

Abstract
The androgen receptor (AR) undergoes nuclear import in response to ligand. AR also undergoes nuclear export by a mechanism(s) that is poorly understood. We developed a permeabilized cell assay to characterize AR nuclear export in LNCaP prostate cancer cells. We found that nuclear export of endogenous AR can be stimulated by dsDNA oligonucleotides containing an androgen response element (ARE). This AR export pathway is dependent on ATP hydrolysis and is enhanced by phosphatase inhibition with okadaic acid (OA). Fluorescence recovery after photobleaching under the conditions that stimulate AR nuclear export in permeabilized cells suggested that ARE-dependent export does not simply reflect the relief of a nuclear retention-based mechanism. A radiolabeled ligand was used to show that AR can remain androgen-bound during translocation through the nuclear pore complex (NPC). Like the ARE, other synthetic DNAs stimulate AR export and yield phosphorylation of exported AR in the presence of OA. A specific inhibitor to DNA-PK (NU7026) inhibits both AR phosphorylation and AR export. In intact cells NU7026 treatment increases androgen-dependent transcription from endogenous genes that are regulated by AR. We propose that DNA-PK provides AR with a phosphorylation “license” to exit the nucleus.
As a member of the nuclear receptor (NR) superfamily of ligand-responsive transcription factors (1), the androgen receptor (AR) is composed of an N-terminal transcriptional activation domain (NTD), a central DNA binding domain (DBD), a hinge region, and a C-terminal ligand binding domain (LBD). AR is important for proper development of male secondary sexual characteristics and for normal prostate function (2). AR is also thought to be critical for the tumorigenesis of prostate cancer (PCa) and for the progression of PCa from an androgen-sensitive to an androgen-insensitive state (3–8). Notably, in PCa xenografts in mice, elevation of AR mRNA levels was the only cellular index that consistently correlated with PCa progression to hormone-insensitive status (9).
AR transcriptional regulatory functions are dependent on the proper subcellular localization of the receptor. AR is thought to associate with a heat shock protein 90 (hsp90)-based chaperone complex in the cytoplasm (10) until the binding of cognate ligand induces a conformational change in AR, chaperone dissociation, and subsequent AR nuclear import (discussed in (11,12)). Once in the nucleus, AR can bind specific androgen response elements to enhance or repress transcription of the associated androgen-responsive genes. The type of ligand bound to AR is critical for its regulatory activity, as agonist and antagonist are known to differentially affect AR subnuclear localization (11–14) and intranuclear mobility (12,15). AR can also undergo export from the nucleus (11), and at least one pathway of AR export appears to be mediated by its DBD (16). Significantly, AR in mouse PCa xenografts exhibited a predominantly nuclear localization in hormone-responsive tumors, exhibited significant cytoplasmic localization following castration, and exhibited predominantly nuclear localization in hormone-resistant tumors (17). These data suggest that AR localization, and therefore receptor activity, may be modulated during PCa progression.
Multiple signal transduction pathways can impinge on AR. A member of the phosphatidylinositol-3-kinase (PI-3-K) family, the DNA-dependent protein kinase (DNA-PK) has recently been implicated in AR transcriptional regulation in human prostate cancer (LNCaP) cells (18). Comprised of a large (~470 kD) catalytic and two Ku regulatory subunits, DNA-PK is involved in the non-homologous end-joining (NHEJ) response to DNA damage, V(D)J recombination, and retroviral DNA integration (19,20). DNA-PK has been reported to associate with members of the NR superfamily (21–23) and can phosphorylate the glucocorticoid receptor (GR) in vitro (24,25). Further, the Ku subunits of DNA-PK can be localized to the promoter of the endogenous AR-responsive PSA gene by ChIP, and they act as AR transcriptional coactivators in reporter assays (18).
AR undergoes nuclear export mediated by its DBD (11,16). In this study we sought to interrogate further the mechanism of AR nuclear export using an in vitro assay in permeabilized LNCaP cells. We found that short, double-stranded DNA (dsDNA) oligonucleotides containing an androgen response element stimulated robust AR export in an ATP-dependent reaction. The phosphatase inhibitor okadaic acid (OA) enhanced the reaction and promoted the phosphorylation of exported AR. Significantly, both AR export and phosphorylation were blocked by chemical inhibitors of the PI-3-K kinase family, including an inhibitor that is specific for DNA-PK. Further, we found that an export receptor, exportin 5, can also stimulate AR export independently of DNA-PK. We speculate that DNA-PK provides AR with a “license” to leave the nucleus via a receptor-mediated export pathway.
Experimental Procedures
Cell Culture and Transfection:
Human prostatic carcinoma LNCaP cells (26) were cultured in T medium (Gibco) containing phenol red and 5% fetal bovine serum. The GFP-AR plasmid used for FRAP analysis and single-cell export assays encodes an N-terminally GFP-tagged form of human AR in a pEGFP-C1 vector backbone (Clontech). LNCaP cells were transfected using Transfectin (Bio-Rad).
Recombinant Protein Preparation:
Exportin 5 - A pQE60 plasmid (Qiagen) encoding C-terminally His-tagged human exportin 5 (Addgene plasmid 12553) was transformed into the TG1 bacterial strain. Following growth to log phase in LB medium with 2% ethanol, cultures were induced using 300 μM IPTG overnight at 25° C. Cultures were centrifuged and resuspended in 20 mM HEPES pH 8, 500 mM NaCl, 10% glycerol, and 14 mM β-mercaptoethanol. Resuspension and wash buffers contained 1 mM PMSF and 5 μg/ml of aprotinin, leupeptin, and pepstatin. Following 3 rounds of lysis using a French press, lysates were clarified and incubated with Talon beads (Clontech) at 4° C for = 2 h. Talon resin was washed with 20 mM HEPES pH 8, 500 mM NaCl, 10% glycerol, 14 mM β-mercaptoethanol, and 10 mM imidazole, and then with 20 mM HEPES pH 8, 200 mM NaCl, 10% glycerol, 7 mM β-mercaptoethanol, and 10 mM imidazole. Recombinant exportin 5 was eluted using 20 mM HEPES pH 8, 200 mM NaCl, 10% glycerol, 14 mM β-mercaptoethanol, and 250 mM imidazole before dialysis into 1x transport buffer (20 mM HEPES pH 7.4, 110 mM potassium acetate, 2 mM magnesium acetate, 0.5 mM EGTA) supplemented with 10% glycerol, 2 mM DTT, and 1 μg/ml of aprotinin, leupeptin, and pepstatin. Exportin 5 was then snap-frozen in single-use aliquots. GST-DBD-Hinge - A pGEX4T3 vector (Pharmacia) encoding a fragment of AR that includes the DBD and hinge domains (residues 549–671) was transformed into BL21 cells. Following growth to log phase, protein expression was induced with I mM IPTG in the presence of 2% ethanol for 3 h at room temperature. Pelleted cells were resuspended in PBS supplemented with protease inhibitors. Following French press-mediated lysis, lysates were clarified and incubated with glutathione beads for 1.5 h at 4 C. The bound fraction was washed with PBS containing 1% Triton X-100, then washed with PBS containing 500 mM NaCl, and then washed with PBS alone. GST-AR-DBD-Hinge protein was eluted with 100 mM Tris pH 8, 10 mM glutathione, 1 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, aprotinin, leupeptin, and pepstatin. Dialysis into 1x transport buffer (20 mM HEPES pH 7.4, 110 mM potassium acetate, 2 mM magnesium acetate, 0.5 mM EGTA) containing 1 mM DTT preceded snap-freezing in single-use aliquots.
In vitro AR Export Assay:
The assay is a modified version of the one used by Love et al. in their study of HIV-1 Rev nuclear export in vitro (27). LNCaP cells expressing endogenous AR are supplied with 1–10 nM synthetic androgen (R1881; PerkinElmer) for 1–3 hours prior to a wash with phosphate-buffered saline (Gibco). Cells are subjected to a brief trypsinization (0.05% Trypsin-EDTA (Gibco)) that is quenched with serum-containing medium, then subjected to gentle centrifugation (~200×g) before resuspension in ice-cold 1x transport buffer. Cells are permeabilized on ice for 6–8 min with 50 μg/mL high purity digitonin (Calbiochem) in ice-cold complete transport buffer (CTB: 1x TB containing 2 mM DTT, 1 μg/ml aprotinin, and 0.5 μg/ml leupeptin and pepstatin). Cells are then washed with ice-cold CTB and aliquotted into standard polystyrene tubes commonly used for DNA transfection (Falcon #352058). Ice-cold export reaction mixes are added to aliquots of permeabilized cells on ice. Final reaction volume is approximately 125 μl (100 μl reaction mix and 25 μl cell suspension); final permeabilized cell concentration in each reaction is approximately 5*106 cells/ml. The tubes containing the permeabilized cells and reaction mixes are incubated at 30°C for 20–30 min. Following the assay, reaction components are centrifuged (~18,000×g for 12 min) to isolate the reaction supernatant (containing the exported AR fraction) from the cell pellet (containing the non-exported, nuclear AR). Equal volumes of reaction supernatants are then separated on 7.5% acrylamide SDS-PAGE gels. Following SDS-PAGE separation, proteins are transferred onto PVDF membranes (Millipore) and immunoblotted using the polyclonal α-AR antibody PG-21 (directed against the N-terminal 21 amino acid residues of AR (Upstate)), providing a semi-quantitative measure of the amount of AR exported during the assay.
In vitro export assays conducted in the presence of CTB alone serve as negative control for AR export. Our energy regenerating system (ERS) consists of BSA (5 mg/mL final in export reaction), creatine phosphokinase (80 U/mL final), creatine phosphate (1.6 mg/mL final), ATP (1 mM final), GTP (1 mM final), and aprotinin. Non-hydrolyzable nucleotide analogs AMP-PNP and GMP-PNP are also used at 1 mM final concentration. Okadaic acid (Calbiochem) is used at a final concentration of 200 nM. Casodex (10 μM; Fisher), wortmannin (20 μM (MP Biomedicals)), LY294002 (50 μM (Calbiochem)), and NU7026 (2.5 μM (Sigma)) are added to LNCaP cells 1–2 h prior to the start of the in vitro export assay. Chemical inhibitors are routinely maintained in pre-export wash buffers and in export reactions. For export assays in the absence of agonist, cells are washed with phenol-red-free RPMI (Gibco) containing 5% charcoal-stripped serum, and then incubated in this medium for 6–24 hours prior to permeabilization and in vitro export assays.
For phosphatase treatment of exported AR, export reactions were first carried out in the presence of ERS, OA, and ARE (1 μM). Following centrifugation, the export supernatants (36 μL) were treated with Antarctic phosphatase (10 U; NEB) for 2 h at 37° C in the presence of the manufacturer’s supplied buffer (final 1x concentration). Reaction components were subjected to SDS-PAGE and AR was immunoblotted with the PG-21 antibody.
For in vitro export assays using 3H-R1881, LNCaP cells were incubated in the presence of 2 nM 3H-R1881 (PerkinElmer) for 1–3 h prior to washing and cell permeabilization. In vitro export reactions ensued at 30°C for 30 min. Reaction supernatants containing the exported AR fraction were then collected, applied to Whatman paper, dried, and measured by scintillation counting. For immunoprecipitations, reaction supernatants were incubated with control resin or AR-21 anti-AR resin in the presence of 0.1% TX-100 for 4 h at 4°C. Resins were washed five times in complete transport buffer containing 0.1% TX-100 prior to release of bound 3H-R1881 with 2% SDS at 90° C for 10 min. Released fractions were applied to Whatman paper, dried, and measured by scintillation counting.
For in vitro export assays followed by detergent extraction, LNCaP cells were subjected to standard export reactions prior to centrifugation and harvest of reaction supernatants containing the exported AR fraction. Cell pellets containing residual, non-exported AR were then subjected to extraction with 0.5% Triton X-100 in complete transport buffer supplemented with BSA (5 mg/mL final), glycerol (0.5% final), and aprotinin, for 20 min on ice. Subsequent centrifugation yielded supernatant (detergent-sensitive) and pellet (detergent-resistant) fractions. In Fig. 6c (Triton in the Export Rxn samples), 0.5% Triton X-100 was included during standard export reactions to disrupt the nuclear envelope and yielded a single released AR fraction.
Figure 6.
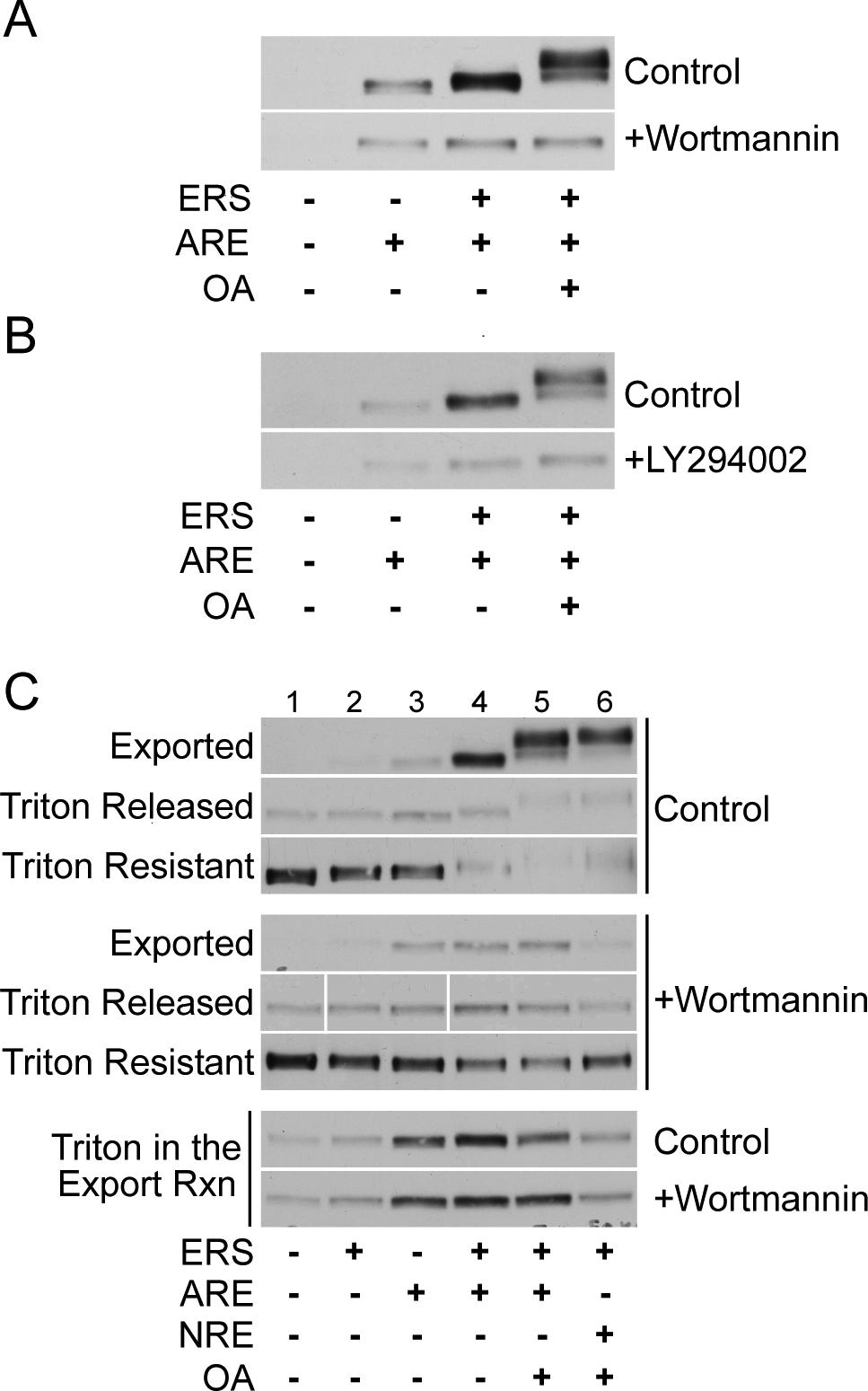
Wortmannin and LY294002 inhibit ARE-dependent AR export and phosphorylation. (A) LNCaP cells were treated with 10 nM R1881 alone or in addition to 20 μM wortmannin prior to standard in vitro export assays in the presence of ERS, OA, and/or ARE as shown. Exported AR fractions were immunoblotted with the PG-21 antibody. (B) LNCaP cells were treated with 10 nM R1881 alone or in addition to 50 μM LY294002 prior to standard in vitro export assays. (C) LNCaP cells were treated with 10 nM R1881 alone or in combination with 20 μM wortmannin. Cells were then subjected to standard in vitro export assays under the conditions shown and reaction supernatants containing the exported AR fraction were harvested. Cell pellets containing residual nuclear, non-exported AR were then subjected to extraction with 0.5% Triton X-100 on ice for 20 min. After centrifugation, the extracted (Triton Released) and non-extracted (Triton Resistant) AR fractions were harvested and subjected to SDS-PAGE along with the exported AR fractions and detected by immunoblotting with the PG-21 antibody. For Triton in the Export Rxn samples, LNCaP cells were subjected to standard in vitro export assays in the presence of 0.5% Triton X-100, i.e. detergent was added at the start of the export reaction rather than at a post-export extraction step.
Recombinant exportin 5 was used at a final concentration of 0.1 (Lo) and 0.2 (Hi) mg/mL. Dialysis buffer obtained from the preparation of recombinant exportin 5 served as negative buffer control in the export reactions.
For in vitro AR export in adherent cells, LNCaP cells were plated on glass coverslips (Fisher) coated with poly-D-lysine (Sigma) and grown in T medium containing phenol red and supplemented with 5% fetal bovine serum. Following treatment with 10 nM R1881, cells were washed three times (4 min/wash) with ice-cold TB, permeabilized with 50 μg/mL digitonin in ice-cold CTB for 6–8 min, and then washed again three times with ice-cold CTB. Export reaction mixes were supplied to the cells before incubation at 30°C for 20–30 min. Cells were washed two more times with ice-cold CTB prior to fixation with 3.7% formaldehyde.
In antibody access experiments to test the integrity of the nuclear envelope during in vitro export assays, an antibody to the nuclear protein RCC1 (Santa Cruz) was included in the export reaction mix (7 μg/mL final). As a positive control for envelope disruption, 0.2% Triton X-100 was also included in the export reaction. Following the export assay, washes, and fixation, cells were then permeabilized with 0.2% Triton X-100 and subjected to standard immunofluorescence processing using an anti-AR primary antibody (AR-21) and secondary antibodies to both the anti-RCC1 and anti-AR primary antibodies. Cells were mounted and viewed using a Nikon Eclipse E800 upright microscope.
DNA Oligonucleotides:
Oligonucleotides used to stimulate AR export in vitro are from Sigma-Genosys (St. Louis). The ARE duplex (Duplex a, see Fig. 7a) contains both half-sites of the androgen response element in the C(3)1 intron of the prostatic binding protein gene (28). The mutant ARE duplex (Duplex b) contains four point mutations within the twelve residue positions comprising the tandem of ARE half-sites (28). Oligonucleotides c and d are the sense and antisense strand components, respectively, of the wild-type ARE duplex (Duplex a). Duplexes f, g, and h are derivatives of the wild-type ARE duplex. In duplex i, the two ARE half-sites are separated by a non-complementary bubble, twelve residues in length on each duplex strand. In oligonucleotide j, the upstream ARE half-site is adjoined to a 13 nucleotide non-complementary loop (see Fig. 7a for schematic representations of these oligonucleotides). The NRE duplex is a derivative of the ARE duplex and harbors an NRE1 sequence (25) instead of an androgen response element. The ARE-NRE duplex contains an NRE1 sequence and an androgen response element in cis. NS1, NS2, and NS3 are intended as nonspecific duplexes that harbor neither an androgen response element nor an NRE1 sequence.
Figure 7.
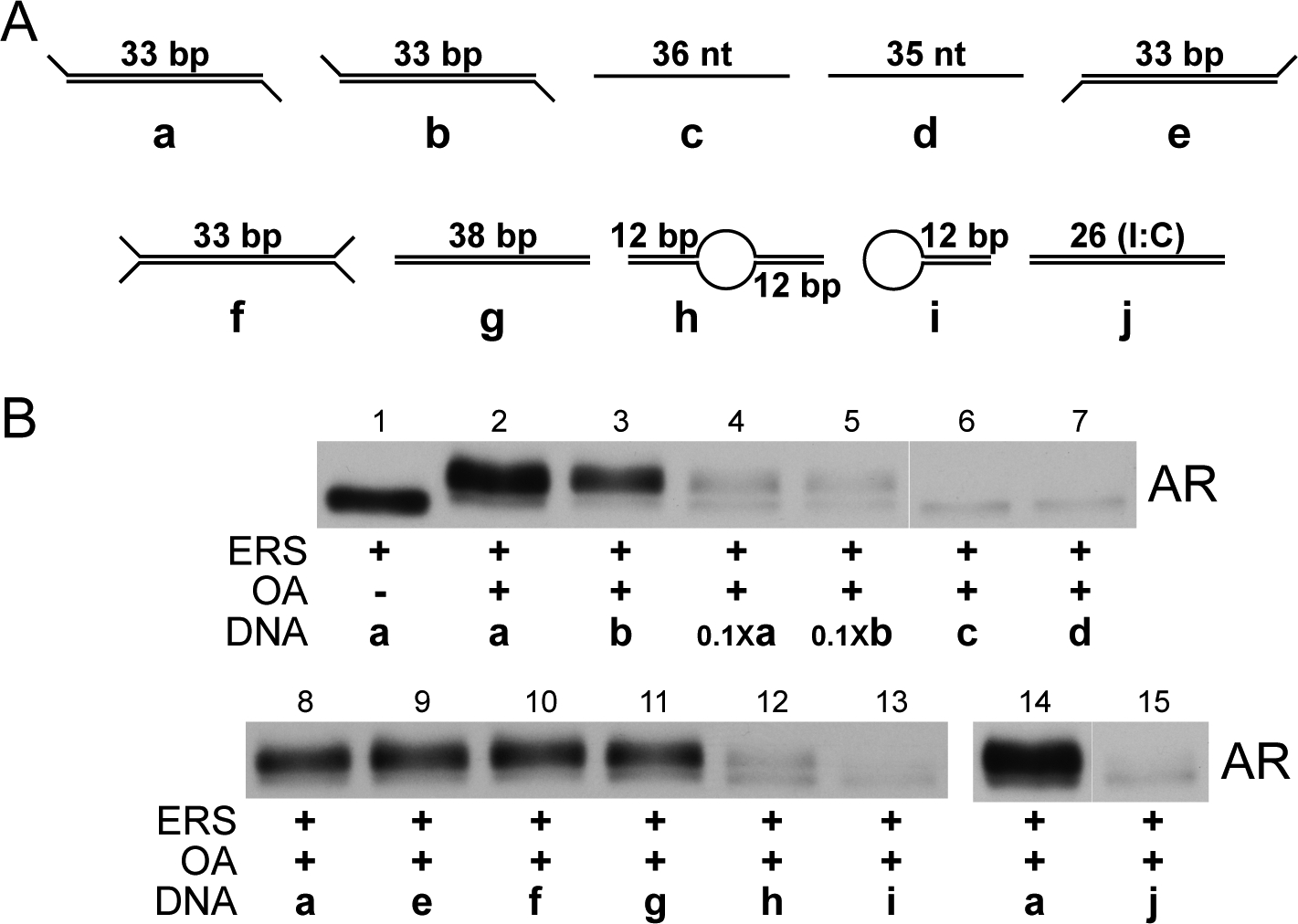
Oligonucleotides with differing sequence and structure can stimulate AR export and phosphorylation. (A) Schema of different DNA oligonucleotides used in (B) to stimulate AR export in vitro. dsDNA oligonucleotide (a) is the standard ARE; (b) is a mutant ARE containing four point mutations within the pair of androgen response element half-sites {Schoenmakers, 2000 #357}; (c) and (d) are the sense and antisense strand components, respectively, of the standard ARE; (e) is a modified ARE possessing two 3’ overhangs; (f) is a modified ARE possessing two 3’ overhangs and two 5’ overhangs; (g) is a modified ARE possessing two blunt ends; (h) is a modified ARE with a non-complementary bubble between both half sites of the androgen response element; (i) is a hairpin oligonucleotide containing within its complementary dsDNA region one of the two half sites of the ARE androgen response element; and (j) is a dsDNA oligonucleotide containing an internal stretch of 26 inosine:cytosine pairs flanked by five C:G pairs on each end of the oligonucleotide. (B) LNCaP cells were pretreated with 10 nM R1881 and subjected to standard in vitro export assays in the presence of the oligonucleotides described in (A). Oligonucleotides were included at 1 μM except in lanes 4 and 5 (100 nM) and lanes 6 and 7 (2 μM). Exported AR fractions were immunoblotted with the PG-21 antibody.
FRAP Analysis and AR Export in Single Cells:
LNCaP cells were grown on Delta T dishes (Fisher) coated with poly-D-lysine. Following expression of GFP-AR, cells were treated with R1881 and subjected to standard permeabilization and washes with complete transport buffer on ice. Cells were then placed on a Bioptechs stage warmer, GFP-AR-expressing cells were identified using the wide field mode of a Zeiss LSM 510 Meta confocal laser scanning microscope, and export reaction mixes were added to cells. For single-cell export assays, nuclear GFP fluorescence intensity was recorded immediately prior to and immediately following a 30 min export reaction at 37° C. The ratio of these two values (plotted in Fig. 4d) provides a measure of nuclear GFP fluorescence lost, and therefore of GFP-AR nuclear export, during the reaction. For FRAP measurements, recovery of GFP fluorescence into the photobleached area was monitored in increments of 2.5 min. Recovery curves represent the ratio of the fluorescence intensity within the photobleached rectangular area to the nuclear fluorescence intensity outside this photobleached area plotted as a function of time. Following addition of an export reaction mix, up to six recovery curves per dish were generated sequentially, each in a separate cell. All recovery curves for a given export reaction condition were averaged, and a representative recovery curve is plotted for each condition in Fig. 4b. The t1/2 value (Table 1) indicates the time at which the half-maximal amount of fluorescence recovery into the photobleached area has occurred, under a given reaction condition.
Figure 4.
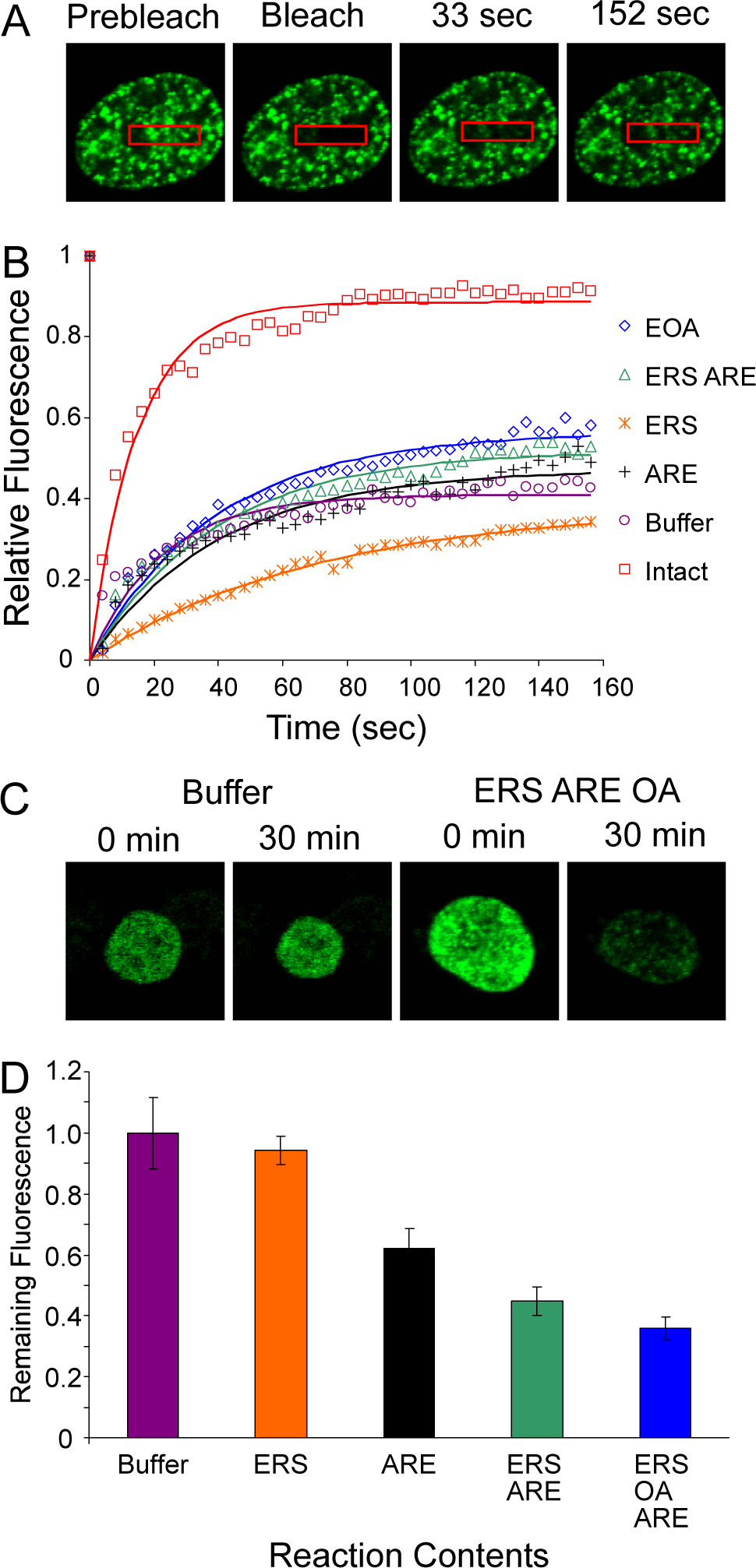
Single-cell analysis of GFP-AR in permeabilized LNCaP cells. (A) LNCaP cells expressing GFP-AR were treated with 10 nM R1881, permeabilized, and subjected to FRAP analysis in the presence of standard export reaction mixes. In the representative cell shown here, recovery of GFP-AR into the bleached zone was visualized in the presence of ERS/OA/ARE (see Materials and Methods for further details). (B) FRAP recovery curves of intra-nuclear GFP-AR were generated using the different export reaction mixes shown. Intact cells were not permeabilized; EOA signifies an export reaction mix of ERS/OA/ARE. Each recovery curve was generated from a single cell that represents the behavior of all cells sampled under a particular reaction condition. See Materials and Methods for further details. (C) LNCaP cells expressing GFP-AR were treated with 10 nM R1881, permeabilized, and subjected to standard in vitro export assays. Individual cells expressing GFP-AR were monitored for the loss of nuclear GFP fluorescence as a measure of GFP-AR nuclear export. Nuclear GFP fluorescence is shown in representative cells before (0 min) and after (30 min) an export reaction in the presence of buffer alone (Buffer) or in the presence of ERS/OA/ARE. (D) LNCaP cells expressing GFP-AR were treated as in (C). The amount of GFP fluorescence remaining in the nucleus after an in vitro export assay, compared to total nuclear GFP fluorescence prior to the assay, was calculated for individual cells in the presence of different export reaction mixes. The average value of this fraction is shown for all cells under a particular reaction condition.
Table 1.
GFP-AR nuclear mobility and nuclear export in permeabilized LNCaP cells. Both the t1/2 and mobile AR fraction values were generated using FRAP analysis (Fig. 4a,b). t1/2 values indicate the time (s) post-bleach at which half-maximal recovery of GFP-AR into the bleached zone has occurred. Mobile AR fraction values reflect the amount of GFP fluorescence recovery by the endpoint of the sampling interval. Exported AR fraction values indicate the amount of GFP fluorescence lost from the nucleus in the course of single-cell export assays (Fig. 4c,d).
| Buffer | 15.8 ± 2.1 | 40 ± 5 | 0 ± 12 |
| ERS | 51.0 ± 8.3 | 36 ± 3 | 6 ± 5 |
| ARE | 25.6 ± 5.1 | 46 ± 4 | 38 ± 7 |
| ERS ARE | 26.3 ± 2.5 | 59 ± 3 | 55 ± 5 |
| ERS OA ARE | 24.7 ± 5.6 | 43 ± 5 | 64 ± 4 |
DNA Oligonucleotides:
Oligonucleotide sequences are as follow: Duplex a, sense: GGGAGCTTACATAGTACGTGATGTTCTCAAGGTCGA, antisense: TCTCGACCTTGAGAACATCACGTACTATGTAAGCT; Duplex b (residues mutated relative to Duplex a are in bold), sense: GGGAGCTTACATTGTTCTTGAGGTTCTCAAGGTCGA, antisense: TCTCGACCTTGAGAACCTCAAGAACAATGTAAGCT; Oligonucleotide c (identical to the sense strand of duplex a): GGGAGCTTACATAGTACGTGATGTTCTCAAGGTCGA; Oligonucleotide d (identical to the antisense strand of duplex a): TCTCGACCTTGAGAACATCACGTACTATGTAAGCT; Duplex e, sense: AGCTTACATAGTACGTGATGTTCTCAAGGTCGACT, antisense: TCGACCTTGAGAACATCACGTACTATGTAAGCTGGG; Duplex f, sense: GGGAGCTTACATAGTACGTGATGTTCTCAAGGTCGACT, antisense: TCTCGACCTTGAGAACATCACGTACTATGTAAGCTGGG; Duplex g, sense: GGGAGCTTACATAGTACGTGATGTTCTCAAGGTCGAGA, antisense: TCTCGACCTTGAGAACATCACGTACTATGTAAGCTCCC; Duplex h, sense: CATAGTACGTGACCCCCCCCCCCCTGATGTTCTCAA, antisense: TTGAGAACATCACCCCCCCCCCCCTCACGTACTATG; Oligonucleotide i: CATAGTACGTGACCCCCCCCCCCCTCACGTACTATG; and Duplex j, sense: CGCGC(I26)CGCGC, antisense: GCGCG(C26)GCGCG; NRE, sense: GGGAGCTAACTGAGAAAGAGAAAGACGA CAGGTCGA, antisense: TCTCGACCTGTCGTCTTTCTCTTTCTCAGTT AGCT; ARE-NRE, sense: AACTGAGAAAGAGAAAGACGAGCTTACATAGTACGTGATGTTCTCAAGGT, antisense: ACCTTGAGAACATCACGTACTATGTAAGCTCGTCTTTCTCTTTCTCAGTT; NS1, sense: TCGAGATGGATTATAAAGCATTTGATAATCTTTAAT, antisense: CTAGATTAAAGATTATCAAATGCTTTATAATCCATC; NS2, sense: GTCCATCTTGTCGTCTTGAGAAATGTTATGAAGCAGGG, antisense: CCCTGCTTCATAACATTTCTCAAGACGACAAGATGGAC; and NS3, sense: AGAAAAAATCCCACATCCTGCTCAGAGCGCTTCTACCAGCTCACC, antisense: GGTGAGCTGGTAGAAGCGCTCTGAGCAGGATGTGGGATTTTTTCT.
The fluorescent ARE duplex used in Figure 1c contains a fluorescein moiety on the 5´ end of the antisense strand of the wild-type ARE duplex (Duplex a).
Figure 1.
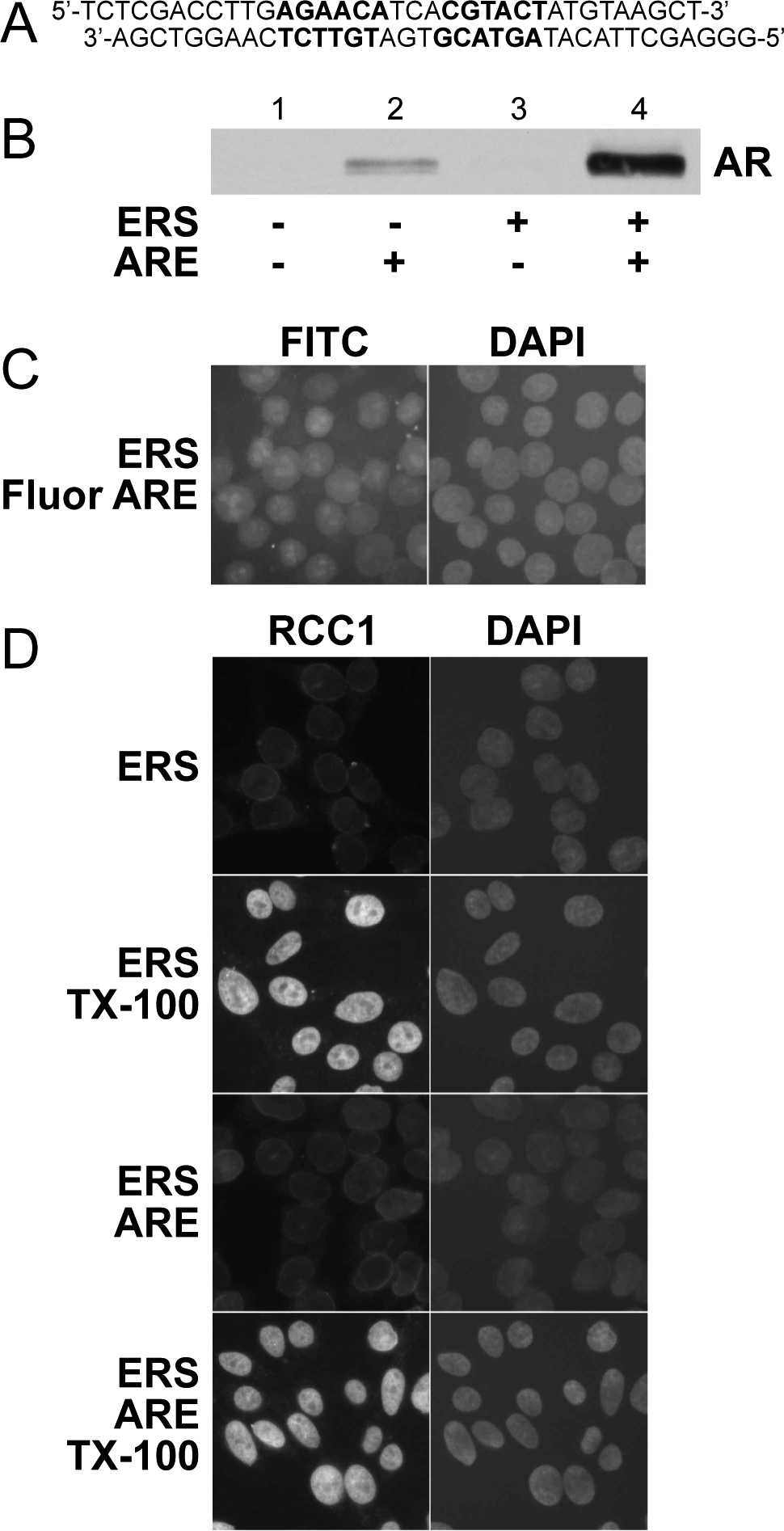
A double-stranded DNA oligonucleotide containing an androgen response element stimulates nuclear export of endogenous AR in permeabilized LNCaP cells. (A) Schematic representation of a 33 bp double-stranded oligonucleotide (ARE) that contains the C3(1) androgen response element {Claessens, 1989 #422} and possesses two 5’ overhangs. (B) ARE (1 μM) and an energy regenerating system (ERS; contains ATP, GTP, creatine phosphate, creatine phosphokinase, and BSA) were added in the combinations shown to permeabilized LNCaP cells to induce AR nuclear export in vitro (as described in Materials and Methods). Reaction supernatants containing the exported AR fraction were subjected to SDS-PAGE (7.5% acrylamide) and immunoblotted using the anti-AR PG-21 antibody (Upstate). (C) Adherent LNCaP cells were permeabilized with digitonin and subjected to standard in vitro export reactions in the presence of a fluorescein-labelled ARE. FITC and DAPI channels show fluorescent ARE and DNA localization, respectively. (D) Standard in vitro assays using adherent LNCaP cells were conducted in the presence or absence of 0.2% Triton X-100. All export reactions contained 7 μg/mL of anti-RCC1 antibody (Santa Cruz). Cells were then washed, fixed, exposed to a secondary antibody against the anti-RCC1 primary antibody, and analyzed using immunofluorescence microscopy.
Real-time RT PCR:
Real-time RT PCR analysis was done on an iCycler optical system (Biorad) using the IQ SYBR Green PCR master mix. LNCaP cells were pre-treated with NU7026 for 16 h and then the cells were untreated or treated with 1 nM R1881 for 24 h. Total RNA was extracted from cells using the RNeasy kit (Qiagen). DNase I treatment was performed directly on the RNeasy mini column with 27 Kunitz units of DNase I (Qiagen) for 15 min at room temperature according to the Qiagen protocol. RNA was quantified using Ribogreen (Molecular Probes). 500 ng of RNA was reverse-transcribed in a 20 μl reaction volume using the iScript cDNA synthesis kit (BioRad) according to the manufacturer’s protocol. The human specific PCR primers used were: forward PSA 5’-TGGTGCATTACCGGAAAGTGGATCA-3’; reverse PSA 5’-GCTTGAGTCTTGGCCTGGTCATTTC-3’; forward S100P 5’-ATGACGGAACTAGAGACAGCC-3’; reverse S100P 5’-AGGAAGCCTGGTAGCTCCTT-3’; forward FKBP51 5’-CATCAAGGCATGGGACATTGG-3’; reverse FKBP51 5’-TCGAGGGAATTTTAGGGAGACT-3’; forward PDF 5’-GGGCAAGAATCTCAGGACGG-3’; reverse PDF 5’-TCTGGAGTCTTCGGAGTGCAA-3’; forward β-glucuronidase (GUS) 5’-CCGACTTCTCTGACAACCGACG’3’; reverse GUS 5’-AGCCGACAAAATGCCGCAGACG-3’. Non-reversed transcribed RNA was subject to PCR as a control; no DNA contamination was observed. Primer annealing temperatures were 66°C (PSA), 58°C (S100P), 60°C (FKBP51), 58°C (PDF), or 68°C (GUS). Mean starting quantity (SQ) of each androgen-induced gene is normalized to mean SQ GUS; this ratio (with standard error for the ratio) is shown in Fig. 9a–d for a representative experiment in which each sample is tested in duplicate.
Figure 9.
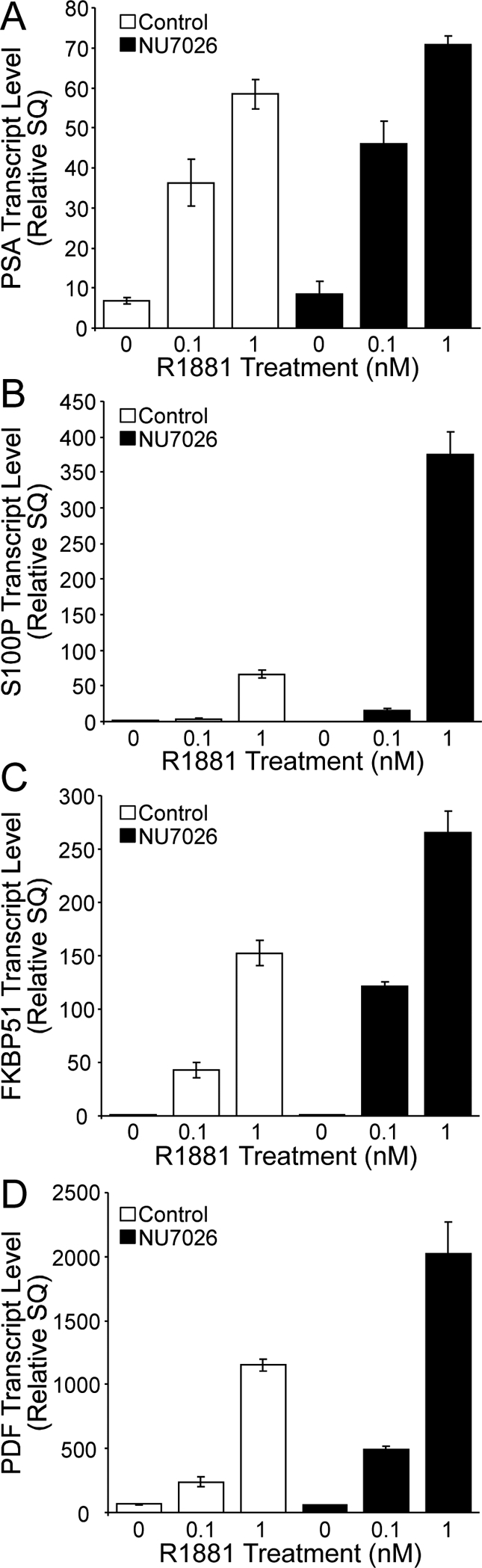
NU7026 enhances transcription of androgen-responsive genes in LNCaP cells. LNCaP cells were treated with 10 μM NU7026 for 16 h and then treated with or without R1881 for an additional 24 h at the concentrations shown. Transcription of PSA (A), S100P (B), FKBP51 (C), and PDF (D) genes was then monitored using real-time RT-PCR. Transcript levels were normalized to those of β-glucuronidase (GUS), and this ratio is shown with standard error. See Materials and Methods for further details.
RESULTS
AR Export in Permeabilized LNCaP Cells.
We developed a permeabilized cell assay to investigate the mechanism of AR export. In this assay, cultured LNCaP cells are pretreated with the synthetic androgen R1881 to induce nuclear import of endogenous AR. The cells are then harvested, washed, and treated with digitonin to selectively permeabilize the plasma membrane. Permeabilized cells are then supplemented with reaction mixes to stimulate nuclear export of AR into the reaction supernatant, which is examined by immunoblotting with affinity-purified AR antibody. In the course of optimizing reaction conditions for the assay, we tested whether a double-stranded DNA (dsDNA) oligonucleotide containing an androgen response element (ARE; Fig. 1a) could improve cytosol-dependent nuclear export by relieving presumed nuclear retention of AR. This approach was used by Kehlenbach and coworkers to improve cytosol-dependent NFAT export in permeabilized cells (29). Unexpectedly, we found that an ARE together with an energy-regenerating system (ERS) stimulated robust AR export without the addition of cytosolic factors (Fig. 1b). Fluorescently-labeled ARE entered the nuclei of digitonin-permeabilized LNCaP cells during the export assay, which is consistent with ARE stimulating AR export from within the nuclei (Fig. 1c). We also verified that the nuclear envelope remains intact under conditions that promote AR export. An antibody to nuclear RCC1 failed to detect its antigen unless the nuclear envelope was disrupted by Triton X-100 treatment (Fig. 1d). Together, these data show that ARE stimulate energy-dependent AR translocation through the NPC.
ARE-dependent AR Export Requires ATP Hydrolysis.
Depending on the export substrate, ATP (29,30) and/or GTP (31) can stimulate nuclear export in permeabilized cells. As our ERS contains both ATP and GTP, we tested whether one or both nucleotides is required for AR export. ATP, but not GTP, stimulated AR export in the presence of ARE (Fig. 2). The non-hydrolyzable ATP analog AMP-PNP did not support AR export, and the non-hydrolyzable GTP analog GMP-PNP did not inhibit export stimulation by ATP (Fig. 2). Our results indicate that ATP hydrolysis, and not GTP hydrolysis, is rate-limiting for ARE-dependent AR export.
Figure 2.

ATP hydrolysis is required for ARE-dependent AR nuclear export. Permeabilized LNCaP cells were subjected to standard AR in vitro export assays in the presence of ARE (1 μM) and the presence (lane 2) or absence (lane 1) of a complete energy regenerating system. ATP, GTP, AMP-PNP, and/or GMP-PNP were added in the combinations shown (each 1 mM final). Reaction supernatants containing the exported AR fraction were immunoblotted using the anti-AR PG-21 antibody (Upstate).
This ATP requirement could reflect the involvement of a kinase(s) or protein chaperones in ARE-stimulated AR export. Because phosphatase activity has been implicated in the regulation of GR nucleocytoplasmic trafficking (32), we tested the phosphatase inhibitor okadaic acid (OA) in our permeabilized cell assay. We found that OA enhanced AR export and caused an upward shift in AR electrophoretic mobility (Fig. 3a, compare lanes 3 and 6). The AR mobility shift was strongly dependent on ERS and ARE addition (Fig. 3a). The upward shift in AR electrophoretic mobility can be reversed by alkaline phosphatase treatment of the exported AR (Fig. 3b). Thus, AR undergoes phosphorylation during the export reaction in the presence of ERS/OA/ARE. By analyzing the time course of AR export under this condition, we obtained data suggesting that there are multiple pools of exported AR (Fig. 3c). The faster-migrating form of AR is exported within four minutes of the start of the assay. Within 8–12 minutes, a slower-migrating form of AR appeared in the exported fraction; the amount of this form of AR increased as a function of time (compare lanes 3–8).
Figure 3.
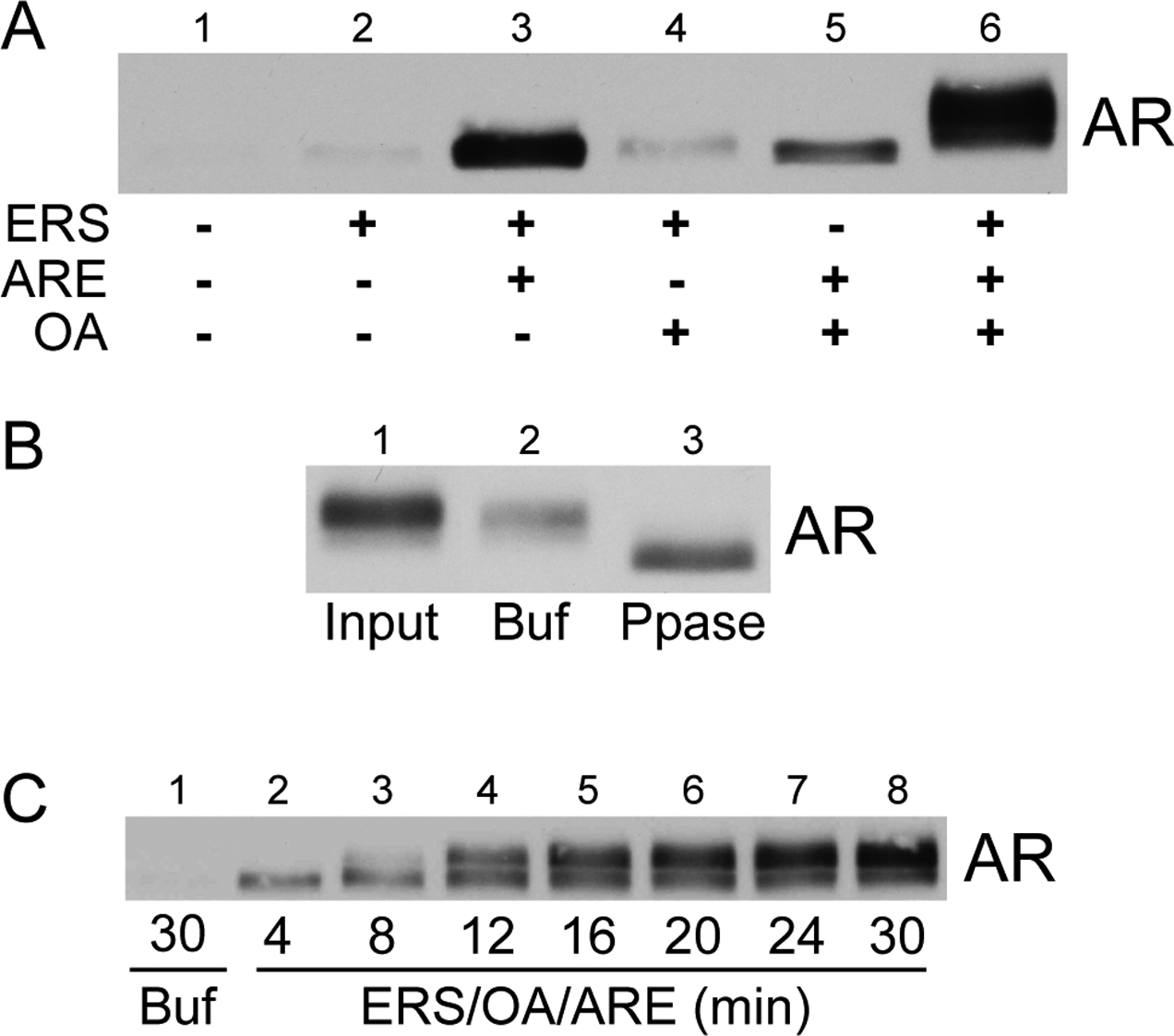
OA enhances ARE-dependent AR nuclear export and stimulates phosphorylation of exported AR. (A) Standard AR in vitro export assays were conducted in the presence of ARE (1 μM), ERS, and/or okadaic acid (OA; 200 nM) as shown. Reaction supernatants containing the exported AR fraction were immunoblotted with the anti-AR PG-21 antibody (Upstate). (B) Permeabilized LNCaP cells were first treated with ERS/OA/ARE to stimulate AR nuclear export. Reaction supernatants containing the exported AR fraction (lane 1) were then incubated in the presence of buffer (lane 2) or phosphatase (lane 3) for 2 h at 37° C. (C) Standard in vitro AR export assays were conducted in the presence of ERS/OA/ARE (lanes 2–8) and harvested at the times indicated. A control reaction in the presence of buffer alone (lane 1) continued for 30 min. Exported AR fractions were immunoblotted using the PG-21 antibody.
FRAP Analysis Suggests that ARE do not Promote Export Simply by Relief of Retention.
We considered two explanations for how ARE could promote AR export: relief of nuclear retention from chromatin binding sites, or induced changes in AR structure and/or compartmentalization. To address whether AR is subject to nuclear retention in our assay, we analyzed the intra-nuclear mobility of GFP-AR by fluorescence recovery after photobleaching (FRAP). LNCaP cells expressing GFP-AR were permeabilized with digitonin, supplemented with different export reaction mixes, subjected to photobleaching, and monitored for fluorescence recovery (Fig. 4b). Single-cell analysis was used to establish that the requirements for GFP-AR export are the same as for unfused, endogenous AR (Fig. 4d, compare with Fig. 3a). The recovery rate (expressed as t1/2 values) of GFP-AR was not correlated with the amount of GFP-AR that underwent nuclear export. For example, maximal intra-nuclear mobility of GFP-AR was observed in the presence of ERS alone; however, this condition resulted in near-background levels of GFP-AR export (Table I). These data suggest that ARE-dependent AR export does not simply reflect mass action-based relief of AR nuclear retention.
Nuclear Export of Androgen-bound AR.
NRs including GR (33), AR (11), and PR (34) undergo ligand-dependent nuclear import. NRs can undergo export following ligand exposure and subsequent withdrawal (11,35–37), but whether ligand dissociation is required for NR nuclear export is unknown. To address this question, we pretreated LNCaP cells in culture with radiolabeled androgen (3H-R1881, 2 nM) to provide a readout for androgen-bound AR. Scintillation counting of the export reaction supernatants revealed that ARE stimulated the release of 3H-R1881 into the export reaction supernatant (Fig. 5a). Further, the amount of 3H-R1881 released into the supernatant under different export conditions was similar to the amount of AR exported from the nucleus in non-radioactive assays (compare Fig. 5a with Fig. 3a). We established by immunoprecipitation and scintillation counting that the 3H-R1881 released into the supernatant was bound to AR (Fig. 5b). These data indicate that androgen dissociation is not a prerequisite for AR nuclear export and that AR remains ligand-bound during export through the nuclear pore complex (NPC).
Figure 5.
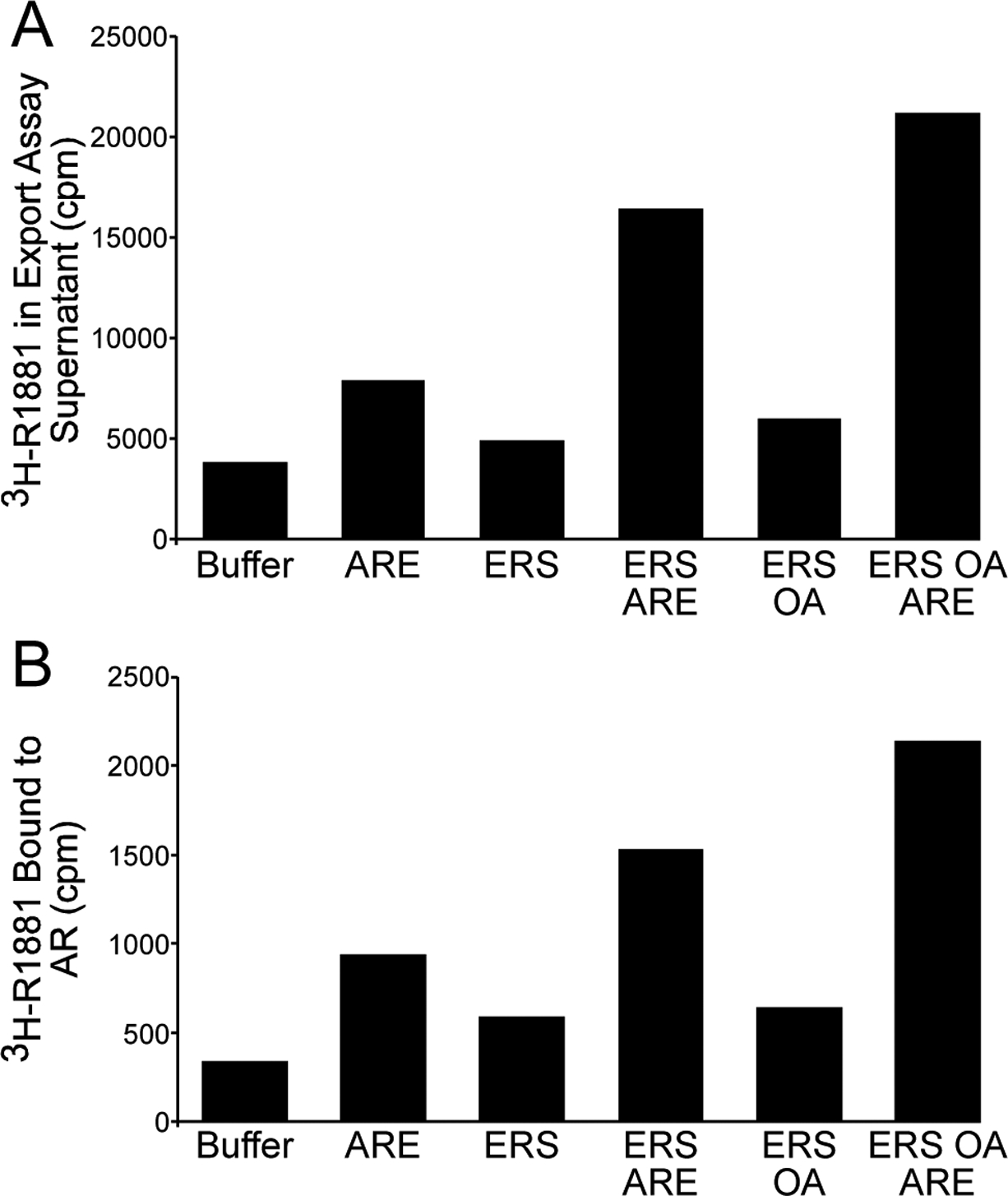
Ligand dissociation is not a prerequisite for AR nuclear export. (A) LNCaP cells were supplied with 3H-R1881 (2 nM) prior to standard in vitro export assays under the reaction conditions shown. Reaction supernatants containing the exported AR fraction were harvested and subjected to scintillation counting to determine the amount of 3H-R1881 liberated from permeabilized cells during the export assay. Values from a representative experiment are shown. (B) LNCaP cells were treated as in (A), except that reaction supernatants containing the exported AR fraction were immunoprecipitated with anti-AR AR-21 antibody. Immunoprecipitates were then washed, extracted (as described in Materials and Methods), and subjected to scintillation counting to determine the amount of 3H-R1881 associated with AR in the exported fraction. Values from a representative experiment, in which background 3H-R1881 binding to control resin has been subtracted to yield corrected values of 3H-R1881 binding to the anti-AR resin, are shown.
Inhibition of PI-3 Kinases Reduces AR Export and Phosphorylation.
ARE stimulation of both AR export and AR phosphorylation prompted us to consider whether a nuclear kinase might be activated by ARE in the permeabilized cell assay. The DNA-dependent protein kinase (DNA-PK) merited consideration for two reasons. DNA-PK kinase activity is stimulated by dsDNA in vitro (38–40), and DNA-PK has been reported to associate with AR in LNCaP cells (18). To address whether DNA-PK is involved in ARE-dependent AR export, we tested wortmannin and LY294002 in the AR export assay. These inhibitors are known to target PI-3 kinase family members including DNA-PK (41,42). In the presence of ERS/OA/ARE, wortmannin and LY294002 each reduced AR export to about the level obtained by ARE alone (Fig. 6a,b, rows 1 and 2, compare lanes 2 and 4). Wortmannin and LY294002 also blocked the upward mobility shift of exported AR, indicating these inhibitors likely block AR phosphorylation as well (Fig. 6a,b).
We next performed biochemical extraction to gain insight into the role of AR compartmentalization in ARE-stimulated AR export. We harvested the supernatants from export reactions (Fig. 6c, Exported) and then treated LNCaP nuclei with Triton X-100 to generate Triton-released and Triton-resistant fractions. In permeabilized cells, AR is initially found in a Triton-resistant compartment (Fig. 6c, lane 1, Control). The amount of AR in the Triton-resistant compartment changes little with the addition of ERS or ARE alone (Fig. 6c, lanes 2 and 3, Control). However, ERS and ARE in combination significantly reduce the amount of AR in the Triton-resistant compartment (lanes 4 and 5, Control). In this assay, wortmannin addition increases the AR level in the Triton-resistant fraction (lanes 4 and 5, compare Control and +Wortmannin), and it blocks the upward electrophoretic mobility shift of AR. These data suggest that a wortmannin-sensitive kinase regulates an early step in the AR export pathway. Finally, wortmannin inhibits AR export stimulated by an oligonucleotide (denoted NRE) that binds the Ku regulatory dimer of DNA-PK (25) (Fig. 6c, lane 6; and see below). These results, together with findings presented below, implicate DNA-PK in the regulation of AR export.
Evidence that DNA-PK Activation Promotes AR Export in vitro.
We used a panel of oligonucleotide duplexes to characterize the structural requirements for DNA stimulation of AR export (Fig. 7). This panel included duplexes similar to ones used by other laboratories to activate DNA-PK in vitro (43) (38–40,44). An ARE synthesized with four point mutations in the ARE half-sites (Fig. 7a, duplex b (28)) stimulated AR export and phosphorylation (Fig. 7b, lane 3), though the level of export was slightly reduced as compared to a WT ARE (duplex a, Fig. 7b, lane 2). The observation that AR export can be stimulated by a mutant ARE suggested the reaction is not sequence-specific, but a duplex synthesized from inosine and cytosine had no activity in the assay (duplex a, Fig. 7b, lane 15). Reducing the concentration of ARE and mutant ARE 10-fold (100 nM final) resulted in significantly less AR export (Fig. 7b, lanes 4, 5). Single-stranded oligonucleotides corresponding to the ARE duplex failed to stimulate AR export and mobility shift (duplexes c and d; lanes 6, 7). As the ARE we used contains 5’ overhangs, we tested whether DNA end structure is important for ARE-stimulated AR export and phosphorylation. ARE duplexes possessing two 5’ and two 3’ overhangs (duplex f, lane 10) or blunt ends (duplex g, lane 11) stimulated AR export and mobility shift similarly to ARE (duplex a, lane 2). Finally, DNA structures containing a bubble in the middle of the ARE (duplex h, lane 12) or on the end of an annealed oligonucleotide (oligo i, lane 13) both failed to stimulate AR export. These results are consistent with ARE stimulation of export by direct binding to AR, activation of DNA-PK, or a combination of both.
As an additional test for the involvement of DNA-PK in ARE-stimulated export, we employed the chemical inhibitor NU7026. When used at a concentration (2.5 μM) to inhibit DNA-PK but not PIKK subfamily kinases ATM or ATR (42,45), NU7026 inhibited AR export and abolished the electrophoretic mobility shift of exported AR induced by ERS/OA/ARE (Fig. 8). We also tested NU7026 in the presence of a modified ARE in which the two half-sites were exchanged for a sequence element that binds the Ku regulatory dimer of the DNA-PK complex (denoted NRE; see (25)). The NRE duplex stimulated AR export in the presence of ERS and OA, and it also induced the electrophoretic mobility shift observed in the presence of ARE (Fig. 8). We next examined whether AR export could be stimulated by an oligonucleotide duplex containing an ARE and an NRE in cis, reasoning that it might function as a scaffold and facilitate the interaction between AR and DNA-PK. The ARE-NRE oligonucleotide stimulated AR export and induced the electrophoretic mobility shift with about the same efficiency as individual ARE and NRE oligonucleotides. One interesting difference was that the NRE oligonucleotide induced export without generating the fastest electrophoretic species of AR routinely observed with other oligonucleotides. Finally, three dsDNA oligonucleotides unrelated to the ARE stimulated AR export, and this was blocked by NU7026 (Fig. 8, see NS1–3 in the presence of ERS/OA/ARE), providing further evidence for DNA-PK involvement in ARE-stimulated AR export.
Figure 8.
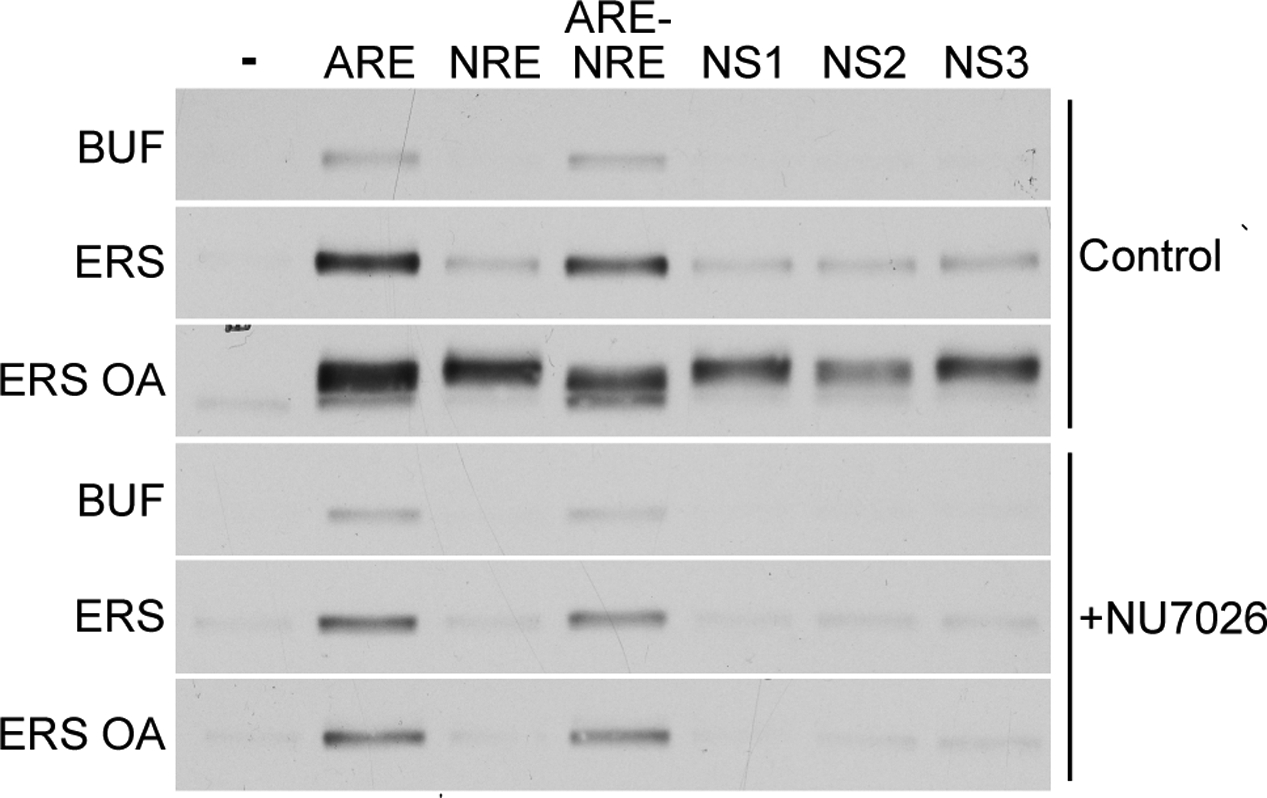
A DNA-PK inhibitor blocks ARE-dependent AR export and phosphorylation. LNCaP cells were treated with 10 nM R1881 alone or in combination with 2.5 μM NU7026 prior to permeabilization and standard in vitro export assays in the presence of ERS, OA, and/or dsDNA as shown. Exported AR fractions were immunoblotted using anti-AR PG-21 (Upstate). In the panel shown, the left-hand column indicates the reaction mix components that are combined with the dsDNA oligonucleotide (1 μM) in the top row to stimulate AR export. NRE is a modified ARE in which the androgen response element has been exchanged for a negative regulatory element-1 sequence {Giffin, 1997 #234}. ARE-NRE is a modified ARE containing a negative regulatory element-1 sequence in cis to the androgen response element. NS1, NS2, and NS3 are dsDNA oligonucleotides that do not contain an androgen response element. All gel lanes shown are from the same film exposure. Therefore, while absolute amounts of exported AR cannot be compared directly between the two cell populations, the amounts of exported AR can be compared between different rows within the same cell population.
NU7026 Enhances Androgen-Dependent Transcription in Prostate Cancer Cells.
To address whether DNA-PK inhibition affects AR transcriptional activity, we treated LNCaP cells with NU7026 and assayed the transcript levels of four genes whose expression is regulated by androgen and AR. Real-time PCR analysis revealed that NU7026 slightly enhances (20% increase) AR-dependent transcription from the PSA gene (Fig. 9A). The effect on androgen-induced transcription of S100P was substantial, as NU7026 increased AR-dependent transcription approximately five-fold. Intermediate effects were measured for the PDF and FKBP51 genes, as NU7026 enhanced the androgen-induced transcript levels of these genes approximately two-fold. These results together with our in vitro biochemical data and previous work showing that DNA-PK binds to AR (18) establish an important link between DNA-PK and AR.
Exportin-Mediated AR Export.
Nuclear import and export of most proteins is mediated by transport receptors belonging to the importin β superfamily (46). We tested 35S-labelled exportins 4, 5, 6, and 7 for binding to the AR DBD. Binding of all four exportins to AR GST-DBD-Hinge protein was stimulated by RanGTP (data not shown). Recombinant exportin 5 stimulated AR export from the nucleus of digitonin-permeabilized LNCaP cells (Fig. 10a), and this export was inhibited by including excess GST-DBD-Hinge in the assay (Fig. 10b). AR export in this setting did not require the addition of recombinant Ran, but our permeabilized cells may retain enough of this factor to facilitate the observed level of exportin 5-mediated AR export. Exportin 5 and DNA-PK appear to function in different AR export pathways, as NU7026 blocked AR phosphorylation but failed to block exportin 5-dependent export in the permeabilized cell setting (Fig. 10c).
Figure 10.
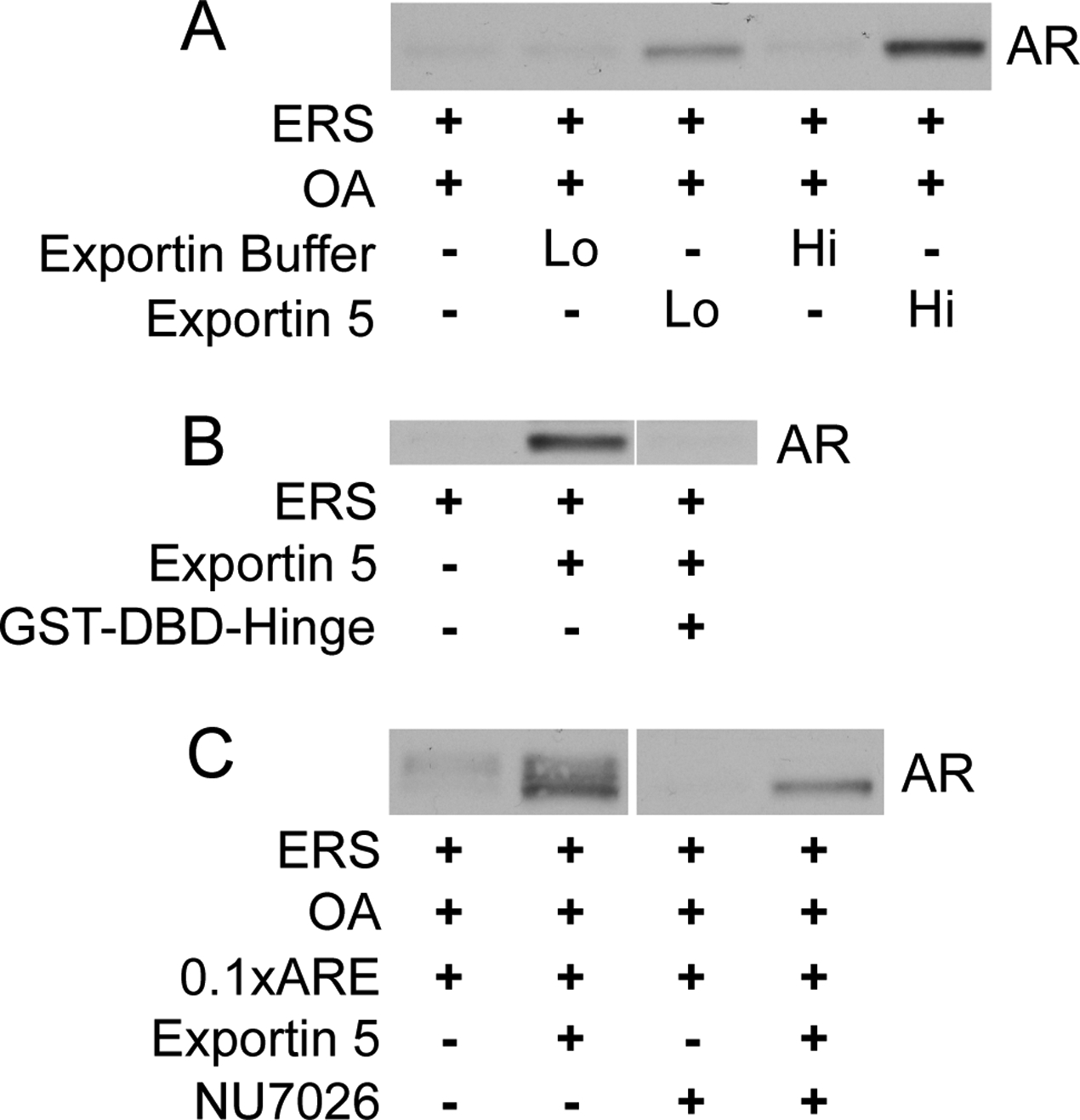
Exportin 5 stimulates AR export in vitro. (A) LNCaP cells were treated with 10 nM R1881 prior to standard in vitro export assays in the presence of ERS, OA, and/or recombinant human exportin 5 (0.1 (Lo) or 0.2 (Hi) mg/mL) as shown. Exported AR fractions were immunoblotted with PG-21. In lanes 2 and 4, dialysate obtained from the purification of recombinant exportin 5 was added to the export reaction mix as a negative (buffer) control. (B) Standard in vitro export reactions were conducted in the presence of ERS, recombinant exportin 5 (1 μM), and recombinant GST-DBD-Hinge (10 μM) as shown. (C) LNCaP cells were treated with 10 nM R1881 alone or in combination with 2.5 uM NU7026 prior to standard in vitro export assays in the presence of ERS, OA, ARE (100 nM), and/or recombinant exportin 5 as shown.
DISCUSSION
We developed a digitonin-permeabilized cell assay to analyze nuclear export of AR from LNCaP cell nuclei. Permeabilized cell assays have been used to purify and characterize cytosolic transport factors that function in a variety of nuclear import and export pathways. Unexpectedly, we found that an energy regenerating system together with DNA duplexes, like those typically used for EMSA, stimulated efficient AR export. The fact that AR export in this assay does not require the addition of cytosol suggests that nuclei of digitonin-permeabilized cells may retain sufficient levels of transport factors needed for this export pathway. A similar observation was made in the context of the in vitro nuclear export of NFAT (29). The permeabilized cell assay enabled us to identify several biochemical features of AR export in vitro. These include: (1) dsDNA oligonucleotides potently stimulate AR export, (2) this AR export pathway requires ATP hydrolysis, (3) dsDNA-dependent export is blocked by the DNA-PK inhibitor NU7026, (4) ligand dissociation is not obligatory for export, and (5) AR export can also be stimulated by recombinant exportin 5.
Energy requirements for ARE-dependent AR export-
ARE-dependent AR nuclear export requires nucleotide hydrolysis because ATP, but not the non-hydrolyzable nucleotide AMP-PNP, supports AR export. In our assay, ATP may serve primarily as a substrate for DNA-PK and/or another nuclear kinase(s). Yang and DeFranco found that ATP hydrolysis is necessary for robust GR export in digitonin-permeabilized cells when tyrosine phosphorylation is stabilized, consistent with an ATP requirement for a nuclear tyrosine kinase involved in GR export (30). ATP is also rate-limiting for export of other cargoes in vitro, including NFAT (29) and the HIV-1 protein Rev (27). However, the identities of the ATP-utilizing enzyme(s) in these systems are undefined. In addition to nuclear kinases, molecular chaperones are appealing candidates. On this note, Elbi and coworkers found that ATP is necessary for restoration of the intra-nuclear mobility of GR and PR by molecular chaperones in permeabilized cells (47), but in that study the relationship between ATP utilization, nuclear receptor mobility, and nuclear export was not examined. Also, GFP-GR recycling on an MMTV promoter array in live cells examined by Stavreva and coworkers is sensitive to energy depletion and to the hsp90 inhibitor geldanamycin (48). These findings with GR imply that ATP may also be required for AR interactions with chaperones prior to AR nuclear export, but the target is not likely to be Hsp90 because geldanamycin does not inhibit AR export in our assay (our unpublished observations).
GTP did not support AR export in permeabilized cells and GMP-PNP did not inhibit AR export. At face value these data suggest that AR export is independent of GTP-utilizing enzymes. GTP hydrolysis by Ran is not required for efficient export of a classical NES-containing cargo (31,49) and Ran-GMP-PNP can stimulate NES export by promoting export complex assembly. Indeed, Ran-GTP is a stochiometric component of numerous export receptor-cargo complexes, and Ran-GTP is predicted to regulate exportin 5-dependent AR export in our assay. It is possible that the level of Ran-GTP remaining in our cells following permeabilization is sufficient to promote formation of an AR export complex. Alternatively, AR could undergo Ran-GTP-independent export. Ran has been reported to bind AR and enhance AR transactivation in luciferase reporter assays (50), but the relationship between these observations and our export data are not clear.
Ligand Occupancy and AR Export-
Nuclear receptor export is often studied in the context of receptor movement from nucleus to cytoplasm after cognate ligand has been added and subsequently withdrawn (11) (35,36,51). Using the permeabilized cell assay we were able to test whether ligand dissociation is a prerequisite for AR export. Using a labeled synthetic androgen, we found that AR can remain agonist-bound during nuclear export. Thus, the agonist-bound form of AR is competent for both import (11,52) and export (this study), and ligand dissociation is not a prerequisite for AR export.
AR Export and Phosphorylation-
Defranco and co-workers observed that the phosphatase inhibitor OA (100 nM) promotes the cytoplasmic accumulation of GR in cultured cells, implying that a kinase/phosphatase cycle influences the nucleocytoplasmic localization of nuclear receptors (32). We applied OA to our permeabilized cell assay to determine if AR export is regulated by a kinase/phosphatase cycle, and observed a modest (approximately 2-fold) enhancement of ERS/ARE-stimulated AR export. Under this condition AR is clearly phosphorylated because it undergoes an electrophoretic mobility shift that can be reversed by alkaline phosphatase treatment. This implies that direct phosphorylation could be one of the rate-limiting steps for AR export in vitro. AR is phosphorylated on multiple serines (53,54) and at least one tyrosine (55). Phosphorylation of Ser650 in the hinge region positively regulates AR internuclear migration in a heterokaryon assay (56). However, Ser650 phosphorylation does not appear to regulate DNA-stimulated export in vitro, because AR containing an alanine substitution at this position still undergoes nuclear export and electrophoretic mobility shift in response to ARE addition in PC-3 cells (data not shown).
Phosphorylation is thought to regulate the nuclear export of nuclear receptors in addition to AR, but thus far a common mechanism has not emerged. GR undergoes export in response to UV irradiation in a pathway that depends on JNK phosphorylation of Ser226 in the AF-1 region (57). GR export in response to UV treatment can be inhibited by leptomycin B (LMB), which provides evidence that Crm1 mediates GR export in this setting. GR lacks a prototypical NES of the type recognized by Crm1, suggesting the interaction is either adaptor-mediated or involves a unique export signal in GR. Because export of GR in non-irradiated cells is LMB-insensitive (51) and can be mediated by the chaperone calreticulin (35), nuclear receptor export probably occurs by more than one pathway (reviewed in (58)). PR export appears to be mediated by Crm1 in a reaction that is dependent on ERK phosphorylation of Ser294 in the AF-1 region and that is linked to protein turnover (59). It has also been reported that cytoplasmic localization of ER is regulated by Thr311 phosphorylation in the AF-1 region (60). It is interesting that AF-1 phosphorylation appears to be a shared feature in these pathways given that AF-1 sequences are variable among nuclear receptors.
DNA Stimulation of AR Export in vitro-
One of the major findings of this study was that a short dsDNA containing an ARE potently stimulates nuclear export of AR. The logic of introducing ARE into the permeabilized cell assay was based on the assumption that AR is subject to nuclear retention through DBD-mediated binding to chromatin, and that titration of this reaction by ARE addition would enhance AR export. The absence of a correlation between GFP-AR nuclear mobility (t1/2 values) and the amount of GFP-AR nuclear export, across different export reaction conditions, suggest that ARE do not merely relieve AR nuclear retention. ARE could bind directly to the DBD of AR and induce changes in AR conformation or sub-nuclear localization to promote AR export. ARE could also stimulate AR export by activating a nuclear kinase that phosphorylates AR or a component of this AR export pathway. Nuclear kinases that can be activated by DNA include the PIKK (phosphoinositide-3-kinase-related protein kinase) subfamily members ATM, ATR, and DNA-PK. We addressed the contributions of these kinases to ARE-induced AR export by pharmacological inhibition. Wortmannin and LY294002 inhibit ATM, ATR, and DNA-PK, and both inhibitors were highly effective at reducing AR export in permeabilized cells. We next utilized the inhibitor NU7026 because the IC50 values of this compound allow discrimination between the involvement of DNA-PK (0.23 mM), ATM (>100 mM), and ATR (>100 mM) (42). NU7026 abolished AR export, as well as AR phosphorylation detected by electrophoretic gel shift, induced by ERS/OA/ARE (Fig. 8), indicating that DNA-PK activity is rate-limiting for the AR export stimulated in this assay. While ARE and the other dsDNA oligonucleotides used in this study provide a means for artificial activation of DNA-PK, it is possible that they are mimicking an activation step that occurs when DNA-PK holoenzyme is bound to chromatin (see below). There is also a pathway for activating DNA-PK end-joining activity that involves inositol phosphate binding to the Ku regulatory dimer (61), an interesting finding in light of the fact that inositol phosphates co-regulate certain export pathways (62,63).
The activity of oligonucleotides of differing sequence and structure in our export assay appeared to corroborate results obtained with NU7026. Neither ssDNA component of the ARE dsDNA oligonucleotide stimulated AR export, consistent with the inability of ssDNA to stimulate DNA-PK activity (38,43,44). Also, dsRNA oligonucleotides also failed to stimulate AR export (data not shown) and do not stimulate DNA-PK activity (40,44). Perhaps the most compelling result linking DNA-PK to AR was our finding that AR export and phosphorylation were stimulated by nonspecific dsDNA that do not contain an androgen response element, including the NRE duplex that binds the Ku regulatory dimer of DNA-PK, and these effects were inhibited by NU7026 (Fig. 8).
DNA-PK is best known for roles in DNA damage response and V(D)J recombination (19), but multiple studies have implicated DNA-PK in pathways of nuclear receptor function. DNA-PK has been shown to physically interact with ERa (21), VDR (22), PR (23), and AR ((18), above). Subunits of the DNA-PK complex have been localized by chromatin immunoprecipitation to promoters regulated by VDR (22) and AR (18). Further, DNA-PK can phosphorylate GR (24) and PR (23). It is interesting to note that DNA-PK phosphorylates PR (23) and GR (24) in the DBD/hinge region, and AR contains a DNA-PK consensus phosphorylation site in the hinge region (Ser656). Also, in early studies of PR phosphorylation it was noted that HeLa nuclear extract contains a kinase that phosphorylates PR in response to addition of plasmid DNA harboring a progesterone response element (64). These data suggest that at least one general mechanism of DNA-PK transcriptional regulation involves direct phosphorylation of nuclear receptors; this function appears to extend to other transcription factors and RNA Pol II as well (65,66). Exactly how DNA-PK regulates transcription has not been elucidated, but it has been proposed to promote recycling of AR by directing re-initiation (18). Knockdown of the Ku70 or Ku80 subunit caused a modest reduction in androgen-dependent PSA transcription (18). Using a pharmacological approach, we found that inhibiting DNA-PK catalytic activity with NU7026 increased AR-dependent transcription from several genes. It is possible that DNA-PK has both positive and negative roles in AR-dependent transcription, and that reducing Ku70/80 levels by knockdown and inhibiting catalytic activity with NU7026 are not equivalent in terms of DNA-PK inactivation.
Exportin 5 Stimulates AR Export-
Virtually all proteins that undergo nuclear transport rely on transport receptors to mediate translocation through the NPC (46). Nuclear localization sequences (NLS) have been defined in most nuclear receptors, and GR undergoes nuclear translocation by contacting any one of several import receptors including importin alpha and importin 7 (67). Our previous work on GR showed that the DBD contains the information that is sufficient to specify export to the cytoplasm (16). This observation, together with the finding that mutations in the DBD of GR and AR interfere with nuclear export (16), lead us to propose that the nuclear export sequence (NES) in nuclear receptors is encoded by the DBD. Using binding assays we found that several exportins bind to a fragment of AR containing its DBD and hinge domains (data not shown). Recombinant exportin 5 can stimulate AR export in the permeabilized cell assay and this can be inhibited by excess GST-DBD-Hinge. These results are consistent with exportin 5 mediation of AR export in permeabilized cells via binding to the DBD.
Model for AR Export-
In the present study we have shown that in a permeabilized cell assay DNA-PK activity is critical for AR export. Our current and previous findings together with data from other laboratories leads us to speculate that phosphorylation provides AR with a “license” to undergo export from the nucleus (Fig. 11, Stage I). Based on the fact that DNA-PK and AR can be localized to the same regions of the PSA promoter by chromatin-immunoprecipitation (18), it seems reasonable to suggest that DNA-PK phosphorylates AR when both proteins are engaged with chromatin. This would provide a means of coupling AR participation in transcription with subsequent AR export. Thus, AR could receive its license to leave the nucleus after a single round or after multiple rounds of transcription, depending on the gene and the influence of OA-sensitive phosphatases. In this model, phosphorylated AR is assembled into an export complex after receiving its license (Stage II). The fact that AR export can be stimulated without adding export factors may indicate that permeabilized cells retain sufficient quantities of these factors such that they are not rate limiting in our assay. On this note, we were unable to directly show that RanGTP is required for AR export in permeabilized cells. Following assembly, an AR:exportin:RanGTP complex would translocate through the central channel of the NPC and enter the cytoplasm (Stage III). The fact that the highly specific DNA-PK inhibitor NU7026 enhances AR-dependent transcription in LNCaP cells can be viewed as evidence that DNA-PK negatively regulates AR activity. A simple interpretation of this result is that inhibition of DNA-PK increases the nuclear concentration of AR and results in additional rounds of transcription. The extent of the NU7026 effect was gene-dependent, which is consistent with our recent finding that the nuclear concentration of AR required for efficient transactivation is not the same for all AR-regulated genes (68). Rigorous evaluation of the export license model will require defining the DNA-PK phosphorylation sites on AR and determining how these modifications influence binding to the nuclear export machinery.
Figure 11.
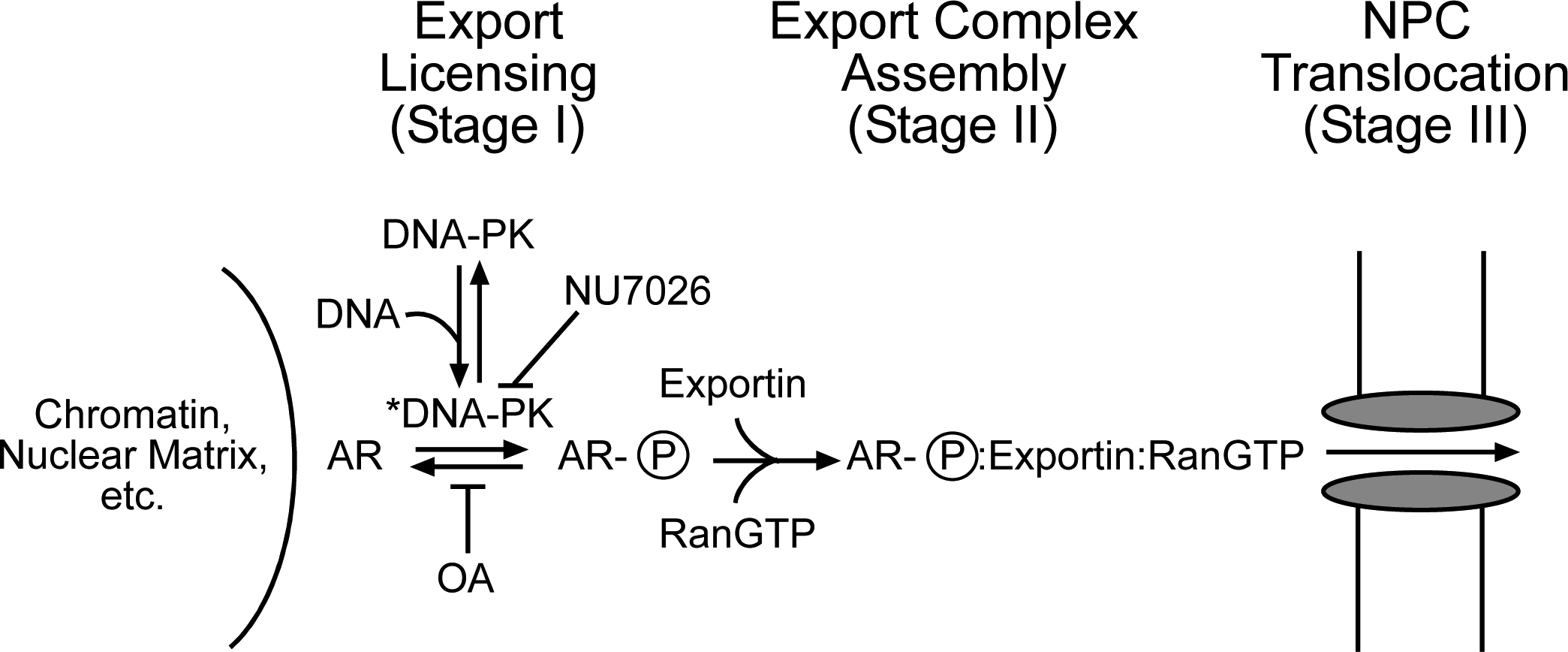
A model of how DNA-PK may regulate AR nuclear export. In this model, DNA-PK becomes activated (DNA-PK*) via interaction with DNA and mediates a phosphorylation licensing of AR associated with nuclear structure. This licensing can be blocked by the DNA-PK inhibitor NU7026 or stabilized by the phosphatase inhibitor okadaic acid. AR licensing could then facilitate engagement with an export receptor and RanGTP, together forming an export complex that is competent for translocation through the nuclear pore complex. See text for further details.
REFERENCES
- 1.Olefsky JM (2001) J Biol Chem 276(40), 36863–36864 [DOI] [PubMed] [Google Scholar]
- 2.Gelmann EP (2002) J Clin Oncol 20(13), 3001–3015 [DOI] [PubMed] [Google Scholar]
- 3.Culig Z, Hobisch A, Bartsch G, and Klocker H (2000) Urol Res 28(4), 211–219 [DOI] [PubMed] [Google Scholar]
- 4.Brinkmann AO, and Trapman J (2000) Nat Med 6(6), 628–629 [DOI] [PubMed] [Google Scholar]
- 5.Balk SP (2002) Urology 60(3 Suppl 1), 132–138; discussion 138–139 [DOI] [PubMed] [Google Scholar]
- 6.Bentel JM, and Tilley WD (1996) J Endocrinol 151(1), 1–11 [DOI] [PubMed] [Google Scholar]
- 7.Brinkmann AO (2001) Mol Cell Endocrinol 179(1–2), 105–109 [DOI] [PubMed] [Google Scholar]
- 8.Heinlein CA, and Chang C (2004) Endocr Rev 25(2), 276–308 [DOI] [PubMed] [Google Scholar]
- 9.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, and Sawyers CL (2004) Nat Med 10(1), 33–39 [DOI] [PubMed] [Google Scholar]
- 10.Prescott J, and Coetzee GA (2006) Cancer Lett 231(1), 12–19 [DOI] [PubMed] [Google Scholar]
- 11.Tyagi RK, Lavrovsky Y, Ahn SC, Song CS, Chatterjee B, and Roy AK (2000) Mol Endocrinol 14(8), 1162–1174 [DOI] [PubMed] [Google Scholar]
- 12.Marcelli M, Stenoien DL, Szafran AT, Simeoni S, Agoulnik IU, Weigel NL, Moran T, Mikic I, Price JH, and Mancini MA (2006) J Cell Biochem 98(4), 770–788 [DOI] [PubMed] [Google Scholar]
- 13.Farla P, Hersmus R, Trapman J, and Houtsmuller AB (2005) J Cell Sci 118(Pt 18), 4187–4198 [DOI] [PubMed] [Google Scholar]
- 14.Tomura A, Goto K, Morinaga H, Nomura M, Okabe T, Yanase T, Takayanagi R, and Nawata H (2001) J Biol Chem 276(30), 28395–28401 [DOI] [PubMed] [Google Scholar]
- 15.Farla P, Hersmus R, Geverts B, Mari PO, Nigg AL, Dubbink HJ, Trapman J, and Houtsmuller AB (2004) J Struct Biol 147(1), 50–61 [DOI] [PubMed] [Google Scholar]
- 16.Black BE, Holaska JM, Rastinejad F, and Paschal BM (2001) Curr Biol 11(22), 1749–1758 [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, Johnson M, Le KH, Sato M, Ilagan R, Iyer M, Gambhir SS, Wu L, and Carey M (2003) Cancer Res 63(15), 4552–4560 [PubMed] [Google Scholar]
- 18.Mayeur GL, Kung WJ, Martinez A, Izumiya C, Chen DJ, and Kung HJ (2005) J Biol Chem 280(11), 10827–10833 [DOI] [PubMed] [Google Scholar]
- 19.Collis SJ, DeWeese TL, Jeggo PA, and Parker AR (2005) Oncogene 24(6), 949–961 [DOI] [PubMed] [Google Scholar]
- 20.Daniel R, Katz RA, and Skalka AM (1999) Science 284(5414), 644–647 [DOI] [PubMed] [Google Scholar]
- 21.Arnold SF, Obourn JD, Yudt MR, Carter TH, and Notides AC (1995) J Steroid Biochem Mol Biol 52(2), 159–171 [DOI] [PubMed] [Google Scholar]
- 22.Okazaki T, Nishimori S, Ogata E, and Fujita T (2003) Biochem Biophys Res Commun 304(4), 632–637 [DOI] [PubMed] [Google Scholar]
- 23.Sartorius CA, Takimoto GS, Richer JK, Tung L, and Horwitz KB (2000) J Mol Endocrinol 24(2), 165–182 [DOI] [PubMed] [Google Scholar]
- 24.Giffin W, Torrance H, Rodda DJ, Prefontaine GG, Pope L, and Hache RJ (1996) Nature 380(6571), 265–268 [DOI] [PubMed] [Google Scholar]
- 25.Giffin W, Kwast-Welfeld J, Rodda DJ, Prefontaine GG, Traykova-Andonova M, Zhang Y, Weigel NL, Lefebvre YA, and Hache RJ (1997) J Biol Chem 272(9), 5647–5658 [DOI] [PubMed] [Google Scholar]
- 26.Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, Mirand EA, and Murphy GP (1983) Cancer Res 43(4), 1809–1818 [PubMed] [Google Scholar]
- 27.Love DC, Sweitzer TD, and Hanover JA (1998) Proc Natl Acad Sci U S A 95, 10608–10613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schoenmakers E, Verrijdt G, Peeters B, Verhoeven G, Rombauts W, and Claessens F (2000) J Biol Chem 275(16), 12290–12297 [DOI] [PubMed] [Google Scholar]
- 29.Kehlenbach RH, Dickmanns A, and Gerace L (1998) J Cell Biol 141(4), 863–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Liu J, and DeFranco DB (1997) J Cell Biol 141(3), 523–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Englmeier L, Olivo JC, and Mattaj IW (1999) Curr Biol 9(1), 30–41 [DOI] [PubMed] [Google Scholar]
- 32.DeFranco DB, Qi M, Borror KC, Garabedian MJ, and Brautigan DL (1991) Mol Endocrinol 5(9), 1215–1228 [DOI] [PubMed] [Google Scholar]
- 33.Savory JG, Hsu B, Laquian IR, Giffin W, Reich T, Hache RJ, and Lefebvre YA (1999) Mol Cell Biol 19(2), 1025–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ylikomi T, Bocquel MT, Berry M, Gronemeyer H, and Chambon P (1992) EMBO Journal. 11(10), 3681–3694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holaska JM, Black BE, Love DC, Hanover JA, Leszyk J, and Paschal BM (2001) J Cell Biol 152(1), 127–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walther RF, Lamprecht C, Ridsdale A, Groulx I, Lee S, Lefebvre YA, and Hache RJ (2003) J Biol Chem 278(39), 37858–37864 [DOI] [PubMed] [Google Scholar]
- 37.Sackey FN, Hache RJ, Reich T, Kwast-Welfeld J, and Lefebvre YA (1996) Mol Endocrinol 10(10), 1191–1205 [DOI] [PubMed] [Google Scholar]
- 38.Leuther KK, Hammarsten O, Kornberg RD, and Chu G (1999) Embo J 18(5), 1114–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hammarsten O, DeFazio LG, and Chu G (2000) J Biol Chem 275(3), 1541–1550 [DOI] [PubMed] [Google Scholar]
- 40.Martensson S, and Hammarsten O (2002) J Biol Chem 277(4), 3020–3029 [DOI] [PubMed] [Google Scholar]
- 41.Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, and Abraham RT (1998) Cancer Res 58(19), 4375–4382 [PubMed] [Google Scholar]
- 42.Veuger SJ, Curtin NJ, Richardson CJ, Smith GC, and Durkacz BW (2003) Cancer Res 63(18), 6008–6015 [PubMed] [Google Scholar]
- 43.Carter TH, Kopman CR, and James CB (1988) Biochem Biophys Res Commun 157(2), 535–540 [DOI] [PubMed] [Google Scholar]
- 44.Carter T, Vancurova I, Sun I, Lou W, and DeLeon S (1990) Mol Cell Biol 10(12), 6460–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, and Smith GC (2004) Bioorg Med Chem Lett 14(24), 6083–6087 [DOI] [PubMed] [Google Scholar]
- 46.Pemberton LF, and Paschal BM (2005) Traffic 6(3), 187–198 [DOI] [PubMed] [Google Scholar]
- 47.Elbi C, Walker DA, Romero G, Sullivan WP, Toft DO, Hager GL, and DeFranco DB (2004) Proc Natl Acad Sci U S A 101(9), 2876–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stavreva DA, Muller WG, Hager GL, Smith CL, and McNally JG (2004) Mol Cell Biol 24(7), 2682–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richards SA, Carey KL, and Macara IG (1997) Science 276(5320), 1842–1844 [DOI] [PubMed] [Google Scholar]
- 50.Hsiao PW, Lin DL, Nakao R, and Chang C (1999) J Biol Chem 274(29), 20229–20234 [DOI] [PubMed] [Google Scholar]
- 51.Liu J, and DeFranco DB (2000) Mol Endocrinol 14(1), 40–51 [DOI] [PubMed] [Google Scholar]
- 52.Zhou ZX, Sar M, Simental JA, Lane MV, and Wilson EM (1994) J Biol Chem 269(18), 13115–13123 [PubMed] [Google Scholar]
- 53.Gioeli D, Ficarro SB, Kwiek JJ, Aaronson D, Hancock M, Catling AD, White FM, Christian RE, Settlage RE, Shabanowitz J, Hunt DF, and Weber MJ (2002) J Biol Chem 277(32), 29304–29314 [DOI] [PubMed] [Google Scholar]
- 54.Yang CS, Xin HW, Kelley JB, Spencer A, Brautigan DL, and Paschal BM (2007) Mol Cell Biol 27(9), 3390–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, Nesheiwat I, Kong X, Melamed J, Handratta VD, Njar VC, Brodie AM, Yu LR, Veenstra TD, Chen H, and Qiu Y (2006) Cancer Cell 10(4), 309–319 [DOI] [PubMed] [Google Scholar]
- 56.Gioeli D, Black BE, Gordon V, Spencer A, Kesler CT, Eblen ST, Paschal BM, and Weber MJ (2006) Mol Endocrinol 20(3), 503–515 [DOI] [PubMed] [Google Scholar]
- 57.Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, and Imai K (2002) Mol Endocrinol 16(10), 2382–2392 [DOI] [PubMed] [Google Scholar]
- 58.Shank LC, and Paschal BM (2005) Crit Rev Eukaryot Gene Expr 15(1), 49–73 [DOI] [PubMed] [Google Scholar]
- 59.Qiu M, Olsen A, Faivre E, Horwitz KB, and Lange CA (2003) Mol Endocrinol 17(4), 628–642 [DOI] [PubMed] [Google Scholar]
- 60.Lee H, and Bai W (2002) Mol Cell Biol 22(16), 5835–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanakahi LA, Bartlet-Jones M, Chappell C, Pappin D, and West SC (2000) Cell 102(6), 721–729 [DOI] [PubMed] [Google Scholar]
- 62.Alcazar-Roman AR, Tran EJ, Guo S, and Wente SR (2006) Nat Cell Biol 8(7), 711–716 [DOI] [PubMed] [Google Scholar]
- 63.Weirich CS, Erzberger JP, Flick JS, Berger JM, Thorner J, and Weis K (2006) Nat Cell Biol 8(7), 668–676 [DOI] [PubMed] [Google Scholar]
- 64.Bagchi MK, Tsai SY, Tsai MJ, and O’Malley BW (1992) Proc Natl Acad Sci U S A 89(7), 2664–2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bannister AJ, Gottlieb TM, Kouzarides T, and Jackson SP (1993) Nucleic Acids Res 21(5), 1289–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peterson SR, Jesch SA, Chamberlin TN, Dvir A, Rabindran SK, Wu C, and Dynan WS (1995) J Biol Chem 270(3), 1449–1454 [DOI] [PubMed] [Google Scholar]
- 67.Freedman ND, and Yamamoto KR (2004) Mol Biol Cell 15(5), 2276–2286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kesler CT, Gioeli D, Conaway MR, Weber MJ, and Paschal BM (2007) Mol Endocrinol 21, In press. [DOI] [PubMed] [Google Scholar]