

Abstract
FLT3 internal tandem duplication (ITD) mutations are present in acute myeloid leukemia (AML) in 30% of patients, most commonly with a normal karyotype, and are associated with short relapse-free survival. in vitro and in vivo studies of FLT3-ITD cell lines have demonstrated reactive oxygen species-mediated DNA double-strand breaks and associated error-prone DNA repair as a mechanism of genomic instability, and we hypothesized that genomic instability might be manifested by cytogenetic changes at relapse of FLT3-ITD AML. We retrospectively reviewed charts of patients with CN FLT3-ITD AML treated at the University of Maryland Greenebaum Cancer Center, with attention to metaphase analysis results at relapse. Cytogenetic data were available from first and, when applicable, subsequent relapses for 15 patients diagnosed with CN FLT3-ITD AML. Among 12 patients with documented FLT3-ITD at first and, when applicable, subsequent relapse, 10 had cytogenetic changes, including 9 with rare structural abnormalities. The high frequency of rare structural chromosome abnormalities at relapse in our case series supports a role of genomic instability in the genesis of relapse, and suggests that ROS-generating and DNA repair pathways might be therapeutic targets in FLT3-ITD AML.
Keywords: Acute myeloid leukemia, FLT3-ITD, relapse, genomic instability, cytogenetics, structural chromosome abnormalities
Introduction
The receptor tyrosine kinase fms-like tyrosine kinase 3 (FLT3) is expressed on malignant blasts in 70 to 100 percent of cases of acute myeloid leukemia (AML) [1, 2] and is mutated, most commonly by internal tandem duplication (ITD), in 20 to 30 percent, with the highest frequency of FLT3-ITD mutations in cytogenetically normal (CN) AML cases [3–7]. FLT3-ITD mutations result in constitutive activation of FLT3, and although complete remission (CR) rates are similar to those of AML without FLT3 mutations, AML with FLT3-ITD is associated withhigh relapse rates and short relapse-free survival (RFS) and overall survival (OS) following chemotherapy [3–8] as well as allogeneic hematopoietic stem cell transplantation (alloSCT) [9].
Regrowth of AML cells following chemotherapy has been attributed to both constitutive FLT3 signaling through signal transducer and activator of transcription (STAT) 5 [10, 11] and stimulation by FLT3 ligand, since FLT3-ITD remains responsive to FLT3 ligand despite constitutive activation [12] and FLT3 ligand levels increase following chemotherapy [13]. In addition, increased levels of reactive oxygen species (ROS), increased DNA double-strand breaks (DSBs), and error-prone DNA repair by an alternative non-homologous end-joining (ALT NHEJ) pathway have been demonstrated in FLT3-ITD AML cells [14, 15], and resulting genomic instability might also contribute to early relapse.
We hypothesized that if genomic instability contributes significantly to early relapse of FLT3-ITD AML, a high frequency of cytogenetic changes, and particularly structural cytogenetic changes, might be evident at relapse. To test this hypothesis, we retrospectively reviewed cytogenetic findings at first relapse and, when applicable, subsequent relapses of patients with CN FLT3-ITD AML treated at the University of Maryland Greenebaum Cancer Center (UMGCC).
Material and Methods
Charts of all patients with CN AML with and without FLT3-ITD mutations diagnosed and treated at UMGCC between February 2004 and December 2013 were reviewed. Patients who received remission induction chemotherapy, achieved CR, relapsed and had cytogenetic data available at relapse were selected for the study. We reviewed presenting characteristics, treatment, RFS, OS and cytogenetic changes in these patients. Relapse-free survival was calculated as time from diagnosis to first relapse, and OS from diagnosis to death. The study was approved by the University of Maryland School of Medicine Institutional Review Board. The primary focus of this review was to examine cytogenetic changes in patients with CN FLT3-ITDAML at diagnosis who relapsed and had FLT3-ITD documented at relapse (Table 1), but the frequency of cytogenetic changes in CN AML without FLT3-ITD at diagnosis was also determined as a control.
Table 1.
Patient characteristics, treatment, outcomes, and karyotype changes
| Patient | Age,Sex | Diagnosis karyotypea | FLT3 status at diagnosis | Therapy | RFS, OSb | Subsequent karyotype | AML status when new karyotype emerged | FLT3 status when new karyotype emerged |
|---|---|---|---|---|---|---|---|---|
| 1 | 53, F | 46,XX | ITD | AraC, Ida: HIDAC, topotecan: sorafenib; alloSCT | 163, 197 | 46,XX,t(1;5) (q25;q13)[5]/46,XX[15] | Relapse 1 | ITD and D835 |
| 2 | 56, F | 46,XX | ITD | AraC, DNR; HIDAC | 408, 966 | 46,XX,t(2;4) (p24;q13)[5]/46,XX,del(16) (q12)[2]/dup(1) (q21q32)[1]/46, XX[12] nucish(1p32×2) (1q21×3)[38/200] |
CR2; Relapse 2 | ITD |
| 3 | 74, F | 46,XX | ITD | AraC, Ida, sorafenib; HIDAC | 183, 394 | 46,XX,t(4;4) (q21;q35)[7]/46,XX[13] | Relapse 1. after chemotherapy | ITD |
| 4 | 58, F | 46,XX | ITD | AraC, Ida: HIDAC | 783, 989 | 46,XX,t(5;13) (q31;q12)[20] | Relapse 1 | ITD |
| 5 | 64, M | 46,XY | ITD | AraC, Ida: HIDAC | 229, 434 | 46,XY,t(9;22) (q34:q11.2)[4]/46,XY[8] | Relapse 1, after chemotherapy | ITD |
| 6 | 48, M | 46,XY | ITD | AraC, DNR. VP16; alloSCT | 227, 243 | 46,XY,inv(10) (p11.2q21.2)[12]/46,XY[8] | Relapse 1 | ITD |
| 7 | 64, M | 46,XY | ITD | AraC, Ida, sorafenib; alloSCT | 177, 726+ | 46,XY,t(10;13) (p11.1q11)[9]/46,XY[9]// 46,XX[2] |
Relapse 1 | ITD |
| 8 | 53, F | 46,XX | ITD | AraC, DNR. VP16; vorinostat, AraC, VP16; alloSCT | 330, 435 | 46,X,t(X;10) (q13;q24),t(3;14) (p21;q11.2), t( 12:17) (p12;q12) [15]//46,XY[5] |
Relapse 1 | ITD |
| 9 | 43, F | 46,XX | ITD | AraC, DNR. VP16; HIDAC | 179, 413 | 46,XX,der(2)t(2;?) (q21;?),del(3) (q12), der(6)t(6;?) (p22;?), der(7)t(7;?) (p15;?), ?del(11) (p12p15), −17,−22,−2mar(19]/46,XX[1] |
Relapse 1 | ITD |
| 10 | 49, F | 46,XX | ITD | AraC, Ida; HIDAC | 175, 567+ | 49,XX,+6,+8, +13[2]/46,XX[18] | Relapse 1 | ITD |
Abbreviations: M, male; F, female; AraC, cytarabine; Ida, idarubicin; DNR, daunorubidn; VP16, etoposide; HIDAC, high dose cytarabine
20 metaphases.
Days.
The FLT3-ITD mutation was detected in bone marrow or blood (one patient) by multiplex polymerase chain reaction and capillary electrophoresis, as previously described [16].
Cytogenetic analysis consisted of analysis of at least 20 Giemsa-banded metaphases from cultures of bone marrow, or peripheral blood in one patient, at diagnosis and at first and, when applicable, subsequent time points. Karyotypes were described according to International System for Human Cytogenetic Nomenclature (ISCN) recommendations [17].
Fluorescence in situ hybridization (FISH) for BCR-ABL1 rearrangement was performed in Patient 5 (Table 1) with a BCR/ABL1 dual fusion probe set (Rainbow Scientific, Inc, Windsor, CT), according to the manufacturer’s instructions. FISH was also performed in a follow-up study in Patient 2 (Table 1) using a 1p32/1q21 probe set (Cytocell, Cambridge, UK) according to the manufacturer’s instructions.
Chromosomal microarray analysis (CMA) was performed on paired DNA samples collected from Patient 9 (Table 1) at diagnosis and at relapse. Peripheral blood mononuclear cells were isolated by density gradient centrifugation. DNA extraction was performed using the Puregene Blood DNA isolation kit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. DNA was processed and hybridized to high-density Genome-Wide Human CytoScan HD arrays with 2.7 million markers including 750,000 single nucleotide polymorphic (SNP) and 1,900,000 non-polymorphic copy number variant (CNV) markers (Affymetrix, Santa Clara, CA), according to the manufacturer’s instructions. The raw data were analyzed using the Genotyping Console software (Affymetrix, Santa Clara, CA) to reveal copy number changes with > 25 markers and 100 kb. A genomic imbalance is reported when deletions/duplications are greater than 1 Mb, unless they represent a region clearly associated with a benign copy number polymorphism in multiple independent studies. Copy number-neutral loss of heterozygosity (CNN-LOH)/acquired uniparental disomy (UPD) is reported when they are greater than 10 Mb. Chromosomal aberrations (CNN-LOH/UPD and copy number variation) were annotated based on the hg19 human genome assembly.
Results
Forty-one patients with CN AML and FLT3-ITD mutations were treated at UMGCC between 2004 and 2013. Twenty-six achieved CR after induction therapy and 11 were in ongoing first CR following chemotherapy or alloSCT at the time of this analysis. Cytogenetic data were available from first relapse and, when applicable, subsequent time points for all 15 patients with CN AML with FLT3-ITD mutations at diagnosis who relapsed. These included 12 with FLT3-ITD also documented at relapse (two did not have FLT3-ITD at relapse, and one was not studied). Among these 12 patients, 10 (83%) had cytogenetic changes at relapse. Demographic data, treatment, RFS, OS and cytogenetic and FLT3 mutation data on these 10 patients are summarized in Table 1. Cytogenetic changes occurred at first (9 patients) or second (one patient, Patient 2 in Table 1) relapse. Of note, as is further detailed below, Patient 2 was initially noted to have an abnormal karyotype in second remission, and then relapsed shortly thereafter, with additional structural cytogenetic abnormalities. Two of the patients who developed new cytogenetic changes in first relapse (Patients 3 and 5 in Table 1) did so after initial reinduction chemotherapy. The two patients with FLT3-ITD mutations at diagnosis and at relapse who did not evolve new cytogenetic changes at relapse received therapy similar to that of the patients who did develop cytogenetic changes. Allelic burden data were not consistently available for the patients in our series and relapse and diagnosis allelic burden could not be compared.
Among the 12 AML patients with FLT3-ITD at diagnosis and at relapse, who are the focus of this report, 10 acquired cytogenetic changes at relapse. Cytogenetic changes were structural only in eight patients, numerical only in one (Patient 10 in Table 1), and both structural and numerical in one (Patient 9). Many of the structural abnormalities were rare rearrangements (Table 1). These included t(1;5)(q25;q13); dup(1)(q21q32); t(2;4)(p24;q13); der(2)t(2;?)(q21;?); del(3)(q12); t(3;14)(p21;q11.2); t(4;4)(q21;q35); t(5;13)(q31;q12); der(6)t(6;?)(p22;?); der(7)t(7;?)(p15;?); t(9;22)(q34;q11.2); inv(10)(p11.2q21.2); t(10;13)(p11.1;q11); t(X;10)(q13;q24); t(12;17)(p12;q12); and del(16)(q12). Three representative karyotypes are shown in Figure 1.
Figure 1. Representative relapse metaphase karyotypes.

Representative metaphase karyotypes from three of the FLT3-ITD patients in Table1, Patients 3, 4, and 7, labeled A, B and C, respectively. Panel D, contains enlarged views of the structural abnormalities in each of these patients.
It is notable that Patient 2 acquired a dup(1)(q21q32) in 5 of 19 metaphase cells during second remission. Shortly afterwards, she developed second relapse, with additional structural abnormalities. Although the previously identified 1q duplication was observed in only one metaphase cell in the relapse sample, FISH using a 1p32/1q21 probe demonstrated the presence of this abnormality in 19% of the interphase cells. In addition to the previously identified dup(1)(q21q32), this patient acquired new DNA breaks, with a t(2;4)(p24;q13) in five cells, and a del(16)(q12) in two. Significantly, the additional changes were not seen in the metaphase with the 1q duplication, suggesting that the new aberrations were in new clones, rather than representing clonal evolution.
Patient 9 had a normal karyotype at diagnosis and complex chromosome abnormalities at relapse (179 days after diagnosis) (Figure 2A). The CMA of the diagnosis sample had three polymorphic duplications, three polymorphic deletions, and no >10 Mb CNN-LOH regions, supporting a normal copy number, without any pathogenic deletions or duplications (Fig 2B). The CMA of the relapse sample revealed the same deletions and duplications as found in the diagnosis sample, and also two additional changes acquired at relapse, including a 19.2 Mb deletion of the 11p15.1-p12 segment and a 95.7 Mb CNN-LOH of the entire 13q (Fig 2B).
Figure 2. Chromosome and SNP microarray results of Patient 9.
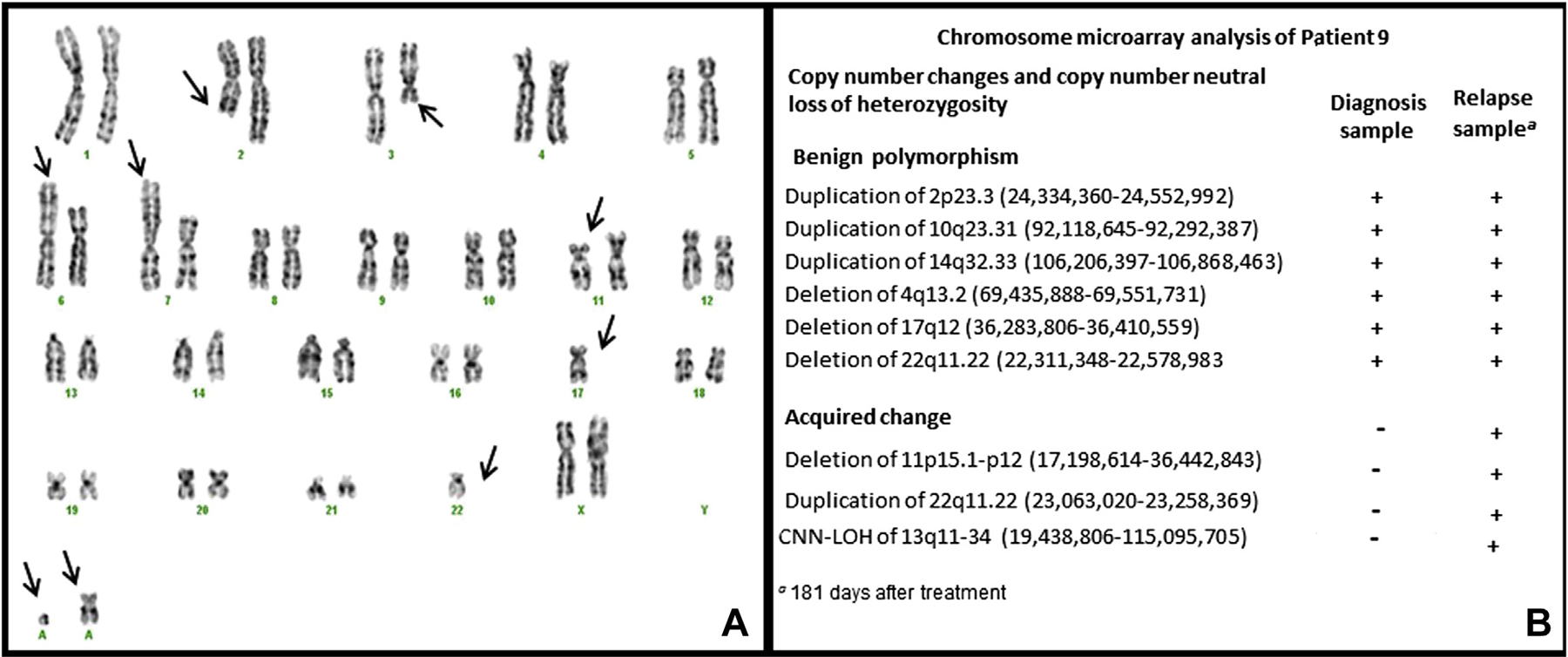
A. G-band karyogram in the relapse sample. Arrows point to abnormal chromosomes including monosomy 17 and 22, rearrangements of 2q21, 3q12, 6p22 and 7p15, possible deletion of 11p, and gain of two marker chromosomes. B. SNP microarray profiles from the diagnosis and relapse samples.
Each cytogenetic change demonstrated at relapse of CN AML with FLT3-ITD in our series was reviewed in the Atlas of Genetics and Cytogenetics in Oncology and Hematology [18] as well as the Mitelman database [19]. Five patients (Patients 1, 2, 3, 4,and 7 in Table 1) developed inversions, translocations, deletions and a duplication with breakpoints previously undescribed in myeloid malignancies. Two patients (Patients 8and 9 in Table 1) evolved complex karyotypes with multiple structural abnormalities, including some with unspecified breakpoints.One patient (Patient 6in Table 1) developed inv(10)(p11.2q21.2), which has been reported in papillary thyroid carcinoma, but not in AML. Another patient (Patient 5 in Table 1) developed t(9;22)(q34;q11.2), with breakpoints in BCR and ABL1 confirmed by FISH. The BCR-ABL1 rearrangement is rare in AML; it has been reportedly previously in association with class II mutations including t(8;21)/RUNX1-RUNX1T1, inv(16)/CBFB-MYH11 and NPM1, but not in association with FLT3-ITD [20].
During the same period, 43 patients with CN AML without FLT3 mutations at diagnosis were treated. Thirty-five of these patients achieved CR and 21 remained in first CR at the time of this analysis. Among the 14 patients who achieved CR and subsequently relapsed, cytogenetic analysis was not performed in two and was inadequate in one. Among the 11 patients who had available cytogenetic results at relapse, five (45%) had cytogenetic changes at relapse, including four with structural changes, and six maintained a normal karyotype. The frequency of cytogenetic changes was lower than for our patients with FLT3-ITD at diagnosis and at relapse (p = 0.05; chi square test), albeit with very small numbers.
Discussion
Several mechanisms are thought to underlie the short RFS that characterizes AML with FLT3-ITD. Among these is genomic instability resulting from DNA damage due to high levels of ROS, and highly error-prone repair of DNA damage due to increased activity of an ALT NHEJ repair pathway for DNA DSBs [14, 15]. We hypothesized that genomic instability could be manifested by new structural chromosome abnormalities at relapse of CN AML with FLT3-ITD. To test this hypothesis, we reviewed our institution’s experience with CN AML with FLT3-ITD mutations to look for cytogenetic evidence of genomic instability in this patient population at relapse. We found that 10 of 12 patients (83%) with CN AML with FLT3-ITD who also had FLT3-ITD at first and, when applicable, subsequent relapse developed new cytogenetic abnormalities; this compares to 5 of 11 patients (45%) with CN AML without FLT3 mutation at diagnosis who developed cytogenetic changes at relapse (p=0.05). Our sample size was very small. Nevertheless, our findings support the concept of a possible role for genomic instability in the short DFS of AML patients with FLT3-ITD.
Strikingly, structural cytogenetic changes at relapse of CN AML with FLT3-ITD included a high proportion of rare translocations, inversions, and duplications. Rare changes included t(1;5)(q25;q13); dup(1)(q21q32); t(2;4)(p24;q13); der(2)t(2;?)(q21;?); del(3)(q12); t(3;14)(p21;q11.2); t(4;4)(q21;q35); t(5;13)(q31;q12); der(6)t(6;?)(p22;?); der(7)t(7;?)(p15;?); t(9;22)(q34;q11.2); inv(10)(p11.2q21.2); t(10;13)(p11.1;q11); t(X;10)(q13;q24); t(12;17)(p12;q12); and del(16)(q12). These structural abnormalities involved many DNA DSBs in various regions of multiple chromosomes, likely representing genomic instability.
The incidence of new cytogenetic abnormalities at relapse was high in our FLT3-ITD patients. Moreover, albeit with a very small sample size, it was higher than that reported in the literature for CN AML cases. Four previous reports addressed cytogenetic changes in CN AML at relapse without regard for FLT3 mutation status, as these four studies antedated routine study of FLT3 mutations [21–24]. In these four series, clonal cytogenetic changes were seen at relapse in 25, 51, 30 and 36 percent of 28, 43, 111 and 56 AML cases that were CN at diagnosis. More recently, Wang et al.[25] reviewed 17 patients with CN AML with an abnormal karyotype at relapse, including 15 characterized with regard to FLT3 mutations at diagnosis. Eight patients had FLT3-ITD, and 7 had wild-type FLT3. New rare structural abnormalities were found at relapse in all 8 cases with FLT3-ITD, and also in 5 of 7 cases with wild-type FLT3. Finally, McCormick et al. [26] found that cytogenetic evolution was more frequent among FLT3-mutated than FLT3-non-mutated AML cases (10 of 14 versus 7 of 21, p= .04). In our series, new structural chromosome changes were found in 11 of 15 patients with CN FLT3-ITD AML at relapse, including 10 of 12 with documented FLT3-ITD at relapse, compared to four of 11 CN AML patients without FLT3 mutations. Confirmation of features unique to relapse of FLT3-ITD AML will require a large series with a large control group. Nevertheless, the finding of a high frequency of structural abnormalities involving various regions of multiple chromosomes supports the hypothesis that increased DNA DSBs and error-prone repair of DNA DSBs result in genomic instability that may contribute to relapse in patients with AML with FLT3-ITD.
Patient 9 in our series (Table 1) presented with neither cytogenetic nor chromosomal microarray alterations at diagnosis, and her relapse sample gained nine cytogenetic aberrations by routine chromosome analysis and two acquired changes by CMA. The acquired changes detected by SNP-microarray in the relapse sample are limited to a 19.2 Mb deletion in 11p and a 95.7 Mb CNN-LOH of 13q, suggesting that most of the complex abnormalities acquired in the relapse karyotype (Fig 2A) represent balanced structural changes. Although microarrays cannot reveal any balanced chromosome structural anomalies, as found by routine chromosome analysis in this patient, SNP microarrays are a useful technique to identify both cryptic chromosomal imbalances and CNN-LOH/UPD. As compared with chromosome banding analysis, SNP microarray results provided more precise breakpoints of an interstitial deletion of 11p. The two breakpoints of the 11p deletion were localized within the PRR5L gene on 11p12 and between the NUCB2 and PIK3C2A genes on 11p15. In addition, SNP microarray analysis revealed the presence of CNN-LOH of 13q, which cannot be recognized by routine chromosome and FISH studies. CNN-LOH of 13q has been found to be the most common abnormality in AML samples at relapse, occurring in 40% of samples [27]. In most forms of cancer, LOH is generally associated with loss of a copy of a tumor suppressor gene. CNN-LOH is the occurrence of LOH in the absence of allelic loss (copy number ≥ 2). CNN-LOH has been shown to be important in cancer biology, as it can lead to tumor suppressor gene inactivation with two mutant alleles (homozygous mutation) possibly through mitotic recombination or a nondisjunction event leading to acquired isodisomy [28]. The FLT3 gene is located at 13q. CNN-LOH of 13q may have led to a change from heterozygosity to homozygosity for FLT3-ITD in this patient. Homozygosity for FLT3-ITD has been found in AML samples at relapse [27], with homozygous mutations likely resulting in a competitive advantage and outgrowth of the clone.
All of the FLT3-ITD AML patients in our series received standard cytarabine and anthracycline induction chemotherapy. Three patients also received the tyrosine kinase inhibitor sorafenib, and one received the histone deacetylase inhibitor vorinostat. Four patients had undergone alloSCT before relapsing with new cytogenetic changes. Nine of the 10 patients with FLT3-ITD AML at diagnosis and at relapse who developed new karyotype abnormalities did so in first relapse, with Patients 3 and 5 (Table 1) evolving cytogenetic changes after initial chemotherapy for AML in first relapse. Patient 2 developed cytogenetic changes while still in morphologic second remission after achieving a second CR with cytarabine and mitoxantrone therapy. She then developed additional cytogenetic changes at the time of second relapse.
The fact that the most of our patients had FLT3-ITD at relapse suggests that this FLT3 mutation was a driver of relapse. Two patients with FLT3-ITD at diagnosis did not have FLT3 mutations identified at relapse, and the possibility that these two patients could actually have had new therapy-related AML (t-AML) rather than relapse of their initial AML cannot be totally excluded. However none of the cytogenetic abnormalities seen in any of our patients at relapse, including these two, were those typically seen in t-AML. Notably, they did not involve 11q23 chromosome [29] nor chromosome 7 [30] breakpoints. Additionally, time to onset was shorter than expected in t-AML [31].
It is clear that more effective treatment is needed for AML with FLT3-ITD. The high frequency of structural cytogenetic changes that we have demonstrated at relapse of AML with FLT3-ITD supports a role of genomic instability in disease progression. Laboratory data [14, 15] support the contribution of ALT-NHEJ, an error-prone pathway for repair of DNA DSBs that has been found to be upregulated in AML cells with FLT3-ITD. This error-prone DNA repair pathway appears to render AML cells with FLT3-ITD more susceptible to genetic instability at baseline. The possibility that cytotoxic chemotherapy potentiates this instability has not been evaluated in this FLT3-ITD AML model, but has been suggested in other tumor types [32]. The contribution of cytotoxic chemotherapy to genomic instability in FLT3-ITD AML should be a point of future investigation. The DNA repair protein poly(ADP)-ribose polymerase 1 (PARP1) is an essential component of the alternative repair pathway, as is DNA ligase III [15]. PARP1 inhibitors are in clinical trials [33] and DNA ligase III inhibitors are in development [34]. Down-regulation of DNA ligase III expression levels in a FLT3-ITD cell line using siRNA oligonucleotides decreased the frequency of DNA repair errors, supporting further investigation of DNA ligase III inhibitors as a therapeutic approach [15]. Our data may support a possible therapeutic role for these inhibitors in AML with FLT3-ITD.
Acknowledgements
Grant support: Leukemia and Lymphoma Society Translational Research Award (MRB), University of Maryland, Baltimore UMMG Cancer Research Grant #CH 649 CRF, State of Maryland Department of Health and Mental Hygiene (DHMH) under the Cigarette Restitution Fund Program, and NCI Cancer Center Support Grant P30 CA134274 (UMGCC)
Footnotes
Conflict of Interest: None of the authors of this manuscript have any undisclosed conflicts of interest to report.
References
- [1].Carow CE, Levenstein M, Kaufmann SH, Chen J, Amin S, Rockwell P, et al. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood. 1996;87:1089–96. [PubMed] [Google Scholar]
- [2].Rosnet O, Bühring HJ, Marchetto S, Rappold I, Lavagna C, Sainty D, et al. Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia. 1996;10:238–48. [PubMed] [Google Scholar]
- [3].Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–9. [DOI] [PubMed] [Google Scholar]
- [4].Fröhling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100:4372–80. [DOI] [PubMed] [Google Scholar]
- [5].Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100:59–66. [DOI] [PubMed] [Google Scholar]
- [6].Schlenk RF, Döhner K, Krauter J, Fröhling S, Corbacioglu A, Bullinger L, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–18. [DOI] [PubMed] [Google Scholar]
- [7].Whitman SP, Maharry K, Radmacher MD, Becker H, Mrozek K, Margeson D, et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010;116:3622–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sengsayadeth SM, Jagasia M, Engelhardt BG, Kassim A, Strickland SA, Goodman S, et al. Allo-SCT for high-risk AML-CR1 in the molecular era: impact of FLT3/ITD outweighs the conventional markers. Bone Marrow Transplantation. 2012;47:1535–7. [DOI] [PubMed] [Google Scholar]
- [10].Spiekermann K, Bagrintseva K, Schwab R, Schmieja K, Hiddemann W. Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin Cancer Res. 2003;9:2140–50. [PubMed] [Google Scholar]
- [11].Rocnik JL, Okabe R, Yu JC, Lee BH, Giese N, Schenkein DP, et al. Roles of tyrosine 589 and 591 in STAT5 activation and transformation mediated by FLT3-ITD. Blood. 2006;108:1339–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zheng R, Bailey E, Nguyen B, Yang X, Piloto O, Levis M, et al. Further activation of FLT3 mutants by FLT3 ligand. Oncogene. 2011;30:4004–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sato T, Yang X, Knapper S, White P, Smith BD, Galkin S, et al. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood. 2011;117:3286–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sallmyr A, Fan J, Datta K, Kim KT, Grosu D, Shapiro P, et al. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 2008;111:3173–82. [DOI] [PubMed] [Google Scholar]
- [15].Fan J, Li L, Small D, Rassool F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: implications for genomic instability and therapy. Blood. 2010;116:5298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Murphy KM, Levis M, Hafez MJ, Geiger T, Cooper LC, Smith BD, et al. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn. 2003;5:96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shaffer L, McGowan-Jordan J, M S. ISCN 2013: An International System for Human Cytogenetic Nomenclature. Recommendations of the International Standing Committee on Human Cytogenetic Nomenclature. Basel: Karger; 2013. [Google Scholar]
- [18].Atlas of Genetics and Cytogenetics in Oncology and Hematology.
- [19].Mitelman F, Johannson B, Mertens F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer.
- [20].Bacher U, Haferlach T, Alpermann T, Zenger M, Hochhaus A, Beelen DW, et al. Subclones with the t(9;22)/BCR-ABL1 rearrangement occur in AML and seem to cooperate with distinct genetic alterations. Br J Haematol. 2011;152:713–20. [DOI] [PubMed] [Google Scholar]
- [21].Testa JR, Rowley JD. Cytogenetic patterns in acute nonlymphocytic leukemia. Virchows Archiv B: Cell Pathology. 1978;29:65–72. [DOI] [PubMed] [Google Scholar]
- [22].Garson OM, Hagemeijer A, Sakurai M, Reeves BR, Swansbury GJ, Williams GJ, et al. Cytogenetic studies of 103 patients with acute myelogenous leukemia in relapse. Cancer Genetics and Cytogenetics. 1989;40:187–202. [DOI] [PubMed] [Google Scholar]
- [23].Estey E, Keating MJ, Pierce S, Stass S. Change in karyotype between diagnosis and first relapse in acute myelogenous leukemia. Leukemia. 1995;9:972–6. [PubMed] [Google Scholar]
- [24].Kern W, Haferlach T, Schnittger S, Ludwig WD, Hiddemann W, Schoch C. Karyotype instability between diagnosis and relapse in 117 patients with acute myeloid leukemia: implications for resistance against therapy. Leukemia. 2002;16:2084–91. [DOI] [PubMed] [Google Scholar]
- [25].Wang ES, Sait SN, Gold D, Mashtare T, Starostik P, Ford LA, et al. Genomic, immunophenotypic, and NPM1/FLT3 mutational studies on 17 patients with normal karyotype acute myeloid leukemia (AML) followed by aberrant karyotype AML at relapse. Cancer Genetics and Cytogenetics. 2010;202:101–7. [DOI] [PubMed] [Google Scholar]
- [26].McCormick SR, McCormick MJ, Grutkoski PS, Ducker GS, Banerji N, Higgins RR, et al. FLT3 mutations at diagnosis and relapse in acute myeloid leukemia: cytogenetic and pathologic correlations, including cuplike blast morphology. Arch Pathol Lab Med. 2010;134:1143–51. [DOI] [PubMed] [Google Scholar]
- [27].Raghavan M, Smith LL, Lillington DM, Chaplin T, Kakkas I, Molloy G, et al. Segmental uniparental disomy is a commonly acquired genetic event in relapsed acute myeloid leukemia. Blood. 2008;112:814–21. [DOI] [PubMed] [Google Scholar]
- [28].Bacolod MD, Schemmann GS, Giardina SF, Paty P, Notterman DA, Barany F. Emerging paradigms in cancer genetics: some important findings from high-density single nucleotide polymorphism array studies. Cancer Res. 2009;69:723–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bloomfield CD, Archer KJ, Mrozek K, Lillington DM, Kaneko Y, Head DR, et al. 11q23 balanced chromosome aberrations in treatment-related myelodysplastic syndromes and acute leukemia: report from an international workshop. Genes, Chromosomes andCancer. 2002;33:362–78. [DOI] [PubMed] [Google Scholar]
- [30].Arana-Yi C, Block AW, Sait SN, Ford LA, Barcos M, Baer MR. Therapy-related myelodysplastic syndrome and acute myeloid leukemia following treatment of acute myeloid leukemia: possible role of cytarabine. Leukemia Research. 2008;32:1043–8. [DOI] [PubMed] [Google Scholar]
- [31].Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102:43–52. [DOI] [PubMed] [Google Scholar]
- [32].Fonseca FL, Sant Ana AV, Bendit I, et al. Systemic chemotherapy induces microsatellite instability in the peripheral blood mononuclear cells of breast cancer patients. Breast Cancer Res. 2005;7:R28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kummar S, Ji J, Morgan R, Lenz HJ, Puhalla SL, Belani CP, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res. 2012;18:1726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tobin LA, Robert C, Rapoport AP, Gojo I, Baer MR, Tomkinson AE, et al. Targeting abnormal DNA double-strand break repair in tyrosine kinase inhibitor-resistant chronic myeloid leukemias. Oncogene. 2013;32:1784–93. [DOI] [PMC free article] [PubMed] [Google Scholar]