

Abstract
Over activity of Glycogen synthase kinase-3β (GSK-3β), a serine/threonine-protein kinase has been implicated in a number of diseases including stroke, type II diabetes and Alzheimer disease (AD). This study aimed to find novel inhibitors of GSK-3β from phyto-constituents of Melissa officinalis with the aid of computational analysis. Molecular docking, induced-fit docking (IFD), calculation of binding free energy via the MM-GBSA approach and Lipinski’s rule of five (RO5) were employed to filter the compounds and determine their druggability. Most importantly, the compounds pIC50 were predicted by machine learning-based model generated by AutoQSAR algorithm. The generated model was validated to affirm its predictive model. The best model obtained was Model kpls_desc_38 (R2 = 0.8467 and Q2 = 0.8069), and this external validated model was utilized to predict the bioactivities of the lead compounds. While a number of characterized compounds from Melissa officinalis showed better docking score, binding free energy alongside adherence to RO5 than co-cystallized ligand, only three compounds (salvianolic acid C, ellagic acid and naringenin) showed more satisfactory pIC50. The results obtained in this study can be useful to design potent inhibitors of GSK-3β.
Keywords: Melissa officinalis, Glycogen synthase kinase-3β, AutoQSAR, MM-GBSA, Induced-fit docking (IFD)
Introduction
Glycogen synthase kinase-3β (GSK-3β) is a serine/threonine-protein kinase, primarily located in cytosol demonstrates important roles in different disease molecular pathophysiology (Mancinelli et al. 2017). Owing to its role in type 2 diabetes and obesity as determined by in vitro and in vivo studies, GSK-3β has gained popularity as a possible drug target (Gum et al. 2003; Ring et al. 2003). It’s also related to Alzheimer's disease (AD) and mood disorders (Hsiung et al. 2003), osteoporosis (Smith and Frenkel 2005), arteriosclerosis (Robertson et al. 2006), and cancer (Inoki et al. 2006). Unlike other protein kinases, GSK-3β under normal conditions is constitutively active and undergoes a rapid and temporary inhibition in response to a variety of external signals (Dorm 2005).
GSK-3β has been explored as a therapeutic target for a range of human diseases including cancer due to its diverse cellular functions (Yuki and Chikashi 2015). It is thus regarded as an obvious target for disease drug development, including neurodegenerative diseases such as Alzheimer's diseases, diabetes mellitus, and cancer (Takahashi and Sasaguri 2009; Gao et al. 2013). This target is unique in that it is constitutively active in cells and its inhibition is liable for cell signalling (Inoki et al. 2006). GSK-3β plays significant roles in numerous signalling pathways that regulate a variety of cellular processes (Xu et al. 2009; Cheng et al. 2011).
Melissa officinalis L. also known as lemon balm, a perennial herb in the family Lamiaceae (Fig. 1) occurs naturally in the Mediterranean and West Asia but is widely cultivated in Europe and North America (Moradkhani et al. 2010). Its leaf contains several phyto-compounds, such as flavonoids, polyphenolic compounds, monoterpenoid aldehyde, tannins, monoterpene glycosides, triterpenesesquiterpenes and essential oils (Sofowora et al. 2013). The usage of M. Officinalis as a supplement ingredient and functional food has increased over time due to its many medicinal properties including sedative, carminative and antispasmodic effects (Ożarowski et al. 2016). Lemon balm leaf, plant, and essential oil are used in herbal medicine (Senderski 2009).
Fig. 1.

Aerial parts of Melissa officnialis (Zarei et al. 2015)
A number of researches have been geared towards the discovery and design of selective GSK-3β inhibitors. Some identified GSK-3 inhibitors include small molecules isolated from organic and aquatic sources or obtained from chemical synthesis. They can function through numerous mechanisms, including competitive or non-competitive ATP inhibition. Attempts have been made to evolve and develop novel GSK-3β inhibitors in academia and industry (Xie et al. 2017). Many chemical families are known to emerge as GSK-3 inhibitors, with great structural diversity. Therefore GSK-3 is regarded as an ideal target for new drug discovery.
Molecular docking is a very fitting and low-cost method to understand the reaction mechanism of proteins or enzymes with ligands with high accuracy for rational drug design and discovery by analyzing the conformation and orientation of molecules into a molecular target binding site (Liu et al. 2018; Kitchen et al. 2004). As a result of the development of the first algorithms in the 1980s, molecular docking is now an important tool in drug discovery (Meng et al. 2011), and the most frequently used technique to predict the binding orientation of potential drugs against protein targets (Kapetanovic 2008). Hence, this study aimed at the use of molecular docking to predict the most potent Glycogen synthase 3-beta (GSK-3β) inhibitors with drug-like properties from M. officinalis.
Materials and methods
Preparation of crystal protein
The crystal structure of GSK-3β (PDB ID-1UV5) was downloaded via the protein preparation wizard of maestro v11.8. The protocol described in our previous studies was used to prepare crystal structure of the protein (Iwaloye et al. 2020a, b). The protein was preprocessed by creating zero bonds to metals, deleting waters from 5.0 Å of het groups, adjusting bond orders and setting the het states at pH 7.0 ± 2.0 (Schrödinger Suite 2012). The protein was refined by optimizing the H-bond network using PROPKA and removing water molecules with less than 3 H-bonds to non-waters (Olsson et al. 2011). The retrained minimization was carried out using the OPLS3 force field to avoid steric clashes that may exist in the structure. The minimization was terminated while the RMSD of non-hydrogen atoms reached 0.30 Å.
Preparation of the phyto-compounds
The library of compounds was built by drawing characterized compounds of Melissa officinalis using Marvinsketch (version 19.26) as documented in a different literature review (Triantaphyllou et al. 2001; Patora and Klimek 2002; Heitz et al. 2000; Tagashira and Ohtake 1998). The compounds were prepared using Ligprep. The Ligprep panel enables the conversion of structures; generate variations of structures and elimination of unwanted structures. After stereoisomer computation was left to generate at most 32 per ligand and the output format was left as maestro, the OPLS3 force field was left at pH 7.0 ± 2.0 using epik (Schrödinger Suite 2012).
Receptor grid generation
The receptor grid file was generated using a receptor grid generation panel, which represents the active sites of the receptor for glide ligand docking jobs. The ligand-binding site was defined by picking the co-crystallized ligand of the protein structure on the workspace. The van der Waals radii of the receptor atoms with partial atomic charge was set scaling factor of 1.0 and partial cutoff of 0.25 to soften the potential for non-polar parts of the receptor. The receptor grid box resolution was centered at coordinates 93.91, 68.0 and 9.8 in respect to x, y, and z-axis.
Glide extra precision docking
The prepared library of compounds was docked into the active site of the protein crystal using extra precision with the ligand sampling set generated as flexible. The choice of the best-docked structure for each ligand was made using model energy score (emodel) that combines glide score, the non-bonded interaction energy and the excess internal energy of the generated ligand conformation.
Induced fit docking (flexible docking)
To accurately predict the binding affinity of the novel inhibitors to the prepared protein crystal, Induced fit docking (IFD) was implemented. IFD is an in silico approach that uses Glide and the Refinement module in Prime that accurately predicts ligand binding modes and concomitant structural changes in the receptor (Sherman et al. 2006).
Calculation of binding free energy
The Prime MM-GBSA panel was used to calculate binding free energy for the ligand-receptor complex using the MM-GBSA technology available with Prime (2018). MMGBSA quantifies the difference in energy between the free and the complex state of both the ligand and the protein after energy minimization. In the prime MM-GBA panel, the OPLS3 force field was selected and VSGB was used as the continuum solvent model. Other options were set to default.
The equations for calculating binding energy are as follows.
| 1 |
| 2 |
where Ecomplex, Eprotein, and Eligand indicate the minimized energies for protein-inhibitor complex, protein, and inhibitor, respectively.
| 3 |
| 4 |
where ∆GSA is the non-polar contribution to the solvation energy due to the surface area. GSA (complex), GSA (protein) and GSA (ligand) are the surface energies of complex, protein and ligand respectively.
Drug-likeness test of the phytochemicals
Lipinski's rule of five was used to determine the drug-likeness of the compounds and this parameter was predicted by Canvas (Duan et al. 2010a, b).
Validation of molecular docking results
The protocol for docking in this study was validated by docking the prepared inhibitors of GSK-3β downloaded from database server of the CHEMBL. Extra precision (XP) docking score of selected compounds were plot against their pChEMBL value to obtain r2 spearman correlation. The plotted graph is illustrated in Fig. 2. The docking protocol was further validated by docking native ligand (co-crystal ligand) with the prepared crystal structure of GSK-3β to determine the root mean square deviation (RMSD). A RMSD value of 0.39 Å (Fig. 3) showed the docking procedure is reproducible (Elekofehinti et al. 2018).
Fig. 2.
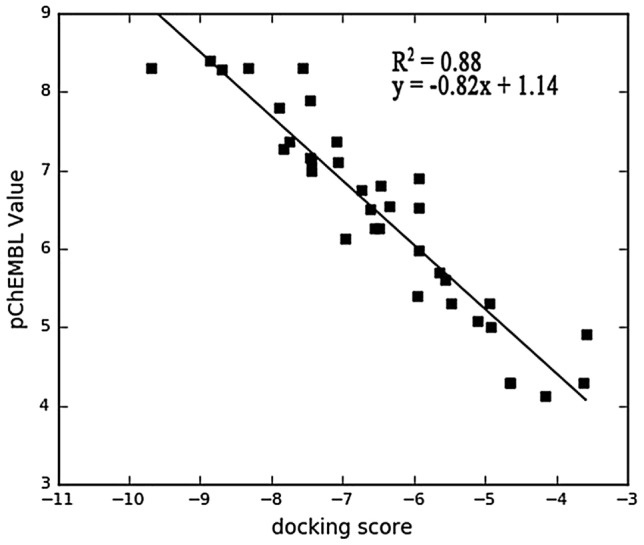
The correlation coefficient graph between the experimentally determined pIC50 of GS3K-3β their docked score with R2 of 0.88 indicating that the docking experiment can replicate the experimentally determined values of the inhibitors
Fig. 3.
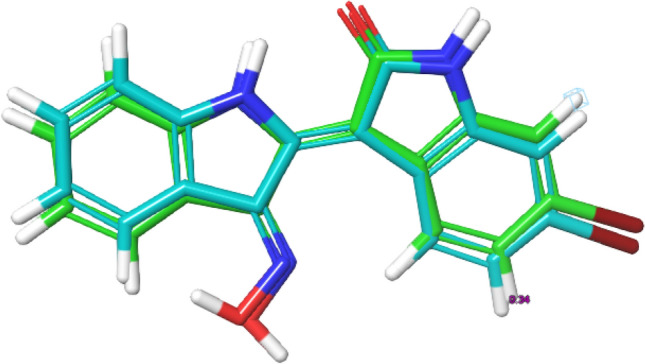
Superposition of the co-crystal ligand with its docked pose
Machine learning principles using automated QSAR
Generation and preparation of dataset
The experimental dataset containing GSK-3β inhibitors were retrieved from CHEMBL database online server (www.ebi.ac.uk/chembl/), by blasting the FASTA sequence of the GSK-3β with online sever (CHEMBL262). Bioactivities of 116 inhibitors of GSK-3β were retrieved with their respective pIC50 were compiled from literatures Olesen et al. (2003), Terrence et al. (2006), Saitoh et al. (2009), Luo et al. (2016) and Cociova et al. (2017). Compounds without pIC50 were deleted from excel sheet before conversion to sdf (structure data file) format using data-warrior package (v.2) (Sander et al. 2005). The sdf format was exported to the workspace of maestro for preparation by ligprep (Schrödinger Suite 2012). The prepared compounds were eventually exported to Canvas cheminformatics (Duan et al. 2010a, b). Canvas clusters the inhibitors based on their Tanimoto similarity between sets of Hashed linear binary fingerprint descriptors, to determine the structural diversity among the inhibitor, and to select representatives from each resulting cluster. This computational study generated a total of 49 clusters and one representative was selected from each cluster to develop our QSAR model.
Principle of autoQSAR
AutoQSAR is a machine-learning algorithm provided by Schrödinger suite that builds and applies QSAR models through automation (Dixon et al. 2016). In order to build a predictive model, AutoQSAR takes the 1D, 2D and 3D structural data of a molecule along with a property (eg: IC50) to be modeled, as an input. It will then compute the fingerprints and descriptors using machine-learning statistical methods for creating a predictive QSAR model. The predictive accuracy of the model is evaluated using various parameters such as ranking score, Root Mean Square Error (RMSE), Standard Deviation (SD), Q2 and R2 values (de Oliveira and Katekawa 2017).
Validation methods for QSAR models
External validation
The predictive power of a QSAR model can be estimated by the following statistical characteristics of the test set which was recommended by Golbraikh and Tropsha (2002):
-
i.
Correlation coefficient R between the predicted and observed activities
-
ii.
Coefficients of determination (R2) (predicted vs. observed activities , and observed vs. predicted activities ) (Sachs 1984).
-
iii.
Slopes k and k′ of the regression lines through the origin.
A model is considered robust if it meets the following criteria (Golbraikh and Tropsha 2002):
Results and discussion
Molecular docking studies
In the context of therapeutic application, GSK-3β has become an interesting drug target due to its exclusive and central role in the pathogenesis of varieties of disease. Several studies have documented that over-expression of Glycogen synthase kinase-3β accounts for memory impairment/increased β-amyloid production, diabetic type II, stroke, cancer and chronic inflammatory disease (Eldar-Finkelman 2002; Beurel 2011; Liu 2014).
An extensive literature survey was done to identify and select phyto-compounds against GSk-3β based on their medicinal properties for drug designing using molecular docking studies. Melissa officinalis became the ideal plant due to its medicinal strength (Kamdem et al. 2013; Ammon et al. 2006) and the small molecular weight of its photo-constituents. The present study utilized glide XP docking and induced fit docking to painstakingly and accurately predict the binding affinity and docking score of the compounds with GSk-3β, thereby denoting compounds with favourable interaction. Table 1 presented the molecular docking results and the interacting residues of the respective ligand–protein complex. The docking score of the hit compounds ranged from − 7.289 to − 17.284 kcal/mol. Luteoin 3′-O-β-d-glucoronopyranoside(VI) attained the highest binding affinity with a score of − 17.284 kcal/mol. The next ranked compounds are luteoin 7-O-β-d-glucoronopyranoside(V), chlorogenic acid and salvianolic acid F with docking scores of − 17.199 kcal/mol, − 15.650 kcal/mol and − 14.285 kcal/mol respectively. However, the co-crystallized, our ligand choice of comparison attained the least docking score (− 4.638 kcal/mol). This suggests that the hit compounds derived from Melissa officnialis are promising agents as GSk-3β inhibitors. Induced fit docking (IFD) offered a more accurate prediction of binding affinity by allowing the protein to undergo rotation on binding to a ligand. The results of the IFD score of the hit compounds did not follow the same trend with the docking score. While luteoin 3′-O-b-d-glucoronopyranoside(VI) still retained the most favourable interaction with the IFD score of − 716.889 kcal/mol, Naringenin and apigenin have more favourable interaction than the other compounds by recording IFD score of − 716.819 kcal/mol and − 715.304 kcal/mol respectively.
Table 1.
Molecular docking results and Interacting residues of the respective ligand–protein complex
| Compounds | Docking score (kcal/mol) | Induced fit docking score (kcal/mol) | H-bond | H-bonding distance (Å) | Predicted pIC50 |
|---|---|---|---|---|---|
| Luteoin 3′-O-β-d-glucoronopyranoside(VI) | − 17.284 | − 716.889 | VAL135, ASN64, 2[LYS183], GLY68 |
2.50, 1.68 1.78, 2.91 2.72 |
6.452 |
| Luteoin 7-O-β-d-glucoronopyranoside(V) | − 17.199 | − 713.219 |
PRO136, ILE62 2[LYS183] ASN64, ASN186 |
2.21, 2.02 4.42, 4.51 1.95, 1.75 |
6.415 |
| Chlorogenic acid | − 15.650 | − 709.342 |
ASP200, LYS183, ASN64 2[VAL135] |
2.02, 4.15, 2.29 1.82, 2.28 |
6.643 |
| Salvianolic acid F | − 14.285 | − 708.102 | PHE67, VAL135 | 1.77, 2.22 | 6.453 |
| Salvianolic acid C | − 14.140 | − 711.207 |
2[LYS183], ASN186, 2[VAL135] ARG141 |
1.66, 3.76, 2.23 2.01, 2.09 2.41 |
7.770 |
| Salvianolic acid A | − 13.213 | − 709.096 | ILE62, PRO136 | 2.09, 2.16 | 6.744 |
| Melitric acid A | − 12.445 | − 710.219 |
VAL135, GLN185 2[LYS183] 2[LYS85], ASN64 |
2.08, 2.99, 3.64 2.02, 2.65 4.85 |
6.427 |
| Yunnaneic acid | − 12.173 | − 709.805 |
ASN64, LYS183 2[GLN185] LYS85 |
1.77, 2.95 1.95, 1.86 3.22 |
6.190 |
| Ellagic acid | − 10.439 | − 705.418 | LYS85, VAL135 | 2.27, 1.83 | 8.169 |
| Naringenin | − 8.182 | − 716.819 | ASP133, PRO136 | 1.99, 1.93 | 7.850 |
| Decadienal | − 7.289 | − 705.512 | VAL135 | 1.94 | 4.178 |
| Rosmarinic acid | − 9.465 | − 712.618 | ASN64, VAL135, ASP133 | 2.92, 2.61, 2.22 | 6.855 |
| Apigenin | − 9.671 | − 715.304 | ASP133, PRO136 | 1.88, 1.99 | 6.811 |
| Co-cyrstallized ligand | − 4.638 | − 704.432 | ASN64, LYS183, ASN186, GLN185 | 2.24, 5.38, 1.80, 1.84 | 7.487 |
Hydrogen bonding interaction
The binding site of GSk-3β is known to contain a group of polar residues such as LYS85, ASP200, and GLU51 that play a leading role in the ligand-ATP recognition; ASP200 specifically interacts with the phosphate group of ATP (De Bondt et al. 1993). The interacting residues of the protein with lead compounds were listed in Table 1. Our results revealed that few of the inhibitors were involved in hydrogen bond interaction with LYS85 and ASP200. Yunnaneic acid, ellagic acid, and melitric acid A formed hydrogen interaction with LYS85. Consequently, chlorogenic acid appeared to form interaction with ASP200. A similar interaction with potential inhibitors was also reported by Padavala et al. (2010). Several studies have recognized VAL135 and ASP133 as key residues for H-bond interaction with a diverse range of GSk-3β inhibitors (Smith et al. 2001; Padavala et al. 2010), in addition to GLN185, LYS183, ILE62, ASN186 and ARG141 (Witherington et al. 2003; Bertrand et al. 2003; Buescher and Phiel 2010). Interestingly all the lead compounds showed interaction with most of these amino acid residues. Figures 4, 5, 6 depicted the docking conformation of the compounds with the three most favorable interactions with GSk-3β. The residues present in the active site of GSk-3β interacted with luteoin 3′-O-β-d-glucoronopyranoside(VI) were VAL135, ASN64, 2[LYS183], and GLY68, with the hydrogen bonding distance between the compound and GSk-3β were found to be 2.50 Å, 1.68 Å, 1.78 Å, 2.91 Å and 2.72 Å. Naringenin showed interaction with ASP133 and PRO136 using the hydroxyl group attached to its phenyl rings. The hydrogen-bonding distance between naringenin and GSk-3β is estimated to be 1.99 Å and 1.93 Å.
Fig. 4.

Binding pose of luteoin 3′-O-β-d-glucoronopyranoside(VI) with GS3K-3β revealing interacting amino acid residues within the active site of GS3K-3β in 2D
Fig. 5.

Binding pose of Naringenin with GS3K-3β revealing interacting amino acid residues within the active site of GS3K-3β in 2D
Fig. 6.

Binding pose of Apigenin with GS3K-3β revealing interacting amino acid residues within the active site of GS3K-3β in 2D
Binding energy assessment
To validate the docking scores, the free energy of binding was calculated via the MMGBSA post docking program, which predicts binding free energies for compounds/ligands by utilizing the combination of molecular mechanics calculations and salvation models. It has been demonstrated in many studies that the MMGBSA post docking method is the most reliable for rating the affinity of a ligand on binding to its protein target (Maffucci et al. 2018; Sun et al. 2014) since results obtained through MMGBSA for binding energies calculations were found to be highly reproducible (Genheden and Ryde 2015). The accuracy of the docking was affirmed by examining the lowest energy poses predicted by the scoring function. The evaluation of binding free energy is listed in Table 2. Interestingly, compounds with the good docking score showed favorable binding energy. In terms of binding free energy, the major energy contributors were identified as van der Waals (∆Gvdw), Coulomb interaction (∆GColulomb), Hydrogen bond (ΔGHbond) and lipophilic energy (∆GsolLipo) that enhance the binding affinity of the compounds towards the binding pocket of the protein.
Table 2.
Calculation of binding free energy
| Compounds | ΔGBinda | ΔGBindbcoulomb | ΔGBindcvdw | ΔGBinddlipo | ΔGBindeHbond |
|---|---|---|---|---|---|
| Luteoin 3′-O-β-d-glucoronopyranoside(VI) | − 36.58 | − 169.83 | − 28.09 | − 12.62 | − 3.64 |
| Luteoin 7-O-β-d-glucoronopyranoside(V) | − 55.08 | − 193.71 | − 46.00 | − 14.57 | − 3.26 |
| Chlorogenic acid | − 59.06 | − 184.44 | − 21.64 | − 14.12 | − 4.40 |
| Salvianolic acid F | − 42.75 | − 156.21 | − 30.74 | − 15.54 | − 2.19 |
| Salvianolic acid C | − 40.79 | − 317.24 | − 48.20 | − 14.76 | − 3.57 |
| Salvianolic acid A | − 58.63 | − 181.40 | − 42.68 | − 13.74 | − 2.12 |
| Melitric acid A | − 31.41 | − 157.46 | − 46.40 | − 14.60 | − 3.97 |
| Yunnaneic acid | − 38.46 | − 209.02 | − 47.16 | − 14.92 | − 2.78 |
| Ellagic acid | − 51.04 | − 23.18 | − 35.39 | − 15.55 | − 2.45 |
| Naringenin | − 45.00 | − 26.40 | − 37.05 | − 9.82 | − 1.12 |
| Decadienal | − 37.36 | − 11.50 | − 20.55 | − 16.15 | − 0.97 |
| Rosmarinic acid | − 7.04 | − 107.38 | − 32.52 | − 16.55 | − 1.91 |
| Apigenin | − 44.98 | − 26.40 | − 37.05 | − 9.82 | − 1.12 |
| Co-cyrstallized Ligand | − 31.11 | − 66.33 | − 31.46 | − 11.73 | − 1.72 |
aMM-GBSA free energy (kcal/mol) of binding
bContribution to the MM-GBSA free energy of binding (kcal/mol) from the Coulomb energy
cContribution to the MMGBSA free energy of binding (kcal/mol) from the van der Waals energy
dContribution to the MM-GBSA free energy of binding (kcal/mol) from lipophilic binding
eContribution to the MM-GBSA free energy of binding (kcal/mol) from hydrogen bonding
ADME studies
The predicted ADME properties (Table 3) include a number of rotatable bonds, the molecular weight of the molecule, number of hydrogen bond acceptors, prediction of binding to human serum albumin, number of hydrogen bond donors, predicted octanol–water partition coefficient and number of violations of Lipinski's rule of five (RO5). Lipinski's RO5 helps to evaluate the drug-likeness, and determine the prospect of small molecules in becoming an orally active drug for humans. The rule permits a molecular weight < 500 Da, octanol–water partition coefficient < 5, hydrogen bond donor ≤ 5 and hydrogen bond acceptor ≤ 10 (Lipinski et al. 2001). Prospective drug candidate that obey the Ro5 tend to have lower attrition rates at the stage of clinical trials and for this reason, it has an increased chance of becoming and staying marketable (Gombar et al. 2003). Compounds of Melissa officinalis have shown excellent results and are in accordance with this rule. In view of this, they can be developed as a promising lead in the design of GSk-3β inhibitors.
Table 3.
Prediction of ADME properties
| Compounds name | aROF | bMW | cAlogP | dHBA | eHBD | fRB | gPSA |
|---|---|---|---|---|---|---|---|
| Luteoin 3′-O-β-d-glucoronopyranoside(VI) | 0 | 460.344 | − 0.359 | 9 | 5 | 4 | 213.010 |
| Luteoin 7-O-β-d-glucoronopyranoside(V) | 1 | 461.353 | 0.318 | 10 | 6 | 4 | 210.180 |
| Chlorogenic acid | 0 | 353.301 | − 1.200 | 7 | 5 | 5 | 167.580 |
| Salvianolic acid F | 0 | 329.324 | 1.172 | 4 | 4 | 5 | 121.050 |
| Salvianolic acid C | 0 | 476.389 | 2.552 | 7 | 4 | 7 | 183.550 |
| Salvianolic acid A | 0 | 493.439 | 2.704 | 7 | 5 | 9 | 190.640 |
| Melitric acid A | 1 | 536.441 | 2.187 | 8 | 5 | 11 | 216.940 |
| Yunnaneic acid | 0 | 359.307 | 1.795 | 6 | 4 | 7 | 147.350 |
| Ellagic acid | 0 | 301.185 | 0.744 | 8 | 4 | 0 | 141.340 |
| Naringenin | 0 | 272.253 | 1.620 | 4 | 2 | 1 | 89.820 |
| Decadienal | 0 | 150.218 | 3.243 | 1 | 1 | 1 | 20.230 |
| rosmarinic acid | 0 | 359.307 | 1.795 | 6 | 4 | 7 | 147.350 |
| Apigenin | 0 | 271.245 | 1.620 | 4 | 2 | 1 | 89.820 |
| Co-cyrstallized ligand | 0 | 376.332 | − 1.167 | 2 | 4 | 1 | 85.710 |
aLipinski rule of five
bMolecular weight
cPredictedoctanol/water partition coefficient
dHydrogen bond acceptor
eHydrogen bon donor
fRotatable bond
gPolar surface area
Automated QSAR analysis
The screening process further continued with the aid of machine learning-based predictive model (pIC50 Calculation) generated by AutoQSAR panel of Schrodinger. Given a learning set of chemical structures and an activity property from CHEMBL database, a total of 497 physicochemical and topological descriptors are computed, together with a variety of Canvas fingerprints (Dixon et al. 2016), giving out a large pool of independent variables from which to build models. The automated module split the dataset randomly into 80% training set, and 20% test set. Models are built on each training set from all possible combination of machine learning method, and sets of independent variables that are supported by each machine learning methods. The observed activities and predicted activities of training set and test set in negative logarithm of inhibitor concentration(pIC50) was represented in Table 4. The algorithm generated 10 best models and the results of the top 5 models are shown in Table 5. The best model Model kpls_desc_38 recorded a standard deviation (S.D) of 0.5505, R2 of 0.8467, root mean square error (RMSE) 0.5366 and Q2 of 0.8069. The best model was computed from kernel based partial least square regression (KPLS), which supports the use of descriptors and fingerprints as independent variables, and fingerprint desc_38, a model code generated from a combination of the machine learning method (MLM). The scatter plot depicting predicted pIC50 versus experimental pIC50 for best generated model is shown in Fig. 7. The Predicted pIC50 using the best model for the data set of the lead compounds and co-ligand are tabulated in Table 1. It is worth noting that three of the compounds are observed to have better predicted pIC50 than co-ligand.
Table 4.
Details of AutoQSAR predicted activities compared with the observed activities
| S/n | CHEMBL CID | Set | Observed pIC50 | Predicted pIC50 | Residue error |
|---|---|---|---|---|---|
| 1 | CHEMBL68397 | Train | 7.4000 | 6.6281 | − 0.7719 |
| 2 | CHEMBL67799 | Train | 7.8000 | 7.6181 | − 0.1819 |
| 3 | CHEMBL76427 | Train | 4.7500 | 4.3437 | − 0.4063 |
| 4 | CHEMBL76427 | Train | 4.7500 | 4.3437 | − 0.4063 |
| 5 | CHEMBL250714 | Train | 6.0600 | 5.9896 | − 0.0704 |
| 6 | CHEMBL109944 | Train | 7.3500 | 7.7096 | 0.3596 |
| 7 | CHEMBL111620 | Train | 7.5800 | 7.5083 | − 0.0717 |
| 8 | CHEMBL112564 | Train | 5.1600 | 5.9676 | 0.8076 |
| 9 | CHEMBL1957099 | Train | 7.2000 | 6.2389 | − 0.9611 |
| 10 | CHEMBL1957078 | Train | 6.5000 | 6.1836 | − 0.3164 |
| 11 | CHEMBL1957942 | Train | 5.5500 | 6.0766 | 0.5266 |
| 12 | CHEMBL326208 | Train | 7.8500 | 7.6141 | − 0.2359 |
| 13 | CHEMBL497398 | Train | 4.6000 | 5.0189 | 0.4189 |
| 14 | CHEMBL188938 | Train | 8.3000 | 7.3258 | − 0.9742 |
| 15 | CHEMBL576540 | Train | 7.0900 | 7.2598 | 0.1698 |
| 16 | CHEMBL183171 | Train | 6.2600 | 6.6905 | 0.4305 |
| 17 | CHEMBL1834116 | Train | 5.4700 | 5.6292 | 0.1592 |
| 18 | CHEMBL1834122 | Train | 4.7700 | 4.8932 | 0.1232 |
| 19 | CHEMBL1957096 | Train | 4.5000 | 5.5342 | 1.0342 |
| 20 | CHEMBL1957089 | Test | 5.6000 | 5.9919 | 0.3919 |
| 21 | CHEMBL1940983 | Train | 6.7500 | 7.0294 | 0.2794 |
| 22 | CHEMBL1801622 | Train | 5.8200 | 6.2720 | 0.4520 |
| 23 | CHEMBL1801637 | Train | 8.3000 | 8.5850 | 0.2850 |
| 24 | CHEMBL399078 | Test | 5.3400 | 6.1300 | 0.7900 |
| 25 | CHEMBL398670 | Train | 6.0400 | 5.5198 | − 0.5202 |
| 26 | CHEMBL249141 | Train | 5.4500 | 5.6634 | 0.2134 |
| 27 | CHEMBL426587 | Train | 5.3200 | 5.8617 | 0.5417 |
| 28 | CHEMBL461139 | Train | 6.5200 | 6.6372 | 0.1172 |
| 29 | CHEMBL1801625 | Train | 5.7000 | 5.6008 | − 0.0992 |
| 30 | CHEMBL251886 | Test | 6.5000 | 5.7403 | − 0.7597 |
| 31 | CHEMBL179725 | Test | 7.8900 | 7.3075 | − 0.5825 |
| 32 | CHEMBL458210 | Train | 6.2700 | 6.2182 | − 0.0518 |
| 33 | CHEMBL270265 | Train | 9.1500 | 10.1193 | 0.9693 |
| 34 | CHEMBL156987 | Train | 7.1200 | 7.2798 | 0.1598 |
| 35 | CHEMBL408564 | Train | 8.1500 | 6.9164 | − 1.2336 |
| 36 | CHEMBL270473 | Train | 8.8200 | 8.0888 | − 0.7312 |
| 37 | CHEMBL576165 | Train | 7.1600 | 6.6797 | − 0.4803 |
| 38 | CHEMBL573933 | Test | 6.8000 | 6.4804 | − 0.3196 |
| 39 | CHEMBL334084 | Train | 6.6200 | 6.8859 | 0.2659 |
| 40 | CHEMBL1081616 | Test | 7.5100 | 7.1326 | − 0.3774 |
| 41 | CHEMBL471430 | Test | 8.3100 | 7.7477 | − 0.5623 |
| 42 | CHEMBL470566 | Train | 7.0300 | 6.7009 | − 0.3291 |
| 43 | CHEMBL513576 | Test | 6.1700 | 6.5270 | 0.3570 |
| 44 | CHEMBL26241 | Train | 5.1500 | 5.6899 | 0.5399 |
| 45 | CHEMBL26101 | Train | 4.7000 | 4.5452 | − 0.1548 |
| 46 | CHEMBL142327 | Train | 4.1200 | 4.0730 | − 0.0470 |
| 47 | CHEMBL359135 | Test | 4.3000 | 3.8430 | − 0.4570 |
| 48 | CHEMBL596364 | Train | 4.1200 | 4.7567 | 0.6367 |
| 49 | CHEMBL405759 | Train | 7.5200 | 7.0731 | − 0.4469 |
Table 5.
Parameters corresponds to five best model generated by AutoQSAR
| Model code | Score | SD | R2 | RMSE | Q2 |
|---|---|---|---|---|---|
| Kpls_desc_38 | 0.8224 | 0.5505 | 0.8467 | 0.5366 | 0.8069 |
| Pls_38 | 0.7846 | 0.6662 | 0.7756 | 0.5444 | 0.8013 |
| Kpls_radial_31 | 0.7652 | 0.6530 | 0.7631 | 0.6276 | 0.7458 |
| Kpls_dendritic | 0.7444 | 0.6250 | 0.7830 | 0.6427 | 0.7334 |
| Kpla_linear_31 | 0.7058 | 0.6033 | 0.7978 | 0.6639 | 0.7156 |
Fig. 7.

Scatter plot analysis of best model predicted from AutoQSAR
External validation of QSAR model
Validation methods are required to affirm the robustness of a model on unseen data. The method of root mean-squared error (RMSE) is one of the internal methods of validating a model (Wold and Ericksson 1998; Yasri and Hartsough 2001).
Strategies for external validation are important and it is of great interest to adopt all available validation strategies to check robustness of the model. All the parameters for external validation of structure based pharmacophore model are presented in Table 6. Cross validation (Q2) value of 0.8069, the correlation coefficient (R2) value of 0.8467. The slopes of regression lines through origin (K and K0 value) and substantial values of correlation coefficients ( and ) were obtained from Observed pIC50 and Predicted pIC50 activity of the dataset. The predictive ability of the selected model was also confirmed by external. A value of is greater than 0.6 may be taken as an indicator of good external predictability. All these values met the necessary criteria for a robustness of QSAR model.
Table 6.
External validation parameters for QutoQSAR
| External validation parameters | Model kpls_desc_38 | Limitations |
|---|---|---|
| Q2a | 0.8069 | Q^2 > 0.5 |
| R2b | 0.8467 | R^2 close to 1 |
| K valuec | 0.9444 | 0.85 ≤ k ≤ 1.15 |
| R02d | 0.8412 | Close to R^2 |
| K’ valuee | 1.0040 | 0.85 ≤ k ≤ 1.15 |
| R0′2f | 0.8412 | Close to R^2 |
| Rm(Loo)2 g | 0.7763 | Rm(Loo)2 > 0.5 |
| rpred2h | 0.6979 | > 0.5 |
aCross-validated coefficient
bCorrelation coefficient between actual and predicted values
cSlope values of regression lines
dCoefficient for regression through origin values
eSlope values of regression lines
fCoefficient for regression through origin values g. modified squared correlation coefficient using LOO method
hPredictive correlation coefficient value
Conclusion
Glycogen synthase kinase-3β is a drug target for Alzheimer's, type II diabetes and other diseases. This study shows the binding ability of a library of compounds generated from Melissa officinalisas potential inhibitors of GSk-3β. It is important to emphasize that three compounds which are salvianolic acid C, ellagic acid and naringenin are found to have better docking score, binding free energy and predicted pIC50 values alongside satisfactory drug likeness than co-crystallized native compounds. Therefore, we recommend data provided in this study should further be validated by in vivo and in vitro studies.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Ammon HP, Kelber O, Okpanyi SN. Spasmolytic and tonic effect of Iberogast (STW 5) in intestinal smooth muscle. Phytomed Suppl. 2006;5:67–74. doi: 10.1016/j.phymed.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Bertrand JA, Thieffine S, Vulpetti A, et al. Structural characterization of the GSK-β active site using selective and non-selective ATP-mimetic inhibitors. J Mol Bio. 2003;333(2):393–407. doi: 10.1016/j.jmb.2003.08.031. [DOI] [PubMed] [Google Scholar]
- Beurel E. Regulation by glycogen synthase kinase-3 of inflammation and T cells in CNS diseases. Front Mol Neurosci. 2011;31(4):1–8. doi: 10.3389/fnmol.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher JL, Phiel CJ. A non catalytic domain of glycogen synthase kinase-3 (GSK-3) is essential for activity. J Biol Chem. 2010;285:7957–7963. doi: 10.1074/jbc.M109.091603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Woodgett J, Maamari M, Force T. Targeting GSK-3 family members in the heart: a very sharp double-edged sword. J Mol Cell Cardiol. 2011;51:607–613. doi: 10.1016/j.yjmcc.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cociova OM, Li B, NomanBhoy T, Li Q, Nakamura A, Nomura M, Okada K, et al. Synthesis and structure–activity relationship of 4-quinolone-3-carboxylic acid based inhibitors of glycogen synthase kinase-3β. Bioorg Mel Chem Lett. 2017;27(16):3733–3738. doi: 10.1016/j.bmcl.2011.07.073. [DOI] [PubMed] [Google Scholar]
- De Bondt HL, Rosenblatt J, Jancaric J, Jones HD, Morgan DO, Kim SH. Crystal structure of cyclin-dependendkinase 2. Nature. 1993;363:595–602. doi: 10.1038/363595a0. [DOI] [PubMed] [Google Scholar]
- de Oliveira MT, Katekawa E. On the virtues of automated QSAR—the new kid on the block. Future Med Chem. 2017 doi: 10.4155/FMC-2017-0170. [DOI] [PubMed] [Google Scholar]
- Dixon SL, Duan J, Smith E, Von Bargen CD, Sherman W, Repasky MP. AutoQSAR: an automated machine learning tool for best-practice quantitative structure–activity relationship modeling. Future Med Chem. 2016;8:1825–1839. doi: 10.4155/fmc-2016-0093. [DOI] [PubMed] [Google Scholar]
- Dorm GW. 2nd, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Investig. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, Dixon SL, Lowrie JF, Sherman W. Analysis and comparison of 2D fingerprints: insights into database screening performance using eight fingerprint methods. J Mol Graph Model. 2010;29:157–170. doi: 10.1016/j.jmgm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Duan J, Dixon SL, Lowrie JF, Sherman W. Analysis and comparison of 2D fingerprints; insights into database screening performance using eight fingerprint method. J Mol Graph Model. 2010;29:157–170. doi: 10.1016/j.jmgm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med. 2002;8(1):26–32. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- Elekofehinti OO, Ejelonu O, Iwaloye O, et al. Discovery of potential visfatin activators using in silico docking and ADME predictions as therapy for type 2 diabetes. Beni Suef Univ J Basic Appl Sci. 2018;7(2):241–249. [Google Scholar]
- Gao X, Wang JY, Gao LM, Yin XF, Liu L. Identification and analysis of glycogen synthase kinase 3 beta1 interactome. Cell Biol Int. 2013;37:768–779. doi: 10.1002/cbin.10095. [DOI] [PubMed] [Google Scholar]
- Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Exp Opin Drug Discov. 2015;10:49–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golbraikh A, Tropsha AJ. Beware of q2! Mol Graph Mod. 2002;20:269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- Gombar VK, Silver IS, Zhao Z. Role of ADME characteristics in drug discovery and their in silico evaluation: in silico screening of chemicals for their metabolic stability. Curr Top Med Chem. 2003;3(11):1205–1225. doi: 10.2174/1568026033452014. [DOI] [PubMed] [Google Scholar]
- Gum RJ, Gaede LL, Koterski SL, Heindel M, Clampit JE, Zinker BA, et al. Reduction of protein tyrosine phosphatase 1B increases insulin-dependent signaling in ob/ob mice. Diabetes. 2003;52:21–28. doi: 10.2337/diabetes.52.1.21. [DOI] [PubMed] [Google Scholar]
- Heitz A, Carnat A, Fraisse D, Carnat AP, Lamaison JL. Luteolin 3′-glucuronide, the major flavonoid from Melissa officinalis subsp. officinalis. Fitoterapia. 2000;71(2):201–202. doi: 10.1016/s0367-326x(99)00118-5. [DOI] [PubMed] [Google Scholar]
- Hsiung SC, Adlersberg M, Arango V, Mann JJ, Tamir H, Liu KP. Attenuated 5-HT1A receptor signaling in brains of suicide victims: involvement of adenylyl cyclase, phosphatidylinositol 3-kinase, Akt and mitogen-activated protein kinase. J Neurochem. 2003;87:182–194. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3b to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- Iwaloye O, Elekofehinti OO, Babatomiwa K, Fadipe TM, Akinjiyan MO, et al. Discovery of TCM derived compounds as wild type and mutant Plasmodium falciparum dihydrofolate reductase inhibitors: induced fit docking and ADME studies. Curr Drug Discov Technol. 2020 doi: 10.2174/1570163817999200729122753. [DOI] [PubMed] [Google Scholar]
- Iwaloye O, Elekofehinti OO, Oluwarotimi EA, Babatomiwa K, Momoh IA. In silico molecular studies of natural compounds as possible anti-Alzheimer’s agents: ligand-based design. Netw Model Anal Health Inform Bioinform. 2020;9:54. doi: 10.1007/s13721-020-00262-7. [DOI] [Google Scholar]
- Kamdem JP, Adeniran A, BoligonAA KCV, Elekofehinti OO, Hassane W, Ibrahimg M, Pansera WE, Meinerza DF, Athayde ML. Antioxidant activity, genotoxicity and cytotoxicity evaluation of lemon balm (Melissa officinalis L.) ethanolic extract: its potential role in neuroprotection. Ind Crops Prod. 2013;51:26–34. [Google Scholar]
- Kapetanovic IM. Computer-aided drug discovery and development (CADDD): in silico-chemicobiological approach. Chem Biol Interact. 2008;171:165–176. doi: 10.1016/j.cbi.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Liu X. Overstimulation can create health problems due to increases of PI/3K/Akt/GSK3 insensitivity and GSK3 activity. SpringerPlus. 2014;3:356. doi: 10.1186/2193-1801-3-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Liu Y, Zeng G, Shao B, Chen M, Li Z, Jiang Y, Liu Y, Zhang Y, Zhong H. Application of molecular docking for the degradation of organic pollutants in the environmental remediation: a review. Chemosphere. 2018;203:139–150. doi: 10.1016/j.chemosphere.2018.03.179. [DOI] [PubMed] [Google Scholar]
- Luo G, Chen L, Burton CR, Xiao H, Sivaprakasam P, Krause CM, Cao Y. Discovery of isonicotinamides as highly selective, brain penetrable, and orally active glycogen synthase kinase-3 inhibitors. J Med Chem. 2016;59(3):1041–1051. doi: 10.1021/acs.jmedchem.5b01550. [DOI] [PubMed] [Google Scholar]
- Maffucci I, Hu X, Fumagalli V, Contini A. An efficient implementation of the Nwat-MMGBSA method to rescore docking results in medium-throughput virtual screenings. Front Chem. 2018;6:43. doi: 10.3389/fchem.2018.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancinelli R, Capino G, Petrungaro S, Mammola CL, Tomaipitinca L, Filippini A, Facchiano A, Ziparo E, Giampietri C. Multifaceted roles of GSK-3 in cancer and autophagy-related diseases. Oxidative Med Cell Longev. 2017;2017:629495. doi: 10.1155/2017/4629495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradkhani H, Sargsyan E, Bibak H, Naseri B, Sadat-Hosseini M, Fayazi-Barjin A, Meftahizade H. Melissa officinalis L., a valuable medicine plant: a review. J Med Plants Res. 2010;4:2753–2759. [Google Scholar]
- Olesen PH, Sørensen AR, Ursø B, Kurtzhals P, Bowler AR, Ehrbar U, Hansen BF. Synthesis and in vitro characterization of 1-(4-Aminofurazan-3-yl)-5-dialkylaminomethyl-1 H-[1,2,3]triazole-4-carboxylic acid derivatives. A new class of selective GSK-3 inhibitors. J Med Chem. 2003;46(15):3333–3341. doi: 10.1021/jm021095d. [DOI] [PubMed] [Google Scholar]
- Olsson MHM, Søndergard CR, Rostkowski M, Jensen JH. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theor Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Ożarowski M, Mikolajczak PL, Piasecka A, Kachlicki P, Kujawski R, Bogacz A, Bartkowiak-Wieczorek J, Szulc M, Kaminska E, Kujawska M, JodynisLiebert J, Gryszczynska A, Opala B, Lowicki Z, Seremak-Mrozikiewicz A, Czerny B. Influence of the Melissa officinalisleaf extract on long-term memory in scopolamine animal model with assessment of mechanism of action. Evid Based Complement Altern Med. 2016 doi: 10.1155/2016/9729818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padavala A, Chitti S, Rajesh B, Vinukonda V, Jayanti R, Vali R. In silico based ligand design and docking studies of GSK-3β inhibitors. Chem Bioinform J. 2010;10:1–12. doi: 10.1273/cbij.10.1. [DOI] [Google Scholar]
- Patora J, Klimek B. Flavonoids from lemon balm (Melissa officinalis L., Lamiaceae) Acta Pol Pharm. 2002;59(2):139–143. [PubMed] [Google Scholar]
- Prime (2018) version 3.9, Schrodinger, LLC, New York
- Ring DB, Johnson KW, Henriksen EJ, Nuss JM, Goff D, Kinnick TR. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes. 2003;52:588–595. doi: 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- Robertson LA, Kim AJ, Werstuck GH. Mechanisms linking diabetes mellitus to the development of atherosclerosis: a role for endoplasmic reticulum stress and glycogen synthase kinase-3. Can J Pharmacol. 2006;84(1):39–48. doi: 10.1139/Y05-142. [DOI] [PubMed] [Google Scholar]
- Sachs L. Applied statistics: a handbook of techniques. Berlin: Springer; 1984. [Google Scholar]
- Saitoh M, Kunitomo J, Kimura E, Washita HI, et al. 2-{3-[4-(Alkylsulfinyl)phenyl]-1 -benzofuran-5-yl}-5-methyl-1,3,4-oxadiazole derivatives as novel inhibitors of glycogen synthase kinase-3β with good brain permeability. J Med Chem. 2009;52(20):6270–6286. doi: 10.1021/jm900647e. [DOI] [PubMed] [Google Scholar]
- Sander T, Freyss J, von Korff M, Rufener C. Datawarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2005;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- Schrödinger Suite (2012) Protein Preparation Wizard; Epik version 2.3, Schrödinger, LLC, New York, NY, 2012; Impact version 5.8, Schrödinger, LLC, New York, NY, 2012; Prime version 3.1, Schrödinger, LLC, New York, NY, 2012.
- Senderski ME (2009) Melisa lekarska, in: Zioła. Praktycznyporadnik o ziołachiziołolecznictwie. Wydawnictwo K.E. Liber. Warszawa, pp 422–426
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Smith E, Frenkel B. Glucocorticoids inhibit the transcriptional activity of LEF/TCF in differentiating osteoblasts in a glycogen synthase kinase-3beta-dependent and -independent manner. J Biol Chem. 2005;280:2388–2394. doi: 10.1074/jbc.M406294200. [DOI] [PubMed] [Google Scholar]
- Smith DG, Buffet M, Fenwick AE, Haigh D, Ife RJ, Saunders M, Slingsby BP, Stacey R, Ward RW. 3-Anilino-4-arylmaleimides: potent and selective inhibitors of glycogen synthase kinase-3 (GSK-3) Bioorg Med Chem Lett. 2001;11:635–639. doi: 10.1016/s0960-894x(00)00721-6. [DOI] [PubMed] [Google Scholar]
- Sofowora A, Ogunbodede E, Onayade A. The role and place of medicinal plants in the strategies for disease prevention. Afr J Tradit Complement Altern Med. 2013;10:210–229. doi: 10.4314/ajtcam.v10i5.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Li Y, Shen M, Tian S, Xu L, Pan P, et al. Assessing the performance of the MM/PBSA and MM/GBSA methods. 5, improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys Chem Chem Phys. 2014;1(6):22035–22045. doi: 10.1039/c4cp03179b. [DOI] [PubMed] [Google Scholar]
- Tagashira M, Ohtake Y. New antioxidative 1,3-benzodioxole from Melissa officinalis. Planta Med. 1998;64(6):555–558. [Google Scholar]
- Takahashi YF, Sasaguri T. Drug development targeting the glycogen synthasekinase-3beta (GSK-3beta)-mediated signal transduction pathway: inhibitors of the Wnt/beta-catenin signaling pathway as novel anticancer drugs. J Pharmacol Sci. 2009;109:179–183. doi: 10.1254/jphs.08r28fm. [DOI] [PubMed] [Google Scholar]
- Terrence LS, Andrew JP, Joyce AB, Scott D, et al. Synthesis and evaluation of novel heterocyclic inhibitors of GSK-3. Bioorg Mel Chem Lett. 2006;16(18):2091–2094. doi: 10.1016/j.bmcl.2006.01.057. [DOI] [PubMed] [Google Scholar]
- Triantaphyllou K, Blekas G, Boskou D. Antioxidative properties of water extracts obtained from herbs of the species Lamiaceae. Int J Food Sci Nutr. 2001;52(4):313–317. doi: 10.1080/09637480120057512. [DOI] [PubMed] [Google Scholar]
- Witherington J, Bordas V, Gaiba A, Naylor A, Rawlings AD, Slingsby BP, Smith DG, TakleAK WRW. 6-heteroaryl-pyrazolo[3,4-b]pyridines: potent and selective inhibitors of glycogen synthase kinase-3 (GSK-3) Bioorg Med Chem Lett. 2003;13:3059–3062. doi: 10.1016/s0960-894x(03)00646-2. [DOI] [PubMed] [Google Scholar]
- Wold S, Ericksson L. Partial least squares projections to latent structures (PLS) in chemistry. In: Ragu P, Schleyer P, editors. Encyclopedia of computational chemistry, vol 3. Chichester: Wiley; 1998. pp. 2006–2021. [Google Scholar]
- Xie H, Wen H, Zhang D, et al. Designing of dual inhibitors for GSK-3beta and CDK5: virtual screening and in vitro biological activities study. Oncotarget. 2017;8(11):18118–18128. doi: 10.18632/oncotarget.15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Kim NG, Gumbiner BM. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle. 2009;8:4032–4039. doi: 10.4161/cc.8.24.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasri A, Hartsough DJ. Toward an optimal procedure for variable selection and QSAR modeling building. Chem Inf Comput Sci. 2001;41:1218–1227. doi: 10.1021/ci010291a. [DOI] [PubMed] [Google Scholar]
- Yuki Y, Chikashi I. Inhibition of glycogen synthase kinase-3 beta induces apoptosis and mitotic catastrophe by disrupting centrosome regulation in cancer cells. Nat Sci Rep. 2015;5:13249. doi: 10.1038/srep13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarei A, Ashtiyani SC, Taheri S, Hosseini N. A brief overview of the effects of Melissa officinalis L. extract on the function of various body organs. J Res Med Sci. 2015;17(7):1–6. [Google Scholar]