

Abstract
Molecular glue compounds induce protein-protein interactions that, in the context of a ubiquitin ligase, lead to protein degradation.1 Unlike traditional enzyme inhibitors, such molecular glue degraders act sub-stoichiometrically to catalyse rapid depletion of previously inaccessible targets.2 They are clinically effective and highly sought-after, but have thus far only been discovered serendipitously. Through systematic mining of databases for correlations between the cytotoxicity of 4,518 clinical and pre-clinical small molecules and E3 ligase expression levels across hundreds of human cancer cell lines,3–5 we identified CR8, a cyclin-dependent kinase (CDK) inhibitor,6 as a compound that acts as a molecular glue degrader. A solvent-exposed pyridyl moiety of CR8, in its CDK-bound form, induces CDK12-cyclin K complex formation with DDB1, the CUL4 adaptor protein, bypassing the requirement for a substrate receptor and presenting cyclin K (cycK) for ubiquitination and degradation. Our studies demonstrate that chemical alteration of surface-exposed moieties can confer gain-of-function glue properties to an inhibitor, and we propose this as a broader strategy to turn target binders into molecular glues.
Molecular glues are a class of small molecule drugs that induce or stabilise protein-protein interactions1. In the context of a ubiquitin ligase, drug-induced interactions can lead to protein degradation, which is an emerging strategy for the inactivation of therapeutic targets intractable by conventional pharmacological means2. Known molecular glue degraders bind to substrate receptors of E3 ubiquitin ligases and recruit target proteins for their ubiquitination and subsequent degradation by the proteasome.
Thalidomide analogues and aryl sulphonamides are two classes of drugs that act as molecular glue degraders. Widely used in the clinic, thalidomide analogues have proven to be an effective treatment for multiple myeloma, other B cell malignancies, and myelodysplastic syndrome with a deletion in chromosome 5q7. Thalidomide analogues recruit zinc-finger transcription factors and other targets to CRBN8–11, the substrate receptor of the cullin-RING E3 ubiquitin ligase CUL4A/B-RBX1-DDB1-CRBN (CRL4CRBN)12. Similarly, aryl sulphonamides degrade the essential RNA-binding protein RBM39 by engaging DCAF15, the substrate receptor of the CRL4DCAF15 E3 ubiquitin ligase13–15. In these examples, the degraders are not dependent on a ligandable pocket on the target protein, but instead leverage complementary protein-protein interfaces between the receptor and the target. By reprogramming ubiquitin ligase selectivity, these molecules divert the ligase to drive multiple rounds of target ubiquitination in a catalytic manner16. Such compounds can thus circumvent limitations of classical inhibitors, expanding the repertoire of “druggable” proteins. Although highly sought-after, molecular glue degraders have only been found serendipitously, and there are currently limited strategies available for identifying or designing such compounds.
CR8 induces proteasomal cycK degradation
To identify small molecules that mediate protein degradation through an E3 ubiquitin ligase, we correlated drug sensitivity data for 4,518 clinical and pre-clinical drugs tested against 578 cancer cell lines3,4 with the mRNA expression levels for 499 E3 ligase components5 (Extended Data Fig. 1a). DCAF15 gene expression correlated with indisulam and tasisulam toxicity, consistent with its known function as a degrader of the essential protein RBM39 by the CRL4DCAF15 E3 ubiquitin ligase, thus demonstrating the potential of the approach (Extended Data Fig. 1b, c). We sought to validate the high-scoring ligase-drug correlations by examining whether CRISPR-mediated inactivation of the identified E3 ligase component would rescue the respective drug-induced toxicity (Extended Data Fig. 1d). These experiments confirmed that sgRNAs targeting DCAF15 confer resistance to indisulam and tasisulam. In addition, we observed a correlation between cytotoxicity of the CDK-inhibitor R-CR86 and mRNA expression levels of the CUL4 adaptor DDB1 (Fig. 1a and Extended Data Fig. 1e). Consistently, sgRNAs targeting DDB1 conferred resistance to R-CR8 (Fig. 1b).
Figure 1 |. CR8-induced degradation of cycK depends on DDB1 and CDK12.
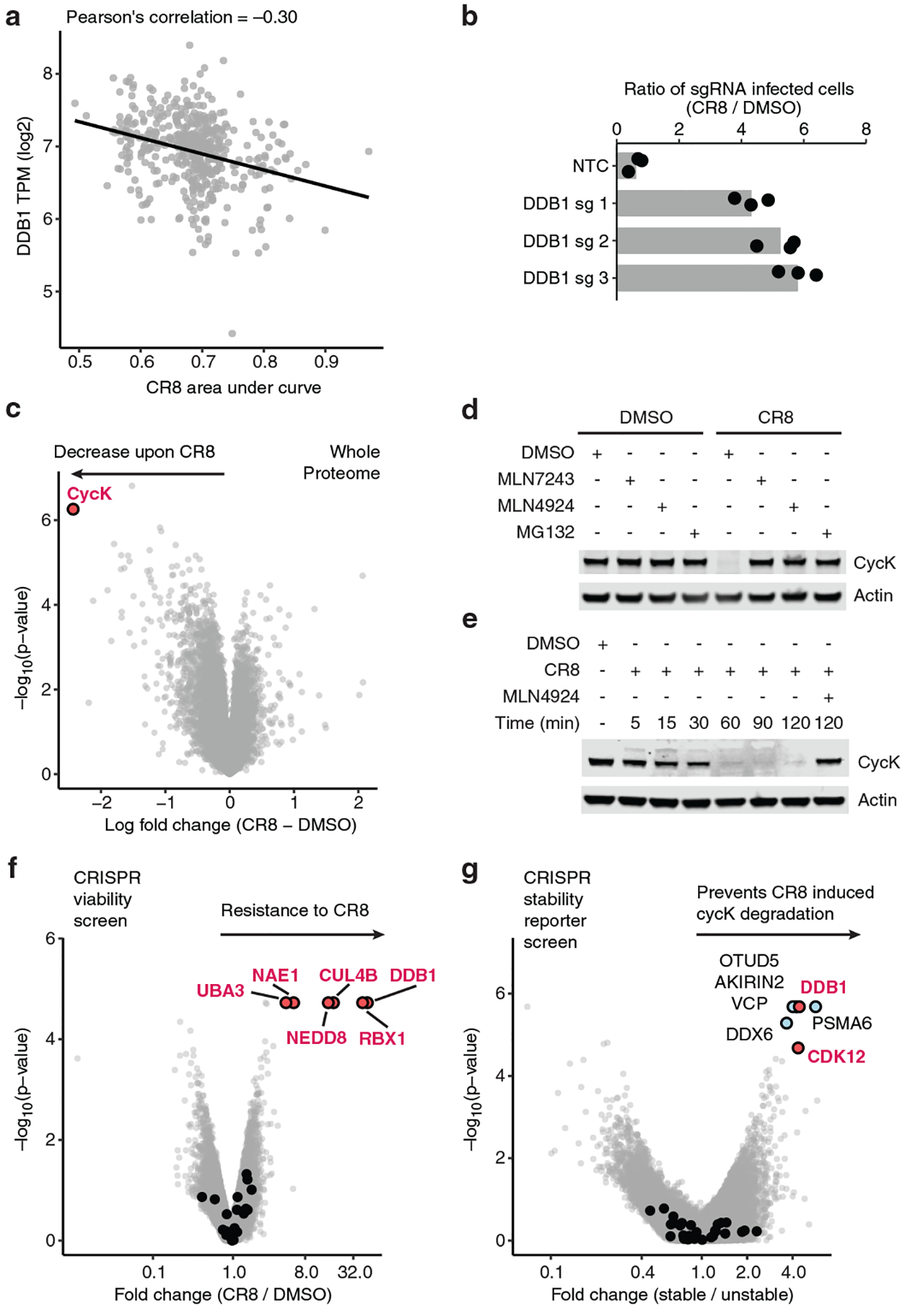
a, Pearson correlation between CR8 toxicity and mRNA expression of DDB1. Dots represent cancer cell lines. Smaller area under the curve (AUC) corresponds to higher drug toxicity. TPM, transcripts per million (n=471). b, Flow analysis of HEK293TCas9 cells expressing sgRNAs and a BFP marker (blue fluorescent protein) after a 3-day treatment with 1 μM CR8 (bars represent mean, n=3). c, Whole proteome quantification of Molt-4 cells treated with 1 μM CR8 (n=1) or DMSO (n=3) for 5 hours (two-sided moderated t-test, n=3). d, Immunoblots of CycK degradation in HEK293TCas9 cells pre-treated with 0.5 μM MLN7243, 1 μM MLN4924, or 10 μM MG132 for 4 hours followed by exposure to 1 μM CR8 for 2 hours (n=3). e, Immunoblots of CycK degradation time course in HEK293TCas9 cells treated with 1 μM CR8 (n=3). f, Genome-wide CRISPR/Cas9 viability screen for CR8 resistance in HEK293TCas9 cells. Guide counts were collapsed to gene-level (n=4 guides/gene; two-sided empirical rank-sum test-statistics). Black dots denote DCAF substrate receptors. g, Genome-wide CRISPR/Cas9 reporter screen for cycKeGFP stability upon 1μM CR8 treatment in HEK293TCas9 cells. Guide counts were collapsed to gene-level (n=4 guides/gene; two-sided empirical rank-sum test-statistics). Black dots denote DCAF substrate receptors.
As DDB1-dependent cytotoxicity of R-CR8 implicated ubiquitin ligase-mediated degradation of one or more essential proteins, we performed quantitative proteome-wide mass spectrometry to evaluate protein abundance following compound treatment. Of the >8,000 quantified proteins, cycK was the only protein that consistently showed decreased abundance following R-CR8 addition (Fig. 1c, Extended Data Fig. 1f, g). As expected, R-CR8 did not alter cycK mRNA levels (Extended Data Fig. 1h) and compound-induced cycK degradation could be rescued by inhibition of the E1 ubiquitin-activating enzyme (MLN7243), cullin neddylation (MLN4924) and the proteasome (MG132) (Fig. 1d). Together, these results suggest that R-CR8 triggers rapid proteasomal degradation of cycK (Fig. 1e) through the activity of a DDB1-containing cullin-RING ubiquitin ligase.
To dissect the molecular machinery required for R-CR8 toxicity, we performed genome-wide and E3 ubiquitin ligase-focused CRISPR-Cas9 resistance screens (Fig. 1f, Extended Data Fig. 2a, b). SgRNAs targeting DDB1, CUL4B, RBX1, the cullin-RING activator NEDD8, and the NEDD8-activating enzyme (NAE1/UBA3) were significantly enriched in the R-CR8-resistant cell population. As all of these proteins are required for CRL activity, our results provide genetic evidence for the involvement of a functional CUL4-RBX1-DDB1 ubiquitin ligase complex in mediating R-CR8 cytotoxicity.
Thus far, all known cullin-RING ligases engage their substrates through specific substrate receptors, and DDB1 serves as an adaptor protein able to bind over 20 such receptors (also known as DDB1-CUL4-associated-factors, DCAFs)17,18 to recruit them to the CUL4-RBX1 ligase core. As no DCAF was identified in our viability screens, we constructed a fluorescent reporter of cycK stability (Extended Data Fig. 2c), in which R-CR8-mediated degradation of endogenous cycK could be recapitulated with a cycKeGFP fusion protein (Fig. 1d, e, Extended Data Fig. 2d–f). Using the stability reporter, in which the extent of degradation can be determined by measuring cycKeGFP levels normalised to mCherry expression, we found that S- and R-CR8 facilitated cycKeGFP degradation to the same extent (Extended Data Fig. 2g; henceforth, CR8 refers to R-CR8). We then performed a genome-wide CRISPR-Cas9 screen for genes involved in cycK reporter stability and validated the involvement of DDB1 in CR8-mediated cycK degradation (Fig. 1g, Extended Data Fig. 2h), but not in compound-independent cycK degradation (Extended Data Fig. 2i). In addition, we identified cyclin-dependent kinase 12 (CDK12), which is a known target of CR819 and whose activity depends on the interaction with cycK20, as a crucial component for CR8-induced cycKeGFP destabilisation (Fig. 1g, Extended Data Fig. 2h–k).
As neither the cycKeGFP stability reporter screen nor the CR8 resistance screen identified a substrate receptor, we performed additional CRISPR screens targeting 29 genes encoding known DCAFs or DCAF-like candidate proteins in four different cell lines. While sgRNAs targeting the previously identified components of the CUL4-RBX1-DDB1 complex consistently caused resistance to CR8, a DCAF substrate receptor could not be identified (Extended Data Fig. 3).
CR8 directs CDK12 to CRL4 core component
Since none of our genetic screens highlighted a DCAF required for cycK degradation, we tested whether CR8-engaged CDK12-cycK directly binds one of the CUL4-RBX1-DDB1 ligase components in the absence of a substrate receptor. We therefore performed in vitro co-immunoprecipitation experiments using recombinantly purified proteins. The kinase domain of CDK12 (CDK12713−1052) bound to cycK1−267 did not markedly enrich DDB1 over the bead binding control in the absence of CR8, whereas equimolar amounts of the compound led to stoichiometric complex formation (Fig. 2a). DDB1 β-propeller domains A (BPA) and C (BPC)17, which are otherwise involved in DCAF binding, were sufficient for drug-induced CDK12-cycK recruitment. DDB1 β-propeller B (BPB), which binds CUL4 and is not involved in DCAF binding, was dispensable for the interaction (Fig. 2a). In vitro ubiquitination assays confirmed that the CUL4A-RBX1-DDB1 ligase core alone is sufficient to drive robust cycK ubiquitination (Fig. 2b). Quantification of the interaction showed that CR8 stimulated binding between CDK12-cycK and DDB1 in the range of 100–500 nM depending on the experimental setup (Fig. 2c, Extended Data Fig. 4). While weak CDK12-cycK-DDB1 interaction was still detectable in the absence of the compound in vitro, CR8 strengthened complex formation 500- to 1000-fold as estimated by isothermal titration calorimetry (ITC) (Extended Data Fig. 4f–k). Thus, our data indicate that CR8-engaged CDK12-cycK is recruited to the CUL4-RBX1-DDB1 ligase core through DDB1, and the compound tightens the complex sufficiently to drive CR8-induced cycK degradation in the absence of a canonical DCAF substrate receptor.
Figure 2 |. CR8-bound CDK12 binds DDB1 in a DCAF-like manner.
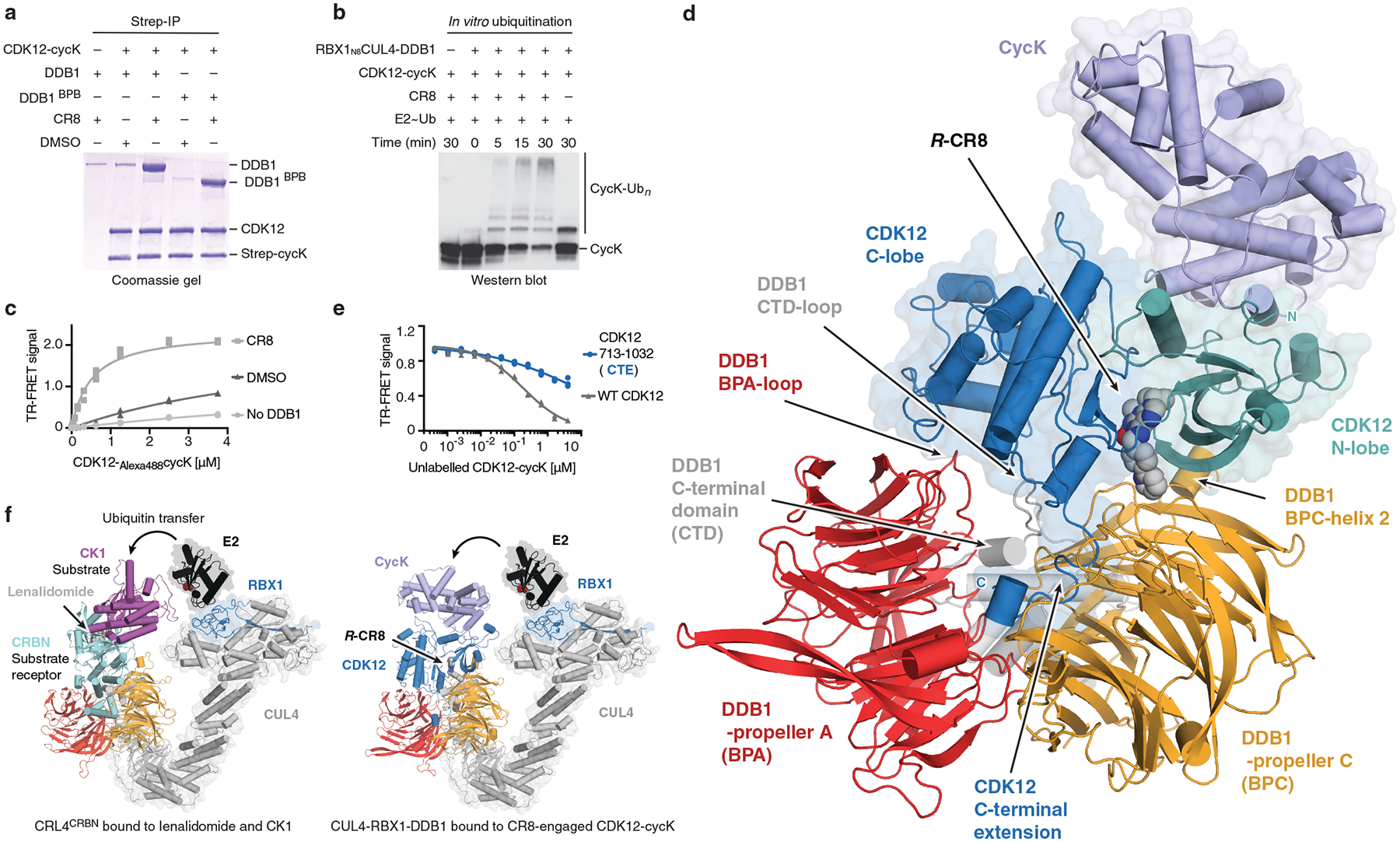
a, Co-immunoprecipitation (IP) experiments with recombinant proteins (n=3). b, In vitro ubiquitination of cycK by the RBX1N8CUL4-DDB1 ubiquitin ligase core (n=2). c, TR-FRET signal for CDK12-Alexa488cycK titrated to TerbiumDDB1 in DMSO or 10 μM CR8 (n=3). No DDB1 only contains streptavidin-terbium and shows concentration-dependent fluorophore effects. d, Cartoon representation of the DDB1ΔBPB-R-CR8-CDK12-cycK crystal structure. e, TR-FRET counter titration of unlabelled wild-type or mutant CDK12-cycK (0–10 μM) into pre-assembled TerbiumDDB1-CR8-CDK12-Alexa488cycK complex (n=3). f, Structural models of CRL4CRBN bound to lenalidomide and CK1α and RBX1-CUL4-DDB1 (CRL4) bound to the R-CR8-CDK12-cycK complex (bottom). The E2 active site cysteine (red spheres) binds ubiquitin through a thioester bond.
We then crystallised CDK12713−1052-cycK1−267 bound to CR8 and DDB1ΔBPB and determined the 3.5 Å resolution structure of this complex (Fig. 2d, Extended Data Table 1). In the structure, CDK12 forms extensive protein-protein interactions (~2100 Å2) with DDB1. CR8 binds the active site of CDK12 and bridges the CDK12-DDB1 interface, while cycK binds CDK12 on the opposite site and does not contact DDB1. The N- and C-lobes of CDK12 are proximal to DDB1 residues located in a loop of the BPA domain (amino acid (aa) 111–114), BPC-helix 2 (aa 986–990), and a loop in the C-terminal domain (aa 1078–1081) that are otherwise involved in DCAF binding (Extended Data Fig. 5). In addition, the C-terminal extension of CDK12 binds the cleft between the DDB1 domains BPA and BPC, a hallmark binding site of DDB1-DCAF interactions (Extended Data Fig. 5a–d, i). The density for this region could only be tentatively assigned, likely due to the presence of multiple conformations, but the CDK12 C-terminal tail clearly engages with DDB1 and assumes a conformation different from those seen in isolated CDK12-cycK structures (Extended Data Fig. 6a, b, d)19,21. Structure-guided mutational analyses combined with time-resolved fluorescence resonance energy transfer (TR-FRET) assays were used to assess the contribution of these interactions to CR8-dependent CDK12-DDB1 complex formation (Fig. 2e, Extended Data Fig. 5e). Taken together, our data demonstrates that CDK12 assumes the role of a glue-induced substrate receptor and places cycK in a position typically occupied by CRL4 substrates (Fig. 2f). This renders CDK12-cycK binding to DDB1 mutually exclusive with that of DCAFs and provides a structural framework for why a canonical substrate receptor is dispensable for cycK ubiquitination.
CDK12-DDB1 interface imparts selectivity
CR8 is a pleiotropic CDK inhibitor reported to bind CDK1/2/3/5/7/9/126,19, yet in cells we observed selective cycK destabilization in the presence of the drug. As cycK is reported to associate with CDK9, CDK12, and CDK1319, we tested whether the other cycK-dependent kinases are also recruited to DDB1. The closely related CDK13 (90.8 % sequence identity), but not the more divergent CDK9 (45.5 % sequence identity) (Extended Data Fig. 7a–c), was recruited to DDB1 in the presence of CR8, albeit with a lower binding affinity (Extended Data Fig. 7d–f). Analogously, less productive in vitro cycK ubiquitination was observed for CDK13 compared to CDK12 (Extended Data Fig. 7g). The key difference between CDK9 and CDK12/13 primary sequence lies in the C-terminal extension (Extended Data Fig. 7a, b), which in our structure nestles up against DDB1 BPA and BPC propellers (Fig. 2d, Extended Data Fig. 5i). Mutations in, or truncation of, the CDK12 C-terminal extension (Extended Data Fig. 5c) abolished basal binding between CDK12 and DDB1, whereas complex formation could still be facilitated by CR8 to a varying extent (Extended Data Fig. 7h, i). Hence, our data show that the pan-selective CDK inhibitor CR8 induces specific protein-protein interactions between CDK12/13 and DDB1 and suggest that the C-terminal extension, while contributing to binding, is not essential for drug-dependent kinase recruitment.
CR8 phenylpyridine confers glue activity
CR8 occupies the ATP binding pocket of CDK12 and forms discrete contacts with residues in the BPC domain of DDB1 (~150 Å2) through its hydrophobic phenylpyridine ring system (Fig. 3a, b). Mutation of DDB1 residues Ile909, Arg928, and Arg947 each diminished drug-induced recruitment of the kinase (Extended Data Fig. 5f), highlighting the contribution of the phenylpyridine moiety to complex formation. To evaluate the structure-activity relationship underlying the gain-of-function activity of CR8, we probed other CDK inhibitors for their ability to drive complex formation between DDB1 and CDK12. DRF05322, a CR8-related inhibitor that carries a differently linked phenylpyridine ring system (Fig. 3a, c), induced binding with two-fold lower affinity than CR8 (Extended Data Fig. 8a). Roscovitine23, the parent compound of CR8 that lacks the 2-pyridyl substituent but retains the phenyl ring proximal to Arg928 (Fig. 3a, c), also facilitated complex formation, albeit with a three-fold lower apparent affinity (Extended Data Fig. 8a). The affinity rank-order observed in our TR-FRET assay correlated with the degree of cycK ubiquitination in vitro, in which DRF053 and roscovitine showed less processive ubiquitination (Fig. 3d). As neither DRF053 nor roscovitine induced degradation of the cycKeGFP reporter in cells (Fig. 3e), our results demonstrate that the presence and correct orientation of the 2-pyridyl on the surface of CDK12 confer the gain-of-function activity of CR8 leading to cycK degradation.
Figure 3 |. A surface-exposed 2-pyridyl moiety of CR8 confers glue degrader activity.
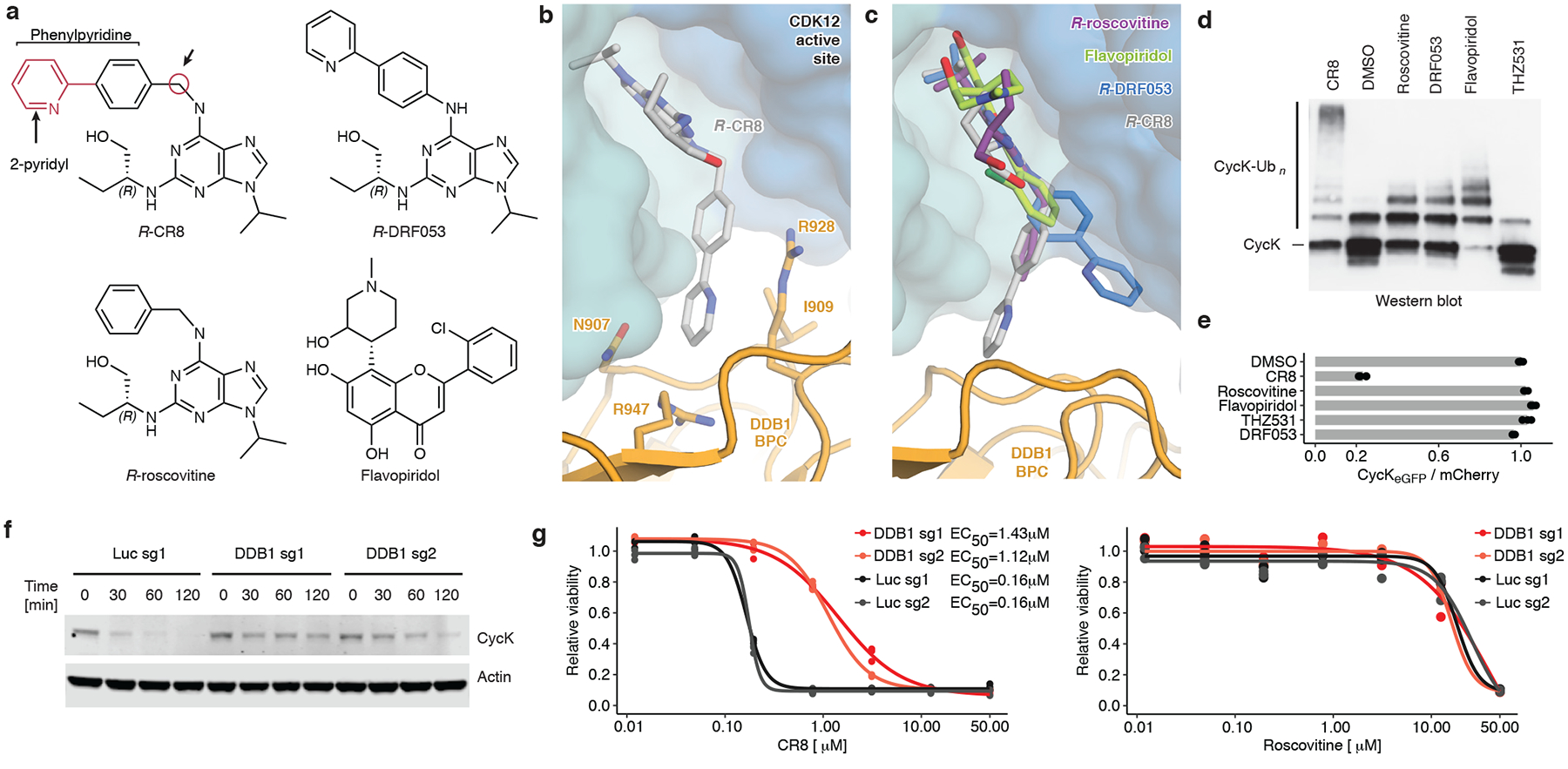
a, Chemical structures of CDK inhibitors. Arrows indicate differences between R-CR8, R-DRF053 and R-roscovitine. b, Close-up of the CDK12-CR8-DDB1 interface. The phenylpyridine moiety of CR8 contacts DDB1 residues. c, R-roscovitine (PDB entry 2A4L), R-DRF053 and flavopiridol (3BLR) in the active site of CDK12 in the DDB1-CR8-CDK12-cycK complex through superposition of kinase domains or the purine moiety (for DRF053). d, In vitro ubiquitination of CDK12-cycK complex by RBX1N8CUL4-DDB1 in the absence (DMSO) or presence of 2 μM compound (n=2). e, Flow analysis of CycKeGFP degradation in HEK293TCas9 cells treated with 1 μM compound for 2 hours (n=3). f, Immunoblots of CycK in HEK293TCas9 cells transfected with the indicated sgRNAs and treated with 1 μM CR8 (n=2). g, Drug sensitivity of sgRNA-transfected HEK293TCas9 cells after a 3-day exposure to CR8 or roscovitine (n=3).
To probe whether any ligand could in principle drive the interaction of CDK12 with DDB1, we tested the endogenous CDK nucleotide cofactor ATP for its ability to promote complex formation. ATP neither facilitated nor abrogated the interaction over basal binding observed in the presence of DMSO (Extended Data Fig. 6c), suggesting that although the nucleotide-bound conformation of CDK12 seems incompatible with approaching DDB1 (Extended Data Fig. 6b), its C-terminal extension is free to adopt multiple conformations21. THZ53124, a bulky covalent CDK12/13 inhibitor predicted to clash with DDB1 (Extended Data Fig. 6d–f), locks the CDK12 C-terminal extension in a conformation incompatible with DDB1 recruitment (Extended Data Fig. 6d). Consistently, THZ531 further decreased the TR-FRET signal and diminished cycK ubiquitination in vitro below DMSO control levels (Fig. 3d, Extended Data Fig. 6c, f)24. Flavopiridol25, a natural product-derived inhibitor structurally distinct from CR8 (Fig. 3a, c), also stimulated the binding of CDK12-cycK to DDB1 (Extended Data Fig. 8a). While flavopiridol gave rise to moderate cycK ubiquitination in vitro (Fig. 3d), it failed to degrade cycK in cells (Fig. 3e). Our results thus show that the DDB1-compound interactions display significant plasticity and that structurally diverse surface-exposed moieties in CR8, DRF053, roscovitine and flavopiridol can facilitate CDK12-cycK recruitment. Small differences in their ability to stabilise the DDB1-CDK12 complex translate, in an almost binary fashion, into cellular degradation of cycK or lack thereof. This behaviour is reminiscent of CRL4CRBN and thalidomide analogues11,26, where an apparent affinity threshold must be overcome to drive drug-induced target degradation.
CycK degradation adds to CR8 toxicity
Finally, to delineate the contribution of CRL4-mediated cycK degradation to CR8 cytotoxicity over non-degradative CDK inhibition, we compared compound toxicity in wild-type HEK293TCas9 cells to cells that were pre-treated with MLN4924 (NEDD8-activating enzyme inhibitor), genetically-depleted for DDB1, or subject to DCAF overexpression. Global inhibition of CRL activity by MLN4924 had only minor effects on cell viability (Extended Data Fig. 9a), but resulted in decreased sensitivity to CR8 (Extended Data Fig. 9b), showing that CRL neddylation significantly contributes to CR8 toxicity. Overexpression of the substrate receptor CRBN also affected sensitivity to CR8 and decreased cycK degradation (Extended Data Fig. 9c–g), presumably by reducing the free pool of DDB1. As expected, CR8-induced endogenous cycK degradation was dependent on DDB1 (Fig. 3f) and, consistently, we found that cytotoxicity of CR8, but not that of the other CDK inhibitors, was ten-fold lower in cells depleted for DDB1 (Fig. 3g, Extended Data Fig. 9h). Together, the data demonstrate that the CRL4-dependent gain-of-function glue degrader activity of CR8 strongly contributes to its cellular potency and provides an additional layer of ortholog-specific CDK inactivation through cycK degradation.
Kinase inhibitors have long been suspected to have a degradation component to their mode of action27,28, and our work provides the first characterization and structural dissection of how a kinase inhibitor scaffold acquires degrader properties. Molecular glue degraders have thus far only been shown to engage substrate-recruiting E3 ligase modules. CDK12 is not a constitutive E3 ligase component, but rather serves as a drug-induced substrate receptor, linking DDB1 to the ubiquitination target. CR8 thus bypasses the requirement for a canonical DCAF and instead hijacks the essential adaptor protein DDB1. Although cycK is the primary ubiquitination target, CDK12 may become subject to autoubiquitination upon prolonged compound exposure similar to canonical DCAFs29,30.
While previously reported molecular glue degraders engage a ligandable pocket on the ligase to recruit target proteins, CR8 instead binds the active site of CDK12 and does not rely on an independent ligand binding site on DDB1 (Extended Data Fig. 4h). This suggests that the repertoire of targets and ubiquitin ligases accessible to targeted protein degradation can be expanded by target-binding small molecules that induce de novo contacts with a ligase or strengthen existing weak protein-protein interactions. Kinase inhibitors in particular often show poor selectivity and small molecule-induced kinase inactivation that leverages complementary protein-protein interfaces offers a path towards improved drug selectivity, which might, for example, facilitate selective inactivation of CDK12, an emerging therapeutic target31.
The gain-of-function glue degrader activity of CR8 is attributed to a 2-pyridyl moiety exposed on the kinase surface. Surface-exposed single residue mutations have been shown to promote the formation of higher-order protein complexes, as the haemoglobin Glu to Val mutation, for example, induces polymerization in sickle cell anaemia32. Accordingly, single residue mutations designed to increase surface hydrophobicity give rise to ordered protein assemblies33. Bound compounds, such as enzyme inhibitors, can in principle mimic such amino acid changes with dramatic effects on the protein interaction landscape, suggesting that compound-induced protein-protein interactions may be more common than previously recognised. Taken together, our results suggest that modifications of surface-exposed regions in target-bound small molecules offer a rational strategy to develop molecular glue degraders for a given protein target.
Methods
Mammalian cell culture
The human HEK293T cell lines were provided by the Genetic Perturbation Platform, Broad Institute and K562Cas9, THP1Cas9, P31FUJCas9 cell lines were provided by Zuzana Tothova (Broad Institute), MOLT-4 were purchased from ATCC and HEK293TCas926 and MM1SCas934 were previously published. HEK293T cells were cultured in DMEM (Gibco) and all other cell lines in RPMI (Gibco), with 10% FBS (Invitrogen), glutamine (Invitrogen) and penicillin–streptomycin (Invitrogen) at 37 °C and 5% CO2.
Compounds
R-CR8 (3605) was obtained from Tocris, S-CR8 (ALX-270–509-M005), flavopiridol (ALX-430–161-M005) from Enzo Life Sciences, roscovitine (HY-30237), THZ531 (HY-103618), LDC00067 (HY-15878) from MedChem Express and DRF053 (D6946) from Sigma.
Antibodies
The following antibodies were used in this study: anti-cycK (Bethyl Laboratories, A301–939A for full length cycK), anti-cycK (abcam, ab251652, for cycK1−267), anti-beta-actin (Cell Signaling, #3700), anti-CRBN (Sigma prestige, HPA045910), anti-mouse 800CW (LI-COR Biosciences, 926–32211), anti-rabbit 680LT (LI-COR Biosciences, 925–68021), anti-rabbit IgG antibodies (abcam, ab6721).
Reporter vectors
The following reporter were used in this study: Artichoke (SFFV.BsmBICloneSite-17aaRigidLinker-eGFP.IRES.mCherry.cppt.EF1α.PuroR, Addgene #73320 for Genome wide screen and validation experiments), Cilantro 2 (PGK.BsmBICloneSite-10aaFlexibleLinker-eGFP.IRES.mCherry. cppt.EF1α.PuroR, Addgene #74450 for degradation kinetics), sgBFP (sgRNA.SFFV.tBFP, for Validation of drug-E3 ligase pairs), sgRFP657, (sgRNA.EFS.RFP657 for Validation of drug-E3 ligase pairs), sgPuro, (pXPR003, Addgene #52963, for drug sensitivity assays).
Oligos
List of all oligonucleotides used in this study can be found in Supplementary Oligo Table 1.
Bioinformatic screen
We computed Pearson correlations of viability of PRISM repurposing compounds in 8 doses and 578 cell lines4 with gene expression (GE) and copy number variation (CN) of all detectable protein-coding genes of matched cell lines from The Cancer Cell Line Encyclopedia (CCLE)5. Z score was computed for each pair of compounds, dose viability, and genomic feature (GE or CN) across all cell lines. For each compound-genomic feature pair, the most extreme correlations are ranked from negative to positive. To focus on novel compound-gene relationships, we restricted genes to a curated list of 499 E3 ligase components and compounds that are not one of “EGFR inhibitor”, ”RAF inhibitor“ or “MDM inhibitor” based on PRISM repurposing annotation5. Hit compounds were selected if either the Z score was less than −6 or ranked in the top 15 with Z score less than −4. The resulting list of 158 E3 gene-compound pairs was further curated and shortened manually to 96 E3 gene-compound pairs, which included 95 unique E3 ligases and 85 unique compounds.
Cloning and lentiviral packaging of sgRNAs targeting 95 E3 ligases
sgRNAs targeting E3 ligases were selected from the human Brunello CRISPR library35. 170 oligo pairs (IDT) targeting 95 E3 ligases were annealed and cloned into the sgRNA.SFFV.tBFP (Guide ID A) or sgRNA.EFS.RFP657 (Guide ID B) fluorescent vectors in a 96-well format using previously published protocols36. Briefly, vectors were linearized with BsmBI (New England Biolabs) and gel purified with the Spin Miniprep Kit (Qiagen). Annealed oligos were phosphorylated with T4 Polynucleotide Kinase (New England Biolabs) and ligated into the linearized and purified vector backbones with T4 DNA Ligase (New England Biolabs). Constructs were transformed into XL10-Gold ultracompetent Escherichia coli (Stratagene/Agilent Technologies), plasmids were purified using MiniPrep Kit (Qiagen), and guide sequence confirmed by Sanger sequencing. For validation of the primary screen, virus was produced in a 96-well format. Briefly, 11,000 HEK293T cells were seeded per well in 100 μL DMEM medium supplemented with 10% FBS and Penicillin-Streptomycin-Glutamine. The next day a Packaging Mix was prepared in a 96-well plate consisting of 500 ng psPAX2, 50 ng pVSV-G and 17 ng sgRNA backbone in 5 μL OptiMem (Invitrogen) and incubated for 5 minutes at room temperature. This mix was combined with 0.1 μL TransIT-LT1 (Mirus) in 5 μL of OptiMem, incubated for 30 minutes at room temperature, and then applied to cells. Two days post-transfection, dead cells were removed by centrifugation and lentivirus containing medium was collected stored at −80°C prior to use.
Validation of drug-E3 ligase pairs from the bioinformatic screen
K562Cas9, OVK16Cas9, A564Cas9, ES2Cas9 and MOLM13Cas9 cell lines were individually transduced with 192 sgRNAs targeting 95 E3 ligases in a 96-well plate format. 3000 cells/well were plated in 100 μL RPMI supplemented with 10% FCS and Penicillin-Streptomycin-Glutamine and 30μL/well of virus supernatant was added. 24 hours post infection the medium was changed. After three days, the percentage of sgRNA transduced cells was determined by flow cytometry. If more than 60% of cells were transduced, un-transduced cells were added to bring the level below 60%. Eight days post-infection cell density was measured and adjusted to 1.5 × 105 cells/mL with RPMI. For treatment, 50 μL of sgRNA transduced cells were seeded into each well of a 384 well plate with pre-plated DMSO or cognate drug in three concentrations (0.1 μM, 1 μM, 10 μM) with Agilent BRAVO Automated Liquid Handling Platform. Plates were sealed with White Rayon adhesive sealing tape (Thermo Fisher Scientific) and grown for three days. Adherent cell lines were trypsinised and re-suspended in 50 μL of RPMI with Matrix WellMate (Thermo Scientific). Suspension cells were directly subjected to analysis. 10 μL of cell suspension was subjected to the flow analysis with FACSCanto equipped with High Throughput Sampler (BD Bioscience). The percentage of sgRNA transduced cells in the drug treatment wells was normalised to the DMSO control. Wells with fluorescent drug and samples with less than 120 viable cells events or less than 6% fluorescent cells were removed from analysis. All E3-drug pairs were ranked based on the number of experimental conditions (cell line and drug dose) with more than 50% of sgRNA transduced cells in drug treatment wells in comparison to corresponding DMSO control.
Validation of DDB1 resistance phenotype
For validation experiments, virus was produced in a 6-well plate format, as described above with the following adjustments: 2.5 × 105 HEK293T cells/well in 2 mL DMEM medium, 3 μL/well of TransIT-LT1, 15 μL/well of OPTI-MEM, 500 ng/well of the desired plasmid, 500 ng/well psPAX2, and 50 ng/well pVSV-G in 32.5 μL/well OPTI-MEM. After collecting the virus, 10 × 103 HEK293TCas9 cells in 100 μL DMEM medium were transduced with 10 μL of virus supernatant. The transduced HEK293TCas9 cells were then mixed with untransduced control cells at a 1:9 ratio. Nine days after sgRNA transduction, cells were treated for 3 days with DMSO or 1 μM CR8 and analysed by flow cytometry to determine the percentage of BFP+ cells. sgRNAs targeting DDB1 provide partial depletion of DDB1 (50% DDB1 alleles modified, reducing DDB1 levels by roughly 50%), which suggests selection towards heterozygous or hypomorphic clones.
Whole proteome quantification using tandem mass tag mass spectrometry
10 × 106 Molt-4 cells were treated with DMSO (triplicate) or 1 μM CR8 (single replicate) for 1 or 5 hours and later were harvested by centrifugation. Samples were processed, measured and analysed as described before37. Data are available in the PRIDE repository (PXD016187 and PXD016188).
Quantitative PCR
HEK293TCas9 cells were treated with DMSO or 1 μM CR8 for 2 hours, collected by centrifugation, washed with PBS, and snap-frozen at −80°C. mRNA was isolated using a QIAGEN RNA kit (Qiagen, 74106). For cDNA synthesis, total RNA was reverse transcribed using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fischer) before qPCR analysis with TaqMan Fast Advanced Master Mix (ThermoFisher Scientific, 4444557) for CCNK (TaqMan, Hs00171095_m1, Life Technologies) and GAPDH (TaqMan, Hs02758991_g1). Reactions were run and analysed on a CFX96 Real Time system (Bio-Rad).
Immunoblots for whole protein lysate
Cells were washed with phosphate buffered saline (PBS) and lysed (150 mM NaCl, 50 mM Tris (pH 7.5), 1% NP-40, 1% glycerol, 1x Halt Cocktail protease and phosphatase inhibitors) for 20 minutes on ice. The insoluble fraction was removed by centrifugation, protein concentration was quantified using a BCA protein assay kit (Pierce), and equal amount of lysate was run on SDS-PAGE 4–12% Bis-Tris Protein Gels (NuPAGE, Thermo Fisher) and subsequently transferred to nitrocellulose membrane with Trans-Blot Turbo System (BIO-RAD). Membranes were blocked in Odyssey Blocking Buffer/PBS (LI-COR Biosciences) and incubated with primary antibodies overnight at 4°C. The membranes were then washed in TBS-T, incubated for 1 hour with secondary IRDye-conjugated antibodies (LI-COR Biosciences), and washed three times in TBS-T for 5 minutes prior to Near-Infrared Western blot detection on an Odyssey Imaging System (LI-COR Biosciences).
CycK stability reporter analysis
HEK293TCas9 expressing the cycKeGFP degradation reporter were transduced with experimental sgRNAs. Nine days after infection the cells were dosed for 2 hours with DMSO or 1 μM CR8 and fluorescent signal quantified by flow cytometry (CytoFLEX, Beckman or LSR Fortessa flow cytometer BD Biosciences). Using FlowJo (flow cytometry analysis software, BD), the geometric mean of eGFP and mCherry fluorescent signal for round and mCherry positive cells was calculated. The ratio of eGFP to mCherry was normalised to the average of three DMSO-treated controls.
Genome wide CRISPR – CR8 resistance screen
5% (v/v) of the human genome-wide CRISPR-KO Brunello library with 0.4 μL Polybrene/mL (stock of 8 mg/mL) was added to 1.5 × 108 HEK293TCas9 in 75 mL of medium and transduced (2400 rpm, 2 hours, 37°C). 24h after infection sgRNA transduced cells were selected with 2 μg/mL of Puromycin for two days. On the ninth day post-infection, cells were treated with either DMSO (n=1) or 1 μM CR8 (n=1) and cultured for an additional 3 days. Resistant live cells were selected by gently washing away detached dead cells from the medium. Cell pellets were resuspended in multiple direct lysis buffer reactions (1 mM CaCl2, 3 mM MgCl2, 1 mM EDTA, 1% Triton X-100, Tris pH 7.5 - with freshly supplemented 0.2 mg/mL Proteinase) with 1×106 cells per 100 μL reaction. The sgRNA sequence was amplified in a first PCR reaction with eight staggered forward primers. 20 μL of direct lysed cells was mixed with 0.04U Titanium Taq (Takara Bio 639210), 0.5 x Titanium Taq buffer, 800 μM dNTP mix, 200 nM SBS3-Stagger-pXPR003 forward primer, 200 nM SBS12-pXPR003 reverse primer in a 50 μL reaction (cycles: 5 minutes at 94°C, 15 x (30 sec at 94°C, 15 sec at 58°C, 30 sec at 72°C), 2 minutes at 72°C). 2 μL of the first PCR reaction was used as the template for 15 cycles of the second PCR, where Illumina adapters and barcodes were added (0.04U Titanium Taq, 1 x Titanium Taq buffer, 800 μM dNTP mix, 200 nM P5-SBS3 forward primer, 200 nM P7-barcode-SBS12 reverse primer). An equal amount of all samples was pooled and subjected to preparative agarose electrophoresis followed by gel purification (Qiagen). Eluted DNA was further purified by NaOAc and isopropanol precipitation. Amplified sgRNAs were quantified using Illumina NextSeq platform (Genomics Platform, Broad Institute). Read counts for all guides targeting the same gene were used to generate p-values. Hits enriched in resistance population with False Discovery Rate (FDR) < 0.05 and enriched > 5-fold, are labelled on the plot (Fig. 1f)38.
BISON CRISPR – CR8 resistance screen
The BISON CRISPR library targets 713 E1, E2, and E3 ubiquitin ligases, deubiquitinases, and control genes and contains 2,852 guide RNAs. It was cloned into the pXPR003 as previously described35 by the Broad Institute Genome Perturbation Platform (GPP). The virus for the library was produced in a T-175 flask format, as described above with the following adjustments: 1.8 × 107 HEK293T cells in 25 mL complete DMEM medium, 244 μL of TransIT-LT1, 5 mL of OPTI-MEM, 32 μg of library, 40 μg psPAX2, and 4 μg pVSV-G in 1 mL OPTI-MEM. 10% (v/v) of BISON CRISPR library was added to 6 × 106 HEK293TCas9 cells in triplicates and transduced. Samples (n=3) were processed as describe above for the genome wide resistance screen.
Genome wide CRISPR – cycK stability reporter screen
A single clone of cycKeGFP HEK293TCas9 was transduced with the genome wide Brunello library as described above with the following modification: 4.5 × 108 cycKeGFP HEK293TCas9 cells in 225 mL of medium. Nine days later cells were treated with CR8 (n=3) or DMSO (n=3) for at least 2 hours and the cycK stable population was separated using fluorescence activated cell sorting. Four populations were collected (top 5%, top 5–15%, lowest 5–15% and lowest 5%) based on the cycKeGFP to mCherry mean fluorescent intensity (MFI) ratio on MA900 Cell Sorter (Sony). Sorted cells were harvested by centrifugation and subjected to direct lysis as described above. The screen was analysed as described below by comparing stable populations (top 5% eGFP/mCherry expression) to unstable populations (lowest 15% eGFP/mCherry expression). Hits enriched in cycK stable population with FDR < 0.05 are labelled on the plot (Fig. 1g).
Pooled CRISPR screen data analysis
The data analysis pipeline comprised the following steps: (1) Each sample was normalised to the total number of reads. (2) For each guide, the ratio of reads in the stable vs. unstable sorted gate was calculated, and sgRNAs were ranked. (3) The ranks for each guide were summed for all replicates. (3) The gene rank was determined as the median rank of the four guides targeting it. (4) P-values were calculated by simulating a distribution with guide RNAs that had randomly assigned ranks over 100 iterations. R scripts can be found in the Supplementary Information.
DCAF arrayed screen
An arrayed DCAF library (targeting DCAFs substrate receptors, DCAF-like and control genes) was constructed as described above with the appropriate oligos (Supplementary Oligo Table 1). K562Cas9, P31FUJCas9, THP1Cas9 and MM1SCas9 were individually transduced and treated with DMSO or 1 μM CR8 (K562Cas9, P31FUJCas9, THP1Cas9) or 0.1 μM CR8 (MM1SCas9). The analysis was performed as described above for validation of DDB1 resistance phenotype.
Protein purification
Human wild-type and mutant versions of DDB1 (Uniprot entry Q16531), CDK12 (Q9NYV4, K965R) and CCNK (O75909) were subcloned into pAC-derived vectors39 and recombinant proteins were expressed as N-terminal His6, His6-Spy, StrepII or StrepII-Avi fusions in Trichoplusia ni High Five insect cells using the baculovirus expression system (Invitrogen)40.
Wild-type or mutant forms of full-length or beta-propeller B domain deletion (ΔBPB: aa 396–705 deleted) constructs of His6-DDB1 and StrepII-Avi-DDB1 were purified as previously described for DDB1-DCAF complexes12. High Five insect cells co-expressing truncated versions of wild-type or mutant His6-CDK12 (aa 713–1052 or 713–1032) and His6- or His6-Spy-tagged cycK (aa 1–267) were lysed by sonication in 50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 10% (v/v) glycerol, 10 mM MgCl2, 10 mM imidazole, 0.25 mM tris(2-carboxyethyl)phosphine (TCEP), 0.1% (v/v) Triton X-100, 1 mM phenylmethylsulfonylfluoride (PMSF), and 1 x protease inhibitor cocktail (Sigma). Following ultracentrifugation, the soluble fraction was passed over HIS-Select Ni2+ affinity resin (Sigma), washed with 50 mM Tris-HCl (pH 8.0), 1 M NaCl, 10% (v/v) glycerol, 0.25 mM TCEP, 10 mM imidazole and eluted in 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 10% (v/v) glycerol, 0.25 mM TCEP, 250 mM imidazole. When necessary, affinity tags were removed by overnight TEV protease treatment. In case of HIS-Select Ni2+ affinity purified CDK12-cycK that was not subjected to TEV cleavage, the pH of the eluate was adjusted to 6.8 before ion exchange chromatography. StrepII-tagged versions of CDK12-cycK were affinity purified using Strep-Tactin Sepharose (IBA) omitting imidazole in lysis, wash and elution buffers, supplementing the elution buffer with 2.5 mM desthiobiotin (IBA GmbH), and using 50 mM Tris-HCl (pH 6.8) throughout.
For ion exchange chromatography, affinity purified proteins were diluted in a 1:1 ratio with buffer A (50 mM Tris-HCl (pH 6.8), 10 mM NaCl, 2.5% (v/v) glycerol, 0.25 mM TCEP) and passed over an 8 mL Poros 50HQ column. The flow through was again diluted in a 1:1 ratio with buffer A and passed over an 8 mL Poros 50HS column. Bound proteins were eluted by a linear salt gradient mixing buffer A and buffer B (50 mM Tris-HCl (pH 6.8), 1 M NaCl, 2.5% (v/v) glycerol, 0.25 mM TCEP) over 15 column volumes to a final ratio of 80% buffer B. Poros 50HS peak fractions containing the CDK12-cycK complex were concentrated and subjected to size exclusion chromatography in 50 mM HEPES (pH 7.4), 200 mM NaCl, 2.5% (v/v) glycerol and 0.25 mM TCEP. The concentrated proteins were flash frozen in liquid nitrogen and stored at −80°C.
Co-immunoprecipitation assay
The purified His6-CDK12/StrepII-cycK complex was mixed with equimolar concentrations of full-length His6-DDB1 or TEV-cleaved DDB1ΔBPB (5 μM) in the presence 5 μM R-CR8 or DMSO in IP buffer (50 mM HEPES (pH 7.4), 200 mM NaCl, 0.25 mM TCEP, 0.05% (v/v) Tween-20) containing 1 mg/mL bovine serum albumin. The solution was added to Strep-Tactin MacroPrep beads (IBA GmbH) preequilibrated in IP buffer and incubated for 1 hour at 4°C on an end-over-end shaker. The beads were extensively washed with IP buffer, and the bound protein was eluted with IP buffer containing 2.5 mM desthiobiotin for 1 hour at 4°C on an end-over-end shaker. Eluted proteins were separated by SDS-PAGE stained with Coomassie Brilliant Blue.
Crystallization and data collection
The protein solution for crystallization contained 70 μM TEV-cleaved DDB1ΔBPB, 80 μM R-CR8 and 80 μM TEV-cleaved CDK12-cycK in 50 mM HEPES (pH 7.4), 200 mM NaCl, 0.25 mM TCEP. Crystals were grown by vapour diffusion in drops containing 1 μL DDB1ΔBPB-R-CR8-CDK12713–1052-cycK1−267 complex solution mixed with 1 μL of reservoir solution containing 0.9 M ammonium citrate tribasic (pH 7.0) in two-well format sitting drop crystallization plates (Swissci). Plates were incubated at 19°C and crystals appeared 5–13 days after setup. Crystals were flash cooled in liquid nitrogen in reservoir solution supplemented with 25% (v/v) glycerol as a cryoprotectant prior to data collection. Diffraction data were collected at the Swiss Light Source (beamline PXI) with an Eiger 16M detector (Dectris) at a wavelength of 1 Å and a crystal cooled to 100 K. Data were processed with DIALS, scaled with AIMLESS supported by other programs of the CCP4 suite41, and converted to structure factor amplitudes with STARANISO42, applying a locally weighted CC1/2 = 0.3 resolution cutoff.
Structure determination and model building
The DDB1ΔBPB–R-CR8-CDK12713−1052-cycK1−267 complex formed crystals belonging space group P3121, with three complexes in the crystallographic asymmetric unit (ASU). Their structure was determined using molecular replacement (MR) in PHASER43 with a search model derived from PDB entry 6H0F for DDB1ΔBPB, and PDB entry 4NST for CDK12-cycK. The initial model was improved by iterative cycles of building with COOT44, and refinement using phenix.refine45 or autoBUSTER46, with ligand restraints generated using eLBOW through phenix.ready_set47. The final model was produced by refinement with autoBUSTER. Analysis with MOLPROBITY48. indicates that 93.9% of the residues in final model are in favourable regions of the Ramachandran plot, with 0.6% outliers. Data processing and refinement statistics are in Extended Data Table 1. Interface analysis was performed using PISA49.
Biotinylation of DDB1
Purified full-length StrepII-Avi-DDB1 was biotinylated in vitro at a concentration of 8 μM by incubation with final concentrations of 2.5 μM BirA enzyme and 0.2 mM D-biotin in 50 mM HEPES (pH 7.4), 200 mM NaCl, 10 mM MgCl2, 0.25 mM TCEP and 20 mM ATP. The reaction was incubated for 1 hour at room temperature and stored at 4°C for 14–16 hours. Biotinylated DDB1 (biotinDDB1) was purified by gel filtration chromatography and stored at −80°C (~20 μM).
Time-resolved fluorescence resonance energy transfer (TR-FRET)
Increasing concentrations of Alexa488-SpyCatcher-labelled26 His6-Spy-cycK/His6-CDK12 (Alexa488cycK-CDK12) were added to a mixture of biotinylated DDB1 (biotinDDB1) at 50 nM, terbium-coupled streptavidin at 4 nM (Invitrogen) and kinase inhibitors at 10 μM (final concentrations) in 384-well microplates (Greiner, 784076) in a buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 0.1% pluronic acid and 0.5% DMSO (see also figure legends). CR8 titrations were carried out by adding increasing concentrations CR8 (0–25 μM) into premixed 500 μM Alexa488cycK-CDK12, 50 nM biotinDDB1, and 4 nM terbium-coupled streptavidin. Before TR-FRET measurements, reactions were incubated for 15 minutes at room temperature. After excitation of terbium (Tb) fluorescence at 337 nm, emissions at 490 nm (Tb) and 520 nm (Alexa488) were measured with a 70 μs delay to reduce background fluorescence and the reactions were followed by recording 60 data points of each well over 1 hours using a PHERAstar FS microplate reader (BMG Labtech). The TR-FRET signal of each data point was extracted by calculating the 520:490 nm ratio. Data were analysed with Prism 7 (GraphPad) assuming equimolar binding of biotinDDB1 to Alexa488cycK-CDK12 using the equations described previously8.
Counter-titrations with unlabelled proteins were carried out by mixing 500 μM Alexa488cycK-CDK12 with 50 nM biotinDDB1 in the presence of 4 nM terbium-coupled streptavidin and 1 μM compound for DDB1 titrations or 12.5 μM compound for CDK12 titrations. After incubation for 15 minutes at room temperature, increasing amounts of unlabelled cycK-CDK12 or DDB1 (0–10 μM) were added to the preassembled Alexa488cycK-CDK12/biotinDDB1 complexes in a 1:1 volume ratio and incubated for 15 minutes at room temperature. TR-FRET data were acquired as described above. The 520/490 nm ratios were plotted to calculate the half maximal inhibitory concentrations (IC50) assuming a single binding site using Prism 7 (GraphPad). IC50 values were converted to the respective Ki values as described previously50. Three technical replicates were carried out per experiment.
DDB1-CUL4-RBX1 reconstitution and in vitro CUL4 neddylation
In vitro CRL4 reconstitution and CUL4 neddylation were performed as described11. His6-CUL4A/His6-RBX1 at 3.5 μM was incubated with His6-DDB1 at 3 μM in a reaction mixture containing 3.8 μM NEDD8, 50 nM NAE1/UBA3 (E1), 30 nM UBC12 (E2), 1 mM ATP, 50 mM Tris (pH 7.5), 100 mM NaCl, 2.5 mM MgCl2, 0.5 mM DTT and 5% (v/v) glycerol for 1.5 hours at room temperature. Neddylated and gel filtration-purified DDB1-CUL4-RBX1 (N8DDB1-CUL4-RBX1) was concentrated to 7.6 μM, flash frozen and stored at −80°C.
In-vitro ubiquitination assays
In vitro ubiquitination was performed by mixing N8DDB1-CUL4-RBX1 at 70 nM with a reaction mixture containing kinase inhibitors at 2 μM, CDK12-cycK at 500 nM, E1 (UBA1, BostonBiochem) at 50 nM, E2 (UBCH5a, BostonBiochem) at 1 μM, and ubiquitin at 20 μM. Reactions were carried out in 50 mM Tris (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 0.2 mM CaCl2, 1 mM ATP, 0.1% Triton X-100 and 0.1 mg/mL BSA, incubated for 0–30 minutes at 30°C and analysed by western blot using anti-cycK and anti-rabbit IgG antibodies. Blots were scanned on an Amersham 600 CCD-based imaging system (GE Life Sciences).
Isothermal titration calorimetry (ITC)
ITC experiments were performed at 25°C on a VP-ITC isothermal titration calorimeter (Microcal Inc.). Purified and TEV-cleaved CDK12-cycK and DDB1ΔBPB were exhaustively dialysed in 50 mM HEPES (pH 7.4), 150 mM NaCl, 0.25 mM TCEP, 0.5% DMSO and loaded into the sample cell at a final concentration of 10–50 μM. Kinase inhibitors (CR8 or roscovitine) were diluted from a 100 mM DMSO stock solution to 100–500 μM in buffer containing 50 mM HEPES (pH 7.4), 150 mM NaCl, 0.25 mM TCEP. The final DMSO concentration was 0.5%. Titrations with 100–500 μM compound were performed typically through about 30 injections of 6–10 μL at 210 second intervals from a 300 μL syringe rotating at 300 rpm. An initial injection of the ligand (4 μL) was made and discarded during data analysis. For probing DDB1-CDK12-cycK complex formation, DDB1ΔBPB (100 μM, in the syringe) was titrated into the cell containing CDK12-cycK (10 μM) or CDK12-cycK (10 μM) pre-incubated with CR8 (30 μM). The heat change accompanying the titration was recorded as differential power by the instrument and determined by integration of the peak obtained. Titrations of ligand to buffer only and buffer into protein were performed to allow baseline corrections. The heat change was fitted using nonlinear least-squares minimization to obtain the dissociation constants, Kd, the enthalpy of binding, ΔH, and the stoichiometry, n. Between one and three replicates were performed per titration.
Bioluminescence Resonance Energy Transfer (BRET) analyses
Bioluminescence resonance energy transfer (BRET) experiments were using a NanoBRET PPI starter kit (Promega N1821) according to the manufacturer’s instructions and as previously described51.
Drug sensitivity assays
HEK293TCas9 cells were resuspended at 0.15 × 106 per mL and plated on a 384 well plate with 50 μl per well and MLN4924, MLN7243 or MG132 with or without CR8 serially diluted with D300e Digital Dispenser (Tecan Inc.).
HEK293TCas9 cells (0.625 × 106 cells/6 well plate format) were seeded the day before transfection. The following day, 2.5 ug of pRSF91-GFP or pRSF91-CRBN9 plasmid DNA was mixed with 250 μl OptiMem and 7.5 μl TransIT-LT1 (Mirus Bio) according to manufacture protocol. 48 hours post transfection cells were resuspended at 0.15 × 106 cells /mL and plated on a 384 well plate with 50 μl per well.
HEK293TCas9 cells were transduced with sgRNAs targeting either DDB1 or Luciferase in pXPR003 backbone (GPP) (Supplementary Oligo Table 1). After nine days of puromycin selection, cells were re-plated into a 96-well format with 2 × 104 cells per well and CR8 and Roscovitine were serially diluted with D300e Digital Dispenser (Tecan Inc.).
After 3 days of drug exposure, cell viability was assessed using the CellTiter-Glo luminescent assay (Promega, #G7572) on an EnVision Multilabel Plate Reader (Perkin Elmer) or CLARIOstar Plus, MARS 3.4 (BMG LabTech). Cell viabilities were calculated relative to DMSO controls.
CycK stability reporter analysis with CRBN overexpression
HEK293TCas9 cells expressing the cycKeGFP degradation reporter were transiently transfected with pLX307-Luc or pLX307-CRBN (for flow experiment) as described above and 48 hours post infection treated with CR8 for 2 hours and analysed by flow cytometry.
Extended Data
Extended Data Figure 1 |. CR8-induced degradation of cycK correlates with DDB1 expression.
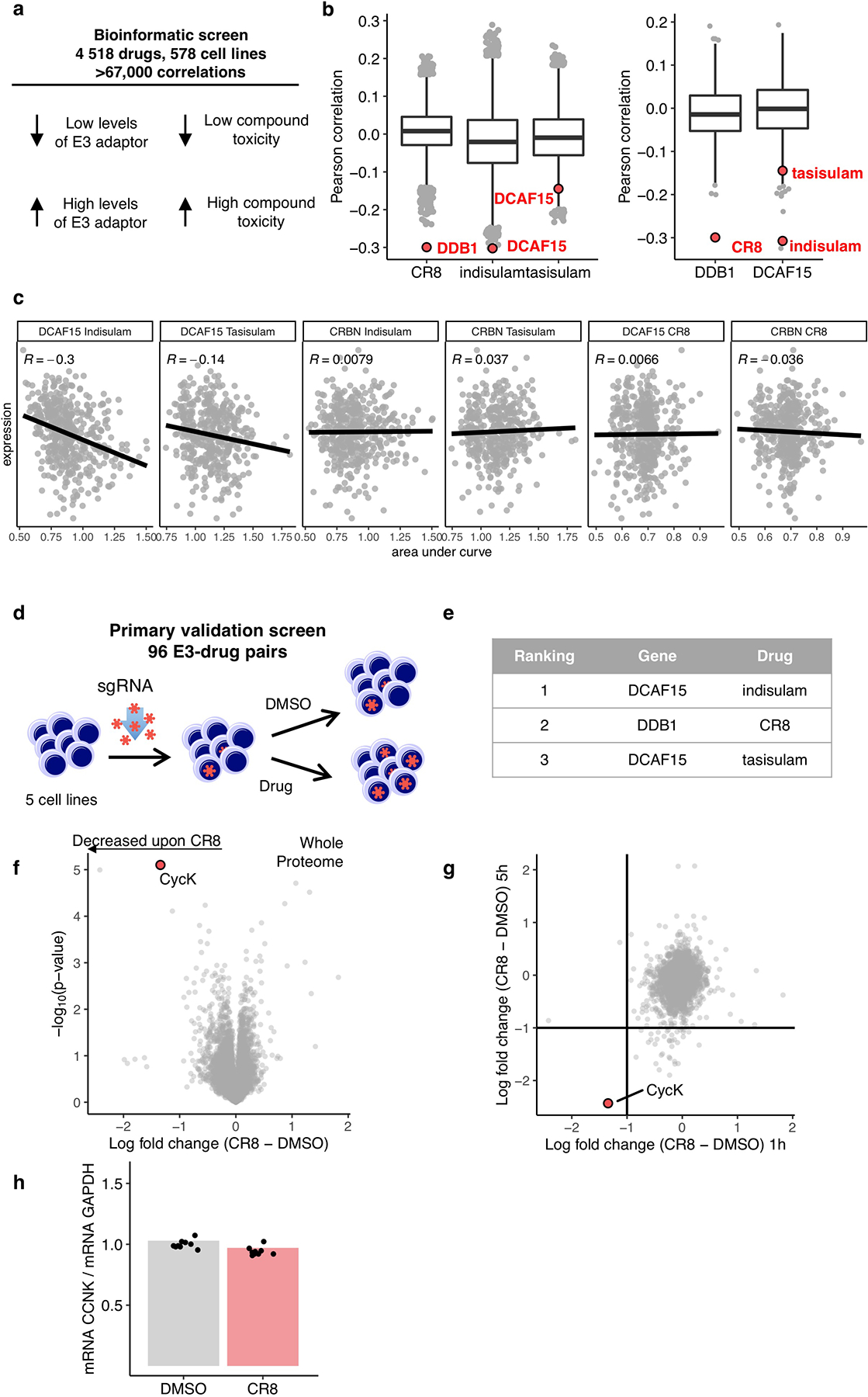
a, Schematic of bioinformatic screen for drug-E3 pairs. b, Box plot (centre, median; box, interquartile range (IQR); whiskers, 1.5 × IQR; outliers, points) for expression–sensitivity correlations (CR8 n=19110; indisulam, tasisulam n= 19109, DDB1, DCAF15 n=1618). c, Example Pearson correlation of selected drug-E3 pairs: positive controls (Indisulam-DCAF15; Tasisualm-DCAF15) and no correlation controls (others), (Indisulam n=452, Tasisulam n=418, CR8 n=471) d, Schematic of flow-based primary validation screen. e, Top three hits from the primary validation screen in 5 cell lines, performed according to the schematic in d. f, Whole proteome quantification of Molt-4 cells treated with 1 μM CR8 (n=1) or DMSO (n=3) for 1 hour (two-sided moderated t-test, n=3). g, The log2 fold changes in whole proteome quantification after 1 and 5 hours of exposure to CR8 plotted against each other. h, mRNA levels quantified by qPCR in HEK293TCas9 cells following 1 μM CR8 for 2 hours. Bars represent the mean (n=9).
Extended Data Figure 2 |. CDK12 is required for CR8-induced cycK degradation.
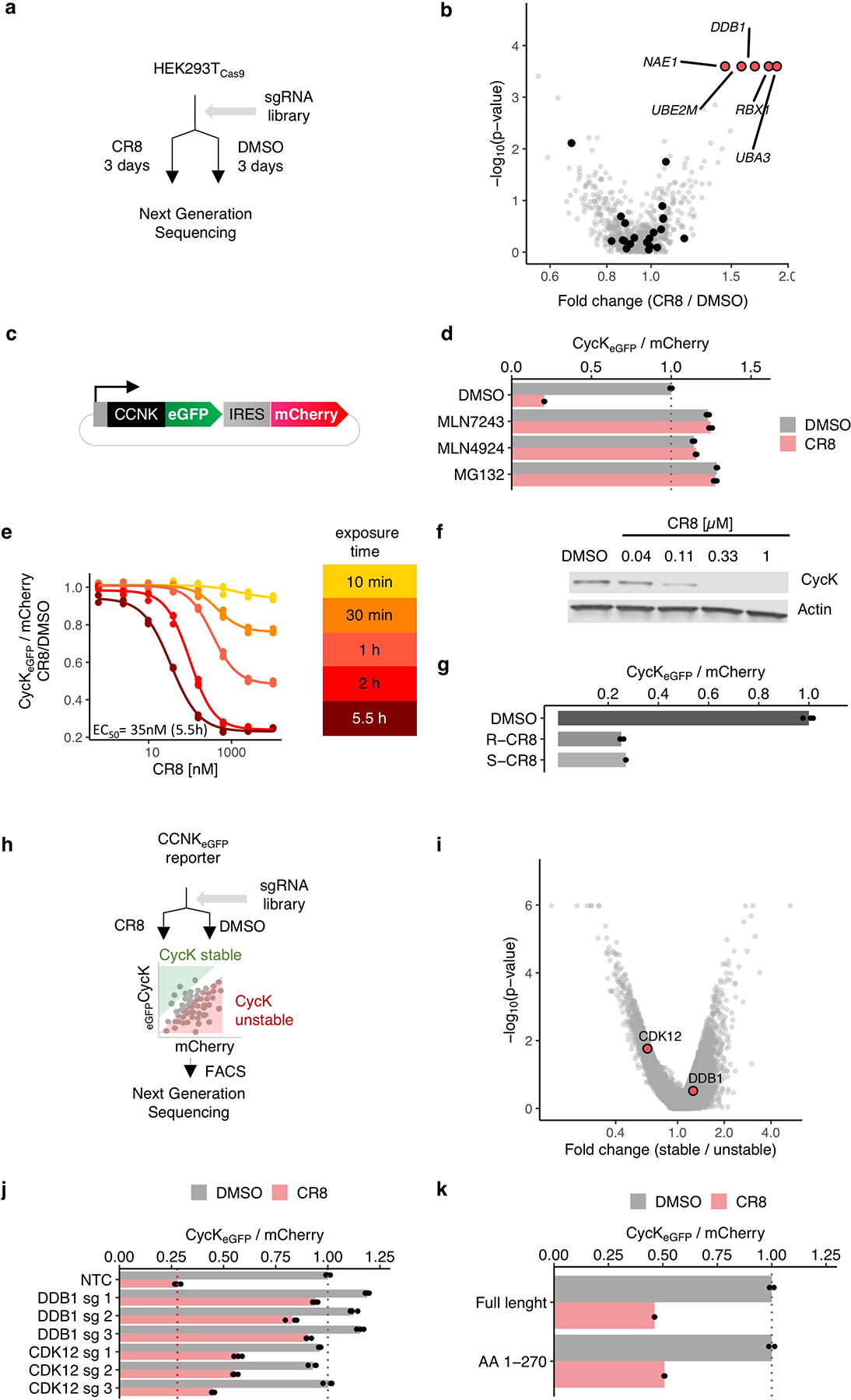
a, Schematic of the genome-wide CRISPR-Cas9 resistance screen. b, `BISON` CRISPR/Cas9 viability screen for CR8 resistance. Guide counts were collapsed to gene-level (n = 4 guides/gene; two-sided empirical rank-sum test-statistics). c, Schematic of the cycK (CCNK) stability reporter. eGFP, enhanced green fluorescent protein, IRES, internal ribosome entry site. d, Flow analysis of CycKeGFP degradation in HEK293TCas9 cells pre-treated with 0.5 μM MLN7243, 1 μM MLN4924, or 10 μM MG132 for 4 hours followed by exposure to 1 μM CR8 for 2 hours (n=3). e, Flow analysis of CycKeGFP degradation in HEK293TCas9 cells treated with CR8 (n=3). f, Immunoblots of CycK degradation in HEK293TCas9 cells treated with CR8 for 2 hours (n=2). g, Flow analysis of CycKeGFP degradation in HEK293TCas9 cells treated with 1μM compound for 2 hours (n=3). h, Schematic of the genome-wide CRISPR-Cas9 cycK stability reporter screen. i, Genome-wide CRISPR/Cas9 reporter screen for cycKeGFP stability with DMSO treatment in HEK293TCas9 cells. Guide counts were collapsed to gene-level (n = 4 guides/gene; two-sided empirical rank-sum test-statistics). j, Flow analysis of CycKeGFP degradation in HEK293TCas9 cells following 1μM CR8 for 2 hours (n=3). k, Flow analysis of CycKeGFPFull Length or CycKeGFPAA1−270 in HEK293TCas9 following 1 μM CR8 for 2 hours (n=3). Bars represent the mean in d, g, j and k.
Extended Data Figure 3 |. CR8-induced cycK degradation is not dependent on a canonical DCAF substrate receptor.
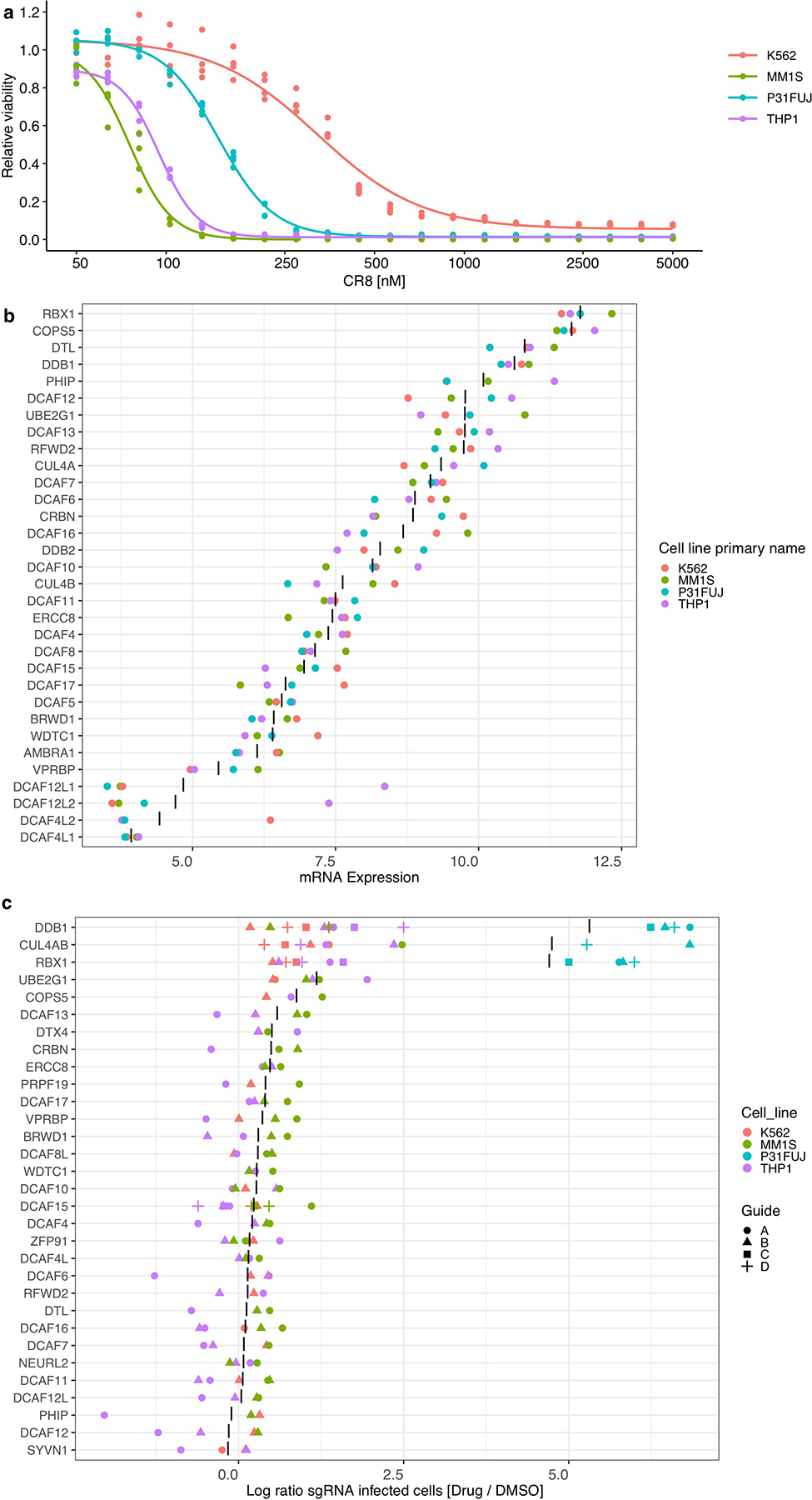
a, Drug sensitivity of K562Cas9, P31FUJCas9, THP1Cas9 and MM1SCas9 cells exposed to CR8 for 3 days (n=3). b, mRNA expression levels for genes in DCAF library. “|” represents mean (n=4). c, Flow analysis of K562Cas9, P31FUJCas9, THP1Cas9 and MM1SCas9 cells expressing sgRNAs and a BFP marker (blue fluorescent protein) after a 3-day treatment with 1 μM CR8. “|” represents mean (n>2).
Extended Data Figure 4 |. Characterization of DDB1-CDK12-cycK complex formation.
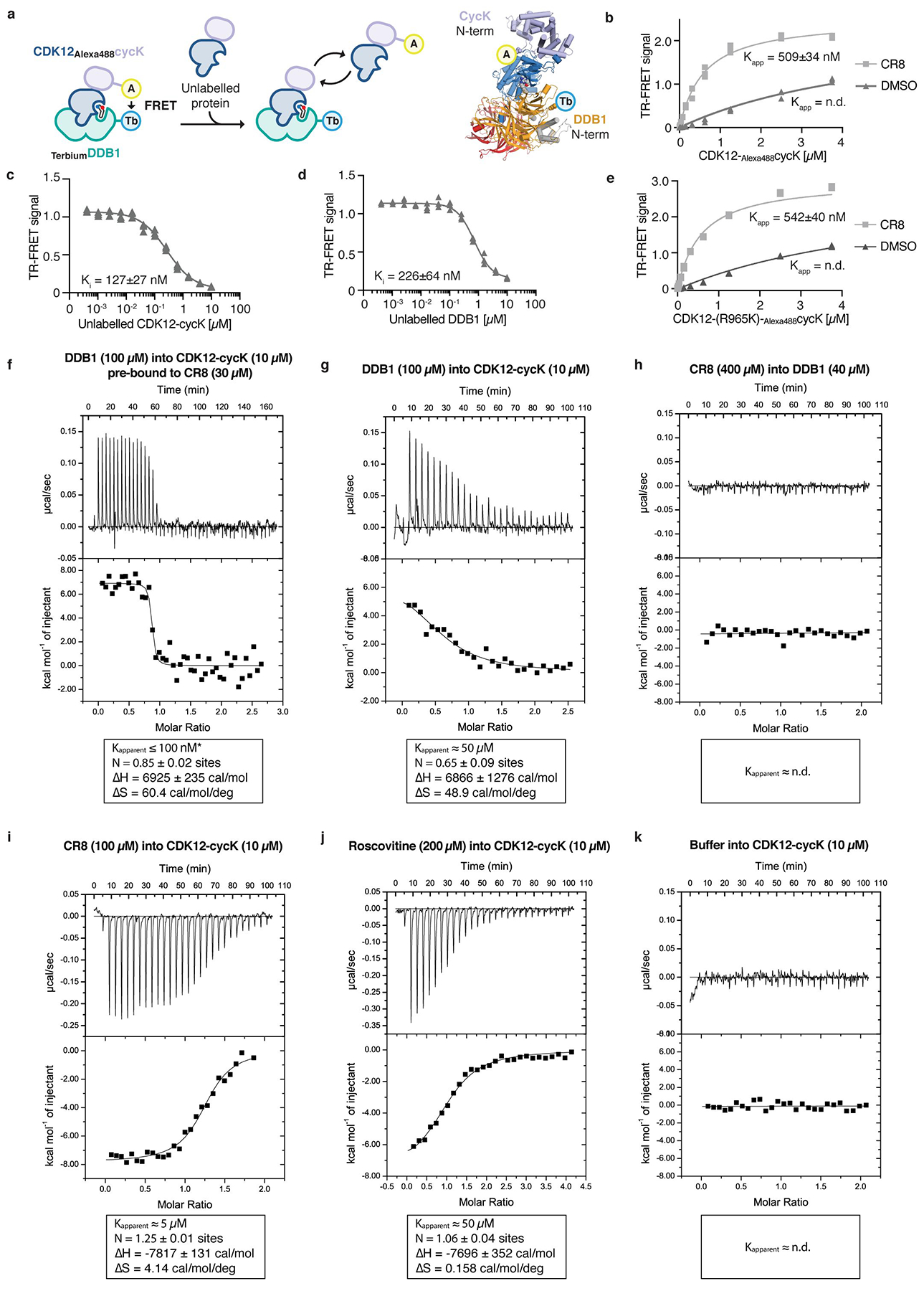
a, Schematic of the TR-FRET setup. Positions of the FRET donor (terbium-streptavidin (Tb)) and acceptor (Alexa488SpyCatcher (A)) are indicated in the structural model. b, Titration of CDK12-Alexa488cycK (0–3.75 μM) to 50 nM terbiumDDB1 and 5 μM CR8 or DMSO (n=3). c, Counter-titration of unlabelled wild-type CDK12-cycK (0–10 μM) to 50 nM terbiumDDB1, 500 nM CDK12-Alexa488cycK and 12.5 μM CR8 (n=3). d, Counter-titration of unlabelled wild-type DDB1 (0–10 μM) to 50 nM terbiumDDB1, 500 nM CDK12-Alexa488cycK and 1 μM CR8 (n=3). e, Titration of CDK12(R965K)-Alexa488cycK (wild-type sequence of canonical isoform of CDK12; 0–3.75 μM) to 50 nM terbiumDDB1 and 5 μM CR8 or DMSO (n=3). The CDK12 K965R variant, which was used throughout our in vitro studies (see Methods), shows a binding affinity indistinguishable from that of wild-type CDK12 (residue distal from the interface with DDB1 and cycK). f, Isothermal titration calorimetry (ITC) experiment (n=2, additional replicates for this and following panels are provided in Supplementary ITC Data). Specifications of the titration are given in the panel. Asterisk marking the approximate Kapparent value denotes that the binding affinity was too high to allow confident fitting of the binding curve. g, ITC experiment as in f (n=2). h, ITC experiment as in f (n=1). i, ITC experiment as in f (n=3). j, ITC experiment as in f (n=3). k, ITC experiment as in f (n=1).
Extended Data Figure 5 |. CDK12 contacts on DDB1 otherwise implicated in DCAF binding.
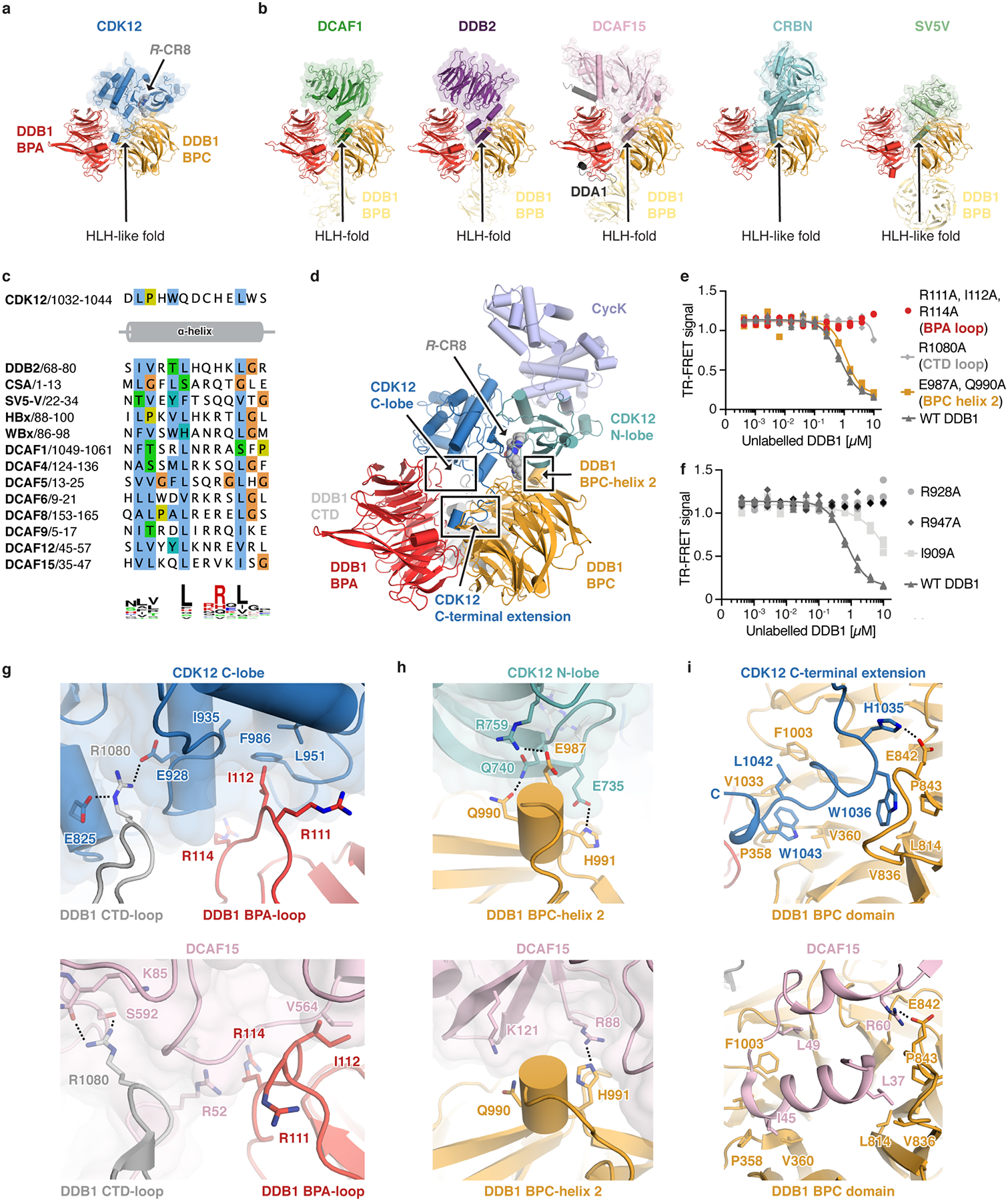
a, Structure of the CDK12-R-CR8-DDB1ΔBPB complex. The CDK12 C-terminal domain binds a cleft between the DDB1 BPA and BPC domains (arrow) and adopts an helix-loop-helix (HLH)-like fold. b, Diverse DCAFs bind DDB1 through HLH- or HLH-like folds. c, Sequence alignment of identically positioned helices of different HLH-domains. d, Overview of protein-protein interaction hotspots. e, Counter-titration of unlabelled wild-type or mutant DDB1 (0–10 μM) into pre-assembled terbiumDDB1-CR8-CDK12-Alexa488cycK complex (n=3). f, Counter titration of unlabelled wild-type or mutant DDB1 (0–10 μM) into pre-assembled terbiumDDB1-CR8-CDK12-Alexa488cycK complex (n=3). g-i, Close-up of DDB1 residues contacted by CDK12 (top) that are also otherwise involved in DCAF binding (bottom).
Extended Data Figure 6 |. CDK12 C-terminal extension adopts different conformations.
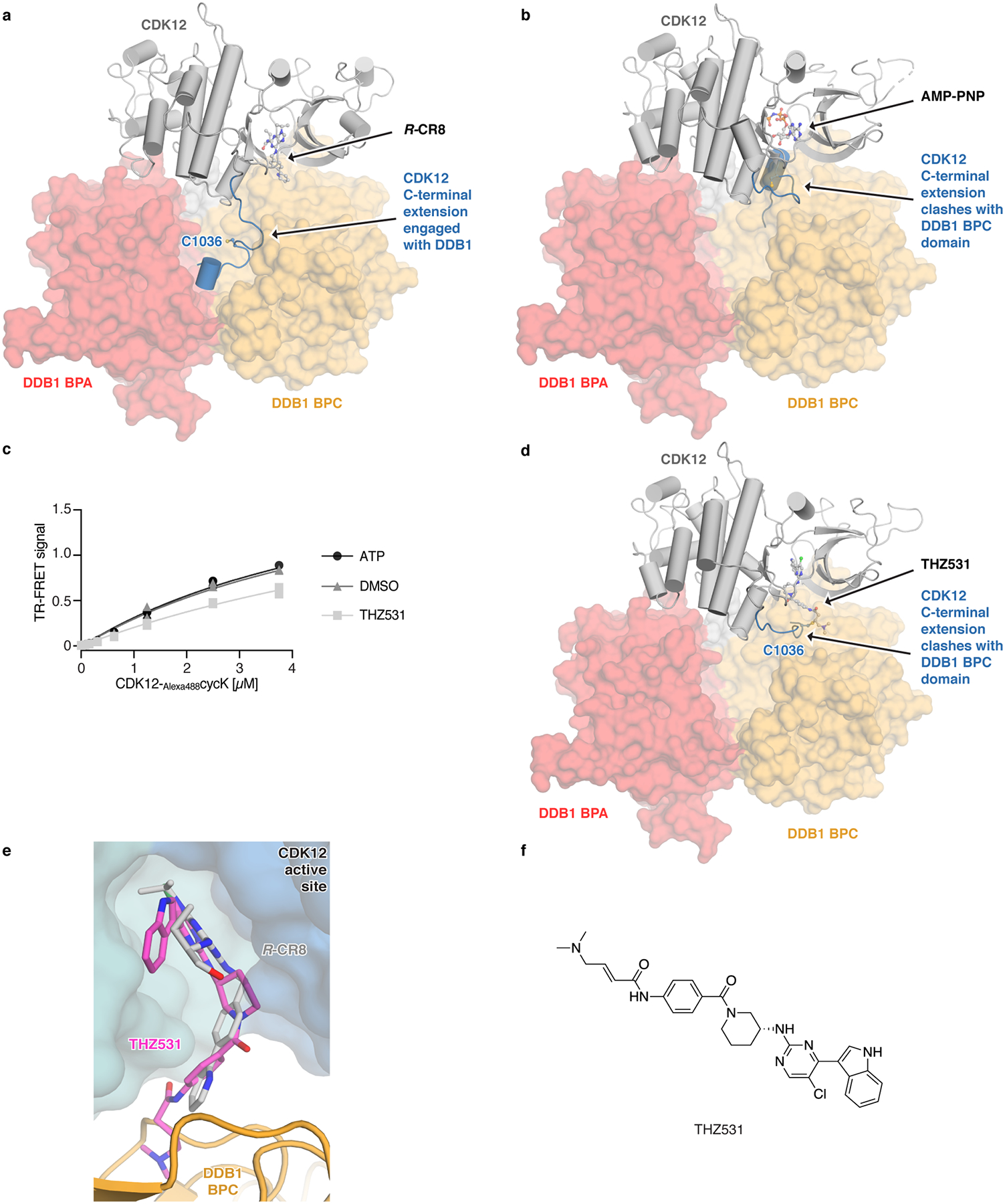
a, Conformation of the C-terminal extension in the structure of the CDK12-CR8-DDB1ΔBPB complex. b, Structure of CDK12 bound to AMP-PNP (PDB entry 4CXA) superimposed onto CDK12 in the CDK12-CR8-DDB1ΔBPB complex. c, Titration of CDK12-Alexa488cycK (0–3.75 μM) to 50 nM terbiumDDB1 in the presence of 5 μM THZ531, ATP or DMSO (n=3). d, Structure of CDK12 bound to THZ531 (PDB entry 5ACB) superimposed onto CDK12 in the CDK12-CR8-DDB1ΔBPB complex. e, THZ531 binding pose in the active site of CDK12 as in d. f, Chemical structure of THZ531.
Extended Data Figure 7 |. Differences between CDK12 and other CDKs highlight residues involved in CR8-induced DDB1 recruitment.
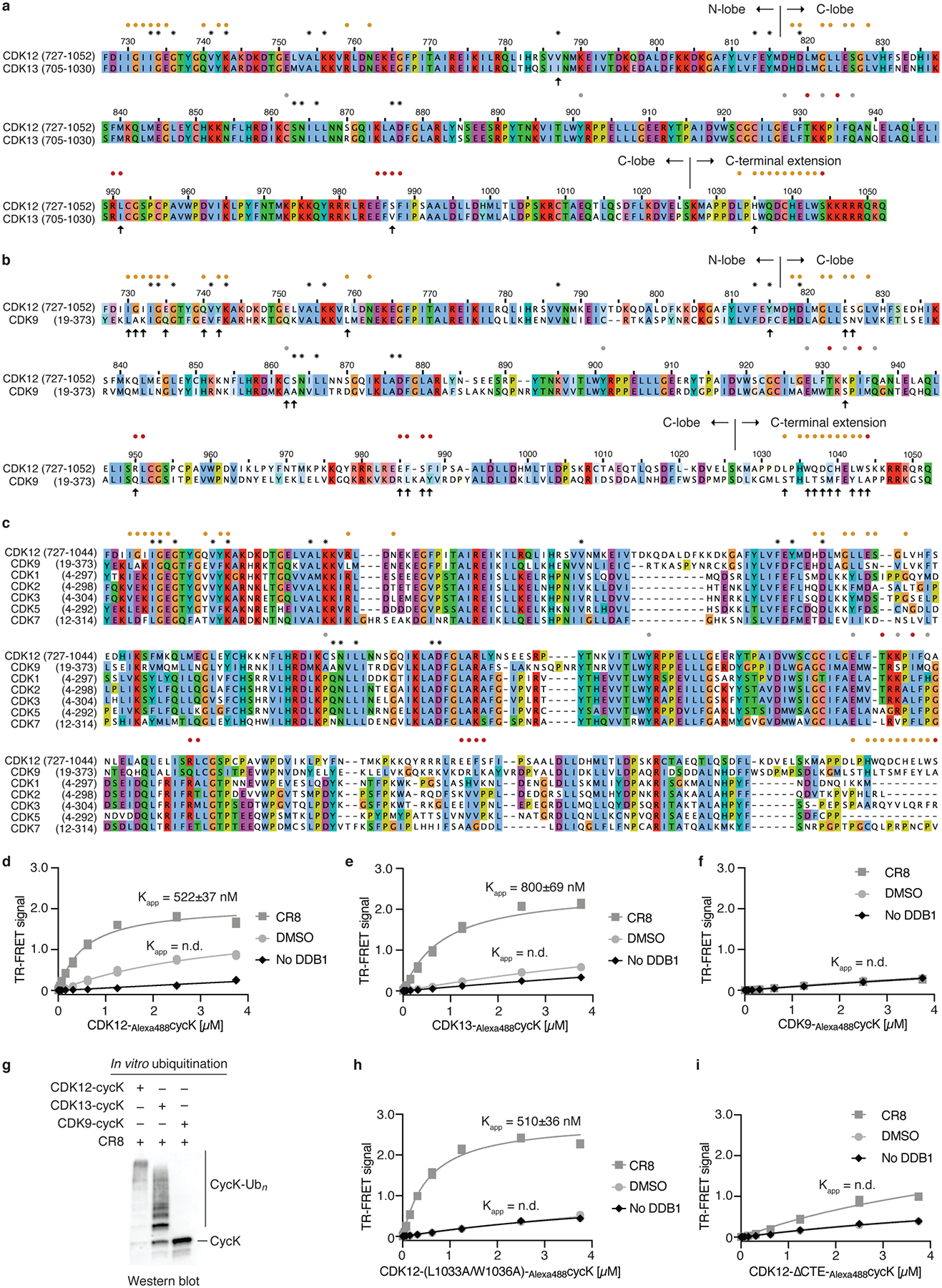
a, Sequence alignment of CDK12 and CDK13. In this and later panels asterisks denote contacts with CR8 and circles indicate contacts with DDB1 (coloured according to DDB1 domains, see Fig. 2). Arrows mark differences at the DDB1-CR8-CDK interface. b, Sequence alignment of CDK12 and CDK9. c, Multiple sequence alignment of different human CDKs. d, Titration of CDK12-Alexa488cycK (0–3.75 μM) to 50 nM terbiumDDB1 and 5 μM CR8 or DMSO (n=3). No DDB1 only contains streptavidin-terbium and shows concentration-dependent fluorophore effects. e, Titration of CDK13-Alexa488cycK (0–3.75 μM) to 50 nM terbiumDDB1 and 5 μM CR8 or DMSO (n=3). f, Titration of CDK9-Alexa488cycK (0–3.75 μM) to 50 nM terbiumDDB1 and 5 μM CR8 or DMSO (n=3). g, RBX1N8CUL4-DDB1 in vitro ubiquitination of cycK bound to CDK12, CDK13 or CDK9 (n=2). h, Titration of CDK12-Alexa488cycK (CDK12 mutant (L1033A, W1036A); 0–3.75 μM) to 50 nM terbiumDDB1 and 5 μM CR8 or DMSO (n=3). i, Titration of CDK12-Alexa488cycK (CDK12 tail truncation (713–1032); 0–3.75 μM) to 50 nM terbiumDDB1 and 5 μM CR8 or DMSO (n=3).
Extended Data Figure 8 |. CDK inhibitors block CR8-induced cycK degradation.
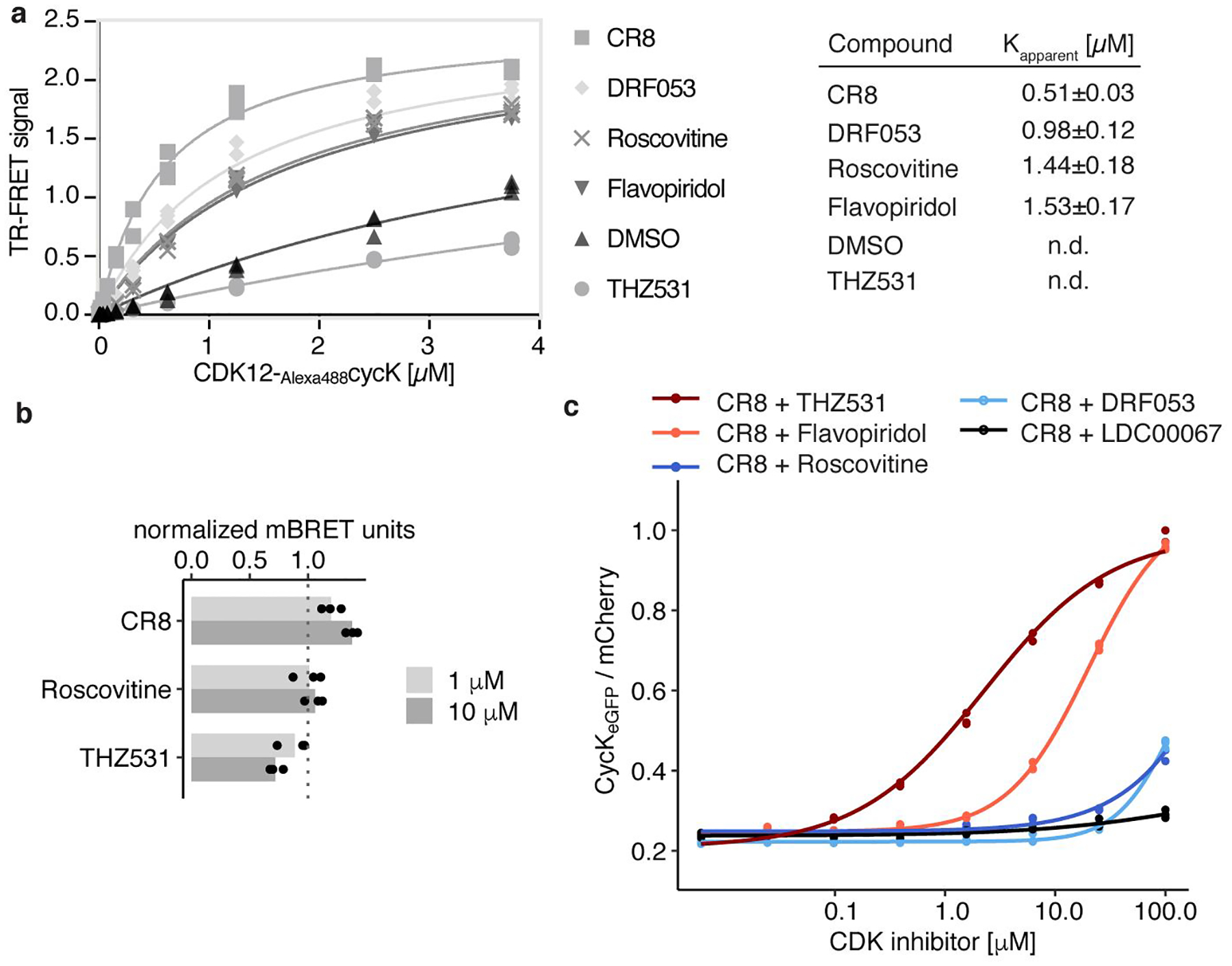
a, Titration of CDK12-Alexa488cycK into terbiumDDB1 in the presence of 10 μM compound (n=3). b, NanoBRET of HEK293T cells transfected with NanoLucCDK12713−1052 and HaloTagDDB1ΔBPB constructs and treated with compound for 2 hours. Bars represent the mean (n=3). c, Flow analysis of CycKeGFP degradation in HEK293TCas9 cells treated with 1 μM CR8 and competitive CDK inhibitor (n=3).
Extended Data Figure 9 |. Cytotoxicity of CR8 analogues does not depend on CRL4 components.
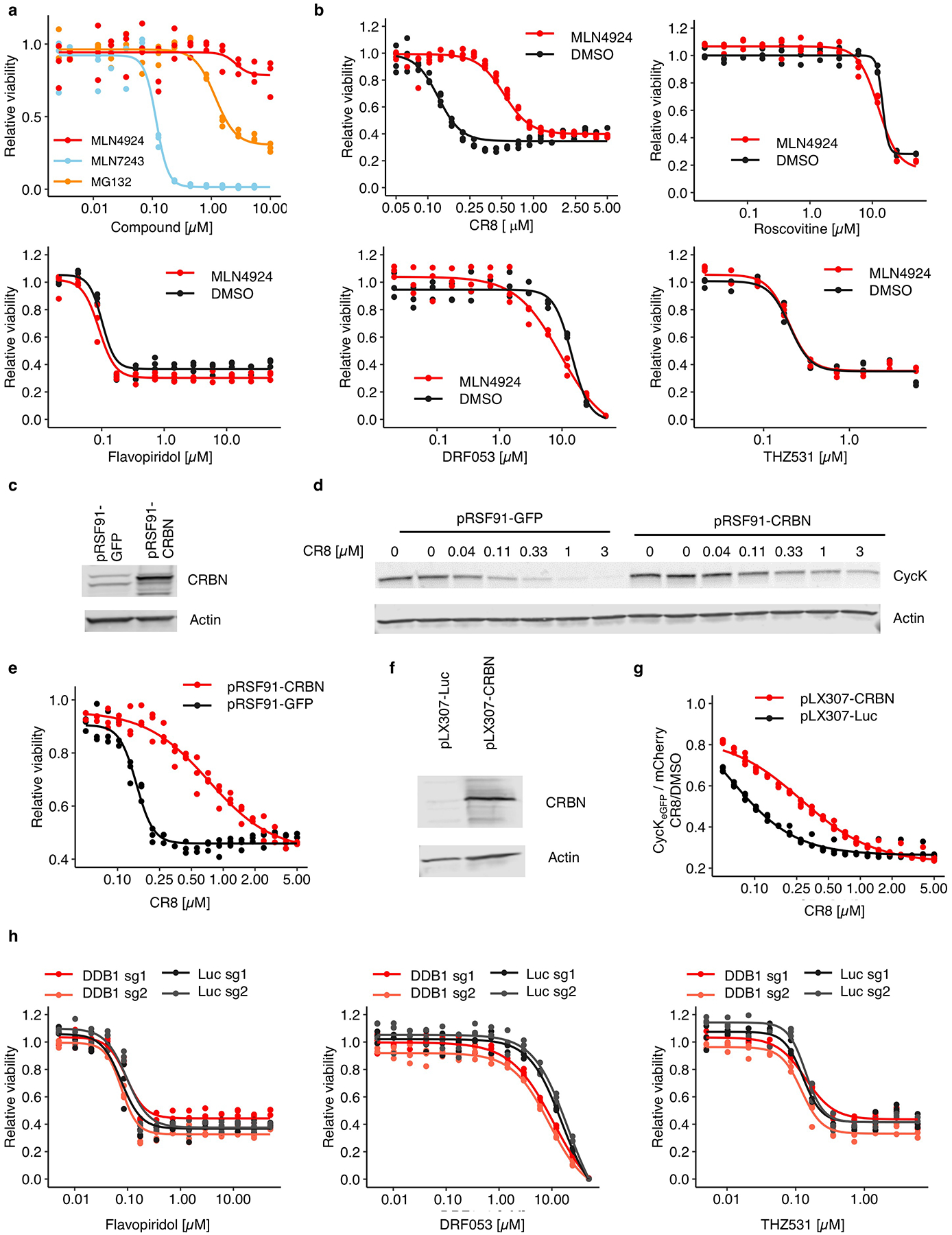
a, Drug sensitivity of HEK293TCas9 cells exposed to inhibitors for 3 days (n=3). b, Drug sensitivity of HEK293TCas9 cells exposed to 100 nM MLN4924 or DMSO in combination with the indicated compound for 3 days (n=3). c, Immunoblots of HEK293TCas9 cells transfected with control (pRSF91-GFP) or CRBN overexpression vectors (pRSF91-CRBN) (n=2). d, Immunoblots of HEK293TCas9 cells expressing pRSF91-GFP or pRSF91-CRBN and exposed to CR8 for 3 days (n=2). e, Drug sensitivity of HEK293TCas9 cells expressing pRSF91-GFP or pRSF91-CRBN and exposed to CR8 for 3 days (n=3). f, Immunoblots of HEK293TCas9 cells transfected with control (pLX307-Luc) or CRBN overexpression vectors (pLX307-CRBN) (n=2). g, Flow analysis of CycKeGFP degradation in HEK293TCas9 cells expressing pLX307-Luc or pLX307-CRBN and treated with CR8 for 2 hours (n=3). h, Drug sensitivity of HEK293TCas9 cells expressing sgRNAs targeting DDB1 or Luc and exposed to inhibitor for 3 days (n=3).
Extended Data Table 1 |.
Data collection and refinement statistics.
| DDBlΔBPB-CR8-CDK12713–1052-cycK1–267 | |
|---|---|
| Data collection* | |
| Space group | P3121 |
| Cell dimensions | |
| a, b, c (Å) | 250.75,250.75,217.92 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 54–3.46 (3.63–3.46) † |
| Rmeas | 0.318 (>4.00) |
| I / σI | 7.2 (0.9) |
| Completeness (%) | 95.1 (68.3)‡ |
| Redundancy | 12.0(11.6) |
| Refinement | |
| Resolution (Å) | 54–3.46 |
| No. reflections | 89,183 |
| Rwork / Rfree | 0.1934/0.220 |
| No. atoms | |
| Protein | 33,781 |
| Ligand/ion | 96 |
| Water | 0 |
| B-factors | |
| Protein | 59.9 |
| Ligand/ion | 39.6 |
| Water | n/a |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.01 |
Footnotes:
Data collected from a single crystal
Values in parentheses are for the highest-resolution shell
From STARANISO42 assuming a local weighted CC1/2 = 0.3 resolution cut-off
Supplementary Material
{kind=link}
Acknowledgements
We thank Simone Cavadini and Andreas Schenk for support during EM data collection and processing and Maxim Kolesnikov for help with ITC experiments. We acknowledge the Paul Scherrer Institute for provision of synchrotron radiation beam time at beamline PXI of the SLS and would like to thank Takashi Tomizaki for assistance. We thank the Broad Institute PRISM, particularly Mustafa Kocak, Compound Management, Cancer Data Science, Walk-up Sequencing, Genetic Perturbation Platform, and Flow Facility teams (particularly Patricia Rogers and Kat DeRuff). We thank James Kennedy for providing sgRNA.SFFV.tBFP and sgRNA.EFS.RFP657 backbones. We are grateful to all member of the Ebert group, particularly Roger Belizaire, Sebastian Koochaki, Peter Miller, Charles Zou, as well as Radosław Nowak and Peter Tsvetkov, for discussions.
This work was supported by the NIH (R01HL082945, P01CA108631, and P50CA206963), the Howard Hughes Medical Institute, the Edward P. Evans Foundation, and the Leukaemia and Lymphoma Society to B.L.E., funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program grant agreement no. 666068, from the Gebert Rüf Stiftung (GRS-057/14) and from the Novartis Research Foundation to N. H. T. as well as NIH grants NCI R01CA214608 and R01CA218278 and a Mark Foundation Emerging Leader Award to E.S.F. M.S. received funding from the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement no. 702642. Z.K. was supported by a European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement no. 765445. G.P. was supported by the Human Frontier Science Program (HFSP Long-Term Fellowship LT000210/2014) and the European Molecular Biology Organization (EMBO Advanced Fellowship aALTF 761-2016). A.S. was supported by a DF/HCC K12 grant, a Conquer Cancer Foundation Young Investigator Award and an award from the Wong Family Foundation. S.M.C. received funding from grants KL2 TR002542 and K08 CA230220.
Footnotes
Accession codes
Protein Data Bank
6TD3
PRIDE
PXD016187 and PXD016188
Data Availability
Structural data have been deposited in the PDB under the accession code 6TD3. Proteome quantification data are available in the PRIDE repository (PXD016187 and PXD016188).
Additional ITC data are shown in Supplementary Figure 1. Uncropped gel and Western blot source data are shown in Supplementary Figure 2. Flow cytometry gating strategy is displayed in Supplementary Figure 3.
Code Availability
Code necessary to reproduce statistical analysis is included in Supplementary Materials.
Competing interests
B.L.E. has received research funding from Celgene and Deerfield. He has received consulting fees from GRAIL, and he serves on the scientific advisory boards for and holds equity in Skyhawk Therapeutics and Exo Therapeutics. E.S.F. is a founder and/or member of the scientific advisory board (SAB), and equity holder of C4 Therapeutics and Civetta Therapeutics and a consultant to Novartis, AbbVie and Pfizer. N.H.T receives funding from the Novartis Research Foundation and is an SAB member of Monte Rosa Therapeutics. The Fischer lab receives or has received research funding from Novartis, Deerfield and Astellas. S.F. has had a consulting or advisory role, received honoraria, research funding, and/or travel/accommodation expenses funding from the following for-profit companies: Bayer, Roche, Amgen, Eli Lilly, PharmaMar, AstraZeneca, and Pfizer. R.B. is now an employee of Monte Rosa Therapeutics. S.M.C and T.R.G receive research funding from Bayer HealthCare. T.R.G. was formerly a consultant and equity holder in Foundation Medicine (acquired by Roche). T.R.G. also is a consultant to GlaxoSmithKline and is a founder of Sherlock Biosciences.
Supplementary Information is available for this paper.
Main References
- 1.Stanton BZ, Chory EJ & Crabtree GR Chemically induced proximity in biology and medicine. Science 359, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chopra R, Sadok A & Collins I A critical evaluation of the approaches to targeted protein degradation for drug discovery. Drug Discov. Today Technol 31, 5–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu C et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol 34, 419–423 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corsello SM et al. Non-oncology drugs are a source of previously unappreciated anti-cancer activity. Biorxiv (2019). doi: 10.1101/730119 [DOI] [Google Scholar]
- 5.Ghandi M et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bettayeb K et al. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 27, 5797–5807 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Fink EC & Ebert BL The novel mechanism of lenalidomide activity. Blood 126, 2366–2369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu G et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science 343, 305–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krönke J et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matyskiela ME et al. A novel cereblon modulator recruits GSPT1 to the CRL4 CRBN ubiquitin ligase. Nature 535, 252–257 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Petzold G, Fischer ES & Thomä NH Structural basis of lenalidomide-induced CK1α degradation by the CRL4 CRBN ubiquitin ligase. Nature 532, 127–130 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Fischer ES et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han T et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356, 3755 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Uehara T et al. Selective degradation of splicing factor CAPERα by anticancer sulfonamides. Nat. Chem. Biol 13, 675–680 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Faust TB et al. Structural complementarity facilitates E7820-mediated degradation of RBM39 by DCAF15. Nat. Chem. Biol (2019). doi: 10.1038/s41589-019-0378-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bondeson DP et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol 11, 611–617 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Angers S et al. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590–593 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Lee J & Zhou P DCAFs, the Missing Link of the CUL4-DDB1 Ubiquitin Ligase. Mol. Cell 26, 775–780 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Bösken CA et al. The structure and substrate specificity of human Cdk12/Cyclin K. Nat. Commun 5, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng S-WG et al. Interaction of Cyclin-Dependent Kinase 12/CrkRS with Cyclin K1 Is Required for the Phosphorylation of the C-Terminal Domain of RNA Polymerase II. Mol. Cell. Biol 32, 4691–4704 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dixon-Clarke SE, Elkins JM, Cheng SWG, Morin GB & Bullock AN Structures of the CDK12/CycK complex with AMP-PNP reveal a flexible C-terminal kinase extension important for ATP binding. Sci. Rep 5, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oumata N et al. Roscovitine-Derived, Dual-Specificity Inhibitors of Cyclin-Dependent Kinases and Casein Kinases 1. J. Med. Chem 51, 5229–5242 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Meijer L et al. Biochemical and Cellular Effects of Roscovitine, a Potent and Selective Inhibitor of the Cyclin-Dependent Kinases cdc2, cdk2 and cdk5. Eur. J. Biochem 243, 527–536 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Zhang T et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol 12, 876–884 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sedlacek HH et al. Flavopiridol (L86 8275; NSC 649890), a new kinase inhibitor for tumor therapy. Int. J. Oncol 9, 1143–1168 (1996). [DOI] [PubMed] [Google Scholar]
- 26.Sievers QL et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones LH Small-Molecule Kinase Downregulators. Cell Chem. Biol 25, 30–35 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Schreiber SL A chemical biology view of bioactive small molecules and a binder-based approach to connect biology to precision medicines. Israel Journal of Chemistry 59, 52–59 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ito T et al. Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Fischer ES et al. The molecular basis of CRL4DDB2/CSAubiquitin ligase architecture, targeting, and activation. Cell 147, 1024–1039 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Johnson SF et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep (2016). doi: 10.1016/j.celrep.2016.10.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dykes GW, Crepeau RH & Edelstein SJ Three-dimensional reconstruction of the 14-filament fibers of hemoglobin S. J. Mol. Biol 130, 451–72 (1979). [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Seisdedos H, Empereur-Mot C, Elad N & Levy ED Proteins evolve on the edge of supramolecular self-assembly. Nature 548, 244–247 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Sievers QL, Gasser JA, Cowley GS, Fischer ES & Ebert BL Genome-wide screen identifies cullin-RING ligase machinery required for lenalidomide-dependent CRL4CRBN activity. Blood 132, 1293–1303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Donovan KA et al. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane radial ray syndrome. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995). [Google Scholar]
- 39.Abdulrahman W et al. A set of baculovirus transfer vectors for screening of affinity tags and parallel expression strategies. Anal. Biochem 385, 383–5 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Li T, Pavletich NP, Schulman BA & Zheng N High-level expression and purification of recombinant SCF ubiquitin ligases. Methods in Enzymology 398, 125–142 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr 67, 235–42 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tickle IJ, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G STARANISO. (2018).
- 43.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Afonine PV et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr 68, 352–67 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bricogne GBE BUSTER. (2011).
- 47.Moriarty NW, Grosse-Kunstleve RW & Adams PD electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D. Biol. Crystallogr 65, 1074–80 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen VB et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krissinel E & Henrick K Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Cer RZ, Mudunuri U, Stephens R & Lebeda FJ IC50-to-Ki: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res 37, 441–5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sperling AS et al. Patterns of substrate affinity, competition, and degradation kinetics underlie biological activity of thalidomide analogs. Blood 134, 160–170 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.