

Abstract
Background
The links between microbial environmental exposures and asthma are well documented, but no study has combined deep sequencing results from pulmonary and indoor microbiomes of patients with asthma with spirometry, clinical, and endotype parameters.
Objective
The goal of this study was to investigate the links between indoor microbial exposures and pulmonary microbial communities and to document the role of microbial exposures on inflammatory and clinical outcomes of patients with severe asthma (SA).
Methods
A total of 55 patients with SA from the national Cohort of Bronchial Obstruction and Asthma cohort were enrolled for analyzing their indoor microbial flora through the use of electrostatic dust collectors (EDCs). Among these patients, 22 were able to produce sputum during “stable” or pulmonary “exacerbation” periods and had complete pairs of EDC and sputum samples, both collected and analyzed. We used amplicon targeted metagenomics to compare microbial communities from EDC and sputum samples of patients according to type 2 (T2)-asthma endotypes.
Results
Compared with patients with T2-low SA, patients with T2-high SA exhibited an increase in bacterial α-diversity and a decrease in fungal α-diversity of their indoor microbial florae, the latter being significantly correlated with fraction of exhaled nitric oxide levels. The β-diversity of the EDC mycobiome clustered significantly according to T2 endotypes. Moreover, the proportion of fungal taxa in common between the sputum and EDC samples was significantly higher when patients exhibited acute exacerbation.
Conclusion
These results illustrated, for the first time, a potential association between the indoor mycobiome and clinical features of patients with SA, which should renew interest in deciphering the interactions between indoor environment, fungi, and host in asthma.
Key words: Indoor environment, mycobiome, microbiome, severe asthma, Feno, type 2 asthma
Abbreviations used: COBRA, Cohort of Bronchial Obstruction and Asthma; EDC, Electrostatic dust collector; Feno, Fraction of exhaled nitric oxide; ICS, Inhaled corticosteroid; ITS2, Internal transcribed spacer 2; LefSe, Linear discriminant analysis effect size; NGS, Next-generation sequencing; OTU, Operational taxonomic unit; qPCR, Quantitative PCR; SA, Severe asthma; T2, Type 2; V3-V4, Variable 3–variable 4 of the 16S rRNA encoding gene
Graphical abstract

Asthma is a complex, chronic inflammatory disease of the airways that affects 339 million people worldwide.1 , 2 Despite recent advances in new treatments and notable efforts to elucidate asthma pathophysiology,2 patients with severe asthma (SA) remain at high risk for complications, exacerbations, poor quality of life, and increased mortality and morbidity.1 , 2 According to the definition of SA proposed by the international European Respiratory Society/ American Thoracic Society Task Force,3 SA is still recognized as a major unmet need, having an overall prevalence estimated at 5% to 10%1, 2, 3 and accounting for approximately 50% of asthma-associated health care costs.1
Similar to nonsevere asthma, SA is currently accepted as a heterogeneous disease comprising multiple endotypes that combine clinical characteristics with several identifiable mechanistic pathways.3 Developments in biologic medications to treat asthma have allowed for targeting of specific endotypes, such as allergic asthma, eosinophilic asthma, and more recently, type 2 (T2)-high asthma. It is now standard practice to differentiate patients with SA in terms of having T2-high or T2-low asthma based on the levels of both fraction of exhaled nitric oxide (Feno) and blood eosinophil count.4
SA is also associated with microbial lung dysbiosis, which activates the inflammasome and other induced pathways.5, 6, 7 In agreement with the hygiene hypothesis, recent findings have demonstrated that exposure to environmental microbes significantly decreases the incidence of wheezing illnesses in young children with a genetic susceptibility at chromosome 17q21.8 Moreover, the ubiquitous distribution of fungal spores leads to their regular inhalation as an inevitable consequence of breathing.9 , 10 Exposure to environmental microbes, especially the fungal indoor environment, is associated with a wide range of adverse health effects, including asthma, and it can influence asthma severity.8 , 11, 12, 13, 14, 15
In the present pilot study, our goal was to evaluate the relationships between bacteria and fungi in sputum samples and dust particles from patients with T2-high and T2-low SA. To this end, we examined whether the indoor fungal and bacterial flora (exogenous mycobiome and microbiome, respectively) are associated with clinical parameters in patients with SA and how the exogenous and endogenous mycobiomes and/or microbiomes are connected.
Methods
Patients
A total of 55 patients from the National COhort of BRonchial obstruction and Asthma (COBRA [ethics committee approval no. 2008-A00294-51/1])16 who had SA based on the European Respiratory Society/American Thoracic Society guidelines and were followed at the University Hospital of Bordeaux were included in the COBRA-ENV (Indoor environment analysis of COBRA cohort) study. The COBRA-ENV protocol (no. 2017/68) was approved by the ethics committee of CPP du Sud-Ouest et Outre Mer III.
Demographic features, clinical characteristics, lung function, and induced sputum test results were determined at COBRA visits as previously described.16 Patients with T2-high SA were defined according to either Feno level (>25 ppb) or blood eosinophil count (>300 cells/mm3).7 In contrast, patients with T2-low SA were defined on the basis of an Feno level lower than 25 ppb and a blood eosinophil count lower than 300 cell/mm3.
Sputum and indoor dust samples
Induced sputa were successfully collected from 22 patients, as previously described.17 Indoor dust samples were collected by using an electrostatic dust collector (EDC). Briefly, each EDC consisted of a 20 × 17-cm textile surface used to catch fungi and bacteria during the exposure period.18 This textile device was mounted in a plastic folder that was left open for 10 weeks in a horizontal position with the textile exposed to the room air such that the settling dust was passively collected.18 , 19 With use of the same protocol, EDCs were placed in patient bedrooms, opened on the same date (ie, December 21, 2017) and exposed for the same duration (ie, 10 weeks), as described previously.19
DNA extraction, library preparation, deep sequencing, and taxonomic assignation
DNA was extracted from the EDC and sputum samples by using a DNeasy PowerSoil Kit (Qiagen, Les Ulis, France). Negative extraction controls (250 μL of DNA-free water, as an extraction blank) were processed using the same protocol. The microbial diversity and taxonomic composition of samples were assessed by using the variable 3–variable 4 of the 16S rRNA encoding gene (V3-V4) region of the bacterial 16S rRNA gene and the internal transcribed spacer 2 (ITS2) region of the fungal rDNA, with adherence to the optimized and standardized library preparation protocols from Metabiote (Genoscreen, Lille, France).20 The respective primers used to amplify the V3-V4 and ITS2 loci were as follows: 16S-forward, TACGGRAGGCAGCAG; 16S-reverse, CTACCNGGGTATCTAAT; ITS2-forward, GTGARTCATCGAATCTTT; and ITS2-reverse, GATATGCTTAAGTTCAGCGGGT. In addition to the extraction blanks, 2 negatives controls (1 library blank and 1 unexposed EDC, but analyzed following the same process as the exposed EDCs) and 2 positive controls (artificial bacterial and fungal communities) were used to valid the experimental procedures. Briefly, PCR amplification was performed by using barcoded primers (at a final concentration of 0.2 μM), with an annealing temperature of 50°C for 30 cycles. PCR products were purified by using magnetic beads, quantified according to the protocol provided by GenoScreen, and mixed in equimolar amounts. Next-generation sequencing (NGS) was performed by using 250-bp paired-end technology on the MiSeq platform (Illumina, San Diego, Calif) at GenoScreen (GenoScreen, Lille, France).
The resulting raw sequences were subjected to a cleaning process as follows: (1) sorting of the sequences according to the indexes and the 16S and ITS2 primers using CutAdapt, with no mismatch allowed within the primers sequences; (2) quality filtration using the PRINSEQ-lite PERL script by truncating bases at the 3' end with Phred quality scores less than 30; and (3) generation of a paired-end read assembly using FLASH with a minimum overlap of 30 bases and greater than 97% identity. The bioinformatic analysis was performed on a fully automated (Metabiote OnLine, version 2.0) pipeline using QIIME, version 1.9.1, software.21 Following the preprocessing steps, the full-length 16S and ITS2 amplicons were assessed for chimeric sequences by using an in-house method based on Usearch 6.1. Then, a clustering step was performed to group similar sequences with a defined nucleic identity threshold (97% identity for an affiliation at least at the genus level for the V3-V4 locus and at the species level for the ITS2 locus) by using Uclust version 1.2.22q22 through an open-reference operational taxonomic unit (OTU) picking process and a complete-linkage method to generate groups of sequences or OTUs. An OTU cleaning step was performed to eliminate singletons. For each OTU, the most abundant sequence was used as a reference sequence and was taxonomically compared with reference databases, including the Greengenes database (release 13_8) for bacterial OTUs and the Unite database (release 7.2) for fungal OTUs using the RDP classifier method, version 2.2.23 A final table with all of the identified OTUs was generated; it comprised the number of sequences for each sample related to each OTU and their respective taxonomic affiliation. α-Diversity metrics within samples (Chao1, Simpson, and Shannon indexes) were generated from this OTU table by using QIIME version 1. The sequence information has been deposited in the National Center for Biotechnology Information Sequence Read Archive under the accession number PRJNA635002.
Estimation of the EDC fungal load using a qPCR assay
The fungal load for each EDC was estimated with a universal quantitative PCR (qPCR) assay that quantifies rDNA (in pg per EDC). Briefly, previously published primers and probes,24 Taqman Gene Expression Master Mix (Applied Biosystems, Life Technologies, Carlsbad, Calif) and a 7900HT Sequence Detection System were used to perform qPCR. The protocol is detailed in the Methods section of the Online Repository (at www.jacionline.org).
Data analysis
The R programming environment and packages were used for statistical analyses. Nonparametric tests (the Mann-Whitney test for quantitative variables) and the Fisher exact test (for qualitative variables) were used when appropriate. Linear relationships between Feno levels and Chao1 fungal diversity were estimated by using the Pearson correlation coefficient. Differences were considered statistically significant at P values less than .05.
Both α-diversity and β-diversity were determined to identify changes in microbial community structures between EDCs and sputa, according to the clinical data. The principal coordinates analysis (after a Bray-Curtis distance analysis) method was used to visualize overall microbial differences among groups. The microbial core was defined by using the Venn diagram function in the RAM package.
To compare α-diversity estimates across patients' clinical parameters, Mann-Whitney tests were performed; Wilcoxon tests were used when sputum and EDC pairs were compared. After a cumulative sum scaling normalization, sample clustering hypotheses were tested by using permutational multivariate ANOVA, with a dispersion among groups assessed by using the Betadisper function to complete the β-diversity analysis. Finally, differentially abundant OTUs between groups were identified by using linear discriminant analysis effect size (LEfSe) analysis, as previously described.25 , 26 According to Segata et al,25 we applied LEfSe analysis with the default parameters (the α value for Wilcoxon tests was set at 0.05, the logarithmic linear discriminant analysis score threshold was set at 2.0, and bootstrapping was performed over 30 cycles) to identify indoor taxonomic biomarkers that characterize the differences between patients with normal and elevated Feno levels by using T2 endotypes as covariables.
Additional details of patient characteristics and sampling, as well as methodologic procedures are provided in the Methods section of the Online Repository.
Results
Study population
A total of 55 patients with SA were enrolled, but only 22 patients with SA had complete pairs of EDC and sputum samples collected and analyzed (see Fig E1 in this article's Online Repository at www.jacionline.org). Patient characteristics from the T2-high and T2-low groups are summarized in Table I . Except for Feno levels, the characteristics of patients with T2-high SA were not significantly different from those of patients with T2-low SA. Similarly, sputum microbial cultures and indoor fungal loads measured by qPCR were not significantly different between the 2 SA groups (Table I).
Fig E1.
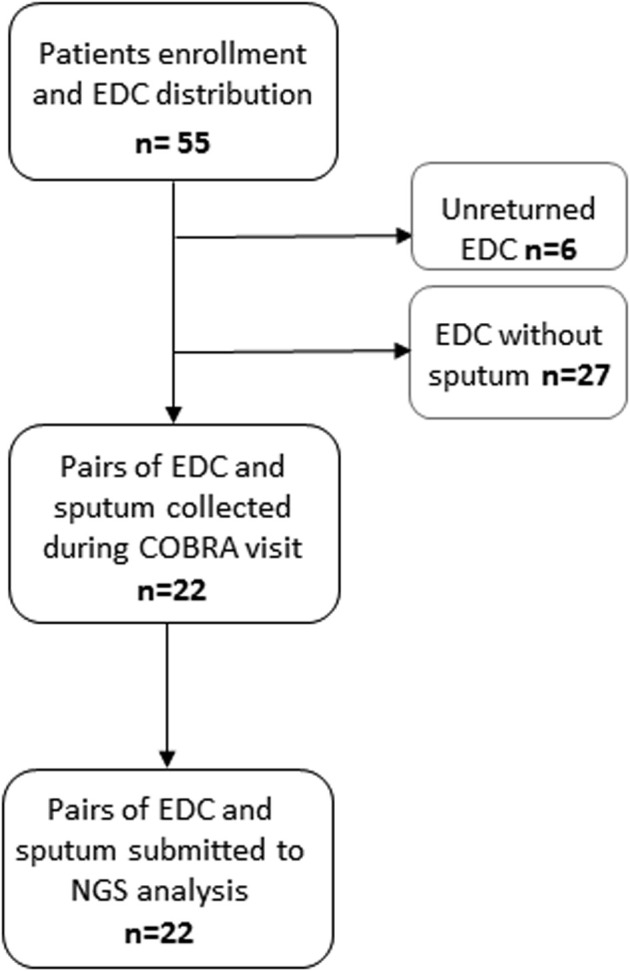
Flowchart of the COBRA-ENV (indoor environment analysis of COhort of BRonchial obstruction and Asthma) study and samples distribution across time.
Table I.
Clinical and biologic characteristics of patients with SA and complete NGS analysis
| Variables | Overall (n = 22) | Type 2–low SA (n = 6) | Type 2–high SA (n = 16) | P values |
|---|---|---|---|---|
| Demographic features | ||||
| Age (y) | 59.09 (16.84) | 52.33 (19.69) | 61.62 (15.58) | .259∗ |
| Male sex, no. (%) | 10 (45.5) | 2 (33.3) | 8 (50.0) | .827 |
| BMI (kg/m2), mean (SD) | 26.96 (4.73) | 27.31 (2.98) | 26.83 (5.32) | .838 |
| Respiratory data, mean (SD) | ||||
| FEV1 (% predicted) | 75.36 (27.60) | 79.32 (33.83) | 73.77 (25.89) | .689 |
| FVC (% predicted) | 91.20 (21.99) | 89.18 (28.75) | 92.01 (19.83) | .798 |
| FEV1/FVC (% predicted) | 68.25 (17.04) | 74.84 (15.82) | 65.62 (17.31) | .274 |
| FEF25-75 (% predicted) | 49.24 (31.30) | 61.83 (36.35) | 44.21 (28.85) | .254 |
| Feno (ppb) | 43.32 (32.74) | 15.86 (6.26) | 53.62 (32.75) | .012 |
| Clinical features at COBRA visit (no.) | ||||
| Very poorly controlled asthma† | 13 | 4 | 9 | .788 |
| SAFS | 10 | 3 | 7 | 1.000 |
| ABPA | 3 | 0 | 3 | .657 |
| Asthma medication used during the last 6 mo† | ||||
| ICS | 22 | 6 | 16 | NA‡ |
| LABA | 22 | 6 | 16 | NA |
| LAMA | 16 | 6 | 10 | .222 |
| Continuous OCS | 9 | 3 | 6 | .965 |
| Biotherapy | 9 | 3 | 6 | .965 |
| Gastroesophageal reflux treatment | 8 | 3 | 5 | .752 |
| Azole treatment | 1 | 0 | 1 (VRC) | 1.000 |
| Exacerbations during COBRA-ENV period, mean (SD), no (%) | 6.45 (5.29) | 9.67 (5.32) | 5.25 (4.91) | .081 |
| OCS courses during COBRA-ENV period, mean (SD), no (%) | 5.45 (5.09) | 8.50 (4.89) | 4.31 (4.81) | .085 |
| ATB courses during COBRA-ENV period, mean (SD), no (%) | 4.68 (4.87) | 6.83 (3.87) | 3.88 (5.07) | .213 |
| Biologic data at COBRA visit | ||||
| Blood eosinophil count (/mm3), mean (SD) | 526.82 (1184.46) | 12.00 (90.77) | 679.38 (1367.61) | .336 |
| Total serum IgE (kUI/L), mean (SD) | 308.23 (264.80) | 236.67 (268.59) | 335.06 (267.03) | .451 |
| Sputum microbial cultures (no.) | 19 | 6 | 13 | .657 |
| Pseudomonas aeruginosa | 1 | 1 | 0 | .601 |
| Escherichia coli | 1 | 1 | 0 | .601 |
| Haemophilus influenzae, parainfluenzae | 2 | 1 | 1 | 1.000 |
| Non-pneumoniae Streptococcus | 2 | 1 | 1 | 1.000 |
| Candida albicans | 14 | 6 | 8 | .094 |
| Other yeast | 5 | 1 | 4 | 1.000 |
| Aspergillus fumigatus | 5 | 3 | 2 | .194 |
| Other Aspergillus | 3 | 2 | 1 | .342 |
| Other mold | 3 | 1 | 2 | 1.000 |
| Indoor fungal contamination | ||||
| Total fungal load (pg/EDC), mean (SD) | 11.39 (9.82) | 11.68 (12.76) | 11.28 (8.98) | .934 |
ABPA, Allergic bronchopulmonary aspergillosis; ATB, antibiotic; BMI, body mass index; COBRA-ENV, indoor environment analysis of COhort of BRonchial obstruction and Asthma; FEF25-75, forced expiratory flow between the 25% and 75% of the FVC; FVC, forced vital capacity; ICS, inhaled corticosteroid; LABA, long-acting β2 agonist; LAMA, long-acting muscarinic antagonist; NA, not available; OCS, oral corticosteroid; SAFS, SA with fungal sensitization; VRC, Voriconazole.
P value with the Mann-Whitney test used for quantitative variables analysis and the Fisher exact test used for qualitative variables.
Asthma control as per the 7-Item Asthma Control Questionnaire.
NA because all samples were in the same condition regarding these variables.
Indoor mycobiomes and microbiomes differed significantly according to T2 endotypes
Patients with T2-high SA showed an increase in the 3 α-diversity metrics for the EDC bacterial communities compared with those observed for the patients with T2-low SA, but only the Shannon and Simpson indexes were statistically significant (Fig 1 , A). In contrast, the same 3 α-diversity metrics for the EDC fungal communities were lower for the patients with T2-high SA than those observed for the patients with T2-low SA, but only the Shannon indexes were statistically significant (Fig 1, B). Feno levels were significantly correlated with the α-diversity of the mycobiome as measured by Chao1 index values (Fig 1, C). The β-diversity of the EDC fungal communities clustered significantly according to the T2 endotypes (Fig 1, D).
Fig 1.

The α- and β-diversities of the EDCs from patients with SA according to T2 endotypes (n = 22). The α-diversity of bacterial (A) or fungal (B) communities from EDCs is shown using Chao1, Shannon, and Simpson indexes. C, Correlation between Feno levels and Chao1 fungal diversities. D, The β-diversity of fungal communities. Patients were split according to whether than had T2-high SA (dark gray, circles) or T2-low SA (light gray, triangles).
Indoor mycobiomes and microbiomes of patients with T2-high SA were significantly enriched for medically relevant fungi and bacteria
The bacterial composition of the indoor microbiome was dominated at the phylum level by Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria, with the most abundant OTUs annotated as Paracoccus spp, Acinetobacter spp, and Streptococcus spp (see Fig E2 in this article's Online Repository at www.jacionline.org). The fungal community was represented by the 2 major phyla, Ascomycota and Basidiomycota, with the most abundant OTUs annotated as Epicoccum nigrum, Cladosporium cladosporioides, Cladosporium sphaerospermum, Malassezia restricta, and Trametes versicolor (see Fig E3 in this article's Online Repository at www.jacionline.org). All bacterial and fungal OTUs were then subjected to the LEfSe analysis method with T2 endotypes used as covariables.
Fig E2.

Histograms representing the relative abundance at the genus level of the EDC microbiome.
Fig E3.

Histograms representing the relative abundance at the genus or species level of the EDC mycobiome.
The LEfSe analysis results indicated significant community enrichment of EDCs from patients with an Feno level higher than 25 ppb and T2-high SA as a covariable; this group of patients consisted of 7 families and 10 genera of bacterial OTUs (Fig 2 , A) as well as 2 genera plus 5 species of fungal OTUs (Fig 2, B). Whereas enrichments of EDCs from patients with low Feno levels were primarily composed of environmental Basidiomycota OTUs (Fig 2, B), those from patients with an Feno level greater than 25 ppb and T2-high SA as a covariable were composed of fungal and bacterial OTUs known to be medically relevant in allergies or asthma (ie, Cladosporium, Aspergillus, Epicoccum, Candida, Rhodotorula, Pseudomonas, Pedobacter, Erwinia, Sphingomonas, Sphingomonadaceae, or Methylocystaceae).1 , 9 , 10
Fig 2.

Taxonomic differences between Feno groups with T2 endotypes used as covariables across the 22 EDCs show significant enrichment of specifics taxa at the OTU level. Linear discriminant analysis (LDA) and effect size analysis identified bacterial (A) and fungal (B) taxa differentially enriched according to an LDA score cutoff at 2. The colors (dark and gray) represent each Feno group (level cutoff at 25 ppb), and the length represents the LDA score, which is the degree of significant difference (in log scale) for a given taxon. As the order of the numerator and denominator when calculating the effect size is determined by alphabetical order, it is possible to obtain an interpretation of the scale of the difference between 2 groups by using the absolute values of the effect size even though the LDA score is negative for a given taxon. Thus, any taxa colored according to the Feno variable may be interpreted as taxa being significantly increased in abundance compared with the other Feno group.
Indoor and sputum samples of patients with asthma exacerbation share a mycobiome core composed of medically relevant fungi
In all, 22 patients were able to produce induced sputum during clinically stable periods (n = 10, including 9 patients with T2-high SA and 1 patient with T2-low SA) or exacerbated periods (n = 12, including 7 patients with T2-high SA and 5 patients with T2-low SA) defined at COBRA visit. Both the bacterial and fungal diversities of these sputa were lower than those of the corresponding EDCs (Fig 3 , A). The taxonomic composition of these 2 types of samples was significantly different (Fig 3, B-D, and see Figs E2-E5 in this article's Online Repository at www.jacionline.org). These results revealed the highly individual signature of sputum mycobiomes and microbiomes compared with that observed for the EDCs, which exhibited a very closely spaced clustering of samples (Fig 3, C and D). For instance, the proportion of Basidiomycota, a phylum primarily composed of environmental fungi, compared with the proportion of Ascomycota, which includes medically relevant fungi, was higher in the EDCs than in the sputa (Fig 3, B), highlighting the complex microbial ecology of the human respiratory tract.
Fig 3.

Comparison of diversity and composition of both sputum and EDC samples. The bacterial (left) and fungal diversities (right) based on Shannon indexes are presented for sputum and EDC samples (A). The proportion of Basidiomycota (a phylum primarily composed of environmental fungi) was higher in EDCs than in sputa (B). Principal coordinates analysis based on Bray-Curtis dissimilarity between sputum (dark gray, squares) and EDC (light gray, diamonds) samples for bacterial (C) and fungal (D) communities showed the highly individual signature of sputa compared with that of EDCs. Proportion of common bacterial (E) or fungal (F) taxa between EDCs and sputa collected at a stable state (STB) or during an exacerbation (EXA). For each OTU, the proportion was expressed as a percentage of the number of reads (ie, relative abundance). PERMANOVA, Permutational multivariate ANOVA.
We did not observe any differences in the bacterial and fungal diversities of these sputum samples according to T2 endotypes (data not shown). In contrast, the proportion of common bacterial taxa between the EDCs and sputa was similar when sputa were collected during exacerbation or at a stable state (Fig 3, E), whereas the proportion of common fungal taxa between EDCs and sputa was significantly higher when sputa were collected during the exacerbation period than the stable period (Fig 3, F). The Venn diagram results confirmed a limited bacterial core composed of 5 OTUs (Fig 4 , A) and a large fungal core composed of 27 OTUs (Fig 4, B).
Fig 4.

Microbial core shared between the 22 pairs of indoor and respiratory samples. Venn diagrams show the proportion of common bacterial (A) and fungal (B) taxa (at the OTU level) between EDCs (green section) and sputa (SPU) collected either during an exacerbation (EXA) (the SPU were collected during an EXA [red section]) or during a stable state (STB) period (the SPU were collected during an STB [blue section]). Only OTUs present at least in 20% of each group of SPU (clustered according the presence or absence of an EXA when the SPU were collected) and in at least 90% of EDCs were taken into account and represented in the Venn diagrams. The bacterial core (A) showed only 1 OTU corresponding to Sphingomonas spp shared between EDCs and SPU collected during an EXA and 4 OTUs belonging to Streptococcus, Paracoccus, and Haemophilus species were shared independently from the clinical state of the patients. The fungal core (B) was larger than the bacterial core and was composed of 9 OTUs associated with SPU collected during an EXA, 1 OTU (ie, Alternaria brassicae) associated with SPU collected at STB, and 17 OTUs (including several medically relevant fungi, such as Malassezia, Aspergillus, and Cladosporium species) belonging to the fungal core independent from the clinical state.
We identified the core microbes between pairs of sputa and EDCs according to the absence or presence of an asthma exacerbation and confirmed that only 1 OTU was shared between the bacterial core and the clinical states (ie, Sphingomonas [Fig 4, A]). Surprisingly, 9 fungal OTUs (ie, Hyphodontia radula, Resinicium bicolor, Phlebia radiate, Penicillium brevicombactum, Vishniacozyma victoriae [synonym Cryptococcus victoriae], Sistrotremastrum spp, Cryptococcus magnus, Bjerkandera spp, and Botryotinia fuckeliana [Fig 4, B]) were identified as the fungal core and were specifically shared between EDCs and sputa collected during exacerbation. In addition, 17 other OTUs were identified as members of the fungal core independent of the clinical status, which included medically relevant yeasts (ie, Saccharomyces cerevisiae, Debaryomyce hansenii, and Malassezia species) and molds (Aspergillus and Cladosporium species) or environmental Basidiomycota and Ascomycota. Only 1 Alternaria brassicae OTU was shared with sputa collected at a stable state (Fig 4, B). Among these OTUs, several were both microbial core members and significantly enriched OTUs in EDC microbial communities, including C sphaerospermum, T versicolor, and Sphingomonadaceae (Figs 2, B and 4, B).
Discussion
To the best of our knowledge, this pilot study is the first to report that the indoor mycobiomes and microbiomes of patients with SA exhibit distinct signatures according to T2 endotypes. On the basis of pairs of EDC and sputum samples, we compared exogenous and endogenous microbial communities and identified a fungal core comprising medically relevant fungi that was significantly more pronounced when sputa were collected during asthma exacerbations.
Regarding the indoor environment, we clearly demonstrated that a higher bacterial α-diversity together with a lower fungal α-diversity was associated with patients with T2-high SA and that the fungal β-diversity of EDC communities was clustered according to T2 endotypes. Moreover, the indoor mycobiomes and microbiomes of patients with an Feno value higher than 25 ppb and T2-high SA were significantly enriched for medically relevant fungi and bacteria. Among them, we identified changes in the relative abundances of Sphingomonadaceae, Methylocystaceae, Erwinia, Sphingomonas, Pseudomonas, and Candida, which is in agreement with previously published data on the gut microbiota and allergy and/or asthma.27 Although not exclusively focused on T2 endotypes, several studies have previously investigated the respiratory microbiomes or mycobiomes of patients with varying degrees of asthma severity.28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42 Among these studies, an association between Firmicutes and Actinobacter with SA has been reported.33 , 35 In addition, an excess of Proteobacteria was shown to be associated with moderately severe to severe asthmatics,33 , 35 whereas an excess of Streptococcus was shown to be associated with eosinophilic asthma.42 However, little is known regarding the lung mycobiome in chronic respiratory diseases. Malassezia yeasts have been shown to be significantly associated with asthma and more recently with exacerbation in cystic fibrosis.26 , 29 The mycobiomes of patients with SA were shown to harbor higher loads of fungi than were patients with nonsevere asthma or healthy individuals,28 with a lower diversity observed in patients with T2-high asthma.7 A low indoor fungal diversity has been proposed to be a causal factor in the development of childhood asthma,11 and although a few studies considered adult patients with asthma with respect to its severity and control, none of them referred to T2 endotypes and combined exogenous to endogenous mycobiome and microbiome analyses.
The first studies of indoor microbial ecology used culture-based techniques,43 but such studies have more recently been performed by using NGS-based approaches. Very few studies have performed NGS analyses of the indoor mycobiome and/or microbiome in asthma.11 , 14 , 15 , 37, 38, 39 , 44 Overall, sampling the indoor microbiome by a passive collector such as an EDC allows for the recovery of house dust particles, which are a complex mixture of inorganic and organic substances. The indoor mycobiome appears to be a large fungal community assemblage10 , 44, 45, 46, 47, 48, 49, 50 that is primarily derived from the outdoor environment38 but also depends on the inhabitants’ mycobiomes under selective pressures.10 , 45 As previously reported,45 , 47 , 48 , 50 we sampled patient bedrooms, which are known to accurately estimate the level of indoor fungal pollution.43 Our NGS results for the EDC mycobiome and microbiome were congruent with these previous studies. Briefly, we identified Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria as the most abundant bacterial phyla in agreement with recently published data.51 , 52 Within these phyla, Pseudomonas and Sphingomonas were significantly enriched in the EDCs of patients with T2-high SA. Regarding the mycobiome, Aspergillus versicolor and C sphaerospermum have been identified as indoor species correlated to asthma.43 , 44 , 51 Using NGS, Dannemiller et al identified numerous fungi that were significantly associated with SA.44 Our results confirmed that several Cryptococcus species and E nigrum were associated with SA and that the genus Epicoccum was significantly enriched in the indoor mycobiome of patients with T2-high SA. Moreover, our indoor data are congruent with the recent identification of Cladosporium and Aspergillus as dominant enriched genera of respiratory samples from patients with T2-high asthma.7
In addition to viral infections, fungal sensitization and fungal exposure have been shown to be highly associated with asthma exacerbations and were recently reviewed.45 We confirmed that fungal exposure may be a key player during exacerbation, as the fungal OTU proportion shared by EDCs and sputa was significantly higher when sputa were collected during asthma exacerbation. These results support the idea that microbial analysis should not be restricted to a specific type of microorganism (viruses, bacteria, or fungi) but rather should involve analyses of interkingdom interactions that may be involved in promoting exacerbations.7 , 26
Feno is an endogenous gaseous molecule incorporated into the clinical management of chronic respiratory diseases and is currently recognized as a biomarker of T2 airway inflammation.53 Through use of conventional mycologic methods (ie, cultures and PCR), Feno levels of patients with asthma have been related to environmental exposure54 whereas the indoor isolation of A versicolor and Cladosporium have been associated with higher Feno levels,55 , 56 which is in agreement with our results. Feno levels were shown to be positively correlated with T2 cytokine levels in patients with asthma and with serum IL-17A levels in patients with SA.57 As adaptive antifungal immunity includes both TH2- and TH17-type CD4+ T cells, we hypothesize that a number of the fungi isolated from the indoor mycobiome of patients with SA (such as Aspergillus and Cladosporium species) should contribute to a detrimental immunopathology in patients with SA. Once fungi are inhaled and colonize the respiratory airways of patients, they participate in complex microbial-host interactions by producing secondary metabolites that are well known to be involved in this immune response, such as cell wall components (β-glucan and/or chitin) or secreted enzymes (proteases and/or glycosidases, or toxins).58
Although our work is novel, it does have several limitations, including its cross-sectional design and the limited number of fully analyzed pairs of EDC plus sputum, which reflects difficulties in sampling the respiratory tract. Indeed, the small number of patients limits the generalizability of our results, especially regarding LEfSe analysis results. For instance, the absence of difference between the group with T2-low SA and that with T2-high SA with respect to the exacerbation rate is probably underpowered. We used induced sputa to analyze the endogenous microbiomes and mycobiomes, a sampling method recently proposed as an acceptable and less invasive alternative compared with bronchoalveolar lavage samples but with several biases.59, 60, 61 This approach can be considered to be a bias of selection as only patients with higher than 50% predicted postbronchodilator FEV1 value were allowed to achieve induced sputum. As most of the published studies on this topic have focused on asthma-associated microbiomes and mycobiomes, this study suffers from small size limitations and a lack of longitudinal data. Furthermore, amplicon deep sequencing represents a promising method, but the protocols need to be improved and standardized. Indeed, there are many potential biases, ranging from the primer, amplification protocol, and NGS machine used to the pipeline and databases selected for analysis.10 , 62 Thus, further studies are warranted to confidently determine the relationships between the indoor environment, the lung mycobiome and microbiome, the inflammatory response, and the development and severity of asthma.
In summary, in the present study, we identified a correlation between the indoor mycobiome and Feno levels of patients with SA (Fig 1, C), which should renew interest in deciphering the interactions between the indoor environment, fungi, and the host to facilitate the development of novel therapeutic managements.14 Given the role of fungi as potent inducers of airway inflammation,58 an increase in Feno level may serve as an indicator of respiratory inflammation related to exposure to specific molds that the indoor mycobiome analysis will characterize.
Key messages.
-
•
The indoor mycobiomes and microbiomes of patients with SA harbor a distinct signature according to T2 endotypes, with lower fungal diversity but higher bacterial diversity associated with T2-high SA.
-
•
The indoor environments of patients with T2-high have significant enrichment of medically relevant fungi and bacteria, such as Aspergillus, Candida, Sphingomonas, and Pseudomonas.
-
•
The respiratory mycobiome of patients with SA shares more fungal taxa with the indoor mycobiome during exacerbation than during stable periods.
Acknowledgments
We thank Isabelle Goasdoue, Virginie Niel, and Marine Servat from the clinical investigation center for technical assistance. The authors would like to thank American Journal Experts for editing the English language of the text.
Footnotes
This work was supported by the GenoScreen society (which provided a grant to L.-E.V.) and Fondation de l’Université de Bordeaux (Fonds FGLMR/AVAD pour les maladies chroniques), in which L.D. was a laureate (recipient of Bronch-Asp project grant).
Disclosure of potential conflict of interest: L.-E. Vandenborght, C. Urien, and S. Ferreira are employees of GenoScreen. P.-O. Girodet reports personal fees and nonfinancial support from AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Novartis, and Sanofi outside the submitted work. P. Berger is the medical coordinator of COBRA, which received grants from AstraZeneca, GlaxoSmithKine, Roche, Chiesi, Novartis, and Legs Poix foundation. Moreover, P. Berger reports grants and personal fees from Novartis; personal fees and nonfinancial support from Chiesi, AstraZeneca, and Sanofi; grants, personal fees, and nonfinancial support from Boehringer Ingelheim; and personal fees from Menarini and TEVA outside the submitted work. In addition, P. Berger has 3 patents: (1) New Compositions and Methods of Treating and/or Preventing Chronic Obstructive Pulmonary Disease (EP no.15152886.6; pending); (2) Use of Plerixafor for Treating and/or Preventing Acute Exacerbations of Chronic Obstructive Pulmonary Disease (EP no. 3050574; delivered); and (3) New Compositions and Methods of Treating COVID-19 Disease), (EP no. 20173595.8; pending). L. Delhaes was invited to the European Respiratory Society International Congress 2019 as a speaker (PG 41000/45500-14200). The rest of the authors declare that they have no relevant conflicts of interest.
Methods
Study patients
A diagnosis of SA with fungal sensitization was considered in patients with SA who were sensitized to 1 or more fungi (as demonstrated by skin prick tests or serum specific IgE) but had a normal serum total IgE level (<150 kUI/mL) and were negative for allergic bronchopulmonary aspergillosis.E1, E2 Asthma control was established by using the 7-Item Asthma Control Questionnaire, with a score higher than 1.5 reflecting very poorly controlled asthma.E3, E4 The numbers of moderate asthma exacerbations (ie, loss of asthma control requiring oral steroid and/or antibiotic courses) were recorded within the year preceding the EDC exposure (ie, from December 2016 to December 2017), during the EDC exposure time (ie, from December 2017 to March 2018), and during the year following EDC exposure (ie, from March 2018 to March 2019). Spirometry was performed according to American Thoracic Society standard. The Feno level (in ppb) was assessed, as previously described.E5
Sputum samples and EDC
Induced sputa were successfully collected from 22 patients as previously described.E5, E6 Briefly, after oral rinse to prevent excessive salivary contamination, inhalation of increasing concentrations of nebulized saline solutions at 3%, 4%, and 5%, respectively, for a 7-minute session each was used. For patients with an FEV1 value of at least 50% predicted but less than 60% predicted, induction was performed with normal/isotonic saline (0.9%). Sputa from subjects with a postbronchodilator FEV1 value less than 50% predicted were not induced. Sputa treated with dithiothreitol were subjected to microbial cultures and stored at –20°C until DNA extraction.
All the patients included into COBRA-ENV (indoor environment analysis of COhort of BRonchial obstruction and Asthma) study received guidelines during their COBRA visit to deploy an EDC at home (in the patient's bedroom) according to the same protocol. The EDCs were then sent to the mycology laboratory, where each EDC was processed as described previously.E7 Each collected solution was stored at –20°C until DNA extraction. As a negative control, 1 unexposed EDC was subjected to the same laboratory protocol.
DNA extraction, library preparation, deep sequencing, and taxonomic assignation
DNA extraction was carried out on the EDC and sputum samples by using a DNeasy PowerSoil Kit (Qiagen). Negative extraction controls (250 μL of DNA-free water) were processed using the same protocol. The microbial diversity and taxonomic composition of samples were assessed by using the V3-V4 of bacterial 16S gene and the ITS2 region of the fungal rDNA, according to an optimized and standardized library preparation protocol Metabiote (Genoscreen, Lille, France).E8 Two positive qualitative controls comprising an artificial bacterial community and a fungal mock community and 2 negatives controls were used to valid the experimental processes. NGS sequencing was performed by using a 250-bp paired-end sequencing protocol on the Illumina MiSeq platform, as described previously.E8 Taxonomic analysis was performed with the Metabiote Online version 2.0 pipeline (GenoScreen, Lille, France), as previously described.E8 Each OTU was then taxonomically compared with reference databases as follows: the Greengenes database (release 13_8) for bacterial OTUs and the Unite database (release 7.2) for fungal OTUs.
Estimation of EDC fungal load using qPCR assay
The fungal load of each EDC was estimated with a universal qPCR assay quantifying 18S rDNA (in pg per EDC).E9 Briefly, the Taqman Gene Expression Master Mix (Applied Biosystem, Life Technologies) kit was used according to the manufacturer's instructions. qPCR was performed using the 7900HT Sequence Detection System and the following published primersE9: FungiQuanΔt-F, 5-GGRAAACTCACCAGGTCCAG-3; FungiQuant-R, 5-GSWCTATCCCCAKCACGA-3’; and FungiQuant-Prb, (6FAM)5-TGGTGCATGGCCGTT-3(MGBNFQ). The cycling conditions were 15 seconds at 95°C and 40 repeats of 15 seconds at 92°C (denaturation step) and 1 minute at 60°C (annealing and extension steps). Each fungal load was quantified on the basis of a standard comprising 11 genomic DNAs extracted from different fungal strains (the artificial fungal community MetaBiote Control) by using the ΔΔCt method. Each sample was analyzed in triplicate; the Ct mean was finally compared with the standard curve to deduce the quantity of fungal 18S DNA of each EDC.
Data analysis
Measurement of α-diversity (within-sample) included computation of the Shannon index, Simpson index, and number of observed OTUs (Chao1 index). For β-diversity (between-sample) measurements, we used the vegan package to compute the Bray-Curtis distances.E10 The principal coordinates analysis method was used to visualize group overall microbial differences with use of the phyloSeq package.E11 To determine and analyze the microbial core, OTUs not contained in at least 20% of sputa per group and 90% of EDCsE12 were removed and the Venn diagram function of the RAM package was used.E13
To compare α-diversity estimates across patients' clinical parameters, Mann-Whitney tests were performed; the Wilcoxon test was used when pairs of sputum and EDC samples were compared. To analyze β-diversity between samples, OTU tables normalized via cumulative sum scaling by metagenomeSeq package were used.E14 Sample clustering hypotheses were tested by using permutational multivariate ANOVA. Homogeneity of dispersion among sample groups was assessed by using the Betadisper function (vegan package). To detect differentially abundant OTUs between groups, LEfSe analysis was performed.E15 It induced the ranking of biomarker relevance with a substantial low false-positive rate (mainly in <0.5% of the cases) and a high false-negative rate.E15
Fig E4.

Histograms representing the relative abundance at the genus level of the sputum microbiome.
Fig E5.

Histograms representing the relative abundance at the genus or species level of the sputum mycobiome.
References
- 1.Global Initiative for Asthma GINA annual report. http://ginasthma.org/archived-reports/ Accessed June 2019.
- 2.Martinez F.D. The state of asthma research: considerable advances, but still a long way to go. Am J Respir Crit Care Med. 2019;199:397–399. doi: 10.1164/rccm.201901-0013ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung K.F., Wenzel S.E., Brozek J.L., Bush A., Castro M., Sterk P.J., et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J. 2014;43:343–373. doi: 10.1183/09031936.00202013. [DOI] [PubMed] [Google Scholar]
- 4.Li X., Hastie A.T., Peters M.C., Hawkins G.A., Phipatanakul W., Li H., et al. Investigation of the relationship between IL-6 and type 2 biomarkers in patients with severe asthma. J Allergy Clin Immunol. 2020;145:430–433. doi: 10.1016/j.jaci.2019.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li N., Qiu R., Yang Z., Li J., Chung K.F., Zhong N., et al. Sputum microbiota in severe asthma patients: relationship to eosinophilic inflammation. Respir Med. 2017;131:192–198. doi: 10.1016/j.rmed.2017.08.016. [DOI] [PubMed] [Google Scholar]
- 6.Chung K.F. Potential role of the lung microbiome in shaping asthma phenotypes. Ann Am Thorac Soc. 2017;14(suppl 5):S326–S331. doi: 10.1513/AnnalsATS.201702-138AW. [DOI] [PubMed] [Google Scholar]
- 7.Sharma A., Laxman B., Naureckas E.T., Hogarth D.K., Sperling A.I., Solway J., et al. Associations between fungal and bacterial microbiota of airways and asthma endotypes. J Allergy Clin Immunol. 2019;144:1214–1227. doi: 10.1016/j.jaci.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loss G.J., Depner M., Hose A.J., Genuneit J., Karvonen A.M., Hyvärinen A., et al. The early development of wheeze. Environmental determinants and genetic susceptibility at 17q21. Am J Respir Crit Care Med. 2016;193:889–897. doi: 10.1164/rccm.201507-1493OC. [DOI] [PubMed] [Google Scholar]
- 9.Denning D.W., Pashley C., Hartl D., Wardlaw A., Godet C., Del Giacco S., et al. Fungal allergy in asthma-state of the art and research needs. Clin Transl Allergy. 2014;4:14. doi: 10.1186/2045-7022-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox M.J., Ege M.J., Mutius E. von, editors. Vol. 83. European Respiratory Society; Lausanne, Switzerland: 2019. (The lung microbiome. ERS Monographs). [Google Scholar]
- 11.Dannemiller K.C., Mendell M.J., Macher J.M., Kumagai K., Bradman A., Holland N., et al. Next-generation DNA sequencing reveals that low fungal diversity in house dust is associated with childhood asthma development. Indoor air. 2014;24:236–247. doi: 10.1111/ina.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharpe R.A., Bearman N., Thornton C.R., Husk K., Osborne N.J. Indoor fungal diversity and asthma: a meta-analysis and systematic review of risk factors. J Allergy Clin Immunol. 2015;135:110–122. doi: 10.1016/j.jaci.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Cavaleiro Rufo J., Madureira J., Paciência I., Aguiar L., Pereira C., Silva D., et al. Indoor fungal diversity in primary schools may differently influence allergic sensitization and asthma in children. Pediatr Allergy Immunol. 2017;28:332–339. doi: 10.1111/pai.12704. [DOI] [PubMed] [Google Scholar]
- 14.O’Connor G.T., Lynch S.V., Bloomberg G.R., Kattan M., Wood R.A., Gergen P.J., et al. Early-life home environment and risk of asthma among inner-city children. J Allergy Clin Immunol. 2018;141:1468–1475. doi: 10.1016/j.jaci.2017.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stein M.M., Hrusch C.L., Gozdz J., Igartua C., Pivniouk V., Murray S.E., et al. Innate immunity and asthma risk in Amish and Hutterite farm children. N Engl J Med. 2016;375:411–421. doi: 10.1056/NEJMoa1508749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pretolani M., Soussan D., Poirier I., Thabut G., Aubier M., COBRA Study Group, et al. Clinical and biological characteristics of the French COBRA cohort of adult subjects with asthma. Eur Respir J. 2017;50 doi: 10.1183/13993003.00019-2017. [DOI] [PubMed] [Google Scholar]
- 17.Silkoff P.E., Laviolette M., Singh D., FitzGerald J.M., Kelsen S., Backer V., et al. Longitudinal stability of asthma characteristics and biomarkers from the Airways Disease Endotyping for Personalized Therapeutics (ADEPT) study. Respir Res. 2016;17:43. doi: 10.1186/s12931-016-0360-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scherer E., Rocchi S., Reboux G., Vandentorren S., Roussel S., Vacheyrou M., et al. qPCR standard operating procedure for measuring microorganisms in dust from dwellings in large cohort studies. Sci Total Environ. 2014;466-67:716–724. doi: 10.1016/j.scitotenv.2013.07.054. [DOI] [PubMed] [Google Scholar]
- 19.Rocchi S., Valot B., Reboux G., Millon L. DNA metabarcoding to assess indoor fungal communities: electrostatic dust collectors and Illumina sequencing. J Microbiol Methods. 2017;139:107–112. doi: 10.1016/j.mimet.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 20.Hermon L., Hellal J., Denonfoux J., Vuilleumier S., Imfeld G., Urien C., et al. Functional genes and bacterial communities during organohalide respiration of chloroethenes in microcosms of multi-contaminated groundwater. Front Microbiol. 2019;10:89. doi: 10.3389/fmicb.2019.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K., et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edgar R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 23.Wang Q., Garrity G.M., Tiedje J.M., Cole J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu C.M., Kachur S., Dwan M.G., Abraham A.G., Aziz M., Hsueh P.-R., et al. FungiQuant: a broad-coverage fungal quantitative real-time PCR assay. BMC Microbiol. 2012;12:255. doi: 10.1186/1471-2180-12-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soret P., Vandenborght L.-E., Francis F., Mucofong Investigation Group. Avalos M., Berger P., et al. Respiratory mycobiome and suggestion of inter-kingdom network during acute pulmonary exacerbation in cystic fibrosis. Sci Rep. 2020;10:3589. doi: 10.1038/s41598-020-60015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buttó L.F., Haller D. Functional relevance of microbiome signatures: the correlation era requires tools for consolidation. J Allergy Clin Immunol. 2017;139:1092–1098. doi: 10.1016/j.jaci.2017.02.010. [DOI] [PubMed] [Google Scholar]
- 28.Fraczek M.G., Chishimba L., Niven R.M., Bromley M., Simpson A., Smyth L., et al. Corticosteroid treatment is associated with increased filamentous fungal burden in allergic fungal disease. J Allergy Clin Immunol. 2018;142:407–414. doi: 10.1016/j.jaci.2017.09.039. [DOI] [PubMed] [Google Scholar]
- 29.van Woerden H.C., Gregory C., Brown R., Marchesi J.R., Hoogendoorn B., Matthews I.P. Differences in fungi present in induced sputum samples from asthma patients and non-atopic controls: a community based case control study. BMC Infect Dis. 2013;13:69. doi: 10.1186/1471-2334-13-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuo C.-H.S., Pavlidis S., Loza M., Baribaud F., Rowe A., Pandis I., et al. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J. 2017;49 doi: 10.1183/13993003.02135-2016. [DOI] [PubMed] [Google Scholar]
- 31.Hauptmann M., Schaible U.E. Linking microbiota and respiratory disease. FEBS Lett. 2016;590:3721–3738. doi: 10.1002/1873-3468.12421. [DOI] [PubMed] [Google Scholar]
- 32.Carpagnano G.E., Malerba M., Lacedonia D., Susca A., Logrieco A., Carone M., et al. Analysis of the fungal microbiome in exhaled breath condensate of patients with asthma. Allergy Asthma Proc. 2016;37:41–46. doi: 10.2500/aap.2016.37.3943. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Q., Cox M., Liang Z., Brinkmann F., Cardenas P.A., Duff R., et al. Airway microbiota in severe asthma and relationship to asthma severity and phenotypes. PLoS ONE. 2016;11 doi: 10.1371/journal.pone.0152724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Venkataraman A., Bassis C.M., Beck J.M., Young V.B., Curtis J.L., Huffnagle G.B., et al. Application of a neutral community model to assess structuring of the human lung microbiome. mBio. 2015;6 doi: 10.1128/mBio.02284-14. e02284-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y.J., Nariya S., Harris J.M., Lynch S.V., Choy D.F., Arron J.R., et al. The airway microbiome in patients with severe asthma: associations with disease features and severity. J Allergy Clin Immunol. 2015;136:874–884. doi: 10.1016/j.jaci.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bassis C.M., Erb-Downward J.R., Dickson R.P., Freeman C.M., Schmidt T.M., Young V.B., et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio. 2015;6 doi: 10.1128/mBio.00037-15. e00037-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma A., Gilbert J.A. Microbial exposure and human health. Curr Opin Microbiol. 2018;44:79–87. doi: 10.1016/j.mib.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 38.Shan Y., Wu W., Fan W., Haahtela T., Zhang G. House dust microbiome and human health risks. Int Microbiol. 2019;22:297–304. doi: 10.1007/s10123-019-00057-5. [DOI] [PubMed] [Google Scholar]
- 39.Caillaud D., Leynaert B., Keirsbulck M., Nadif R., mould ANSES working group Indoor mould exposure, asthma and rhinitis: findings from systematic reviews and recent longitudinal studies. Eur Respir Rev. 2018;27 doi: 10.1183/16000617.0137-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sverrild A., Kiilerich P., Brejnrod A., Pedersen R., Porsbjerg C., Bergqvist A., et al. Eosinophilic airway inflammation in asthmatic patients is associated with an altered airway microbiome. J Allergy Clin Immunol. 2017;140:407–417.e11. doi: 10.1016/j.jaci.2016.10.046. [DOI] [PubMed] [Google Scholar]
- 41.Durack J., Lynch S.V., Nariya S., Bhakta N.R., Beigelman A., Castro M., et al. Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J Allergy Clin Immunol. 2017;140:63–75. doi: 10.1016/j.jaci.2016.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor S.L., Leong L.E.X., Choo J.M., Wesselingh S., Yang I.A., Upham J.W., et al. Inflammatory phenotypes in patients with severe asthma are associated with distinct airway microbiology. J Allergy Clin Immunol. 2018;141:94–103.e15. doi: 10.1016/j.jaci.2017.03.044. [DOI] [PubMed] [Google Scholar]
- 43.Reboux G., Rocchi S., Laboissière A., Ammari H., Bochaton M., Gardin G., et al. Survey of 1012 moldy dwellings by culture fungal analysis: threshold proposal for asthmatic patient management. Indoor Air. 2019;29:5–16. doi: 10.1111/ina.12516. [DOI] [PubMed] [Google Scholar]
- 44.Dannemiller K.C., Gent J.F., Leaderer B.P., Peccia J. Indoor microbial communities: influence on asthma severity in atopic and nonatopic children. J Allergy Clin Immunol. 2016;138:76–83.e1. doi: 10.1016/j.jaci.2015.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richardson M., Bowyer P., Sabino R. The human lung and Aspergillus: you are what you breathe in? Med Mycol. 2019;57(suppl 2):S145–S154. doi: 10.1093/mmy/myy149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yashiro E., Savova-Bianchi D., Niculita-Hirzel H. Major Differences in the diversity of mycobiomes associated with wheat processing and domestic environments: significant findings from high-throughput sequencing of fungal barcode ITS1. Int J Environ Res Public Health. 2019;16:2335. doi: 10.3390/ijerph16132335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hegarty B., Dannemiller K.C., Peccia J. Gene expression of indoor fungal communities under damp building conditions: implications for human health. Indoor Air. 2018;28:548–558. doi: 10.1111/ina.12459. [DOI] [PubMed] [Google Scholar]
- 48.Coombs K., Taft D., Ward D.V., Green B.J., Chew G.L., Shamsaei B., et al. Variability of indoor fungal microbiome of green and non-green low-income homes in Cincinnati, Ohio. Sci Total Environ. 2018;610-611:212–218. doi: 10.1016/j.scitotenv.2017.07.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tong X., Leung M.H.Y., Wilkins D., Lee P.K.H. City-scale distribution and dispersal routes of mycobiome in residences. Microbiome. 2017;5:131. doi: 10.1186/s40168-017-0346-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanson B., Zhou Y., Bautista E.J., Urch B., Speck M., Silverman F., et al. Characterization of the bacterial and fungal microbiome in indoor dust and outdoor air samples: a pilot study. Environ Sci-Proc Imp. 2016;18:713–724. doi: 10.1039/c5em00639b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richardson M., Gottel N., Gilbert J.A., Gordon J., Gandhi P., Reboulet R., et al. Concurrent measurement of microbiome and allergens in the air of bedrooms of allergy disease patients in the Chicago area. Microbiome. 2019;7:82. doi: 10.1186/s40168-019-0695-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adams R.I., Bateman A.C., Bik H.M., Meadow J.F. Microbiota of the indoor environment: a meta-analysis. Microbiome. 2015;3:49. doi: 10.1186/s40168-015-0108-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miskoff J.A., Dewan A., Chaudhri M. Fractional exhaled nitric oxide testing: diagnostic utility in asthma, chronic obstructive pulmonary disease, or asthma-chronic obstructive pulmonary disease overlap syndrome. Cureus. 2019;11:e4864. doi: 10.7759/cureus.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Kampen V., Brüning T., Merget R. Serial fractional exhaled nitric oxide measurements off and at work in the diagnosis of occupational asthma. Am J Ind Med. 2019;62:663–671. doi: 10.1002/ajim.22996. [DOI] [PubMed] [Google Scholar]
- 55.Norbäck D., Hashim J.H., Hashim Z., Cai G.-H., Sooria V., Ismail S.A., et al. Respiratory symptoms and fractional exhaled nitric oxide (FeNO) among students in Penang, Malaysia in relation to signs of dampness at school and fungal DNA in school dust. Sci Total Environ. 2017;577:148–154. doi: 10.1016/j.scitotenv.2016.10.148. [DOI] [PubMed] [Google Scholar]
- 56.Tham R., Erbas B., Dharmage S.C., Tang M.L., Aldakheel F., Lodge C.J., et al. Outdoor fungal spores and acute respiratory effects in vulnerable individuals. Environ Res. 2019;178:108675. doi: 10.1016/j.envres.2019.108675. [DOI] [PubMed] [Google Scholar]
- 57.Chien J.-W., Lin C.-Y., Yang K.D., Lin C.-H., Kao J.-K., Tsai Y.-G. Increased IL-17A secreting CD4+ T cells, serum IL-17 levels and exhaled nitric oxide are correlated with childhood asthma severity. Clin Exp Allergy. 2013;43:1018–1026. doi: 10.1111/cea.12119. [DOI] [PubMed] [Google Scholar]
- 58.Bartemes K.R., Kita H. Innate and adaptive immune responses to fungi in the airway. J Allergy Clin Immunol. 2018;142:353–363. doi: 10.1016/j.jaci.2018.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loverdos K., Bellos G., Kokolatou L., Vasileiadis I., Giamarellos E., Pecchiari M., et al. Lung microbiome in asthma: current perspectives. J Clin Med. 2019;8:1967. doi: 10.3390/jcm8111967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tangedal S., Aanerud M., Grønseth R., Drengenes C., Wiker H.G., Bakke P.S., et al. Comparing microbiota profiles in induced and spontaneous sputum samples in COPD patients. Respir Res. 2017;18:164. doi: 10.1186/s12931-017-0645-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Whiteson K.L., Bailey B., Bergkessel M., Conrad D., Delhaes L., Felts B., et al. The upper respiratory tract as a microbial source for pulmonary infections in cystic fibrosis. Parallels from island biogeography. Am J Respir Crit Care Med. 2014;189:1309–1315. doi: 10.1164/rccm.201312-2129PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nilsson R.H., Anslan S., Bahram M., Wurzbacher C., Baldrian P., Tedersoo L. Mycobiome diversity: high-throughput sequencing and identification of fungi. Nat Rev Microbiol. 2019;17:95–109. doi: 10.1038/s41579-018-0116-y. [DOI] [PubMed] [Google Scholar]
References
- Denning D.W., Pashley C., Hartl D., Wardlaw A., Godet C., Del Giacco S., et al. Fungal allergy in asthma-state of the art and research needs. Clin Transl Allergy. 2014;4:14. doi: 10.1186/2045-7022-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal R., Chakrabarti A., Shah A., Gupta D., Meis J.F., Guleria R., et al. Allergic bronchopulmonary aspergillosis: review of literature and proposal of new diagnostic and classification criteria. Clin Exp Allergy. 2013;43:850–873. doi: 10.1111/cea.12141. [DOI] [PubMed] [Google Scholar]
- Pretolani M., Soussan D., Poirier I., Thabut G., Aubier M., COBRA Study Group, et al. Clinical and biological characteristics of the French COBRA cohort of adult subjects with asthma. Eur Respir J. 2017;50 doi: 10.1183/13993003.00019-2017. [DOI] [PubMed] [Google Scholar]
- Nordon C., Grimaldi-Bensouda L., Pribil C., Nachbaur G., Amzal B., Thabut G., et al. Clinical and economic burden of severe asthma: a French cohort study. Respir Med. 2018;144:42–49. doi: 10.1016/j.rmed.2018.10.002. [DOI] [PubMed] [Google Scholar]
- Silkoff P.E., Laviolette M., Singh D., FitzGerald J.M., Kelsen S., Backer V., et al. Longitudinal stability of asthma characteristics and biomarkers from the Airways Disease Endotyping for Personalized Therapeutics (ADEPT) study. Respir Res. 2016;17:43. doi: 10.1186/s12931-016-0360-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozier A., Girodet P.-O., Bara I., Tunon de Lara J.-M., Marthan R., Berger P. Control maintenance can be predicted by exhaled NO monitoring in asthmatic patients. Respir Med. 2011;105:989–996. doi: 10.1016/j.rmed.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Rocchi S., Valot B., Reboux G., Millon L. DNA metabarcoding to assess indoor fungal communities: electrostatic dust collectors and Illumina sequencing. J Microbiol Methods. 2017;139:107–112. doi: 10.1016/j.mimet.2017.05.014. [DOI] [PubMed] [Google Scholar]
- Hermon L., Hellal J., Denonfoux J., Vuilleumier S., Imfeld G., Urien C., et al. Functional genes and bacterial communities during organohalide respiration of chloroethenes in microcosms of multi-contaminated groundwater. Front Microbiol. 2019;10:89. doi: 10.3389/fmicb.2019.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.M., Kachur S., Dwan M.G., Abraham A.G., Aziz M., Hsueh P.-R., et al. FungiQuant: a broad-coverage fungal quantitative real-time PCR assay. BMC Microbiol. 2012;12:255. doi: 10.1186/1471-2180-12-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14:927–930. [Google Scholar]
- McMurdie P.J., Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8 doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza R.S.C., Okura V.K., Armanhi J.S.L., Jorrín B., Lozano N., da Silva M.J., et al. Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Sci Rep. 2016;6:28774. doi: 10.1038/srep28774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAM citation information. https://cran.r-project.org/web/packages/RAM/citation.html Available at: Accessed May 15, 2018.
- Paulson J.N., Stine O.C., Bravo H.C., Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10:1200–1202. doi: 10.1038/nmeth.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]