

Abstract
Objective:
Autoimmune responses to DNA topoisomerase-I (TOP1) are found in a subset of patients with scleroderma at high risk for interstitial lung disease (ILD) and mortality. Anti-TOP1 antibodies (ATA) are associated with specific HLA-DRB1 alleles, and the frequency of HLA-DR-restricted TOP1-specific CD4+ T cells is associated with the presence, severity, and progression of ILD. Although this strongly implicates the presentation of TOP1 peptides by HLA-DR in scleroderma pathogenesis, the processing and presentation of TOP1 has not been studied.
Methods:
We developed a natural antigen processing assay (NAPA) to identify putative CD4+ T cell epitopes of TOP1 presented by monocyte-derived dendritic cells (mo-DCs) from six ATA-positive patients with scleroderma. Mo-DCs were pulsed with TOP1 protein, HLA-DR:peptide complexes were isolated, and eluted peptides were analyzed by mass spectrometry. We then examined the ability of these naturally presented peptides to induce CD4+ T cell activation in 11 ATA-positive and 11 ATA-negative scleroderma patients.
Results:
We found that a common set of 10 TOP1 epitopes was presented by mo-DCs from scleroderma patients with diverse HLA-DR variants. Sequence analysis revealed shared peptide-binding motifs within the HLA-DRβ chains of ATA-positive patients and a subset of TOP1 epitopes with distinct sets of anchor residues capable of binding to multiple different HLA-DR variants. The NAPA-derived epitopes elicited robust CD4+ T cell responses in 73% (8/11) ATA-positive patients, and the number of epitopes recognized correlated with ILD severity (p=0.025).
Conclusion:
These findings mechanistically implicate presentation of a convergent set of TOP1 epitopes in the development of scleroderma.
Introduction
Scleroderma, also called systemic sclerosis, is a complex autoimmune rheumatic disease of unknown etiology, characterized by widespread vasculopathy, fibrosis of the skin and internal organs (1), and immunological derangements, including the production of autoantibodies to specific nuclear antigens (2). Anti-topoisomerase-I (TOP1) autoantibodies (ATA; also known as anti-Scl-70) are present in 20–45% of patients with scleroderma and are associated with diffuse skin involvement, pulmonary fibrosis and high mortality (3). ATA production in patients with scleroderma is associated with specific HLA-DRB1 alleles (4), implicating CD4+ T helper cells in driving immune responses to TOP1. TOP1-specific CD4+ T cells have been detected in the peripheral blood of patients with scleroderma (5–8), are HLA-DR restricted, and their frequency is associated with the presence, severity, and progression of lung fibrosis (9). Previous attempts to define TOP1 epitopes in patients with scleroderma using overlapping peptide/protein fragment libraries or peptides derived from in silico prediction have resulted in the identification of scattered epitopes with unspecified biological and clinical significance (5–8). Therefore, although ATA-associated HLA-DRB1 alleles have been defined and TOP-I-specific CD4+ T cells have been detected in ATA-positive patients, the identification of the TOP1 epitopes driving the autoimmune process has remained elusive.
To overcome current limitations related to the identification and study of autoreactive T cell epitopes, we developed a method for identifying the repertoire of naturally processed TOP1 peptides presented by antigen presenting cells (APCs) obtained from patients with scleroderma. This approach has innovative features compared to traditional epitope-discovery methods, as it harnesses the cellular MHC class II antigen processing machinery present in an individual to identify naturally presented peptides, without requiring pre-defined knowledge about the target peptide length or HLA status. Our analysis of naturally presented TOP1 peptides has revealed unexpected insights into the pathogenesis of scleroderma and identified a discrete set of clinically relevant TOP1-specific CD4+ T cell epitopes that may inform the future development of antigen-specific diagnostic and therapeutic tools.
Materials and Methods
Human Subjects
Patients in this study were drawn from an observational cohort of patients with scleroderma followed at the Johns Hopkins Scleroderma Center. All patients participating in this study met the 2013 Classification Criteria for Systemic Sclerosis or met at least 3 out of 5 criteria for CREST syndrome (12,13). The Johns Hopkins institutional review board approved the study and all patients signed written informed consent. Detailed information about this cohort is provided in the supplementary materials and methods. Clinical data are also summarized in Table S1.
Natural antigen processing assay (NAPA)
Monocyte-derived dendritic cell differentiation and antigen-loading.
Peripheral blood mononuclear cells (PBMCs) were isolated by density-gradient centrifugation (Ficoll-Paque Plus, GE Healthcare) from 40mL of whole blood obtained from 6 ATA-positive patients. CD14 MicroBeads were used to positively select CD14+ monocytes and cells were differentiated for 7 days into monocyte-derived dendritic cells (mo-DCs), using Mo-DC Differentiation Medium (Miltenyi Biotec). Mo-DCs (2–5 × 106) were incubated overnight at 37°C in 5% CO2 with or without 400 ug of custom-made human recombinant TOP1 protein purified from baculovirus-infected insect cells (GenScript), to allow the mo-DC to internalize the protein, process it and present TOP1 peptides onto HLA-DR molecules.
Isolation of HLA-DR- peptide complexes.
Antigen-pulsed mo-DCs were collected and washed with cold buffer A (PBS, 2 mM EDTA; pH 7.4–7.6). The cells were then lysed in cold buffer B (1% CHAPS, 1 mM EDTA, protease inhibitor cocktail) and incubated at 4° C on a rocking table for 15 minutes. This lysis step was repeated, insoluble material removed by centrifugation, and the supernatant was incubated with Protein G Dynabeads (Thermo Fisher) coated with 20 ug of anti-HLA-DR (clone L243, BioLegend) or isotype control (clone MOPC-173, BioLegend) antibodies at 4° C for one hour with inversion. The beads were then washed 4-times with lysis buffer B, and 3-times with buffer C (20 mM Tris, 150 mM NaCl, pH 7.4). The HLA-DR-bound peptides were eluted by incubating the beads with 0.1% TFA for 1 minute at room temperature and harvesting the eluate. This step was repeated once and the pooled eluate was centrifuged through a 10kD centrifugal filter (Microcon, Millipore). The filter was washed with 0.1 % TFA and pooled flow-through containing the eluted peptides was lyophilized by vacuum centrifugation. The peptides were then analyzed by mass spectrometry, as described in detail in the online supplementary material and methods.
HLA-DRB1 genotyping and sequence alignment
Whole blood was collected using PAXgene Blood DNA tubes and DNA was extracted according to manufacturer’s instructions (PreAnalytiX). High-resolution HLA-DRB1 typing was performed by next generation sequencing at the Johns Hopkins University Immunogenetics Laboratory, as previously described (12). Amino acid sequences encoded for by the HLA-DRB1 alleles of interest were obtained using the online IMGT/HLA submission tools (Immune Polymorphism Database) (13). Peptide contact and pocket residues in each HLA-DRβ1 chain were identified using the online MHC motif viewer (14). Sequence alignment and consensus sequence visualization was performed using Jalview version 2.10.5 software (15). Affinity and register of peptides for HLA-DR variants were predicted using the online NetMHCII 2.3 algorithm (16).
Protein Structure Images
Linear mapping of TOP1 epitopes was plotted using R (R Core Team, 2018). The three-dimensional location of TOP1 peptides within the TOP1 protein crystal structure (1K4T) and location of consensus peptide binding motifs within HLA-DR11 (6CPN) was visualized using the PyMOL Molecular Graphics System, version 2.3 (Schrödinger, LLC).
T cell stimulation
PBMCs from ATA-positive (n=11) and ATA-negative (n=11) patients with scleroderma were isolated by Ficoll density-gradient centrifugation (Ficoll-Paque Plus, GE Healthcare). The cells were resuspended to 1.5 × 106 cells/well in RPMI medium supplemented with 5% autologous serum, as previously described (9). Briefly, PBMCs were pre-incubated with anti-human CD40 blocking antibody (1 ug/ml; G28.5, BioLegend) prior to stimulation for 18 hours with 2.5 uM of each candidate TOP1 peptide (Table 1; >95% purity, Genscript), or 1.45 ug/mL human recombinant TOP1 protein (Genscript). Following stimulation, cells were stained with BV510-conjugated anti-CD3 (OKT3, BioLegend), Pacific Blue-conjugated anti-CD4 (RPA-T4, BD Pharmingen), APC-H7-conjugated anti-CD8 (SK1, BD Biosciencies), PE-conjugated anti-CD154 (TRAP1, BD Pharmingen), and Live/Dead Fixable Stain (Molecular Probes). The percentage of live CD4+ T cells that upregulated CD154 was quantified by flow cytometry (FACSAria, BD Biosciences; FCS Express software, De Novo Software). A cutoff of greater than the 95th percentile of the responses observed amongst the ATA-negative patients (%CD154+CD4+Tcells > 0.015%) was used to define a positive CD4+ T cell response to antigen stimulation.
Table 1.
TOP1 peptides corresponding to the TOP1 regions identified by NAPA
| Peptide | TOP1 RESIDUES | TOP1 PEPTIDE SEQUENCE |
|---|---|---|
| 1 | 127–145 | VPPKEDIKPLKRPRDEDDA |
| 2 | 279–292 | KDWRKEMTNEEKN |
| 3 | 304–318 | TQMSQYFKAQTEARKQ |
| 4 | 422–438 | GSIKYIMLNPSSRIKGEK |
| 5 | 517–536 | DGQEYVVEFDFLGKDSIRY |
| 6 | 585–606 | TAKVFRTYNASITLQQQLKELTA |
| 7 | 606–627 | APDENIPAKILSYNRANRAVAI |
| 8 | 659–676 | DARRDLKSAKADAKVMK |
| 9 | 676–696 | KDAKTKKVVESKKKAVQRLEE |
| 10 | 696–713 | EQLMKLEVQATDREENKQ |
Statistical analyses
Naturally presented peptides were analyzed using the PANTHER 14.1 gene list analysis tool statistical overrepresentation test for “biological processes” to identify if presented peptides were enriched in proteins belonging to specific biological processes (17). The “GO biological processes complete” annotation set was queried and the FDR and Fisher’s exact p-value for each pathway was calculated. The median number of peptides eliciting positive responses in ATA-positive versus ATA-negative patients was compared using a Mann-Whitney test. The association between minimum FVC and the number of stimulatory TOP1 epitopes was analyzed using the Spearman’s rho test. The association between minimum FCV and the percentage of TOP1-specific CD154+CD4+ T cells was analyzed using the Pearson’s r. Statistical analyses were performed using Prism version 7.03 (Graphpad) and Stata version 14.2 (Stata Corp) software. Statistical significance was defined as a two-sided p-value ≤0.05 throughout.
Results
Identification of naturally presented TOP1 peptides
We developed a natural antigen processing assay (NAPA) to identify TOP1 peptides presented by MHC class II molecules expressed on APCs from patients with scleroderma. As a source of patient-specific APCs, monocytes were isolated from the peripheral blood of 6 ATA-positive patients with scleroderma and cultured in the presence of GM-CSF and IL-4 to generate monocyte-derived dendritic cells (mo-DCs). Mo-DCs were then incubated withwhole TOP1 protein overnight, to allow for internalization and processing of the protein and presentation of TOP1 peptides by the HLA-DR molecules present on these patient-specific APCs (Figure 1). HLA-DR-peptide complexes were isolated by anti-HLA-DR immunoprecipitation, and presented peptides were eluted and sequenced by mass spectrometry.
Figure 1. Natural Antigen Processing Assay (NAPA).
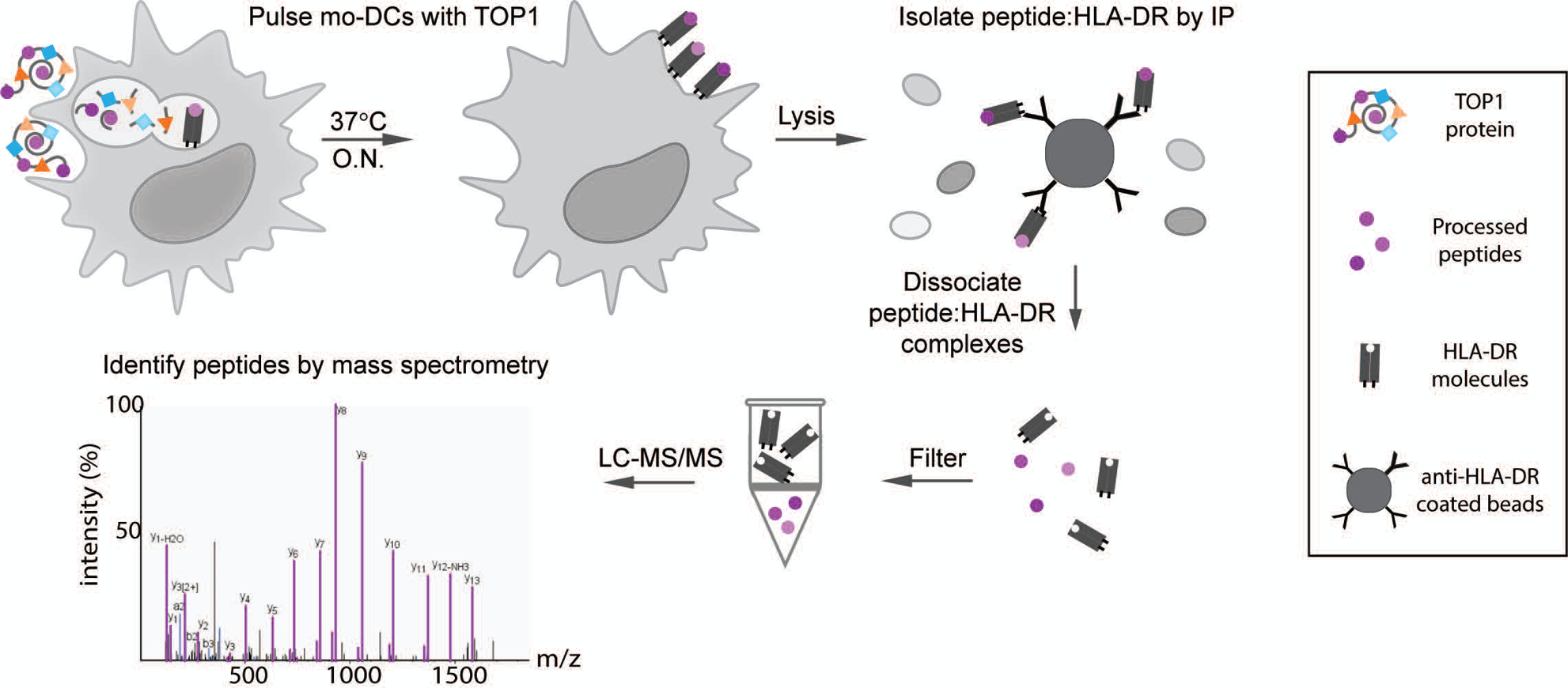
MoDCs from 6 donors with ATA-positive scleroderma were incubated with whole TOP1 protein overnight (O.N.) at 37°C. Cells were then lysed and the peptide:HLA-DR complexes isolated by immunoprecipitation using magnetic beads coated with anti-HLA-DR antibodies (clone L243). The presented peptides were dissociated from the HLA-DR molecules using 0.1% TFA and separated from HLA-DR molecules using a 10kDa spin filter, which allows the smaller molecular weight peptides to pass through. The filtered peptides were then identified and sequenced by liquid chromatography with tandem mass spectrometry (LC-MS/MS).
NAPA identified hundreds of naturally processed peptides derived from the exogenously added TOP1 protein, as well as from other endogenous self-proteins derived from the mo-DCs (Figure S1, Table S2). The median length of these peptides was 15 amino acids, and clustering of variable length peptides into nested sets around a common core sequence was observed (Table S2). These are features typical of naturally processed MHC class II peptides, which tend to be 13–25 amino acids long and exist in nested sets (18,19). As expected, the naturally processed peptides were enriched in those derived from proteins expected to traffic through the secretory pathway, where MHC class II antigen processing occurs (Figure S1C). This includes a global enrichment in peptides derived from proteins involved in antigen processing itself (Figure S1B). Analysis of the peptides presented between individuals revealed common presentation of peptides derived from membrane-associated (e.g. ITGAM and MRC1) and secreted proteins (e.g. TGFBI and TIMP3), as well as other MHC molecules. Importantly, the only antigen commonly presented by all 6 patients was the pulsed antigen, TOP1, demonstrating the ability of NAPA to detect peptides from endocytosed proteins. The specificity of NAPA for detecting HLA-DR-bound peptides, including those derived from TOP1, was confirmed using unpulsed mo-DCs and isotype control antibodies for immunoprecipitation (Tables S3 and S4). In these experimental conditions, no TOP1 peptides were detected.
Remarkably, we observed substantial overlap in the TOP1 peptides presented by ATA-positive scleroderma patients, with all subjects presenting peptides originating from only 10 discrete regions of the TOP1 molecule (Figure 2A). The median number of putative regions presented by each individual patient was 4 (range 1–7), and 70% were presented by at least two subjects. The 10 regions from which TOP1 peptides were presented are from diverse domains of TOP1 and largely reside within structural elements, most dominantly alpha helices flanked by unstructured loops on the surface of the TOP1 protein (Figure 2A and B). The localization of putative TOP1 epitopes within structural elements adjacent to flexible loops is similar to what has been observed for CD4+ T cell epitopes from other infectious and self-antigens (20,21). Using NAPA, we successfully identified a common set of TOP1 peptides derived from the whole TOP1 protein, which were naturally processed and presented by HLA-DR molecules in mo-DCs generated ex vivo from patients with ATA-positive scleroderma.
Figure 2. A common set of naturally processed TOP1 peptides was presented by scleroderma patients with diverse HLA-DR variants.
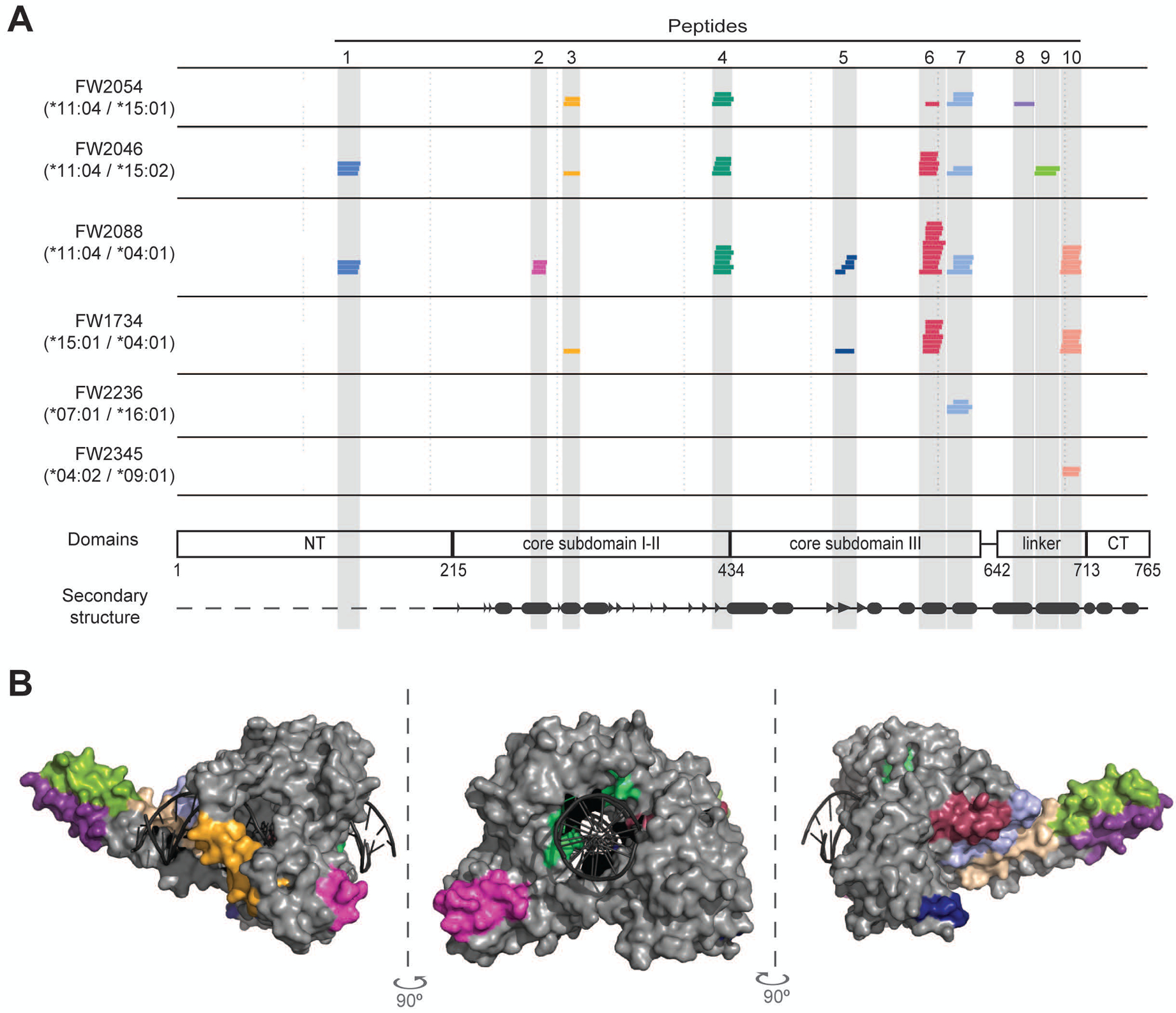
(A) The HLA-DRB1 haplotype for each patient is given and each colored line represents a unique TOP1 peptide sequence identified by mass spectrometry. The location of the TOP1 peptides within the linear TOP1 protein sequence is indicated. TOP1 protein domains and secondary structure elements are shown (β-strands and α-helices are represented by arrow heads and ovals, respectively). (B) The location of the naturally processed TOP1 peptides within the three-dimensional crystal structure of the TOP1 molecule bound to DNA (1K4T) is indicated. The peptides are colored-coded and correspond to the colors used in panel A.
Shared motifs present in HLA-DRβ chains of patients with ATA-positive scleroderma
We next performed HLA typing to determine if the presentation of common TOP1 peptides identified in our patients was due to the presence of shared HLA-DRB1 alleles. Despite the known association of ATA with specific HLA-DRB1 alleles (i.e. HLA-DRB1*11:01, *11:04, *08:02, *08:04, or *15:02) (4), only 3 subjects (50%) harbored a previously defined risk allele. While these 3 patients concurrently presented peptides from more regions of the TOP1 molecule compared to those who did not carry a known ATA-associated allele (median 6 vs. 1), common peptides were presented regardless of the allele status.
Given this unexpected ability of diverse HLA-DR variants to present common TOP1 peptides, we sought to determine if similar features were present in the HLA-DRβ chains of ATA-positive scleroderma patients. To explore this, we performed amino acid sequence alignment of the HLA-DRβ variants linked to the development of ATA-positive scleroderma (4) and compared them to the HLA-DRβ chains in these patients. This analysis revealed two conserved motifs within the DRβ-chain encoded for by the HLA-DRB1 alleles most strongly linked to ATA (22–25) (HLA-DRB1*08:02, *08:04, *11:01, *11:04): 9E-Y-S-T-S/G-E14 and 67F-L-E-D-R-R-A-A/L74 (Figure 3A). HLA-DRβ1*15:02, associated with ATA in Asian populations (4), had a similar 67I-L-E-Q-A-R-A-A74 motif but a distinct sequence at position 9–14 (9W-Q-P-K-R-E14) (Figure 3A). Importantly, these two motifs contain key peptide contact residues at positions 9, 11, 13, 14 and 67, 70, 71, 74 respectively (Figure 3B).
Figure 3. Presentation of common TOP1 peptides linked to shared ATA-associated motifs in the HLA-DRβ chain.
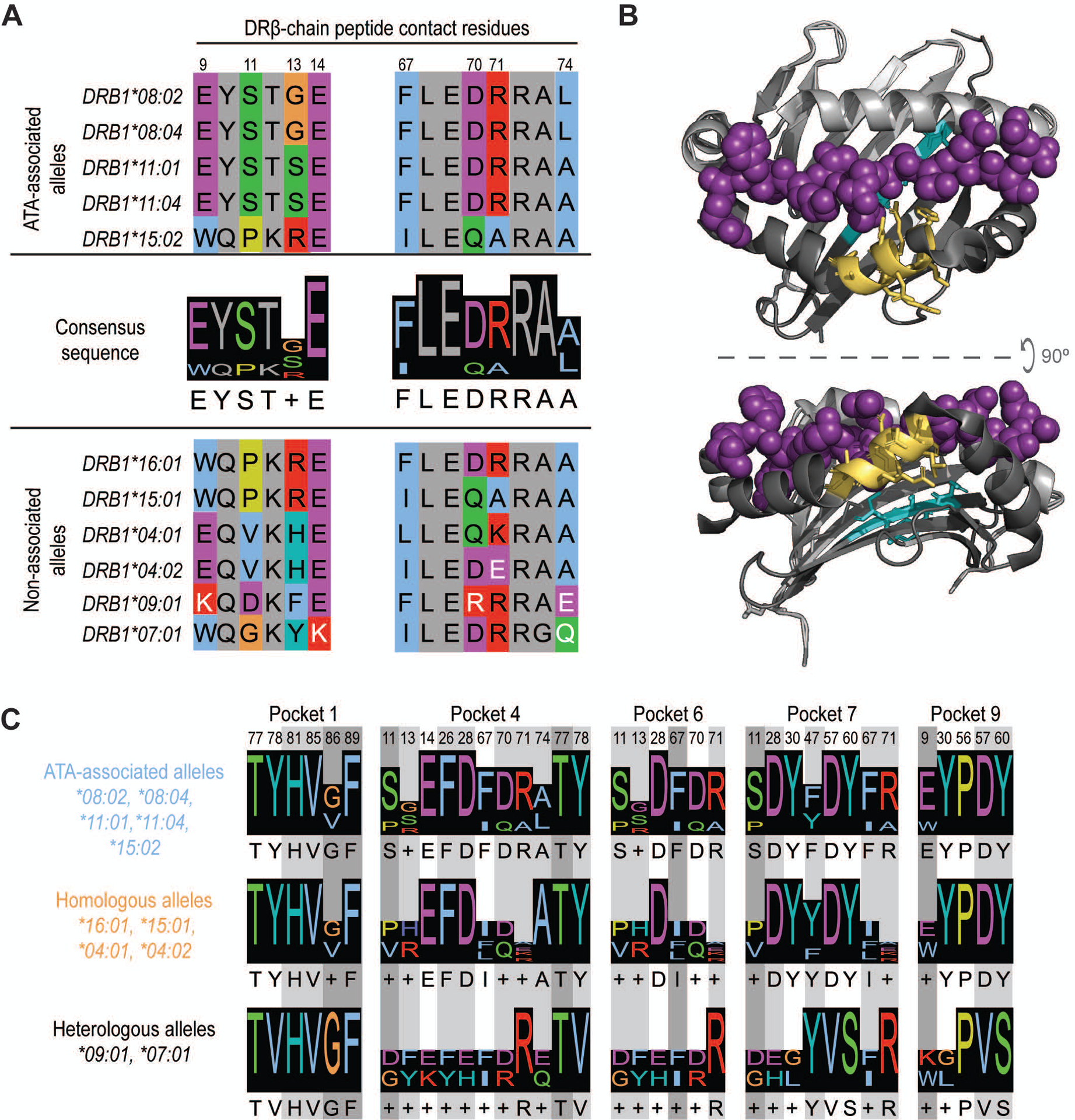
(A) Amino acid sequence alignment of ATA-associated (top panel) and non-associated alleles (bottom panel) is shown. The two consensus sequences identified in the peptide-binding groove of ATA-associated alleles are shown (middle panel). Peptide contact residues in the DRβ chain are shown (hydrophobic=blue, negative=purple, positive=red, polar=green, glycine=orange, prolines=yellow, and aromatic=cyan) and their position within the sequence is numbered, while intervening residues are shown in gray. The white text indicates non-conserved residues. (B) A depiction of HLA-DR11 crystalized with a peptide (6CPN) is shown and features of the peptide binding groove have been color-coded: DR α-chain residues (light gray), DR β-chain (dark gray) and peptide (magenta). The 9E-Y-S-T-S/G13 and 67F-L-E-D-R-R-A-A/L74 motifs are shown in cyan and yellow, respectively. (C) Peptide contact residues for each MHC binding pocket and the relevant consensus sequence for ATA-associated (light blue), homologous (orange) and heterologous (black) alleles are represented. Different shades of gray indicate the importance of each residue to peptide binding, with dark gray being the most critical.
Analysis of the HLA-DRβ chains present in patients who did not have previously defined ATA-associated HLA-DRB1 alleles, but were able to present common TOP1 peptides, revealed several conserved features. HLA-DRβ1*15:01 and *16:01 contained motifs at positions 9–14 and 67–74 that were identical to known ATA-associated alleles (Figure 3A). HLA-DRβ1*04:01 and *04:02 were less similar, but had conserved amino acids at known peptide contact positions (i.e. 9, 14, and 67, 70, 74). A more detailed analysis of β-chain residues that comprise the five major peptide binding pockets in the HLA-DR groove (denoted as P1, P4, P6, P7, and P9) confirmed that ATA-associated and homologous alleles have highly similar peptide binding pockets (Figure 3C). Interestingly, HLA- DRβ1*09:01 and *07:01 were divergent from ATA-associated alleles at several critical peptide contact residues (Figure 3, A and C), and HLA- DRβ1*07:01 has been reported to be protective against scleroderma development (26). While all the ATA-positive patients in our study possessed at least one allele that contained ATA-associated motifs, those who harbored HLA- DRβ1*09:01 and *07:01 presented the fewest number of TOP1 peptides (Figure 2A). These data reveal shared features in the peptide binding grooves of seemingly unrelated HLA- DRβ1 variants present in ATA-positive patients with scleroderma, providing a mechanism for the shared presentation of common TOP1 peptides.
Promiscuous binding of TOP1 peptides to multiple HLA-DR variants
To examine the intermolecular interactions between the commonly presented TOP1 peptides identified using NAPA and the HLA-DR variants detected in these patients, we used the NetMHCII 2.3 algorithm to predict the peptide binding affinity and register (16). Since all of the patients were heterozygous at the HLA-DRB1 locus, we examined the predicted binding of peptides to both HLA-DR variants present in a given individual. This analysis revealed peptides that could be categorized into two distinct groups, termed group A and B, based on their predicted binding register (Figure 4). Group A peptides (peptides 1, 2, 8, 9, and 10) were predicted to bind in the same register to all associated HLA-DR variants (Figure 4A). This is in contrast to group B peptides, which were predicted to bind in different registers to multiple different HLA-DR variants (Figure 4B). Group A peptides were also predicted to bind with significantly lower affinity to their respective HLA-DR molecules than Group B peptides (4421 ± 1184 vs. 535 ± 276 nM, respectively; p=0.013, Table S5), and were presented by fewer individuals than group B peptides (median 1 vs. 3). This suggests that group A peptides are relatively weak-binding peptides that contain a single core epitope (e.g. LMKLEVQAT in peptide 10) whose anchor residues interact with shared features present in the HLA-DR binding grooves of patients with ATA. In contrast, group B peptides appear to contain multiple distinct epitopes, each possessing a unique set of anchor residues that enable high-affinity interaction with a more diverse set of HLA-DR binding pockets (e.g. LSYNRANRA, ILSYNRANR, and YNRANRAVA in peptide 7). Taken together, these data support a model of convergent TOP1 peptide presentation in patients with scleroderma driven by shared peptide-binding motifs in the HLA-DR groove and a promiscuous group of TOP1 peptides.
Figure 4. Allele-specific TOP1 peptide binding registers may contribute to promiscuous presentation by diverse HLA-DR variants.
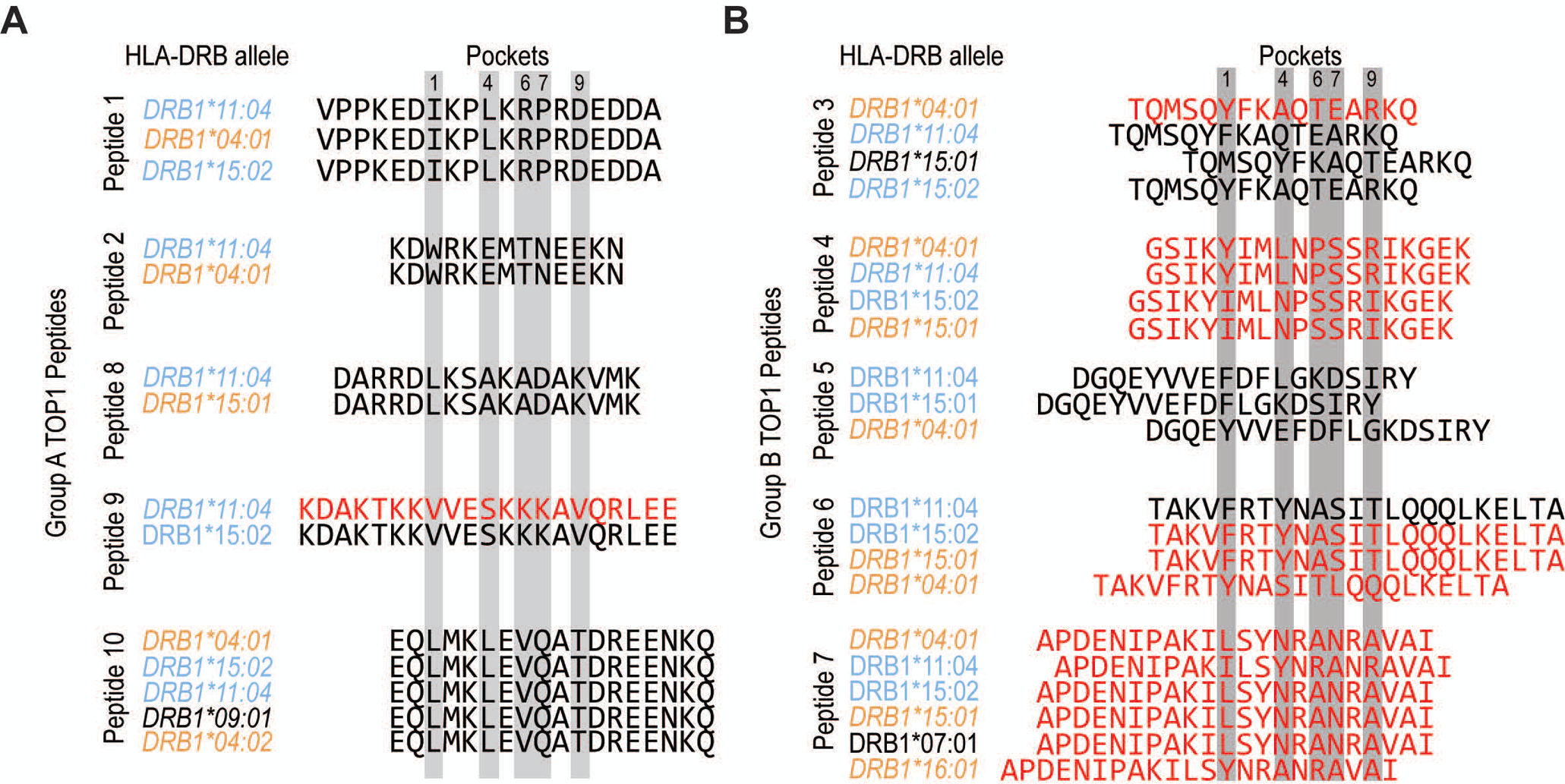
(A) Group A TOP1 peptides (peptides 1, 2, 8, 9 and 10) and (B) group B TOP1 peptides (peptides 3, 4, 5, 6 and 7) are aligned according to their predicted binding registers as per NetMHCII algorithm with relevant anchor residues highlighted in gray. The HLA-DRB1 alleles are color-coded as ATA-associated (light blue), homologous (orange) and heterologous (black) alleles, and ordered from highest to lowest predicted affinity for each peptide. TOP1 peptides with a predicted binding affinity of <500 nM are indicated with red text, and are expected to represent moderate to high affinity peptides.
Robust recognition of commonly presented TOP1 epitopes by CD4+ T cells in scleroderma
The discovery of a shared mechanism for the presentation of a core set of TOP1 peptides suggests that these epitopes may be central to the disease process in patients with scleroderma. To determine if these epitopes are immunologically relevant in ATA-positive patients, we synthesized 10 peptides corresponding to the putative TOP1 epitopes identified using NAPA (Figure 2A; Table 1) and examined their ability to stimulate CD4+ T cells from 11 ATA-positive and 11 ATA-negative scleroderma patients (clinical variables are shown in Table S1). CD4+ T cell activation, as measured by upregulation of the early activation marker CD154 (9,27), was observed in response to at least one TOP1 peptide in 73% (8/11) of ATA-positive patients compared to 27% (3/11) of ATA-negative individuals (Figure 5A, Table S6). ATA-positive patients also responded to significantly more TOP1 epitopes (median 2 vs. 0; p=0.02) (Figure S2A). This was most pronounced in ATA-positive subjects with known disease-associated HLA-DRB1 alleles, compared to those without (median 7 vs. 1.5 epitopes, respectively), and recognition of group A and B peptides was observed regardless of ATA-associated allele status. Importantly, the increased T cell responses observed in ATA-positive compared to ATA-negative patients were not due to differences in exposure to immunosuppressive medications (Table S1).
Figure 5. Distribution and clinical significance of CD4+ T cell responses to naturally presented TOP1 epitopes in scleroderma.
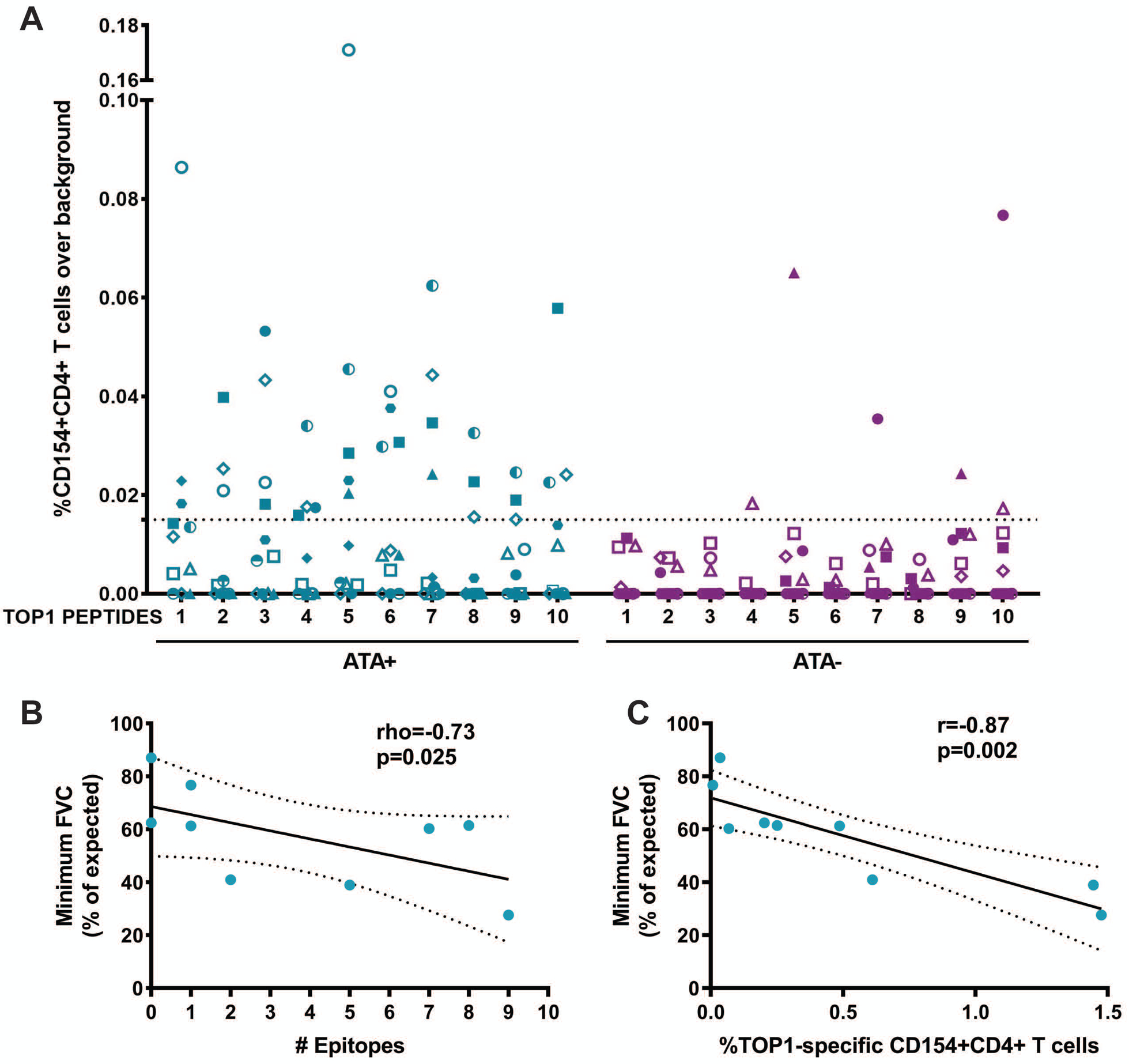
(A) %CD154+CD4+ T cell responses over background observed with media alone for 11 ATA-positive and 11 ATA-negative scleroderma patients for each of the 10 naturally processed TOP1 peptides identified using NAPA are shown. The dotted line represents the cutoff for positivity that was defined as >95th percentile of the T cell responses observed in ATA-negative patients (95th percentile=0.015%). Each unique colored symbol represents a different patient. The minimum FVC (% of expected) was plotted against (B) the number of TOP1 epitopes eliciting a T cell response and (C) the percentage of CD154+CD4+ T cells responses elicited against the whole TOP1 protein in ATA-positive scleroderma patients with clinically documented ILD (n=9). The correlation between minimum FVC and the number of stimulatory TOP1 epitopes was analyzed using a Spearman’s rho test, and the correlation with the magnitude of the T cell responses to the TOP1 protein was analyzed using a Pearson’s r test. A p-value <0.05 was considered significant throughout.
For 6 out of the 11 ATA-positive patients (54%), the combined frequency of CD154+ cells observed in response to the individual TOP1 peptides (Figure S2, B and C) was comparable to that measured in response to the whole TOP1 protein. This suggests the immunodominance of this set of 10 naturally processed TOP1 peptides in these individuals. Robust recognition of a core set of TOP1 epitopes in immunogenetically diverse ATA-positive patients supports our observation that features on both sides of the peptide-MHC interaction contribute to the development of anti-TOP1 immune responses in patients with scleroderma.
Diversity of TOP1-specific T cell responses associated with ILD severity
ATA-positive scleroderma is associated with diffuse skin involvement and ILD (3). Therefore, we explored whether the presence of TOP1-specific T cells is associated with either of these clinical characteristics. We evaluated the responses to both the whole TOP1 protein and the number of TOP1 epitopes recognized by T cells in relation to the type of skin involvement, but no significant association was found (Figure S3). We then sought to investigate if there is any correlation with scleroderma lung disease. We evaluated the lowest forced vital capacity (FVC) recorded during the course of clinical follow-up for each patient as a measure of the severity of lung fibrosis. We focused this analysis on the 9 ATA-positive patients with radiographically defined scleroderma-associated ILD. A significant negative association was observed between the minimum FVC and the number of TOP1 epitopes that elicited a T cell response (p=0.025; Figure 5B). While this relationship was also observed in response to whole TOP1 protein (p=0.002; Figure 5C), it is remarkable that these 10 commonly presented epitopes were sufficient to identify T cell responses associated with more severe lung disease. This implicates CD4+ T cells targeting commonly presented TOP1 epitopes in the pathogenesis of scleroderma-ILD.
Discussion
This investigation sought to evaluate one of the most proximal events in the stimulation of autoreactive TOP1-specific T cells, the presentation of autoantigenic epitopes by ATA-associated HLA-DR variants. Our study supports two parallel and complementary mechanisms for how unique features present on both sides of the “peptide-HLA-DR interaction” synergize to promote the presentation of common epitopes, which drive the development of convergent immune responses and clinical characteristics. Together with the observation that the breadth of the CD4+ T cell responses to these TOP1 epitopes is associated with presence and severity of lung fibrosis, these findings offer novel insights into the pathogenesis of scleroderma.
The identification of common TOP1 epitopes enabled the definition of two distinct groups of TOP1 peptides that differ in their interaction with HLA-DR. Group A TOP1 peptides contain a single core epitope that is bound in the same register to multiple HLA variants, likely interacting with shared features in the peptide binding groove. Group B TOP1 peptides, however, contain multiple distinct epitopes that utilize different anchor residues to bind promiscuously to diverse HLA variants. This suggests that features intrinsic to the peptide binding groove are responsible for presenting of Group A peptides, while presentation of Group B peptides is largely driven by peptide-intrinsic features.
Analysis of the peptide binding grooves of seemingly unrelated HLA-DR variants indeed demonstrated the existence of shared motifs(9E-Y-S-T-S/G-E14 and 67F-L-E-D-R-A-A/L74) found in patients with ATA-positive scleroderma, which enabled the presentation of a common set of TOP1 epitopes. Importantly, the 67F-L-E-D-R71 motif was previously identified by Kuwana et al. in 1993 and hypothesized to be a common feature of alleles associated with ATA in different ethnicities (23,24,28), but the mechanism for this was unknown. Our data provide direct evidence that the presence of HLA-DR variants containing these or homologous sequences is associated with the increased presentation and recognition of TOP1 epitopes. Our finding of shared motifs in seemingly diverse HLA-DRB1 alleles in scleroderma is analogous to the “shared epitope hypothesis” in rheumatoid arthritis (29,30), in which a conserved amino acid sequence (70R/Q-R/K-R-A-A74) present in the HLA-DRβ chain of a group of HLA-DR variants is hypothesized to lead to the presentation of common “arthritogenic” self-peptides (31), though this has not been directly demonstrated in patients. While the collective analysis of alleles carrying a common motif has not yet been widely adopted for genetic studies in scleroderma, our data provides strong evidence for a shared epitope-like motif in scleroderma and bolsters the rationale for the use of such analyses in future studies.
While this mechanism may be dominant in patients who harbor ATA-associated risk alleles, scleroderma can also develop in patients who lack these HLA-DRB1 alleles. Our discovery of a group of promiscuous TOP1 peptides provides a potential mechanism for how the development of immune responses to TOP1 could by-pass the need for these risk alleles. While promiscuous peptides have been described in the setting of cancer, infection, and mouse models of autoimmunity (32–36), to our knowledge such peptides have not been described to be naturally presented in patients with autoimmunity. Further biophysical studies are required to define the precise anchor residues responsible for presentation of promiscuous TOP1 peptides in scleroderma, and to determine if this mechanism is operative in other autoimmune diseases.
In addition to our study, other approaches have been taken to define TOP1 specific T cell epitopes in patients with scleroderma. Using 6 large overlapping fragments spanning the entire TOP1 protein, Kuwana et al. was the first to define DR-restricted immunologic hot-spots within the TOP1 protein (37,37). Through a series of experiments, they determined that amino acids 276–386 contained an epitope that could stimulate T cell proliferation in healthy controls and patients with scleroderma irrespective of HLA-DR allele or ATA status, and additional fragments (amino acids 363–563 and 541–657) were also able to stimulate robust responses in individuals with specific HLA alleles (5,37). Interestingly, these three regions contain the group B peptides defined in our study (peptides 3, 4, 5, 6, and 7), as well as one group A peptide (peptide 2). Consistent with our results, TOP1 fragments of the N- (amino acids 1–93) and C-termini (amino acids 658–765) were less broadly stimulatory. Building on this work, Veeraraghavan et al. found that 3 in silico predicted TOP1 peptides derived from these hot-spots were able to stimulate T cell proliferation in patients with scleroderma, one corresponding to peptide 6 and two others not identified in our study (amino acids 373–387 and 641–656) (7). Using a different approach, Hu et al. created an overlapping peptide library of 15mer peptides spanning the entire TOP1 protein sequence and used this to define the epitope specificity of T cell clones derived from patients with scleroderma and healthy controls (8). They found 13 distinct epitopes identified to cause clonal T cell expansion, three of which were also identified in our study (peptides 4, 8, and 9).
There are several important points that emerge from these comparisons. The first is that while T cell responses were observed to TOP1 peptides in all studies, ours is the only one to have found an association with clinical indices of ATA-positive scleroderma. This could, in part, be due to differences in the assays used to study T cell responses: upregulation of the early activation marker CD154 in our study and cell proliferation in the other three studies. Upregulation of CD154 is highly specific for TCR engagement with cognate antigens and not influenced by non-specific stimuli, whereas cell proliferation can have high background depending on the background proliferative capacity of the donor and length of stimulation (38). This may also explain why we found T cell responses to TOP1 peptides to be highly enriched in ATA-positive vs. -negative patients, whereas the other studies did not. Lastly, while our study identified clinically meaningful set of TOP1 epitopes, this does not represent all of the TOP1 epitopes capable of driving pathogenic anti-TOP1 immune responses in scleroderma. The finding of T cell responses to TOP1 peptides in other studies not detected using NAPA may reflect a finite sensitivity of the assay, but may also suggest additional TOP1 epitopes could be revealed by studying the processing of TOP1 by other APCs. While dendritic cells are most effective at initiating T cell responses, other APCs like macrophages and B cells express MHC class II and have the ability to present CD4+ T cell epitopes (39). Differences in the presented peptide repertoire may differ by the type or anatomical site of the APC (40,41), suggesting that additional TOP1 epitopes could be revealed by studying the processing of TOP1 by other APCs.
The pathogenic relevance of convergent TOP1 epitope presentation in patients with scleroderma is illustrated by the detection of TOP1-specific CD4+ T cell responses to this core set of epitopes in >70% of patients with ATA-positive scleroderma. In our previous work, we have shown that the frequency of TOP1-specific CD4+ T cells is associated with the severity of lung fibrosis (9). This new study extends that observation by defining the epitope-specificity of these autoreactive T cells and demonstrating that the number of TOP1 epitopes recognized by a patient inversely associates with lung function. While unique HLA-DR variants have been linked to scleroderma development, ATA positivity, and ILD (42), the mechanism for this has not been directly demonstrated. Importantly, HLA-DRB1*11:01 was shown to be significantly associated with ILD only in the presence of ATA (43). Together with the evidence provided by our study, this suggests a model where specific HLA variants are mechanistically linked to the development of scleroderma ILD via activation of TOP1-specific autoreactive T cells, which provide help to autoreactive B cells and drive the development of ATA.
Tetramers made from MHC molecules bound to antigenic peptides can be used to identify, track, and study antigen-specific immune responses over time in peripheral blood, as well as target tissues (44). TOP1 tetramers containing epitopes identified in our study could therefore be developed as a precise tool to study the frequency and phenotype of TOP1 specific T cells ex vivo. Longitudinal studies in early scleroderma cohorts are needed to define whether the repertoire of TOP1 epitope-specific CD4+ T cells changes over time and predicts onset or progression of fibrotic lung disease. By leveraging recent advances in medical bioengineering (45), these TOP1 epitopes may also be utilized for the future development of novel antigen-specific nanoparticle therapies aimed at downregulating TOP1-specific CD4+ T cell responses in patients with scleroderma. Precise definition of autoantigenic epitopes is therefore critical to understanding the primordial mechanisms for the loss of tolerance, studying disease-propagating autoreactive T cells, and developing antigen-specific clinical tools.
Supplementary Material
Acknowledgments:
We would like to thank the research coordinators (Adrianne Woods, Margaret Sampedro, and Gwendolyn Leatherman), physicians and rheumatology fellows of the Johns Hopkins Scleroderma Center for their support in obtaining the patient samples and data; the patients of the Johns Hopkins Scleroderma Center who participated in this study; Scheherazade Sadegh-Nasseri, Ph.D. for critical review of the data; and Felipe Andrade M.D., Ph.D. for critical review of the manuscript.
Funding: This research was supported by the Jerome L. Greene Foundation, Rheumatology Research Foundation, Scleroderma Research Foundation and National Institutes of Health (grants K23AR071473, R01AR073208, and P30AR070254 [Bayview Immunocomics Core Facility]).
Footnotes
Competing interests: The authors have declared that no conflict of interest exists.
References and Notes:
- 1.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. New England Journal of Medicine 2009;360:1989–2003. [DOI] [PubMed] [Google Scholar]
- 2.Boin F, Rosen A. Autoimmunity in systemic sclerosis: current concepts. Curr Rheumatol Rep 2007;9:165–172. [DOI] [PubMed] [Google Scholar]
- 3.Walker UA, Tyndall A, Czirják L, Denton C, Farge-Bancel D, Kowal-Bielecka O, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials And Research group database. Ann Rheum Dis 2007;66:754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chairta P, Nicolaou P, Christodoulou K. Genomic and genetic studies of systemic sclerosis: A systematic review. Hum Immunol 2017;78:153–165. [DOI] [PubMed] [Google Scholar]
- 5.Kuwana M, Medsger TA, Wright TM. T cell proliferative response induced by DNA topoisomerase I in patients with systemic sclerosis and healthy donors. J Clin Invest 1995;96:586–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuwana M, Feghali CA, Medsger TA, Wright TM. Autoreactive T cells to topoisomerase I in monozygotic twins discordant for systemic sclerosis. Arthritis Rheum 2001;44:1654–1659. [DOI] [PubMed] [Google Scholar]
- 7.Veeraraghavan S, Renzoni EA, Jeal H, Jones M, Hammer J, Wells AU, et al. Mapping of the immunodominant T cell epitopes of the protein topoisomerase I. Ann Rheum Dis 2004;63:982–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu PQ, Oppenheim JJ, Medsger TA, Wright TM. T cell lines from systemic sclerosis patients and healthy controls recognize multiple epitopes on DNA topoisomerase I. J Autoimmun 2006;26:258–267. [DOI] [PubMed] [Google Scholar]
- 9.Fava A, Cimbro R, Wigley FM, Liu Q-R, Rosen A, Boin F. Frequency of circulating topoisomerase-I-specific CD4 T cells predicts presence and progression of interstitial lung disease in scleroderma. Arthritis Res Ther 2016;18:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoogen F van den, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis 2013;72:1747–1755. [DOI] [PubMed] [Google Scholar]
- 11.LeRoy EC, Medsger TA. Criteria for the classification of early systemic sclerosis. J Rheumatol 2001;28:1573–1576. [PubMed] [Google Scholar]
- 12.Cappelli LC, Konig MF, Gelber AC, Bingham CO, Darrah E. Smoking is not linked to the development of anti-peptidylarginine deiminase 4 autoantibodies in rheumatoid arthritis. Arthritis Res Ther 2018;20:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SGE. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res 2015;43:D423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rapin N, Hoof I, Lund O, Nielsen M. MHC motif viewer. Immunogenetics 2008;60:759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009;25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018;154:394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, et al. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nature Protocols 2019;14:703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Appella E, Padlan EA, Hunt DF. Analysis of the structure of naturally processed peptides bound by class I and class II major histocompatibility complex molecules. EXS 1995;73:105–119. [DOI] [PubMed] [Google Scholar]
- 19.Godkin AJ, Smith KJ, Willis A, Tejada-Simon MV, Zhang J, Elliott T, et al. Naturally processed HLA class II peptides reveal highly conserved immunogenic flanking region sequence preferences that reflect antigen processing rather than peptide-MHC interactions. J Immunol 2001;166:6720–6727. [DOI] [PubMed] [Google Scholar]
- 20.Darrah E, Kim A, Zhang X, Boronina T, Cole RN, Fava A, et al. Proteolysis by Granzyme B Enhances Presentation of Autoantigenic Peptidylarginine Deiminase 4 Epitopes in Rheumatoid Arthritis. J Proteome Res 2017;16:355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mirano-Bascos D, Tary-Lehmann M, Landry SJ. Antigen structure influences helper T-cell epitope dominance in the human immune response to HIV envelope glycoprotein gp120. Eur J Immunol 2008;38:1231–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnett FC, Gourh P, Shete S, Ahn CW, Honey RE, Agarwal SK, et al. Major histocompatibility complex (MHC) class II alleles, haplotypes and epitopes which confer susceptibility or protection in systemic sclerosis: analyses in 1300 Caucasian, African-American and Hispanic cases and 1000 controls. Ann Rheum Dis 2010;69:822–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeuchi F, Nakano K, Yamada H, Hong GH, Nabeta H, Yoshida A, et al. Association of HLA-DR with progressive systemic sclerosis in Japanese. J Rheumatol 1994;21:857–863. [PubMed] [Google Scholar]
- 24.Kang SH, Park MH, Song EY, Kang SJ, Lee EB, Song YW, et al. Association of HLA class II genes with systemic sclerosis in Koreans. J Rheumatol 2001;28:1577–1583. [PubMed] [Google Scholar]
- 25.Kuwana M, Kaburaki J, Okano Y, Inoko H, Tsuji K. The HLA-DR and DQ genes control the autoimmune response to DNA topoisomerase I in systemic sclerosis (scleroderma). J Clin Invest 1993;92:1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gladman DD, Kung TN, Siannis F, Pellett F, Farewell VT, Lee P. HLA markers for susceptibility and expression in scleroderma. J Rheumatol 2005;32:1481–1487. [PubMed] [Google Scholar]
- 27.Joseph CG, Darrah E, Shah AA, Skora AD, Casciola-Rosen LA, Wigley FM, et al. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science 2014;343:152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furukawa H, Oka S, Kawasaki A, Shimada K, Sugii S, Matsushita T, et al. Human Leukocyte Antigen and Systemic Sclerosis in Japanese: The Sign of the Four Independent Protective Alleles, DRB1*13:02, DRB1*14:06, DQB1*03:01, and DPB1*02:01. PLoS ONE 2016;11:e0154255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 1987;30:1205–1213. [DOI] [PubMed] [Google Scholar]
- 30.Almeida DE de, Ling S, Holoshitz J. New insights into the functional role of the rheumatoid arthritis shared epitope. FEBS Lett 2011;585:3619–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wucherpfennig KW, Strominger JL. Selective binding of self peptides to disease-associated major histocompatibility complex (MHC) molecules: a mechanism for MHC-linked susceptibility to human autoimmune diseases. J Exp Med 1995;181:1597–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Panina-Bordignon P, Tan A, Termijtelen A, Demotz S, Corradin G, Lanzavecchia A. Universally immunogenic T cell epitopes: promiscuous binding to human MHC class II and promiscuous recognition by T cells. Eur J Immunol 1989;19:2237–2242. [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Chow I-T, Sosinowski T, Torres-Chinn N, Greenbaum CJ, James EA, et al. Autoreactive T cells specific for insulin B:11–23 recognize a low-affinity peptide register in human subjects with autoimmune diabetes. Proc Natl Acad Sci USA 2014;111:14840–14845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corper AL, Stratmann T, Apostolopoulos V, Scott CA, Garcia KC, Kang AS, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science 2000;288:505–511. [DOI] [PubMed] [Google Scholar]
- 35.Shi Y, Kaliyaperumal A, Lu L, Southwood S, Sette A, Michaels MA, et al. Promiscuous presentation and recognition of nucleosomal autoepitopes in lupus: role of autoimmune T cell receptor alpha chain. J Exp Med 1998;187:367–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seamons A, Sutton J, Bai D, Baird E, Bonn N, Kafsack BFC, et al. Competition between two MHC binding registers in a single peptide processed from myelin basic protein influences tolerance and susceptibility to autoimmunity. J Exp Med 2003;197:1391–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuwana M, Medsger TA, Wright TM. Highly restricted TCR-alpha beta usage by autoreactive human T cell clones specific for DNA topoisomerase I: recognition of an immunodominant epitope. J Immunol 1997;158:485–491. [PubMed] [Google Scholar]
- 38.Chattopadhyay PK, Yu J, Roederer M. A live-cell assay to detect antigen-specific CD4+ T cells with diverse cytokine profiles. Nat Med 2005;11:1113–1117. [DOI] [PubMed] [Google Scholar]
- 39.Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol 2015;15:203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Viner NJ, Nelson CA, Deck B, Unanue ER. Complexes generated by the binding of free peptides to class II MHC molecules are antigenically diverse compared with those generated by intracellular processing. J Immunol 1996;156:2365–2368. [PubMed] [Google Scholar]
- 41.Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol 2010;11:350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stock CJW, Renzoni EA. Genetic predictors of systemic sclerosis-associated interstitial lung disease: a review of recent literature. Eur J Hum Genet 2018;26:765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fanning GC, Welsh KI, Bunn C, Du Bois R, Black CM. HLA associations in three mutually exclusive autoantibody subgroups in UK systemic sclerosis patients. Br J Rheumatol 1998;37:201–207. [DOI] [PubMed] [Google Scholar]
- 44.Altman JD, Davis MM. MHC-Peptide Tetramers to Visualize Antigen-Specific T Cells. Curr Protoc Immunol 2016;115:17.3.1–17.3.44. [DOI] [PubMed] [Google Scholar]
- 45.Serra P, Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nat Biotechnol 2019;37:238–251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.