculopathic processes characterize kawasaki disease: A light and transmission electron microscopic study. PloS one. 2012;7:e38998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elia L, Kunderfranco P, Carullo P, Vacchiano M, Farina FM, Hall IF, Mantero S, Panico C, Papait R, Condorelli G, Quintavalle M. Uhrf1 epigenetically orchestrates smooth muscle cell plasticity in arterial disease. J Clin Invest. 2018;128:2473–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clement M, Chappell J, Raffort J, Lareyre F, Vandestienne M, Taylor AL, Finigan A, Harrison J, Bennett MR, Bruneval P, Taleb S, Jorgensen HF, Mallat Z. Vascular smooth muscle cell plasticity and autophagy in dissecting aortic aneurysms. Arteriosclerosis, thrombosis, and vascular biology. 2019;39:1149–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao G, Fu Y, Cai Z, Yu F, Gong Z, Dai R, Hu Y, Zeng L, Xu Q, Kong W. Unspliced xbp1 confers vsmc homeostasis and prevents aortic aneurysm formation via foxo4 interaction. Circ Res. 2017;121:1331–1345 [DOI] [PubMed] [Google Scholar]
- 18.Zeng Z, Xia L, Fan X, Ostriker AC, Yarovinsky T, Su M, Zhang Y, Peng X, Xie Y, Pi L, Gu X, Chung SK, Martin KA, Liu R, Hwa J, Tang WH. Platelet-derived mir-223 promotes a phenotypic switch in arterial injury repair. J Clin Invest. 2019;129:1372–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirschbaum M, Karimian G, Adelmeijer J, Giepmans BN, Porte RJ, Lisman T. Horizontal rna transfer mediates platelet-induced hepatocyte proliferation. Blood. 2015;126:798–806 [DOI] [PubMed] [Google Scholar]
- 20.Lee Y, Wakita D, Dagvadorj J, Shimada K, Chen S, Huang G, Lehman TJ, Fishbein MC, Hoffman HM, Crother TR, Arditi M. Il-1 signaling is critically required in stromal cells in kawasaki disease vasculitis mouse model: Role of both il-1alpha and il-1beta. Arteriosclerosis, thrombosis, and vascular biology. 2015;35:2605–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee Y, Schulte DJ, Shimada K, Chen S, Crother TR, Chiba N, Fishbein MC, Lehman TJ, Arditi M. Interleukin-1beta is crucial for the induction of coronary artery inflammation in a mouse model of kawasaki disease. Circulation. 2012;125:1542–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehman TJ, Walker SM, Mahnovski V, McCurdy D. Coronary arteritis in mice following the systemic injection of group b lactobacillus casei cell walls in aqueous suspension. Arthritis Rheum. 1985;28:652–659 [DOI] [PubMed] [Google Scholar]
- 23.Elgheznawy A, Shi L, Hu J, Wittig I, Laban H, Pircher J, Mann A, Provost P, Randriamboavonjy V, Fleming I. Dicer cleavage by calpain determines platelet microrna levels and function in diabetes. Circ Res. 2015;117:157–165 [DOI] [PubMed] [Google Scholar]
- 24.Laffont B, Corduan A, Ple H, Duchez AC, Cloutier N, Boilard E, Provost P. Activated platelets can deliver mrna regulatory ago2*microrna complexes to endothelial cells via microparticles. Blood. 2013;122:253–261 [DOI] [PubMed] [Google Scholar]
- 25.Michael JV, Wurtzel JGT, Mao GF, Rao AK, Kolpakov MA, Sabri A, Hoffman NE, Rajan S, Tomar D, Madesh M, Nieman MT, Yu J, Edelstein LC, Rowley JW, Weyrich AS, Goldfinger LE. Platelet microparticles infiltrating solid tumors transfer mirnas that suppress tumor growth. Blood. 2017;130:567–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wakita D, Kurashima Y, Crother TR, Noval Rivas M, Lee Y, Chen S, Fury W, Bai Y, Wagner S, Li D, Lehman T, Fishbein MC, Hoffman HM, Shah PK, Shimada K, Arditi M. Role of interleukin-1 signaling in a mouse model of kawasaki disease-associated abdominal aortic aneurysm. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:886–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roth L, Rombouts M, Schrijvers DM, Martinet W, De Meyer GR. Cholesterol-independent effects of atorvastatin prevent cardiovascular morbidity and mortality in a mouse model of atherosclerotic plaque rupture. Vascul Pharmacol. 2016;80:50–58 [DOI] [PubMed] [Google Scholar]
- 28.Ruan J, Luo M, Wang C, Fan L, Yang SN, Cardenas M, Geng H, Leonard JP, Melnick A, Cerchietti L, Hajjar KA. Imatinib disrupts lymphoma angiogenesis by targeting vascular pericytes. Blood. 2013;121:5192–5202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by pdgf inhibition. J Clin Invest. 2005;115:2811–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu QB, Wan MY, Wang PY, Zhang CX, Xu DY, Liao X, Sun HJ. Chicoric acid prevents pdgf-bb-induced vsmc dedifferentiation, proliferation and migration by suppressing ros/nfkappab/mtor/p70s6k signaling cascade. Redox Biol 2018;14:656–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li FF, Shang XK, Du XL, Chen S. Rapamycin treatment attenuates angiotensin ii -induced abdominal aortic aneurysm formation via vsmc phenotypic modulation and down-regulation of erk1/2 activity. Curr Med Sci. 2018;38:93–100 [DOI] [PubMed] [Google Scholar]
- 32.Carestia A, Mena HA, Olexen CM, Ortiz Wilczynski JM, Negrotto S, Errasti AE, Gomez RM, Jenne CN, Carrera Silva EA, Schattner M. Platelets promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell reports. 2019;28:896–908.e895 [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Ding YY, Chen Y, Xu QQ, Qian GH, Qian WG, Cao L, Zhou WP, Hou M, Lv HT. Mir-223–3p alleviates vascular endothelial injury by targeting il6st in kawasaki disease. Frontiers in pediatrics. 2019;7:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sunderland N, Skroblin P, Barwari T, Huntley RP, Lu R, Joshi A, Lovering RC, Mayr M. Microrna biomarkers and platelet reactivity: The clot thickens. Circ Res. 2017;120:418–435 [DOI] [PubMed] [Google Scholar]
- 35.Shan Z, Qin S, Li W, Wu W, Yang J, Chu M, Li X, Huo Y, Schaer GL, Wang S, Zhang C. An endocrine genetic signal between blood cells and vascular smooth muscle cells: Role of microrna-223 in smooth muscle function and atherogenesis. Journal of the American College of Cardiology. 2015;65:2526–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abou Sherif S, Ozden Tok O, Taskoylu O, Goktekin O, Kilic ID. Coronary artery aneurysms: A review of the epidemiology, pathophysiology, diagnosis, and treatment. Frontiers in cardiovascular medicine. 2017;4:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tremoulet AH, Jain S, Jone PN, Best BM, Duxbury EH, Franco A, Printz B, Dominguez SR, Heizer H, Anderson MS, Glode MP, He F, Padilla RL, Shimizu C, Bainto E, Pancheri J, Cohen HJ, Whitin JC, Burns JC. Phase i/iia trial of atorvastatin in patients with acute kawasaki disease with coronary artery aneurysm. The Journal of pediatrics. 2019;215:107–117.e112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui RR, Li SJ, Liu LJ, Yi L, Liang QH, Zhu X, Liu GY, Liu Y, Wu SS, Liao XB, Yuan LQ, Mao DA, Liao EY. Microrna-204 regulates vascular smooth muscle cell calcification in vitro and in vivo. Cardiovascular research. 2012;96:320–329 [DOI] [PubMed] [Google Scholar]
- 39.Nagalla S, Shaw C, Kong X, Kondkar AA, Edelstein LC, Ma L, Chen J, McKnight GS, Lopez JA, Yang L, Jin Y, Bray MS, Leal SM, Dong JF, Bray PF. Platelet microrna-mrna coexpression profiles correlate with platelet reactivity. Blood. 2011;117:5189–5197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tong L, Qi G. Crocin prevents plateletderived growth factor bbinduced vascular smooth muscle cells proliferation and phenotypic switch. Mol Med Rep. 2018;17:7595–7602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gavin PJ, Crawford SE, Shulman ST, Garcia FL, Rowley AH. Systemic arterial expression of matrix metalloproteinases 2 and 9 in acute kawasaki disease. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:576–581 [DOI] [PubMed] [Google Scholar]
- 42.Chu M, Wu R, Qin S, Hua W, Shan Z, Rong X, Zeng J, Hong L, Sun Y, Liu Y, Li W, Wang S, Zhang C. Bone marrow-derived microrna-223 works as an endocrine genetic signal in vascular endothelial cells and participates in vascular injury from kawasaki disease. J Am Heart Assoc 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Y, Ding YY, Ren Y, Cao L, Xu QQ, Sun L, Xu MG, Lv HT. Identification of differentially expressed micrornas in acute kawasaki disease. Mol Med Rep. 2018;17:932–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elgheznawy A, Fleming I. Platelet-enriched micrornas and cardiovascular homeostasis. Antioxidants & redox signaling. 2018;29:902–921 [DOI] [PubMed] [Google Scholar]
- 45.Moriya N, Shibasaki S, Karasaki M, Iwasaki T. The impact of microrna-223–3p on il-17 receptor d expression in synovial cells. PloS one. 2017;12:e0169702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vidal-Folch N, Gavrilov D, Raymond K, Rinaldo P, Tortorelli S, Matern D, Oglesbee D. Multiplex droplet digital pcr method applicable to newborn screening, carrier status, and assessment of spinal muscular atrophy. Clin Chem. 2018;64:1753–1761 [DOI] [PubMed] [Google Scholar]
- 47.Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res. 2017;120:229–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H, Wang H, Cheng P. Kawasaki disease on pdgf expression and vsmc proliferation. J Tongji Med Univ. 1998;18:243–246 [DOI] [PubMed] [Google Scholar]
- 49.Paniagua RT, Robinson WH. Imatinib for the treatment of rheumatic diseases. Nature clinical practice. Rheumatology. 2007;3:190–191 [DOI] [PubMed] [Google Scholar]
- 50.Frost AE, Barst RJ, Hoeper MM, Chang HJ, Frantz RP, Fukumoto Y, Galie N, Hassoun PM, Klose H, Matsubara H, Morrell NW, Peacock AJ, Pfeifer M, Simonneau G, Tapson VF, Torres F, Dario Vizza C, Lawrence D, Yang W, Felser JM, Quinn DA, Ghofrani HA. Long-term safety and efficacy of imatinib in pulmonary arterial hypertension. J Heart Lung Transplant. 2015;34:1366–1375 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.