

Abstract
A dynamic mucosal layer shields the epithelial cells lining the body cavities and is made up of high molecular weight, heavily glycosylated, multidomain proteins called mucins. Mucins, broadly grouped into transmembrane and secreted mucins, are the first responders to any mechanical or chemical insult to the epithelia and help maintain tissue homeostasis. However, their intrinsic properties to protect and repair the epithelia are exploited during oncogenic processes, where mucins are metamorphosed to aid the tumor cells in their malignant journey. Diverse domains, like the variable number tandem repeats (VNTR), sea urchin sperm protein enterokinase and agrin (SEA), adhesion-associated domain (AMOP), nidogen-like domain (NIDO), epidermal growth factor-like domain (EGF), von Willebrand factor-like domain (vWD) on mucins, including MUC1, MUC4, MUC5AC, MUC5B, MUC16 have been shown to facilitate cell-to-cell and cell-to-matrix interactions, and cell-autonomous signaling to promote tumorigenesis and distant dissemination of tumor cells. Several obstacles have limited the study of mucins, including technical difficulties in working with these huge glycoproteins, the dearth of scientific tools, and lack of animal models; thus, the tissue-dependent and domain-specific roles of mucins during mucosal protection, chronic inflammation, tumorigenesis and hematological dissemination of malignant cells are still unclear. Future studies should try to integrate information on the rheological, molecular, and biological characteristics of mucins to comprehensively delineate their pathophysiological role and evaluate their suitability as targets in future diagnostic and therapeutic strategies.
Keywords: Mucins, metastasis, tumorigenesis, mucin domains, cancer
Introduction
A mucosal lining covers the body cavities exposed to environmental insults, including the gastrointestinal, respiratory, and urogenital tracts. The external lining made by epithelial cells is studded with highly glycosylated proteins called mucins [1]. Broadly, gel-forming and transmembrane mucins, expressed by the goblet cells and the epithelial cells, respectively, are the structural components of the mucus layer that act as armors against pathogenic attack, chemical insult, and mechanical abrasion [2]. The synthesis, processing, and secretion of these mucins are highly coordinated and evolutionarily conserved, which attests to their indispensable role as a barrier between the environment and the parenchyma. The mucus layers on different epithelial surfaces in various organs not only differ in their thickness but also in their mucin composition. For example, the lower gastrointestinal tract, duodenum, ileum, and colon mostly express MUC2, among the secreted mucins. In contrast, MUC5AC is predominantly expressed on the gastric mucosa [3], and MUC5AC and MUC5B form the thick viscoelastic mucus covering the upper respiratory tract [4]. The transmembrane mucins, which are thought to have evolved from their secreted partners [5–7], consist of membrane-associated domains and intracellular cytoplasmic tails along with glycan-studded extended extracellular domains. While the glycosylated domains of transmembrane mucins support barrier function like their secreted partners, their cytoplasmic domains contribute to intracellular signaling. The carbohydrate moieties on the glycosylated regions of the mucins perform a multitude of functions, including trapping invading bacteria, acting as decoy receptors via shedding, interacting with immune cells, and sequestering soluble factors. In the face of a pathogenic infection or chronic insult, the cytoplasmic tails of transmembrane mucins can initiate intracellular signals and gene regulation leading to inflammation, damage repair, and epithelial maintenance [2,8].
Apart from their role in normal epithelia, mucins are overexpressed by malignant epithelia and have been shown to play a multifaceted role during tumor initiation, progression, and metastasis [9,5,10–14]. Cancer hijacks the normal physiological and developmental mechanisms to establish at the primary site and systemically spread to the secondary sites. Mucins are integral to this process, as growing tumors exploit the homeostasis and protection functions of mucins for sustenance and metastatic dissemination. The cytoplasmic domains of the transmembrane mucins interact with intracellular signal mediators and facilitate oncogenic signaling [15], while the secreted mucins interact with cell adhesion molecules, receptor tyrosine kinases (RTKs) and other cell-surface molecules to amplify growth, survival, differentiation, and metastatic signals by the tumor cells [16]. Furthermore, circulatory tumor cells utilize the shielding mechanism of mucins for longer survival in circulation, successful arrest, and subsequent colonization at a secondary site [17].
Although several studies have detected the shed part of the transmembrane mucins in the circulation of cancer patients, the mechanistic link of mucin shedding and intracellular signaling in carcinogenesis and the metastatic cascade remain poorly understood. While the multidimensional functions of mucin domains have been documented concerning mucosal protection, epithelial homeostasis, malignant transformation, and metastasis, substantial gaps in the literature remain. In this review article, we aimed to assimilate the knowledge from these fields of mucin research to better understand the involvement of mucins’ functional domains, which can further bolster the design of therapeutic strategies against specific cancers.
1. Mucins as epithelial guardians
Epithelial cells form an organized layer at the interface of complex parenchyma of multicellular organisms and their external environment. Depending on the nature of the tissue, they primarily manifest as specialized single-layered cells with apical-basal polarity, secretory nature, and high turnover rates [18,19]. Most epithelial lining, including those at the surfaces of alimentary, respiratory, reproductive, urinary tracts, and eye, are covered by biological hydrogels known as mucus. This substance helps it protect an enormous surface area exposed to the harsh physical and chemical insults from the environment. Owing to its lubricating, hydrating, and cleansing functions, the mucus barrier has been long presumed to be an inert protective layer over the epithelial cells. However, this protective layer of mucus actively participates in a multitude of biological processes that shape the physiology of the underlying tissue during acute and chronic insult. The abundance, nature, and function of the mucus layer differ by tissue, which is exemplified by the fact that the eye has a thin layer of mucus (10 μM) in order to keep it hydrated, whereas the colon and lung epithelia are lined with thick mucosal layers (300–700 μM) that ensure either entrapment or the clearance of microbes to maintain homeostasis [20]. High molecular-weight glycoproteins, called mucins, produced by specialized cells called goblets cells of the epithelium and submucosal glands, form the structural and functional unit of the mucus barrier. The mucin family comprises 21 members that are broadly divided into two groups: secreted mucins and the membrane-associated mucins. The secreted mucins include the gel-forming (MUC2, MUC5AC, MUC5B, MUC6) and the non-gel-forming (MUC 7) mucins; the gel-forming mucins play a critical role in barrier function at mucosal surfaces. The transmembrane mucins (MUC1, MUC3A/B, MUC4, MUC12, MUC13, MUC15, MUC17, MUC20, and MUC21) form integral components of the cell-surface glycocalyx; their extracellular domains and the associated glycans act as sensors for environmental cues, and their cytoplasmic tails (CT) mediate intracellular signaling [8]. The structural signature of the mucin family members is the proline-threonine-serine (PTS)-rich domain. This domain contains repeated serine and threonine residues that form glycosidic bonds with GalNAc, the first sugar added during the synthesis of O-glycans, the hallmark of mucins [21]. Hence, the PTS domains, expressed as tandem repeats, carry a large number of glycans and makeup to 70% of the mass of these complex glycoproteins. Other than the PTS domains, numerous unique and mutually exclusive domains belong to members of the secreted and transmembrane and are enumerated in Table 1A, B. Each secreted mucin molecule with its extended protein core studded with a huge number of glycans forms the typical “bottle-brush” structure; however, mucins must also assume a macromolecular organization to form the dense mucus layer on exposed epithelial surfaces. Secreted mucin oligomerization is a multistep process starting with dimerization during biogenesis in the endoplasmic reticulum, followed by glycosylation in the Golgi, and finally multimerization of dimers [7,22,23]. While the mucin core determines the biochemical pattern, the extended and gel-like conformation formed by dense glycosylation and oligomerization of the mucin units guides their rheological properties and thereby guards the underlying epithelia against intruding pathogens, abrasive chemicals or other cells.
Table 1: Unique domains and their associated pathological roles of (A) Transmembrane and (B) Secreted mucins.
A) Domains present in MUC1, MUC3A, MUC4, MUC12, MUC13, MUC15, MUC16, MUC17, and MUC21 mucins. While the interacting partners and downstream signaling of several mucin domains have been studied, many yet remain unknown (marked as *). Apart from the domains enumerated in the table, variable number tandem repeats (VNTRs) and transmembrane (TM) domains exist on each of these transmembrane mucins. VNTRs are the sites for glycosylation, and TM domains anchor these glycoproteins to the cell membrane. B) Several domains present in secreted mucins are characterized (107). However, the mechanistic contribution of these domains in cancer progression and metastasis is still elusive (marked as *) and may form the basis for future research.
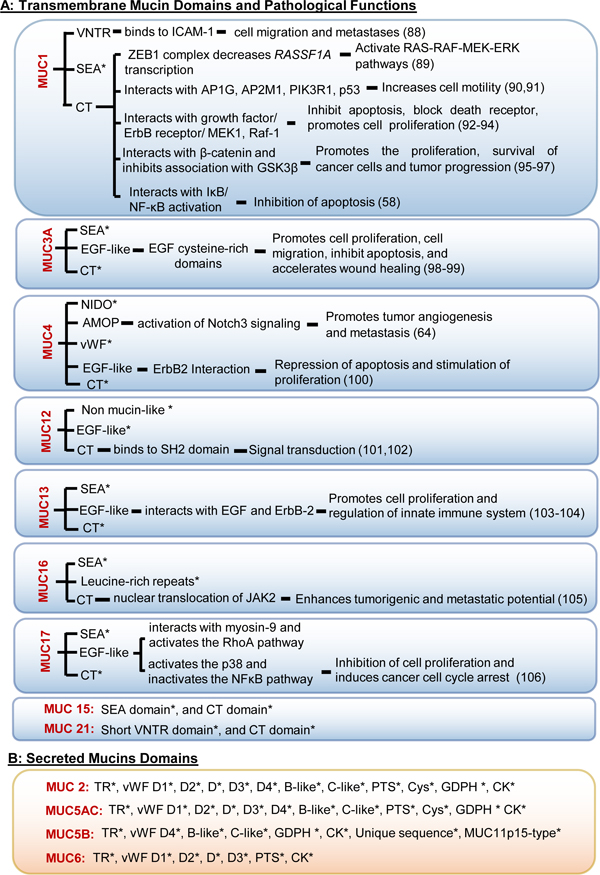 |
Abbreviations: AMOP: Adhesion-associated domain; CYS rich: Cysteine-rich domain; EGF repeats: EGF-like repeat region; NIDO: Nidogen-like domain; SEA: Sea urchin sperm receptor enterokinase agrin domain; vWF D1,2,3,4, B, C: Von Willebrand Factor type D1,2,3,4, B, C domains; PTS domain: Proline, threonine, serine-rich domains; CK domain: Cysteine Knot domain; GDPH: glycine-aspartic acid-proline-histidine.
2. Mucins as the vulnerable link between mucosal immunity, inflammation, and tumorigenesis
The sugary PTS tandem repeat (TR) domain of transmembrane mucins serves as the first attachment point for invading pathogens, once they manage to destabilize the secreted mucin layer by secreting mucus-dissolving enzymes like glycosulfatases, sialidases, N-acetyl neuraminate lyases, O-acetylesterases, and mucinases. Bacterial adhesins not only exploit the mucins for attachment to the epithelium but also utilize their oligosaccharides for energy metabolism [2]. Upon interaction with the microbes, transmembrane mucins often shed their extracellular domains as decoys for microbes anchored to the glycocalyx, while the cytoplasmic domains initiate intracellular signaling to shape the cellular response during potential infection. For example, during intestinal C. jejuni and gastric H. pylori infections, the well-studied membrane-associated mucin MUC1 protects the underlying epithelia from pathogenic toxins as well as releases receptor decoys to prevent colonization [24]. The absence of MUC1 in transgenic mice increased the severity of H. pylori infection and the associated gastritis [25]. A recent study demonstrated a similar role of MUC16 during S. aureus infection of corneal cells [26].
In response to microbial products (lipopolysaccharides, etc.), infiltrating immune effector cells release inflammatory mediators, such as interleukins (IL1β, IL-4, IL-6, IL-9, IL-13), reactive oxygen species (ROS), reactive nitrogen species (RNS), interferons, tumor necrosis factor-α (TNF-α), neutrophil elastase [27–31,16,12]. The resulting inflammatory milieu upregulates secreted and cell-surface mucins in the epithelial mucosa, which is accompanied by hydration and volume expansion of the mucus layer. The expanded layer, in turn, maintains the local inflammation by sequestering the anti-pathogenic immune modulators. Apart from their synthesis and release, mucins can also bolster their protective role via alteration in their glycosylation and sialylation patterns. During an ongoing infection/inflammation (like cystic fibrosis, bronchitis, and H. pylori infection), immune cells (e.g. CD4-T cells) and their soluble mediators (e.g. TNF-α) alter the mucin glycans and thereby prevent the pathogens from threatening the mucus barrier [32–34]. Hence, the regulation of mucin expression and glycosylation serves as important component of host innate and adaptive immune responses against mucosal infection.
The amount of metabolic energy invested for mucin regulation, synthesis, processing, and release at the epithelial interface exemplifies their importance in epithelial barrier protection and repair. However, overexpression of mucins can aggravate inflammatory conditions [35–37]. Specifically, during inflammatory bowel disease, MUC1 expression is upregulated by inflammatory cytokines; however, in IL10−/− mice, in the absence of a counteractive anti-inflammatory signal, MUC1 hyperactivation leads to chronic inflammation and colon cancer [38]. MUC1 has also been shown to activate the inhibitor of nuclear factor-κB kinase-β (IKKβ)-nuclear factor-κB (NF-κB) pathway, which in turn can lead to inflammation-induced tumorigenesis [39]. During H. pylori infection, allelic variations and deregulated cellular localization of MUC1 lead to chronic gastritis (inflammation) followed by gastric cancer [40]. MUC1 hyperexpression associated with Epstein Barr virus (EBV) infection disrupts cell-matrix interactions, thereby driving cellular invasiveness and tumorigenesis [41].
Epithelial cells under normal physiological conditions have a defined polarity where the mucins are organized on the apical interface, and the RTKs are present on the basolateral membranes. Cell-cell and cell-matrix junctional complexes are essential for maintaining epithelial integrity. During chronic inflammation and associated stress-induced signaling, epithelial cells lose their tight junctions, which activates reversible repair and morphogenesis pathways [42,43]. Owing to the loss of polarity, the mucins are distributed along both sides of the epithelial membrane, resulting in an aberrant mucin interactome with homo and heterodimers of RTKs and membrane-associated members of junctional complexes. This phenomenon leads to the enrichment of the mesenchymal phenotype. It promotes survival programming in the inflamed tissue, which in turn leads to mucin hypersecretion, as observed in the case of goblet cell hyperplasia [44]. Accumulating evidence suggests that transmembrane mucins, including MUC1 and MUC4, by virtue of their unique domains, can disrupt tight junctions and adherens junctions during oncogenesis. For instance, the CT domain of MUC1 (MUC1-CT) can sequester β-catenin and thereby reduce its availability to the membrane-associated E-cadherin/β-catenin/α-catenin complex, hence disrupting the complex [45]. In addition, the MUC1-CT and β-catenin interaction promote the upregulation of Wnt signaling [46], which has been well associated with tumorigenesis, stemness, and cancer aggressiveness. Similarly, MUC1 and MUC4 mediate RTK signaling upon loss of apical-basal polarity due to their interaction and association with ERBB2. MUC1 mediates constitutive ERBB2 signaling, which leads to disruption of the partitional complex known as the PAR3-PAR6-atypical protein kinase C (aPKC) complex [47]. By contrast, MUC4 sequesters ERBB2 with its EGF-like domains, stabilizes ERBB2 signaling, and hinders its interaction with ERBB3, causing loss of tight junctions and driving the tumorigenic cascade [48–50]. Although the role of secreted mucins in tumorigenic onset is still elusive, studies indicate that hypersecretion of MUC5AC can lead to steric hindrance within E-cadherin/β-catenin complexes during cancer progression [51,52]. In summary, mucin overexpression, as a consequence of chronic inflammation and the epithelial stress response pathway, results in a transient loss of cell polarity, cell-cell, and cell-matrix interactions; these events ensure the activation of repair, differentiation, and survival pathways. However, malignant cells exploit mucin-mediated restoration pathways for constitutive growth and proliferation signaling, resulting in tumorigenesis. The cancer-associated functions of the various mucins’ domains and the signaling pathways involved during the progression of several malignancies are enlisted in Table 1A, B.
3. Ubiquitous role of mucins in the metastatic cascade
Metastasis accounts for the majority of cancer-related deaths; however, the molecular players and their contribution in this cascade are less understood. Although a significant number of malignant cells leave the primary tumor, only a few metastatic lesions are clinically detected, indicating that metastasis is an incompetent process. For the malignant cells to colonize a distant organ, they need to undergo a complex sequence of events, including dissociation from the primary site by breaching the basement membrane barrier; survival in the bloodstream; homing to the secondary site; extravasation via transendothelial migration; and survival and proliferation at the metastatic organ. Tumor cells that successfully form a metastatic lesion require autonomous gene-regulatory mechanisms and/or support from acellular contributors like inflammation, ROS, RNS, mechanical forces; and cellular partners like immune cells, platelets, stromal cells, mesenchymal stem cells at the primary site, in circulation as well as at the pre-metastatic niche of the secondary site [53].
Aberrant expression of mucins characterizes the specific molecular signature during tumorigenesis and metastasis in multiple malignancies. Multiple studies have reported the involvement of diverse domains (e.g. VNTR, SEA, AMOP, NIDO, EGF, and CT) in transmembrane mucins, including MUC1, MUC4, and MUC16 in various homo and heterotypic interactions that promote distant dissemination of tumor cells [54,55]. Furthermore, accumulating evidence suggests that glycans on the mucin extracellular domains mediate interactions with the tissue microenvironment and immune cells in the circulation, and thereby relay outside-in signals [56,17]. While several literature reviews from our group and others [55,17,9,57,8,5] have compiled the studies on several mucins in facilitating tumor progression and metastasis, it is important to assimilate the findings with respect to the unique domains that attribute to the phenotype. Hence, in the following sections and in Table 1A and B, we have specifically focused on the mucin domains and their interactome or associated signaling axis in the various steps of the metastatic event.
3.1. Dissociation from the primary tumor
During the early stages of metastasis, cancer cells acquire features that promote motility and invasion of the cancer cells through the tissue microenvironment. These include an altered transcriptome, decreased cell-cell adhesion, and altered matrix interaction, and in fact, mucins can participate in each of these processes. In pancreatic cancer, MUC1 upregulates the transcription of mesenchymal genes, including Snail and Slug. Mutation of the MUC1-CT reversed the phenotype by abrogating the ability of MUC1 to bind to β-catenin and promote transcription of epithelial-to-mesenchymal-transition (EMT)-associated genes. In the same study, MUC1-knockout mice demonstrated a decrease in EMT and metastatic propensity as compared to wild-type mice [58]. Another study reported that ectopic expression of a truncated MUC1 construct, marked by high levels of transmembrane and secreted MUC1 and devoid of a cytoplasmic domain, facilitates EMT and proliferation of breast cancer cells via extracellular-signal-regulated-kinase (ERK) and phosphatidyl inositol-3 kinase (PI3K), respectively [59]. Furthermore, the interaction between the MUC1-CT and the DNA-binding domain of the androgen receptor promoted EMT and invasiveness of prostate cancer cells [60]. Another transmembrane mucin, MUC4, has been shown to enhance the mesenchymal phenotype by activation of FAK signaling and upregulation of N-cadherin in pancreatic [61] and ovarian [62] cancer. The mechanism involves interaction with HER2, potentially stabilizing it through three juxta-membrane EGF-like domains in MUC4. In pancreatic cancer cells, the silencing of MUC4 resulted in a downregulation of fibroblast growth factor receptor 1 (FGFR1) with a subsequent decrease in mesenchymal markers, vimentin and N-cadherin, and upregulation of epithelial markers, including E-cadherin, occludin, and cytokeratins [63]. A recent study demonstrated the role of the AMOP domain of MUC4 in facilitating invasion propensity of pancreatic cancer cells via activation of Notch signaling and subsequent upregulation of MMPs [64]. Moreover, the role of MUC4 in promoting EMT and invasiveness in cancer cells was also observed in breast [65] as well as in lung cancer [66] models. Another membrane-tethered mucin, MUC16 have been shown to facilitate pancreatic cancer metastasis via FAK-mediated upregulation of mesenchymal markers [67] [68]. On the other hand, conflicting functions of MUC16 have been observed during the EMT process in ovarian cancer. While depletion of MUC16 from the cell surface led to the internalization of E-cadherin, causing enhanced expression of mesenchymal markers vimentin and N-cadherin [69], overexpression of the MUC16-CT increased the motility and invasiveness of ovarian cancer cells by repressing E-cadherin expression [70]. These observations further suggest unique and mutually exclusive functions for the mucin domains. Apart from the transmembrane mucins, secreted mucin MUC5AC has been associated with the metastasis process in the lung, colon, and pancreatic cancers [16]. MUC5AC was found to interact with integrin β4, leading to induction of FAK signaling and enhanced lung cancer metastasis [71]. A recent study showed that MUC5AC facilitates colon cancer progression and metastasis via interaction with CD44 [72]. Unpublished work from our group suggests that overexpression of MUC5AC potentiates the cancer stem cells and hence enhances the metastatic phenotype of pancreatic cancer cells. Other groups have shown that MUC5AC overexpression (mediated by GLI1) interfered with E-cadherin- based homotypic interactions, leading to the loss of cell-cell contact and an EMT phenotype in pancreatic cancer [52]. MUC5AC was also found to induce invasiveness and motility of pancreatic cancer cells by upregulation of integrins and matrix metalloproteinases [73]. Transmembrane and secreted mucins may cooperate in inducing cancer cell metastasis, as anti-MUC1 and anti-MUC5AC antibodies were observed to abrogate the invasiveness of colon carcinoma HT-29 cells by the restoration of E-cadherin expression [74].
Apart from adopting a migratory phenotype, cancer cells need to breach the basement membrane (BM) as an important initial step for successful metastasis in epithelial malignancies. The BM is a dense layer of extracellular matrix proteins comprising laminin, collagen, nidogen, proteoglycan, and growth factors. The coordinated action of proteases, immune mediators, and oxygen radicals that are associated with mucin expression allows cancer cells to breach the BM, undergo transmigration, and step into the circulation [53]. However, under physiological conditions of immune surveillance and during inflammation-associated pathologies, immune cells and other specialized cells such as mesenchymal stem cells cross the BM independently of proteases, relying primarily on cell-matrix adhesion [75]. Molecular studies of the mucin domains have shown that transmembrane mucins MUC1, MUC4, and MUC16 share structural similarities with extracellular matrix proteins that constitute the BM. For example, the sea urchin sperm protein-enterokinase-agrin (SEA) domain of MUC1 is thought to have evolved from perlecan or heparin sulfate proteoglycan (HSPG2), while the NIDO domain of MUC4 evolved from a common ancestor of nidogen, and MUC16 bears multiple SEA domains [54,15,8]. Mucins, by their specialized domains, can interact with different components of the BM and thereby facilitate the BM transmigration of cancer cells. The MUC4-NIDO domain has been shown to interact with fibulin-2 and contribute to pancreatic cancer metastasis [76]. Another study showed that MUC4-devoid of tandem repeats (MUC4/X) increases the adhesion of cancer cells to the extracellular matrix proteins by activation of the integrinβ1/FAK pathway, and thereby enhances their metastatic propensity [77]. However, the N and C terminals of MUC6, a secreted mucin, were observed to inhibit adhesion of cancer cells to matrix components including collagen, fibronectin, and laminin [78]. Further molecular studies are required for a comprehensive understanding of the involvement of different mucin domains in the metastatic cascade.
3.2. Survival in the circulation
Once the malignant cells enter into circulation, they encounter circulating immune cells and mechanical stress caused by blood flow. To cope with this hostile environment, the circulating tumor cells (CTCs) acquire traits for evading immune surveillance and shielding against the physical turbulence of the bloodstream. CTCs acquire these by initiating platelet activation and the coagulation cascade, using mucins to facilitate attachment to platelets [79]. CTCs express altered glycans, including T, sTn, sLea, and sLex, predominantly associated with the VNTR domains of the tumor-associated mucins, MUC1, MUC4, and MUC16. The carbohydrate moieties on the mucins act as ligands for binding P-selectin on the platelets, resulting in platelet attachment to the tumor cells and thrombin formation. Platelet activation and subsequent platelet-tumor cell aggregates have been shown to enhance metastatic dissemination [17]. This process is facilitated by fibrin, which can interact with tumor cells and platelets through integrins, leading to the formation of thick fibrin coats on the CTC-platelet aggregates [80]. Such aggregates help the tumor cells survive the fluid shear stress in circulation and physically shield the tumor epitopes from immune surveillance, primarily by the natural killer (NK) cells. Besides interaction with platelets, transmembrane mucin MUC4, by its bulky glycosylated extended structure, can shield the cancer-associated antigens and hence protect the CTCs from immune recognition [81]. The coating of the CTC surfaces with the MHC-I molecules of the platelets also favors evasion from the innate immune system. Besides providing physical protection, CTC-platelet-fibrin clots have been shown to contribute to CTC survival by impairing the functionality and anti-tumor activity of NK cells in circulation [82].
3.3. Transendothelial migration and establishment of a metastatic niche
The CTCs that survive the hostilities of circulation extravasate through the endothelium to establish colonies at a secondary organ, a process that closely resembles transendothelial migration of immune cells. The intrinsic characteristics of the migratory cancer cells as well as the extrinsic factors of the pre-metastatic niche, including inflammation, exosomes, tissue-resident fibroblasts, and mesenchymal stem cells cause the initial arrest of the CTCs and subsequent extravasation to form metastatic colonies [83]. During inflammation and cancer progression, the endothelial cells express E-selectin, which interacts with sialylated glycans (SLeX, SLeA) of the mucins expressed on the tumor cells; this marks the first step of docking of the tumor cells on the endothelial cells [84]. After the glycan-based docking, CTCs undergo a rolling process that upregulates their integrin expression and induces a strong interaction with the intercellular adhesion molecule-1 (ICAM-1) on the endothelial cells, leading to extravasation [85]. Several mucins, including MUC1, has been shown to upregulate integrin expression and interaction with ICAM-1, enhancing the invasiveness and extravasation ability of the tumor cells. Furthermore, the extracellular domain shed from MUC1 serves as a decoy receptor to bait leukocytes, thereby compromising immune surveillance during metastatic establishment [86].
Extravasation and initial seeding of the tumor cells at the secondary site is facilitated by an activated endothelium and a primed pre-metastatic niche. Cytokines and chemokines secreted by the tissue resident stromal cells and immune cells primarily contribute to this phenomenon of priming the endothelium and thereby capture incoming CTCs by upregulation of selectins and cell adhesion molecules on the endothelial cells. During chronic inflammation, several immune modulators, including IL1β, IL-2, IL-6, IL-8, IL-17, IL-33, and TNF-α, can upregulate the expression of mucins. Likewise, the local immune milieu at the metastatic site may enhance the production of mucins, which further undergo altered glycosylation, allowing them to interact with siglecs and lectins of immune cells [17], especially neutrophils. Neutrophils, in turn, help cancer cells to colonize and survive in the metastatic tissue by forming neutrophil extracellular traps (NETs) [87]. Platelets also contribute to the formation of the pre-metastatic niche, primarily by releasing TGF-β. Recent studies have demonstrated the role of MUC4 in the interaction with macrophages and platelets during the extravasation of cancer cells and subsequent survival in the metastatic lesions [81]. Hence, tumor cells not only hijack the protective and shielding functions of mucin glycans during the transit through the bloodstream but also employ the survival and proliferative signaling imparted by the various mucin domains to establish as successful metastatic colonies.
As explained above, mucins utilize their multifaceted domains and carbohydrate moieties to facilitate different steps of the metastatic cascade. Until now, studies have mostly focused on the role of transmembrane mucins in promoting outside-in signaling in cancer cells in autocrine fashion. However, it is not known whether mucins can mediate a paracrine signaling by direct interaction with the other cells in the microenvironment, like, stromal cells in the primary tumor and endothelial cells at the port of entry to the metastatic sites. In this context, understanding the role of shed extracellular domains of transmembrane mucins or the secreted mucins may be crucial to evaluate. These cleaved mucin fragments, shed from the primary tumor, may entrap exosomes, inflammatory modulators, and immune cells at the distant sites, even before the cellular migration, thereby priming the endothelium and the tissue microenvironment to capture the CTCs. Multiple studies have demonstrated a strong positive correlation between the clinical stage of the patients and the level of mucins in their serum; however, the mechanistic relevance and the underlying biology remain elusive. Findings from our group and others have shown the sustained expression of mucins in the metastatic lesions; however, their mechanistic role in facilitating the colonization event is largely unexplored. Looking at the similarity between mucin domains and certain matrix proteins, it is tempting to speculate that mucin backbones can modulate the tissue microenvironment and subsequently serve as scaffolds for the incoming tumor cells at the metastatic sites, thereby, contributing towards preparing the soil for the incoming seed.
Conclusion and future perspectives
Mucins are epithelial guardians that perform multiple functions in physiologic and pathologic conditions. They can prevent pathogenic infection, modulate inflammation, promote repair, and facilitate cell survival. Broadly, most studies have identified the secreted mucins as dimeric or multimeric layers of protective sheet that can shelter the commensal bacteria in the colon or clear pathogenic intruders through ciliary mechanisms from the upper respiratory tract. Transmembrane mucins, on the other hand, have been mostly described as cell-surface sensors, which can undergo outside-in signaling by their extracellular domains and phospho-active intracellular domain (cytoplasmic tail). During a pathogenic intrusion, the transmembrane mucins shed their extracellular domains as “mucin decoys”, which can help to clear the pathogen and prime the intracellular domain for inflammatory signaling and gene regulation. To preserve epithelial integrity, these mucins can detect chronic stress and inflammation cues in the environment and initiate signals for damage repair, differentiation, survival, and proliferation. At the crossroads of chronic inflammation and carcinogenesis, overexpression and altered localization of these mucins play a crucial role.
During malignant transformation, the otherwise apically localized mucins get dispersed all along the neoplastic cell surface, allowing them to interact with multiple cell-adhesion molecules and onco-signaling partners. Malignant cells exploit the restitution properties of mucins to stimulate aberrant growth and survival; thus, mucins are crucial drivers in the initiation and progression of multiple malignancies. Interestingly, mucins can exhibit both adhesive and anti-adhesive properties during different stages of tumorigenesis and metastasis. During the breaching of the basement membrane, they can sterically break cell-cell homotypic interactions while facilitating cell-matrix interactions to initiate the metastatic cascade. The mucin glycans also harbor ligands for binding the selectins and siglecs present on immune cells, platelets, and endothelial cells. By virtue of mucin’s adhesive properties, circulating cancer cells can form fibrin-platelet aggregates, which allow them to withstand the turbulent shock in the bloodstream, evade immune surveillance, and extravasate the endothelial lining during metastasis. The high concentration of mucins in the serum during malignant progression has not only facilitated biomarker research but also presents an opportunity to predict therapy response and disease relapse. However, the mechanistic understanding behind mucin shedding during tumorigenesis and its correlation with intracellular signaling is still elusive and needs further study. Owing to the technical difficulties in working with these glycoproteins and dearth of animal models, most of our knowledge on transmembrane mucins is based on studies in the cancer field; similarly, information about secreted mucins is mostly derived from the field of mucosal infection and host-pathogen interaction. Mucin survival and repair functions during mucosal protection share similarities with the events of carcinogenesis and metastatic spread; thus, future studies should consider the rheological (mechanical pressure, viscoelasticity), molecular (domain organization, splice variants), chemical (glycan structures, intramolecular linkages) and biological (homotypic/heterotypic cell-cell/cell-matrix interactions, downstream signaling) characteristics of mucins to comprehensively delineate their pathophysiological role. This information can guide new strategies for improved diagnostics and therapeutics in cancer.
Figure 1: Mucins at the crossroads of mucosal protection, inflammation and tumorigenesis.
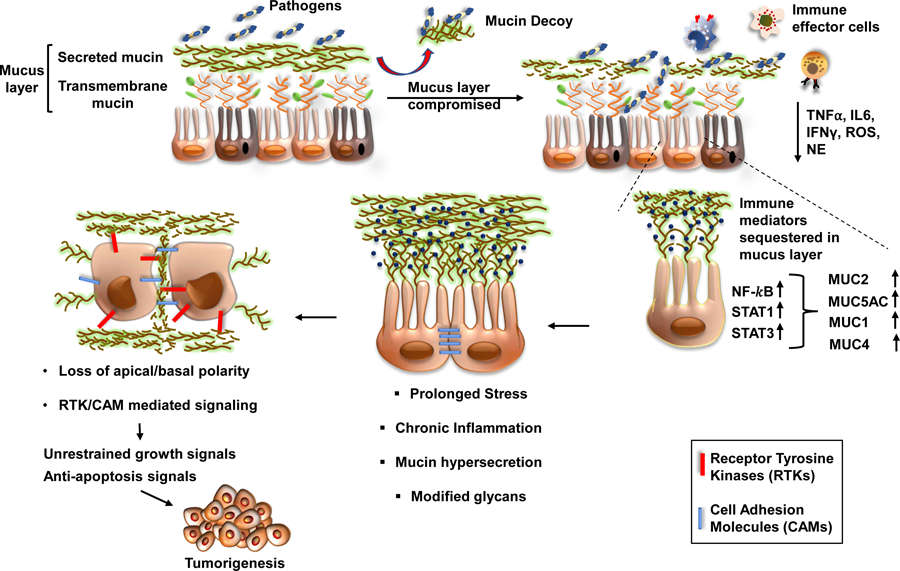
The protective mucus layer on the epithelial lining is comprised of transmembrane and secreted mucins. The invading pathogens can compromise the secreted mucus layer and gain access to the transmembrane mucins, which are apically arranged close to the epithelial surface. Upon binding of the pathogens, mucins shed their extracellular domains as decoy receptors, thereby facilitating pathogen clearance. Subsequently, the cytoplasmic domain of the mucins like MUC1 and MUC16 can transduce intracellular signals in response to local inflammatory milieu containing TNFα, IL6, IFNγ, ROS, and RNS. The localized enrichment of immune mediators leads to an activation of transcription factors such as NF-κβ, STAT1, and STAT3, which upregulate the expression of mucins. In response to infection and subsequent inflammation, the mucin-producing goblet cells become hyperplastic (commonly called goblet cell hyperplasia) and secrete mucins with altered glycans to obscure pathogenic adhesion and prevent mucus degradation. Chronic inflammation and stress, however, cause the underlying epithelial cells to lose their polarity and activate signaling pathways for repair, cellular differentiation, and survival. Due to loss of apical-basal polarity, mucins can interact with various receptor tyrosine kinases and cell-adhesion molecules to mediate sustained growth signals, thereby facilitating the events of tumorigenesis.
Figure 2: Mucins provide dynamic shield to the tumor cells during their metastatic journey.
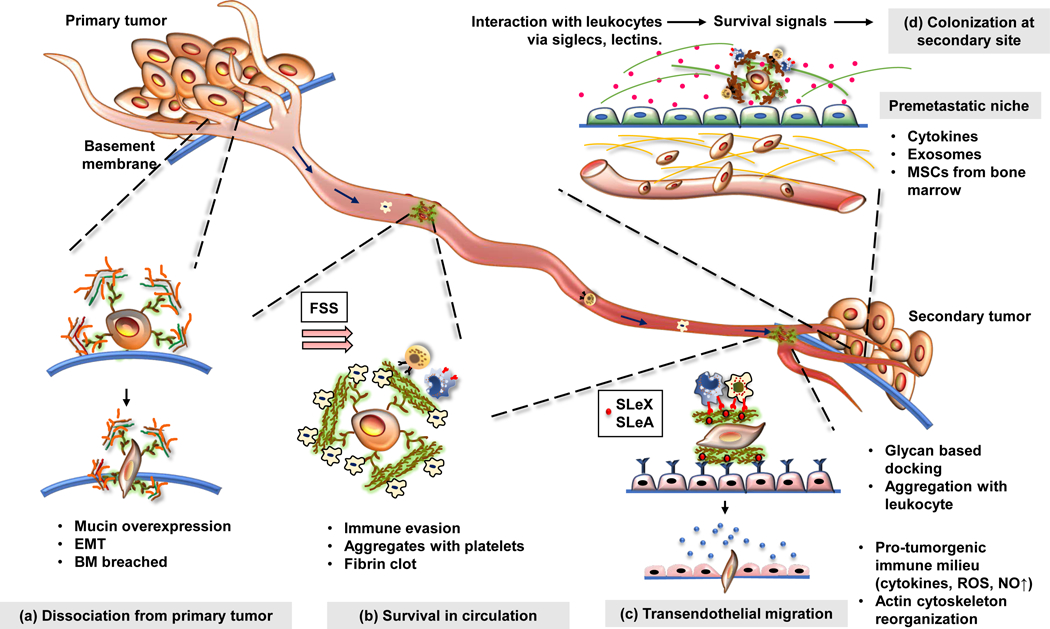
Mucin overexpression is associated with most malignancies. (a) At the primary tumor, secreted and transmembrane mucins facilitate epithelial to mesenchymal transition of the cancer cells through a plethora of signaling pathways and steric hindrance of cell-cell interactions. Furthermore, cancer cells utilize multiple cell-autonomous and paracrine mechanisms to breach the basement membrane. Mucins facilitate the synthesis of matrix-degrading proteases, maintain a pro-tumorigenic immune milieu, and establish interaction between cancer cells and the basement membrane via their extracellular domains which resemble matrix proteins. (b) Once the cancer cells exit the primary tumor, they face the fluid shear stress (FSS) and, circulatory immune cells in the bloodstream. Mucin glycans bear ligands that can bind to selectins on platelets and subsequently form a cancer cell-platelet-fibrin aggregate that protects cancer cells from the mechanical stress in circulation. Meanwhile, heavily glycosylated extracellular domains of mucins can mask tumor-associated antigens on the cancer cells, helping them evade immune surveillance. (c) While circulating through the bloodstream, the cancer cells dock on the endothelium lining (through selectins), undergo rolling, and subsequently extravasate to reach the secondary organ. Mucins help the cancer cells to attach to the endothelium via sialylated ligands for selectins (SLeX, SLeA) and help them extravasate using various adhesion molecules (integrins, ICAMs). (d) At the secondary site, mucins may facilitate the formation of a pre-metastatic niche and provide survival signals to the cancer cells. Together, these steps culminate in the establishment of a successful metastatic lesion.
Acknowledgements:
This authors/work was partly supported by funding from the National Institutes of Health (P01CA217798, U01 CA210240, U01 CA200466, R01 CA206444, R01 CA228524 and R01 CA195586). We thank Dr. Jessica Mercer for editorial contribution to the manuscript.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Diagnostics and Therapeutics, Inc. The other authors have no conflicts of interest to declare.
Conflicts of Interest: SKB is one of the co-founders of the Sanguine Diagnostic and Therapeutics, Inc.
References
- 1.Peterson LW, & Artis D (2014). Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nature reviews immunology, 14(3), 141–153. [DOI] [PubMed] [Google Scholar]
- 2.Linden S, Sutton P, Karlsson N, Korolik V, & McGuckin M (2008). Mucins in the mucosal barrier to infection. Mucosal immunology, 1(3), 183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johansson ME, Sjövall H, & Hansson GC (2013). The gastrointestinal mucus system in health and disease. Nature reviews Gastroenterology & hepatology, 10(6), 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lillehoj EP, Kato K, Lu W, & Kim KC (2013). Cellular and molecular biology of airway mucins In International review of cell and molecular biology (Vol. 303, pp. 139–202): Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kufe DW (2009). Mucins in cancer: function, prognosis and therapy. Nature Reviews Cancer, 9(12), 874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desseyn J-L, Buisine M-P, Porchet N, Aubert J-P, Degand P, & Laine A (1998). Evolutionary history of the 11p15 human mucin gene family. Journal of molecular evolution, 46(1), 102–106. [DOI] [PubMed] [Google Scholar]
- 7.Duraisamy S, Ramasamy S, Kharbanda S, & Kufe D (2006). Distinct evolution of the human carcinoma-associated transmembrane mucins, MUC1, MUC4 AND MUC16. Gene, 373, 28–34. [DOI] [PubMed] [Google Scholar]
- 8.van Putten JP, & Strijbis K (2017). Transmembrane mucins: signaling receptors at the intersection of inflammation and cancer. Journal of innate immunity, 9(3), 281–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lakshmanan I, Ponnusamy MP, Macha MA, Haridas D, Majhi PD, Kaur S, et al. (2015). Mucins in lung cancer: diagnostic, prognostic, and therapeutic implications. Journal of Thoracic Oncology, 10(1), 19–27. [DOI] [PubMed] [Google Scholar]
- 10.Hollingsworth MA, & Swanson BJ (2004). Mucins in cancer: protection and control of the cell surface. Nature Reviews Cancer, 4(1), 45–60. [DOI] [PubMed] [Google Scholar]
- 11.Kaur S, Kumar S, Momi N, Sasson AR, & Batra SK (2013). Mucins in pancreatic cancer and its microenvironment. Nature reviews Gastroenterology & hepatology, 10(10), 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pothuraju R, Krishn SR, Gautam SK, Pai P, Ganguly K, Chaudhary S, et al. (2020). Mechanistic and Functional Shades of Mucins and Associated Glycans in Colon Cancer. Cancers, 12(3), 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mukhopadhyay P, Chakraborty S, Ponnusamy MP, Lakshmanan I, Jain M, & Batra SK (2011). Mucins in the pathogenesis of breast cancer: implications in diagnosis, prognosis and therapy. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer, 1815(2), 224–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh AP, Senapati S, Ponnusamy MP, Jain M, Lele SM, Davis JS, et al. (2008). Clinical potential of mucins in diagnosis, prognosis, and therapy of ovarian cancer. The lancet oncology, 9(11), 1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bafna S, Kaur S, & Batra SK (2010). Membrane-bound mucins: the mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene, 29(20), 2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishn SR, Ganguly K, Kaur S, & Batra SK (2018). Ramifications of secreted mucin MUC5AC in malignant journey: a holistic view. Carcinogenesis, 39(5), 633–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhatia R, Gautam SK, Cannon A, Thompson C, Hall BR, Aithal A, et al. (2019). Cancer-associated mucins: role in immune modulation and metastasis. Cancer and Metastasis Reviews, 38(1–2), 223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cavey M, & Lecuit T (2009). Molecular bases of cell–cell junctions stability and dynamics. Cold Spring Harbor perspectives in biology, 1(5), a002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tepass U, Tanentzapf G, Ward R, & Fehon R (2001). Epithelial cell polarity and cell junctions in Drosophila. Annual review of genetics, 35(1), 747–784. [DOI] [PubMed] [Google Scholar]
- 20.Wagner C, Wheeler K, & Ribbeck K (2018). Mucins and their role in shaping the functions of mucus barriers. Annual review of cell and developmental biology. [DOI] [PubMed]
- 21.Silverman HS, Parry S, Sutton-Smith M, Burdick MD, McDermott K, Reid CJ, et al. (2001). In vivo glycosylation of mucin tandem repeats. Glycobiology, 11(6), 459–471. [DOI] [PubMed] [Google Scholar]
- 22.Sheehan JK, Brazeau C, Kutay S, Pigeon H, Kirkham S, Howard M, et al. (2000). Physical characterization of the MUC5AC mucin: a highly oligomeric glycoprotein whether isolated from cell culture or in vivo from respiratory mucous secretions. Biochemical Journal, 347(1), 37–44. [PMC free article] [PubMed] [Google Scholar]
- 23.Sheehan JK, Howard M, Richardson PS, Longwill T, & Thornton DJ (1999). Physical characterization of a low-charge glycoform of the MUC5B mucin comprising the gel-phase of an asthmatic respiratory mucous plug. Biochemical Journal, 338(2), 507–513. [PMC free article] [PubMed] [Google Scholar]
- 24.McAuley JL, Linden SK, Png CW, King RM, Pennington HL, Gendler SJ, et al. (2007). MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. The Journal of clinical investigation, 117(8), 2313–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGuckin MA, Every AL, Skene CD, Linden SK, Chionh YT, Swierczak A, et al. (2007). Muc1 mucin limits both Helicobacter pylori colonization of the murine gastric mucosa and associated gastritis. Gastroenterology, 133(4), 1210–1218. [DOI] [PubMed] [Google Scholar]
- 26.Blalock TD, Spurr-Michaud SJ, Tisdale AS, Heimer SR, Gilmore MS, Ramesh V, et al. (2007). Functions of MUC16 in corneal epithelial cells. Investigative ophthalmology & visual science, 48(10), 4509–4518. [DOI] [PubMed] [Google Scholar]
- 27.Ahmad R, Raina D, Joshi MD, Kawano T, Ren J, Kharbanda S, et al. (2009). MUC1-C oncoprotein functions as a direct activator of the nuclear factor-κB p65 transcription factor. Cancer research, 69(17), 7013–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindén SK, Florin TH, & McGuckin MA (2008). Mucin dynamics in intestinal bacterial infection. PloS one, 3(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmad R, Raina D, Trivedi V, Ren J, Rajabi H, Kharbanda S, et al. (2007). MUC1 oncoprotein activates the IκB kinase β complex and constitutive NF-κB signalling. Nature cell biology, 9(12), 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smirnova MG, Guo L, Birchall JP, & Pearson JP (2003). LPS up-regulates mucin and cytokine mRNA expression and stimulates mucin and cytokine secretion in goblet cells. Cellular immunology, 221(1), 42–49. [DOI] [PubMed] [Google Scholar]
- 31.Fischer BM, Cuellar JG, Diehl ML, deFreytas AM, Zhang J, Carraway KL, et al. (2003). Neutrophil elastase increases MUC4 expression in normal human bronchial epithelial cells. American Journal of Physiology-Lung Cellular and Molecular Physiology, 284(4), L671–L679. [DOI] [PubMed] [Google Scholar]
- 32.Delmotte P, Degroote S, Lafitte J-J, Lamblin G, Perini J-M, & Roussel P (2002). Tumor necrosis factor α increases the expression of glycosyltransferases and sulfotransferases responsible for the biosynthesis of sialylated and/or sulfated Lewis x epitopes in the human bronchial mucosa. Journal of Biological Chemistry, 277(1), 424–431. [DOI] [PubMed] [Google Scholar]
- 33.Lindén SK, Wickström C, Lindell G, Gilshenan K, & Carlstedt I (2008). Four modes of adhesion are used during Helicobacter pylori binding to human mucins in the oral and gastric niches. Helicobacter, 13(2), 81–93. [DOI] [PubMed] [Google Scholar]
- 34.Schulz BL, Sloane AJ, Robinson LJ, Prasad SS, Lindner RA, Robinson M, et al. (2007). Glycosylation of sputum mucins is altered in cystic fibrosis patients. Glycobiology, 17(7), 698–712. [DOI] [PubMed] [Google Scholar]
- 35.Xavier R, & Podolsky D (2007). Unravelling the pathogenesis of inflammatory bowel disease. Nature, 448(7152), 427–434. [DOI] [PubMed] [Google Scholar]
- 36.Feagins LA, Souza RF, & Spechler SJ (2009). Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nature reviews Gastroenterology & hepatology, 6(5), 297. [DOI] [PubMed] [Google Scholar]
- 37.Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, et al. (2008). Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS medicine, 5(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwerbrock NM, Makkink MK, van der Sluis M, Büller HA, Einerhand AW, Sartor RB, et al. (2004). Interleukin 10-deficient mice exhibit defective colonic Muc2 synthesis before and after induction of colitis by commensal bacteria. Inflammatory bowel diseases, 10(6), 811–823. [DOI] [PubMed] [Google Scholar]
- 39.Karin M, & Greten FR (2005). NF-κB: linking inflammation and immunity to cancer development and progression. Nature reviews immunology, 5(10), 749–759. [DOI] [PubMed] [Google Scholar]
- 40.Vinall LE, King M, Novelli M, Green CA, Daniels G, Hilkens J, et al. (2002). Altered expression and allelic association of the hypervariable membrane mucin MUC1 in Helicobacter pylori gastritis. Gastroenterology, 123(1), 41–49. [DOI] [PubMed] [Google Scholar]
- 41.Kondo S, Yoshizaki T, Wakisaka N, Horikawa T, Murono S, Jang KL, et al. (2007). MUC1 induced by Epstein-Barr virus latent membrane protein 1 causes dissociation of the cell-matrix interaction and cellular invasiveness via STAT signaling. Journal of virology, 81(4), 1554–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vermeer PD, Einwalter LA, Moninger TO, Rokhlina T, Kern JA, Zabner J, et al. (2003). Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature, 422(6929), 322–326. [DOI] [PubMed] [Google Scholar]
- 43.Shin K, Fogg VC, & Margolis B (2006). Tight junctions and cell polarity. Annu. Rev. Cell Dev. Biol, 22, 207–235. [DOI] [PubMed] [Google Scholar]
- 44.Polyak K, & Weinberg RA (2009). Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nature Reviews Cancer, 9(4), 265–273. [DOI] [PubMed] [Google Scholar]
- 45.Huang L, Chen D, Liu D, Yin L, Kharbanda S, & Kufe D (2005). MUC1 oncoprotein blocks glycogen synthase kinase 3β–mediated phosphorylation and degradation of β-catenin. Cancer research, 65(22), 10413–10422. [DOI] [PubMed] [Google Scholar]
- 46.Huang L, Ren J, Chen D, Li Y, Kharbanda S, & Kufe D (2003). MUC1 cytoplasmic domain coactivates Wnt target gene transcription and confers transformation. Cancer biology & therapy, 2(6), 702–706. [PubMed] [Google Scholar]
- 47.Aranda V, Haire T, Nolan ME, Calarco JP, Rosenberg AZ, Fawcett JP, et al. (2006). Par6–aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nature cell biology, 8(11), 1235–1245. [DOI] [PubMed] [Google Scholar]
- 48.Ren J, Bharti A, Raina D, Chen W, Ahmad R, & Kufe D (2006). MUC1 oncoprotein is targeted to mitochondria by heregulin-induced activation of c-Src and the molecular chaperone HSP90. Oncogene, 25(1), 20–31. [DOI] [PubMed] [Google Scholar]
- 49.Chaturvedi P, Singh AP, Chakraborty S, Chauhan SC, Bafna S, Meza JL, et al. (2008). MUC4 mucin interacts with and stabilizes the HER2 oncoprotein in human pancreatic cancer cells. Cancer research, 68(7), 2065–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Funes M, Miller JK, Lai C, Carraway KL, & Sweeney C (2006). The mucin Muc4 potentiates neuregulin signaling by increasing the cell-surface populations of ErbB2 and ErbB3. Journal of Biological Chemistry, 281(28), 19310–19319. [DOI] [PubMed] [Google Scholar]
- 51.Pokutta S, & Weis WI (2007). Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu. Rev. Cell Dev. Biol, 23, 237–261. [DOI] [PubMed] [Google Scholar]
- 52.Inaguma S, Kasai K, & Ikeda H (2011). GLI1 facilitates the migration and invasion of pancreatic cancer cells through MUC5AC-mediated attenuation of E-cadherin. Oncogene, 30(6), 714–723. [DOI] [PubMed] [Google Scholar]
- 53.Labelle M, & Hynes RO (2012). The initial hours of metastasis: the importance of cooperative host–tumor cell interactions during hematogenous dissemination. Cancer discovery, 2(12), 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar S, Cruz E, Joshi S, Patel A, Jahan R, Batra SK, et al. (2017). Genetic variants of mucins: unexplored conundrum. Carcinogenesis, 38(7), 671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.P Ponnusamy M, Seshacharyulu P, Lakshmanan I, P Vaz A, Chugh S, & K Batra S (2013). Emerging role of mucins in epithelial to mesenchymal transition. Current cancer drug targets, 13(9), 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chugh S, Gnanapragassam VS, Jain M, Rachagani S, Ponnusamy MP, & Batra SK (2015). Pathobiological implications of mucin glycans in cancer: Sweet poison and novel targets. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer, 1856(2), 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Byrd JC, & Bresalier RS (2004). Mucins and mucin binding proteins in colorectal cancer. Cancer and Metastasis Reviews, 23(1–2), 77–99. [DOI] [PubMed] [Google Scholar]
- 58.Roy LD, Sahraei M, Subramani DB, Besmer D, Nath S, Tinder TL, et al. (2011). MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene, 30(12), 1449–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horn G, Gaziel A, Wreschner DH, Smorodinsky NI, & Ehrlich M (2009). ERK and PI3K regulate different aspects of the epithelial to mesenchymal transition of mammary tumor cells induced by truncated MUC1. Experimental cell research, 315(8), 1490–1504. [DOI] [PubMed] [Google Scholar]
- 60.Rajabi H, Ahmad R, Jin C, Joshi MD, Guha M, Alam M, et al. (2012). MUC1-C oncoprotein confers androgen-independent growth of human prostate cancer cells. Prostate, 72(15), 1659–1668, 10.1002/pros.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaturvedi P, Singh AP, Chakraborty S, Chauhan SC, Bafna S, Meza JL, et al. (2008). MUC4 mucin interacts with and stabilizes the HER2 oncoprotein in human pancreatic cancer cells. Cancer Res, 68(7), 2065–2070, 10.1158/0008-5472.CAN-07-6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ponnusamy MP, Singh AP, Jain M, Chakraborty S, Moniaux N, & Batra SK (2008). MUC4 activates HER2 signalling and enhances the motility of human ovarian cancer cells. Br J Cancer, 99(3), 520–526, 10.1038/sj.bjc.6604517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rachagani S, Macha MA, Ponnusamy MP, Haridas D, Kaur S, Jain M, et al. (2012). MUC4 potentiates invasion and metastasis of pancreatic cancer cells through stabilization of fibroblast growth factor receptor 1. Carcinogenesis, 33(10), 1953–1964, 10.1093/carcin/bgs225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang J, Zhu Y, Xie K, Zhang X, Zhi X, Wang W, et al. (2016). The role of the AMOP domain in MUC4/Y-promoted tumour angiogenesis and metastasis in pancreatic cancer. Journal of Experimental & Clinical Cancer Research, 35(1), 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mukhopadhyay P, Lakshmanan I, Ponnusamy MP, Chakraborty S, Jain M, Pai P, et al. (2013). MUC4 overexpression augments cell migration and metastasis through EGFR family proteins in triple negative breast cancer cells. PloS one, 8(2), e54455, 10.1371/journal.pone.0054455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Majhi PD, Lakshmanan I, Ponnusamy MP, Jain M, Das S, Kaur S, et al. (2013). Pathobiological implications of MUC4 in non-small-cell lung cancer. J Thorac Oncol, 8(4), 398–407, 10.1097/JTO.0b013e3182829e06. [DOI] [PubMed] [Google Scholar]
- 67.Muniyan S, Haridas D, Chugh S, Rachagani S, Lakshmanan I, Gupta S, et al. (2016). MUC16 contributes to the metastasis of pancreatic ductal adenocarcinoma through focal adhesion mediated signaling mechanism. Genes & cancer, 7(3–4), 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haridas D, Ponnusamy MP, Chugh S, Lakshmanan I, Seshacharyulu P, & Batra SK (2014). MUC16: molecular analysis and its functional implications in benign and malignant conditions. FASEB J, 28(10), 4183–4199, 10.1096/fj.14-257352. [DOI] [PubMed] [Google Scholar]
- 69.Comamala M, Pinard M, Theriault C, Matte I, Albert A, Boivin M, et al. (2011). Downregulation of cell surface CA125/MUC16 induces epithelial-to-mesenchymal transition and restores EGFR signalling in NIH:OVCAR3 ovarian carcinoma cells. Br J Cancer, 104(6), 989–999, 10.1038/bjc.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thériault C, Pinard M, Comamala M, Migneault M, Beaudin J, Matte I, et al. (2011). MUC16 (CA125) regulates epithelial ovarian cancer cell growth, tumorigenesis and metastasis. Gynecologic oncology, 121(3), 434–443. [DOI] [PubMed] [Google Scholar]
- 71.Lakshmanan I, Rachagani S, Hauke R, Krishn SR, Paknikar S, Seshacharyulu P, et al. (2016). MUC5AC interactions with integrin beta4 enhances the migration of lung cancer cells through FAK signaling. Oncogene, 35(31), 4112–4121, 10.1038/onc.2015.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pothuraju R, Rachagani S, Krishn SR, Chaudhary S, Nimmakayala RK, Siddiqui JA, et al. (2020). Molecular implications of MUC5AC-CD44 axis in colorectal cancer progression and chemoresistance. Mol Cancer, 19(1), 37, 10.1186/s12943-020-01156-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamazoe S, Tanaka H, Sawada T, Amano R, Yamada N, Ohira M, et al. (2010). RNA interference suppression of mucin 5AC (MUC5AC) reduces the adhesive and invasive capacity of human pancreatic cancer cells. J Exp Clin Cancer Res, 29, 53, 10.1186/1756-9966-29-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Truant S, Bruyneel E, Gouyer V, De Wever O, Pruvot FR, Mareel M, et al. (2003). Requirement of both mucins and proteoglycans in cell-cell dissociation and invasiveness of colon carcinoma HT-29 cells. Int J Cancer, 104(6), 683–694, 10.1002/ijc.11011. [DOI] [PubMed] [Google Scholar]
- 75.Chang J, & Chaudhuri O (2019). Beyond proteases: Basement membrane mechanics and cancer invasion. J Cell Biol, 218(8), 2456–2469, 10.1083/jcb.201903066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Senapati S, Gnanapragassam VS, Moniaux N, Momi N, & Batra SK (2012). Role of MUC4-NIDO domain in the MUC4-mediated metastasis of pancreatic cancer cells. Oncogene, 31(28), 3346–3356, 10.1038/onc.2011.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jahan R, Macha MA, Rachagani S, Das S, Smith LM, Kaur S, et al. (2018). Axed MUC4 (MUC4/X) aggravates pancreatic malignant phenotype by activating integrin-beta1/FAK/ERK pathway. Biochim Biophys Acta Mol Basis Dis, 1864(8), 2538–2549, 10.1016/j.bbadis.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leir SH, & Harris A (2011). MUC6 mucin expression inhibits tumor cell invasion. Exp Cell Res, 317(17), 2408–2419, 10.1016/j.yexcr.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 79.Erpenbeck L, & Schon MP (2010). Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood, 115(17), 3427–3436, 10.1182/blood-2009-10-247296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hook P, Litvinov RI, Kim OV, Xu S, Xu Z, Bennett JS, et al. (2017). Strong Binding of Platelet Integrin alphaIIbbeta3 to Fibrin Clots: Potential Target to Destabilize Thrombi. Sci Rep, 7(1), 13001, 10.1038/s41598-017-12615-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rowson-Hodel AR, Wald JH, Hatakeyama J, O’Neal WK, Stonebraker JR, VanderVorst K, et al. (2018). Membrane Mucin Muc4 promotes blood cell association with tumor cells and mediates efficient metastasis in a mouse model of breast cancer. Oncogene, 37(2), 197–207, 10.1038/onc.2017.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bambach SK, & Lammermann T (2017). Platelets, On Your Marks, Get Set, Migrate! Cell, 171(6), 1256–1258, 10.1016/j.cell.2017.11.026. [DOI] [PubMed] [Google Scholar]
- 83.Joyce JA, & Pollard JW (2009). Microenvironmental regulation of metastasis. Nat Rev Cancer, 9(4), 239–252, 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McEver RP (2015). Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res, 107(3), 331–339, 10.1093/cvr/cvv154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kam JL, Regimbald LH, Hilgers JH, Hoffman P, Krantz MJ, Longenecker BM, et al. (1998). MUC1 synthetic peptide inhibition of intercellular adhesion molecule-1 and MUC1 binding requires six tandem repeats. Cancer Res, 58(23), 5577–5581. [PubMed] [Google Scholar]
- 86.von Mensdorff-Pouilly S, Snijdewint FG, Verstraeten AA, Verheijen RH, & Kenemans P (2000). Human MUC1 mucin: a multifaceted glycoprotein. Int J Biol Markers, 15(4), 343–356. [DOI] [PubMed] [Google Scholar]
- 87.Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. (2016). Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med, 8(361), 361ra138, 10.1126/scitranslmed.aag1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hayashi T, Takahashi T, Motoya S, Ishida T, Itoh F, Adachi M, et al. (2001). MUC1 mucin core protein binds to the domain 1 of ICAM-1. Digestion, 63 Suppl 1, 87–92, 10.1159/000051917. [DOI] [PubMed] [Google Scholar]
- 89.Rajabi H, Hata T, Li W, Long MD, Hu Q, Liu S, et al. (2019). MUC1-C represses the RASSF1A tumor suppressor in human carcinoma cells. Oncogene, 38(47), 7266–7277, 10.1038/s41388-019-0940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kinlough CL, Poland PA, Bruns JB, Harkleroad KL, & Hughey RP (2004). MUC1 membrane trafficking is modulated by multiple interactions. Journal of Biological Chemistry, 279(51), 53071–53077. [DOI] [PubMed] [Google Scholar]
- 91.Kinlough CL, McMahan RJ, Poland PA, Bruns JB, Harkleroad KL, Stremple RJ, et al. (2006). Recycling of MUC1 is dependent on its palmitoylation. Journal of Biological Chemistry, 281(17), 12112–12122. [DOI] [PubMed] [Google Scholar]
- 92.Singh PK, & Hollingsworth MA (2006). Cell surface-associated mucins in signal transduction. Trends in cell biology, 16(9), 467–476. [DOI] [PubMed] [Google Scholar]
- 93.Schroeder JA, Thompson MC, Gardner MM, & Gendler SJ (2001). Transgenic MUC1 interacts with epidermal growth factor receptor and correlates with mitogen-activated protein kinase activation in the mouse mammary gland. Journal of Biological Chemistry, 276(16), 13057–13064. [DOI] [PubMed] [Google Scholar]
- 94.Hattrup CL, & Gendler SJ (2006). MUC1 alters oncogenic events and transcription in human breast cancer cells. Breast Cancer Research, 8(4), R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schroeder JA, Adriance MC, Thompson MC, Camenisch TD, & Gendler SJ (2003). MUC1 alters β-catenin-dependent tumor formation and promotes cellular invasion. Oncogene, 22(9), 1324–1332. [DOI] [PubMed] [Google Scholar]
- 96.Bitler BG, Menzl I, Huerta CL, Sands B, Knowlton W, Chang A, et al. (2009). Intracellular MUC1 peptides inhibit cancer progression. Clinical Cancer Research, 15(1), 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li Y, Kuwahara H, Ren J, Wen G, & Kufe D (2001). The c-Src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3β and β-catenin. Journal of Biological Chemistry, 276(9), 6061–6064. [DOI] [PubMed] [Google Scholar]
- 98.Ho SB, Dvorak LA, Moor RE, Jacobson AC, Frey MR, Corredor J, et al. (2006). Cysteine-rich domains of muc3 intestinal mucin promote cell migration, inhibit apoptosis, and accelerate wound healing. Gastroenterology, 131(5), 1501–1517. [DOI] [PubMed] [Google Scholar]
- 99.Gum JR, Ho JJ, Pratt WS, Hicks JW, Hill AS, Vinall LE, et al. (1997). MUC3 human intestinal mucin analysis of gene structure, the carboxyl terminus, and a novel upstream repetitive region. Journal of Biological Chemistry, 272(42), 26678–26686. [DOI] [PubMed] [Google Scholar]
- 100.Kargı A, Dinç ZA, Başok O, & Üçvet A (2006). MUC4 expression and its relation to ErbB2 expression, apoptosis, proliferation, differentiation, and tumor stage in non-small cell lung cancer (NSCLC). Pathology-Research and Practice, 202(8), 577–583. [DOI] [PubMed] [Google Scholar]
- 101.Songyang Z, Shoelson S, McGlade J, Olivier P, Pawson T, Bustelo X, et al. (1994). Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Molecular and cellular biology, 14(4), 2777–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Williams SJ, McGuckin MA, Gotley DC, Eyre HJ, Sutherland GR, & Antalis TM (1999). Two novel mucin genes down-regulated in colorectal cancer identified by differential display. Cancer research, 59(16), 4083–4089. [PubMed] [Google Scholar]
- 103.Burgel P, & Nadel J (2008). Epidermal growth factor receptor-mediated innate immune responses and their roles in airway diseases. European Respiratory Journal, 32(4), 1068–1081. [DOI] [PubMed] [Google Scholar]
- 104.Chauhan SC, Ebeling MC, Maher DM, Koch MD, Watanabe A, Aburatani H, et al. (2012). MUC13 mucin augments pancreatic tumorigenesis. Molecular cancer therapeutics, 11(1), 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Das S, Rachagani S, Torres-Gonzalez MP, Lakshmanan I, Majhi PD, Smith LM, et al. (2015). Carboxyl-terminal domain of MUC16 imparts tumorigenic and metastatic functions through nuclear translocation of JAK2 to pancreatic cancer cells. Oncotarget, 6(8), 5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang B, Wu A, Hu Y, Tao C, Wang JM, Lu Y, et al. (2019). Mucin 17 inhibits the progression of human gastric cancer by limiting inflammatory responses through a MYH9-p53-RhoA regulatory feedback loop. Journal of Experimental & Clinical Cancer Research, 38(1), 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dhanisha SS, Guruvayoorappan C, Drishya S, & Abeesh P (2018). Mucins: structural diversity, biosynthesis, its role in pathogenesis and as possible therapeutic targets. Critical reviews in oncology/hematology, 122, 98–122. [DOI] [PubMed] [Google Scholar]