

Abstract
It is increasingly accepted that early cognitive impairment in Alzheimer’s disease results in considerable part from synaptic dysfunction caused by the accumulation of a range of oligomeric assemblies of amyloid β-protein (Aβ). Most studies have used synthetic Aβ peptides to explore the mechanisms of memory deficits in rodent models, but recent work suggests that Aβ assemblies isolated from human (AD) brain tissue are far more potent and disease-relevant. Although reductionist experiments show Aβ oligomers to impair synaptic plasticity and neuronal viability, the responsible mechanisms are only partly understood. Glutamatergic receptors, GABAergic receptors, nicotinic receptors, insulin receptors, the cellular prion protein, inflammatory mediators and diverse signaling pathways have all been suggested. Studies using AD brain-derived soluble Aβ oligomers suggest that only certain bioactive forms (principally small, diffusible oligomers) can disrupt synaptic plasticity, including by binding to plasma membranes and changing excitatory-inhibitory balance, perturbing mGluR, PrP and other neuronal surface proteins, downregulating glutamate transporters, causing glutamate spillover and activating extrasynaptic GluN2B-containing NMDA receptors. We synthesize these emerging data into a mechanistic hypothesis for synaptic failure in Alzheimer’s disease that can be modified as new knowledge is added and specific therapeutics are developed.
Keywords: Alzheimer’s disease, amyloid β-protein, synaptic plasticity, long-term potentiation, long-term depression, soluble Aβ oligomers
Introduction
Memory and cognitive deficits in the prodromal and mild cognitive impairment (MCI) stages of Alzheimer’s disease may primarily result from synaptic dysfunction (Arendt, 2009; Selkoe, 2002). However, the precise molecular and cellular mechanisms for how synaptic failure occurs in AD remain unclear. Growing but not definitive evidence suggests that AD is precipitated in considerable part by the progressive cerebral accumulation of a range of oligomeric assemblies of amyloid β-protein (Aβ) (Selkoe & Hardy, 2016). Clinical PET imaging indicates that synaptic density is significantly reduced in the hippocampus in Aβ-positive patients on the AD spectrum, especially in its early symptomatic stages (Bastin et al., 2019). The severity of cognitive deficits in AD correlates more strongly with the levels of soluble forms of Aβ than with insoluble amyloid plaque load (Lue et al., 1999; McLean et al., 1999). Similarly, impairment of hippocampal synaptic plasticity can be detected in vivo before the formation of insoluble Aβ plaques in APP transgenic mouse models of AD (Oddo et al., 2006). Experimentally, soluble Aβ oligomers (oAβ) have consistently been found to block long-term hippocampal potentiation (LTP), an electrophysiological correlate of learning and memory, in vivo and in vitro (Gulisano et al., 2018; Li et al., 2018; Li, Liu, & Selkoe, 2019; Shankar et al., 2008; Walsh et al., 2002). In line with these findings, Aβ immunotherapy has been confirmed to protect against the cognitive deficits observed in amyloid precursor protein (APP) transgenic mice (Carrera et al., 2015; Hartman et al., 2005; Sevigny et al., 2016) and prevent the LTP impairment induced by Aβ oligomers (Klyubin et al., 2008; Klyubin et al., 2005; Li et al., 2018). Although there are extensive studies on the Aβ-mediated disruption of synaptic plasticity, the detailed mechanisms appear highly complex. Understanding the neurobiological basis for how Aβ impairs synaptic plasticity could help develop more efficacious therapeutics for AD.
Aβ peptides are generated via proteolytic cleavages of APP by the β- and γ-secretases. Aβ monomers can apparently self-associate to form oligomers, protofibrils and fibrils. Accumulation and further aggregation of protofibrils and fibrils lead to the formation of insoluble amyloid plaques, a universal feature of AD neuropathology. The aggregation state of Aβ peptides, based in part on the specific amino acid sequences of Aβ, is a key factor in determining their cytotoxicity (Li et al., 2018; Scopes et al., 2012). Furthermore, the aggregation rate is strongly correlated with the ratio of Aβ42:Aβ40 peptides in cellular and animal models (Pauwels et al., 2012; Terrill-Usery, Colvin, Davenport, & Nichols, 2016). However, the aggregation state of a pure synthetic Aβ peptide of a single length may not closely mimic the natural aggregation states of the heterogeneous Aβ peptides in vivo. Furthermore, aggregated synthetic Aβ species may differ significantly from the soluble Aβ oligomers isolated from the cerebral cortex of humans dying with clinically and neuropathologically confirmed AD, because AD brain-derived Aβ contains multiple forms of various Aβ species (Brinkmalm et al., 2019). Here, we review recent findings regarding Aβ oligomer-mediated impairment of synaptic plasticity and discuss potential mechanisms as to how Aβ alters synapse form and function.
The difference between human brain-derived Aβ and other sources of Aβ
Most studies of Aβ-mediated neurotoxicity have used pure, synthetic Aβ peptides such as Aβ1–40, Aβ1–42 and Aβ25–35. Two broad and not mutually exclusive types of synthetic Aβ aggregates are referred to as ADDLs (Aβ-derived diffusible ligands) (Krafft & Klein, 2010) and protofibrils (Hartley et al., 1999; Shankar & Walsh, 2009). Inserting cysteine in place of serine at residue 26 of synthetic Aβ1–40 can produce disulfide-crosslinked dimers ([AβS26C]2) which are prone to aggregate further and mimic features of the natural human oligomers; they are more potent in inhibiting hippocampal LTP than are wild-type synthetic Aβ aggregates (Hu, Smith, Walsh, & Rowan, 2008; Li, Feig, & Hartley, 2007; Li et al., 2011; Shankar et al., 2008). It well accepted that Aβ monomers do not interrupt normal synaptic function, whereas small (low-n Aβ oligomers) and large Aβ aggregates (protofibrils) impair synaptic plasticity (Klyubin, Cullen, Hu, & Rowan, 2012). For example, the Arctic mutant Aβ peptide which has a high propensity to form protofibrils, showed a high potency to inhibit LTP (Klyubin et al., 2004; Nilsberth et al., 2001). A second source of Aβ species is from the culture medium of certain cells that secrete monomers and some forms of soluble Aβ oligomers. For example, a Chinese hamster ovary (CHO) cell line (called 7PA2) that stably expresses human mutant APP has been shown to release biochemically-characterized monomeric and low-n oligomeric Aβ species (Kittelberger, Piazza, Tesco, & Reijmers, 2012; Li et al., 2011; Podlisny et al., 1995; Walsh et al., 2002) but also certain N-terminally extended Aβ-containing monomeric fragments (i.e., beyond the conventional Aβ Asp1 start site) with LTP-blocking properties; these extended monomeric APP fragments arise from protease cleavages creating an N-terminus other than from classical β-secretase processing followed by γ-secretase cleavage (Welzel et al., 2014). A third general source of Aβ peptides and oligomers is from highly disease-relevant human (AD) brain tissue itself. Such species can be obtained from homogenates of postmortem AD cerebral cortex made in Tris- or phosphate-buffered saline (TBS or PBS) or artificial cerebrospinal fluid (ACSF). These human brain extracts contain highly heterogeneous Aβ monomers and detergent-stable dimers, trimers and higher oligomers (Hong et al., 2018; Li et al., 2018; Sebollela et al., 2017; Shankar et al., 2008; Yang, Li, Xu, Walsh, & Selkoe, 2017). Studies examining such human brain Aβ-rich extracts indicate that their synaptotoxic and neuritotoxic potency is perhaps ~100-fold greater than that of synthetic Aβ aggregates (Jin et al., 2011; Zhang, Mably, Walsh, & Rowan, 2017). In accord, AD brain-derived Aβ can induce seeding activity upon intracerebral injection into APP transgenic mice that is at least 100-fold more potent than synthetic Aβ or Aβ species obtained from human CSF (Meyer-Luehmann et al., 2006; Stohr et al., 2012). As regards insoluble amyloid fibrils isolated from human brain, cryo-electron microscopy (cryo-EM) has demonstrated that brain-derived amyloid fibrils are right-hand twisted β-sheets and their peptide fold differs sharply from in vitro formed synthetic Aβ fibrils that are left-hand twisted (Kollmer et al., 2019).
We recently demonstrated that not all buffer-soluble AD brain extracts can cause neurotoxic activity in rodent brain slices or human iPSC neurons, at least not in their initial state of isolation (Li et al., 2018; Yang et al., 2017). In this context, the total Aβ levels in pieces of AD brain tissue measured by ELISA do not correlate directly with induced LTP deficits. These results may help explain the long-noted inconsistency between brain amyloid plaque levels and the degree of cognitive decline in AD patients (e.g., Dubois et al., 2018; Esparza et al., 2013). Different synthetic or human AD brain extract Aβ preparations (based on use of various detergents and buffers or aggregation stimulation) may yield different species or fractions of oAβ (Hayden & Teplow, 2013; Nagel-Steger, Owen, & Strodel, 2016). These and other experimental observations suggest that only certain forms of soluble Aβ assemblies in the AD brain, perhaps a small minority (Hong et al., 2018), actually play an active role in Aβ-mediated bioactivity.
Bioactive human brain-derived Aβ species
Several different forms of Aβ, including monomers and dimers of Aβ1–40 and Aβ1–42, low-n oligomers, and other non-fibrillar forms of water-soluble Aβ have been found in CSF, blood and brain extract from AD patients (Brinkmalm et al., 2019; Lewczuk et al., 2017; Shankar et al., 2008; Yang et al., 2015). While pure synthetic Aβ monomers can form low-molecular-weight oligomers, ADDLs, ‘globulomers”, protofibrils and/or ‘annular assemblies’, and plaque-like amyloid fibrils in vitro (Walsh & Selkoe, 2007), it has been difficult to confirm that analogous assembles actually arise naturally in the human brain. The isolation and bioactivity analyses of a series of Aβ-rich extracts from neuropathologically typical AD brains has suggested that ~4 kDa Aβ monomers as well as highly insoluble amyloid plaque cores do not inhibit hippocampal LTP (Shankar et al., 2008), while some buffer-soluble fractions (~16–60 kDa) and small oligomers (7–16 kDa) can significantly impair LTP. Interestingly, large soluble assemblies isolated from cortical homogenates on a size exclusion chromatography (SEC) column, i.e., in the void volume, did not significantly alter LTP, but when these large Aβ assemblies were dissociated in vitro incubation into smaller, readily diffusible Aβ dimers and oligomers, they could now inhibit hippocampal LTP (Li et al., 2018; Shankar et al., 2008; Yang et al., 2017).
Studies that using synthetic Aβ have demonstrated that oligomeric Aβ1–28 peptides (Lambert et al., 2007; Sebollela et al., 2014) or peptides of the Aβ16–21 hydrophobic core (Sebollela et al., 2017; Velasco et al., 2012) may play a key role in AD-relevant neurotoxicity, and their antibodies (NU4 or NUsc1, respectively) or other antibodies, such as A11 (Knight et al., 2016) that preferentially target soluble oAβ can neutralize Aβ-mediated neurotoxicity in experimental systems. It was also reported that synthetic ADDLs (Gong et al., 2003), synthetic globulomers (Hillen et al., 2010) or the APP transgenic mouse brain-derived Aβ*56 oligomers (Reed et al., 2011) are all Aβ species that can induce synaptic dysfunction and cognitive impairment.
It is well accepted that the Aβ1–42 fragment plays an initiating role in neurotoxic activity because of its relative hydrophobicity (i.e., having an extra alanine and isoleucine) and its consequent aggregation propensity. Other peptide lengths of Aβ, such as Aβ37, Aβ38, Aβ39, Aβ40, Aβ43, and heterogeneous N-terminally truncated Aβs can also be found in AD brain and human cerebrospinal fluid (Kakuda et al., 2012; Soares et al., 2016; Wirths et al., 2017). Hippocampal slice LTP experiments have demonstrated that longer Aβ peptides (Aβ42, Aβ43) but not the shorter forms (Aβ37, Aβ38, Aβ39) can significantly inhibit LTP. Moreover, certain N-terminal anti-Aβ antibodies such as 3D6 (mouse) or bapineuzumab (humanized), but not C-terminal antibodies such as 2G3 (anti- Aβ40) and 21F12 (Aβ42), can fully prevent slice LTP impairment by soluble Aβ-rich extracts of AD cortex (Li et al., 2018). The C-terminal region, which is abundant in hydrophobic residues, may form a hydrophobic core with a central cavity that stabilizes the oligomer structure (Kawai et al., 2020). A recent study suggested that synthetic Aβ soluble oligomers containing α-sheet secondary structure are strongly correlated with neurotoxicity (Shea et al., 2019). These results suggest that the long-form (>40 residue) Aβ peptides preferentially aggregate into oligomers, and the N-terminus of Aβ may play a role in targeting cell membranes and execute bioactivity.
Soluble Aβ oligomers effect on short-term plasticity
Neurons communicate with each other by transmission via chemical synapses that can undergo a variety of short-lasting processes to regulate the dynamic course of synaptic transmission. Short-term plasticity (STP) is a use-dependent change in synaptic strength on the timescale of a millisecond to seconds and can be observed in almost every synapse of the central nervous system (Motanis, Seay, & Buonomano, 2018). Several forms of STP have been described, including post-tetanic potentiation (PTP), synaptic augmentation, paired-pulse facilitation (PPF), and synaptic depression, which are each distinguished by their decay kinetics. PPF is an increase in the postsynaptic response to the second presynaptic action potential over the response to the first one. It is well studied in the involvement of presynaptic neurotransmitter release.
Exogenous application of Aβ oligomers onto hippocampal slices or their injection into the lateral ventricle has been found not to alter PPF or PTP(Cerpa et al., 2010; Li et al., 2009; Schmid, Freir, & Herron, 2008; Shankar et al., 2008; Talantova et al., 2013). On the other hand, soluble Aβ from human (AD) brain extracts can significantly impair the burst stimulation form of STP (Wang et al., 2017) (Fig.1). Interestingly, the second pulse of bursts did not produce change in the response, as the PPF indicated. PPF may reflect the probability of neurotransmitter vesicle release, with reduced ratios implying an increased probability of synaptic vesicle release. However, the precise mechanism of burst stimulation as to its involvement in short-term plasticity remains unclear. Whether this alternation is due to the extracellular Aβ oligomers regulating presynaptic function by promoting APP-APP interactions through activation of APP homodimers in hippocampus, as has been suggested (Fogel et al., 2014; Wang et al., 2017), needs further investigation.
Figure 1. Human (AD) brain-derived Aβ oligomers impair short-term plasticity.
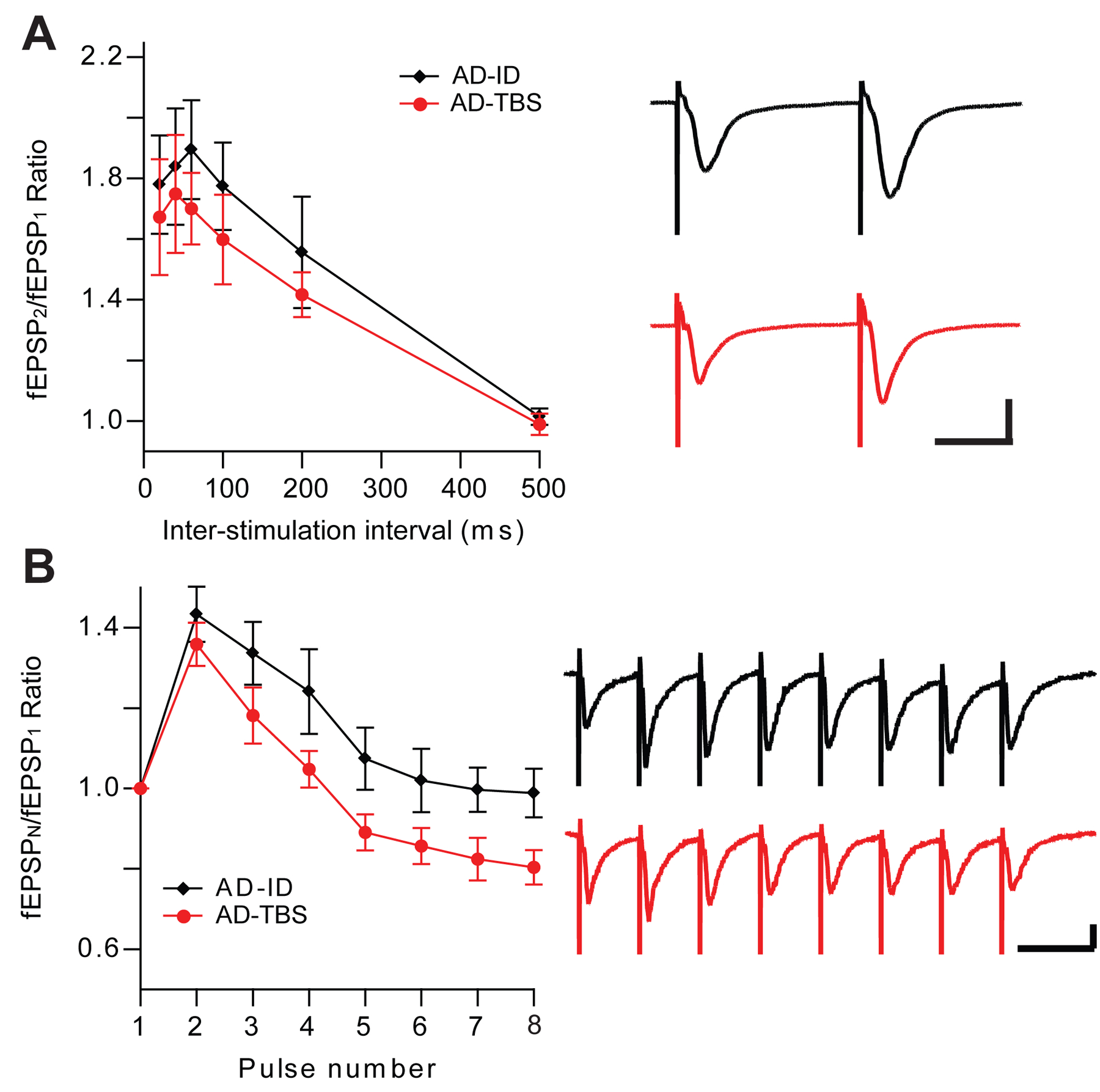
(A) AD brain oAβ effect on the paired-pulse facilitation (PPF), Horizontal bars: 20 ms; vertical bars: 0.5 mV; (B) AD brain oAβ effect on the burst stimulation form of short-term plasticity, Horizontal bars: 40 ms; vertical bars: 0.25 mV
Human Aβ inhibits hippocampal LTP
Although there is some disagreement in the literature as regards oAβ effects on short-term plasticity or long-term synaptic depression (LTD), there are highly consistent reports that exogenously applied soluble Aβ oligomers or endogenous elevated Aβ levels can each impair hippocampal LTP, a well-accepted electrophysiological correlate of learning and memory. Soluble oAβ can alter synaptic function, including plasticity, within minutes, much more quickly than inflammatory and other cellular responses require. More importantly, electrophysiological recording is a sensitive technique for studying synaptic transmission in real-time; therefore, it becomes a useful physiological readout for synaptic changes caused by Aβ oligomers and thus the study of therapeutic interventions.
1. AD brain-derived Aβ disrupts E/I balance
LTP in the hippocampus is commonly induced by electrical tetanic or high-frequency stimulation (HFS) of presynaptic axons, thereby causing activation of postsynaptic AMPA and NMDA receptors. Increasing activity of excitatory glutamatergic receptors mediates the LTP. Soluble Aβ oligomers, including those isolated from AD brain, have been shown to induce hyperexcitability in individual neurons and neural circuits (Del Vecchio, Gold, Novick, Wong, & Hyde, 2004; Lei et al., 2016; Li et al., 2011; Zott et al., 2019). These experimental results may help explain the clinical observation of a significantly higher incidence of epilepsy in AD patients (up to 10–22% higher) than in age-matched control subjects (<1%) (Amatniek et al., 2006; Vossel, Tartaglia, Nygaard, Zeman, & Miller, 2017).
Neuronal activity in the brain depends on the regulation of a complex equilibrium between excitatory and inhibitory neurotransmission. An imbalance of excitation/inhibition (E/I) in favor of excitation could result in epileptiform activity. Through application of AD brain-derived oAβ to brain slices, we found that neuronal excitability was increased around 3 times while inhibitory tone decreased 50%; thus, the change of the E/I balance was as much as 6-fold (Wang et al., 2017). Some studies have suggested that the loss of balance of excitatory vs. inhibitory neurotransmitter systems may underline early cognitive deficits and occasional epileptiform activity in AD (Goutagny et al., 2013; Petrache et al., 2019; Stoiljkovic, Kelley, Horvath, & Hajos, 2018; Vossel et al., 2016). The imbalance of glutamatergic and GABAergic neurotransmission may also be interrupted by oAβ (Quevenco et al., 2019). This and other evidence supports an imbalance between excitatory and inhibitory transmission that may contribute to cognitive deficits in AD patients (Selten, van Bokhoven, & Nadif Kasri, 2018; Vico Varela, Etter, & Williams, 2019).
The increased neuronal excitability by AD brain-derived oAβ further supports previous results from synthetic Aβ oligomers that increase EPSPs, mEPSC, membrane depolarizations, and action potentials (Gilbert et al., 2016; Hartley et al., 1999; Kurudenkandy et al., 2014; Minkeviciene et al., 2009). Mechanistically, the levels of the AMPA receptor subunit GluA1, which is a critical mediator of LTP expression in CA1 (Terashima, Suh, & Isaac, 2019), have been shown to be increased in AD hippocampus compared to healthy controls (Marcello et al., 2012). Further studies confirmed that GluA1 phosphorylation (Megill et al., 2015) and GluA3 levels (Cantanelli et al., 2014) are both increased in young AD model mice, but GluA1 and GluA3 levels are significantly decreased in old AD model mice in comparison to age-matched WT mice. Aβ might initially target GluA2-lacking AMPAR (also called calcium-permeable AMPAR, or CP-AMPAR) to induce synaptic alterations (Gilbert et al., 2016; Whitcomb et al., 2015). Once the Aβ oligomers activate the CP-AMPARs, other subunits of AMPAR will be involved in the excess synaptic activity. In this regard, it has been demonstrated that GluA3-containing AMPARs are required for Aβ oligomer-mediated LTP deficits (Reinders et al., 2016).
An impairment of GABAergic inhibition could also induce hyperexcitability and epileptiform activity, and it could alter the hippocampal theta and gamma rhythmicity (Goutagny et al., 2013). Such abnormal inhibitory circuit activity could also disrupt normal hippocampal LTP induction. GABAergic neurotransmission has long been considered relatively well-preserved in AD (Mitew, Kirkcaldie, Dickson, & Vickers, 2013; Rissman, De Blas, & Armstrong, 2007). An early study of the LTP impairment induced by Aβ showed that it was independent of GABAA receptor-mediated synaptic inhibition, since the Aβ-mediated LTP inhibition was not affected by application of the GABA inhibitor picrotoxin (Raymond, Ireland, & Abraham, 2003). We reported that conventional fEPSP recordings (from striatum radiatum) of CA1 did not suggest that decreased LTP induced by oAβ related to GABAergic inhibition, whereas population spike recordings (from pyramidal cell layer) demonstrated that the involvement of GABAA receptors (Lei et al., 2016). This study confirmed other findings that exogenous Aβ application can cause GABAergic interneuron loss (Villette et al., 2012) and dysfunction (Wu, Guo, Gearing, & Chen, 2014) in AD model mice. The Aβ-induced GABAergic synaptic inhibition may be due to Aβ-triggered GABA receptor internalization (Ulrich, 2015) and/or a suppression endocannabinoid-mediated peri-tetanic disinhibition (Orr et al., 2014). Therefore, an increase of glutamatergic excitatory activity and/or a decrease of GABAergic inhibitory activities could promote neuronal hyperexcitability and impair hippocampal LTP, which is in line with the known deficits in LTP after status epilepticus (Kryukov, Kim, Magazanik, & Zaitsev, 2016; Postnikova et al., 2017; Zaitsev et al., 2015)
2. AD brain-derived Aβ binds in part to cellular prion protein
Cellular prion protein (PrPC) is a glycoprotein highly expressed in the brain. The role of PrPC in Aβ oligomer-induced synaptic impairment is a matter of interest and some controversy. It has been reported that the impairment of LTP by Aβ oligomers isolated from the brains of AD patients was rescued by pretreatment with an anti-PrPC antibody (Barry et al., 2011; Freir et al., 2011; Klyubin et al., 2014) and that oAβ failed to inhibit LTP in PrP null mice (Freir et al., 2011). These results suggest a role of PrPC in synaptotoxicity mediated by soluble Aβ. In contrast, three independent studies did not observe the effects of PrPc in the LTP alterations and memory deficits of a different mouse model of AD (Balducci et al., 2010; Calella et al., 2010; Kessels, Nguyen, Nabavi, & Malinow, 2010). These conflicting results may come from differences in experimental conditions such as Aβ species applied and AD mouse models.
Despite the disagreement over whether PrPc mediates Aβ synaptotoxicity per se, the specific binding of Aβ42 oligomers to PrPC has been reported in AD brain tissue but the PrPC-interacting Aβ oligomers are absence in healthy control brain tissue (Dohler et al., 2014; Kostylev et al., 2015). The soluble oAβ binding to PrP were also confirmed in mouse studies (Balducci et al., 2010; Calella et al., 2010; Lauren, Gimbel, Nygaard, Gilbert, & Strittmatter, 2009). Several studies confirmed that the so-called N1 fragment, the equivalent of that released by α- cleavage of endogenous PrPC, bound synthetic Aβ oligomers to a similar degree as did full-length PrP and thus blocked oAβ-mediated synaptotoxicity (Fluharty et al., 2013; Nicoll et al., 2013; Scott-McKean et al., 2016) . The high-affinity binding of the N1 fragment of PrPc to Aβ oligomers apparently requires both residues 23–27 at the N-terminus as well as the segment ~95–110 at the C-terminus (Fluharty et al., 2013; Zhang, Qi, et al., 2017), and this could explain that certain PrPc antibodies and the N1 fragment can prevent oAβ-induced inhibition of LTP in hippocampal slices. It is also reported that PrPC eliminates glutamate-mediated neuronal excitotoxicity caused by Aβ by inhibiting NMDA receptors (Biasini, Turnbaugh, Unterberger, & Harris, 2012; Khosravani et al., 2008). Interestingly, Aβ oligomers, but not monomers, were found to increase the localization of PrPC to the cell surface in several cell lines and hippocampal neurons (Caetano et al., 2011). We speculate that soluble Aβ oligomers, which are highly hydrophobic, first bind to lipid membranes and interrupt membrane fluidity and thereby trigger altered trafficking of several membrane-spanning receptors (NMADR, AMPAR, insulin) and other surface proteins (PrPC).
3. AD brain-derived Aβ increases inflammatory microglial activities.
Although the loss of synapses is putatively an early and eventually clinically important feature of Alzheimer’s disease, activated microglia and astrocytes are abundantly present in the vicinity of plaques, suggesting that glial cells also play a dynamic role in the pathogenesis of the amyloid aspect of AD. Aβ oligomers are found to interact with various receptors of microglial cells and increase the levels of inflammatory cytokines like tumor necrosis factor-alpha (TNFα), interleukins (IL-1, IL-6), interferon-γ (IFNγ) and chemokines such as CCL2, CXCL8, CXCL10, CCL5 and CCL3 (Domingues, da Cruz, & Henriques, 2017; Lyons, Griffin, Costelloe, Clarke, & Lynch, 2007). These changes likely boost microglial activation and make microglia more susceptible to secondary stimuli. Specifically, it has been shown that Intracerebroventricular (ICV) injection of AD brain-derived oAβ significantly increases Ccl3, Ccl4, and Tnf mRNA levels in wild-type adult mice (Xu, Rajsombath, Weikop, & Selkoe, 2018). Several cytokines such as TNFα, IL‐1β, IFN‐γ or IL‐6 that are released from microglia and astroglia during neuroinflammation in AD have been reported to help accelerate AD progression in both AD patients and animal model of AD (Heneka et al., 2015; Heppner, Ransohoff, & Becher, 2015) and impair hippocampal LTP (Hoshino, Hasegawa, Kamiya, & Morimoto, 2017; Maggio & Vlachos, 2018; Prieto, Tong, Smith, & Cotman, 2019). The LTP deficit in transgenic mouse models of AD has also been associated with increased expression of IL-1β (Gallagher et al., 2012; Kelly et al., 2013). In line with these findings, inactivating microglia by minocycline prevents Aβ inhibited LTP (Lyons et al., 2007; Riazi et al., 2015). These results suggest that proinflammatory cytokines help mediate the molecular pathway involved in synaptic dysfunction during Aβ-driven synaptotoxicity in AD (Kinney et al., 2018; Minter, Taylor, & Crack, 2016).
Soluble Aβ oligomers facilitate hippocampal LTD
A principal neuropathological finding in AD patients is cortical atrophy marked by neurite degeneration, dendritic spine density decreases and frank neuronal loss (Pozueta, Lefort, & Shelanski, 2013). Morphological studies in rodents suggest that the induction of LTP is associated with increased spine volume and spine formation. In contrast, the induction of LTD results in decreased spine volume and spine elimination (Hasegawa, Sakuragi, Tominaga-Yoshino, & Ogura, 2015; Wiegert & Oertner, 2013). Thus, the phenomenon of LTD may more closely represent AD neuropathological events. Mechanistically, the induction of LTD requires activation of NMDAR and/or mGluR in the CA1 region of the hippocampus, depending on the specific stimulation protocol and recording conditions (Collingridge, Peineau, Howland, & Wang, 2010; Connor & Wang, 2016). An increase in intracellular calcium can trigger signaling cascades involved in LTD induction, including calcineurin (protein phosphatase 2B, PP2B), p38 MAPK and glycogen synthase kinase-3 (GSK3) (Connor & Wang, 2016).
In contrast with oAβ effects on hippocampal LTP, only a few studies have examined LTD induction, and they have yielded inconsistent results. Earlier studies showed synthetic Aβ did not alter LTD induction and magnitude when performed by standard low-frequency stimulation (LFS, 900 pulses at 1 Hz). Interestingly, when a dose of 50 μM AP5, which fully blocked the 900-pulse (LFS) LTD, if added to Aβ oligomer-treated slices, the magnitude of LTD does not change and this LTD requires much higher concentration of AP5, suggesting oAβ causes increased extracellular glutamate levels (Lei et al., 2016; Li et al., 2009; Talantova et al., 2013). To further explore the oAβ effect on hippocampal LTD, we first used a weak-LFS (1 Hz, 300-pulse LFS) that usually has no significant effect on synaptic transmission, but it could induce significant and stable LTD in oAβ-exposed hippocampal slices (Li et al., 2009; Shankar et al., 2008). This phenomenon has been replicated by several other groups (Hu et al., 2014; Ma et al., 2012; O’Riordan, Hu, & Rowan, 2018a; Olsen & Sheng, 2012; Salgado-Puga, Rodriguez-Colorado, Prado-Alcala, & Pena-Ortega, 2017). We conclude that soluble Aβ oligomers can induction of facilitate hippocampal LTD (Fig.2).
Figure 2. Soluble Aβ facilitates long-term depression in the CA1 region of hippocampal slices.
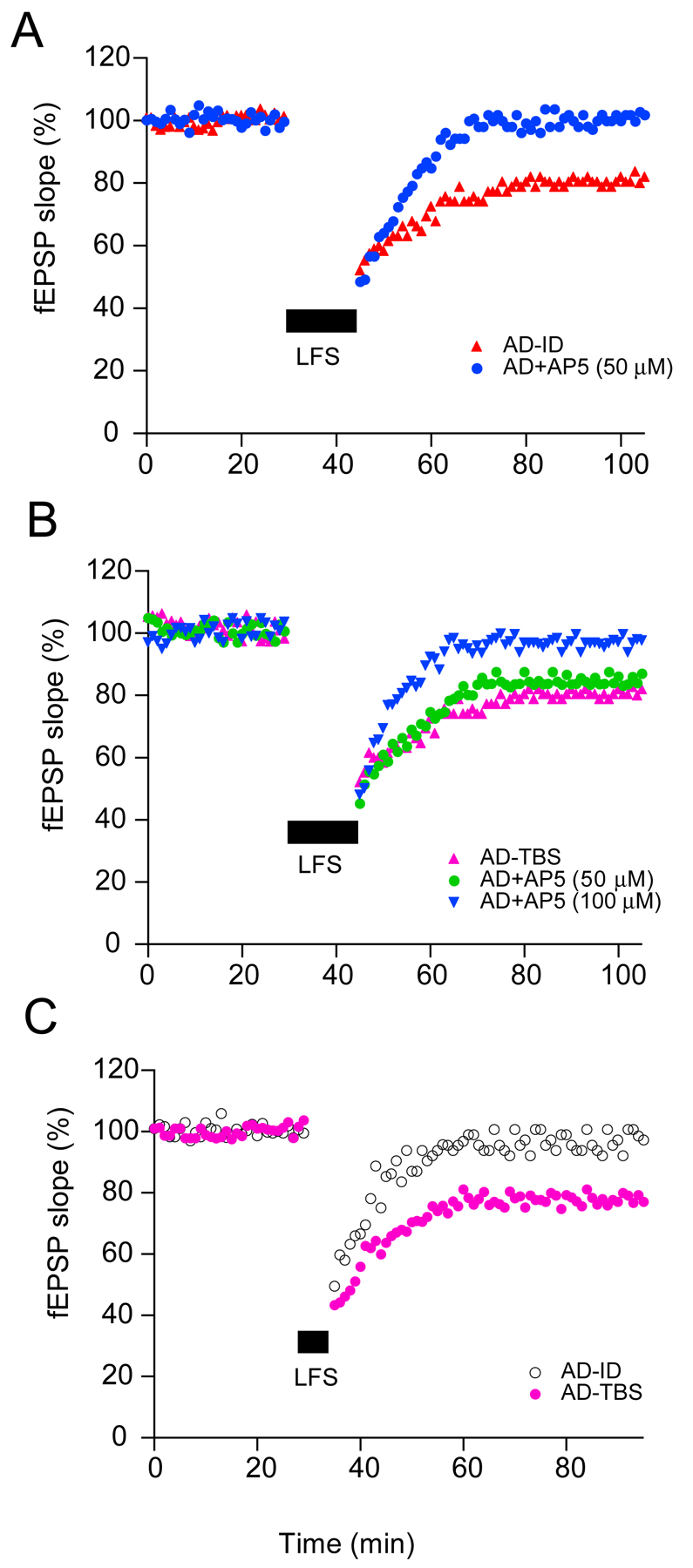
(A) The LTD induced by standard 900-pulse (1 Hz, 15 min, black bar) in hippocampal slices after control (Aβ-immunodepleted AD extracts) treatment (red triangles) was blocked by co-administering the NMDAR antagonist, AP5 (50 μM, blue circles). (B) The 900-pulse LTD in AD brain extracts-treated slices (pink triangles) was not blocked upon coperfusing AP5 (50 μM, green circles), whereas the LTD in AD brain extracts-treated slices was blocked by treatment with D-AP5 at 100 μM (blue triangles). (C) A train of 300 single pulses at 1 Hz (5 min; small black bar) did not induce LTD in acute mouse hippocampal slices in the presence of (Aβ-immunodepleted AD extracts (open circles) but induced a significant LTD in the presence of AD brain extract (solid pink circles).
1. Metabotropic glutamate receptors
Metabotropic glutamate receptors (mGluRs) are coupled to GTP-binding proteins that link the receptors to downstream signaling pathways. They have been classified into three groups by pharmacological properties, sequence similarities, and intracellular signal transduction mechanisms (Ribeiro, Vieira, Pires, Olmo, & Ferguson, 2017). Activation of the mGluR has yields a diverse range of electrophysiological effects, such as potentiation of AMPAR and NMDAR synaptic responses, inhibition or activation of potassium and calcium currents, mediation of slow excitatory postsynaptic potentials, presynaptic inhibition of transmitter release, and involvement in the generation of oscillatory and epileptiform activity (Anwyl, 1999).
From the application of AD brain-derived Aβ to wild-type mouse hippocampal slices or ICV injection into intact rat brain, it has been shown that the facilitation of LTD by soluble Aβ oligomers is mediated in part by activation of mGluR5(Hu et al., 2014; Kessels, Nabavi, & Malinow, 2013; Li et al., 2009; O’Riordan, Hu, & Rowan, 2018b; Shankar et al., 2008). The Aβ oligomer perturbation of synaptic function may include their binding to mGluR5, thus limiting the mobility of mGluR5 receptors, and eventually causing an aberrant increase in intracellular Ca2+ and the removal of NMDAR from synapses (Renner et al., 2010). Although Aβ oligomers have been shown to have high affinity of binding with PrPC in vitro (Lauren et al., 2009), several studies suggest that the mGluR5 is required for this and may initiate the formation of oAβ-PrPC-mGluR5 complexes that mediate multiple changes in synaptic homeostasis, leading to synaptic loss (Beraldo et al., 2016; Haas et al., 2016; Hu et al., 2014; Um et al., 2013). These findings may help explain why mGluR group I antagonists can prevent oAβ-induced enhancement of LTD (Hu et al., 2014; Li et al., 2009; Shankar et al., 2008). Similarly, inhibition of mGluR5 can prevent oAβ-induced hippocampal LTP impairment (Hu et al., 2014; Rammes, Hasenjager, Sroka-Saidi, Deussing, & Parsons, 2011; Zhang, Qi, et al., 2017), and application of expressed β-CTF generated Aβ leads to a “mGluR-like” LTD of AMPARs (Hsieh et al., 2006). Therefore, soluble Aβ oligomers may bind to or interact with mGluR or depolarize the membrane through mGluR, thereby activate subsequent cascades (including PrPc, GluN2B, Fyn, Arc/Arg3.1 signaling) which facilitate LTD induction or elicit “LTD-like” changes.
2. Glutamate transporters
Early AD studies focused on frank Aβ-induced glutamatergic excitotoxicity. The physiological clearance of glutamate from the synaptic cleft via glutamate transporters can prevent the excitotoxicity mediated by excessive NMDA receptor activation. Alteration of glutamate transporters has been documented in postmortem AD brain tissue (Jacob et al., 2007; Scott, Gebhardt, Mitrovic, Vandenberg, & Dodd, 2011; Woltjer et al., 2010) and APP transgenic mice (Mookherjee et al., 2011; Schallier et al., 2011). Synthetic Aβ aggregates have been shown to inhibit glutamate uptake and decrease glutamatergic transporter expression in various experimental systems (Lanznaster et al., 2017; Li et al., 2009; Matos, Augusto, Oliveira, & Agostinho, 2008; Piermartiri et al., 2010; Tong et al., 2017). Using the glutamate uptake inhibitor TBOA, we and others have mimicked and even occluded the known oAβ effects of increasing extracellular glutamate levels to impair LTP and enhance LTD in the hippocampus (Lei et al., 2016; Li et al., 2009; Li et al., 2011; Varga et al., 2015; Zhang, Mably, et al., 2017). Application of extracellular glutamate scavengers to remove excessive glutamate was able to prevent Aβ-mediated synaptic dysfunction (Li et al., 2009; Varga et al., 2015; Zhang, Mably, et al., 2017).
The disruption of synaptic plasticity by glutamate is dependent on its concentration. Exogenous application of low doses of NMDA inhibits hippocampal LTP, while high doses induce LTD (Li et al., 2011) and can eliminate synapses (Henson, Tucker, Zhao, & Dudek, 2017). An increase of glutamate levels has been reported in the CSF of AD patients (Csernansky, Bardgett, Sheline, Morris, & Olney, 1996; Jimenez-Jimenez et al., 1998; Kaiser et al., 2010)and in the hippocampus of rodent brain after oAβ administration (Lanznaster et al., 2017; Lei et al., 2016; O’Shea et al., 2008). Synthetic Aβ-induced rises in extracellular glutamate levels may result from several different mechanisms, such as decreasing glutamate uptake (Lanznaster et al., 2017; Li et al., 2009; Matos et al., 2012), inducing rapid glutamate transporter mislocalization and internalization in astrocytes, thus reducing glutamate clearance (Scimemi et al., 2013), or increasing glutamate release (Hascup & Hascup, 2016; Talantova et al., 2013). The increased extracellular glutamate levels could be a key upstream factor that activates extrasynaptic GluN2B receptors and leads to LTP impairment, LTD enhancement and synapse loss. The evidence that soluble Aβ oligomers may interrupt various glutamate receptors (NMADRs, AMPARs and mGluRs) has suggested that oAβ may impair their upstream regulators, including glutamate transporters. The pro-inflammatory cytokines potentially released from activated microglial/glial cells in pathological conditions have been shown to alter the ability for glial glutamate uptake (Carmen, Rothstein, & Kerr, 2009; Dumont, Goursaud, Desmet, & Hermans, 2014; Sulkowski, Dabrowska-Bouta, Kwiatkowska-Patzer, & Struzynska, 2009; Tong et al., 2017; Verma, Mishra, Sasmal, & Raghubir, 2010). Thus, soluble Aβ oligomers may activate glia to release cytokines, further impairing the function of glutamate transporters, leading to changes in a series of neuronal signaling cascades.
Towards a unified oAβ mechanism through future studies
Although Aβ oligomer accumulation has been defined as characteristic of AD and widely recognized as probably contributing directly to gradual cognitive decline, the exact mechanism of how Aβ interrupts the synaptic function in the earlier stages of AD is incompletely understood. As reviewed above, soluble Aβ oligomers can apparently affect several cell-surface neuronal receptors (NMDAR, AMPAR, mGluR, GABAR) and alter numerous signaling pathways. Aβ has been reported in in vitro experiments to potentially interact with PrP, Insulin receptors, EphB2, nAChRs, and β-adrenergic receptors (Ferreira, Lourenco, Oliveira, & De Felice, 2015; Mroczko, Groblewska, Litman-Zawadzka, Kornhuber, & Lewczuk, 2018; Smith & Strittmatter, 2017), all of which could modulate NMDAR activity. Specifically, it has been reported that oAβ bound predominantly to postsynaptic dendritic arbors of excitatory neurons (Lacor et al., 2007); and a striking shrinkage of PSDs and synapse loss could be found within a halo of oligomeric Aβ surrounding the plaque from AD model mice (Koffie et al., 2009). It is likely that the hydrophobic Aβ oligomers actually interact with lipid binding sites on plasma membranes that act upstream of these various receptors, including dynamic Aβ oligomer clusters around these receptors, thus perturbing their function secondarily.
Our previous findings suggested that soluble Aβ oligomers activate extrasynaptic GluN2B-containing NMDA receptors; however, we do not have specific evidence that the soluble Aβ oligomers bind directly to GluN2B or to any other partially hydrophilic cell-surface receptor. Oligomer-mediated perturbation of the fine structure of the neuronal plasma membrane could then lead to secondary biophysical effects on the structure and function of a variety of transmembrane receptors (e.g., the NMDA, AMPA, mGlu, insulin, α7AChRs and/or β-adrenoceptors), the cellular prion protein, and glutamate transporters. When oAβ increases or is exogenously applied to neuronal culture medium, brain slices or intact brain, the hydrophobic oligomers will stick on to the neuronal and glial lipid membrane surfaces, thereby interrupting its normal biophysical properties, activating cytokines and associated inflammatory mediators. These multifaceted actions can result in an increase of the extracellular glutamate concentration by reducing neuronal and astrocytic glutamate reuptake. Of particular concern, excessive release of glutamate by astrocytes can allow access to extrasynaptic NMDA receptors, induce neuronal excitotoxicity, impair LTP, enhance LTD (Fig.3) and influence the genesis and strength of seizure-like events (Barker-Haliski & White, 2015). Although exploring the exact binding partners of natural Aβ oligomers will be labor-intensive, it would be valuable for the investigation of the mechanism of oAβ-induced neurotoxicity. If we find specific oAβ binding partners, we would have a chance to study the exact mechanisms of Aβ interrupt synaptic function and develop potential compounds to treat AD.
Figure 3. Schematic hypothesis of how Aβ may interrupt synaptic plasticity based on current evidence reviewed herein.
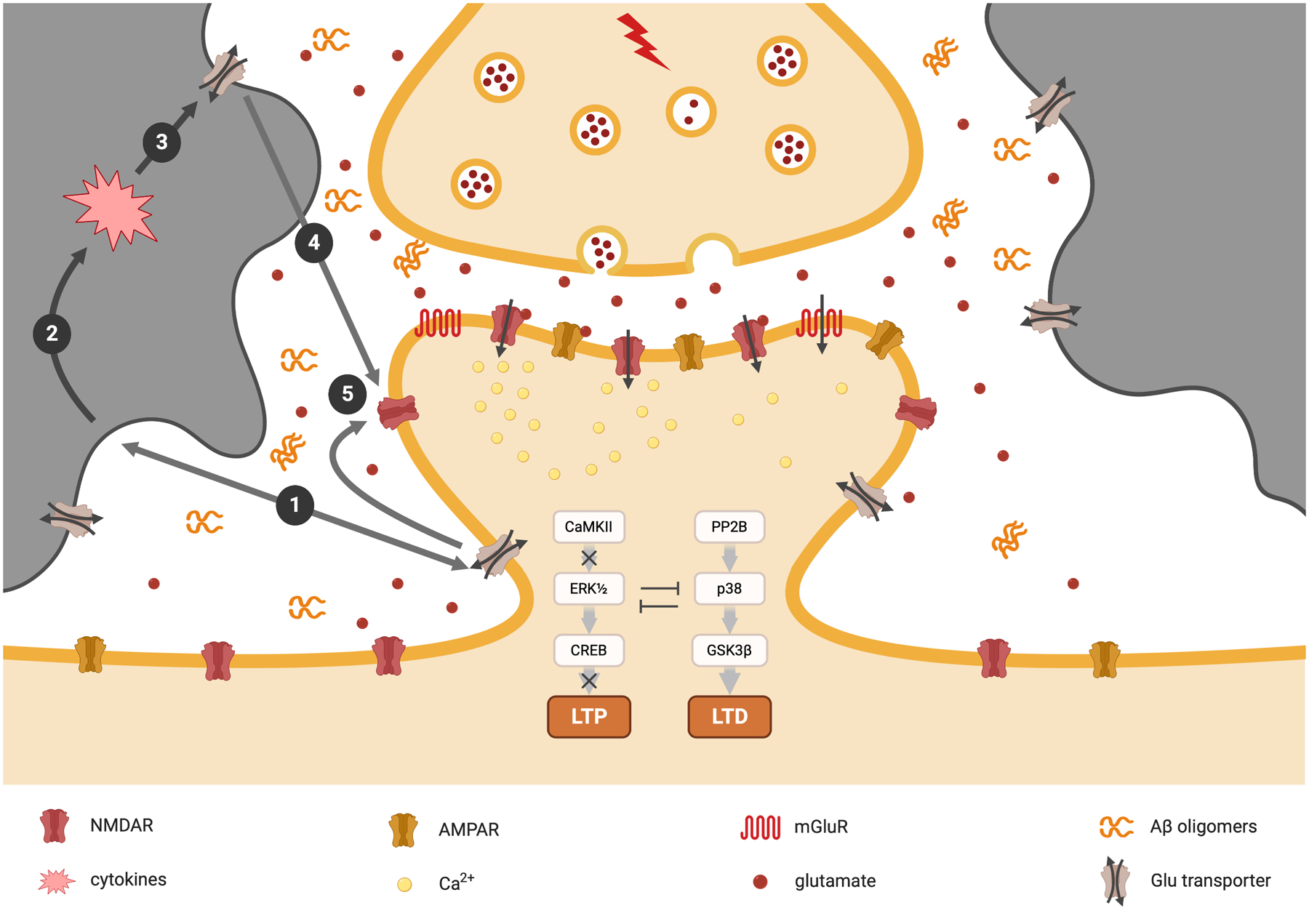
When oAβ increases or is exogenously applied to the culture medium, brain slices, or intact brain, the hydrophobic oligomers will likely stick on the neuronal or glial lipid membrane surface ①, therefore interrupting its normal functions, stimulating glial cells to release cytokines and associated inflammatory mediators ②, reducing astrocytic and/or neuronal glutamate reuptake ③; therefore increasing the extracellular glutamate concentration ④; the high concentrations of extracellular glutamates further activate the extrasynaptic GluN2B-containing NMDA receptors ⑤; therefore inducing LTP impairment and LTD enhancement and other neuronal harmful effects.
Synaptic dysfunction induced by oAβ may represent in part a relatively acute form of Aβ neurotoxicity, as it has been observed in a few minutes after application in vitro or in vivo, and the levels of heterogeneous brain proteins can then change within 3 hours, with morphological changes of neurons seen by ~16 hours after application (Li et al., 2011). Whether the alteration of synaptic plasticity contributes directly to prodromal or very early symptomatic AD is worthy of further investigation. As oAβ (including the high molecular weight fraction) but not Aβ monomers or insoluble amyloid plaques from AD brain extracts significantly impair synaptic plasticity, and immunodepletion of the former bioactive species prevents the synaptic dysfunction, the development of effective small molecules and antibodies that specifically target soluble Aβ oligomers (monoclonal antibodies could help prevent oAβ formation and enhance clearance of existing oAβ) represents an attractive therapeutic strategy for the AD. Interestingly, other misfolded proteins such as α-synuclein oligomers (the key pathogen in Parkinson’s disease) (Ghiglieri, Calabrese, & Calabresi, 2018; van Diggelen et al., 2019) may also disrupt hippocampal and striatal synaptic plasticity, suggesting that oligomer-mediated synaptic dysfunction may be an early stage in the cytotoxicity of several protein-mediated neurodegenerative diseases.
Further studies using dual recordings (Lei et al., 2016; Zhao et al., 2018) could provide more details of oAβ effects on synaptic plasticity and neural networks. Clinically, using noninvasive brain stimulation techniques, such as visual evoked potentials (Spriggs, Cadwallader, Hamm, Tippett, & Kirk, 2017), transcranial direct current stimulation (tDCS) and repetitive transcranial magnetic stimulation (rTMS) (Cirillo et al., 2017) could induce LTP-like and LTD-like phenomena in the human subjects and AD patients. Such recordings at different stages of AD patients could show that LTP/LTD-like changes are significantly related to cognitive behavior, thus providing an earlier biomarker for diagnosis and therapeutic approaches.
Acknowledgements
Supported by NIH grants AG 027443 and AG 036694 (to D.J.S.) and a grant (to S.L.) from the Massachusetts Alzheimer’s Disease Research Center (5P50 AG 005134).
Alphabetical list of abbreviations
- ACSF
artificial cerebrospinal fluid
- AD
Alzheimer’s disease
- ADDL
Aβ-derived diffusible ligand
- APP
β-amyloid precursor protein
- Aβ
amyloid β-protein
- CHO
Chinese Hamster Ovary
- CP-AMPAR
calcium-permeable AMPAR
- CSF
cerebrospinal fluid
- E/I
excitation/inhibition
- fEPSP
field excitatory postsynaptic potential
- GSK3
glycogen synthase kinase-3
- HFS
high-frequency stimulation
- ICV
intracerebroventricular
- IFNγ
interferon-γ
- IL-1
interleukin-1
- LFS
low-frequency stimulation
- LTD
long-term depression
- LTP
long-term potentiation
- MCI
mild cognitive impairment
- mGluRs
metabotropic glutamate receptors
- oAβ
soluble Aβ oligomers
- PBS
phosphate-buffered saline
- PP2B
protein phosphatase 2B
- PPF
paired-pulse facilitation
- PrPC
Cellular prion protein
- PTP
post-tetanic potentiation
- rTMS
repetitive transcranial magnetic stimulation
- SEC
size exclusion chromatography
- STP
Short-term plasticity
- TBS
Tris-buffered saline
- tDCS
transcranial direct current stimulation
- TNFα
tumor necrosis factor-alpha
Footnotes
Conflict of interest: DJS is a director of and consultant to Prothena Biosciences. SL has no conflicts to declare.
References
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, … Stern Y (2006). Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia, 47(5), 867–872. doi: 10.1111/j.1528-1167.2006.00554.x [DOI] [PubMed] [Google Scholar]
- Anwyl R (1999). Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev, 29(1), 83–120. doi: 10.1016/s0165-0173(98)00050-2 [DOI] [PubMed] [Google Scholar]
- Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, Chmielewska N, … Quiroz YT (2019). Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med, 25(11), 1680–1683. doi: 10.1038/s41591-019-0611-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T (2009). Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol, 118(1), 167–179. doi: 10.1007/s00401-009-0536-x [DOI] [PubMed] [Google Scholar]
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, … Forloni G (2010). Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A, 107(5), 2295–2300. doi: 10.1073/pnas.0911829107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker-Haliski M, & White HS (2015). Glutamatergic Mechanisms Associated with Seizures and Epilepsy. Cold Spring Harb Perspect Med, 5(8), a022863. doi: 10.1101/cshperspect.a022863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, … Rowan MJ (2011). Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci, 31(20), 7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastin C, Bahri MA, Meyer F, Manard M, Delhaye E, Plenevaux A, … Salmon E (2019). In vivo imaging of synaptic loss in Alzheimer’s disease with [18F]UCB-H positron emission tomography. Eur J Nucl Med Mol Imaging. doi: 10.1007/s00259-019-04461-x [DOI] [PubMed] [Google Scholar]
- Beraldo FH, Ostapchenko VG, Caetano FA, Guimaraes AL, Ferretti GD, Daude N, … Prado MA (2016). Regulation of Amyloid beta Oligomer Binding to Neurons and Neurotoxicity by the Prion Protein-mGluR5 Complex. J Biol Chem, 291(42), 21945–21955. doi: 10.1074/jbc.M116.738286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini E, Turnbaugh JA, Unterberger U, & Harris DA (2012). Prion protein at the crossroads of physiology and disease. Trends Neurosci, 35(2), 92–103. doi: 10.1016/j.tins.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmalm G, Hong W, Wang Z, Liu W, O’Malley TT, Sun X, … Walsh DM (2019). Identification of neurotoxic cross-linked amyloid-beta dimers in the Alzheimer’s brain. Brain, 142(5), 1441–1457. doi: 10.1093/brain/awz066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano FA, Beraldo FH, Hajj GN, Guimaraes AL, Jurgensen S, Wasilewska-Sampaio AP, … Prado MA (2011). Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J Neurochem, 117(3), 538–553. doi: 10.1111/j.1471-4159.2011.07225.x [DOI] [PubMed] [Google Scholar]
- Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, … Aguzzi A (2010). Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med, 2(8), 306–314. doi: 10.1002/emmm.201000082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantanelli P, Sperduti S, Ciavardelli D, Stuppia L, Gatta V, & Sensi SL (2014). Age-Dependent Modifications of AMPA Receptor Subunit Expression Levels and Related Cognitive Effects in 3xTg-AD Mice. Front Aging Neurosci, 6, 200. doi: 10.3389/fnagi.2014.00200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmen J, Rothstein JD, & Kerr DA (2009). Tumor necrosis factor-alpha modulates glutamate transport in the CNS and is a critical determinant of outcome from viral encephalomyelitis. Brain Res, 1263, 143–154. doi: 10.1016/j.brainres.2009.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrera I, Etcheverria I, Fernandez-Novoa L, Lombardi VR, Lakshmana MK, Cacabelos R, & Vigo C (2015). A comparative evaluation of a novel vaccine in APP/PS1 mouse models of Alzheimer’s disease. Biomed Res Int, 2015, 807146. doi: 10.1155/2015/807146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerpa W, Farias GG, Godoy JA, Fuenzalida M, Bonansco C, & Inestrosa NC (2010). Wnt-5a occludes Abeta oligomer-induced depression of glutamatergic transmission in hippocampal neurons. Mol Neurodegener, 5, 3. doi: 10.1186/1750-1326-5-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo G, Di Pino G, Capone F, Ranieri F, Florio L, Todisco V, … Di Lazzaro V (2017). Neurobiological after-effects of non-invasive brain stimulation. Brain Stimul, 10(1), 1–18. doi: 10.1016/j.brs.2016.11.009 [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, & Wang YT (2010). Long-term depression in the CNS. Nat Rev Neurosci, 11(7), 459–473. doi: 10.1038/nrn2867 [DOI] [PubMed] [Google Scholar]
- Connor SA, & Wang YT (2016). A Place at the Table: LTD as a Mediator of Memory Genesis. Neuroscientist, 22(4), 359–371. doi: 10.1177/1073858415588498 [DOI] [PubMed] [Google Scholar]
- Csernansky JG, Bardgett ME, Sheline YI, Morris JC, & Olney JW (1996). CSF excitatory amino acids and severity of illness in Alzheimer’s disease. Neurology, 46(6), 1715–1720. doi: 10.1212/wnl.46.6.1715 [DOI] [PubMed] [Google Scholar]
- Del Vecchio RA, Gold LH, Novick SJ, Wong G, & Hyde LA (2004). Increased seizure threshold and severity in young transgenic CRND8 mice. Neurosci Lett, 367(2), 164–167. doi: 10.1016/j.neulet.2004.05.107 [DOI] [PubMed] [Google Scholar]
- Dohler F, Sepulveda-Falla D, Krasemann S, Altmeppen H, Schluter H, Hildebrand D, … Glatzel M (2014). High molecular mass assemblies of amyloid-beta oligomers bind prion protein in patients with Alzheimer’s disease. Brain, 137(Pt 3), 873–886. doi: 10.1093/brain/awt375 [DOI] [PubMed] [Google Scholar]
- Domingues C, da Cruz ESOAB, & Henriques AG (2017). Impact of Cytokines and Chemokines on Alzheimer’s Disease Neuropathological Hallmarks. Curr Alzheimer Res, 14(8), 870–882. doi: 10.2174/1567205014666170317113606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Epelbaum S, Nyasse F, Bakardjian H, Gagliardi G, Uspenskaya O, … Hampel H (2018). Cognitive and neuroimaging features and brain beta-amyloidosis in individuals at risk of Alzheimer’s disease (INSIGHT-preAD): a longitudinal observational study. Lancet Neurol, 17(4), 335–346. doi: 10.1016/S1474-4422(18)30029-2 [DOI] [PubMed] [Google Scholar]
- Dumont AO, Goursaud S, Desmet N, & Hermans E (2014). Differential regulation of glutamate transporter subtypes by pro-inflammatory cytokine TNF-alpha in cortical astrocytes from a rat model of amyotrophic lateral sclerosis. PLoS One, 9(5), e97649. doi: 10.1371/journal.pone.0097649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, & Brody DL (2013). Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol, 73(1), 104–119. doi: 10.1002/ana.23748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ST, Lourenco MV, Oliveira MM, & De Felice FG (2015). Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front Cell Neurosci, 9, 191. doi: 10.3389/fncel.2015.00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C, … Harris DA (2013). An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J Biol Chem, 288(11), 7857–7866. doi: 10.1074/jbc.M112.423954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel H, Frere S, Segev O, Bharill S, Shapira I, Gazit N, … Slutsky I (2014). APP homodimers transduce an amyloid-beta-mediated increase in release probability at excitatory synapses. Cell Rep, 7(5), 1560–1576. doi: 10.1016/j.celrep.2014.04.024 [DOI] [PubMed] [Google Scholar]
- Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, … Collinge J (2011). Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun, 2, 336. doi: 10.1038/ncomms1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JJ, Finnegan ME, Grehan B, Dobson J, Collingwood JF, & Lynch MA (2012). Modest amyloid deposition is associated with iron dysregulation, microglial activation, and oxidative stress.. J Alzheimers Dis, 28(1), 147–161. doi: 10.3233/JAD-2011-110614 [DOI] [PubMed] [Google Scholar]
- Ghiglieri V, Calabrese V, & Calabresi P (2018). Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front Neurol, 9, 295. doi: 10.3389/fneur.2018.00295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J, Shu S, Yang X, Lu Y, Zhu LQ, & Man HY (2016). beta-Amyloid triggers aberrant over-scaling of homeostatic synaptic plasticity. Acta Neuropathol Commun, 4(1), 131. doi: 10.1186/s40478-016-0398-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, … Klein WL (2003). Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A, 100(18), 10417–10422. doi: 10.1073/pnas.1834302100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutagny R, Gu N, Cavanagh C, Jackson J, Chabot JG, Quirion R, … Williams S (2013). Alterations in hippocampal network oscillations and theta-gamma coupling arise before Abeta overproduction in a mouse model of Alzheimer’s disease. Eur J Neurosci, 37(12), 1896–1902. doi: 10.1111/ejn.12233 [DOI] [PubMed] [Google Scholar]
- Gulisano W, Melone M, Li Puma DD, Tropea MR, Palmeri A, Arancio O, … Puzzo D (2018). The effect of amyloid-beta peptide on synaptic plasticity and memory is influenced by different isoforms, concentrations, and aggregation status. Neurobiol Aging, 71, 51–60. doi: 10.1016/j.neurobiolaging.2018.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas LT, Salazar SV, Kostylev MA, Um JW, Kaufman AC, & Strittmatter SM (2016). Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain, 139(Pt 2), 526–546. doi: 10.1093/brain/awv356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, … Selkoe DJ (1999). Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci, 19(20), 8876–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman RE, Izumi Y, Bales KR, Paul SM, Wozniak DF, & Holtzman DM (2005). Treatment with an amyloid-beta antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer’s disease. J Neurosci, 25(26), 6213–6220. doi: 10.1523/JNEUROSCI.0664-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascup KN, & Hascup ER (2016). Soluble Amyloid-beta42 Stimulates Glutamate Release through Activation of the alpha7 Nicotinic Acetylcholine Receptor. J Alzheimers Dis, 53(1), 337–347. doi: 10.3233/JAD-160041 [DOI] [PubMed] [Google Scholar]
- Hasegawa S, Sakuragi S, Tominaga-Yoshino K, & Ogura A (2015). Dendritic spine dynamics leading to spine elimination after repeated inductions of LTD. Sci Rep, 5, 7707. doi: 10.1038/srep07707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden EY, & Teplow DB (2013). Amyloid beta-protein oligomers and Alzheimer’s disease. Alzheimers Res Ther, 5(6), 60. doi: 10.1186/alzrt226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, … Kummer MP (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol, 14(4), 388–405. doi: 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson MA, Tucker CJ, Zhao M, & Dudek SM (2017). Long-term depression-associated signaling is required for an in vitro model of NMDA receptor-dependent synapse pruning. Neurobiol Learn Mem, 138, 39–53. doi: 10.1016/j.nlm.2016.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner FL, Ransohoff RM, & Becher B (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci, 16(6), 358–372. doi: 10.1038/nrn3880 [DOI] [PubMed] [Google Scholar]
- Hillen H, Barghorn S, Striebinger A, Labkovsky B, Muller R, Nimmrich V, … Ebert U (2010). Generation and therapeutic efficacy of highly oligomer-specific beta-amyloid antibodies. J Neurosci, 30(31), 10369–10379. doi: 10.1523/JNEUROSCI.5721-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W, Wang Z, Liu W, O’Malley TT, Jin M, Willem M, … Walsh DM (2018). Diffusible, highly bioactive oligomers represent a critical minority of soluble Abeta in Alzheimer’s disease brain. Acta Neuropathol, 136(1), 19–40. doi: 10.1007/s00401-018-1846-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K, Hasegawa K, Kamiya H, & Morimoto Y (2017). Synapse-specific effects of IL-1beta on long-term potentiation in the mouse hippocampus. Biomed Res, 38(3), 183–188. doi: 10.2220/biomedres.38.183 [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, & Malinow R (2006). AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron, 52(5), 831–843. doi: 10.1016/j.neuron.2006.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu NW, Nicoll AJ, Zhang D, Mably AJ, O’Malley T, Purro SA, … Rowan MJ (2014). mGlu5 receptors and cellular prion protein mediate amyloid-beta-facilitated synaptic long-term depression in vivo. Nat Commun, 5, 3374. doi: 10.1038/ncomms4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu NW, Smith IM, Walsh DM, & Rowan MJ (2008). Soluble amyloid-beta peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain, 131(Pt 9), 2414–2424. doi: 10.1093/brain/awn174 [DOI] [PubMed] [Google Scholar]
- Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, … Grunblatt E (2007). Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimers Dis, 11(1), 97–116. doi: 10.3233/jad-2007-11113 [DOI] [PubMed] [Google Scholar]
- Jimenez-Jimenez FJ, Molina JA, Gomez P, Vargas C, de Bustos F, Benito-Leon J, … Arenas J (1998). Neurotransmitter amino acids in cerebrospinal fluid of patients with Alzheimer’s disease. J Neural Transm (Vienna), 105(2–3), 269–277. doi: 10.1007/s007020050056 [DOI] [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, & Selkoe DJ (2011). Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A, 108(14), 5819–5824. doi: 10.1073/pnas.1017033108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser E, Schoenknecht P, Kassner S, Hildebrandt W, Kinscherf R, & Schroeder J (2010). Cerebrospinal fluid concentrations of functionally important amino acids and metabolic compounds in patients with mild cognitive impairment and Alzheimer’s disease. Neurodegener Dis, 7(4), 251–259. doi: 10.1159/000287953 [DOI] [PubMed] [Google Scholar]
- Kakuda N, Shoji M, Arai H, Furukawa K, Ikeuchi T, Akazawa K, … Ihara Y (2012). Altered gamma-secretase activity in mild cognitive impairment and Alzheimer’s disease. EMBO Mol Med, 4(4), 344–352. doi: 10.1002/emmm.201200214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai R, Chiba S, Okuwaki K, Kanada R, Doi H, Ono M, … Okuno Y (2020). Stabilization Mechanism for a Nonfibrillar Amyloid beta Oligomer Based on Formation of a Hydrophobic Core Determined by Dissipative Particle Dynamics. ACS Chem Neurosci, 11(3), 385–394. doi: 10.1021/acschemneuro.9b00602 [DOI] [PubMed] [Google Scholar]
- Kelly RJ, Minogue AM, Lyons A, Jones RS, Browne TC, Costello DA, … Lynch MA (2013). Glial Activation in AbetaPP/PS1 Mice is Associated with Infiltration of IFNgamma-Producing Cells. J Alzheimers Dis, 37(1), 63–75. doi: 10.3233/JAD-130539 [DOI] [PubMed] [Google Scholar]
- Kessels HW, Nabavi S, & Malinow R (2013). Metabotropic NMDA receptor function is required for beta-amyloid-induced synaptic depression. Proc Natl Acad Sci U S A, 110(10), 4033–4038. doi: 10.1073/pnas.1219605110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Nguyen LN, Nabavi S, & Malinow R (2010). The prion protein as a receptor for amyloid-beta. Nature, 466(7308), E3–4; discussion E4–5. doi: 10.1038/nature09217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, … Zamponi GW (2008). Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol, 181(3), 551–565. doi: 10.1083/jcb.200711002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, & Lamb BT (2018). Inflammation as a central mechanism in Alzheimer’s disease.. Alzheimers Dement (N Y), 4, 575–590. doi: 10.1016/j.trci.2018.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittelberger KA, Piazza F, Tesco G, & Reijmers LG (2012). Natural amyloid-beta oligomers acutely impair the formation of a contextual fear memory in mice. PLoS One, 7(1), e29940. doi: 10.1371/journal.pone.0029940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, … Rowan MJ (2008). Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci, 28(16), 4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Cullen WK, Hu NW, & Rowan MJ (2012). Alzheimer’s disease Abeta assemblies mediating rapid disruption of synaptic plasticity and memory. Mol Brain, 5, 25. doi: 10.1186/1756-6606-5-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Nicoll AJ, Khalili-Shirazi A, Farmer M, Canning S, Mably A, … Collinge J (2014). Peripheral administration of a humanized anti-PrP antibody blocks Alzheimer’s disease Abeta synaptotoxicity. J Neurosci, 34(18), 6140–6145. doi: 10.1523/JNEUROSCI.3526-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Cullen WK, Fadeeva JV, Anwyl R, Selkoe DJ, & Rowan MJ (2004). Soluble Arctic amyloid beta protein inhibits hippocampal long-term potentiation in vivo. Eur J Neurosci, 19(10), 2839–2846. doi: 10.1111/j.1460-9568.2004.03389.x [DOI] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, … Rowan MJ (2005). Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med, 11(5), 556–561. doi: 10.1038/nm1234 [DOI] [PubMed] [Google Scholar]
- Knight EM, Kim SH, Kottwitz JC, Hatami A, Albay R, Suzuki A, … Steele JW (2016). Effective anti-Alzheimer Abeta therapy involves depletion of specific Abeta oligomer subtypes. Neurol Neuroimmunol Neuroinflamm, 3(3), e237. doi: 10.1212/NXI.0000000000000237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, … Spires-Jones TL (2009). Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A, 106(10), 4012–4017. doi: 10.1073/pnas.0811698106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmer M, Close W, Funk L, Rasmussen J, Bsoul A, Schierhorn A, … Fandrich M (2019). Cryo-EM structure and polymorphism of Abeta amyloid fibrils purified from Alzheimer’s brain tissue. Nat Commun, 10(1), 4760. doi: 10.1038/s41467-019-12683-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostylev MA, Kaufman AC, Nygaard HB, Patel P, Haas LT, Gunther EC, … Strittmatter SM (2015). Prion-Protein-interacting Amyloid-beta Oligomers of High Molecular Weight Are Tightly Correlated with Memory Impairment in Multiple Alzheimer Mouse Models. J Biol Chem, 290(28), 17415–17438. doi: 10.1074/jbc.M115.643577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krafft GA, & Klein WL (2010). ADDLs and the signaling web that leads to Alzheimer’s disease. [Review]. Neuropharmacology, 59(4–5), 230–242. doi: 10.1016/j.neuropharm.2010.07.012 [DOI] [PubMed] [Google Scholar]
- Kryukov KA, Kim KK, Magazanik LG, & Zaitsev AV (2016). Status epilepticus alters hippocampal long-term synaptic potentiation in a rat lithium-pilocarpine model. Neuroreport, 27(16), 1191–1195. doi: 10.1097/WNR.0000000000000656 [DOI] [PubMed] [Google Scholar]
- Kurudenkandy FR, Zilberter M, Biverstal H, Presto J, Honcharenko D, Stromberg R, … Fisahn A (2014). Amyloid-beta-induced action potential desynchronization and degradation of hippocampal gamma oscillations is prevented by interference with peptide conformation change and aggregation. J Neurosci, 34(34), 11416–11425. doi: 10.1523/JNEUROSCI.1195-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, … Klein WL (2007). Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci, 27(4), 796–807. doi: 10.1523/JNEUROSCI.3501-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, … Klein WL (2007). Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem, 100(1), 23–35. doi: 10.1111/j.1471-4159.2006.04157.x [DOI] [PubMed] [Google Scholar]
- Lanznaster D, Mack JM, Coelho V, Ganzella M, Almeida RF, Dal-Cim T, … Tasca CI (2017). Guanosine Prevents Anhedonic-Like Behavior and Impairment in Hippocampal Glutamate Transport Following Amyloid-beta1–40 Administration in Mice. Mol Neurobiol, 54(7), 5482–5496. doi: 10.1007/s12035-016-0082-1 [DOI] [PubMed] [Google Scholar]
- Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, & Strittmatter SM (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature, 457(7233), 1128–1132. doi: 10.1038/nature07761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, Xu H, Li Z, Wang Z, O’Malley TT, Zhang D, … Li S (2016). Soluble Abeta oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol Dis, 85, 111–121. doi: 10.1016/j.nbd.2015.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewczuk P, Matzen A, Blennow K, Parnetti L, Molinuevo JL, Eusebi P, … Fagan AM (2017). Cerebrospinal Fluid Abeta42/40 Corresponds Better than Abeta42 to Amyloid PET in Alzheimer’s Disease. J Alzheimers Dis, 55(2), 813–822. doi: 10.3233/JAD-160722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Feig LA, & Hartley DM (2007). A brief, but repeated, swimming protocol is sufficient to overcome amyloid beta-protein inhibition of hippocampal long-term potentiation. Eur J Neurosci, 26(5), 1289–1298. doi: 10.1111/j.1460-9568.2007.05760.x [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, & Selkoe D (2009). Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron, 62(6), 788–801. doi: 10.1016/j.neuron.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, & Selkoe DJ (2011). Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci, 31(18), 6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Liu L, Dang Y, Ostaszewski BL, & Selkoe DJ (2018). Decoding the synaptic dysfunction of bioactive human AD brain soluble Abeta to inspire novel therapeutic avenues for Alzheimer’s disease. Acta Neuropathol Commun, 6(1), 121. doi: 10.1186/s40478-018-0626-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Liu L, & Selkoe D (2019). Verubecestat for Prodromal Alzheimer’s Disease. N Engl J Med, 381(4), 388. doi: 10.1056/NEJMc1906679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, … Rogers J (1999). Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol, 155(3), 853–862. doi: 10.1016/s0002-9440(10)65184-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A, Griffin RJ, Costelloe CE, Clarke RM, & Lynch MA (2007). IL-4 attenuates the neuroinflammation induced by amyloid-beta in vivo and in vitro. J Neurochem, 101(3), 771–781. doi: 10.1111/j.1471-4159.2006.04370.x [DOI] [PubMed] [Google Scholar]
- Ma T, Du X, Pick JE, Sui G, Brownlee M, & Klann E (2012). Glucagon-like peptide-1 cleavage product GLP-1(9–36) amide rescues synaptic plasticity and memory deficits in Alzheimer’s disease model mice. J Neurosci, 32(40), 13701–13708. doi: 10.1523/JNEUROSCI.2107-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio N, & Vlachos A (2018). Tumor necrosis factor (TNF) modulates synaptic plasticity in a concentration-dependent manner through intracellular calcium stores. J Mol Med (Berl), 96(10), 1039–1047. doi: 10.1007/s00109-018-1674-1 [DOI] [PubMed] [Google Scholar]
- Marcello E, Epis R, Saraceno C, Gardoni F, Borroni B, Cattabeni F, … Di Luca M (2012). SAP97-mediated local trafficking is altered in Alzheimer disease patients’ hippocampus. Neurobiol Aging, 33(2), 422 e421–410. doi: 10.1016/j.neurobiolaging.2010.09.015 [DOI] [PubMed] [Google Scholar]
- Matos M, Augusto E, Machado NJ, dos Santos-Rodrigues A, Cunha RA, & Agostinho P (2012). Astrocytic adenosine A2A receptors control the amyloid-beta peptide-induced decrease of glutamate uptake. J Alzheimers Dis, 31(3), 555–567. doi: 10.3233/JAD-2012-120469 [DOI] [PubMed] [Google Scholar]
- Matos M, Augusto E, Oliveira CR, & Agostinho P (2008). Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience, 156(4), 898–910. doi: 10.1016/j.neuroscience.2008.08.022 [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, … Masters CL (1999). Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol, 46(6), 860–866. doi: [DOI] [PubMed] [Google Scholar]
- Megill A, Tran T, Eldred K, Lee NJ, Wong PC, Hoe HS, … Lee HK (2015). Defective Age-Dependent Metaplasticity in a Mouse Model of Alzheimer’s Disease. J Neurosci, 35(32), 11346–11357. doi: 10.1523/JNEUROSCI.5289-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, … Jucker M (2006). Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science, 313(5794), 1781–1784. doi: 10.1126/science.1131864 [DOI] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fulop L, … Tanila H (2009). Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci, 29(11), 3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter MR, Taylor JM, & Crack PJ (2016). The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J Neurochem, 136(3), 457–474. doi: 10.1111/jnc.13411 [DOI] [PubMed] [Google Scholar]
- Mitew S, Kirkcaldie MT, Dickson TC, & Vickers JC (2013). Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol Aging, 34(10), 2341–2351. doi: 10.1016/j.neurobiolaging.2013.04.010 [DOI] [PubMed] [Google Scholar]
- Mookherjee P, Green PS, Watson GS, Marques MA, Tanaka K, Meeker KD, … Cook DG (2011). GLT-1 loss accelerates cognitive deficit onset in an Alzheimer’s disease animal model. J Alzheimers Dis, 26(3), 447–455. doi: 10.3233/JAD-2011-110503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motanis H, Seay MJ, & Buonomano DV (2018). Short-Term Synaptic Plasticity as a Mechanism for Sensory Timing. Trends Neurosci, 41(10), 701–711. doi: 10.1016/j.tins.2018.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroczko B, Groblewska M, Litman-Zawadzka A, Kornhuber J, & Lewczuk P (2018). Cellular Receptors of Amyloid beta Oligomers (AbetaOs) in Alzheimer’s Disease. [Review]. Int J Mol Sci, 19(7). doi: 10.3390/ijms19071884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel-Steger L, Owen MC, & Strodel B (2016). An Account of Amyloid Oligomers: Facts and Figures Obtained from Experiments and Simulations. Chembiochem, 17(8), 657–676. doi: 10.1002/cbic.201500623 [DOI] [PubMed] [Google Scholar]
- Nicoll AJ, Panico S, Freir DB, Wright D, Terry C, Risse E, … Collinge J (2013). Amyloid-beta nanotubes are associated with prion protein-dependent synaptotoxicity. Nat Commun, 4, 2416. doi: 10.1038/ncomms3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, … Lannfelt L (2001). The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat Neurosci, 4(9), 887–893. doi: 10.1038/nn0901-887 [DOI] [PubMed] [Google Scholar]
- O’Riordan KJ, Hu NW, & Rowan MJ (2018a). Ass Facilitates LTD at Schaffer Collateral Synapses Preferentially in the Left Hippocampus. Cell Rep, 22(8), 2053–2065. doi: 10.1016/j.celrep.2018.01.085 [DOI] [PubMed] [Google Scholar]
- O’Riordan KJ, Hu NW, & Rowan MJ (2018b). Physiological activation of mGlu5 receptors supports the ion channel function of NMDA receptors in hippocampal LTD induction in vivo. Sci Rep, 8(1), 4391. doi: 10.1038/s41598-018-22768-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea SD, Smith IM, McCabe OM, Cronin MM, Walsh DM, & O’Connor WT (2008). Intracerebroventricular Administration of Amyloid beta-protein Oligomers Selectively Increases Dorsal Hippocampal Dialysate Glutamate Levels in the Awake Rat. Sensors (Basel), 8(11), 7428–7437. doi: 10.3390/s8117428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, & LaFerla FM (2006). Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem, 281(3), 1599–1604. doi: 10.1074/jbc.M507892200 [DOI] [PubMed] [Google Scholar]
- Olsen KM, & Sheng M (2012). NMDA receptors and BAX are essential for Abeta impairment of LTP. Sci Rep, 2, 225. doi: 10.1038/srep00225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr AL, Hanson JE, Li D, Klotz A, Wright S, Schenk D, … Madison DV (2014). beta-Amyloid inhibits E-S potentiation through suppression of cannabinoid receptor 1-dependent synaptic disinhibition. Neuron, 82(6), 1334–1345. doi: 10.1016/j.neuron.2014.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels K, Williams TL, Morris KL, Jonckheere W, Vandersteen A, Kelly G, … Broersen K (2012). Structural basis for increased toxicity of pathological abeta42:abeta40 ratios in Alzheimer disease. J Biol Chem, 287(8), 5650–5660. doi: 10.1074/jbc.M111.264473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache AL, Rajulawalla A, Shi A, Wetzel A, Saito T, Saido TC, … Ali AB (2019). Aberrant Excitatory-Inhibitory Synaptic Mechanisms in Entorhinal Cortex Microcircuits During the Pathogenesis of Alzheimer’s Disease. Cereb Cortex, 29(4), 1834–1850. doi: 10.1093/cercor/bhz016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piermartiri TC, Figueiredo CP, Rial D, Duarte FS, Bezerra SC, Mancini G, … Tasca CI (2010). Atorvastatin prevents hippocampal cell death, neuroinflammation and oxidative stress following amyloid-beta(1–40) administration in mice: evidence for dissociation between cognitive deficits and neuronal damage. Exp Neurol, 226(2), 274–284. doi: 10.1016/j.expneurol.2010.08.030 [DOI] [PubMed] [Google Scholar]
- Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, & Selkoe DJ (1995). Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem, 270(16), 9564–9570. doi: 10.1074/jbc.270.16.9564 [DOI] [PubMed] [Google Scholar]
- Postnikova TY, Zubareva OE, Kovalenko AA, Kim KK, Magazanik LG, & Zaitsev AV (2017). Status Epilepticus Impairs Synaptic Plasticity in Rat Hippocampus and Is Followed by Changes in Expression of NMDA Receptors. Biochemistry (Mosc), 82(3), 282–290. doi: 10.1134/S0006297917030063 [DOI] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, & Shelanski ML (2013). Synaptic changes in Alzheimer’s disease and its models. [Review]. Neuroscience, 251, 51–65. doi: 10.1016/j.neuroscience.2012.05.050 [DOI] [PubMed] [Google Scholar]
- Prieto GA, Tong L, Smith ED, & Cotman CW (2019). TNFalpha and IL-1beta but not IL-18 Suppresses Hippocampal Long-Term Potentiation Directly at the Synapse. Neurochem Res, 44(1), 49–60. doi: 10.1007/s11064-018-2517-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevenco FC, Schreiner SJ, Preti MG, van Bergen JMG, Kirchner T, Wyss M, … Unschuld PG (2019). GABA and glutamate moderate beta-amyloid related functional connectivity in cognitively unimpaired old-aged adults. Neuroimage Clin, 22, 101776. doi: 10.1016/j.nicl.2019.101776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammes G, Hasenjager A, Sroka-Saidi K, Deussing JM, & Parsons CG (2011). Therapeutic significance of NR2B-containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of beta-amyloid oligomers on long-term potentiation (LTP) in murine hippocampal slices. Neuropharmacology, 60(6), 982–990. doi: 10.1016/j.neuropharm.2011.01.051 [DOI] [PubMed] [Google Scholar]
- Raymond CR, Ireland DR, & Abraham WC (2003). NMDA receptor regulation by amyloid-beta does not account for its inhibition of LTP in rat hippocampus. Brain Res, 968(2), 263–272. doi: 10.1016/s0006-8993(03)02269-8 [DOI] [PubMed] [Google Scholar]
- Reed MN, Hofmeister JJ, Jungbauer L, Welzel AT, Yu C, Sherman MA, … Cleary JP (2011). Cognitive effects of cell-derived and synthetically derived Abeta oligomers. Neurobiol Aging, 32(10), 1784–1794. doi: 10.1016/j.neurobiolaging.2009.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders NR, Pao Y, Renner MC, da Silva-Matos CM, Lodder TR, Malinow R, & Kessels HW (2016). Amyloid-beta effects on synapses and memory require AMPA receptor subunit GluA3. Proc Natl Acad Sci U S A, 113(42), E6526–E6534. doi: 10.1073/pnas.1614249113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, & Triller A (2010). Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron, 66(5), 739–754. doi: 10.1016/j.neuron.2010.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazi K, Galic MA, Kentner AC, Reid AY, Sharkey KA, & Pittman QJ (2015). Microglia-dependent alteration of glutamatergic synaptic transmission and plasticity in the hippocampus during peripheral inflammation. J Neurosci, 35(12), 4942–4952. doi: 10.1523/JNEUROSCI.4485-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro FM, Vieira LB, Pires RG, Olmo RP, & Ferguson SS (2017). Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol Res, 115, 179–191. doi: 10.1016/j.phrs.2016.11.013 [DOI] [PubMed] [Google Scholar]
- Rissman RA, De Blas AL, & Armstrong DM (2007). GABA(A) receptors in aging and Alzheimer’s disease. J Neurochem, 103(4), 1285–1292. doi: 10.1111/j.1471-4159.2007.04832.x [DOI] [PubMed] [Google Scholar]
- Salgado-Puga K, Rodriguez-Colorado J, Prado-Alcala RA, & Pena-Ortega F (2017). Subclinical Doses of ATP-Sensitive Potassium Channel Modulators Prevent Alterations in Memory and Synaptic Plasticity Induced by Amyloid-beta. J Alzheimers Dis, 57(1), 205–226. doi: 10.3233/JAD-160543 [DOI] [PubMed] [Google Scholar]
- Schallier A, Smolders I, Van Dam D, Loyens E, De Deyn PP, Michotte A, … Massie A (2011). Region- and age-specific changes in glutamate transport in the AbetaPP23 mouse model for Alzheimer’s disease. J Alzheimers Dis, 24(2), 287–300. doi: 10.3233/JAD-2011-101005 [DOI] [PubMed] [Google Scholar]
- Schmid AW, Freir DB, & Herron CE (2008). Inhibition of LTP in vivo by beta-amyloid peptide in different conformational states. Brain Res, 1197, 135–142. doi: 10.1016/j.brainres.2007.11.056 [DOI] [PubMed] [Google Scholar]
- Scimemi A, Meabon JS, Woltjer RL, Sullivan JM, Diamond JS, & Cook DG (2013). Amyloid-beta1–42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J Neurosci, 33(12), 5312–5318. doi: 10.1523/JNEUROSCI.5274-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scopes DI, O’Hare E, Jeggo R, Whyment AD, Spanswick D, Kim EM, … Treherne JM (2012). Abeta oligomer toxicity inhibitor protects memory in models of synaptic toxicity. Br J Pharmacol, 167(2), 383–392. doi: 10.1111/j.1476-5381.2012.01973.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-McKean JJ, Surewicz K, Choi JK, Ruffin VA, Salameh AI, Nieznanski K, … Surewicz WK. (2016). Soluble prion protein and its N-terminal fragment prevent impairment of synaptic plasticity by Abeta oligomers: Implications for novel therapeutic strategy in Alzheimer’s disease. Neurobiol Dis, 91, 124–131. doi: 10.1016/j.nbd.2016.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott HA, Gebhardt FM, Mitrovic AD, Vandenberg RJ, & Dodd PR (2011). Glutamate transporter variants reduce glutamate uptake in Alzheimer’s disease. Neurobiol Aging, 32(3), 553 e551–511. doi: 10.1016/j.neurobiolaging.2010.03.008 [DOI] [PubMed] [Google Scholar]
- Sebollela A, Cline EN, Popova I, Luo K, Sun X, Ahn J, … Klein WL, (2017). A human scFv antibody that targets and neutralizes high molecular weight pathogenic amyloid-beta oligomers. J Neurochem, 142(6), 934–947. doi: 10.1111/jnc.14118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebollela A, Mustata GM, Luo K, Velasco PT, Viola KL, Cline EN, … Klein WL (2014). Elucidating molecular mass and shape of a neurotoxic Abeta oligomer. ACS Chem Neurosci, 5(12), 1238–1245. doi: 10.1021/cn500156r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2002). Alzheimer’s disease is a synaptic failure. Science, 298(5594), 789–791. doi: 10.1126/science.1074069 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, & Hardy J (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med, 8(6), 595–608. doi: 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selten M, van Bokhoven H, & Nadif Kasri N (2018). Inhibitory control of the excitatory/inhibitory balance in psychiatric disorders. [Review]. F1000Res, 7, 23. doi: 10.12688/f1000research.12155.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, … Sandrock A (2016). The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature, 537(7618), 50–56. doi: 10.1038/nature19323 [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, … Selkoe DJ (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med, 14(8), 837–842. doi: 10.1038/nm1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, & Walsh DM (2009). Alzheimer’s disease: synaptic dysfunction and Abeta. Mol Neurodegener, 4, 48. doi: 10.1186/1750-1326-4-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea D, Hsu CC, Bi TM, Paranjapye N, Childers MC, Cochran J, … Daggett V (2019). alpha-Sheet secondary structure in amyloid beta-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc Natl Acad Sci U S A, 116(18), 8895–8900. doi: 10.1073/pnas.1820585116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, & Strittmatter SM (2017). Binding Sites for Amyloid-beta Oligomers and Synaptic Toxicity. [Review]. Cold Spring Harb Perspect Med, 7(5). doi: 10.1101/cshperspect.a024075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares HD, Gasior M, Toyn JH, Wang JS, Hong Q, Berisha F, … AbuTarif M (2016). The gamma-Secretase Modulator, BMS-932481, Modulates Abeta Peptides in the Plasma and Cerebrospinal Fluid of Healthy Volunteers. [Randomized Controlled Trial]. J Pharmacol Exp Ther, 358(1), 138–150. doi: 10.1124/jpet.116.232256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs MJ, Cadwallader CJ, Hamm JP, Tippett LJ, & Kirk IJ (2017). Age-related alterations in human neocortical plasticity. Brain Res Bull, 130, 53–59. doi: 10.1016/j.brainresbull.2016.12.015 [DOI] [PubMed] [Google Scholar]
- Stohr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, … Giles K (2012). Purified and synthetic Alzheimer’s amyloid beta (Abeta) prions. Proc Natl Acad Sci U S A, 109(27), 11025–11030. doi: 10.1073/pnas.1206555109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoiljkovic M, Kelley C, Horvath TL, & Hajos M (2018). Neurophysiological signals as predictive translational biomarkers for Alzheimer’s disease treatment: effects of donepezil on neuronal network oscillations in TgF344-AD rats. Alzheimers Res Ther, 10(1), 105. doi: 10.1186/s13195-018-0433-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski G, Dabrowska-Bouta B, Kwiatkowska-Patzer B, & Struzynska L (2009). Alterations in glutamate transport and group I metabotropic glutamate receptors in the rat brain during acute phase of experimental autoimmune encephalomyelitis. Folia Neuropathol, 47(4), 329–337. [PubMed] [Google Scholar]
- Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, … Lipton SA (2013). Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A, 110(27), E2518–2527. doi: 10.1073/pnas.1306832110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima A, Suh YH, & Isaac JTR (2019). The AMPA Receptor Subunit GluA1 is Required for CA1 Hippocampal Long-Term Potentiation but is not Essential for Synaptic Transmission. Neurochem Res, 44(3), 549–561. doi: 10.1007/s11064-017-2425-3 [DOI] [PubMed] [Google Scholar]
- Terrill-Usery SE, Colvin BA, Davenport RE, & Nichols MR (2016). Abeta40 has a subtle effect on Abeta42 protofibril formation, but to a lesser degree than Abeta42 concentration, in Abeta42/Abeta40 mixtures. Arch Biochem Biophys, 597, 1–11. doi: 10.1016/j.abb.2016.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong H, Zhang X, Meng X, Xu P, Zou X, & Qu S (2017). Amyloid-beta peptide decreases expression and function of glutamate transporters in nervous system cells. Int J Biochem Cell Biol, 85, 75–84. doi: 10.1016/j.biocel.2017.01.017 [DOI] [PubMed] [Google Scholar]
- Ulrich D (2015). Amyloid-beta Impairs Synaptic Inhibition via GABA(A) Receptor Endocytosis. J Neurosci, 35(24), 9205–9210. doi: 10.1523/JNEUROSCI.0950-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, … Strittmatter SM (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron, 79(5), 887–902. doi: 10.1016/j.neuron.2013.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Diggelen F, Hrle D, Apetri M, Christiansen G, Rammes G, Tepper A, & Otzen DE (2019). Two conformationally distinct alpha-synuclein oligomers share common epitopes and the ability to impair long-term potentiation. PLoS One, 14(3), e0213663. doi: 10.1371/journal.pone.0213663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga E, Juhasz G, Bozso Z, Penke B, Fulop L, & Szegedi V (2015). Amyloid-beta1–42 Disrupts Synaptic Plasticity by Altering Glutamate Recycling at the Synapse. J Alzheimers Dis, 45(2), 449–456. doi: 10.3233/JAD-142367 [DOI] [PubMed] [Google Scholar]
- Velasco PT, Heffern MC, Sebollela A, Popova IA, Lacor PN, Lee KB, … Klein WL (2012). Synapse-binding subpopulations of Abeta oligomers sensitive to peptide assembly blockers and scFv antibodies. ACS Chem Neurosci, 3(11), 972–981. doi: 10.1021/cn300122k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Mishra V, Sasmal D, & Raghubir R (2010). Pharmacological evaluation of glutamate transporter 1 (GLT-1) mediated neuroprotection following cerebral ischemia/reperfusion injury. Eur J Pharmacol, 638(1–3), 65–71. doi: 10.1016/j.ejphar.2010.04.021 [DOI] [PubMed] [Google Scholar]
- Vico Varela E, Etter G, & Williams S (2019). Excitatory-inhibitory imbalance in Alzheimer’s disease and therapeutic significance. Neurobiol Dis, 127, 605–615. doi: 10.1016/j.nbd.2019.04.010 [DOI] [PubMed] [Google Scholar]
- Villette V, Poindessous-Jazat F, Bellessort B, Roullot E, Peterschmitt Y, Epelbaum J, … Dutar P (2012). A new neuronal target for beta-amyloid peptide in the rat hippocampus. Neurobiol Aging, 33(6), 1126 e1121–1114. doi: 10.1016/j.neurobiolaging.2011.11.024 [DOI] [PubMed] [Google Scholar]
- Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, … Nagarajan SS (2016). Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann Neurol, 80(6), 858–870. doi: 10.1002/ana.24794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, & Miller BL (2017). Epileptic activity in Alzheimer’s disease: causes and clinical relevance. Lancet Neurol, 16(4), 311–322. doi: 10.1016/S1474-4422(17)30044-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, … Selkoe DJ (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature, 416(6880), 535–539. doi: 10.1038/416535a [DOI] [PubMed] [Google Scholar]
- Walsh DM, & Selkoe DJ (2007). A beta oligomers - a decade of discovery. J Neurochem, 101(5), 1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x [DOI] [PubMed] [Google Scholar]
- Wang Z, Jackson RJ, Hong W, Taylor WM, Corbett GT, Moreno A, … Walsh DM (2017). Human Brain-Derived Abeta Oligomers Bind to Synapses and Disrupt Synaptic Activity in a Manner That Requires APP. J Neurosci, 37(49), 11947–11966. doi: 10.1523/JNEUROSCI.2009-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welzel AT, Maggio JE, Shankar GM, Walker DE, Ostaszewski BL, Li S, … Selkoe DJ (2014). Secreted amyloid beta-proteins in a cell culture model include N-terminally extended peptides that impair synaptic plasticity. Biochemistry, 53(24), 3908–3921. doi: 10.1021/bi5003053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcomb DJ, Hogg EL, Regan P, Piers T, Narayan P, Whitehead G, … Cho K (2015). Intracellular oligomeric amyloid-beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci Rep, 5, 10934. doi: 10.1038/srep10934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegert JS, & Oertner TG (2013). Long-term depression triggers the selective elimination of weakly integrated synapses. Proc Natl Acad Sci U S A, 110(47), E4510–4519. doi: 10.1073/pnas.1315926110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, Walter S, Kraus I, Klafki HW, Stazi M, Oberstein TJ, … Weggen S (2017). N-truncated Abeta4-x peptides in sporadic Alzheimer’s disease cases and transgenic Alzheimer mouse models. Alzheimers Res Ther, 9(1), 80. doi: 10.1186/s13195-017-0309-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woltjer RL, Duerson K, Fullmer JM, Mookherjee P, Ryan AM, Montine TJ, … Cook DG (2010). Aberrant detergent-insoluble excitatory amino acid transporter 2 accumulates in Alzheimer disease. J Neuropathol Exp Neurol, 69(7), 667–676. doi: 10.1097/NEN.0b013e3181e24adb [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Guo Z, Gearing M, & Chen G (2014). Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat Commun, 5, 4159. doi: 10.1038/ncomms5159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Rajsombath MM, Weikop P, & Selkoe DJ (2018). Enriched environment enhances beta-adrenergic signaling to prevent microglia inflammation by amyloid-beta. EMBO Mol Med, 10(9). doi: 10.15252/emmm.201808931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Li S, Xu H, Walsh DM, & Selkoe DJ (2017). Large Soluble Oligomers of Amyloid beta-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J Neurosci, 37(1), 152–163. doi: 10.1523/JNEUROSCI.1698-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, O’Malley TT, Kanmert D, Jerecic J, Zieske LR, Zetterberg H, … Selkoe DJ (2015). A highly sensitive novel immunoassay specifically detects low levels of soluble Abeta oligomers in human cerebrospinal fluid. Alzheimers Res Ther, 7(1), 14. doi: 10.1186/s13195-015-0100-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitsev AV, Kim K, Vasilev DS, Lukomskaya NY, Lavrentyeva VV, Tumanova NL, … Magazanik LG (2015). N-methyl-D-aspartate receptor channel blockers prevent pentylenetetrazole-induced convulsions and morphological changes in rat brain neurons. J Neurosci Res, 93(3), 454–465. doi: 10.1002/jnr.23500 [DOI] [PubMed] [Google Scholar]
- Zhang D, Mably AJ, Walsh DM, & Rowan MJ (2017). Peripheral Interventions Enhancing Brain Glutamate Homeostasis Relieve Amyloid beta- and TNFalpha- Mediated Synaptic Plasticity Disruption in the Rat Hippocampus. Cereb Cortex, 27(7), 3724–3735. doi: 10.1093/cercor/bhw193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Qi Y, Klyubin I, Ondrejcak T, Sarell CJ, Cuello AC, … Rowan MJ (2017). Targeting glutamatergic and cellular prion protein mechanisms of amyloid beta-mediated persistent synaptic plasticity disruption: Longitudinal studies. Neuropharmacology, 121, 231–246. doi: 10.1016/j.neuropharm.2017.03.036 [DOI] [PubMed] [Google Scholar]
- Zhao J, Li A, Rajsombath M, Dang Y, Selkoe DJ, & Li S (2018). Soluble Abeta Oligomers Impair Dipolar Heterodendritic Plasticity by Activation of mGluR in the Hippocampal CA1 Region. iScience, 6, 138–150. doi: 10.1016/j.isci.2018.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zott B, Simon MM, Hong W, Unger F, Chen-Engerer HJ, Frosch MP, … Konnerth A (2019). A vicious cycle of beta amyloid-dependent neuronal hyperactivation. Science, 365(6453), 559–565. doi: 10.1126/science.aay0198 [DOI] [PMC free article] [PubMed] [Google Scholar]