

Abstract
SARS-CoV-2, the virus causing COVID-19, has infected millions and has caused hundreds of thousands of fatalities. Risk factors for critical illness from SARS-CoV-2 infection include male gender, obesity, diabetes, and age >65. The mechanisms underlying the susceptibility to critical illness are poorly understood. Of interest, these comorbidities have previously been associated with increased signaling of Th17 cells. Th17 cells secrete IL-17A and are important for clearing extracellular pathogens, but inappropriate signaling has been linked to acute respiratory distress syndrome. Currently there are few treatment options for SARS-CoV-2 infections. This review describes evidence linking risk factors for critical illness in COVID-19 with increased Th17 cell activation and IL-17 signaling that may lead to increased likelihood for lung injury and respiratory failure. These findings provide a basis for testing the potential use of therapies directed at modulation of Th17 cells and IL-17A signaling in the treatment of COVID-19. The Journal of Immunology, 2020, 205: 892–898.
Severe acute respiratory syndrome coravirus 2 (SARS-CoV-2), the cause of COVID-19, has infected over 8 million people globally and caused over 400,000 deaths. The mechanisms underlying susceptibility to critical illness with COVID-19 are still poorly understood. Roughly 80% of people infected with SARS-CoV-2 are either asymptomatic or mildly symptomatic (1), suggesting that in most individuals, the immune system is able to control viral replication and eliminate the virus appropriately. However, in up to 20% of infections, patients manifest more severe illness with respiratory involvement requiring oxygen support in ~14% of those infected and critical illness requiring intensive care unit (ICU) admission in ~5% of those infected (2). Those who develop critical illness most often have severe hypoxemia and may also develop shock requiring vasopressors and acute renal failure requiring hemodialysis (3). Risk factors for developing these severe manifestations of COVID-19 are age >65, hypertension, diabetes, obesity, and male sex, among others (4, 5). Many of these risk factors have been previously associated with increased Th17 cell numbers and/or increased IL-17A signaling. Considerable evidence demonstrates that Th17 cells/IL-17A signaling are important for bacterial and fungal host responses and may also be involved in the pathogenesis of acute respiratory distress syndrome (ARDS). This review aims to describe the current state of knowledge of Th17 cells and IL-17A signaling in both lung pathophysiology and COVID-19 comorbidities and discusses the rationale for therapeutic inhibition of Th17 cells and/or IL-17A signaling in COVID-19.
SARS-coronavirus infections: pathogenesis
Although the clinical characteristics of SARS-CoV, the cause of SARS, and SARS-CoV-2 differ, particularly in terms of transmission (higher for SARS-CoV-2) and mortality (higher for SARS-CoV), they do exhibit similarities in terms of mechanism of viral entry and host immune evasion. Evidence shows that SARS-CoV can avoid detection in most cell types through the avoidance of a type-I IFN response, which is necessary for inhibition of viral replication in host cells (6). A notable exception are plasmacytoid dendritic cells that have been shown to secrete type-I IFNs in response to ssRNA via TLR-7 signaling in endosomes (6). As the virus evades immune recognition, there is a lag in immune response, allowing time for viral replication. Siddiqi and Mehra (7) have suggested that patients with COVID-19 start to manifest symptoms at the time of immune system activation during initial cellular recognition and initiation of a viral elimination program in host cells and then progresses to more severe symptoms as immune cell recognition and activation/hyperactivation occurs. Hyperactivation can be seen in critically ill patients with COVID-19 in a process that has been termed “cytokine storm” leading to profound hypotension, severe hypoxemia, and multiorgan system failure (7). Therapies directed at neutralization of IL-6 (anti–IL-6 or anti–IL-6R–neutralizing Abs) have been proposed as a treatment for “cytokine storm,” but randomized clinical trials demonstrating efficacy have not emerged. Uncontrolled studies have suggested a survival benefit in in patients with COVID-19 and ARDS treated with high dose rIL-1R antagonist (rIL-1RA) therapy (8). Notably, these proinflammatory pathways may be upregulated as a result of activation of Th17 cells and increased IL-17A– induced signaling in response to SARS-CoV-2 infection.
SARS-CoV-2 infection of the lower respiratory tract causes pulmonary inflammation leading to cough, shortness of breath, and hypoxemia. COVID-19 commonly manifests as hypoxemia and ARDS in patients admitted to the ICU (3). Histologic findings in the lungs from COVID-19 autopsies differ by phase of disease ranging from edema and pneumocyte hyperplasia in infections found incidentally to diffuse alveolar damage and exudates with robust mononu-clear, predominantly lymphocytic, inflammation in critically ill patients (9).
A case for targeting Th17 cells and IL-17A signaling
Several groups have identified high levels of Th17 cells in the periphery of patients infected with SARS-CoV-2 (10, 11). This may have been expected given that prior to the COVID-19 pandemic, evidence supported a relationship between IL-17A and severe respiratory infection and respiratory failure. In patients with ARDS, we have shown that there exists a strong relationship between alveolar IL-17A levels and alveolar protein, an important correlate of severity of lung injury (12). We also demonstrated an association between IL-17A levels and the degree of organ dysfunction (12). Increased IL-17A signaling or increased Th17/ T regulatory cell (Treg) ratios have also been correlated with disease severity in mouse models of LPS-induced acute lung injury (13) and in respiratory syncytial virus (RSV) infections in children (14). In contrast, we have also reported that higher levels of Th17 cells in alveolar fluid from patients being invasively mechanically ventilated is associated with protection from ventilator associated pneumonias (15) and Almansa et al. (16) reported that detectable circulating IL-17A within the first 24 h of 2009 pandemic influenza infections predicted better outcomes. These somewhat conflicting relationships between IL-17A and susceptibility and severity of respiratory failure might be explained by differences in pathogen (viral versus bacterial) or timing (early versus late) of sampling. An understanding of the relationship between circulating IL-17A or Th17 activation in host response to SARS-CoV-2 is still emerging, but given its prior demonstrated association with poor outcomes in critically ill patients with Middle East respiratory syndrome (12, 17), we hypothesize that increased activity of Th17 cells early in the course of infection with SARS-CoV-2 and subsequent signaling through IL-17A leads to worse critical illness, either directly via action of IL-17A signaling or indirectly by inhibiting a Th1 or Treg response that would have led to viral clearance. The mechanisms driving the pathology in COVID-19 pneumonia are currently poorly understood and will require further study, however our hypothesis of Th17 cell and IL-17A involvement is based on animal models of acute lung injury as well as human studies of cytokine signaling in the setting of ARDS.
Th17 and susceptibility to infection
Th17 cells are a subset of Th cells that are important for host defense against fungal and extracellular bacterial infections (18). Th17 cell signaling leads to secretion of proinflammatory cytokines such as IL-17A. In the lungs, Th17 cell signaling has also been associated with release of antibacterial peptides, chemokines that lead to neutrophil recruitment, and increased expression of cell adhesion molecules such as ICAM-1 (19). Patients that lack functional Th17 cells (hyper-IgE syndrome or “Job syndrome”) are at increased risk for recurrent and often severe pulmonary infections, pneumatoceles, eczema, staphylococcal abscesses, mucocutaneous candidiasis, and abnormalities of bone and connective tissue (18). In murine models, IL-17A is necessary for Klebsiella pneumoniae and Mycoplasma pneumoniae clearance (20, 21); in HIV-infected rhesus macaques, Th17 cells are preferentially depleted from the gastrointestinal tract, which is thought to place them at risk for increased mucosal permeability, resulting in bacterial translocation (22). Following influenza infections, there is a high risk for secondary bacterial pneumonia with Staphylococcus aureus which may be a result of increased type-I IFN signaling inhibiting Th17 cell signaling as has been shown in mice (23). An important consideration is that although Th17 cells are a major source of IL-17A, it is also produced by CD8+ T cells (24), NK T cells (25), γδ-T cells (26), innate lymphoid cells (27), mucosa-associated invariant T cells (28), and neutrophils (29, 30). Production of IL-17A by these other cell types may, in part, explain the fact that there does not appear to be a strong correlation between Th17 cell numbers and IL-17A levels in bronchoalveolar lavage fluid as we have shown in patients with nosocomial pneumonia (15). There is a paucity of evidence that fully explores the compartment-specific aspects to Th17 cells and IL-17A production (e.g., alveolar versus circulating). However, we have found that levels of IL-17A in the periphery correlated well with levels of IL-17A in bronchoalveolar lavage fluid (31). There is no current published information regarding the relative number and function of Th17 cells in the lung and intravascular spaces during respiratory infections or lung injury, nor are there studies that investigate how levels of IL-17A in the periphery correlate to numbers of Th17 cells in the airways.
Th17 and the inflammatory response
Th17 cells differentiate in the setting of a proinflammatory cytokine milieu and secrete several proinflammatory cytokines, including IL-17A, IL-17F, and IL-22 (19). In mice, TGF-β and IL-6 increase Th17 cell differentiation via activation of the transcription factor STAT3 (32). In humans, the exact combination of cytokines necessary for initiation of Th17 cell differentiation is not established, but IL-1β and IL-23 both contribute to Th17 cell differentiation with possible contributions from TGF-β and IL-6 (19, 32) (Fig. 1). STAT3 activates transcription of retinoic acid receptor–related orphan receptor γt (RORγt). RORγt, also known as the “master regulator” of Th17 cell differentiation, activates transcription of genes such as IL-17A, IL-17F, and IL-22 (19). In humans, TGF-β has dose-dependent effects on T cell differentiation. At low levels, TGF-β increases RORγt transcription, but at higher levels, TGF-β promotes transcription of FOXP3, a regulator of differentiation of Tregs, leading to inhibition of RORγt transcription (19) and suppression of Th17 cell differentiation. Studies suggest that there is inhibition between different Th cell pathways as Th1 cytokines (IFN-γ) and Th2 cytokines (IL-4 and IL-12) are able to inhibit Th17 cell differentiation (32, 33) and TGF-β inhibits differentiation of Th1 and Th2 cells (33).
FIGURE 1.
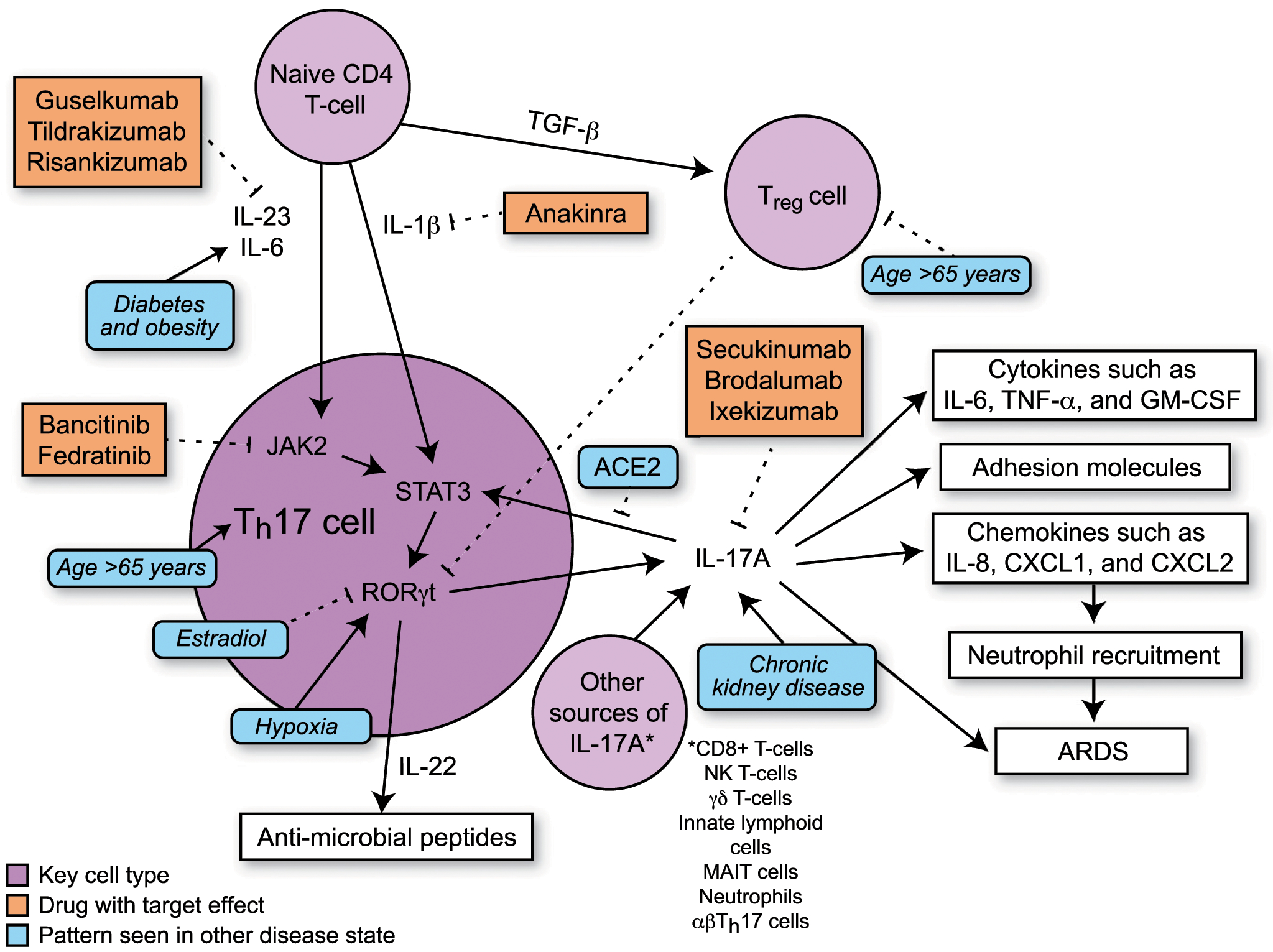
Schematic showing Th17 differentiation and downstream signaling. Comorbidities of critical illness in COVID19 and their effects on Th17 signaling are depicted in blue. Inhibitors of Th17 signaling are depicted in orange.
There are also links between IL-17A and angiotensin-converting enzyme (ACE) 2, the receptor used by SARS-CoV-2 to enter cells. Sodhi et al. (34) reported that in a murine model of severe bacterial pneumonia, ACE2 regulates neutrophilic infiltration in the lungs in an IL-17A–dependent manner. These investigators found that recombinant ACE2 inhibited IL-17A–induced activation of STAT3, leading to reduced neutrophil infiltration (34) and they proposed a model in which ACE2 expression is downregulated early in infection and then upregulated during resolution of infection to assist with recovery. Furthermore, they suggested that if ACE2 expression is interrupted during recovery, there is a tendency toward prolonged inflammation (34). Glowacka et al. (35) showed that in vitro infection with SARS-CoV-2 downregulates the expression of ACE2 after 24–72 h. This suggests a potential mechanism whereby SARS-CoV-2 interrupts ACE2 expression in the lung, leading to disinhibition of IL-17A expression, resulting in prolonged lung inflammation as has been observed in severe COVID-19. Th17 cell differentiation may also be regulated by hypoxia that occurs during the course of severe lung injury and IL-17A has been shown in vitro to induce human lung microvascular cell migration leading to angiogenesis (36). Hypoxia inducible factor (HIF)–1α, a transcription factor upregulated by hypoxia, can induce RORγt transcription, which tips the balance of Th17/Treg differentiation to favor increased Th17 activity (37). Hypoxia can also induce expression of microRNA (miR)-155 (38), a miR that upregulates Th17 cell differentiation and activity (39, 40). These studies suggest that Th17 cell differentiation may be potentiated by reduced ACE2 levels and the extremely hypoxic environment seen in severe COVID-19.
IL-17A and ARDS
ARDS is a major driver of mortality in the setting of many infections, including influenza, SARS-CoV-2, and bacterial sepsis. ARDS is characterized by noncardiogenic pulmonary edema as a result of increased alveolar endothelial and epithelial damage with resultant hypoxemia (41). IL-17A has been implicated as a possible driver of the disease process in ARDS via neutrophil-dependent and -independent mechanisms. Neutrophils are recruited in the setting of lung injury during infection as part of pathogen clearance but may cause tissue damage by secreting oxidants and proteases (13, 42). In a murine model of LPS-induced acute lung injury, Ag-dependent αβTh17 cells were identified as a source of IL-17A. In mice with engineered deficiency in αβTh17 cells, there was decreased permeability of the epithelial barrier in experimental lung injury (13). Similar results were observed in mice deficient in IL-17RC (13) or that were treated with anti–IL-17A (43). There was no difference in lung neutrophils after LPS challenge in the αβTh17-deficient mice who were protected against lung injury (13). Another study showed that a genetic deficiency of IL-17A is also protective in murine LPS-induced lung injury but found that the primary source of IL-17A was innate lymphoid cells and, in contrast with the prior study, found that this protection was strongly associated with reduced lung neutrophil recruitment (44). As mentioned above, we have shown that circulating and alveolar levels of IL-17A are associated with increased numbers of alveolar neutrophils, alveolar permeability, and organ dysfunction in patients with ARDS (31). Others have reported that increased circulating IL-17A levels measured early in the course of sepsis-related ARDS is associated with death by day 7, and increased IL-17A levels were also associated with a lower PaO2/FiO2 ratio (43). These data strongly link the IL-17 pathway to more severe lung injury, although the mechanism by which this occurs is not completely elucidated.
Th17 cell signaling and IL-17A involvement in the pathogenesis of other viral infections
There is evidence supporting a role for the IL-17 pathway in ARDS in the setting of influenza A. Li et al. (45) used a mouse model of H1N1 infection to show that IL-17A–neutralizing Abs, but not TNF-α–neutralizing Abs, greatly decreased acute lung injury. Similarly, mice genetically deficient in IL-17A had less weight loss and higher survival after challenge with influenza A (46). In humans, patients hospitalized with H1N1 Influenza A, had elevated circulating Th17 (IL-8, IL-9, IL-17A, IL-6) cytokines compared with patients infected with H1N1 that did not require hospitalization (47); however, as mentioned previously, others have shown that detection of IL-17 early in the disease course correlated to better outcomes (16). Although there are clearly differences in the epidemiologic risk factors for pandemic influenza (48) and COVID-19, these data support a role for the IL-17 pathway in viral-induced ARDS.
Outside of influenza, Th17 cells and the IL-17 pathway have been implicated in several other forms of viral infections. Th17 cells have been implicated in the pathogenesis of RSV infections in children who do not commonly develop ARDS but can develop respiratory distress as a result of severe bronchiolitis (14). Studies have shown increased levels of IL-17A in tracheal aspirates and plasma of children infected with RSV (14). However, it appears that the imbalance of Th17 cells to Tregs is most highly associated with clinical severity of RSV infection (14). IL-17A has also been reported to be a mediator of hepatic tissue injury in murine models of HSV and dengue infections, potentially via its role in recruiting neutrophils to the liver (49). These findings provide evidence supporting the clinical relevance of the IL-17 pathway in severe lower respiratory viral infections.
Risk factors associated with critical illness in SARS-CoV-2 infections
Risk for COVID-19 is strongly associated with sex, obesity, diabetes, hypertension, and chronic kidney disease (CKD), and prior evidence suggests all of these characteristics are associated with higher Th17 cell or IL-17A signaling activity. Case series from Italy (4) and the United States (3) show that 60–80% of the patients admitted with critical illness from COVID-19 were men. Evidence from mice suggests that reduced IL-17 pathway activity, due to estradiol-induced impairment of dendritic cell production of IL-23 and, in turn, Th17 activity, may lead to susceptibility to Candida albicans (50). Notably, estrogen deficiency can lead to increased differentiation of Th17 cells and downregulation of Tregs (51). This is thought to occur by the release of estradiol-mediated inhibition of RORγt induction of Th17 cell differentiation (52). In a murine model of coxsackievirus B3–induced myocarditis, an inflammatory disease that exhibits a male predominance, male mice show augmented induction of Th17 cells relative to females, and the male-specific increase in myocarditis can be ameliorated by an IL-17A–neutralizing Ab (53). These data suggest a relative suppression of Th17 responses in females compared with males and support the hypothesis that an increased Th17 response might contribute to the increased risk for severe COVID-19 illness in men.
Obesity is another risk factor for critical illness in COVID-19 that has potential links to IL-17. Winer et al. (54) have shown that obesity predisposes to a Th17 bias in mice via increased signaling of IL-6 without an effect on Th1, Th2, or Treg cell populations. In humans, it has been shown that obese women have higher circulating levels of IL-17A, but not IFN-γ or IL-12, compared with lean women (55). These data suggest increased activity of the IL-17 pathway in the setting of obesity which could predispose to worse outcomes in COVID19.
Two other common comorbidities linked to risk for COVID19 are CKD and hypertension. Both have been linked to altered IL-17 pathway activity. Patients on hemodialysis have been reported to have increased levels of IL-17A as well as an imbalance in Th17/Treg cell subsets with increased Th17 cell activity and decreased Treg activity (56). Presence of CKD has been linked to higher IL-17RA protein levels and individuals with CKD have higher levels of Th17 cell subsets (57). In murine models, Th17 cells and IL-17A have been associated with hypertension in the setting of angiotensin II signaling. Studies have shown that in an experimental mouse model of angiotensin II induced hypertension, genetic deficiency in IL-17A leads to lower blood pressures (58), and an imbalance in Th17/Tregs has also been associated with increased blood pressure (59). In humans, increased levels of IL-17A are observed in the serum of hypertensive diabetic patients compared with nonhypertensive diabetic controls (58) and in patients with hypertension compared with healthy controls (60). These studies suggest that individuals with either CKD or hypertension may be predisposed to increased Th17 cell and/or IL-17 pathway activity that may be relevant to COVID-19 outcomes.
Patients with diabetes may also have altered Th17 activity. Higher levels of Th17 cell activity are observed in individuals with type-I and type II diabetes compared with controls (61, 62) and evidence suggests that this increase may be a result of signaling via miRs such as miR-146a. miR-146a negatively regulates the expression of proinflammatory cytokines such as IL-1R–associated kinase 1 (IRAK1), TNF receptor–associated factor 6 (TRAF6), IL-21, and IL-6 (63). Li et al (64) have shown that miR-146a regulates Th17 cell differentiation via negative regulation of IL-6 and IL-21. Lower levels of miR-146a are present in the serum of individuals with diabetes and are associated with increased proinflammatory cytokine signaling and poor glycemic control (63, 65). Thus, lower miR-146a levels and, in turn, higher Th17 activity might be a characteristic of diabetes that might influence risk for severe COVID-19.
Increased age (>65) is a strong risk factor for critical illness in COVID-19, and evidence exists that the IL-17A/Th17 pathway is altered in the elderly. Several groups have shown increased percentages of Th17 cells or IL-17A signaling in PBMCs in healthy elderly individuals compared with healthy middle aged and young controls [>65 compared with <65 (66); 70–80 compared with 20–30 y olds (67)]. Others have shown that percentages of circulating Th17 cells are decreased in the elderly (>65), but the ratio of Th17 cells to Tregs also changed with age to favor increased signaling through Th17 cells relative to Tregs (68). Serrano et al. (69) investigated responses to Helicobacter pylori infections in adults and children and they found that whereas healthy gastric mucosa had equal levels of Th17 cells, in the setting of an H. pylori infection, adults had higher levels of Th17 cell activation and increased IL-17A in the infected mucosa relative to children. They also found that Th1 cell activity was increased whereas Treg activity was lower in the gastric mucosa of adults (69). These findings suggest older adults may be predisposed to increased Th17 activity via increased cell numbers or in the dysregulation of the balance between Th17 and Treg cell activity.
Possible therapeutics
Based on this body of evidence, several therapies that target Th17 cell signaling warrant consideration as potential treatments for SARS-CoV-2 infections. There are three IL-17A inhibitors that are currently approved by the Food and Drug Administration for treatment of dermatologic and rheumatologic diseases: secukinumab, a human anti–IL-17A Ab; brodalumab, a human anti–IL-17RA mAb; and ixekizumab, a humanized IgG4 mAb that neutralizes IL-17A (70). There are also several IL-23 inhibitors including risankizumab, guselkumab, and gildrakizumab, which are all selective human mAbs targeting the p19 subunit of IL-23 (71). Guselkumab and tildrakizumab are currently approved for the treatment of psoriasis and risankizumab is currently undergoing clinical trials to treat psoriasis, ulcerative colitis, and Crohn disease. There are several case reports from Italy of patients on IL-17A or IL-23 inhibitors for dermatologic conditions that have had either mild or asymptomatic COVID-19 (72–74). In a retrospective multicenter observational study of 5206 patients prescribed immunomodulatory medications for psoriasis (including inhibitors of IL-17A, IL-12/23, or IL-23), there were only four hospitalizations and no deaths from COVID-19 despite an overrepresentation of risk factors for severe disease, such as age and male sex, and high prevalence in the surrounding population (75). These studies suggest that being on an IL-17A inhibitor may be safe or even protective in the setting of exposure to SARS-CoV-2. A related therapeutic target is the JAK/STAT pathway. JAK2 activates STAT3, which then induces RORγt expression and Th17 cell differentiation and can block IL-23–induced IL-17A production in vitro (14). There is currently an ongoing clinical trial of the drug baricitinib, which is a JAK1/JAK2 inhibitor (76). This drug was chosen by an artificial intelligence algorithm for its ability to inhibit viral entry into alveolar cells and its potential to decrease inflammation in patients (77). It should be noted that several other biopharmaceuticals are being tested that target mediators up and downstream of the IL-17 pathway. As mentioned, treatment with anakinra, a recombinant modified IL-1R antagonist, was reported to be associated with improved outcomes in COVID-19 in a small, uncontrolled trial (8). Inhibitors of cytokines/chemokines that are associated with Th17 cell signaling, such as TNF-α and IL-8 are currently being studied in the setting of SARS-CoV-2 infections. There are also small molecules that inhibit RORγt that are being investigated as an alternative strategy for Th17 modulation in animal models (78).
Of course, there are concerns regarding the numerous side effects observed with immunomodulatory drugs. Baricitinib has been associated with development of infections, malignancies, and thrombosis (79). IL-17A and IL-23 inhibitors also place patients at risk for infections and hematologic and gastrointestinal side effects. It is important to note that these adverse events have been observed during chronic administration of the drugs. None of these medications have been tested for safety with acute administration in critically ill patients, so extra care will need to be taken if applied to treat severe COVID-19. Timing of administration will also be an important consideration. Early administration might be preferable to maximize the chances of avoiding severe or fatal COVID-19 but with discontinuation of the intervention before risk of opportunistic infection becomes excessive.
Conclusions
Several comorbidities have been consistently associated with poor outcomes in SARS-CoV-2 infections, including sex, diabetes, obesity, hypertension, and CKD. Evidence suggests that patients with these comorbidities may have pre-existing alterations in the Th17/IL-17A axis that might be relevant to the pathology seen in COVID-19. It is possible that increased Th17 activity, IL-17A signaling, or dysregulation in Th17/Treg cell balance may play a causal role in the chronic inflammation seen in these conditions. We hypothesize that the Th17/IL-17A axis may be “primed” for an overexuberant Th17 response after SARS-CoV-2 infection, leading to both increased inflammation and impaired adaptive immune responses. Thus, delivery of a therapy targeting the Th17/IL-17A axis early in COVID-19 may be efficacious in dampening the disease course and improving outcomes such as severe respiratory failure. It will be important to think carefully about the timing and specificity of the intervention so as to avoid adverse effects such as opportunistic infections. On the plus side, there is considerable evidence that these candidates can be given safely in the outpatient and acute care setting. However, care must be taken to consider the impact of these medications in the critically ill. Nonetheless, given the ongoing high morbidity and mortality seen in critically ill patients with COVID-19, new candidates for therapy are badly needed. The Th17/IL-17A axis is a plausible therapeutic target for COVID-19 that should be considered for investigation in clinical trials.
Abbreviations used in this article:
- ACE
angiotensin-converting enzyme
- ARDS
acute respiratory distress syndrome
- CKD
chronic kidney disease
- ICU
intensive care unit
- miR
microRNA
- RORγt
retinoic acid receptor–related orphan receptor γt
- RSV
respiratory syncytial virus
- SARS
severe acute respiratory syndrome
- SARS-CoV-2
severe acute respiratory syndrome coravirus 2
- Treg
T regulatory cell
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Li R, Pei S, Chen B, Song Y, Zhang T, Yang W, and Shaman J. 2020. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2). Science 368: 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Z, and McGoogan JM. 2020. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA DOI: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 3.Bhatraju PK, Ghassemieh BJ, Nichols M, Kim R, Jerome KR, Nalla AK, Greninger AL, Pipavath S, Wurfel MM, Evans L, et al. 2020. Covid-19 in critically ill patients in the Seattle region - case series. N. Engl. J. Med 382: 2012–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, Cereda D, Coluccello A, Foti G, Fumagalli R, et al. ; COVID-19 Lombardy ICU Network. 2020. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy region, Italy. JAMA 323: 1574–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrilli CM, Jones SA, Yang J, Rajagopalan H, O’Donnell L, Chernyak Y, Tobin KA, Cerfolio RJ, Francois F, and Horwitz LI. 2020. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ 369: m1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiel V, and Weber F. 2008. Interferon and cytokine responses to SARS-coronavirus infection. Cytokine Growth Factor Rev. 19: 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siddiqi HK, and Mehra MR. 2020. COVID-19 illness in native and immunosuppressed states: a clinical-therapeutic staging proposal. J. Heart Lung Transplant 39: 405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, Oltolini C, Castiglioni B, Tassan Din C, Boffini N, et al. 2020. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2: e325–e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanley B, Lucas SB, Youd E, Swift B, and Osborn M. 2020. Autopsy in suspected COVID-19 cases. J. Clin. Pathol 73: 239–242. [DOI] [PubMed] [Google Scholar]
- 10.Wu D, and Yang XO. 2020. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor Fedratinib. J. Microbiol. Immunol. Infect 53: 368–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, Liu S, Zhao P, Liu H, Zhu L, et al. 2020. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med 8: 420–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahallawi WH, Khabour OF, Zhang Q, Makhdoum HM, and Suliman BA. 2018. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine 104: 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li JT, Melton AC, Su G, Hamm DE, LaFemina M, Howard J, Fang X, Bhat S, Huynh K-M, O’Kane CM, et al. 2015. Unexpected role for adaptive αβTh17 cells in acute respiratory distress syndrome. J. Immunol 195: 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mangodt TC, Van Herck MA, Nullens S, Ramet J, De Dooy JJ, Jorens PG, and De Winter BY. 2015. The role of Th17 and Treg responses in the pathogenesis of RSV infection. Pediatr. Res 78: 483–491. [DOI] [PubMed] [Google Scholar]
- 15.Orlov M, Dmyterko V, Wurfel MM, and Mikacenic C. 2017. Th17 cells are associated with protection from ventilator associated pneumonia. PLoS One 12: e0182966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Almansa R, Socias L, Ramirez P, Martin-Loeches I, Vallés J, Loza A, Rello J, Kelvin DJ, León C, Blanco J, et al. 2011. Imbalanced pro- and anti-Th17 responses (IL-17/granulocyte colony-stimulating factor) predict fatal outcome in 2009 pandemic influenza. Crit. Care 15: 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faure E, Poissy J, Goffard A, Fournier C, Kipnis E, Titecat M, Bortolotti P, Martinez L, Dubucquoi S, Dessein R, et al. 2014. Distinct immune response in two MERS-CoV-infected patients: can we go from bench to bedside? PLoS One 9: e88716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, et al. 2008. Impaired T(H) 17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452: 773–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai H-C, Velichko S, Hung L-Y, and Wu R. 2013. IL-17A and Th17 cells in lung inflammation: an update on the role of Th17 cell differentiation and IL-17R signaling in host defense against infection. Clin. Dev. Immunol 2013: 267971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, Schwarzenberger P, Shellito JE, and Kolls JK. 2001. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am. J. Respir. Cell Mol. Biol 25: 335–340. [DOI] [PubMed] [Google Scholar]
- 21.Wu Q, Martin RJ, Rino JG, Breed R, Torres RM, and Chu HW. 2007. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 9: 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milner JD, Sandler NG, and Douek DC. 2010. Th17 cells, Job’s syndrome and HIV: opportunities for bacterial and fungal infections. Curr. Opin. HIV AIDS 5: 179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, Khader SA, Dubin PJ, Enelow RI, Kolls JK, and Alcorn JF. 2011. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol 186: 1666–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Happel KI, Zheng M, Young E, Quinton LJ, Lockhart E, Ramsay AJ, Shellito JE, Schurr JR, Bagby GJ, Nelson S, and Kolls JK. 2003. Cutting edge: roles of toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J. Immunol 170: 4432–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michel M-L, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, Wong C-H ,Schneider E, Dy M, and Leite-de-Moraes MC. 2007. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J. Exp. Med 204: 995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, and Ley K. 2005. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 22: 285–294. [DOI] [PubMed] [Google Scholar]
- 27.Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, and O’Shea JJ. 2009. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med 206: 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meierovics A, Yankelevich W-JC, and Cowley SC. 2013. MAIT cells are critical for optimal mucosal immune responses during in vivo pulmonary bacterial infection. Proc. Natl. Acad. Sci. USA 110: E3119–E3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferretti S, Bonneau O, Dubois GR, Jones CE, and Trifilieff A. 2003. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J. Immunol 170: 2106–2112. [DOI] [PubMed] [Google Scholar]
- 30.Cai S, Batra S, Langohr I, Iwakura Y, and Jeyaseelan S. 2016. IFN-γ induction by neutrophil-derived IL-17A homodimer augments pulmonary antibacterial defense. Mucosal Immunol. 9: 718–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mikacenic C, Hansen EE, Radella F, Gharib SA, Stapleton RD, and Wurfel MM. 2016. Interleukin-17A is associated with alveolar inflammation and poor outcomes in acute respiratory distress syndrome. Crit. Care Med 44: 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGeachy MJ, and Cua DJ. 2008. Th17 cell differentiation: the long and winding road. Immunity 28: 445–453. [DOI] [PubMed] [Google Scholar]
- 33.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, and Littman DR. 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126: 1121–1133. [DOI] [PubMed] [Google Scholar]
- 34.Sodhi CP, Nguyen J, Yamaguchi Y, Werts AD, Lu P, Ladd MR, Fulton WB, Kovler ML, Wang S, Prindle T Jr, et al. 2019. A dynamic variation of pulmonary ACE2 is required to modulate neutrophilic inflammation in response to Pseudomonas aeruginosa lung infection in mice. J. Immunol 203: 3000–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glowacka I, Bertram S, Herzog P, Pfefferle S, Steffen I, Muench MO, Simmons G, Hofmann H, Kuri T, Weber F, et al. 2010. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol 84: 1198–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pickens SR, Volin MV, Mandelin AM II, Kolls JK, Pope RM, and Shahrara S. 2010. IL-17 contributes to angiogenesis in rheumatoid arthritis. J. Immunol 184: 3233–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dang EV, Barbi J, Yang H-Y, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen H-R, et al. 2011. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146: 772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruning U, Cerone L, Neufeld Z, Fitzpatrick SF, Cheong A, Scholz CC, Simpson DA, Leonard MO, Tambuwala MM, Cummins EP, and Taylor CT. 2011. MicroRNA-155 promotes resolution of hypoxia-inducible factor 1alpha activity during prolonged hypoxia. Mol. Cell. Biol 31: 4087–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma L, Xue H-B, Wang F, Shu C-M, and Zhang J-H. 2015. MicroRNA-155 may be involved in the pathogenesis of atopic dermatitis by modulating the differentiation and function of T helper type 17 (Th17) cells. Clin. Exp. Immunol 181: 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao R, Ma Y-L, Liang W, Li H-H, Ma Z-J, Yu X, and Liao Y-H. 2012. MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PLoS One 7: e46082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lew TWK, Kwek T-K, Tai D, Earnest A, Loo S, Singh K, Kwan KM, Chan Y, Yim CF, Bek SL, et al. 2003. Acute respiratory distress syndrome in critically ill patients with severe acute respiratory syndrome. JAMA 290: 374–380. [DOI] [PubMed] [Google Scholar]
- 42.Sutherland TE, Logan N, Rückerl D, Humbles AA, Allan SM, Papayannopoulos V, Stockinger B, Maizels RM, and Allen JE. 2014. Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat. Immunol 15: 1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding Q, Liu G-Q, Zeng Y-Y, Zhu J-J, Liu Z-Y, Zhang X, and Huang J-A. 2017. Role of IL-17 in LPS-induced acute lung injury: an in vivo study. Oncotarget 8: 93704–93711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muir R, Osbourn M, Dubois AV, Doran E, Small DM, Monahan A, O’Kane CM, McAllister K, Fitzgerald DC, Kissenpfennig A, et al. 2016. Innate lymphoid cells are the predominant source of IL-17A during the early pathogenesis of acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med 193: 407–416. [DOI] [PubMed] [Google Scholar]
- 45.Li C, Yang P, Sun Y, Li T, Wang C, Wang Z, Zou Z, Yan Y, Wang W, Wang C, et al. 2012. IL-17 response mediates acute lung injury induced by the 2009 pandemic influenza A (H1N1) virus. Cell Res. 22: 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, Ross TM, Witztum JL, and Kolls JK. 2009. Critical role of IL-17RA in immunopathology of influenza infection. J. Immunol 183: 5301–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bermejo-Martin JF, Ortiz de Lejarazu R, Pumarola T, Rello J, Almansa R, Ramírez P, Martin-Loeches I, Varillas D, Gallegos MC, Serón C, et al. 2009. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit. Care 13: R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Kerkhove MD, Vandemaele KAH, Shinde V, Jaramillo-Gutierrez G, Koukounari A, Donnelly CA, Carlino LO, Owen R, Paterson B, Pelletier L, et al. ; WHO Working Group for Risk Factors for Severe H1N1pdm Infection. 2011. Risk factors for severe outcomes following 2009 influenza A (H1N1) infection: a global pooled analysis. PLoS Med. 8: e1001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma W-T, Yao X-T, Peng Q, and Chen D-K. 2019. The protective and pathogenic roles of IL-17 in viral infections: friend or foe? Open Biol. 9: 190109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Relloso M, Aragoneses-Fenoll L, Lasarte S, Bourgeois C, Romera G, Kuchler K, Corbí AL, Muñoz-Fernández MA, Nombela C, Rodríguez-Fernández JL, and Diez-Orejas R. 2012. Estradiol impairs the Th17 immune response against Candida albicans. J. Leukoc. Biol 91: 159–165. [DOI] [PubMed] [Google Scholar]
- 51.Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, and Singh D. 2012. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PLoS One 7: e44552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen R-Y, Fan Y-M, Zhang Q, Liu S, Li Q, Ke G-L, Li C, and You Z. 2015. Estradiol inhibits Th17 cell differentiation through inhibition of RORγt transcription by recruiting the ERα/REA complex to estrogen response elements of the RORγt promoter. J. Immunol 194: 4019–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Z, Yue Y, and Xiong S. 2013. Distinct Th17 inductions contribute to the gender bias in CVB3-induced myocarditis. Cardiovasc. Pathol 22: 373–382. [DOI] [PubMed] [Google Scholar]
- 54.Winer S, Paltser G, Chan Y, Tsui H, Engleman E, Winer D, and Dosch H-M. 2009. Obesity predisposes to Th17 bias. Eur. J. Immunol 39: 2629–2635. [DOI] [PubMed] [Google Scholar]
- 55.Sumarac-Dumanovic M, Stevanovic D, Ljubic A, Jorga J, Simic M, Stamenkovic-Pejkovic D, Starcevic V, Trajkovic V, and Micic D. 2009. Increased activity of interleukin-23/interleukin-17 proinflammatory axis in obese women. Int. J. Obes 33: 151–156. [DOI] [PubMed] [Google Scholar]
- 56.Cortvrindt C, Speeckaert R, Moerman A, Delanghe JR, and Speeckaert MM. 2017. The role of interleukin-17A in the pathogenesis of kidney diseases. Pathology 49: 247–258. [DOI] [PubMed] [Google Scholar]
- 57.Coto E, Gómez J, Suárez B, Tranche S, Díaz-Corte C, Ortiz A, Ruiz-Ortega M, Coto-Segura P, Batalla A, and López-Larrea C. 2015. Association between the IL17RA rs4819554 polymorphism and reduced renal filtration rate in the Spanish RENASTUR cohort. Hum. Immunol 76: 75–78. [DOI] [PubMed] [Google Scholar]
- 58.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, and Harrison DG. 2010. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Du Y-N, Tang X-F, Xu L, Chen W-D, Gao P-J, and Han W-Q. 2018. SGK1-FoxO1 signaling pathway mediates Th17/Treg imbalance and target organ inflammation in angiotensin II-induced hypertension. Front. Physiol 9: 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ji Q, Cheng G, Ma N, Huang Y, Lin Y, Zhou Q, Que B, Dong J, Zhou Y, and Nie S. 2017. Circulating Th1, Th2, and Th17 levels in hypertensive patients. Dis. Markers 2017: 7146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ryba-Stanisławowska M, Skrzypkowska M, Myśliwiec M, and Myśliwska J. 2013. Loss of the balance between CD4(+)Foxp3(+) regulatory T cells and CD4(+) IL17A(+) Th17 cells in patients with type 1 diabetes. Hum. Immunol 74: 701–707. [DOI] [PubMed] [Google Scholar]
- 62.Abdel-Moneim A, Bakery HH, and Allam G. 2018. The potential pathogenic role of IL-17/Th17 cells in both type 1 and type 2 diabetes mellitus. Biomed. Pharmacother 101: 287–292. [DOI] [PubMed] [Google Scholar]
- 63.Balasubramanyam M, Aravind S, Gokulakrishnan K, Prabu P, Sathishkumar C, Ranjani H, and Mohan V. 2011. Impaired miR-146a expression links subclinical inflammation and insulin resistance in Type 2 diabetes. Mol. Cell. Biochem 351: 197–205. [DOI] [PubMed] [Google Scholar]
- 64.Li B, Wang X, Choi IY, Wang Y-C, Liu S, Pham AT, Moon H, Smith DJ, Rao DS, Boldin MP, and Yang L. 2017. miR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J. Clin. Invest 127: 3702–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baldeón RL, Weigelt K, de Wit H, Ozcan B, van Oudenaren A, Sempértegui F, Sijbrands E, Grosse L, Freire W, Drexhage HA, and Leenen PJ. 2014. Decreased serum level of miR-146a as sign of chronic inflammation in type 2 diabetic patients. PLoS One 9: e115209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li Q, Ding S, Wang YM, Xu X, Shen Z, Fu R, Liu M, Hu C, Zhang C, Cao Q, and Wang Y. 2017. Age-associated alteration in Th17 cell response is related to endothelial cell senescence and atherosclerotic cerebral infarction. Am. J. Transl. Res 9: 5160–5168. [PMC free article] [PubMed] [Google Scholar]
- 67.Ouyang X, Yang Z, Zhang R, Arnaboldi P, Lu G, Li Q, Wang W, Zhang B, Cui M, Zhang H, et al. 2011. Potentiation of Th17 cytokines in aging process contributes to the development of colitis. Cell. Immunol 266: 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmitt V, Rink L, and Uciechowski P. 2013. The Th17/Treg balance is disturbed during aging. Exp. Gerontol 48: 1379–1386. [DOI] [PubMed] [Google Scholar]
- 69.Serrano C, Wright SW, Bimczok D, Shaffer CL, Cover TL, Venegas A, Salazar MG, Smythies LE, Harris PR, and Smith PD. 2013. Downregulated Th17 responses are associated with reduced gastritis in Helicobacter pylori-infected children. Mucosal Immunol. 6: 950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wasilewska A, Winiarska M, Olszewska M, and Rudnicka L. 2016. Interleukin-17 inhibitors. A new era in treatment of psoriasis and other skin diseases. Postepy Dermatol. Alergol 33: 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dong J, and Goldenberg G. 2017. New biologics in psoriasis: an update on IL-23 and IL-17 inhibitors. Cutis 99: 123–127. [PubMed] [Google Scholar]
- 72.Conti A, Lasagni C, Bigi L, and Pellacani G. 2020. Evolution of COVID-19 infection in four psoriatic patients treated with biological drugs. J. Eur. Acad. Dermatol. Venereol DOI: 10.1111/jdv.16587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Messina F, and Piaserico S. 2020. SARS-CoV-2 infection in a psoriatic patient treated with IL-23 inhibitor. J. Eur. Acad. Dermatol. Venereol 34: e254–e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Balestri R, Rech G, and Girardelli CR. 2020. SARS-CoV-2 infection in a psoriatic patient treated with IL-17 inhibitor. J. Eur. Acad. Dermatol. Venereol DOI: 10.1111/jdv.16571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gisondi P, Facheris P, Dapavo P, Piaserico S, Conti A, Naldi L, Cazzaniga S, Malagoli P, and Costanzo A. 2020. The impact of the COVID-19 pandemic on patients with chronic plaque psoriasis being treated with biological therapy: the Northern Italy experience. Br. J. Dermatol DOI: 10.1111/bjd.19158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.National Institute of Mental Health; Hospital of Prato. 2020. Baricitinib in symptomatic patients infected by COVID-19: an open-label, pilot study In ClinicalTrials.gov. National Library of Medicine (US), Bethesda, MD: NLM Identifier: NCT04320277. Available at https://clinicaltrials.gov/ct2/show/study/NCT04320277. Accessed: April 21, 2020. [Google Scholar]
- 77.Richardson P, Griffin I, Tucker C, Smith D, Oechsle O, Phelan A, Rawling M, Savory E, and Stebbing J. 2020. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 395: e30–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Solt LA, Kumar N, Nuhant P, Wang Y, Lauer JL, Liu J, Istrate MA, Kamenecka TM, Roush WR, Vidović D, et al. 2011. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 472: 491–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baricitinib: drug information. 2020. UpToDate. Available at: https://www.uptodate.com/contents/baricitinib-drug-information?search=baricitinib&source=panel_search_result&selectedTitle=1~19&usage_type=panel&kp_tab=drug_general&display_rank=1. Accessed: June 5, 2020. [Google Scholar]